

Abstract
In normal neurons, neurofilament (NF) proteins are phosphorylated in the axonal compartment. However, in neurodegenerative disorders such as Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS), NF proteins are aberrantly hyperphosphorylated within the cell bodies. The aberrant hyperphosphorylation of NF accumulations found in neurodegeneration could be attributable to either deregulation of proline-directed Ser/Thr kinase(s) activity or downregulation of protein phosphatase(s) activity. In this study, we found that protein phosphatase 2A (PP2A) expression is high in neuronal cell bodies and that inhibition of PP2A activity by okadaic acid (OA), microcystin LR (mLR), or fostriecin (Fos) leads to perikaryal hyperphosphorylation of NF. Peptidyl-prolyl isomerase Pin1 inhibits the dephosphorylation of NF by PP2A in vitro. In cortical neurons, Pin1 modulates the topographic phosphorylation of the proline-directed Ser/Thr residues within the tail domain of NF proteins by inhibiting the dephosphorylation by PP2A. Inhibition of Pin1 inhibits OA-induced aberrant perikaryal phosphorylation of NF. Treatment of cortical neurons with OA or Fos prevents the general anterograde transport of transfected green fluorescent protein–high-molecular-mass (NF-H) into axons caused by hyperphosphorylation of NF-H, and inhibition of Pin1 rescues this effect. Furthermore, inhibition of Pin1 inhibits the OA- or Fos-induced neuronal apoptosis. We show that OA-induced hyperphosphorylation of NF is a consequence of dephosphorylation of NF and is independent of c-Jun N-terminal protein kinase, extracellular signal-regulated kinase, and cyclin-dependent kinase-5 pathways. This study highlights a novel signaling role of PP2A by Pin1 and implicates Pin1 as a therapeutic target to reduce aberrant phosphorylation of NF proteins in neurodegenerative disorders such as AD, PD, and ALS.
Introduction
Neuronal cytoskeletal proteins including neurofilaments (NFs) are extensively phosphorylated on the proline-directed Ser/Thr residues selectively within the axonal compartment under normal physiological conditions (Jaffe et al., 1998). Under pathological conditions such as Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS), NFs are aberrantly hyperphosphorylated within the cell bodies (Sternberger et al., 1985; Goldman and Yen, 1986; Lee et al., 1988; Manetto et al., 1988; Veeranna et al., 1995). The underlying mechanisms of this compartment-specific phosphorylation are still unclear. The aberrant hyperphosphorylation of NFs observed in these neurological disorders might be attributable to increased kinase(s) activity, decreased phosphatase(s) activity, or both. NFs are composed of triplet proteins of low (NF-L), medium (NF-M), and high (NF-H) molecular mass. The Lys-Ser-Pro (KSP) motif is represented 52 times in the rat NF-H tail domain (Chin and Liem, 1990). In vivo, the tail domain of NF can be extensively phosphorylated on almost all the SP repeats (Eagles et al., 1990; Jaffe et al., 1998; Veeranna et al., 1998).
Protein kinases that can phosphorylate NF proteins have been characterized, but much less is known about the protein phosphatases (PPs) in the nervous system (Sim, 1991), especially those acting on NFs. Previous studies demonstrated the presence of a protein phosphatase 2A (PP2A) activity in high-salt extracts of NF preparations from bovine and rat spinal cord that can dephosphorylate NF proteins (Guru et al., 1991; Shetty et al., 1992). Phosphate turnover occurs on NFs during axonal transport (Nixon et al., 1987). The number of phosphorylated epitopes of NF proteins at the nodes of Ranvier decreases relative to that in internodal myelinated regions (de Waegh et al., 1992; Mata et al., 1992), demonstrating the physiological significance of PP(s) in regulating the state of NF phosphorylation. Previous studies from our laboratory have reported that PP2A from both rat spinal cord and rabbit skeletal muscle can dephosphorylate NF-M/H tail domain protein after phosphorylation by cyclin-dependent kinase-5 (Cdk5) (Veeranna et al., 1995).
The prolyl isomerase Pin1 recognizes and induces cis-trans isomerization of pSer/Thr-Pro bonds, conferring phosphorylation-dependent conformational changes relevant for protein function (Lu and Zhou, 2007). The multiple repeats of the KSP motif suggest that reconfiguration of the NF-M/H may involve peptidyl-prolyl isomerization by Pin1, which has a specificity for phosphorylated S/T-P dipeptides (Yaffe et al., 1997). Recently, we have shown that Pin1 regulates the oxidative stress-induced phosphorylation of NF-H by proline-directed kinases such as Cdk5, mitogen-activated protein kinase (MAPK), and c-Jun N-terminal protein kinase 3 (JNK3) (Rudrabhatla et al., 2008).
In this study, we show that PP2A expression is robust in neuronal cell bodies, and inhibition of its activity results in aberrant and hyperphosphorylation of NF on S/T-P residues. Inhibition of Pin1 inhibits okadaic acid (OA)-induced aberrant hyperphosphorylation of NF/M-H in the cell bodies and rescues the general anterograde transport of NF in OA- and fostriecin (Fos)-treated neurons. Furthermore, inhibition of Pin1 inhibits OA- and Fos-induced neuronal cell death. We also show that Pin1 can directly modulate the NF dephosphorylation mediated by PP2A, independent of JNK, extracellular signal-regulated kinase (ERK), and Cdk5 pathways.
Materials and Methods
Materials.
We obtained the following antibodies commercially: polyclonal rabbit and goat PP2A antibodies (Santa Cruz Biotechnology), mouse monoclonal PP2A antibodies (Millipore), monoclonal antibodies to β-actin and β-tubulin (Sigma), and SMI31, SMI32, and SMI34 against phospho-S/T-P epitopes of NF-M/H tail domain (Covance). The RT97 monoclonal antibody clone was a kind gift from Dr. Brian Anderton (Institute of Psychiatry, London, UK). The protein phosphatase inhibitors OA, microcystin LR, Fos, and cyclosporine A (cyA) were purchased from Calbiochem. All cell culture reagents were purchased from Invitrogen.
