

Abstract
The design, synthesis, and evaluation of the potency of new isoform-selective inhibitors of sphingosine kinases 1 and 2 (SK1 and SK2), the enzyme that catalyzes the phosphorylation of d-erythro-sphingosine to produce the key signaling lipid, sphingosine 1-phosphate, are described. Recently, we reported that 1-(4-octylphenethyl)piperidin-4-ol (RB-005) is a selective inhibitor of SK1. Here we report the synthesis of 43 new analogues of RB-005, in which the lipophilic tail, polar headgroup, and linker region were modified to extend the structure–activity relationship profile for this lead compound, which we explain using modeling studies with the recently published crystal structure of SK1. We provide a basis for the key residues targeted by our profiled series and provide further evidence for the ability to discriminate between the two isoforms using pharmacological intervention.
Introduction
The lipid kinase, sphingosine kinase (SK), plays a myriad of roles in regulating cell survival, growth, and migration of mammalian cells through its product, sphingosine 1-phosphate (S1P). S1P is a ligand for five cell-surface G-protein-coupled receptors and for several intracellular targets, such as histone deacetylases 1 and 2 (HDAC1/2, which regulate gene expression).1 There are two isoforms of sphingosine kinase, SK1 and SK2, that are encoded by different genes and that exhibit distinct biochemical properties, substrate and inhibitor sensitivities, and subcellular distribution. SK1 and SK2 have redundant roles to some extent as knockout of both genes is embyronically lethal in mice, whereas animals survive when either gene is removed alone.2 Moreover, there is evidence that both SK1 and SK2 play a similar role in certain cancers.3,4 Surprisingly, SK1 and SK2 may have opposing roles in inflammation, being largely proinflammatory and antiinflammatory, respectively. On the other hand, a proinflammatory role for SK2 in other cell types has also been reported.3 The differing roles of SK1 and SK2 in inflammatory disease and supporting data from knockout mice make a compelling case for the development of isoform-selective inhibitors in order to elucidate the functions and roles of each isozyme and for designing drugs for therapeutic intervention in pathophysiological processes such as inflammatory diseases and cancer.
Various SK inhibitors have been identified (see Figure 1 for examples). Enzyme kinetic studies show that many of these are competitive with sphingosine (Sph). The initial SK inhibitors were Sph analogues, such as d,l-threo-dihydrosphingosine which has a Ki of ∼5 μM.3N,N-Dimethylsphingosine also inhibits both SK isoforms. The first SK1-selective inhibitor to be reported was the water-soluble Sph analogue SK1-I (also known as BML-258; Ki ∼ 10 μM).5 The most potent nanomolar SK1-selective inhibitor is PF-543.6 However, this compound is also a substrate for SK1, thereby compromising data interpretation. Analogues of the immunosuppressive agent FTY720 (Gilenya) have been prepared using a synthetic route starting with 4-octylphenethyl alcohol and were found to be SK1-selective inhibitors (e.g., RB-005).7 FTY720 is an inhibitor of SK18 and also inhibits other sphingolipid-metabolizing enzymes, such as ceramide synthase and S1P lyase.9,10 We also recently showed that SK1 contains an allosteric site.11 Replacement of the amino group in (S)-FTY720-vinylphosphonate with an azido group changes this compound from an allosteric inhibitor to an activator of SK1.12 Therefore, allosteric inhibitors of SK1 are also an exciting option for future study. With regard to SK2-selective inhibitors, ABC294640,13 (R)-FTY720 methyl ether (ROMe),14K145 (3-(2-amino-ethyl)-5-[3-(4-butoxylphenyl)propylidene]thiazolidine-2,4-dione),15 and SLR080811 ((S)-2-[3-(4-octylphenyl)-1,2,4-oxadiazol-5-yl]pyrrolidine-1-carboximidamide)16 have Ki values in the range of ∼1–10 μM. Taken together, it is clear that there is still a need to develop new SK1 and SK2 inhibitors, both to increase the number of selective tools that can be used to interrogate the biology of these enzymes and to increase the possibility of having useful new therapeutic agents to treat disease.
Figure 1.

Structures of selected SK inhibitors.
In a recent study, we showed that RB-005 is a highly selective SK1 inhibitor.7 Herein, we describe the synthesis of analogues of the known SK1 inhibitors RB-005, FTY720,8SKi,17 compounds 36a(18,19) and 82,20BML-258,5CB5468139,21 and SLR080811(16) (Figure 2). These new analogues, which were designed to possess some degree of structural similarity to the known inhibitors, were evaluated through enzyme activity studies as inhibitors of SK1 and SK2. Structure–activity relationship (SAR) studies are discussed, which are supported by an analysis of molecular modeling poses of the inhibitors in the active site of human SK1.
Figure 2.

Modifications introduced into the known SK inhibitors RB-005, SKi, SLR080811, 36a (VPC96091), and 82.
Results and Discussion
The structures of the new compounds and their key structural modifications are shown in Tables 1–4.
Table 1. Piperidyl Analogues of RB-005 Synthesized for Evaluation as SK Inhibitors.
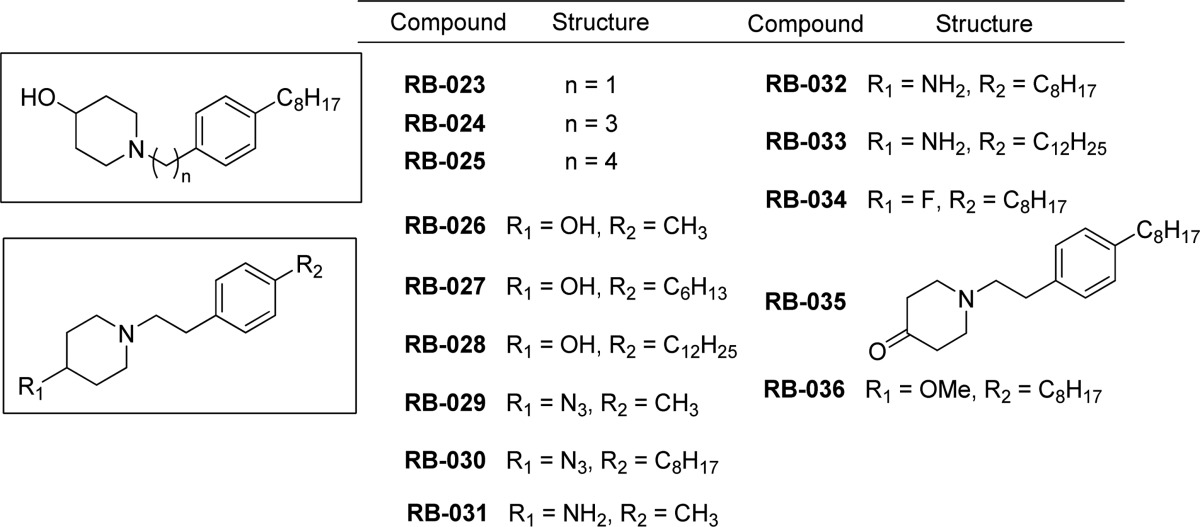
Table 4. Pyridine, Pyridinium, and Triazole Analogues of RB-005 Synthesized for Evaluation as SK Inhibitors.
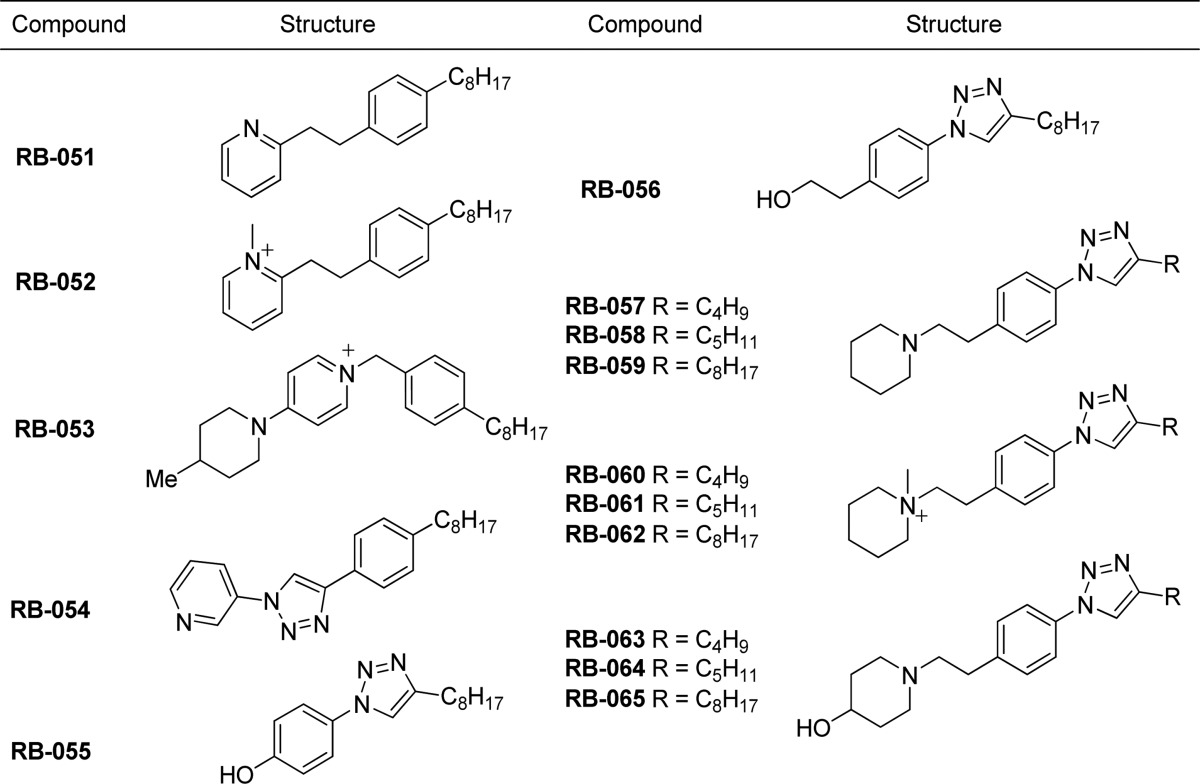
Table 2. Pyrrolidine Analogues of RB-005 Synthesized for Evaluation as SK Inhibitors.
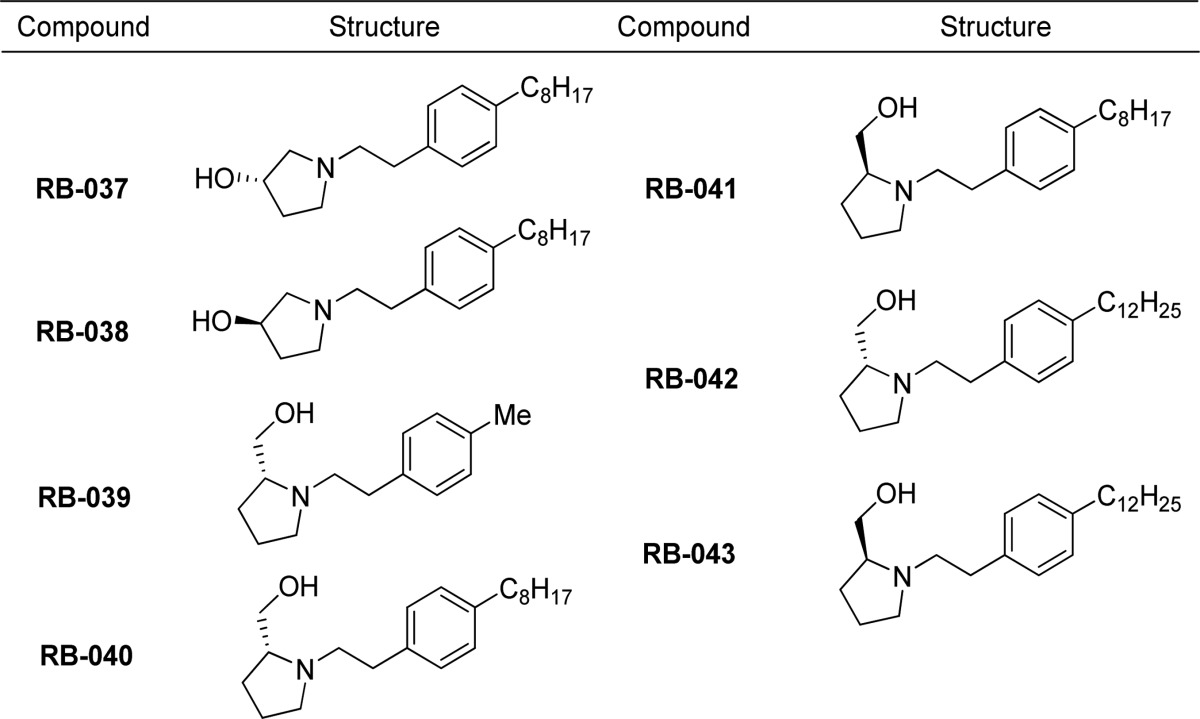
Table 3. Benzamide Analogues of RB-005 Synthesized for Evaluation as SK Inhibitors.
Chemical Synthesis
Linker Length
The synthetic routes we employed to prepare compounds with one-, three-, and four-carbon tethers are displayed in Scheme 1. The sole carbon atom of the hydroxymethyl group of 4-iodobenzyl alcohol provided the tether in RB-023, whereas the three carbons of propargyl alcohol were the source of the tether in RB-024. In each of these compounds, a Sonogashira reaction followed by catalytic hydrogenation of the alkyne intermediate (1 and 3) and a SN2 reaction of a mesylate intermediate derived from 2 and 4 with 4-hydroxypiperidine gave the desired compounds. We used 4-(4-octylphenyl)butan-1-ol (5)22 as the starting material for the preparation of RB-025.
Scheme 1. Synthesis of RB-023–RB-025.

Reagents and conditions: (a) 1-octyne, Pd(PPh3)4, CuI, Et3N, 50 °C, 12 h; (b) Pd/C, H2, EtOAc, rt, 12 h; (c) (i) MsCl, Et3N, CH2Cl2, 3 h, rt; (ii) 4-hydroxypiperidine, MeCN, 50 °C, 12 h; (d) propargyl alcohol, Pd(PPh3)4, CuI, Et3N, 50 °C, 12 h.
Modifications of the 4-Alkylphenyl and the Piperidyl Groups
Scheme 2 shows the synthetic pathways employed to prepare RB-026–RB-033. After a Sonogashira reaction was used to install an alkynyl group with 6–12 carbons, catalytic hydrogenation of 6 and 7 afforded alkyl derivatives 8 and 9, and SN2 displacement as in Scheme 1 gave the desired compounds in good yield. To assess the role of the hydroxyl group in RB-005, RB-026, and RB-028 in inhibition of SK, we replaced this group with an azido group via mesylation of the alcohol and reaction with sodium azide in DMF to obtain 10, RB-029, and RB-030. Reduction of the azide afforded the amino derivatives, RB-031, RB-032, and RB-033.
Scheme 2. Synthesis of RB-026–RB-033.

Reagents and conditions: (a) 1-hexyne or 1-dodecyne, Pd(PPh3)4, CuI, Et3N, 50 °C, 12 h; (b) Pd/C, H2, EtOAc; (c) (i) MsCl, Et3N, CH2Cl2; (ii) 4-hydroxypiperidine, MeCN, 50 °C, 12 h; (d) (i) MsCl, Et3N, CH2Cl2, 3 h, rt; (ii) NaN3, DMF, 80–100 °C, 12 h; (e) Pd/C, H2, CH2Cl2/MeOH (1:3), rt, 12 h.
