

Abstract
We describe a rapid assay for measuring the cellular activity of small GTPases in response to a specific stimulus. Effector functionalized beads are used to quantify in parallel multiple, GTP-bound GTPases in the same cell lysate by flow cytometry. In a biologically relevant example, five different Ras family GTPases are shown for the first time to be involved in a concerted signaling cascade downstream of receptor ligation by Sin Nombre hantavirus.
Keywords: Ras, Rho, Rac, Cdc42 and Rab GTPases, cell signaling, hantavirus, flow cytometry, integrins
Introduction
Members of the ras-related superfamily of small GTPases, including Rho, Ras and Rab subfamilies, serve as critical integrators of cellular functions from cell division and survival to membrane trafficking; making the measurement of their activites of pivotal significance. The GTPases cycle between active GTP-bound and inactive GDP-bound states the balance of which is regulated by guanine nucleotide-exchange factors (GEFs), GTPase-activating proteins (GAPs) and guanine nucleotide-dissociation inhibitors (GDIs) [1; 2]. Generally, there is an overabundance of regulatory factors that reflects in part the pathway and cell type specificity of GTPase activation cascades. For example, over 30 GEFs and GAPS serve to activate and inactivate, respectively, the approximately half as many Rho proteins [3; 4; 5]. Rho specific GDIs inhibit nucleotide cycling and sequester Rho GTPases in the cytosol away from membranes, adding a third layer of regulation [3]. Once activated, GTPases interact with specific downstream effector proteins to yield definitive physiological responses.
The functions of multiple Ras-related GTPases are frequently coopted during infection as well as in acquired and genetic diseases. Additionally, the GTPases are gaining traction as therapeutic targets [6; 7; 8]. Thus, there is a need to pinpoint the pivotal GTPases in each scenario by quantitatively and temporally monitoring changes in activities. Current methods for measuring the activation status of small GTPases rely on GST-bead based effector pulldown assays, and ELISA based effector binding assay kits. The significant shortcomings of all of these methods are that they are labor intensive and require large amounts of cell samples, purified effector proteins and/or expensive kits and reagents. When working with primary cell samples or infectious agents large cell lysate samples are a significant hurdle.
Here we describe a rapid, bead-based effector-binding assay that can monitor the activation status of multiple GTPases from a single cell lysate derived from <100,000 cells. As proof-of-principle we demonstrate the utility of the assay in measuring the previously uncharacterized cascade of GTPases that is activated to allow cellular entry of Sin Nombre hantavirus (SNV) in the course of a productive infection.
Hantaviruses, which cause hemorrhagic fever with renal syndrome (HFRS) and hantavirus cardiopulmonary syndrome (HCPS), are of particular interest because their mechanisms of host cell entry and subsequent pathogenesis are poorly understood (Supplemental note 1) [9]. Viruses attach to cell surface receptors, promote the cytoskeletal remodeling required for breaching inter- and intracellular cellular barriers, and enter the cell through endocytic membrane trafficking pathways; all processes that fundamentally depend on the activation of various GTPase cascades [10]. Parallel measurements of active GTPases downstream of receptor engagement are therefore important for understanding how these signals are regulated and how their consolidation is related to both physiological and pathological contexts.
The active GTPase effector trap flow cytometry assay (G-trap) has significant advantages over standard pulldown techniques including: 1) G-trap enables rapid measurement, sensitivity and analysis with results within 4 h, compared to days required for other assays; 2) uses small samples e.g. <100,000 cells grown in a 48 well plate, well below the requisite minimum of 2×106 cells for single ELISA based measures; 3) can measure multiple GTPases from a single lysate using a multiplex approach (Fig 1A) whereas conventional assays require the entire lysate for measurement of a single GTPase, and 4) it facilitates replicate measures on precious cell samples.
Figure 1. The GTPase effector trap flow cytometry assay (G-trap).

A. Plot of red fluorescence (FL4) versus sidescatter (SSC) of a set 12 of Cyto-Plex™ beads dyed with 12 discrete levels (Li i = 1,…12) of 700 nm fluorescence. B. Li beads are coated with effectors for GTPase bait, some beads (e.g. L4) are not functionalized with effectors and used as controls for non-specific binding. Li GSH beads individually coated with specific effectors for detection of discrete active GTPase populations are added to a sample tube containing cell lysates. As illustrated GTP-bound GTPases (P3 in schema; P2 = GDP) are recruited to their cognate effectors immobilized on beads, while GDP-bound GTPases remain unbound. Bead-bound active GTPase assemblies are quantified using fluorescently labeled antibodies. In the G-trap assay, the letters a, b, c, d, and e identify and link effectors to their cognate GTPases and fluorescently labeled reporter antibodies (FL1 (520 nm emission). C. Prototypical multiplex format, electronic gates on red fluorescence (FL4) versus side scatter (SSC) on the flow cytometer are used to select individual Li beads that bear known analytes, after which bead associated green fluorescence reflecting bound antibody (FL1) in each gate were measured. D. Reporter antibody cross talk was examined to determine the significance of differential non-specific staining of cytoplex beads. Multiplex samples were prepared by exposing GSH beads to multiple antibodies and measuring the aggregate fluorescence of nonspecifically bound antibodies. Fraction samples were prepared by staining, washing and measuring fluorescence of bead populations separately. Mixed fraction samples were prepared as the fraction samples but mixed and measured in multiplex format.
Materials and Methods
Antibodies
Monoclonal rabbit anti-RAP1 (5G7): sc-47695 and rat monoclonal anti H-Ras (259): sc-35 were obtained from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). Monoclonal mouse antibodies: anti-Rho (A, B, C) clone 55 (#05-7788), anti-Rac1, clone 23A8, #05-389, the secondary antibody goat anti-mouse IgG (H+L) conjugated to Alexa Fluor 488, Rabbit Polyclonal anti Cdc42 #07-1466 were obtained from Millipore (Temecula, CA ). Mouse Monoclonal Anti Rac1 #ARC03 was also purchased from Cytoskelton Inc. (Denver, CO). Mouse monoclonal anti-Rab 7 antibody was from Sigma-Aldrich (St. Louis, MO). Mouse monoclonal AP5 β3 anti LIBS antibody was purchased from the Wisconsin Blood Center.
Activators and Inhibitors
Rap1 Activator 8-Cpt-2me-cAMP was used at 50 μM and from R & D Systems (Minneapolis, MN). Rac1 Inhibitor, NSC23766 was used at 100 μM and from Calbiochem Inc. (now www.emdmillipore.com). Calpeptin (Rho activator) was used at 0.3 μM and from Calbiochem Inc. The human recombinant form of the catalytic domain of p50RhoGAP from Cytskeleton Inc. (Denver, CO) was used at 2.50 μg/μl. Recombinant EGF was used at 10 nM and was from Life Technologies (Carlsbad, CA). The novel Cdc42 inhibitor CID2950007 was used at 10 μM synthesized and characterized as previously described [6] and can be purchased commercially as ML141.