Human AD and ALS spinal cord.
Closely matched, age and postmortem time, control, and ALS-affected spinal cord tissues were obtained from the National Institute of Child Health and Human Development (NICHD) Brain and Tissue Bank (Bethesda, MD). The ALS spinal cord tissue corresponds to lumbar region (35–60 years). The spinal cord tissue from controls comprise non-neurological disease (atherosclerotic cardiovascular disease or multiple injuries). Closely matched, age and postmortem time, control, and AD-affected brain tissue, frontal cortex (66–86 years), were obtained from the Harvard University Brain Resource Center (Boston, MA). Frozen human tissue was used in accordance with the National Institutes of Health guidelines.
Primary neuronal cultures and treatment with phosphatase inhibitors.
Primary cortical neurons were established from embryonic day 18 (E18) Sprague Dawley rat embryos (Charles River Laboratories). An 18-d-old timed pregnant rat was killed using CO2, pups were removed and decapitated, and cortex was dissected in Hibernate-E media (Brain Bits). Dissociated cortical neurons were obtained by incubating the cortex in Earle's balanced salt solution containing 15 U/ml papain (Worthington Biochemicals) for 45 min at 37°C before triturating in Neurobasal medium containing 20% fetal bovine serum (Hyclone), DNase (0.2 mg/ml), and 0.1 m MgSO4. Undissociated neurons were removed from the cell suspension by passing the cell suspension through a 40 μm cell strainer (Fisher Scientific). Neurons were centrifuged at 800 × g for 5 min at 20°C, and the pellet was resuspended in Neurobasal medium supplemented with B27, penicillin (100 U/ml), streptomycin (100 U/ml), and l-glutamine (0.5 mm; Invitrogen). Neurons were then plated at a density of 150,000 cells/ml on circular glass coverslips and six-well tissue culture dishes, coated with poly-l-lysine (50 μg/ml; Sigma), and incubated in a humidified atmosphere containing 5% CO2/95% O2 at 37°C. The following PP inhibitors, OA, microcystin LR, Fos, and cyclosporine A, were added to the 7 d in culture (DIC) neurons.
Preparation of the NF proteins.
NF proteins were prepared as described by Tokutake et al. (1983) with some modifications, as follows. Rat spinal cords and sciatic nerve were immediately washed thoroughly in ice-cold buffer after dissection in a buffer A composed of 20 mm Tris-HCI buffer, pH 6.8, containing 1 mm each EDTA, EGTA, and DTT and 100 mm NaCl to remove blood clots and meninges. Tissues were homogenized in the same buffer (1:5 w/v) containing 0.2 mm phenylmethylsulfonyl fluoride, leupeptin (5 μg/ml), and 1% Triton X-100. The homogenate was centrifuged at 20,000 × g at 4°C for 45 min. The pellet was resuspended in the same buffer A containing 1 m sucrose (buffer B) and centrifuged at 20,000 × g for 30 min. The floating myelin and supernatant were discarded. The pellet was resuspended in buffer B and centrifuged as above. These steps were repeated four more times, and the final pellet obtained in the last step is called the NF preparation.
Dephosphorylation of NF proteins.
The dephosphorylation of the NF proteins by PP2A was performed as described previously (Veeranna et al., 1995). In brief, 2 μg of NF preparation was dephosphorylated with 1 unit of PP2A (Millipore) in a PP2A buffer (20 mm HEPES, pH 7.0, 1 mm DTT, 1 mm MnCl2, 100 μg/ml BSA, and 50 μm leupeptin) for 30 min. The dephosphorylation was confirmed by Western blot analysis with phospho-NF antibodies (SMI31/RT97).
Expression and purification of the Pin1 protein.
Wild-type Pin1 was cloned into pGEX-5X-1 obtained from GE Healthcare for glutathione S-transferase (GST) expression studies. The pGEX recombinant plasmid containing Pin1 was expressed and purified as described previously (Rudrabhatla et al., 2008).
Plasmids.
Dominant-negative Pin1 was produced by making a point mutation to produce an alanine at serine 16, using the QuikChange mutagenesis kit (Stratagene) according to the instructions of the manufacturer. Dominant-negative (DN) Pin1 was cloned as a green fluorescent protein (GFP) fusion protein of Pin1 in pCEFL–GFP vector. This was a kind gift from Dr. Silvio Gutkind (National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD). The NF-H was constructed as enhanced GFP (EGFP) fusion. The NF-H tail domain with 52 S/T-P repeats is cloned in pCDNA 3.1 vector for transfection studies.
Pin1 short interfering RNA.
Using the HiPerformance Design Algorithm, synthetic 21-mer short interfering (siRNA) was designed and synthesized (Qiagen). Pin1 siRNA (silencing) sense and antisense sequences were as follows: 5′-r(GCUCAGGCCGAGUGUACUA)dTdT-3′ and 5′-r(UAGUACACUCGGCCUGAGC)dTdT-3′, respectively. All Stars negative control siRNA, a scrambled, nonsilencing control siRNA, was obtained (Qiagen). Sense and antisense sequence for nonsilencing siRNA were as follows: 5′-r(UUCUCCGAACGUGUCACGU)d(TT)-3′ and 5′-r(ACGUGACACGUUCGGAGAA)d(TT)-3′, respectively. To ensure the absence of possible complementary binding sites in mammalian genomes, the siRNA sequences were extensively checked against the GenBank database using the Smith–Waterman algorithm. This strategy was used to exclude significant sequence homology within the genomes of mammalian species, which may interfere with the target specificity of the siRNA and contribute to unwanted off-target effects in later experiments. The sense and antisense strands were annealed to create the double-stranded siRNA at a 20 μm concentration. Control siRNA and Pin1 siRNA were dissolved in suspension buffer to obtain a 20 μm solution and then heated at 90°C for 1 min. The final transfection step was preceded by a 60 min incubation at 37°C. Delivery of siRNA was performed as described previously (Kesavapany et al., 2007; Rudrabhatla et al., 2008).