Fluorination of RB-005 with diethylaminosulfur trifluoride (DAST) gave RB-034 in 90% yield (Scheme 3). The 4-keto derivative RB-035 was synthesized by oxidation of the 4-hydroxyl group of RB-005 with pyridinium chlorochromate. Reaction of mesylate 11(7) with 4-methoxypiperidine provided RB-036. Similarly, reaction of commercially available chiral pyrrolidines containing a 3-hydroxyl or a 2-hydroxymethyl substituent gave RB-037–RB-043.
Scheme 3. Synthesis of RB-034–RB-043.

Reagents and conditions: (a) DAST, CH2Cl2, 0 °C to rt, 5 h, rt; (b) PCC, CH2Cl2, 4 h, rt; (c) cyclic amine, MeCN, 50 °C, 12 h.
Benzamide Derivatives
Benzamide-containing analogues RB-044–RB-050 were prepared from 4-iodobenzoic acid as outlined in Scheme 4. Alkyne intermediate 12 was reduced to carboxylate 13, which was converted to the acyl chloride with thionyl chloride and then treated with the desired cyclic amine in the presence of potassium carbonate.
Scheme 4. Synthesis of RB-044–RB-050 from 4-Iodobenzoic Acid.

Reagents and conditions: (a) 1-octyne, Pd(PPh3)4, CuI, Et3N, 60 °C, 12 h; (b) Pd/C, H2, EtOAc, 12 h, rt; (c) (i) SOCl2, CH2Cl2, reflux, 12 h; (ii) cyclic amine, K2CO3, MeCN, 50 °C, 12 h.
Quaternary Ammonium Derivatives
To synthesize RB-052, which contains a pyridinium ion in the polar headgroup, we used the halogens in 1-bromo-4-iodobenzene to carry out two separate Sonogashira reactions, as shown in Scheme 5. First, we used 1-octyne to prepare alkyne 14, which reacted with 3-ethynylpyridine to give dialkyne 15. Reduction of 15 yielded RB-051, and N-alkylation with methyl iodide (5 equiv) in acetonitrile afforded the N-methylpyridinium salt RB-052.
Scheme 5. Synthesis of RB-051 and RB-052 from 1-Bromo-4-iodobenzene.

Reagents and conditions: (a) 1-octyne, Pd(PPh3)4, CuI, Et3N, 50 °C, 12 h; (b) 3-ethynylpyridine, Pd(PPh3)4, CuI, Et3N, 80 °C, 3 days; (c) Pd/C, H2, EtOAc, 12 h, rt; (d) K2CO3, MeI, MeCN, overnight, rt.
Pyridinium salt RB-053 was prepared by a similar route, as shown in Scheme 6. A Sonogashira reaction of 4-iodobenzyl bromide with 1-octyne provided alkyne 16; displacement of bromide ion with 4-(4-methylpiperidin-1-yl)pyridine (17) afforded alkyne 18, which provided RB-053 on catalytic hydrogenation in MeOH/CH2Cl2 (1:3).
Scheme 6. Synthesis of Pyridinium Salt RB-053 from 4-Iodobenzyl Bromide and of 4-(4-Methylpiperidin-1-yl)pyridine (17) from 4-Chloropyridine Hydrochloride.

Reagents and conditions: (a) 1-octyne, Pd(PPh3)4, CuI, Et3N; (b) 4-methylpiperidine, DIPEA, MeCN, microwave heating, 160 °C, 1 h; (c) 17, 2-butanone, 100 °C, 3 days; (d) Pd/C, H2, MeOH/CH2Cl2 (1:3).
Triazole Derivatives
A Pd/Cu-catalyzed Sonogashira reaction followed by a Cu(I)-catalyzed azide–alkyne 1,3-dipolar addition (click reaction) was employed to prepare RB-054, in which a triazole ring bearing a pyridine substituent is the headgroup (Scheme 7). Thus, Sonogashira coupling of 1-octyne with 4-iodoaniline afforded alkynylaniline derivative 19. After conversion of amine 19 to aryl azide 20, the triazole ring was installed in a click reaction with 3-ethynylpyridine. Finally, catalytic hydrogenation of alkyne 21 gave RB-054.
Scheme 7. Synthesis of RB- 054 from 4-Iodoaniline.
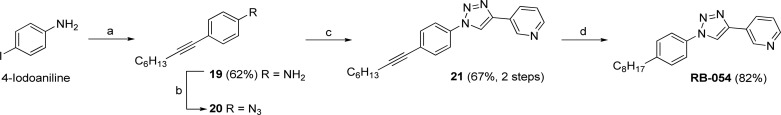
Reagents and conditions: (a) 1-octyne, Pd(PPh3)4, CuI, Et3N, 60 °C, 12 h; (b) (i) 10% aq HCl, NaNO2; (ii) NaN3; (c) 3-ethynylpyridine, sodium ascorbate, CuSO4, t-BuOH/H2O (1:1); (d) Pd/C, H2, EtOAc, rt, 2 days.
We reversed the location of the triazole in analogues RB057–RB-065. As shown in Scheme 8, the headgroup in these analogues is a piperidyl derivative and the lipophilic tail contains an alkyl-substituted triazole. Azides 22 and 23 were prepared from commercially available 4-aminophenol and 2-(4-aminophenyl)ethanol, respectively. The click reaction with terminal alkynes afforded triazoles RB-055, 24, 25, and RB-056. To prepare RB-059–RB-065, alcohols 24, 25, and RB-056 were converted to their corresponding mesylates 26, 27, and 28, which were treated with piperidine, 1-methylpiperidine, or 4-hydroxypiperidine.
Scheme 8. Synthesis of RB-055–RB-065.

Reagents and conditions: (a) (i) 10% aq HCl, NaNO2; (ii) NaN3; (b) 1-decyne, sodium ascorbate, CuSO4, t-BuOH/H2O (1:1), 12 h, rt; (c) acetylenic substrate (24, 1-hexyne; 25, 1-heptyne; RB-056, 1-decyne), sodium ascorbate, CuSO4, t-BuOH, H2O; (d) MsCl, Et3N, CH2Cl2, 0 °C to rt, 5 h; (e) piperidine, MeCN (RB-059–RB-061), 1-methylpiperidine, MeCN (RB-060–RB-062) or 4-hydroxypiperidine, MeCN, 50 °C, 12 h (RB-063–RB-065).
Effects on SK1 and SK2 Activity and SAR
We previously demonstrated the importance of the 4-hydroxypiperidinyl group in the selective inhibition of SK1,7 which was subsequently confirmed by Gustin et al., who generated chiral piperidyl analogues bearing hydroxyl and hydroxymethyl groups.20 To examine SAR among a panel of related compounds, we prepared a series of analogues bearing a 4-hydroxypiperidinyl group but varied the linker length between the aryl group and the piperidine. We also assessed the role of the alkyl substituent in the aryl group. To evaluate compound selectivity against SK1 or SK2, the assays were performed (see Supporting Information) using Sph at concentrations of 3 and 10 μM (the Km values of SK1 and SK2, respectively), which corresponds to 50% substrate saturation and enables a qualitative estimation of selectivity by comparing the % inhibition of each kinase using a fixed concentration of inhibitor. We consider this approach to be an appropriate comparison of selectivity, since both enzymes exhibit 50% occupancy with the substrate. Compounds that were found to be effective inhibitors were then analyzed in more detail by performing dose–response curves.
We have previously shown that RB-005 (the “parent compound”) is a selective inhibitor of SK1 and exhibits an IC50 = 3.6 ± 0.38 μM at 3 μM Sph (which corresponds to the Km of SK1) and reduces SK1 activity by ∼90% at 50 μM RB-005.7 The effect of linker length on potency was assessed by comparing the % inhibition of SK1 and SK2 obtained with RB-023 (which has a one-carbon tether), RB-024 (three-carbon tether), and RB-025 (four-carbon tether). The linker length did not significantly alter the ability of RB-023, RB-024, and RB-025 to inhibit SK1 activity (Figure 3). RB-023–RB-025 also retained selectivity for SK1 over SK2.
Figure 3.

Effect of inhibitors on SK1 or SK2 activity. SK1 activity was measured using 3 μM Sph and 250 μM ATP. SK2 activity was assayed using 10 μM Sph and 250 μM ATP (n = 3 for each compound; results are expressed as % of control ± SD). RB series compounds were used at 50 μM. BML-258 (50 μM) inhibited SK1 activity by 74.5 ± 3.3% (n = 3). The control is 100% and equals activity against Sph alone.
The aliphatic chain at the para position of the benzene ring of FTY720 is C8H17, which is known to be optimal for the action of FTY720 on its targets such as S1P receptors.23 Knott et al.24 reported that the ability of quaternary ammonium salts with a phenyl-substituted cyclohexylamine scaffold to inhibit SK2 was affected by the alkyl chain length. To examine the role of the alkyl substituent on the benzene ring of RB-005, and thus the lipophilicity of the molecule, we compared the inhibitory activity of RB-026 (which has a methyl group as the alkyl substituent), RB-027 (which has a n-hexyl group), RB-005 (which has a n-octyl group), and RB-028 (which has a n-dodecyl group). SK1 inhibition was decreased by more than 6-fold in RB-026 compared with RB-023. The almost complete lack of inhibition displayed by RB-026 against SK1 indicates that a larger alkyl group than a methyl group is required for inhibitory activity. We also evaluated the effect of alkyl chain length in the lipophilic tail of compounds in which the 4-hydroxypiperidinyl group was replaced by a 4-aminopiperidinyl group (see below). Changing the n-octyl group of RB-032 to a methyl or n-dodecyl group gave RB-031 and RB-033, respectively, and eliminated the inhibitory activity toward SK1. These results confirm the critical requirement for the n-octyl group.
Next, we probed the role of the 4-hydroxyl group of RB-005 by replacing it with an azido, amino, fluoro, keto, or methoxy group (RB-029–RB-036). Azido replacement (RB-029, RB-030) reduced SK1 inhibition markedly, while replacement of the 4-hydroxyl group with an amino group (RB-032) diminished the potency of SK1 inhibition (Figure 4). The isoform selectivity of SK1 over SK2 was retained for RB-032, suggesting that the amino group replacement maintains efficient binding to SK1. Replacement of the 4-hydroxyl group of RB-005 with a fluoro (RB-034) or methoxy group (RB-036) eliminated inhibitory activity against SK1, while replacement with a keto group to produce RB-035 increased inhibition of SK2 and maintained inhibition of SK1 but eliminated the isoform selectivity.
Figure 4.
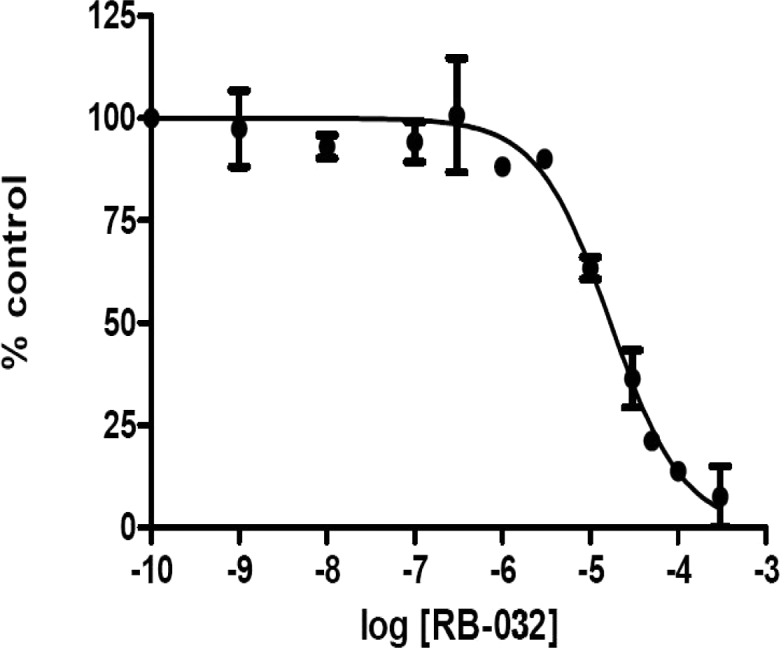
Effect of RB-032 on SK1 activity. Concentration-dependent inhibition of SK1 activity by RB-032 using 3 μM Sph and 250 μM ATP. The results are expressed as % of control ± SD; n = 3. The control is 100% and equals activity against Sph alone. RB-032 inhibits SK1 activity with IC50 = 16.9 ± 1.6 μM. RB-005 inhibits SK1 activity with IC50 = 3.6 ± 0.36 μM.7
To examine the role of the piperidyl group in inhibition of SK, we replaced it with a pyrrolidine ring; the hydroxyl-containing substituent was retained (as either a chiral hydroxyl or a chiral hydroxymethyl group), but its orientation was varied, as shown in compounds RB-037–RB-043. RB-037 and RB-038 retained inhibitory activity against SK1 despite having opposite configurations at C-3 of the pyrrolidin-3-ol group. Stereoisomers RB-040 and RB-042, which differ in the length of the aliphatic chain (C8H17 vs C12H5) but possess the R configuration at C-2 of the 2-hydroxymethylpyrrolidinyl group, were equipotent inhibitors of SK1 and SK2 (Figure 3 and Figure 5). The corresponding S enantiomers RB-041 and RB-043 were much less active (Figure 3). To establish whether RB-041 and RB-043 were capable of inhibiting SK1 and SK2 activity in a concentration-dependent manner, we used a higher concentration of each (100 μM, compared to the 50 μM concentration data shown in Figure 3), and found that the inhibition of SK1 and SK2 with RB-041 was 72.2 ± 5.9% and 45.7 ± 2.6%, respectively, whereas with RB-043 the inhibition of SK1 and SK2 was 49.9 ± 6.2% and 49.7 ± 7%, respectively. These findings indicate that RB-041 and RB-043 can inhibit SK1 and SK2 but that the sensitivity of inhibition compared with RB-040 and RB-042 is considerably reduced. Interestingly, the S enantiomers RB-041 and RB-043 are substrates for SK2 (see Supporting Information, Figure S1).
Figure 5.

Effect of RB-040 and RB-042 on (A) SK1 activity and (B) SK2 activity. Concentration-dependent inhibition of SK activity by RB-040 and RB-042 using 3 μM Sph (SK1) or 10 μM Sph (SK2) and 250 μM ATP. The results are expressed as % of control ± SD (n = 3). The control is 100% and equals activity against Sph alone. RB-040 inhibits SK1 activity with IC50 = 2.2 ± 0.22 μM and SK2 activity with IC50 = 5.2 ± 0.82 μM. RB-042 inhibits SK1 activity with IC50 = 5.3 ± 0.5 μM and SK2 activity with IC50 = 5.0 ± 1.3 μM.7
To further examine the influence of the length of the alkyl substituent on the benzene ring on SK activity, we assessed the extent of SK inhibition afforded by pyrrolidine derivatives RB-039, RB-042, and RB-043. The ability of the compound to inhibit SK1 is abolished in RB-039 and RB-043, which have a methyl and a n-dodecyl group in the lipophilic tail, respectively.
An amidine or proline headgroup incorporated into a benzamide-containing scaffold was shown to provide potent SK1 inhibitors,18,19 as in compound 36a (Figure 1). When we replaced the methylene linker between the aryl group and the heterocycle with a keto group to produce the benzamide analogues RB-044–RB-050, inhibition of SK1 was effectively abolished (Figure 3), as were the pyridine derivatives (RB-048 and RB-051). The report that quaternary ammonium salts are selective SK2 inhibitors25 prompted us to prepare RB-052, RB-053, RB-060, RB-061, and RB-062, which were ineffective as SK inhibitors, although RB-053 demonstrated a moderate selectivity for SK2 (Figure 3). We also prepared aliphatic quaternary ammonium salts (not shown) that were inactive.