Buffers
RIPA buffer: 74 mM CaCl2, 50 mM Tris-HCl pH 7.4, 1% NP-40 (v/v), 0.5% (w/v) deoxycholic acid, 0.1% (w/v) SDS, 1 mM sodium orthovanadate, and protease inhibitors. HHB buffer: 7.98 g/L HEPES (Na salt), 6.43 g/L NaCl, 0.75 g/L KCl, 0.095 g/L MgCl2 and 1.802 g/L glucose.
Cell Culture
HeLa and Vero E6 cells from ATCC were plated at 2×104 cells per well in a 48-well plate, then allowed to grow for ~48 h to medium/low confluence in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS, pen/strep, and L-glutamine in a 37°C incubator with 5% CO2.
Production of Sin Nombre Virus
SNV was propagated and titered in Vero E6 cells under strict standard operating procedures using biosafety level 3 (BSL3) facilities and practices (CDC registration number C20041018-0267) as previously described [11]. For preparation of UV-inactivated SNV, we placed 100 μL of virus stock (typically 1.5–2×106 focus forming units/ml) in each well of a 96-well plate and subjected the virus to UV irradiation at 254 nm for various time intervals (~5 mW/cm2) as described elsewhere [12]. We verified efficiency of virus inactivation by focus assay before removing from the BSL-3 facility.
Fluorecent Labeling of SNV
The envelope membrane of hantavirus particles was stained with the lipophilic lipid probe octadecylrhodamine (R18) and purified as previously described [12]. The typical yield of viral preparation was 1 ± 0.5×108 particles/μl in 300 μl tagged with ~10,000 R18 probes/particle or 2.7 mole % R18 probes in the envelope membrane of each particle of 192 nm diameter average size [12]. Samples were aliquoted and stored in 0.1% HSA HHB buffer, and used within two days of preparation and storage at 4°C. For long-term storage, small aliquots suitable for single use were stored at −80°C.
Effector Proteins
The GST-effector chimeras consisting of the minimal GTPase binding domains (RBD) used for the studies were as follows. PAK-1 RBD, a Rac1 effector and Raf-1 RBD, a RAS effector protein were obtained from Millipore. Rhotekin-RBD, a Rho effector protein was purchased from Cytoskeleton Inc.. RalGDS-RBD, a RAP1 effector protein was expressed and purified from a plasmid kindly provided by Dr Burridge (UNC Chapel Hill) [13]. GST-RILP RBD was purified as described below.
Expression and purification of GST-RILP
A plasmid encoding GST-RILP RBD was prepared by Daniel Cimino as previously described [14]. Protein was expressed in competent E. coli BL21 cells. Cultures were grown at 37°C to an absorbance of 0.5 O.D. measured at 595 nm and protein expression induced by transfer to room temperature and addition of 0.2 mM isopropyl-beta-D-1-thiogalactopyranoside (IPTG) for 16–18 h to maximize the yield of properly folded active fusion protein. Purification of GST-RILP was performed according to standard procedures and as previously described [15] [16]. Briefly, the harvested bacterial cell pellet was resuspended in cold PBS. Cells were lysed using lysozyme and a microtip sonicator (Misonix Inc., Newtown, CT, U.S.A.). The cell lysates were mixed with 10% Triton X-100 and mixed end over end for 30 min at 4°C. Lysates were then centrifuged at 8,000 × g for 10 min to pellet the cellular debris. The supernatant was mixed with freshly prepared Glutathione Sepharose 4B slurry and bound at 4°C for 2 h using gentle agitation to keep the resin suspended in the lysate solution. Glutathione beads with bound protein were settled using low speed centrifugation (500 × g) followed by multiple washes before eluting the bound GST-tagged protein with 10 mM glutathione in 50 mM Tris -HCl elution buffer. Eluted protein was concentrated by passing through Millipore Amplicon Ultra Tubes (MWCO 30,000). The purified protein was quantified using the BCA protein estimation kit (Pierce, City, State). Single use aliquots stored at −80 °C were used in the experiments.
Preparation of GSH Beads
13 μm Superdex peptide beads were derivatized with glutathione (γ-glu-cys-gly or GSH) as previously described [7; 15; 16]. 5 μm Cyto-Plex™ carboxylated beads (FM5CR0L L=1,2,3,…..12) dyed with graded levels (L) of red emission were purchased from Thermo Scientific (City, State). The beads were then functionalized with glutathione as previously described [17]. Briefly, the carboxyl functionalized beads were converted to amine reactive N-hydroxysulfosuccinimide (Sulfo-NHS) esters using 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC or EDAC) chemistry following protocols provided by the manufacturer (Thermo Scientific). The amino derivatized beads were then reacted with a bifunctional chemical crosslinker succinimidyl 4-[N-maleimidomethyl]-cyclohexane-1-carboxylate (SMCC) in order to enable subsequent attachment of GSH. 5×10 beads were suspended in 400 μl of 50mM sodium phosphate buffer (pH 7.5) containing 0.01% Tween-20 (w/v) and mixed with 8 μl of 100 mM SMCC in dimethyl sulfoxide (DMSO) and incubated with mild agitation for 30 min. The beads were centrifuged and resuspended in 360 μL of fresh buffer together with 40 μl of 200mM GSH (pH 7) and 4μl of 100 mM EDTA (pH 7–8). Nitrogen was bubbled slowly through the suspension for 5 min, the tube was capped to exclude oxygen, and the beads were gently mixed for 30 min. The beads were washed four times and stored in single use aliquots in 30 mM HEPES (pH 7.5), 100 mM KCl, 20 mM NaCl, 1 mM EDTA, and 0.02% NaN3 at 4°C.
Assembly of GST Effector proteins on GSH beads and GST pull-down assay
Site occupancy (θ) of GSH sites on beads (limiting reagent) is regulated by the dissociation constant Kd and concentration of free glutathione -S-transferase (GST) fused to effector proteins, according to Equation 1 [18]:
| (1) |
We relied on previous studies wherein the characteristic kinetic and equilibrium binding constants (Kd ~ 80 nM) of GST fused to Green fluorescent protein (GFP) were documented [7; 15; 16; 17; 19], to establish optimal stoichiometric mixtures of GSH beads and specific GST effector fusion proteins for obtaining saturating site occupancies of the GST effector proteins for the present work. Typical site occupancies of the beads at saturation are in the 1–4 ×106 ligand sites/bead range. For example, 10,000 beads present an upper limit of 4×1010 sites or 3.3 nM in 20 μL. Incubating 800 nM (10 × Kd) of GST effector protein with the GSH beads is expected to yield a bead site occupancy, θ, of 0.91 (or 91%). In this way, known quantities of beads were mixed with effector proteins of known concentration at the desired stoichiometry range, incubated with shaking for 2 h at 4 °C, centrifuged, and resuspended in RIPA buffer at 10,000 beads/tube. Effector-bearing beads were prepared fresh for each experiment and kept on ice while cell lysates were prepared.