Electrophoresis and Western blotting.
Electrophoresis and Western blotting were performed as described previously (Rudrabhatla et al., 2008).
Immunocytochemistry of primary cortical neurons.
Cortical neurons fixed in 4% paraformaldehyde were permeabilized for 20 min in 0.2% Triton X-100, blocked in 5% fetal calf serum in PBS containing 1% Triton X-100 for 1 h, and incubated with primary antibodies (diluted in blocking buffer) against phospho-specific monoclonal antibody against NF (SMI31) or with polyclonal antibodies to Pin1 or PP2A (Cell Signaling Technology) for overnight at 4°C. After six washes in PBS, the neurons were incubated with anti-rabbit (Alexa 488) and anti-mouse (Alexa 594) secondary antibodies (Invitrogen). Secondary antibodies were diluted in the same buffer as the primary antibodies and incubated for 2 h at room temperature. Cells were washed and mounted on the coverslips, and images were captured with an oil-immersion 63× objective on a Zeiss LSM510 using LSM Image Software. The images were analyzed using a TCS software program on an Axiovert 200 M microscope (Zeiss).
In situ cell death detection (terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay).
In situ cell death detection was performed as described previously (Rudrabhatla et al., 2008). In brief, after primary cortical neurons were cultured and treated, cells were fixed and prepared for terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining according to the instructions of the manufacturer (In Situ Cell Death Detection kit; Roche Diagnostics). Nuclei were counterstained using 4′ 6-diamidino-2-phenylindole (DAPI). TUNEL staining fluorescent images were captured with a Zeiss LSM510 laser-scanning confocal microscope. Cell counts were performed as described in figure legends.
Protein phosphatase PP2A activity measurements.
Closely matched, age and postmortem time, control, and ALS-affected spinal cord tissues were obtained from the NICHD Brain and Tissue Bank. Homogenates were prepared in duplicate from three ALS and control brains, and four assays were performed with each homogenate. Mean values and SDs were calculated. The amount of released phosphate was determined by measuring the absorbance of a malachite green:phosphate complex at 650 nm. Spinal cord extracts of age-matched control and ALS samples were prepared with a Teflon homogenizer (40 strokes at 100 rpm) in 1× TBS and 1% (v/v) Triton X-100 in the presence of protease inhibitors (CompleteTM with EDTA; Roche Diagnostics). Phosphatase assays were performed with these extracts using the PP2A immunoprecipitation phosphatase assay kit (catalog number 17-313; Millipore) (Guan et al., 2007). Release of phosphate from a chemically synthesized phospho-peptide (K-R-pT-I-R-R, in which pT is phospho-threonine), which is a substrate for PP2A measured over a period of 30 min.
Statistics.
Each experiment was repeated at least four times. The data were expressed as the means ± SD. Student's t tests were used to compare the effects of all treatments. The differences were considered statistically significant as *p < 0.01 and **p < 0.001.
Results
PP2A regulates topographic phosphorylation of NFs
Four phosphatase inhibitors were used in the present study: (1) OA, a marine sponge toxin first isolated from a genus of sponges, Halichondria, reported to directly inhibit the catalytic subunits of multiple phosphatases; (2) mLR, a potent and selective protein phosphatase 1 and 2A inhibitor, hepatotoxin isolated from strains of the blue-green algae Microcystis aeruginosa; (3) Fos is an antitumor antibiotic with inhibitory activity against serine/threonine PP1 and PP2A and is highly selective for PP2A; (4) cyclosporine A, a PP2B (calcineurin) specific inhibitor. To study the involvement of protein phosphatases on perikaryal phosphorylation of NFs, we began with the PP1/PP2A inhibitor okadaic acid (0.25 μm). The E18 7 DIC dissociated rat cortical neurons were stained with phospho-NF antibodies. We used SMI31, a monoclonal antibody that specifically detects phosphorylated Ser/Thr-Pro repeats of NF-M/H tail domain. Figure 1 A represents the NF-M and NF-H tail domain comprising multiple KSP repeats (rat NF-H, 52 KSP repeats). The antibodies used in this study pertaining to phosphorylated and non-phospho-NF antibodies is also shown (SMI31, SMI34, RT97, and SMI32) in Figure 1 A. Figure 1 Ba shows the SMI31 staining of cortical neurons under normal conditions. The phospho-NF stains only the axons and processes, and there is no staining of SMI31 in the perikarya (Fig. 1 Ba). When okadaic acid is added to the cortical neurons for 2.5 h, we observed phospho-NF accumulations in the perikarya (Fig. 1 Bb). To confirm that PP2A is involved, we also tested the PP1/PP2A inhibitor microcystin LR (15 μm), which increased perikaryal phosphorylation of NF in cortical neurons (Fig. 1 Bc). Also, there was a significant increase in the perikaryal hyperphosphorylation of NF with the PP2A-selective inhibitor fostriecin (1 μm) (Fig. 1 Bd). We then tested the effect of the PP2B inhibitor cyA on topographic phosphorylation of NF. We used increasing concentrations of cyA, 0.5 μm (Fig. 1 Cb), 1 μm (Fig. 1 Cc), and 2.5 μm (Fig. 1 Cd), and detected no change in the perikaryal phosphorylation of NF compared with nontreated neurons (Fig. 1 Ca). These data implicate that there was significant phospho-NF-M/H accumulations in neuronal perikarya with the PP2A inhibitors OA, mLR, and Fos but not with the PP2B inhibitor cyclosporine A. To further confirm these results, we have used SMI34 antibodies that specifically detect phospho-NF, under these experimental conditions. At 1:20,000 dilutions, SMI34 does not cross-react with phospho-Tau, further demonstrating the specificity of phospho-NF immunoreactivity. These results are also confirmed by using the phospho-NF-H antibody RT97. The densitometry analysis of perikaryal p-NF-M/H as detected by SMI31, SMI34, and RT97 from untreated and treated (OA, mLR, Fos, and cyA) neurons from four independent experiments is shown in Figure 1 D. We then tested the effect of PP2A inhibitors on NF phosphorylation using the non-phospho-NF antibody SMI32. In normal neurons, there is maximal staining of SMI32 in cell bodies (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Treatment of cortical neurons with OA, mLR, and Fos decreased SMI32 (non-phospho-NF) immunoreactivity in neuronal perikarya, but cyA did not have any effect (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The densitometry analysis of perikaryal p-NF-M/H from untreated and treated neurons using SMI32 is shown in supplemental Figure 1 B (available at www.jneurosci.org as supplemental material).