SK inhibitors containing a central thiazole group have been reported (e.g., SKi and compound 82, Figure 1). 1,2,3-Triazoles are mimics of thiazoles and are easily prepared by Cu(I)-catalyzed azide/alkyne click chemistry. In our series of triazole analogues of RB-005 (RB-054–RB-065), we found that RB-065 was a highly selective SK1 inhibitor, whereas the other 10 triazole analogues, all of which lack the 4-hydroxypiperidinyl group, were inactive (Figure 3).
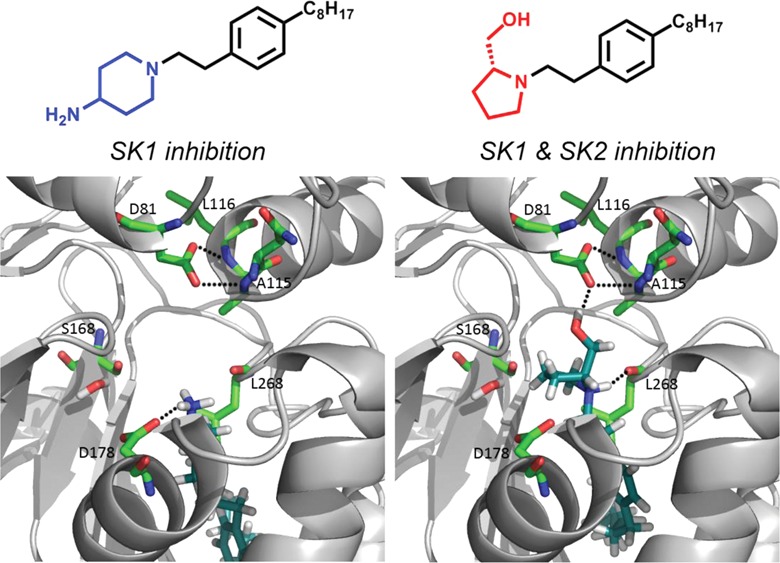
Modeling the Inhibitors in the Atomic Structure of SK1
The crystal structures of human SK1 in a complex with ADP and SKi were determined recently.26 We demonstrate here that the chemical modifications of the highly selective SK1 inhibitor RB-005 produced SAR that can be explained using this crystal structure. Figure 6A displays the result of a modeling analysis of RB-005 in the active site of human SK1; the piperidyl hydroxyl group is hydrogen-bonded to D81, and the protonated amine of the headgroup forms a salt bridge with the carboxylate of D178. Fluoro (RB-034) or methoxy (RB-036) containing compounds do not exhibit inhibitory activity against SK1 (Figure 3), as these groups no longer have hydrogen bond donating capacity, suggesting that the interaction of the 4-hydroxypiperidinyl group of RB-005 is with a hydrogen bond acceptor in the protein. The lack of SK1 inhibitor activity of the azide-containing compounds (RB-029, RB-030) supports this possibility. One of the two oxygens of the carboxylate ion of D81 is hydrogen-bonded to the backbone NH of L116, and the other is hydrogen-bonded to the backbone NH of A115; these interactions prevent the carboxylate of D81 from being catalytic and shift the catalytic role to D178 (Figure 7). The latter carboxylate oxygen also forms a hydrogen bond to the hydroxyl group of the inhibitor. If these compounds formed hydrogen bonds with the side chain of S168, then RB-029, RB-034, and RB-036 could also bind to the donor/acceptor hydroxyl group of S168 or water and therefore act as inhibitors. Since RB-029, RB-034, and RB-036 cannot form hydrogen bonds with D81 and are not inhibitors, we propose that the key interaction of the hydroxyl group of RB-005 (Figure 6A), RB-025 (Figure 6B), and RB-028 (Figure 6C) is with D81 and not with S168. In contrast, modeling of RB-035 (which contains a 4-keto group instead of a 4-hydroxyl group, yet maintains inhibition of SK1) suggests that the carbonyl group can form a hydrogen bond with the hydroxyl group of S168 and water (see Supporting Information, Figure S2).
Figure 6.

Various modeled poses of SK1 inhibitors in the catalytic site of SK1: (A) RB-005, (B) RB-025, (C) RB-028, (D) RB-032, (E) RB-033, (F) RB-040, (G) RB-042, and (H) RB-065. Dotted lines represent hydrogen bonds.
Figure 7.
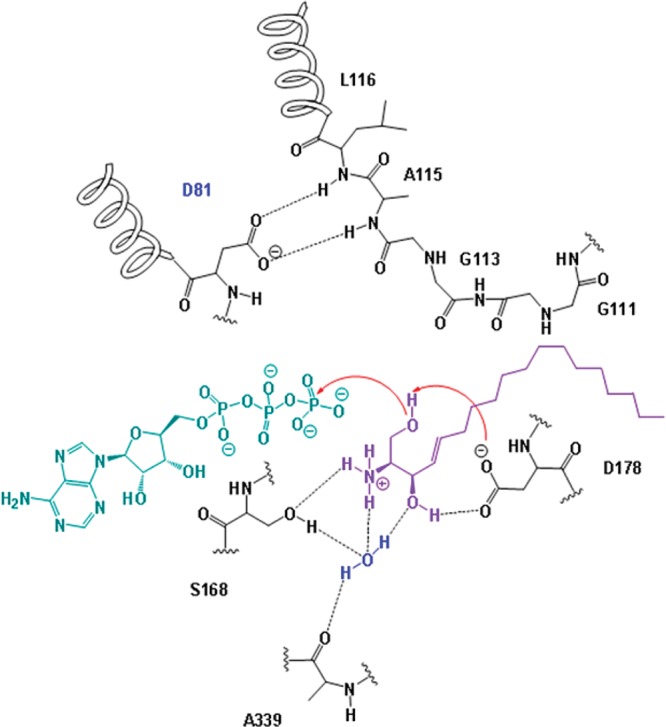
Schematic model of the proposed mechanism of the phosphorylation of Sph catalyzed by SK1.
The inhibitory effect of RB-032 (Figure 6D) and its absence for RB-033 (Figure 6E) can be explained by protonation of the primary amine. When the protonated primary amine rather than the piperidyl group forms a salt bridge with D178, the inhibitors are pushed deeper into the J channel, which was identified by Wang et al.26 as the region that accommodates the alkyl chain of Sph. Since RB-032 has a shorter alkyl chain than RB-033, it can be accommodated in the substrate pocket, whereas the dodecyl group of RB-033 cannot fit into the channel formed when the protonated primary amine forms a salt bridge with D178. Therefore, RB-033, which is inactive, does not bind to D81, D178, S168, or L268, indicating that these are key amino acid residues required for binding SK inhibitors.
The salt bridge between the NH3+ group of RB-032 and D178 (Figure 6D) is the only polar interaction with the protein, which might explain the lower SK1 inhibitor activity when compared with RB-005; the latter forms a salt bridge and hydrogen-bonds with D81. Interestingly, the R-enantiomers RB-040 and RB-042 are equipotent inhibitors of SK1 and SK2, while the S-enantiomers RB-041 and RB-043 are weak substrates for SK2, implying that the spatial orientation of the hydroxyl group in RB-041 and RB-043 required for catalysis is different in SK2 compared with SK1. The protonated amine in RB-040 (Figure 6F) and RB-042 (Figure 6G) can form a salt bridge with D178 and can also form a hydrogen bond with the carbonyl oxygen of L268. Both inhibitors can orientate the hydroxymethyl group of the pyrrolidine (R enantiomer) to also form a hydrogen bond with the side chain of D81. The protonated amino group of RB-041 and RB-043 can form a salt bridge with D178 but, because of the orientation of the hydroxymethyl group of the pyrrolidine (S enantiomer), cannot form a hydrogen bond between their hydroxyl group and D81, as found in our modeling study. Instead, the hydroxymethyl group could form a hydrogen bond to D178. As the experimental evidence shows that RB-041 and RB-043 do not inhibit SK1, this suggests that dynamic factors (accessing the binding site), which are not taken into account by docking studies, prevent the binding of these compounds.
RB-044–RB-050 are ineffective inhibitors of SK1. There are three possible explanations: first, the nitrogen in an amide cannot be protonated, thus preventing salt bridge formation. Second, the link between nitrogen and phenyl is constrained and planar compared with a methylene group, which prevents optimization of the hydrogen bonding network with the hydroxyl group. Third, the carbonyl group of the amide would be proximal to the side chain of D178, which would result in electrostatic repulsion.
The pyridinium salts RB-052 and RB-053 and the quaternary ammonium salts RB-060, RB-061, and RB-062 were also ineffective SK1 inhibitors. The absence of a hydroxyl group in these compounds rules out hydrogen bonding with D81 or D178. The triazole moiety in RB-065 forms a hydrogen bond with T196 (Figure 6H) and, furthermore, adds a kink in the chain that helps orientate the alkyl group into the J channel, which may account for its SK1 inhibitory activity.
Our modeling studies suggest that RB-005 (Figure 6A), RB-025 (Figure 6B), and RB-028 (Figure 6C) interact with D81 and not with S168. These findings are consistent with D81 not acting as a base, because RB-005, RB-025, or RB-028 are not substrates for SK1. As depicted in Figure 7, modeling of Sph into the catalytic site of SK1 suggests that S168 can form hydrogen bonds with the NH3+ group of Sph and, via a water molecule, with the secondary hydroxyl group of Sph. The water molecule also hydrogen-bonds with A339, thereby linking this amino acid residue to the secondary hydroxyl group of Sph (Figure 7). Thus, D178 functions as the deprotonating base in this model to enable nucleophilic attack by Sph on the γ-phosphate group of ATP, with subsequent transfer of this phosphate to Sph.
Conclusion
In this study we have identified a series of SK1-selective inhibitors and have used molecular modeling to define their interactions with the catalytic site of the enzyme. These studies reveal a substantial flexibility in the catalytic site in terms of binding SK1 inhibitors. For instance, RB-005 is proposed to interact with D81 and D178, while RB-025 appears to interact with D81 and L268. The findings obtained from the modeling study fully account for the SAR of the inhibitors and explain why some of these compounds are inactive. These findings also reveal the architecture of the SK1 catalytic site and suggest a major role for D178 as the deprotonating base that facilitates phosphorylation of Sph by ATP. In summary, the novel information presented here should enable development of new SK1 inhibitors with improved potency and selectivity. Similarly, resolution of the atomic structure of SK2 (yet to be achieved) along with information provided herein will enable better insights into the molecular basis of the selectivity of these inhibitors for SK1 over SK2.
Experimental Section
Docking Studies
The crystal structure of SK1 in complex with Sph (PDB entry 3VZB) was used for docking studies. Chain A of the complex was kept along with a single water molecule found to be tightly bound to the complex which hydrogen-bonds to the side chain hydroxyl group of S168, the backbone −NH of G342, and the secondary hydroxyl group of Sph (water number 680). Hydrogen atoms were added to the protein and water using Accelrys Discovery Studio 3.1 (Accelrys Software, San Diego, CA), and all of the inhibitors presented in this study were docked using GOLD 5.1 for Windows (Cambridge Crystallographic Data Centre, Cambridge, U.K.). Default software settings were used, keeping ChemPLP as a scoring function after redocking Sph in place in the 3VZB crystal structure as well as the SKi inhibitor in the 3VZD PDB entry (rmsd of 1.8 and 0.2 Å, respectively) as validation.
Synthesis. General Methods
All chemicals were reagent grade and used as purchased. Reactions were run under nitrogen and were monitored by TLC using silica gel 60 F254 aluminum-backed plates. Flash column chromatography was performed on silica gel grade 60 (230–400 mesh). THF was distilled over sodium/benzophenone immediately prior to use. Dichloromethane was distilled over CaH2, and Et3N was distilled over KOH pellets. All other solvents were of anhydrous quality and were used as received. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance I spectrometer, and chemical shifts are reported in δ units relative to deuterated solvents, which served as internal references, at 400 and 100 MHz, respectively. High-resolution mass spectra were recorded at the CUNY Mass Spectrometry Facility on an Agilent Technologies G6520A Q-TOF mass spectrometer using electrospray ionization (ESI). Microwave reactions were performed in a Biotage Emrys Creator synthesizer. HPLC was carried out using a reverse-phase column with a gradient of acetonitrile/water from 50/50 to 90/10, with detection at 214 and 254 nm. Elemental analysis was performed at Columbia Analytical Services, Tucson, AZ. All compounds were ≥95% pure as determined by examining their HRMS and 1H NMR spectra.
(4-(Oct-1-ynyl)phenyl)methanol (1)
To a solution of 4-iodobenzyl alcohol (200 mg, 0.85 mmol), bis(triphenylphosphine)palladium dichloride (49 mg, 0.040 mmol), and copper(I) iodide (7.6 mg, 0.04 mmol) in anhydrous triethylamine (10 mL) was added 1-octyne (283 mg, 2.56 mmol) at room temperature. After the reaction mixture was heated at 50 °C for 12 h, saturated aqueous ammonium chloride solution was added, and the mixture was extracted with EtOAc. The combined solution was washed with water, brine, and dried. Flash column chromatography with hexanes/EtOAc (5:1) as the eluent gave alkyne 1 (180 mg, 98%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 0.90 (t, J = 6.9 Hz, 3H), 1.28–1.36 (m, 4H), 1.41–1.49 (m, 2H), 1.60 (quin, J = 7.3 Hz, 2H), 1.89 (br s, 1H), 2.40 (t, J = 7.1 Hz, 2H), 4.65 (s, 2H), 7.25 (d, J = 8.1 Hz, 2H), 7.37 (d, J = 8.1 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 19.4, 22.6, 28.6, 28.7, 31.4, 65.0, 80.3, 90.6, 123.4, 126.8, 131.7, 140.1; ESI-HRMS (M + H)+m/z calcd for C15H21O 217.1592, found 217.1588.
(4-Octylphenyl)methanol (2)
Compound 1 (180 mg, 0.83 mmol) was dissolved in EtOAc (10 mL), and 10% Pd/C (90 mg, 50 wt %) was added. The reaction mixture was hydrogenated at room temperature for 12 h. The catalyst was removed by filtration through a pad of Celite, which was rinsed with EtOAc. Product 2 was obtained, without purification, as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 6.6 Hz, 3H), 1.26–1.30 (m, 10H), 1.56–1.63 (m, 2H), 1.78 (br s, 1H), 2.59 (t, J = 7.7 Hz, 2H), 4.64 (s, 2H), 7.16 (d, J = 8.0 Hz, 2H), 7.26 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 29.3, 29.4, 29.5, 31.6, 31.9, 35.7, 65.3, 127.1, 128.6, 138.1, 142.6; ESI-HRMS (M + H)+m/z calcd for C15H25O 221.1905, found 221.1887.