Configurations of the GTPase effector trap flow cytometry assay (G-trap)
Vero E6 or HeLa cells were plated in 48 well plates or T 75 flasks and incubated overnight in culture media. Cells were starved for overnight in serum-free medium followed by stimulation with activator or inhibitor at concentrations and times shown in the Results section. After incubating cells with various inhibitors, activators, and/or SNVR18 for desired times, cells were lysed with 100 μL ice-cold RIPA buffer. For the purposes of standardization, a fraction of the supernatant was collected to measure protein concentrations. Lysates were kept ice cold at all times to limit hydrolysis of active GTPases. Lysates were sonicated briefly, and then centrifuged at 14,000 rpm for 10 min to clear the lysate of any unlysed materials and DNA. For each effector assay, 10,000 beads were added to 50 μg of protein in 100 μl of cell lysate in RIPA buffer. The beads and lysate were allowed to incubate for 1 h at 4°C. After incubation, the beads were pelleted in a cold centrifuge. The bead pellets were resuspended in RIPA buffer and incubated with a primary monoclonal antibody for the target GTPase, with gentle shaking for 1 h. Depending on availability, the residual lysates were collected and re-assayed for a different GTPase by adding the appropriate effector beads. Lysates may be saved for future assays by flash freezing in liquid nitrogen, then storing at −20°C for up to one week. To minimize reporter antibody crosstalk, samples were split into separate fractions after lysate binding to the multiplex bead sets and then individually probed with a reporter antibody specific for a single target GTPase per fraction. The beads were than centrifuged and resuspended in 10% BSA HHB buffer with an Alexa 488 secondary antibody at a 1:200 dilution for 1 h with shaking at 4°C. Finally the beads were centrifuged once more and resuspended in 100 μL RIPA buffer for reading on the flow cytometer.
PI-PLC treatment
To determine whether cytoskeletal remodeling was caused by the specific interaction of SNVR18 with DAF, Vero E6 cells were treated with phosphatidylinositol-specific phospholipase C (PI-PLC), an enzyme known to cleave GPI- anchored proteins from cell surfaces [20; 21]. Monolayers of Vero E6 cells plated in 8-well Lab-Tek chambers were washed with HHB and treated with 1.0 unit of PI-PLC from Bacillus cereus (Life Technologies, Carlsbad, CA). After incubation at 37°C for 30 min, cells were washed and resuspended in HHB buffer and analyzed at the microscope.
GLISA
Colorimetric based, commercial kits (Cytoskeleton Inc., Denver, CO) for measuring Rho GTPase activation were used to measure SNV induced activation of Rac1 and RhoA in Vero E6 cells. About 8×104 Vero E6 cells were plated in a 12-well plate in RPMI 1640 medium. After reaching 50–60 % confluency, the media was replaced with starvation media containing 0.5 % BSA. Cells were then serum starved for 24 h before treatment with ~109 SNV particles at different time points. Media conditions included calcium (Ca2+), and PI-PLC (as described above). Cells were washed with ice cold PBS and then lysed and prepared for protein assays. Positive controls included purified Rac1-GTP and RhoA-GTP provided in the kit and were used to quantify active GTPase levels in the cell lysates. Negative controls included buffer-only controls, and cell lysates prepared from quiescent control cells after serum starvation. Cell lysates were frozen, and active Rac1 levels were quantified based on a p21-activated protein kinase binding assay [22], whereas active RhoA was quantified based on a Rhotekin binding assay [23]. All assays were performed in 96-well microtiter plates.
Stress Fiber Assay for RhoA activation
RhoA activation was independently confirmed by monitoring actin stress fiber formation following calpeptin or SNV stimulation (1 μg/ml) for 30 min. Actin filaments in control and stimulated cells were detected following paraformaldehyde (4%) fixation and Triton X-100 (0.5%) permeabilization using Alexa Fluor® 488 phalloidin (1 unit, or 0.17 μM) (Life Technologies).
Confocal Microscopy of SNVR18
For live cell imaging of virus binding and endocytosis, Vero E6 cells were plated in 8-well Lab-Tek chambers (Nunc) and temperature was maintained at 30°C with an objective heater (Bioscience Tools) in appropriate buffer containing the desired cations, Ca2+ or Mn2+, which respectively confer low and high affinity states of αvβ3 [24]. In situ addition of SNVR18 was performed by micropipeting an aliquot (100μL) of ≈ 1×109 virus particles per well containing ≈ 1.5×105 cells in 200 μl in HBSS and mixing well by trituration with the pipet. Confocal laser scanning microscopy was performed with Zeiss META or LSM 510 systems using 63× 1.4 oil immersion objectives as previously described [24]. Images were collected using 2× line averaging at 3–10 s intervals, depending on imaging time, where longer intervals were necessary to minimize photobleaching.
Results and Discussion
Configuration and General Applicability of G-trap Assay
Measurements of the activity of several GTPases in receptor-stimulated cells makes use of micron-sized glutathione beads that are individually functionalized with GST-tagged cognate effector protein and specific for individual target small GTPases. To simplify bacterial expression and purification of effector proteins, only the GTPase binding domains (RBD) are used. Effector coated beads are incubated with cell lysates that contain active, GTP-bound Ras, Rho and Rab GTPases. The GTPases are selectively recruited to beads that bear the cognate effector and are detected directly using fluorophore conjugated monoclonal antibodies specific for each GTPase or indirectly using secondary antibodies with fluorophore tags. It is important to note that the optimal signal-ratio between site occupancy of effector beads exposed to resting and activated cell lysates is achieved by not using large excesses of cell lysate proteins. This is because the effector beads are nominally used as limiting reagents, and therefore can approach saturation even in the resting cell lysate. This would obscure the accurate detection of increased activity status of GTPases in stimulated cell lysates. Impirically, we found that 10,000 beads and 50 μg protein in 100 μl were optimal for each effector assay (see methods).