Figure 1.

Effect of protein phosphatase inhibitors on perikaryal phosphorylation of NF. A, Structure of rat NF-M/H representing KSP repeats in the tail domain. The KSP repeats are recognized by phospho-NF-M/H antibodies (SMI31, SMI34) and phospho-NF-H antibodies (RT97). The SMI32 antibodies recognize the non-phospho-KSP repeat region of NF-M/H. B, The dissociated E18 rat primary cortical neurons were cultured for 7 d in situ and then treated with or without different protein phosphatase PP2A inhibitors for 2.5 h [OA (b; 0.25 μm), microcystin LR (c; 15 μm), or Fos (d; 1 μm)] and then subjected to immunofluorescence with phospho-NF (SMI31) antibodies. Control (a) shows phosphorylated NF in processes with no phosphorylation in cell bodies. There was a significant increase of somatic phosphorylation in OA-treated (b), mLR-treated (c), and fostriecin-treated (d) neurons. C, The 7 DIC cortical neurons were subjected to the dose-dependent treatment of the PP2B-specific inhibitor cyclosporine A [0.5 μm (b), 1 μm (c), and 2.5 μm (d)]. Scale bar, 10 μm. D, Densitometry analyses of phospho-NF was performed with images captured using NIH Image software. To determine the relative intensity of SMI31 immunoreactivity within perikarya, representative areas of perikarya excluding obvious vesicles and the nucleus were quantified using the freehand selection tool. Similar results were obtained using RT97 and SMI34 antibodies. Intensity of SMI31 (blue), RT97 (red), and SMI34 (yellow) immunoreactivity (p-NF-H) in neuronal cell bodies was analyzed with the NIH ImageJ histogram from 50 to 100 individual cells from at least three experiments. The mean signal intensity (total pixel density per number pixels) is shown. Values in graphs are means ± fluorescence intensity (in arbitrary densitometric units) in perikarya from three different experiments. Mean fluorescence of phosphorylated NF-M/H immunoreactivity. *p < 0.01 and **p < 0.001 of phospho-NF-M/H in perikarya relative to fluorescence intensity values of nontreated neurons (NT).
Colocalization of PP2A and phospho-NF
To examine the cellular localization of PP2A with phospho-NF, immunocytochemical analyses were performed on dissociated E18 rat cortical neurons cultured for 7 d in nontreated and treated with okadaic acid. A rhodamine-conjugated secondary antibody was used to show endogenous phospho-NF expression, and Oregon green secondary antibody was used for endogenous PP2A (Fig. 2). expression is robust in the cell bodies of cortical neurons. The phospho-NF is predominantly localized in the axons in nontreated neurons (Fig. 2, top row) and in cell bodies of OA-treated cortical neurons (Fig. 2, bottom row). In the overlay image, colocalization of PP2A and phospho-NF can be seen in the cell bodies in OA-treated neurons (Fig. 2, bottom right panel).
Figure 2.

Colocalization of PP2A and phospho-NF in neuronal perikarya in OA-treated cortical neurons. Colocalization of PP2A and phospho-NF in the nontreated (top; NT) and OA-treated (bottom) neurons. The E18 dissociated rat cortical neurons were cultured in vitro for 6 d, treated with OA, and subjected to immunofluorescence. The phospho-NF was visualized using the mouse phospho-NF-M/H monoclonal antibody SMI31 (red), and PP2A was visualized using goat polyclonal antibody (green). Scale bar, 10 μm.
Pin1 prevents the dephosphorylation of NF proteins by PP2A in vitro
Tau and NF-H/M are the physiological substrates of PP2A. Previous reports have shown that Pin1 modulates the dephosphorylation of Tau at Thr(231) by PP2A. Rat NF-H has 52 SP repeats in the tail domain in tandem. Therefore, we wanted to study the effect of Pin1 in NF tail domain dephosphorylation by PP2A. The close relationship between phosphorylation and NF structure and function suggests that Pin1 could play a direct role in modulating NF function by influencing phosphorylation/dephosphorylation. We first tested the effect of Pin1 on the ability of PP2A to dephosphorylate the endogenously phosphorylated NF. We prepared rat NF triplet proteins from spinal cord and sciatic nerve. NF proteins are highly phosphorylated in vivo and form an excellent substrate to study dephosphorylation. We purified recombinant human Pin1 as a GST fusion protein from Escherichia coli. Purified NF proteins were dephosphorylated with PP2A in the presence or absence of increasing amounts of GST–Pin1, and the NF dephosphorylation was assayed by Western blotting with a phospho-NF-H antibody (RT97). RT97 is a monoclonal antibody that recognizes the phosphorylated S/T-P residues of NF-H. The results show that PP2A was able to dephosphorylate endogenously phosphorylated NF-H (Fig. 3 A, lane 1, endogenous phospho-NF-H; lane 2, PP2A-treated NF-H). The addition of GST–Pin1 inhibited NF-H dephosphorylation in a dose-dependent manner (Fig. 3, lanes 3, 4). GST alone did not have an effect on NF dephosphorylation by PP2A (data not shown). These results suggest that the Pin1 reduces dephosphorylation of the NF proteins by inhibiting PP2A activity. Figure 3 B represents the effect of Pin1 on NF dephosphorylation with the data obtained from three separate experiments.