1-(4-Octylbenzyl)piperidin-4-ol (RB-023)
To a solution of 2 (37 mg, 0.17 mmol) and triethylamine (0.23 mL, 1.7 mmol) in CH2Cl2 (5 mL) at 0 °C was added methanesulfonyl chloride (40 μL, 0.50 mmol). After being stirred at room temperature for 3 h, the reaction mixture was evaporated, diluted with water, and the product was extracted with EtOAc. The extract was washed with brine, dried, and evaporated. To a solution of the reaction mixture (0.17 mmol) in MeCN (3 mL) was added 4-hydroxypiperidine (86 mg, 0.85 mmol). The reaction mixture was stirred at 50 °C for 12 h and concentrated. Purification by silica gel chromatography, eluting with CH2Cl2/MeOH (5:1), gave 35 mg (69%, two steps) of RB-023 as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.23–1.30 (m, 10H), 1.56–1.73 (m, 4H), 1.97–2.06 (m, 2H), 2.35–2.47 (m, 2H), 2.59 (t, J = 7.7 Hz, 2H), 2.85–2.90 (m, 2H), 3.64 (s, 2H), 3.78–3.81 (m, 1H), 7.15 (d, J = 7.9 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 29.3, 29.4, 29.5, 31.5, 31.9, 33.2, 35.7, 50.1, 62.2, 128.5, 129.7, 137.2, 142.8; ESI-HRMS (M + H)+m/z calcd for C20H34NO 304.2640, found 304.2637.
3-(4-Octylphenyl)prop-2-yn-1-ol (3)
Compound 3 was prepared from 1-bromo-4-n-octylbenzene and propargyl alcohol according to a Sonogashira reaction procedure similar to that described for 1. Yield = 82%; 1H NMR (400 MHz, CDCl3) δ 0.86 (t, J = 6.8 Hz, 3H), 1.22–1.30 (m, 10H), 1.57–1.60 (m, 2H), 2.59 (t, J = 7.7 Hz, 2H), 4.49 (s, 2H), 7.11 (d, J = 7.8 Hz, 2H), 7.34 (d, J = 7.7 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 29.2, 29.4, 31.2, 31.9, 35.9, 51.7, 85.9, 86.5, 119.6, 128.4, 131.6, 143.7; ESI-HRMS (M + H)+m/z calcd for C17H25O 245.1905, found 245.1903.
3-(4-Octylphenyl)propan-1-ol (4)
Compound 4 was prepared from 3 by a catalytic hydrogenation procedure similar to that described for 2. 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.24–1.30 (m, 10H), 1.59 (quin, J = 7.4 Hz, 2H), 1.88 (quin, J = 6.5 Hz, 2H), 2.56 (t, J = 7.7 Hz, 2H), 2.67 (t, J = 7.7 Hz, 2H), 3.66 (t, J = 6.4 Hz, 2H), 7.10 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 29.3, 29.4, 29.5, 31.6, 31.7, 31.9, 34.3, 35.6, 62.4, 128.3, 128.4, 138.9, 140.5; ESI-HRMS (M + H)+m/z calcd for C17H29O 249.2218, found 249.2210.
1-(3-(4-Octylphenyl)propyl)piperidin-4-ol (RB-024)
Compound RB-024 was prepared from 4 according to a procedure similar to that described for RB-023. Yield = 73%; 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 6.8 Hz, 3H), 1.25–1.30 (m, 10H), 1.55–1.65 (m, 4H), 1.79–1.92 (m, 4H), 2.16–2.20 (m, 2H), 2.40–2.43 (m, 2H), 2.53–2.63 (m, 4H), 2.80–2.83 (m, 2H), 3.67–3.69 (m, 1H), 7.08 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 28.5, 29.3, 29.4, 29.5, 31.6, 31.9, 33.3, 34.0, 35.6, 50.9, 57.9, 128.2, 128.4, 138.9, 140.4; ESI-HRMS (M + H)+m/z calcd for C22H38NO 332.2953, found 332.2951.
1-(4-(4-Octylphenyl)butyl)piperidin-4-ol (RB-025)
Compound RB-025 was prepared from 4-(4-octylphenyl)butan-1-ol (5)22 according to a procedure similar to that described for compound RB-023. Yield = 67% (two steps); 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.9 Hz, 3H), 1.24–1.33 (m, 10H), 1.55–1.68 (m, 4H), 1.80–1.88 (m, 4H), 2.21–2.26 (m, 2H), 2.56 (t, J = 7.8 Hz, 2H), 2.61 (t, J = 7.5 Hz, 2H), 2.71–2.89 (m, 4H), 3.11 (t, J = 9.0 Hz, 2H), 3.97–4.01 (m, 1H), 7.05 (d, J = 8.4 Hz, 2H), 7.09 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 28.9, 29.3, 29.4, 29.5, 31.6, 31.9, 34.9, 35.6, 57.5, 128.2, 128.5, 138.6, 140.7; ESI-HRMS (M + H)+m/z calcd for C23H40NO 346.3110, found 346.3107.
2-(4-(Hex-1-ynyl)phenyl)ethanol (6)
Compound 6 was prepared from 2-(4-bromophenyl)ethanol and 1-hexyne according to a Sonogashira procedure similar to that described for 1. Yield = 60%; 1H NMR (400 MHz, CDCl3) δ 0.95 (t, J = 7.3 Hz, 3H), 1.43–1.52 (m, 2H), 1.55–1.62 (m, 2H), 2.40 (t, J = 7.0 Hz, 2H), 2.85 (t, J = 6.5 Hz, 2H), 3.84 (t, J = 6.5 Hz, 2H), 7.14 (d, J = 8.1 Hz, 2H), 7.34 (t, J = 8.1 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.7, 19.1, 22.0, 29.7, 30.9, 39.0, 63.5, 80.3, 90.2, 122.3, 128.9, 131.7, 137.9; ESI-HRMS (M + H)+m/z calcd for C14H19O 203.1436, found 203.143 33.
2-(4-(Dodec-1-ynyl)phenyl)ethanol (7)
Compound 7 was prepared from 2-(4-bromophenyl)ethanol and 1-decyne according to a procedure similar to that described for 1. Yield = 62%; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.23–1.30 (m, 12H), 1.40–1.45 (m, 2H), 1.59 (quin, J = 7.3 Hz, 2H), 2.38 (t, J = 7.1 Hz, 2H), 2.81 (t, J = 6.6 Hz, 2H), 3.79 (t, J = 6.5 Hz, 2H), 7.11 (d, J = 8.2 Hz, 2H), 7.32 (d, J = 8.1 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 19.4, 22.7, 24.9, 28.8, 29.0, 29.2, 29.4, 29.6, 31.9, 39.0, 63.4, 80.4, 90.2, 122.3, 128.6, 131.4, 138.0; ESI-HRMS (M + H)+m/z calcd for C20H31O 287.2375, found 287.2371.
2-(4-Hexylphenyl)ethanol (8)
Compound 8 was prepared from 6 according to a procedure similar to that described for 2. 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.6 Hz, 3H), 1.22–1.37 (m, 6H), 1.55–1.63 (m, 2H), 2.57 (t, J = 7.8 Hz, 2H), 2.84 (t, J = 6.6 Hz, 2H), 3.84 (t, J = 6.5 Hz, 2H), 7.13 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.6, 29.0, 31.5, 31.7, 35.6, 38.8, 63.8, 128.6, 128.9, 135.5, 141.2; ESI-HRMS (M + H)+m/z calcd for C14H23O 207.1749, found 207.1725.
2-(4-Dodecylphenyl)ethanol (9)
Compound 9 was prepared from 7 according to a procedure similar to that described for 2. 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.23–1.31 (m, 18H), 1.57–1.60 (m, 2H), 2.56 (t, J = 7.8 Hz, 2H), 2.81 (t, J = 6.6 Hz, 2H), 3.81 (t, J = 6.6 Hz, 2H), 7.12 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 29.4, 29.6, 29.7, 31.6, 31.9, 35.6, 38.8, 63.7, 128.6, 128.9, 135.5, 141.2; ESI-HRMS (M + Na)+m/z calcd for C20H34ONa 313.2507, found 313.2502.
1-(4-Methylphenethyl)piperidin-4-ol (RB-026)
Compound RB-026 was prepared from 2-(4-methylphenyl)ethanol according to a procedure similar to that described for RB-023. Yield = 69%; 1H NMR (400 MHz, CDCl3) δ 1.69–1.77 (m, 2H), 1.99–2.04 (m, 2H), 2.31 (s, 3H), 2.48–2.52 (2H), 2.72–2.76 (m, 2H), 2.79 (s, 1H), 2.84–2.88 (m, 2H), 2.98–3.03 (m, 2H), 3.78–3.84 (m, 1H), 7.10 (s, 4H); ESI-HRMS (M + H)+m/z calcd for C14H22NO 220.1701, found 220.1699.
1-(4-Hexylphenethyl)piperidin-4-ol (RB-027)
Compound RB-027 was prepared from 8 according to a procedure similar to that described for RB-023. Yield = 79%; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.5 Hz, 3H), 1.26–1.36 (m, 6H), 1.58 (quin, J = 7.4 Hz, 2H), 1.75–1.80 (m, 2H), 2.08–2.13 (m, 2H), 2.56 (t, J = 7.7 Hz, 2H), 2.74–2.77 (m, 2H), 2.90–2.94 (m, 2H), 3.00–3.04 (m, 2H), 3.86–3.90 (m, 1H), 7.11 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.6, 29.0, 31.5, 31.7, 35.6, 50.1, 59.9, 128.5, 128.6, 141.2; ESI-HRMS (M + H)+m/z calcd for C19H32NO 290.2484, found 290.2478.
1-(4-Dodecylphenethyl)piperidin-4-ol (RB-028)
Compound RB-028 was prepared from 9 according to a procedure similar to that described for RB-023. Yield = 75%; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.6 Hz, 3H), 1.23–1.33 (m, 18H), 1.56–1.60 (m, 2H), 1.67–1.72 (m, 2H), 1.98–2.01 (m, 2H), 2.34–2.39 (m, 2H), 2.56 (t, J = 7.4 Hz, 2H), 2.64–2.68 (m, 2H), 2.81–2.84 (m, 2H), 2.91–2.95 (m, 2H), 3.75–3.79 (m, 1H), 7.10 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 29.4, 29.5, 29.6, 29.7, 31.6, 31.9, 32.8, 35.6, 50.6, 60.3, 128.5, 136.7, 140.9; ESI-HRMS (M + H)+m/z calcd for C25H44NO 374.3423, found 374.3414.
4-Azido-1-(4-methylphenethyl)piperidine (RB-029)
To a solution of RB-026 (115 mg, 0.52 mmol) and triethylamine (0.73 mL, 5.24 mmol) in CH2Cl2 (5 mL) at 0 °C was added methanesulfonyl chloride (0.12 mL, 1.57 mmol). After being stirred at room temperature for 4 h, the reaction mixture was evaporated, diluted with water, and the product was extracted with EtOAc. The extract was washed with brine, evaporated, and dried. To a solution of reaction mixture in 5 mL of DMF was added sodium azide (170 mg, 2.62 mmol). The reaction mixture was stirred at 80 °C for 12 h and then concentrated. The residue was dissolved in EtOAc, and the organic phase was evaporated and dried. Purification by silica gel chromatography, eluting with hexane/EtOAc (1/1), gave 79 mg (62%) of RB-029 as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 1.69–1.78 (m, 2H), 1.96–1.99 (m, 2H), 2.30–2.35 (m, 2H), 2.31 (s, 3H), 2.60–2.65 (m, 2H), 2.76–2.80 (m, 2H), 2.86–2.89 (m, 2H), 3.44–3.48 (m, 1H), 7.09 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 21.0, 29.4, 30.5, 33.0, 50.9, 57.3, 60.4, 128.6, 129.2, 132.4, 135.7, 136.7; ESI-HRMS (M + H)+m/z calcd for C14H21N4 245.1766, found 245.1763.
4-Azido-1-(4-octylphenethyl)piperidine (RB-030)
Compound RB-030 was prepared from RB-005(7) according to a procedure similar to that described for RB-029. Yield = 71%; 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 6.8 Hz, 3H), 1.24–1.31 (m, 10H), 1.58 (quin, J = 7.2 Hz, 2H), 1.67–1.76 (m, 2H), 1.93–1.97 (m, 2H), 2.24–2.29 (m, 2H), 2.54–2.61 (m, 4H), 2.75–2.79 (m, 2H), 2.84–2.90 (m, 2H), 3.40–3.46 (m, 1H), 7.10 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 29.3, 29.4, 29.5, 29.7, 30.3, 30.7, 31.6, 31.9, 33.2, 35.6, 51.1, 57.6, 60.5, 128.5, 137.2, 140.8; ESI-HRMS (M + H)+m/z calcd for C21H35N4 343.2862, found 343.2857.
1-(4-Methylphenethyl)piperidin-4-amine (RB-031)
To a solution of RB-029 (30 mg, 0.12 mmol) in MeOH/CH2Cl2 (1/3, 3 mL) was added 10% Pd/C (50 wt %). The reaction mixture was hydrogenated at room temperature for 12 h. The catalyst was removed by filtration through a pad of Celite, which was rinsed with MeOH/CH2Cl2 (3/1). The residue was washed with EtOAc/hexane (1/1), evaporated, and dried. RB-031 was obtained as a white solid. 1H NMR (400 MHz, CD3OD) δ 1.82–1.90 (m, 2H), 2.17 (d, J = 10.2 Hz, 2H), 2.28 (s, 3H), 2.34 (t, J = 10.6 Hz, 2H), 2.68–2.71 (m, 2H), 2.79–2.83 (m, 2H), 3.13 (d, J = 11.2 Hz, 2H), 3.13–3.20 (m, 1H), 7.07 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 21.0, 30.7, 32.4, 48.2, 51.4, 59.8, 128.6, 129.2, 135.8, 136.0; ESI-HRMS (M + H)+m/z calcd for C14H23N2 219.1861, found 219.1858.
1-(4-Octylphenethyl)piperidin-4-amine (RB-032)
Compound RB-032 was prepared from RB-030 according to a procedure similar to that described for RB-031. 1H NMR (400 MHz, CD3OD) δ 0.91 (t, J = 6.8 Hz, 3H), 1.28–1.34 (m, 10H), 1.61 (quin, J = 6.5 Hz, 2H), 2.06–2.15 (m, 2H), 2.32 (d, J = 13.2 Hz, 2H), 2.60 (t, J = 7.52 Hz, 2H), 3.08–3.14 (m, 2H), 3.24 (t, J = 11.5 Hz, 2H), 3.35–3.38 (m, 2H), 3.51–3.58 (m, 1H), 3.76 (d, J = 11.8 Hz, 2H), 7.17 (d, J = 8.0 Hz, 2H), 7.23 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CD3OD) δ 14.4, 23.7, 30.3, 30.4, 30.6, 31.1, 32.7, 33.0, 36.5, 36.9, 129.8, 129.9, 134.7, 143.2; ESI-HRMS (M + H)+m/z calcd for C21H37N2 317.2957, found 317.2951.
1-(4-Dodecylphenethyl)piperidin-4-amine (RB-033)
To a solution of RB-028 (20 mg, 0.050 mmol) and triethylamine (70 μL, 0.54 mmol) in CH2Cl2 (3 mL) at 0 °C was added methanesulfonyl chloride (10 μL, 0.15 mmol). After being stirred at room temperature for 4 h, the reaction mixture was evaporated, diluted with water, and the product was extracted with EtOAc. The extract was washed with brine, evaporated, and dried. To a solution of the reaction mixture in 3 mL of DMF was added sodium azide (10 mg, 0.16 mmol). The reaction mixture was stirred at 100 °C for 12 h and then concentrated. The residue was dissolved in EtOAc, and the organic phase was evaporated and dried. To a solution of residue in MeOH/CH2Cl2 (3/1, 3 mL) was added 10% Pd/C (50 wt %). The reaction mixture was hydrogenated at room temperature for 12 h. The catalyst was removed by filtration through a pad of Celite, which was rinsed with MeOH/CH2Cl2 (1/3). The residue was washed with EtOAc/hexane (1/1), evaporated, and dried, affording RB-033 as a white solid. 1H NMR (400 MHz, CD3OD) δ 0.89 (t, J = 6.9 Hz, 3H), 1.27–1.32 (m, 18H), 1.58–1.62 (m, 2H), 2.00–2.10 (m, 2H), 2.29 (d, J = 13.2, 2H), 2.58–2.62 (m, 2H), 3.05–3.09 (m, 2H), 3.15 (d, J = 13.6 Hz, 2H), 3.28–3.33 (m, 2H), 3.46–3.54 (m, 1H), 3.71 (d, J = 10.9 Hz, 2H), 7.17 (d, J = 7.9 Hz, 2H), 7.22 (d, J = 7.9 Hz, 2H); 13C NMR (100 MHz, CD3OD) δ 14.5, 23.8, 29.9, 30.3, 30.5, 30.6, 30.7, 30.8, 30.9, 31.3, 32.8, 33.1, 36.5, 36.9, 129.9, 130.1, 133.1, 143.1; ESI-HRMS (M + H)+m/z calcd for C25H45N2 373.3583, found 373.3576.