In multiplex configuration distinct effectors were immobilized on Cytoplex™ beads of graded fluorescence intensities of a fluorophore with a fixed red wavelength of 700 nm [19] (Fig 1). An extra set of effector free beads was used as a control for nonspecific binding (e.g. L4 in Fig 1B. To assess the degree of nonspecific reporter antibody crosstalk during the labeling step, we measured the levels of binding to unconjugated GSH beads using a mixture of reporter antibodies for RhoA, Rac, Cdc42, H-Ras and Rap1 GTPases under the following three conditions: a) GSH beads mixed with all antibodies and measurement of the aggregate fluorescence of nonspecifically bound antibodies (multiplex in Fig. 1D), b) individual bead populations were treated with each antibody separately and measured separately (fraction in Fig 1D), c) individual bead populations were treated with each antibody separately, then combined and measured as a single mixture (mixed fraction in Fig. 1D). For the five GTPase antibodies tested, the multiplex sample was comparable to fraction and mixed fraction samples for the Rac1, RhoA, and Rap1 antibodies but higher for Cdc42 and H-Ras antibodies. The degree of non-specific binding depends on antibody type, batch and supplier. Therefore it is important for the user to be familiar with binding characteristics of new antibody batches before using them in multiplex format. In the present application, non-specific binding values shown in Fig. 1D, were significantly lower (≥5 fold to ≥30 fold) than fluorescence signals associated with resting and activated cells, depending on assay conditions (cf Fig 2). In these circumstances errors from non-specific binding are trivial. However, in cases were the fluorescence signals associated with specific GTPase-activity assays are of comparable order of magnitude to nonspecific background readings (e.g. NSC23766 or ML141 treated cells in Fig 2), it is important to modify the multiplex protocol. This can be addressed by adopting the mixed fraction format, where multiplexed samples that are recovered from the lysates are partitioned into fractions that correspond to the number of target analytes, and stained separately with reporter antibodies. In the case of the example shown in Fig 1D, Cdc42 and Rap1, may be stained with reporter antibodies as separate fractions, while Rac1, RhoA and HRas can be treated as a full multiplex. In summary it is important for the user to be familiar with binding characteristics of antibody batches before using them in a format of the G-trap assay that is most suitable for the prevailing conditions.
Figure 2. G-trap Assay Validation.
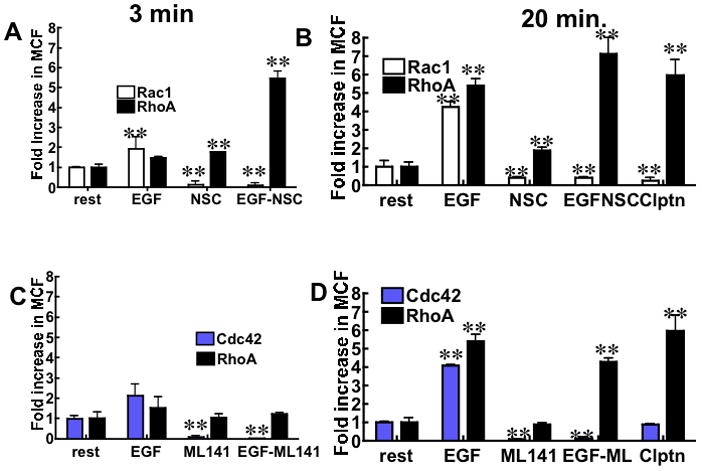
Serum starved Hela cells were stimulated with: 10 nM EGF to activate Rac1, Cdc42, and RhoA, 100 μM NSC23766 to suppress Rac1 activity, 10 μM ML141 to inhibit Cdc42, 1 μM Calpeptin (clptn) to activate RhoA,. Control samples (rest and non-cognate GSH effector beads) were mock-treated with 0.1% DMSO to account for compound solvent. A. Nonspecific binding-corrected plot of fold change (relative to resting cells) in median channel fluorescence (MCF) flow cytometer readings corresponding to changes in active Rac1 and RhoA in cells treated with EGF as indicated in panel. NSC refers to resting cells treated with NSC23766. EGF-NSC refers to cells treated with NSC23766 (1 h) and then EGF stimulated (3 min) before analysis of cell lysates for activated GTPases by flow cytometry. B. Data were collected after 20 min of EGF stimulation where applicable, all other condtions are the same as Panel A. C. Conditions as in Panel A measuring changes in active Cdc42 and RhoA. ML141 or CID2950007 served as the Cdc42 specific inhibitor. D. Conditions as in Panel B with applicable changes for Cdc42. The errors represent standard deviation of 3 independent experiments measured in duplicate each time. ** P<0.001 for all data compared to resting (rest) cells.
Cdc42, Rac1 and RhoA are among the most well characterized members of the Rho subfamily of GTPases. They are known for orchestrating cytoskeletal reorganization dynamics (filamentous actin and myosin 2) and crosstalk to antagonize each other’s activities [25; 26; 27; 28]. The availability of specific regulators of their activity makes these GTPases a suitable platform to test the applicability of our G-trap assay. HeLa cells were treated with the following signaling regulators: 1) NSC23766, a specific inhibitor of Rac1 interaction with its two upstream GEFs Tiam1 and Trio (Trio is also identified as a RhoA target [29; 30]), 2) CID2950007, a novel Cdc42 specific inhibitor [6] commercially sold as ML141, and 3) calpeptin, an upstream activator of RhoA [31]. Cdc42, Rac1 and RhoA were activated by adding 10 nM EGF to cells. Readings were taken at 3 min and 20 min after stimulation. The data were corrected for nonspecific binding by subtracting the fluorescent antibody binding to the GST-conjugated control beads from the antibody binding to GST-effector bearing beads. EGF-stimulated cell samples were normalized to unstimulated resting control cell samples. Data were collected 3 min (Fig. 2A, 2C) and 20 min (Fig. 2B and 2D) post-EGF-stimulation. RhoA data were separately paired with Rac1 and Cdc42 for ease of comparison of the putative antagonists. The activation of RhoA lagged behind both Rac1 and Cdc42 activation, at 3 min and supplanted both Cdc42 and Rac1 at 20 min. At this juncture it is not yet clear whether the time-dependent dominance in the relative amplitudes of Rac1 and RhoA are due to their mutual antagonism. Cell treatment with the Rac1-GEF inhibitor, NSC23766, suppressed Rac1 while RhoA activity was elevated relative to resting cells. Treatment of cells with EGF and NSC23766 in combination resulted in a dramatic activation of RhoA (>5-fold relative to resting cells) that was well above the activation of RhoA observed following EGF-stimulation alone. Calpeptin-mediated activation of RhoA was tested for comparison and found to be accompanied by a concomitant decrease in Rac1 activity. Collectively, these data are consistent with the known mutual antagonism of Rac1 and RhoA [25; 32]; wherein the inhibition of Rac1 is sensed and results in amplified RhoA activity in the absense of a counteracting Rac1 effect. The targeted activity of NSC23766 against Rac1 and not RhoA suggests that Trio is not an important upstream factor for RhoA activation under our experimental conditions. Interestingly, though Rac1 and Cdc42 are believed to have partially overlapping functions in mediating the formation of actin-rich protrusions in migrating cells [2; 33], the total inhibition of Cdc42 had no effect on RhoA activity under our experimental conditions (Fig. 2C and 2D) as previously shown [6]. These results suggest that under our experimental conditions, the up or downstream effector signals associated with Cdc42 do not impinge on or regulate RhoA and vice versa. Together the data demonstrate the power of the G-trap assay in sensitively and temporally dissecting GTPase responses to stimuli and pharmacologic manipulation.