Figure 3.
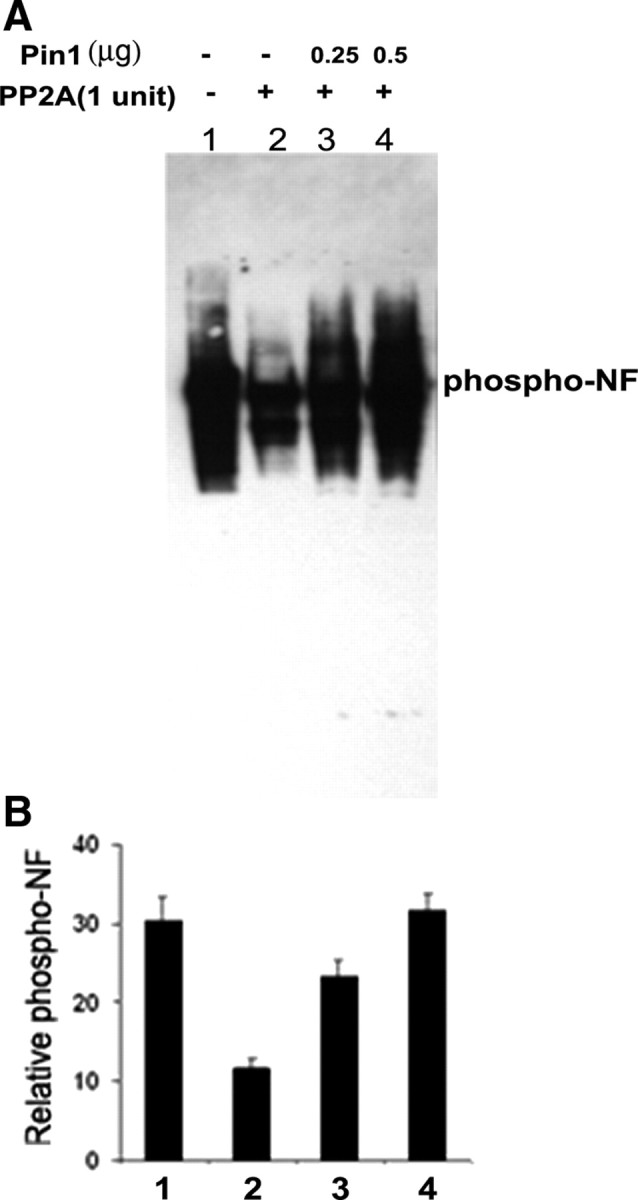
Dephosphorylation of NF by PP2A is inhibited by addition of Pin1 in vitro. A, NF triplet (NF-H/M/L) proteins were prepared from rat spinal cord and sciatic nerve as described in Material and Methods and was subjected to dephosphorylation with 1 U of PP2A (lane 2). The effect of Pin1 on NF-H dephosphorylation was tested by preincubating 2.5 μg of NF-triplet protein with GST–Pin1 (0.25 and 0.5 μg; lanes 3–4) at 30°C for 10 min, followed by addition of 1 U of PP2A. Reaction mixtures were incubated at 30°C for 30 min. Reactions were terminated by addition of SDS loading buffer, and proteins were analyzed by 4–20% SDS-PAGE. Blots were probed with monoclonal antibodies for phosphorylated NF-H (RT97). Data show the effect of Pin1 on NF-H dephosphorylation. PP2A dephosphorylates NF (lane 2). Addition of Pin1 prevents NF dephosphorylation in a dose-dependent manner [lane 3 (0.25 μg of Pin1); lane 4 (0.5 μg of Pin1)]. B, Densitometry analyses of phospho-NF obtained from A.
Pin1 modulates NF dephosphorylation in vivo
The studies described above show that Pin1 prevents dephosphorylation of NF in vitro. We next examined whether Pin1 modulates NF dephosphorylation in vivo. Treatment of 7 DIC cortical neurons with 0.25 μm OA increased the phosphorylation of NF in a time-dependent manner (Fig. 4). Maximal NF phosphorylation was observed 4 h after OA treatment (Fig. 4 A). Figure 4 B represents the densitometry analysis of OA-induced phosphorylation of NF. The cortical neurons treated with OA showed accumulation of phospho-NF-M/H in cell bodies in a time-dependent manner. At 4 h after OA treatment, cortical neurons showed maximum accumulation of phospho-NF in cell bodies (Fig. 4 C).
Figure 4.

OA increases the NF phosphorylation in a time-dependent manner. A, The 7 DIC cortical neurons were treated with OA (0.25 μm) for 1 h (lane 2), 2 h (lane 3), and 4 h (lane 4) and subjected to Western blot analysis with phospho-NF (SMI31) antibodies. The OA induced hyperphosphorylation of NF in a time-dependent manner. B, Densitometry analyses of phospho-NF obtained from A. Mean density, Phospho-NF/actin is plotted on the y-axis. C, Five DIC neurons were treated with OA for 1, 2, and 4 h, and immunofluorescence was performed with SMI31 antibodies. In normal neurons, SMI31 is stained in axonal compartment (a). The phospho-NF accumulates in cell bodies in a time-dependent manner. Scale bar, 10 μm.