4-Fluoro-1-(4-octylphenethyl)piperidine (RB-034)
To a solution of RB-005 (12 mg, 0.040 mmol) in CH2Cl2 (3 mL) at 0 °C was added diethylaminosulfur trifluoride (DAST, 15 μL, 0.12 mmol). After being stirred at room temperature for 5 h, the reaction mixture was diluted with water, and the product was extracted with EtOAc. The extract was washed with brine, evaporated, and dried. Purification by silica gel chromatography, eluting with CH2Cl2/MeOH (10:1), gave 11 mg (90%) of RB-034 as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 6.8 Hz, 3H), 1.25–1.30 (m, 10H), 1.54–1.62 (m, 2H), 1.90–2.00 (m, 4H), 2.48–2.63 (m, 6H), 2.66–2.71 (m, 2H), 2.76–2.80 (m, 2H), 4.61–4.66 (m, 0.5H), 4.74–4.78 (m, 0.5H), 7.10 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 29.3, 29.4, 29.5, 29.7, 33.2, 35.6, 49.4, 60.5, 128.5, 137.2, 140.8; ESI-HRMS (M + H)+m/z calcd for C21H35FN 320.2754, found 320.2751.
1-(4-Octylphenethyl)piperidin-4-one (RB-035)
To a solution of RB-005 (25 mg, 0.080 mmol) in CH2Cl2 (3 mL) at 0 °C was added pyridinium chlorochromate (PCC, 25 mg, 0.12 mmol). After being stirred at room temperature for 4 h, the reaction mixture was diluted with water, and the product was extracted with EtOAc. The extract was washed with brine, evaporated, and dried. Purification by silica gel chromatography, eluting with CH2Cl2/MeOH (3:1), gave 17 mg (70%) of RB-035 as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 6.6 Hz, 3H), 1.22–1.30 (m, 10H), 1.58–1.60 (m, 2H), 2.47–2.52 (m, 4H), 2.57 (t, J = 7.7 Hz, 2H), 2.72–2.74 (m, 2H), 2.81–2.86 (m, 6H), 7.12 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 29.3, 29.4, 29.5, 29.7, 31.6, 31.9, 33.7, 35.6, 36.0, 41.2, 53.1, 59.4, 128.6, 137.0, 141.0, 178.0; ESI-HRMS (M + H)+m/z calcd for C21H34 NO 316.2640, found 316.2635.
4-Methoxy-1-(4-octylphenethyl)piperidine (RB-036)
To a solution of 11(7) (17 mg, 0.050 mmol) in MeCN (3 mL) was added 4-methoxypiperidine (19 mg, 0.16 mmol). The reaction mixture was stirred at 50 °C for 12 h. The solvent was evaporated and the residue was purified by silica gel chromatography, eluting with CH2Cl2/MeOH (5:1), to give 14 mg (79%) of RB-036 as a yellow liquid. 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 6.5 Hz, 3H), 1.23–1.31 (m, 10H), 1.56–1.61 (m, 2H), 1.67–1.72 (m, 2H), 1.95–2.00 (m, 2H), 2.30–2.37 (m, 2H), 2.56 (t, J = 7.7 Hz, 2H), 2.62–2.64 (m, 2H), 2.79–2.85 (m, 4H), 3.25–3.30 (m, 1H), 3.34 (s, 3H), 7.10 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 29.3, 29.4, 29.5, 31.6, 31.9, 35.6, 50.8, 55.6, 60.5, 128.5, 128.6, 140.8; ESI-HRMS (M + H)+m/z calcd for C22H38NO 332.2953, found 332.2948.
(S)-1-(4-Octylphenethyl)pyrrolidin-3-ol (RB-037)
To a solution of 11 (20 mg, 0.064 mmol) in MeCN (4 mL) was added K2CO3 (44 mg, 0.32 mmol) at room temperature. After the suspension was stirred for 10 min, (S)-pyrrolidine-3-ol hydrochloride (79 mg, 0.64 mmol) was added. The reaction mixture was stirred at 50 °C for 12 h. The reaction mixture was diluted with water, and the product was extracted with EtOAc. The extract was washed with brine, dried, and evaporated. Purification by silica gel chromatography, eluting with CH2Cl2/MeOH (3:1), gave 17 mg (86%) of RB-037 as a yellow liquid. 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.22–1.32 (m, 10H), 1.58 (quin, J = 7.3 Hz, 2H), 1.85 (quin, J = 6.7 Hz, 1H), 2.19–2.28 (m, 1H), 2.46–2.54 (m, 2H), 2.56 (t, J = 7.8 Hz, 2H), 2.68 (dd, J = 5.1, 10.4 Hz, 1H), 2.78–2.88 (m, 4H), 2.91 (d, J = 10.4 Hz, 1H), 3.07–3.13 (m, 1H), 4.38–4.41 (m, 1H), 7.12 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 29.3, 29.4, 29.5, 31.6, 31.9, 34.3, 34.7, 35.6, 52.6, 57.8, 62.9, 71.1, 128.5, 128.6, 136.5, 141.0; ESI-HRMS (M + H)+m/z calcd for C20H34NO 304.2640, found 304.2639.
(R)-1-(4-Octylphenethyl)pyrrolidin-3-ol (RB-038)
Compound RB-038 was prepared from 11 according to a coupling procedure similar to that described for RB-036, using (R)-pyrrolidine. Yield = 72%; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.7 Hz, 3H), 1.26–1.29 (m, 10H), 1.56–1.59 (m, 2H), 1.94–2.01 (m, 1H), 2.22–2.31 (m, 1H), 2.56 (t, J = 7.9 Hz, 2H), 2.74–2.76 (m, 1H), 2.91–2.99 (m, 4H), 3.14–3.24 (m, 2H), 3.30–3.35 (m, 1H), 4.47–4.50 (m, 1H), 7.12 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.6, 29.3, 29.4, 29.5, 31.5, 31.9, 33.4, 34.2, 35.6, 52.9, 57.9, 62.7, 70.4, 128.5, 128.7, 135.2, 141.5; ESI-HRMS (M + H)+m/z calcd for C20H34NO 304.2640, found 304.2637.
(R)-(1-(4-Methylphenethyl)pyrrolidin-2-yl)methanol (RB-039)
Compound RB-039 was prepared from 2-(4-methylphenyl)ethanol according to a coupling procedure similar to that described for RB-037, using d-prolinol. Yield = 62%; 1H NMR (400 MHz, CDCl3) δ 1.84–1.91 (m, 1H), 1.97–1.21 (m, 1H), 2.03–2.13 (m, 2H), 2.31 (s, 3H), 2.83–2.89 (m, 1H), 2.96–3.08 (m, 2H), 3.13–3.21 (m, 1H), 3.34–3.41 (m, 1H), 3.49–3.56 (m, 1H), 3.72–3.77 (m, 1H), 3.85 (d, J = 5.3 Hz, 2H), 7.12 (dd, J = 1.9, 8.4 Hz, 4H); 13C NMR (100 MHz, CDCl3) δ 21.0, 23.6, 26.5, 32.0, 39.4, 50.7, 55.0, 57.9, 61.1, 69.5, 128.6, 129.5, 133.7, 136.7; ESI-HRMS (M + H)+m/z calcd for C14H22NO 220.1701, found 220.1698.
(R)-(1-(4-Octylphenethyl)pyrrolidin-2-yl)methanol (RB-040)
Compound RB-040 was prepared from 11 according to a coupling procedure similar to that described for RB-037, using d-prolinol. Yield = 59%; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.22–1.31 (m, 10H), 1.54–1.62 (m, 2H), 1.84–2.05 (m, 4H), 2.56 (t, J = 7.8 Hz, 2H), 2.64–2.70 (m, 1H), 2.85–3.00 (m, 3H), 3.08–3.13 (m, 1H), 3.32–3.36 (m, 1H), 3.57–3.62 (m, 1H), 3.67 (dd, J = 5.5, 13.7, Hz, 1H), 3.78 (dd, J = 3.2, 12.2 Hz, 1H), 7.11 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 23.8, 26.9, 29.3, 29.5, 29.7, 31.0, 31.5, 31.9, 35.6, 54.5, 56.9, 61.4, 66.3, 128.5, 128.7, 135.5, 141.5; ESI-HRMS (M + H)+m/z calcd for C21H36NO 318.2797, found 318.2792.
(S)-(1-(4-Octylphenethyl)pyrrolidin-2-yl)methanol (RB-041)
Compound RB-041 was prepared from 11 according to a coupling procedure similar to that described for RB-037, using l-prolinol. Yield = 54%; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.7 Hz, 3H), 1.22–1.31 (m, 10H), 1.58 (quin, J = 7.3 Hz, 2H), 1.80–1.96 (m, 4H), 2.52–2.55 (m, 1H), 2.56 (t, J = 7.8 Hz, 2H), 2.73 (td, J = 11.6, 2.7 Hz, 1H), 2.86–2.96 (m, 3H), 3.18–3.25 (m, 1H), 3.47 (quin, J = 4.7 Hz, 1H), 3.56 (dd, J = 4.4, 11.7 Hz, 1H), 3.70 (dd, J = 3.2, 11.8 Hz, 1H), 7.11 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 23.8, 27.1, 29.3, 29.4, 29.5, 29.7, 31.5, 31.9, 33.7, 35.6, 54.3, 57.0, 61.5, 66.9, 128.5, 128.8, 135.8, 141.2; ESI-HRMS (M + H)+m/z calcd for C21H36NO 318.2797, found 318.2791.
(R)-(1-(4-Dodecylphenethyl)pyrrolidin-2-yl)methanol (RB-042)
Compound RB-042 was prepared from 9 according to a coupling procedure similar to that described for RB-037, using d-prolinol. Yield = 62%; 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 6.6 Hz, 3H), 1.22–1.31 (m, 18H), 1.56–1.60 (m, 2H), 2.00–2.16 (m, 4H), 2.56 (t, J = 7.7 Hz, 2H), 2.82–2.88 (m, 1H), 3.03–3.09 (m, 2H), 3.22–3.30 (m, 1H), 3.35–3.39 (m, 1H), 3.46–3.53 (m, 1H), 3.75–3.80 (m, 1H), 3.86 (dd, J = 6.5, 12.7 Hz, 1H), 3.95 (dd, J = 2.1, 12.0 Hz, 1H), 7.12 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 24.0, 26.5, 29.3, 29.4, 29.5, 29.6, 29.7, 31.5, 31.8, 31.9, 35.5, 54.7, 58.0, 60.9, 70.2, 128.5, 128.9, 133.6, 142.0; ESI-HRMS (M + H)+m/z calcd for C25H44NO 374.3423, found 374.3418.
(S)-(1-(4-Dodecylphenethyl)pyrrolidin-2-yl)methanol (RB-043)
Compound RB-043 was prepared from 9 according to a coupling procedure similar to that described for RB-037, using l-prolinol. Yield = 55%; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.6 Hz, 3H), 1.23–1.30 (m, 18H), 1.56–1.62 (m, 2H), 1.93–2.11 (m, 4H), 2.56 (t, J = 7.8 Hz, 2H), 2.98–3.06 (m, 2H), 3.14–3.22 (m, 1H), 3.28–3.32 (m, 1H), 3.41–3.49 (m, 1H), 3.65–3.68 (m, 1H), 3.71–3.73 (m, 1H), 3.79–3.83 (m, 1H), 3.89 (dd, J = 2.5, 12.0 Hz, 1H), 7.12 (s, 4H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 23.9, 26.8, 29.4, 29.5, 29.6, 29.7, 31.5, 31.9, 35.6, 54.5, 57.5, 61.1, 70.1, 128.5, 128.8, 134.1, 141.7; ESI-HRMS (M + H)+m/z calcd for C25H44NO 374.3423, found 374.3415.
4-(Oct-1-ynyl)benzoic Acid (12)
To a deaerated solution of 4-iodobenzoic acid (500 mg, 2.02 mmol), bis(triphenylphosphine)palladium dichloride (116 mg, 0.10 mmol), and copper(I) iodide (19 mg, 0.10 mmol) in anhydrous triethylamine (15 mL) was added 1-octyne (0.89 mL, 6.05 mmol) at room temperature. The reaction mixture was heated at 60 °C for 12 h. After saturated aqueous ammonium chloride solution was added, the product was extracted with EtOAc. The combined solution was washed with water, brine, and dried. Purification by silica gel chromatography, eluting with hexane/EtOAc (3:1), gave 395 mg (85%) of alkyne 12 as a yellow liquid. 1H NMR (400 MHz, CDCl3) δ 0.90 (t, J = 6.7 Hz, 3H), 1.31–1.35 (m, 4H), 1.43–1.50 (m, 2H), 1.58–1.66 (m, 2H), 2.43 (t, J = 7.1 Hz, 2H), 7.47 (d, J = 8.4 Hz, 2H), 8.02 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.8, 19.3, 22.3, 28.3, 31.1, 79.8, 94.4, 127.6, 129.6, 129.7, 131.3, 171.8; ESI-HRMS (M – H)−m/z calcd for C15H17O2– 229.1234, found 229.1237.
4-Octylbenzoic Acid (13)
Compound 12 (300 mg, 1.30 mmol) was dissolved in EtOAc (10 mL), and 10% Pd/C (150 mg, 50 wt %) was added. The reaction mixture was hydrogenated at room temperature for 12 h. The catalyst was removed by filtration through a pad of Celite, which was rinsed with EtOAc, affording 13 as a yellow solid without purification. 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.22–1.32 (m, 10H), 1.61–1.64 (m, 2H), 2.64 (t, J = 7.7 Hz, 2H), 7.25 (d, J = 8.1 Hz, 2H), 8.02 (d, J = 8.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 29.3, 29.4, 31.1, 31.9, 36.1, 126.9, 128.6, 130.3, 149.6, 172.6; ESI-HRMS (M–H)−m/z calcd for C15H21O2– 233.1547, found 233.1548.