Monitoring Virus-Induced GTPase Activation: Morphologic and Biochemical Assessment
Exposure of Vero E6 cells to known titers of UV killed and fluorescently labeled SNV particles causes cells to contract, lose cell contacts, and form filopodia, lamellipodia and stress fibers (Fig. 3A–F). Ultimately the virus is internalized and present in Rab7 positive late endosomes (Supplemental Note 3 and Fig. S3). Such morphological changes and membrane trafficking events are anticipated to be associated with the changes in the activities of multiple GTPases. Actin based structure changes are associated with Cdc42 (filopodia), Rac1 (lamellipodia), and RhoA (stress fiber formation and cellular contraction) [34]. Staining for actin with Alexa Fluor 488® phalloidin, identified RhoA induced F-actin stress fibers formed in response of treatment of resting cells with calpeptin (compare Fig. 3D vs. 3E) were similar to the stress fibers formed in response to SNV exposure (Fig. 3F). The ruffling observed in SNV- treated cells is similar to that seen in response to EGF-mediated activation of Rac1-dependent ruffling [35]. The loss of cell adhesion is connected to integrin affinity regulation, which is presumed to be downstream of antagonsitic Rap1 and H-Ras activity [36] [37]. AP-5 monoclonal antibodies recognize the PSI domain of extended conformation activated β3 integrins at the cell surface [38] and enabled detection of the onset of cellular detachment at the cell junctions in virus treated cells; based on poor AP5 staining of the plasma membrane and instead a strong perinuclear substratum staining (arrows in Fig. 3I). See for comparison resting and Mn2+ stimulated (maximal cell surface integrin activation) controls (Fig. 3G-H). All of the described morphologic indicators of changes in GTPase activity prompted us to use conventional GLISAs in comparision with our G-trap assay for monitoring the activation status of individual GTPases in SNV treated cells.
Figure 3.
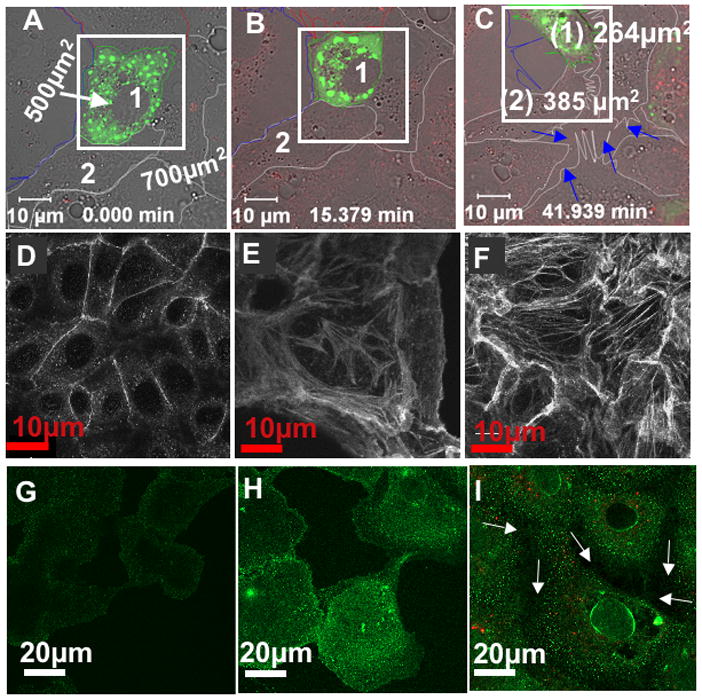
UV-killed SNVR18 induces vigorous remodeling of the actin cytoskeleton of Vero E6 cells, causing loss of cell adhesion. Confocal microscopy images of resting Vero E6 monolayers transiently transfected with the cell adhesion marker, paxillin GFP. Individual cells are outlined. A. Cells designated 1 and 2 before virus exposure are also defined by their surface area. The white square was included as size reference for cell 1. B. After 15 min exposure to 10,000 SNVR18 particles/cell the size of reference cell 1 is decreased relative to the rectangular marker. C. Time lapse frame taken 42 min after cells were exposed SNVR18 shows how cells 1 and 2 shrank in size and lost cell adhesion to each other and the coverslip. Arrows mark broken cell-cell junctions. D. Resting Vero E6 cells stained for actin with Alexa Fluor 488® phalloidin. E. RhoA induced F-actin stress fibers are shown 15 min activation with calpeptin and F. after SNV treatment. G. Resting Vero cells stained with integrin affinity sensitive AP-5 antibodies. H. Increased AP-5 staining in Mn2+ activated cells. I. Detection of differential activation of β3 integrins at the perinuclear and peripheral regions of Vero cells using AP5 antibodies. Arrows mark broken cell-cell junctions (see Fig. S2 for relative quantitative limits of AP-5 staining in resting and integrin activated cells).
We first used GLISA assays to measure Rho GTPase activation as a function of virus exposure time of wild-type Vero cells and cells where surface-expressed DAF was first cleaved with PI-PLC [20] (Fig 4). Virus treatment resulted in a robust activation of first RhoA at 3 min and subsequently Rac1 at 7 min with both falling to baseline levels by 30 min. After 60 min of virus exposue the levels of active GTP-Rho and -Rac fell below baseline levels, most likely because the expression of GTP-bound Rho GTPases is regulated by cell-cell and cell-matrix adhesion [33] [39]. In DAF-cleared cells, Rac1 failed to be activated across all timepoints indicating that virus engagement of a GPI anchored receptor such as DAF was required upstream of Rac1 activation. It is important to note that, the GLISA assays were performed separately on different days using different cell lysates, therefore the relative amplitudes of RhoA and Rac1 can not be accurately correlated to their temporal responses in the same way that the G-trap data shown in Fig 2 was analyzed.
Figure 4. SNV induces increase in GTP-bound Rho GTPase levels in Vero E6 cells.
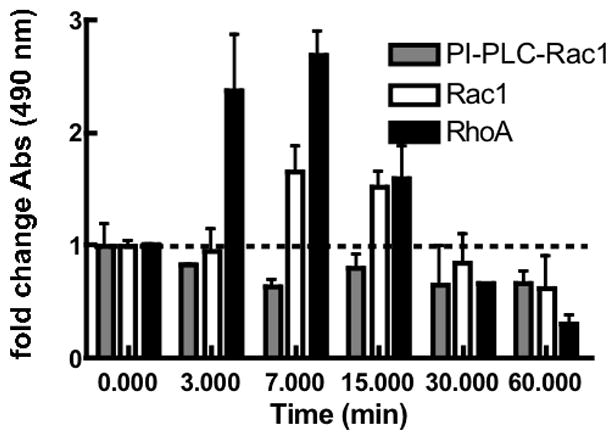
Bar graph shows GLISAs measuring kinetics of adhesion-dependent Rho protein (Rac1 and Cdc42) activation. PI-PLC-treated cells were cleared of DAF surface expression, a receptor for virus binding. Serum starved cells were stimulated with SNV particles as described in the methods and compared with unstimulated cells (0 min). Control cells were mock-treated with media to account for virus diluent. After cell lysis, the amounts of active Rac1 and RhoA were quantified based on p21-activated protein kinase or Rhotekin binding by GLISA respectively. The error bars represent the standard deviation of three independent measurements.