If Pin1 acts by preventing the dephosphorylation of NF, then transfection of DN Pin1 should inhibit the OA-induced increase in NF phosphorylation. Transfection of DN Pin1 inhibited the OA-induced phospho-NF accumulations (Fig. 5 A). The endogenous Pin1 migrates at 18 kDa, whereas transfected GFP DN Pin1 migrates at 50 kDa. In addition, knockdown of Pin1 by Pin1 siRNA inhibited the increase in phospho-NF accumulations by OA. Knockdown of Pin1 by Pin1 siRNA reduced significant levels of Pin1 expression (Fig. 5 A). Figure 5 B represents the densitometry analysis of OA-induced phosphorylation of NF (NF-M/H) by inhibition of Pin1 activity. These results suggest that Pin1 prevents dephosphorylation of NF-M/H in vivo, in cortical neurons.
Figure 5.
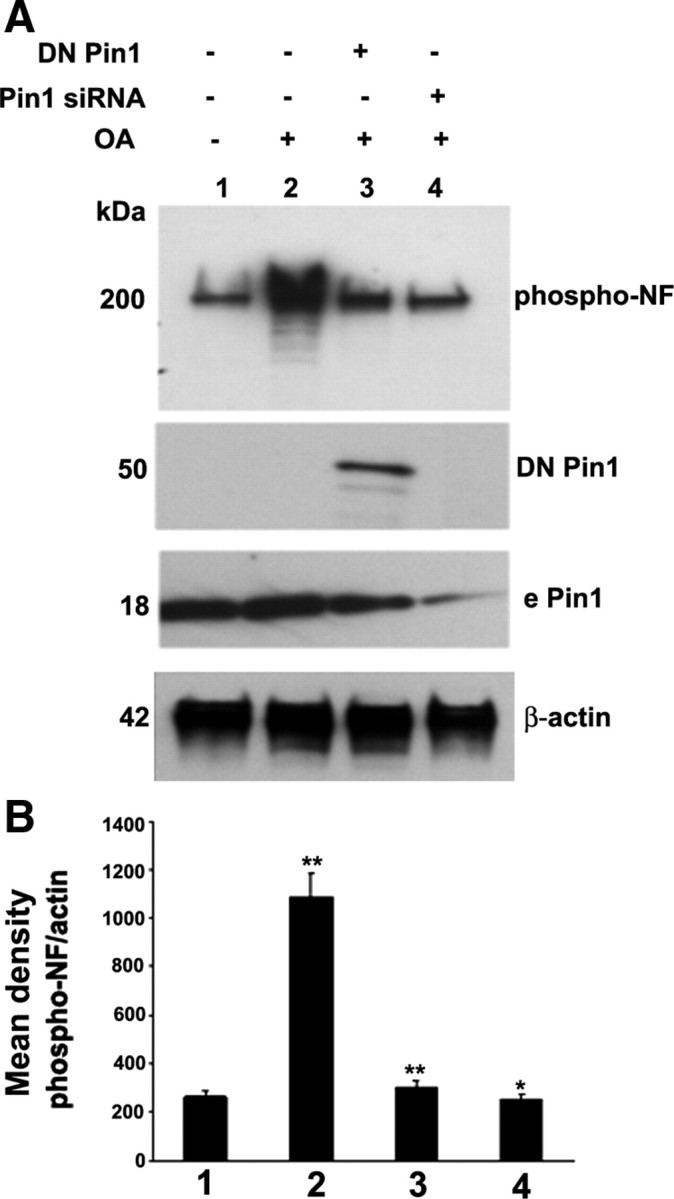
Pin1 expression and activity is critical toward OA-induced aberrant hyperphosphorylation of NF. A, Cortical neurons were subjected to treatment with OA (0.2 μm for 2.5 h), and Western blot analysis was performed with phospho-NF-M/H (SMI31) staining. The OA-induced hyperphosphorylation of NF is inhibited by knockdown of Pin1 by Pin1 siRNA and DN Pin1-transfected neurons. Lanes 1 and 2, Control scrambled siRNA-transfected neurons. Lanes 2–4, OA-treated neurons. Lane 3, Pin1 siRNA-transfected neurons. Lane 4, DN Pin1-transfected neurons. The transfected Pin1 is a GFP fusion protein and migrates at 50 kDa, at which the endogenous Pin1 (e Pin1) is 18 kDa. B, Densitometry analysis of phospho-NF immunoreactivity obtained from A. Mean density, Phospho-NF/actin is plotted on the y-axis. *p < 0.01 and **p < 0.001 of phospho-NF-M/H relative to nontreated neurons.
OA-induced aberrant perikaryal phospho-NF accumulations are inhibited by transfection of DN Pin1 and Pin1 siRNA in cortical neurons
The PP2A activity is downregulated in AD brain (Sontag et al., 2004). In a cellular model of primary cortical cultures, we used OA to inhibit PP2A. Under normal conditions, the cortical neurons show phospho-NF staining in the axonal compartment (Fig. 6 Aa). SMI31 antibodies do not stain phospho-NF-M/H in the neuronal perikarya in normal neurons. However, the cross reactivity of SMI31 antibodies with nuclei is reported (Schilling et al., 1989). The nuclear localization of SMI31 is attributable to the association of phosphorylated histones with nuclear components (Weigum et al., 2003). Pin1 is present in both axons and cell bodies (Fig. 6 Ab). Pin1 colocalizes with phospho-NF only in the axons (Fig. 6 Ac). The cortical neurons treated with OA show phospho-NF accumulations in the perikarya (Fig. 6 Ad), and Pin1 colocalizes with the phospho-NF in the cell bodies (Fig. 6 Af). Knockdown of Pin1 with Pin1 siRNA shows the reduction of Pin1 staining by 80% compared with control scrambled siRNA-transfected neurons (Fig. 6 Ah). Pin1 siRNA-transfected neurons show the phospho-NF staining as observed in control neurons (Fig. 6 Ag–Ai). For comparison purposes, we captured neurons that show both Pin1 siRNA-transfected neurons (indicated in yellow arrows) and nontransfected neurons (white arrows). It should be noted that only the Pin1 siRNA-transfected neurons show the reduction in perikaryal phosphorylation of NF (Fig. 6 Ag–Ai). The quantitation of perikaryal p-NF-M/H from untreated and OA-treated neurons from four independent experiments is shown in Figure 6 B. Figure 6 C shows the effect of Pin1 on perikaryal phosphorylation of NF-M/H by treatment of cortical cultures with Fos. These results suggest that the treatment of cultured neuronal cells with OA induces perikaryal accumulation of the NF phospho-epitopes that are normally confined to axons, and these effects are prevented by inhibition of Pin1.