(R)-(4-Dodecylphenyl)-(2-(hydroxymethyl)pyrrolidin-1-yl)methanone (RB-044)
To a solution of 4-(n-dodecyl)benzoic acid (10 mg, 0.034 mmol) in CH2Cl2 (3 mL) was added thionyl chloride (0.25 mL, 0.34 mmol) at room temperature. The reaction mixture was heated at reflux for 12 h. The reaction mixture was diluted with water, and the product was extracted with EtOAc. The extract was washed with brine, dried, and evaporated. To a solution of residue in MeCN (3 mL) was added K2CO3 (24 mg, 0.17 mmol) at room temperature. After the suspension was stirred for 10 min, d-prolinol (10 mg, 0.10 mmol) was added. The reaction mixture was stirred at 50 °C for 12 h. The reaction mixture was diluted with water, and the product was extracted with EtOAc. The extract was washed with brine, dried, and evaporated. Purification by silica gel chromatography, eluting with CH2Cl2/MeOH (10:1), gave 9 mg (72%) of RB-044 as a yellow liquid. 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.6 Hz, 3H), 1.23–1.34 (m, 18H), 1.59–1.64 (m, 4H), 1.70–1.77 (m, 1H), 1.84–1.89 (m, 1H), 2.14–2.21 (m, 1H), 2.62 (t, J = 7.6 Hz, 2H), 3.46–3.59 (m, 2H), 3.71–3.82 (m, 2H), 4.39–4.44 (m, 1H), 7.20 (d, J = 7.9 Hz, 2H), 7.43 (d, J = 7.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 25.1, 28.6, 29.2, 29.3, 29.4, 29.5, 29.6, 29.7, 31.3, 31.9, 35.8, 51.3, 61.6, 67.6, 127.2, 128.3, 133.8, 145.5, 172.5; ESI-HRMS (M + H)+m/z calcd for C24H40NO2 374.3059, found 374.3055.
(S)-(4-Dodecylphenyl)(2-(hydroxymethyl)pyrrolidin-1-yl)methanone (RB-045)
Compound RB-045 was prepared from 4-(n-dodecyl)benzoic acid according to a coupling procedure similar to that described for RB-044, using l-prolinol instead of d-prolinol. Yield = 65%; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.7 Hz, 3H), 1.23–1.30 (m, 18H), 1.58–1.64 (m, 4H), 1.70–1.77 (m, 1H), 1.84–1.89 (m, 1H), 2.14–2.21 (m, 1H), 2.62 (t, J = 7.7 Hz, 2H), 3.47–3.59 (m, 2H), 3.71–3.82 (m, 2H), 4.38–4.44 (m, 1H), 7.20 (d, J = 8.0 Hz, 2H), 7.43 (d, J = 7.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 25.1, 28.6, 29.2, 29.4, 29.5, 29.6, 29.7, 31.2, 31.9, 35.8, 51.3, 61.6, 67.5, 127.2, 128.3, 133.8, 145.5, 172.5; ESI-HRMS (M + H)+m/z calcd for C24H40NO2 374.3059, found 374.3058.
(4-Hydroxypiperidin-1-yl)(4-octylphenyl)methanone (RB-046)
Compound RB-046 was prepared from 4-(n-dodecyl)benzoic acid according to a coupling procedure similar to that described for RB-044, using 4-hydroxypiperidine. Yield = 70%; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.7 Hz, 3H), 1.26–1.30 (m, 10H), 1.51–1.62 (m, 4H), 1.80–1.97 (m, 2H), 2.63 (t, J = 7.7 Hz, 2H), 3.21–3.36 (m, 2H), 3.67–3.76 (m, 1H), 3.93–4.00 (m, 1H), 4.18–4.26 (m, 1H), 7.19 (d, J = 8.2 Hz, 2H), 7.31 (d, J = 8.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 29.2, 29.3, 29.4, 31.3, 35.8, 67.4, 126.9, 128.5, 130.2, 133.2, 144.8, 170.7; ESI-HRMS (M + H)+m/z calcd for C20H32NO2 318.2428, found 318.2432.
(4-Dodecylphenyl)(4-hydroxypiperidin-1-yl)methanone (RB-047)
Compound RB-047 was prepared from 4-(n-dodecyl)benzoic acid according to a coupling procedure similar to that described for RB-044, using 4-hydroxypiperidine. Yield = 81%; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.7 Hz, 3H), 1.22–1.37 (m, 18H), 1.58–1.63 (m, 4H), 1.84–1.95 (m, 2H), 2.61 (t, J = 7.7 Hz, 2H), 3.21–3.34 (m, 2H), 3.67–3.75 (m, 1H), 3.95 (sep, J = 3.9 Hz, 1H), 4.18–4.23 (m, 1H), 7.19 (d, J = 7.9 Hz, 2H), 7.30 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.2, 22.7, 29.3, 29.4, 29.5, 29.6, 29.7, 31.3, 31.9, 35.8, 67.2, 126.9, 128.5, 133.1, 144.9, 170.8; ESI-HRMS (M + H)+m/z calcd for C24H40NO2 374.3059, found 374.3055.
4-Octyl-N-(pyridin-4-ylmethyl)benzamide (RB-048)
Compound RB-048 was prepared from 4-(n-dodecyl)benzoic acid according to a coupling procedure similar to that described for RB-044, using 4-(aminomethyl)pyridine. Yield = 69%; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.07–1.30 (m, 10H), 1.59–1.63 (m, 2H), 2.65 (t, J = 7.7 Hz, 2H), 4.63 (d, J = 6.0 Hz, 2H), 6.97 (t, J = 5.6 Hz, NH), 7.24 (d, J = 8.0 Hz, 4H), 7.74 (d, J = 8.2 Hz, 2H), 8.51–8.56 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 21.1, 22.7, 29.2, 29.4, 29.7, 31.2, 31.8, 35.8, 42.7, 60.4, 127.1, 128.5, 128.7, 131.2, 147.4, 147.8, 149.9, 167.7, 171.2; ESI-HRMS (M + H)+m/z calcd for C21H29N2O 325.2274, found 325.2277.
N-(4-Hydroxyphenyl)-4-octylbenzamide (RB-049)
Compound RB-049 was prepared from 4-(n-dodecyl)benzoic acid according to a coupling procedure similar to that described for RB-044, using 4-aminophenol. Yield = 55%; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.11–1.29 (m, 10H), 1.54–1.60 (m, 2H), 2.67 (t, J = 7.1 Hz, 2H), 6.83 (d, J = 7.8 Hz, 2H), 7.28 (d, J = 7.8 Hz, 2H), 7.43 (d, J = 6.8 Hz, 2H), 7.80 (d, J = 6.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.3, 22.9, 27.6, 29.6, 29.7, 30.0, 30.4, 31.6, 32.2, 36.2, 123.2, 127.5, 129.0, 132.5, 147.6, 167.8; ESI-HRMS (M + H)+m/z calcd for C21H28NO2 326.2115, found 326.2118.
N-(4-(2-Hydroxyethyl)phenyl)-4-octylbenzamide (RB-050)
Compound RB-050 was prepared from 4-(n-dodecyl)benzoic acid according to a coupling procedure similar to that described for RB-044, using 2-(4-aminophenyl)ethanol. Yield = 73%; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.24–1.32 (m, 10H), 1.51–1.60 (m, 2H), 2.67 (t, J = 7.7 Hz, 2H), 2.87 (t, J = 6.5 Hz, 2H), 3.86 (t, J = 6.5 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 7.29 (d, J = 8.1 Hz, 2H), 7.58 (d, J = 8.4 Hz, 2H), 7.76 (br s, NH), 7.78 (d, J = 8.1 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 27.3, 29.2, 29.4, 29.7, 31.2, 31.9, 38.6, 63.7, 120.5, 127.0, 128.9, 129.7, 132.3, 134.6, 136.5, 147.4, 165.7; ESI-HRMS (M + Na)+m/z calcd for C23H31NO2Na 376.2247, found 376.2251.
1-Bromo-4-(oct-1-ynyl)benzene (14)
Compound 14 was prepared from 1-bromo-4-iodobenzene according to a procedure similar to that described for 1. Yield = 70%; 1H NMR (400 MHz, CDCl3) δ 0.90 (t, J = 7.1 Hz, 3H), 1.29–1.34 (m, 4H), 1.40–1.47 (m, 2H), 1.59 (quin, J = 7.3 Hz, 2H), 2.37 (t, J = 7.1 Hz, 2H), 7.24 (dt, J = 8.3, 2.0 Hz, 2H), 7.39 (dt, J = 8.6, 2.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 19.5, 22.6, 28.8, 31.4, 79.6, 91.8, 121.5, 123.1, 131.4, 133.0; ESI-HRMS (M)+m/z calcd for C14H17Br 264.0514, found 264.0508.
4-(4-Octylphenethyl)pyridine (RB-051)
To a deaerated solution of 14 (100 mg, 0.38 mmol), bis(triphenylphosphine)palladium dichloride (22 mg, 0.010 mmol), and copper(I) iodide (4 mg, 10 μmol) in anhydrous triethylamine (8 mL) was added 3-ethynylpyridine (78 mg, 0.75 mmol) at room temperature. The reaction mixture was heated at 80 °C for 3 days. After saturated ammonium chloride solution was added, the product was extracted with EtOAc. The combined solution was washed with water, brine and dried. The catalyst was removed by filtration through a pad of Celite, which was rinsed with hexanes/EtOAc (3:1). 3-((4-(Oct-1-ynyl)phenyl)ethynyl)pyridine (15) (53 mg, 0.18 mmol) was dissolved in EtOAc (8 mL), and 10% Pd/C (53 mg, 100 wt %) was added. The reaction mixture was hydrogenated at room temperature for 12 h. The catalyst was removed by filtration through a pad of Celite, which was rinsed with EtOAc. Flash column chromatography with hexanes/EtOAc (1:1) as eluent gave RB-051 (48 mg, 88%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.27–1.34 (m, 10H), 1.59 (quin, J = 7.3 Hz, 2H), 2.56 (t, J = 7.8 Hz, 2H), 2.86–2.92 (m, 4H), 7.05 (d, J = 8.1 Hz, 2H), 7.09 (d, J = 8.1 Hz, 2H), 7.17 (dd, J = 4.8, 7.7 Hz, 1H), 7.43 (dt, J = 7.8, 1.8 Hz, 1H), 8.42–8.44 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 14.2, 22.7, 29.3, 29.4, 29.5, 31.6, 31.9, 35.0, 35.6, 37.1, 123.2, 128.3, 128.5, 135.9, 137.0, 140.8, 147.5, 150.0; ESI-HRMS (M + H)+m/z calcd for C21H30N 296.2378, found 296.2374.
1-Methyl-4-(4-octylphenethyl)pyridinium Iodide (RB-052)
To a solution of RB-051 (32 mg, 0.11 mmol) in MeCN (5 mL) was added K2CO3 (75 mg, 0.54 mmol). After the suspension was stirred at room temperature for 10 min, MeI (30 μL, 0.54 mmol) was added. The reaction mixture was stirred overnight at room temperature. The reaction mixture was diluted with water, and the product was extracted with EtOAc. The extract was washed with brine, dried, and evaporated. The residue was washed with hexane to give 38 mg (80%) of RB-052 as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.9 Hz, 3H), 1.26–1.30 (m, 10H), 1.56 (quin, J = 7.3 Hz, 2H), 2.54 (t, J = 7.8 Hz, 2H), 3.02 (t, J = 7.6 Hz, 2H), 3.17 (t, J = 7.6 Hz, 2H), 4.60 (s, 3H), 7.07 (s, 4H), 7.92 (dd, J = 6.2, 7.7 Hz, 1H), 8.06 (d, J = 8.0 Hz, 1H), 9.12 (d, J = 6.5 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 29.3, 29.4, 29.5, 31.6, 31.9, 34.4, 35.5, 35.7, 49.2, 127.6, 128.6, 128.7, 136.1, 141.4, 142.8, 143.0, 145.1, 145.2; ESI-HRMS (M)+m/z calcd for C22H32N+ 310.2535, found 310.2533.
1-(Bromomethyl)-4-(oct-1-ynyl)benzene (16)
Compound 16 was prepared from 4-iodobenzyl bromide according to a procedure similar to that described for 1. Yield = 69%; 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 6.9 Hz, 3H), 1.27–1.30 (m, 8H), 2.38 (t, J = 6.8 Hz, 2H), 4.44 (s, 2H), 7.30 (d, J = 8.0 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 19.3, 22.4, 28.4, 28.7, 31.2, 33.3, 80.0, 91.2, 124.3, 128.9, 131.8, 136.9; ESI-HRMS (M + H)+m/z calcd for C15H20Br 279.0748, found 279.0730.
4-(4-Methylpiperidin-1-yl)-1-(4-(oct-1-ynyl)benzyl)pyridinium Bromide (18)27
To a Pyrex vessel charged with a magnetic stirring bar was added a suspension of 4-chloropyridine hydrochloride (0.20 g, 1.3 mmol) in dry acetonitrile (4 mL). N,N-Diisopropylethylamine (DIPEA, 0.7 mL, 4.0 mmol) was added, followed by 4-methylpiperidine (0.16 mL, 1.3 mmol). The reactor was placed in a microwave apparatus and irradiated at 160 °C for 1 h. After the reaction mixture was cooled to room temperature, EtOAc was added, and the solution was washed with water and brine, dried (Na2SO4), and concentrated in vacuo. Ether (3 mL) was added to the resulting crude oil, and the inorganic precipitate was removed by filtration. Evaporation of the solvent gave 4-(4-methylpiperidin-1-yl)pyridine (17). A solution of 16 (100 mg, 0.35 mmol) and 17 (124 mg, 0.71 mmol) in 5 mL of 2-butanone was placed in a sealed tube. The reaction mixture was stirred at 100 °C for 3 days and concentrated. The residue was washed with EtOAc to give 125 mg (78%) of 18 as a slightly yellow solid. 1H NMR (400 MHz, CDCl3) δ 0.90 (t, J = 6.9 Hz, 3H), 0.98 (d, J = 6.4 Hz, 3H), 1.15–1.32 (m, 6H), 1.40–1.47 (m, 2H), 1.59 (quin, J = 7.28 Hz, 2H), 1.74–1.79 (m, 1H), 1.84 (d, J = 13.2 Hz, 2H), 2.39 (t, J = 7.1 Hz, 2H), 3.12 (td, J = 12.9, 2.1 Hz, 2H), 4.04 (d, J = 13.4 Hz, 2H), 5.60 (s, 2H), 6.95 (d, J = 7.1 Hz, 2H), 7.36 (d, J = 8.1 Hz, 2H), 7.43 (d, J = 8.1 Hz, 2H), 8.56 (d, J = 7.1 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.2, 19.4, 21.3, 22.6, 28.6, 29.7, 30.5, 31.3, 33.5, 47.4, 60.3, 79.8, 92.2, 108.4, 125.3, 128.9, 132.4, 133.1, 143.0, 155.2; ESI-HRMS (M + H)+m/z calcd for C26H36N2+ 376.2873, found 376.2871.
4-(4-Methylpiperidin-1-yl)-1-(4-octylbenzyl)pyridinium Bromide (RB-053)
Compound 18 (10 mg, 0.02 mmol) was dissolved in MeOH/CH2Cl2 (1/3, 3 mL), and 10% Pd/C (10 mg, 100 wt %) was added. The reaction mixture was hydrogenated at room temperature for 12 h. The catalyst was removed by filtration through a pad of Celite, which was rinsed with CH2Cl2 and concentrated. The residue was washed with hexane to give 9 mg (87%) of RB-053 as a white solid. 1H NMR (400 MHz, CDCl3) δ 0.86 (t, J = 7.0 Hz, 3H), 0.98 (d, J = 6.4 Hz, 3H), 1.16–1.33 (m, 10H), 1.55–1.60 (m, 4H), 1.72–1.78 (m, 1H), 1.84 (d, J = 13.8 Hz, 2H), 2.58 (d, J = 7.6 Hz, 2H), 3.13 (td, J = 12.5, 2.4 Hz, 2H), 4.08 (d, J = 13.5 Hz, 2H), 5.47 (s, 2H), 7.02 (d, J = 7.6 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 8.46 (d, J = 7.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 21.3, 22.7, 29.2, 29.3, 29.4, 29.7, 30.3, 30.5, 31.4, 31.9, 33.5, 35.7, 47.5, 60.8, 108.5, 128.8, 129.5, 130.9, 142.8, 144.5, 155.2; ESI-HRMS (M)+m/z calcd for C26H39N2+ 379.3113, found 379.3108.