We next turned to the G-trap assay to examine the induction Rho protein activity in virus treated Vero E6 cells. Using known Rho GTPase activators and inhibitors the assay validation experiments (Fig 2) and results were recapitulated using permissive Vero E6 cells (Fig S1). In virus stimulated cell lysates (Fig 5A) and analogous to the GLISA results, RhoA activity increased ≥ 4-fold above resting levels within 3 min post-treatment. Active Rac1 levels also increased to a lesser extent at the 3 min timepoint. At the 20 min timepoint timepoint, the activity of both GTPases decrease to baseline levels confirming the trend documented by GLISA above (compare Fig. 4 vs. Fig. 5A). The temporal overlap in the activation of RhoA and Rac1 implies that SNV binding induces signaling cascades that lie upstream of RhoA and Rac1 activation, which mirrors what has been observed in a study on Group B Coxsackievirus-activated cells [40]. Rac1 and Rho normally mutually suppress each other’s activity in response to single stimuli as affirmed in Fig. 2 and elsewhere in the literature [2; 25; 41]. Their apparent co-stimulation by the virus may reflect the activation of multiple signaling cascades through sequential events with distinct spatial and temporal characteristics [32]. This notion is further supported by the simultaneous visualization of multiple GTPase biosensors during cell migration where surprisingly RhoA was activated exclusively near the cell edge connected with leading-edge advancement. In contrast, Cdc42 and Rac1 were activated away from the leading edge with a delay of ~ 40 seconds [25].
Figure 5.
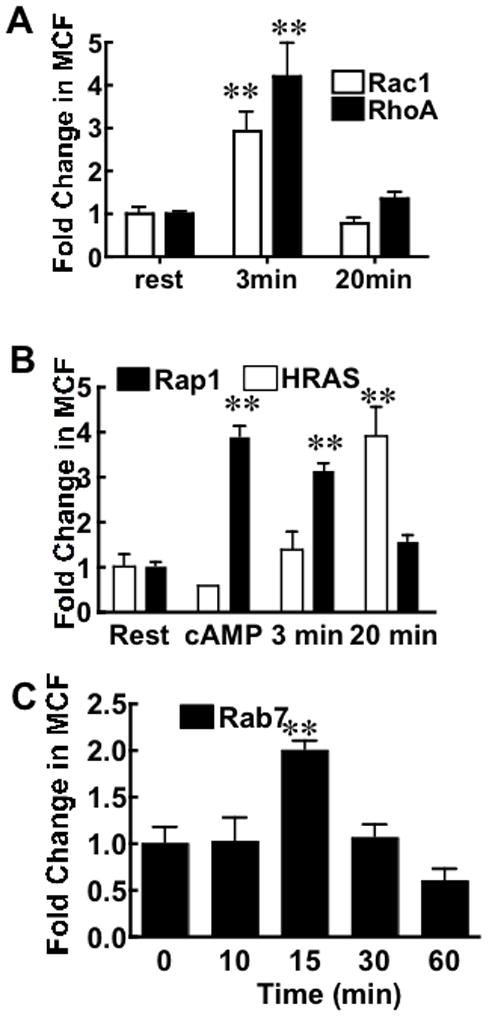
SNV induces the activity of several GTPases in Vero E6 cells. A. Vero E6 cells were serum starved for 24 h and treated with 10,000 SNVR18/cell. Active Rac1 and RhoA were detected in cell lysates at 3 min and 20 min after virus exposure using PAK1 and Rhotekin beads, respectively.. The errors represent standard deviations in 3 separate measurements. C. Rap1 and H-Ras were sequentially activated by SNVR18 in Vero E6 cells. Vero E6 cells were serum starved for 24 h and treated with 10,000 SNVR18/cell. Active Rap1 and H-Ras were measured on Ral and Raf-functionalized beads, respectively, at 3 and 20 min after virus exposure. Cells were treated with the cAMP analog 8-pCPT-2′-O-methyl-cAMP (O-Me-cAMP), which stimulates the Epac/Rap1 pathway as a specificity control. Error bars represent standard deviations 3 separate measurements. D. Rab7 is activated in response to SNV exposure. Cell lysates prepared at varying timepoints (0–60 min) following SNV exposure were analyzed for the presence of active Rab7 using RILP functionalized beads. Error bars represent 3 separate measurements. ** P<0.001 for all data compared to resting (rest) cells.
The potential roles of the mutually antagonistic Rap1 and H-Ras downstream of SNV signals that lead to loss in cell adhesion were also examined by G-trap assay. Beads functionalized with effectors for Rap1 (GST RalGDS-RBD) and H-Ras (Raf-1 RBD) were used to simultaneously assay activated Rap1, and H-Ras GTPases from cell lysates of virus treated cells (Fig. 5B). An activator of Epac/Rap1, 8-Cpt-2m-cAMP, served as a specificity control. Activation of Rap1 by 8-Cpt-2m-cAMP was readily demonstrated and as expected decreased basal levels of H-Ras in the G-trap assay [42]. The activity of Rap1 and H-Ras in virus-activated cell lysates at 3 min and 20 min post-exposure showed that the GTPase pair was activated sequentially, where active Rap 1 peaked within the first 3 min and subsequently declined at 20 min. H-Ras remained low while Rap1 was at its zenith, eventually peaking at 20 min as active Rap1 was in decline. The coincidence between high H-Ras activity and loss of cell-cell adhesion might be linked to the proclivity of H-Ras to suppress integrin activity [43] (cf. Fig 3F).
Finally, the activation status of GTPases involved in the intracellular transport of virus particles following internalization, were assessed by monitoring the levels of active Rab7, a GTPase associated with early to late endosome and lysosomal transport [14; 44; 45]. Active, GTP-bound Rab7 levels peaked 15 min following virus exposure (Fig. 5C). The timing of maximal Rab7 coincides with the timing of fluorescently labeled SNV delivery to perinuclear late endosomes traced by microscopy (Fig S3, Supplemental Note 3). The time frame is also within the range reported for the delivery of other large viral cargoes to Rab7-positive late endosomes [46; 47].