Figure 6.
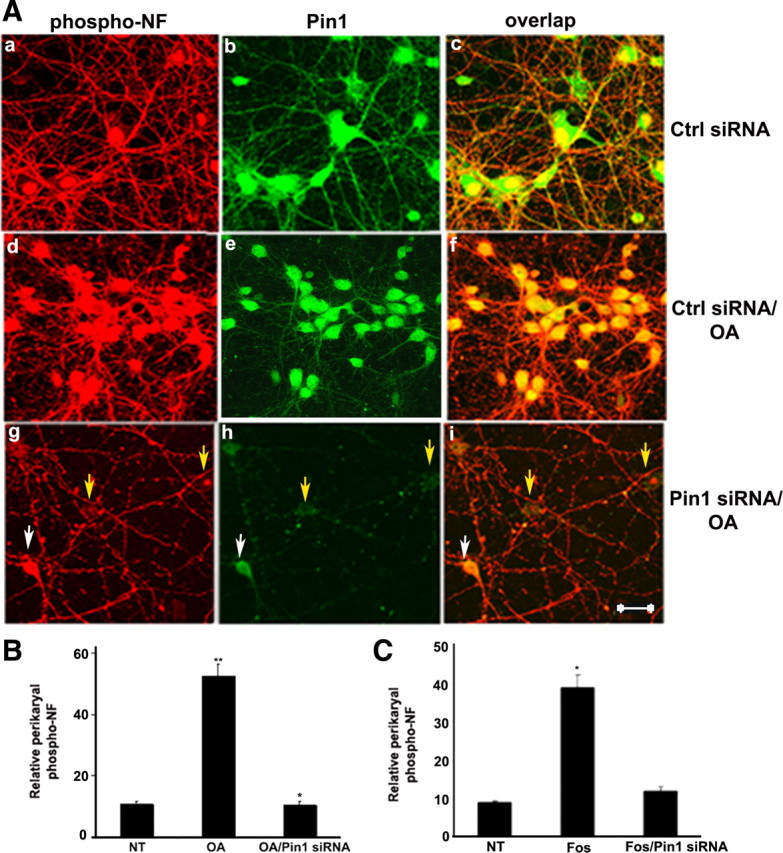
Knockdown of Pin1 by Pin1 siRNA inhibits OA-induced perikaryal phosphorylation of NF-M/H. A, Five DIC cortical neurons were transfected with either control (Ctrl) and scrambled siRNA (a–f) or Pin1 siRNA (g–i) and then treated with 0.25 μm OA (d–i) on day 7 for 2.5 h. Neurons were immunostained, p-NF-H was detected using SMI31 (red), and Pin1 was detected by using Pin1 antibody (green). Neurons transfected with Pin1 siRNA exhibited reduced p-NF-H in the cell body. Nontreated neurons exhibited SMI31 staining in the processes with little or no staining in the cell body (a–c), which is increased during OA treatment (d–f). Cell body SMI31 staining was reduced in Pin1 siRNA-treated neurons (g–i). Scale bar, 20 μm. B, Representative neurons are treated with OA as indicated. Intensity of SMI31 immunoreactivity (phospho-NF) in neuronal cell bodies was analyzed with the NIH ImageJ histogram. The mean signal intensity (total pixel density per number pixels) is shown. Values in graphs are means ± fluorescence intensity (in arbitrary densitometric units) in perikarya from four different experiments. Note the accumulation of phospho-NF in perikarya in OA-treated neurons and restoration of normal levels of p-NF-H in Pin1 siRNA-treated cortical neurons. C, Intensity of SMI31 immunoreactivity (phospho-NF) in neuronal cell bodies was analyzed with the NIH ImageJ histogram after treatment of cortical neurons with Fos. *p < 0.01, phospho-NF-M/H in perikarya relative to fluorescence intensity values of control. NT, Nontreated.
DN Pin1 inhibits the Pin1 activity without changing Pin1 protein levels. Transfection of cortical neurons with GFP–DN Pin1 inhibited the OA-induced hyperphosphorylation of NF in the cell body (Fig. 7). Two neurons are captured, with and without GFP–DN Pin1 transfection. In OA-treated neurons, the densitometry analysis of the relative perikaryal phospho-NF in GFP–DN Pin1-transfected neurons is reduced by ∼3.5-fold compared with nontransfected neurons. These results further demonstrate that inhibition of Pin1 levels (Pin1 siRNA) or Pin1 activity (DN Pin1) inhibits OA-induced aberrant perikaryal hyperphosphorylation of neurons. These results show that Pin1 prevents the dephosphorylation of NF in vivo.
Figure 7.

OA-mediated increase in phosphorylated NF is reduced by overexpression of DN Pin1. Five-day-old cortical neurons were transfected with GFP–DN Pin1 and after 24 h were treated with 0.25 μm OA for 2.5 h. Neurons were immunostained, phospho-NF was detected using SMI31 (red), and DN Pin1 was detected through expression of GFP. Note that only neurons that show DN Pin1 expression exhibited significant reduction of phosphorylated NF in the cell body. Scale bar, 20 μm.