4-(Oct-1-ynyl)aniline (19)
Compound 19 was prepared from 4-iodoaniline according to a procedure similar to that described for 1. Yield = 62%; 1H NMR (400 MHz, CDCl3) δ 0.90 (t, J = 7.0 Hz, 3H), 1.26–1.35 (m, 4H), 1.40–1.47 (m, 2H), 1.58 (quin, J = 7.4 Hz, 2H), 2.37 (t, J = 7.1 Hz, 2H), 6.57 (d, J = 8.6 Hz, 2H), 7.19 (d, J = 8.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 19.5, 22.6, 28.6, 29.0, 31.4, 80.7, 87.9, 113.7, 114.8, 132.7, 145.9; ESI-HRMS (M + H)+m/z calcd for C14H20N 202.1596, found 202.1589.
3-(1-(4-(Oct-1-ynyl)phenyl)-1H-1,2,3-triazol-4-yl)pyridine (21)28
To a solution of 19 (157 mg, 0.78 mmol) in 2 mL of 10% aqueous HCl was added NaNO2 (65 mg, 0.94 mmol) in 1 mL of water at 0 °C. After the solution was stirred for 30 min, NaN3 (61 mg, 0.94 mmol) in 1 mL of water was added at 0 °C, with stirring for another hour. The reaction mixture was warmed to 25 °C, diluted with EtOAc, washed with water and brine, dried (Na2SO4), and concentrated in vacuo, affording 20. Without purification, 20 (43 mg, 0.19 mmol) and 3-ethynylpyridine (39 mg, 0.38 mmol) were dissolved in t-BuOH/H2O (3 mL, 1:1), and CuSO4 (30 mg, 0.19 mmol) and sodium ascorbate (37 mg, 0.19 mmol) were added at room temperature. The reaction mixture was stirred for 2 days and then was diluted with EtOAc and washed with brine. The aqueous layer was extracted with EtOAc. The combined organic layers were dried (MgSO4) and concentrated in vacuo. Purification by silica gel chromatography, eluting with hexanes/EtOAc (1:1), gave 50 mg (67%, two steps) of 21 as a white solid. 1H NMR (400 MHz, CDCl3) δ 0.92 (t, J = 6.9 Hz, 3H), 1.29–1.38 (m, 4H), 1.47 (quin, J = 7.3 Hz, 2H), 1.63 (quin, J = 7.3 Hz, 2H), 2.44 (t, J = 7.1 Hz, 2H), 7.41 (dd, J = 4.8, 7.9 Hz, 1H), 7.57 (d, J = 8.6 Hz, 2H), 7.74 (d, J = 8.6 Hz, 2H), 8.27–8.30 (m, 2H), 8.61–8.63 (m, 1H), 9.08 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.1, 19.5, 22.6, 28.5, 28.6, 31.4, 79.3, 93.0, 117.8, 120.2, 123.1, 123.9, 125.2, 126.4, 133.0, 133.3, 135.6, 139.5, 145.4, 147.1, 149.6, 153.2, ESI-HRMS (M + H)+m/z calcd for C21H23N4 331.1923, found 331.1919.
3-(1-(4-Octylphenyl)-1H-1,2,3-triazol-4-yl)pyridine (RB-054)
Compound RB-054 was prepared from 21 according to a procedure similar to that described for 2. Yield = 82%; 1H NMR (400 MHz, CDCl3) δ 0.89 (t, J = 6.8 Hz, 3H), 1.25–1.35 (m, 10H), 1.62–1.68 (m, 2H), 2.69 (t, J = 7.7 Hz, 2H), 7.36 (d, J = 8.3 Hz, 2H), 7.42 (dd, J = 4.8, 7.8 Hz, 1H), 7.69 (d, J = 8.3 Hz, 2H), 8.25 (s, 1H), 8.30 (d, J = 7.9 Hz, 1H), 8.61–8.63 (m, 1H), 9.08 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.7, 29.2, 29.3, 29.4, 29.7, 31.4, 31.9, 35.5, 118.1, 120.6, 123.9, 126.6, 129.8, 133.2, 134.7, 144.4, 145.2, 147.1, 149.4; ESI-HRMS (M + H)+m/z calcd for C21H27N4 335.2236, found 335.2232.
4-(4-Octyl-1H-1,2,3-triazol-1-yl)phenol (RB-055)
To a solution of 4-aminophenol (100 mg, 0.92 mmol) in 2 mL of 10% aqueous HCl was added NaNO2 (76 mg, 1.10 mmol) in 1 mL of water at 0 °C. After the solution was stirred for 30 min, NaN3 (72 mg, 1.10 mmol) in 1 mL of water was added at 0 °C, with stirring for another hour. The reaction mixture was warmed to 25 °C, diluted with EtOAc, washed with water and brine, dried (Na2SO4), and concentrated in vacuo, affording 4-azidophenol (22). Without purification, 22 (20 mg, 0.15 mmol) and 1-decyne (61 mg, 0.44 mmol) were dissolved in t-BuOH/H2O (5 mL, 1:1), and CuSO4 (35 mg, 0.22 mmol) and sodium ascorbate (44 mg, 0.22 mmol) were added at room temperature. The reaction mixture was stirred for 2 days and then was diluted with EtOAc and washed with brine. The aqueous layer was extracted with EtOAc. The combined organic layers were dried (MgSO4) and concentrated in vacuo. Purification by silica gel chromatography, eluting with CH2Cl2/MeOH (10:1), gave 26 mg (70%, two steps) of RB-055 as a white solid. 1H NMR (400 MHz, CDCl3) δ 0.86 (t, J = 6.7 Hz, 3H), 1.23–1.38 (m, 10H), 1.72 (t, J = 8.6 Hz, 2H), 2.79 (t, J = 8.5 Hz, 2H), 7.11 (d, J = 7.6 Hz, 2H), 7.54 (d, J = 7.6 Hz, 2H), 7.68 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.6, 25.5, 29.2, 29.3, 29.4, 31.8, 116.7, 119.6, 122.3, 129.7, 148.8, 157.7; ESI-HRMS (M + H)+m/z calcd for C16H24N3O 274.1914, found 274.1918.
2-(4-(4-Octyl-1H-1,2,3-triazol-1-yl)phenyl)ethanol (RB-056)
To a solution of 23(26) (200 mg, 1.23 mmol) and 1-decyne (508 mg, 3.68 mmol) in t-BuOH/H2O (6 mL, 1:1) were added CuSO4 (196 mg, 1.23 mmol) and sodium ascorbate (243 mg, 1.23 mmol). The reaction mixture was stirred at room temperature for 12 h and then was diluted with EtOAc and washed with brine. The aqueous layer was extracted with EtOAc. The combined organic layers were dried (MgSO4) and concentrated in vacuo. Purification by silica gel chromatography, eluting with CH2Cl2/MeOH (10:1), gave 296 mg (80%) of RB-056 as a white solid. 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.9 Hz, 3H), 1.27–1.42 (m, 10H), 1.71 (quin, J = 7.5 Hz, 2H), 2.76 (t, J = 7.7 Hz, 2H), 2.92 (t, J = 6.6 Hz, 2H), 3.89 (t, J = 6.6 Hz, 2H), 7.35 (d, J = 8.5 Hz, 2H), 7.61 (d, J = 8.5 Hz, 2H), 7.70 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.6, 25.6, 29.1, 29.2, 29.3, 29.4, 31.8, 38.7, 63.1, 118.9, 120.4, 130.2, 135.6, 139.7, 149.1; ESI-HRMS (M + H)+m/z calcd for C18H28N3O 302.2227, found 302.2230.
4-(4-Butyl-1H-1,2,3-triazol-1-yl)phenethyl Methanesulfonate (26)
To a solution of 23(26) (200 mg, 1.23 mmol) and 1-hexyne (0.42 mL, 3.68 mmol) in tert-BuOH/H2O (6 mL, 1:1) were added CuSO4 (196 mg, 1.23 mmol) and sodium ascorbate (243 mg, 1.23 mmol). The reaction mixture was stirred at room temperature for 12 h and then was diluted with EtOAc and washed with brine. The aqueous layer was extracted with EtOAc. The combined organic layers were dried (MgSO4) and concentrated in vacuo. Compound 24 was obtained, without purification, as a yellow liquid. To a solution of 24 (1.23 mmol) and triethylamine (0.86 mL, 6.15 mmol) in CH2Cl2 (10 mL) at 0 °C was added methanesulfonyl chloride (0.29 mL, 3.69 mmol). After being stirred at room temperature for 5 h, the reaction mixture was evaporated, diluted with water, and the product was extracted with EtOAc. The extract was washed with brine, dried, and evaporated. Purification by silica gel chromatography, eluting with hexanes/EtOAc (1:2), gave 253 mg (66%, two steps) of 26 as a white solid. 1H NMR (400 MHz, CDCl3) δ 0.96 (t, J = 7.3 Hz, 3H), 1.43 (sex, J = 7.5 Hz, 2H), 1.72 (quin, J = 7.6 Hz, 2H), 2.80 (t, J = 7.7 Hz, 2H), 2.92 (s, 3H), 3.12 (t, J = 6.7 Hz, 2H), 4.45 (t, J = 6.7 Hz, 2H), 7.38 (d, J = 8.5 Hz, 2H), 7.69 (d, J = 8.5 Hz, 2H), 7.73 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 13.9, 22.3, 25.3, 31.5, 31.6, 35.1, 37.4, 69.7, 118.8, 120.6, 130.3, 136.3, 136.9, 149.2; ESI-HRMS (M + H)+m/z calcd for C15H22N3O3S 324.1382, found 324.1382.
4-(4-Pentyl-1H-1,2,3-triazol-1-yl)phenethyl Methanesulfonate (27)
Compound 27 was prepared from 23, via 25, according to a coupling procedure similar to that described for 26, using 1-heptyne. Yield = 58% (two steps); 1H NMR (400 MHz, CDCl3) δ 0.91 (t, J = 7.1 Hz, 3H), 1.36–1.40 (m, 4H), 1.74 (quin, J = 7.5 Hz, 2H), 2.79 (t, J = 7.7 Hz, 2H), 2.93 (s, 3H), 3.12 (t, J = 6.7 Hz, 2H), 4.46 (t, J = 6.7 Hz, 2H), 7.38 (d, J = 8.5 Hz, 2H), 7.69 (d, J = 8.5 Hz, 2H), 7.74 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.0, 22.4, 25.6, 29.1, 31.4, 31.6, 35.1, 37.4, 60.4, 69.7, 118.8, 120.6, 130.3, 136.2, 136.9, 149.3; ESI-HRMS (M + H)+m/z calcd for C16H24N3O3S 338.1538, found 338.1536.
1-(4-(4-Butyl-1H-1,2,3-triazol-1-yl)phenethyl)piperidine (RB-057)
To a solution of 26 (50 mg, 0.15 mmol) in 3 mL of acetonitrile was added piperidine (150 μL, 1.54 mmol). The reaction mixture was stirred at 50 °C for 12 h and concentrated. Purification by silica gel chromatography, eluting with CH2Cl2/MeOH (5:1), gave 11 mg (75%) of RB-057 as a slightly yellow waxy solid. 1H NMR (400 MHz, CDCl3) δ 0.96 (t, J = 7.4 Hz, 3H), 1.42 (sex, J = 7.4 Hz, 2H), 1.51–1.55 (m, 2H), 1.72 (quin, J = 7.6 Hz, 2H), 1.80 (quin, J = 5.4 Hz, 4H), 2.74–2.83 (m, 8H), 3.04–3.08 (m, 2H), 7.36 (d, J = 8.4 Hz, 2H), 7.63 (d, J = 8.4 Hz, 2H), 7.71 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 13.9, 22.3, 23.5, 24.7, 25.3, 29.7, 31.5, 31.9, 54.1, 60.0, 118.8, 120.5, 130.0, 135.7, 139.6, 149.1; ESI-HRMS (M + H)+m/z calcd for C19H29N4 313.2392, found 313.2385.
1-(4-(4-Pentyl-1H-1,2,3-triazol-1-yl)phenethyl)piperidine (RB-058)
Compound RB-058 was prepared from 27 according to a coupling procedure similar to that described for RB-057. Yield = 81%; 1H NMR (400 MHz, CDCl3) δ 0.91 (t, J = 6.9 Hz, 3H), 1.36–1.43 (m, 4H), 1.47–1.52 (m, 2H), 1.67–1.77 (m, 6H), 2.53–2.61 (m, 4H), 2.65–2.69 (m, 2H), 2.78 (d, J = 7.7 Hz, 2H), 2.92–2.96 (m, 2H), 7.34 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.4 Hz, 2H), 7.69 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.0, 22.4, 24.0, 25.5, 25.7, 29.1, 31.5, 32.7, 54.4, 60.7, 118.8, 120.5, 129.9, 135.6, 140.6, 149.1; ESI-HRMS (M + H)+m/z calcd for C20H31N4 327.2549, found 327.2543.
1-(4-(4-Octyl-1H-1,2,3-triazol-1-yl)phenethyl)piperidine (RB-059)
To a solution of RB-056 (50 mg, 0.17 mmol) and triethylamine (116 μL, 0.83 mmol) in CH2Cl2 (5 mL) at 0 °C was added methanesulfonyl chloride (39 μL, 0.51 mmol). After being stirred at room temperature for 5 h, the reaction mixture was evaporated, diluted with water, and the product was extracted with EtOAc. The extract was washed with brine, dried, and evaporated to afford 28 as a yellow liquid. To a solution of 65 mg (0.17 mmol) of 28 (without purification) in 3 mL of acetonitrile was added piperidine (168 μL, 1.70 mmol). The reaction mixture was stirred at 50 °C for 12 h and concentrated. Purification by silica gel chromatography, eluting with CH2Cl2/MeOH (5:1), gave 11 mg (66%) of RB-059 as a slightly yellow waxy solid. 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.25–1.39 (m, 10H), 1.58–1.62 (m, 2H), 1.72 (quin, J = 7.3 Hz, 2H), 1.89 (quin, J = 5.1 Hz, 4H), 2.78 (t, J = 7.7 Hz, 2H), 2.96–3.04 (m, 6H), 3.14–3.18 (m, 2H), 7.40 (d, J = 8.1 Hz, 2H), 7.64 (d, J = 8.1 Hz, 2H), 7.67 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.0, 22.6, 22.8, 23.9, 25.6, 29.1, 29.2, 29.3, 30.9, 31.8, 53.8, 59.1, 118.9, 120.6, 130.0, 135.8, 138.5, 149.1, 173.3; ESI-HRMS (M + H)+m/z calcd for C23H37N4 369.3013, found 369.3015; (M + H–N2)+m/z calcd for C23H35N2 341.2951, found 341.2954.
1-(4-(4-Butyl-1H-1,2,3-triazol-1-yl)phenethyl)-1-methylpiperidinium Methanesulfonate (RB-060)
To a solution of 26 (15 mg, 46 μmol) in 3 mL of acetonitrile was added 1-methylpiperidine (23 mg, 0.23 mmol). The reaction mixture was stirred at 50 °C for 12 h and concentrated. The residue was washed with hexane to give 16 mg (82%) of RB-060 as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 0.95 (t, J = 7.3 Hz, 3H), 1.41 (sex, J = 7.5 Hz, 2H), 1.66–1.74 (m, 4H), 1.80–1.87 (m, 4H), 2.77 (s, 3H), 2.74–2.84 (m, 2H), 3.13–3.17 (m, 2H), 3.26 (s, 3H), 3.57 (t, J = 5.5 Hz, 4H), 3.71–3.76 (m, 2H), 7.54 (d, J = 8.5 Hz, 2H), 7.62 (d, J = 8.5 Hz, 2H), 7.76 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 13.9, 20.2, 20.8, 21.5, 22.3, 22.8, 25.3, 27.8, 31.5, 39.6, 44.2, 55.4, 61.0, 119.1, 120.6, 120.7, 136.1, 136.2, 149.2; ESI-HRMS (M)+m/z calcd for C20H31N4+ 327.2549, found 327.2546.