In sum, we have devised a novel, cost effective, flow cytometry-compatible, bead-based effector-binding assay (G-trap) for rapidly monitoring the activation status of multiple members of the Ras GTPase superfamily in cell lysates. We have validated the assay through the use of known agonists and antagonists of individual GTPases. The study provides previously unknown mechanistic detail about SNV induced activation of cellular signaling to promote virus entry and transport. We have recently used a novel inhibitor to define the involvement of Cdc42 GTPase in the lifecycle of Sin Nombre virus infection [6]. Selective pharmacological inhibitors of the active GTPases can be paired with the G-trap assay to further define the signaling pathways that are important for regulating the lifecycles of pathogens that rely on the same processes. In similar vein we have shown that Y27632, the inhibitor of RhoA kinase (ROCK), blocks the loss of cell barrier function in polarized endothelial cells and thus limits viral infection (Buranda, unpublished results). We therefore anticipate that this approach will enable investigations into the interconnection of signaling networks via GTPase cascades in normal cellular functions as compared to viral or bacterial infections or other pathogenic processes.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants (R03AI092130, R21NS066429, 1P50GM085273), a University of New Mexico (UNM) School of Medicine RAC award and the New Mexico State Cigarette Tax to the UNM Cancer Center to TB. NIH R21 NS066435, NSF MCB0956027, DOD OC110514 to AWN. We thank Brian Hjelle for assistance with generation of UV-killed SNV and Dan Cimino for technical assistance with complementary DNA (cDNA) recovery experiments and for preparation of the GST–RILP vector. Images in this article were generated in the University of New Mexico Cancer Center Fluorescence Microscopy Facility P30CA118100-08S2 Sub-Project ID: 6322.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–69. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 2.Guilluy C, Garcia-Mata R, Burridge K. Rho protein crosstalk: another social network? Trends Cell Biol. 2011 doi: 10.1016/j.tcb.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 2013;93:269–309. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- 4.Schiller MR. Coupling receptor tyrosine kinases to Rho GTPases--GEFs what’s the link. Cell Signal. 2006;18:1834–43. doi: 10.1016/j.cellsig.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 5.Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13:13–22. doi: 10.1016/s0962-8924(02)00004-1. [DOI] [PubMed] [Google Scholar]
- 6.Hong L, Kenney SR, Phillips GK, Simpson D, Schroeder CE, Noth J, Romero E, Swanson S, Waller A, Strouse JJ, Carter M, Chigaev A, Ursu O, Oprea T, Hjelle B, Golden JE, Aube J, Hudson LG, Buranda T, Sklar LA, Wandinger-Ness A. Characterization of a cdc42 protein inhibitor and its use as a molecular probe. J Biol Chem. 2013;288:8531–43. doi: 10.1074/jbc.M112.435941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agola JO, Hong L, Surviladze Z, Ursu O, Waller A, Strouse JJ, Simpson DS, Schroeder CE, Oprea TI, Golden JE, Aube J, Buranda T, Sklar LA, Wandinger-Ness A. A competitive nucleotide binding inhibitor: in vitro characterization of Rab7 GTPase inhibition. ACS Chem Biol. 2012;7:1095–108. doi: 10.1021/cb3001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friesland A, Zhao Y, Chen YH, Wang L, Zhou H, Lu Q. Small molecule targeting Cdc42-intersectin interaction disrupts Golgi organization and suppresses cell motility. Proc Natl Acad Sci U S A. 2013;110:1261–6. doi: 10.1073/pnas.1116051110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Safronetz D, Ebihara H, Feldmann H, Hooper JW. The Syrian hamster model of hantavirus pulmonary syndrome. Antiviral Res. 2012;95:282–92. doi: 10.1016/j.antiviral.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grove J, Marsh M. Host-pathogen interactions: The cell biology of receptor-mediated virus entry. J Cell Biol. 2011;195:1071–82. doi: 10.1083/jcb.201108131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bharadwaj M, Lyons CR, Wortman IA, Hjelle B. Intramuscular inoculation of Sin Nombre hantavirus cDNAs induces cellular and humoral immune responses in BALB/c mice. Vaccine. 1999;17:2836–43. doi: 10.1016/s0264-410x(99)00096-1. [DOI] [PubMed] [Google Scholar]
- 12.Buranda T, Wu Y, Perez D, Jett SD, Bondu-Hawkins V, Ye C, Lopez GP, Edwards B, Hall P, Larson RS, Sklar LA, Hjelle B. Recognition of DAF and avb3 by inactivated Hantaviruses, towards the development of HTS flow cytometry assays. Anal Biochem. 2010;402:151–160. doi: 10.1016/j.ab.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wittchen ES, Burridge K. Analysis of low molecular weight GTPase activity in endothelial cell cultures. Methods Enzymol. 2008;443:285–98. doi: 10.1016/S0076-6879(08)02014-4. [DOI] [PubMed] [Google Scholar]
- 14.Colucci AM, Spinosa MR, Bucci C. Expression, assay, and functional properties of RILP. Methods Enzymol. 2005;403:664–75. doi: 10.1016/S0076-6879(05)03057-0. [DOI] [PubMed] [Google Scholar]
- 15.Tessema M, Simons PC, Cimino DF, Sanchez L, Waller A, Posner RG, Wandinger-Ness A, Prossnitz ER, Sklar LA. Glutathione-S-transferase-green fluorescent protein fusion protein reveals slow dissociation from high site density beads and measures free GSH. Cytometry A. 2006;69:326–34. doi: 10.1002/cyto.a.20259. [DOI] [PubMed] [Google Scholar]
- 16.Schwartz SL, Tessema M, Buranda T, Pylypenko O, Rak A, Simons PC, Surviladze Z, Sklar LA, Wandinger-Ness A. Flow cytometry for real-time measurement of guanine nucleotide binding and exchange by Ras-like GTPases. Anal Biochem. 2008;381:258–66. doi: 10.1016/j.ab.2008.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Curpan RF, Simons PC, Zhai D, Young SM, Carter MB, Bologa CG, Oprea TI, Satterthwait AC, Reed JC, Edwards BS, Sklar LA. High-throughput screen for the chemical inhibitors of antiapoptotic bcl-2 family proteins by multiplex flow cytometry. Assay Drug Dev Technol. 2011;9:465–74. doi: 10.1089/adt.2010.0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klasse PJ, Moore JP. Quantitative model of antibody- and soluble CD4-mediated neutralization of primary isolates and T-cell line-adapted strains of human immunodeficiency virus type 1. J Virol. 1996;70:3668–77. doi: 10.1128/jvi.70.6.3668-3677.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simons PC, Young SM, Carter MB, Waller A, Zhai D, Reed JC, Edwards BS, Sklar LA. Simultaneous in vitro molecular screening of protein-peptide interactions by flow cytometry, using six Bcl-2 family proteins as examples. Nat Protoc. 2011;6:943–52. doi: 10.1038/nprot.2011.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krautkramer E, Zeier M. Hantavirus causing hemorrhagic fever with renal syndrome enters from the apical surface and requires decay-accelerating factor (DAF/CD55) J Virol. 2008;82:4257–64. doi: 10.1128/JVI.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mold C, Walter EI, Medof ME. The influence of membrane components on regulation of alternative pathway activation by decay-accelerating factor. J Immunol. 1990;145:3836–41. [PubMed] [Google Scholar]
- 22.Reeder MK, Serebriiskii IG, Golemis EA, Chernoff J. Analysis of small GTPase signaling pathways using p21-activated kinase mutants that selectively couple to Cdc42. J Biol Chem. 2001;276:40606–13. doi: 10.1074/jbc.M103925200. [DOI] [PubMed] [Google Scholar]
- 23.Reid T, Furuyashiki T, Ishizaki T, Watanabe G, Watanabe N, Fujisawa K, Morii N, Madaule P, Narumiya S. Rhotekin, a new putative target for Rho bearing homology to a serine/threonine kinase, PKN, and rhophilin in the rho-binding domain. J Biol Chem. 1996;271:13556–60. doi: 10.1074/jbc.271.23.13556. [DOI] [PubMed] [Google Scholar]
- 24.Chigaev A, Buranda T, Dwyer DC, Prossnitz ER, Sklar LA. FRET detection of cellular alpha 4-integrin conformational activation. Biophysical Journal. 2003;85:3951–3962. doi: 10.1016/S0006-3495(03)74809-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461:99–103. doi: 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–79. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 27.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–9. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 28.Ridley AJ. Rho family proteins: coordinating cell responses. Trends Cell Biol. 2001;11:471–7. doi: 10.1016/s0962-8924(01)02153-5. [DOI] [PubMed] [Google Scholar]
- 29.Medley QG, Serra-Pages C, Iannotti E, Seipel K, Tang M, O’Brien SP, Streuli M. The trio guanine nucleotide exchange factor is a RhoA target. Binding of RhoA to the trio immunoglobulin-like domain. J Biol Chem. 2000;275:36116–23. doi: 10.1074/jbc.M003775200. [DOI] [PubMed] [Google Scholar]
- 30.Bellanger JM, Astier C, Sardet C, Ohta Y, Stossel TP, Debant A. The Rac1- and RhoG-specific GEF domain of Trio targets filamin to remodel cytoskeletal actin. Nat Cell Biol. 2000;2:888–92. doi: 10.1038/35046533. [DOI] [PubMed] [Google Scholar]
- 31.Schoenwaelder SM, Petch LA, Williamson D, Shen R, Feng GS, Burridge K. The protein tyrosine phosphatase Shp-2 regulates RhoA activity. Curr Biol. 2000;10:1523–6. doi: 10.1016/s0960-9822(00)00831-9. [DOI] [PubMed] [Google Scholar]
- 32.Burridge K, Doughman R. Front and back by Rho and Rac. Nat Cell Biol. 2006;8:781–2. doi: 10.1038/ncb0806-781. [DOI] [PubMed] [Google Scholar]
- 33.Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol. 2010;11:633–43. doi: 10.1038/nrm2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–99. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 35.Kurokawa K, Itoh RE, Yoshizaki H, Nakamura YO, Matsuda M. Coactivation of Rac1 and Cdc42 at lamellipodia and membrane ruffles induced by epidermal growth factor. Mol Biol Cell. 2004;15:1003–10. doi: 10.1091/mbc.E03-08-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kinbara K, Goldfinger LE, Hansen M, Chou FL, Ginsberg MH. Ras GTPases: integrins’ friends or foes? Nat Rev Mol Cell Biol. 2003;4:767–76. doi: 10.1038/nrm1229. [DOI] [PubMed] [Google Scholar]
- 37.Mochizuki N, Yamashita S, Kurokawa K, Ohba Y, Nagai T, Miyawaki A, Matsuda M. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature. 2001;411:1065–8. doi: 10.1038/35082594. [DOI] [PubMed] [Google Scholar]
- 38.Honda S, Tomiyama Y, Pelletier AJ, Annis D, Honda Y, Orchekowski R, Ruggeri Z, Kunicki TJ. Topography of ligand-induced binding sites, including a novel cation-sensitive epitope (AP5) at the amino terminus, of the human integrin beta 3 subunit. J Biol Chem. 1995;270:11947–54. doi: 10.1074/jbc.270.20.11947. [DOI] [PubMed] [Google Scholar]
- 39.del Pozo MA, Alderson NB, Kiosses WB, Chiang HH, Anderson RG, Schwartz MA. Integrins regulate Rac targeting by internalization of membrane domains. Science. 2004;303:839–42. doi: 10.1126/science.1092571. [DOI] [PubMed] [Google Scholar]
- 40.Coyne CB, Bergelson JM. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell. 2006;124:119–31. doi: 10.1016/j.cell.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 41.Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol. 2002;39:187–99. doi: 10.1016/s1537-1891(03)00008-9. [DOI] [PubMed] [Google Scholar]
- 42.Remans PH, Gringhuis SI, van Laar JM, Sanders ME, Papendrecht-van der Voort EA, Zwartkruis FJ, Levarht EW, Rosas M, Coffer PJ, Breedveld FC, Bos JL, Tak PP, Verweij CL, Reedquist KA. Rap1 signaling is required for suppression of Ras-generated reactive oxygen species and protection against oxidative stress in T lymphocytes. J Immunol. 2004;173:920–31. doi: 10.4049/jimmunol.173.2.920. [DOI] [PubMed] [Google Scholar]
- 43.Hughes PE, Renshaw MW, Pfaff M, Forsyth J, Keivens VM, Schwartz MA, Ginsberg MH. Suppression of integrin activation: a novel function of a Ras/Raf-initiated MAP kinase pathway. Cell. 1997;88:521–30. doi: 10.1016/s0092-8674(00)81892-9. [DOI] [PubMed] [Google Scholar]
- 44.Feng Y, Press B, Wandinger-Ness A. Rab 7: an important regulator of late endocytic membrane traffic. J Cell Biol. 1995;131:1435–52. doi: 10.1083/jcb.131.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bucci C, Parton RG, Mather IH, Stunnenberg H, Simons K, Hoflack B, Zerial M. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell. 1992;70:715–28. doi: 10.1016/0092-8674(92)90306-w. [DOI] [PubMed] [Google Scholar]
- 46.Lozach PY, Huotari J, Helenius A. Late-penetrating viruses. Curr Opin Virol. 2011;1:35–43. doi: 10.1016/j.coviro.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 47.Lozach PY, Mancini R, Bitto D, Meier R, Oestereich L, Overby AK, Pettersson RF, Helenius A. Entry of bunyaviruses into mammalian cells. Cell Host Microbe. 2010;7:488–99. doi: 10.1016/j.chom.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.