1-Methyl-1-(4-(4-pentyl-1H-1,2,3-triazol-1-yl)phenethyl)piperidinium Methanesulfonate (RB-061)
Compound RB-061 was prepared from 27 according to a coupling procedure similar to that described for RB-060. Yield = 77%; 1H NMR (400 MHz, CDCl3) δ 0.90 (t, J = 7.2 Hz, 3H), 1.34–1.39 (m, 4H), 1.68–1.75 (m, 4H), 1.81–1.86 (m, 4H), 2.76 (s, 3H), 2.74–2.83 (m, 2H), 3.13–3.18 (m, 2H), 3.28 (s, 3H), 3.59 (t, J = 5.5 Hz, 4H), 3.75–3.79 (m, 2H), 7.55 (d, J = 8.5 Hz, 2H), 7.64 (d, J = 8.5 Hz, 2H), 7.76 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.0, 20.2, 20.8, 21.5, 22.4, 22.8, 25.6, 27.9, 29.1, 31.5, 39.7, 44.2, 55.3, 61.0, 119.0, 120.6, 120.8, 130.3, 130.8, 136.0, 136.2, 149.3; ESI-HRMS (M)+m/z calcd for C21H33N4+ 341.2705, found 341.2704.
1-Methyl-1-(4-(4-octyl-1H-1,2,3-triazol-1-yl)phenethyl)piperidinium Methanesulfonate (RB-062)
Compound RB-062 was prepared from 28 according to a coupling procedure similar to that described for RB-060. Yield = 71%; 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 7.1 Hz, 3H), 1.27–1.49 (m, 10H), 1.71–1.78 (m, 4H), 1.85–1.89 (m, 4H), 2.76 (s, 3H), 2.80–2.90 (m, 2H), 3.14–3.18 (m, 2H), 3.35 (s, 3H), 3.48 (t, J = 5.5 Hz, 4H), 3.67–3.85 (m, 2H), 7.60 (d, J = 8.5 Hz, 2H), 7.68 (d, J = 8.5 Hz, 2H), 7.83 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.2, 20.2, 20.5, 20.9, 21.4, 22.7, 23.0, 25.7, 27.9, 28.9, 29.1, 29.3, 29.4, 29.5, 31.9, 39.6, 44.0, 55.0, 61.1, 119.1, 120.4, 120.8, 130.2, 130.8, 136.1, 136.3, 149.3; ESI-HRMS (M)+m/z calcd for C24H39N4+ 383.3169, found 383.3174.
1-(4-(4-Butyl-1H-1,2,3-triazol-1-yl)phenethyl)piperidin-4-ol (RB-063)
Compound RB-063 was prepared from 26 according to a coupling procedure similar to that described for RB-057, using 4-hydroxypiperidine. Yield = 65%; 1H NMR (400 MHz, CDCl3) δ 0.96 (t, J = 7.4 Hz, 3H), 1.42 (sex, J = 7.4 Hz, 2H), 1.60–1.75 (m, 4H), 1.93–1.97 (m, 2H), 2.26 (t, J = 9.4 Hz, 2H), 2.60–2.65 (m, 2H), 2.79 (t, J = 7.7 Hz, 2H), 2.85–2.89 (m, 4H), 3.75 (sep, J = 4.4 Hz, 1H), 7.33 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.4 Hz, 2H), 7.69 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 13.9, 22.3, 25.3, 29.7, 31.5, 33.3, 34.4, 51.1, 60.0, 67.6, 118.8, 120.4, 129.9, 135.5, 141.0, 149.1; ESI-HRMS (M + H)+m/z calcd for C19H29N4O 329.2341, found 329.2336.
1-(4-(4-Pentyl-1H-1,2,3-triazol-1-yl)phenethyl)piperidin-4-ol (RB-064)
Compound RB-064 was prepared from 27 according to a coupling procedure similar to that described for RB-057, using 4-hydroxypiperidine. Yield = 70%; 1H NMR (400 MHz, CDCl3) δ 0.90 (t, J = 7.1 Hz, 3H), 1.34–1.42 (m, 4H), 1.61–1.77 (m, 4H), 1.93–1.97 (m, 2H), 2.26 (t, J = 9.5 Hz, 2H), 2.61–2.65 (m, 2H), 2.78 (t, J = 7.7 Hz, 2H), 2.85–2.89 (m, 4H), 3.75 (sep, J = 4.2 Hz, 1H), 7.33 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.4 Hz, 2H), 7.69 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.0, 22.4, 25.6, 29.1, 31.4, 33.3, 34.4, 51.0, 60.0, 67.6, 118.8, 120.4, 129.9, 135.5, 141.0, 149.1; ESI-HRMS (M + H)+m/z calcd for C20H31N4O 343.2498, found 343.2495.
1-(4-(4-Octyl-1H-1,2,3-triazol-1-yl)phenethyl)piperidin-4-amine (RB-065)
Compound RB-065 was prepared from 28 according to a coupling procedure similar to that described for RB-057, using 4-hydroxypiperidine. Yield = 63%; 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 6.7 Hz, 3H), 1.27–1.39 (m, 12H), 1.71 (quin, J = 7.5 Hz, 2H), 1.77–1.84 (m, 2H), 2.10–2.15 (m, 2H), 2.77 (t, J = 7.7 Hz, 4H), 2.91–2.95 (m, 2H), 3.05–3.07 (m, 2H), 3.09–3.14 (m, 2H), 3.89–3.93 (m, 1H), 7.37 (d, J = 8.4 Hz, 2H), 7.63 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.6, 25.6, 29.1, 29.2, 29.3, 29.4, 31.7, 50.0, 58.9, 118.9, 120.6, 130.0, 135.8, 138.9, 149.2; ESI-HRMS (M + H)+m/z calcd for C23H37N4O 385.2962, found 385.2965.
Acknowledgments
The work was supported by NIH Grant HL-083187 (to R.B.) and British Heart Foundation Grant 29476 (to N.J.P. and S.P.).
Glossary
Abbreviations Used
- SK1
sphingosine kinase 1
- SK2
sphingosine kinase 2
- Sph
sphingosine
Supporting Information Available
Evaluation of compounds as putative substrates of SK1 and SK2 and a molecular docking pose of RB-035 in the SK1 binding pocket. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ D.J.B. and N.M. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hait N. C.; Allegood J.; Maceyka M.; Strub G. M.; Harikumar K. B.; Singh S. K.; Luo C.; Marmorstein R.; Kordula T.; Milstien S.; Spiegel S. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizugishi K.; Yamashita T.; Olivera A.; Miller G. F.; Spiegel S.; Proia R. L. Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol. 2005, 25, 11113–11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne S.; Pyne N. J. Translational aspects of sphingosine 1-phosphate biology. Trends Mol. Med. 2011, 17, 463–472. [DOI] [PubMed] [Google Scholar]
- Xia P.; Gamble J. R.; Wang L.; Pitson S. M.; Moretti P. A.; Wattenberg B. W.; D’Andrea R. J.; Vadas M. A. An oncogenic role of sphingosine kinase. Curr. Biol. 2000, 10, 1527–1530. [DOI] [PubMed] [Google Scholar]
- Paugh S. W.; Paugh B. S.; Rahmani M.; Kapitonov D.; Almenara J. A.; Kordula T.; Milstien S.; Adams J. K.; Zipkin R. E.; Grant S.; Spiegel S. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood 2008, 112, 1382–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnute M. E.; McReynolds M. D.; Kasten T.; Yates M.; Jerome G.; Rains J. W.; Hall T.; Chrencik J.; Kraus M.; Cronin C. N.; Saabye M.; Highkin M. K.; Broadus R.; Ogawa S.; Cukyne K.; Zawadzke L. E.; Peterkin V.; Iyanar K.; Scholten J. A.; Wendling J.; Fujiwara H.; Nemirovskiy O.; Wittwer A. J.; Nagiec M. M. Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 2012, 444, 79–88. [DOI] [PubMed] [Google Scholar]
- Baek D. J.; MacRitchie N.; Pyne N. J.; Pyne S.; Bittman R. Synthesis of selective inhibitors of sphingosine kinase 1. Chem. Commun. 2013, 49, 2136–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonelli F.; Lim K. G.; Loveridge C.; Long J.; Pitson S. M.; Tigyi G.; Bittman R.; Pyne S.; Pyne N. J. FTY720 and (S)-FTY720 vinylphosphonate inhibit sphingosine kinase 1 and promote its proteasomal degradation in human pulmonary artery smooth muscle, breast cancer and androgen-independent prostate cancer cells. Cell. Signalling 2010, 22, 1536–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S.; Park H.; Laviad E. L.; Lu X.; Bittman R.; Futerman A. H. Ceramide synthesis is modulated by the sphingosine analog FTY720 via a mixture of uncompetitive and noncompetitive inhibition in an acyl-CoA chain length-dependent manner. J. Biol. Chem. 2009, 284, 16090–16098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandhuvula P.; Tam Y. Y.; Oskouian B.; Saba J. D. The immune modulator FTY720 inhibits sphingosine-1-phosphate lyase activity. J. Biol. Chem. 2005, 280, 33697–33700. [DOI] [PubMed] [Google Scholar]
- Lim K. G.; Tonelli F.; Li Z.; Lu X.; Bittman R.; Pyne S.; Pyne N. J. FTY720 analogues as sphingosine kinase 1 inhibitors: enzyme inhibition kinetics, allosterism, proteasomal degradation and actin rearrangement in MCF-7 breast cancer cells. J. Biol. Chem. 2011, 286, 18633–18640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; MacRitchie N.; Pyne S.; Pyne N. J.; Bittman R. Synthesis of (S)-FTY720 vinylphosphonate analogues and evaluation of their potential as sphingosine kinase 1 inhibitors and activators. Bioorg. Med. Chem. 2013, 21, 2503–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French K. J.; Zhuang Y.; Maines L. W.; Gao P.; Wang W.; Beljanski V.; Upson J. J.; Green C. L.; Keller S. N.; Smith C. D. Pharmacology and anti-tumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J. Pharmacol. Exp. Ther. 2010, 333, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K. G.; Sun C.; Bittman R.; Pyne N. J.; Pyne S. (R)-FTY720 methyl ether is a specific sphingosine kinase 2 inhibitor: effect on sphingosine kinase 2 expression in HEK 293 cells and actin rearrangement and survival of MCF-7 breast cancer cells. Cell. Signalling 2011, 23, 1590–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.; Guo T. L.; Hait N. C.; Allegood J.; Parikh H. I.; Xu W.; Kellogg G. E.; Grant S.; Spiegel S.; Zhang S. Biological characterization of 3-(2-amino-ethyl)-5-[3-(4-butoxyl-phenyl)-propylidene]-thiazolidine-2,4-dione (K145) as a selective sphingosine kinase-2 inhibitor and anticancer agent. PLoS One 2013, 8, e56471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharel Y.; Raje M.; Gao M.; Gellett A. M.; Tomsig J. L.; Lynch K. R.; Santos W. L. Sphingosine kinase type 2 inhibition elevates circulating sphingosine 1-phosphate. Biochem. J. 2012, 447, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French K. J.; Schrecengost R. S.; Lee B. D.; Zhuang Y.; Smith S. N.; Eberly J. L.; Yun J. K.; Smith C. D. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003, 63, 5962–5969. [PubMed] [Google Scholar]
- Mathews T. P.; Kennedy A. J.; Kharel Y.; Kennedy P. C.; Nicoara O.; Sunkara M.; Morris A. J.; Wamhoff B. R.; Lynch K. R.; Macdonald T. L. Discovery, biological evaluation, and structure–activity relationship of amidine based sphingosine kinase inhibitors. J. Med. Chem. 2010, 53, 2766–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy A. J.; Mathews T. P.; Kharel Y.; Field S. D.; Moyer M. L.; East J. E.; Houck J. D.; Lynch K. R.; Macdonald T. L. Development of amidine-based sphingosine kinase 1 nanomolar inhibitors and reduction of sphingosine 1-phosphate in human leukemia cells. J. Med. Chem. 2011, 54, 3524–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin D. J.; Li Y.; Brown M. L.; Min X.; Schmitt M. J.; Wanska M.; Wang X.; Connors R.; Johnstone S.; Cardozo M.; Cheng A. C.; Jeffries S.; Franks B.; Li S.; Shen S.; Wong M.; Wesche H.; Xu G.; Carlson T. J.; Plant M.; Morgenstern K.; Rex K.; Schmitt J.; Coxon A.; Walker N.; Kayser F.; Wang Z. Structure guided design of a series of sphingosine kinase (SphK) inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 4608–4616. [DOI] [PubMed] [Google Scholar]
- Gao P.; Peterson Y. K.; Smith R. A.; Smith C. D. Characterization of isoenzyme-selective inhibitors of human sphingosine kinases. PLoS One 2012, 7, e44543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.; Sun C.; Valentine W. J.; E S.; Liu J.; Tigyi G.; Bittman R. Chiral vinylphosphonate and phosphonate analogues of the immunosuppressive agent FTY720. J. Org. Chem. 2009, 74, 3192–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiuchi M.; Adachi K.; Kohara T.; Minoguchi M.; Hanano T.; Aoki Y.; Mishina T.; Arita M.; Nakao N.; Ohtsuki M.; Hoshino Y.; Teshima K.; Chiba K.; Sasaki S.; Fujita T. Synthesis and immunosuppressive activity of 2-substituted 2-aminopropane-1,3-diols and 2-aminoethanols. J. Med. Chem. 2000, 43, 2946–2961. [DOI] [PubMed] [Google Scholar]
- Knott K.; Kharel Y.; Raje M. R.; Lynch K. R.; Santos W. L. Effect of alkyl chain length on sphingosine kinase 2 selectivity. Bioorg. Med. Chem. Lett. 2012, 22, 6817–6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raje M. R.; Knott K.; Kharel Y.; Bissel P.; Lynch K. R.; Santos W. L. Design, synthesis and biological activity of sphingosine kinase selective inhibitors. Bioorg. Med. Chem. 2012, 20, 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Min X.; Xiao S. H.; Johnstone S.; Romanow W.; Meininger D.; Xu H.; Liu J.; Dai J.; An S.; Thibault S.; Walker N. Molecular basis of sphingosine kinase 1 substrate recognition and catalysis. Structure 2013, 21, 798–809. [DOI] [PubMed] [Google Scholar]
- Conejo-García A.; Pisani L.; Núñez M. D. C.; Catto M.; Nicolotti O.; Leonetti F.; Campos J. M.; Gallo M.; Espinosa A.; Carotti A. Homodimeric bis-quaternary heterocyclic ammonium salts as potent acetyl- and butyrylcholinesterase inhibitors: a systematic investigation of the influence of linker and cationic heads over affinity and selectivity. J. Med. Chem. 2011, 54, 2627–2645. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Kamlet A. S.; Steinman J. B.; Liu D. R. A biomolecule-compatible visible-light-induced azide reduction from a DNA-encoded reaction-discovery system. Nat. Chem. 2011, 3, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.