Pin1 directly modulates PP2A-mediated dephosphorylation of NF, independent of ERK, JNK, and Cdk5 pathways
There are several reports that Pin1 modulates PP2A-mediated dephosphorylation of target proteins (Zhou et al., 2000). Inhibition of PP2A by OA may activate JNK. To determine whether aberrant hyperphosphorylation of NF in OA-treated neurons in response to Pin1 knockdown is a consequence of kinase activation or a direct consequence of PP2A-mediated dephosphorylation of NF, we tested the NF phosphorylation with kinase inhibitors (JNK, ERK, and roscovitine) before the addition of PP2A inhibitor. Nontreated and kinase inhibitor treatment of cortical neurons with the JNK inhibitor SP600125 (anthra[1,9-cd]pyrazol-6(2H)-one), the ERK inhibitor PD98059 [2-(2-amino-3-methyoxyphenyl)-4H-1-benzopyran-4-one], or the Cdk5 inhibitor roscovitine before the addition of OA did not change the levels of phospho-NF accumulation after the treatment of neurons with OA, suggesting that the aberrant phosphorylation of NF mediated by PP2A is not attributable to the activation of JNK/ERK/Cdk5 kinase pathways (Fig. 8, top). These results suggest that Pin1 directly modulates PP2A-mediated hyperphosphorylation of NF independent of JNK, ERK, or Cdk5 cascades (Fig. 8, bottom). These results corroborate the in vitro studies (Fig. 3) that Pin1 prevents the dephosphorylation of NFs.
Figure 8.

Pin1 modulates PP2A-mediated dephosphorylation of NF independent of ERK, JNK, and Cdk5 pathways. Seven DIC neurons were treated with PD98059 (ERK inhibitor; PD; lanes 2, 6), SP600125 (JNK inhibitor; SP; lanes 3, 7), and roscovitine (Cdk5 inhibitor; Ros; lanes 4, 8) for 1 h. The neurons were then subjected to treatment with OA (lanes 5–8) for 2 h. The phospho-NF was detected by SMI31 antibodies. The equal loading of the protein was confirmed by Western blot analysis with monoclonal β-tubulin antibodies. The bottom shows the densitometry analysis of mean phospho-NF/tubulin obtained from the top. *p < 0.01 and **p < 0.001 of phospho-NF-M/H intensity relative to nontreated neurons (NT).
Pin1 modulates the NF transport in OA-treated neurons
Phosphorylation has long been considered to regulate NF interaction and axonal transport, and, in turn, influences axonal stability and their maturation. Shea et al. (2004) have shown that NF phosphorylation modulates NF dynamics and axonal transport. The above studies show the aberrant distribution of phospho-NF in OA-treated neurons. This prompted us to study the NF axonal transport under these conditions. We transfected GFP–NF-H in primary cortical neurons for 24 h to allow the accumulation of GFP-tagged subunits. Previous studies have demonstrated that 18–24 h were required for transfected cells to accumulate sufficient GFP–NF-M for visualization (Yabe et al., 1999). Cultures were treated for the final 2 h with OA or Fos. Transfection of GFP–NF-H resulted in the transport of exogenously expressed NF-H into the axons (Fig. 9 a–c). We performed staining with polyclonal Tuj1 (red), a neuronal marker to confirm the neuronal identity. When cortical neurons are treated with OA for 2 h, NF translocation is disrupted as observed by the GFP–NF-H in the cell bodies and proximal axons only, and NF proteins are not transported until the distal part of axons (Fig. 9 d–f). Similar results were observed by treatment of cortical neurons with Fos (Fig. 9 g–i). For inhibition of PP2A with OA or Fos, GFP–NF-H was not targeted into the axonal processes of transfected cells and thus reduced anterograde GFP–NF-H axonal transport (Fig. 9 d–i). Then, we asked whether inhibition of Pin1 rescues general axonal transport of GFP–NF-H in OA- and Fos-treated neurons. In Pin1 siRNA-transfected neurons, treatment with OA retained the transport of GFP–NF-H, as observed in nontreated neurons (Fig. 9 j–l). Knockdown of Pin1 rescues the general anterograde transport of GFP–NF-H in OA-treated neurons (Fig. 9 j–l) and Fos-treated neurons (Fig. 9 m–o). The NF translocation is disrupted in OA-treated neurons as a result of the hyperphosphorylation of NF in the cell bodies. These results suggest that NF phosphorylation modulates the NF transport. These results indicate a possible role of Pin1 in NF dynamics. We also transfected C-terminal KSP repeat domain of NF-H (non-GFP-tagged) in primary cortical neurons for 24 h and performed immunofluorescence with total polyclonal NF-H antibodies. Transfected NF-H is translocated into the axonal compartment in normal neurons (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). When cortical neurons are treated with OA for 2 h, NF translocation is disrupted and NF proteins are not transported until the distal part of axons (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Knockdown of Pin1 rescues the NF axonal translocation in OA-treated neurons (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material). These results are consistent with previous studies (Shea et al., 2004) that show that phosphorylation of NF regulates NF transport. To determine the effect of DN Pin1 on NF-H translocation in primary neurons, we cotransfected GFP–DN Pin1 with NF-H construct (non-GFP-tagged) and subjected to treatment with OA. The GFP–DN Pin1-transfected neurons show normal translocation of NF-H in OA-treated neurons (supplemental Fig. 2D, 1–3, available at www.jneurosci.org as supplemental material). These results suggest that the generalized anterograde transport of GFP–NF-H defects are caused by treatment of cortical neurons with OA and Fos, and inhibition of Pin1 rescues the normal transport of cortical neurons.
Figure 9.

Pin1 modulates NF transport of OA-treated neurons. The GFP–NF-H was transfected on 5 DIC cortical neurons. After 24 h, the NF translocation of transfected NF-H was monitored. The colocalization of GFP–NF-H-transfected neurons was observed by staining the cortical neurons with the neuronal marker TuJ1 (red) polyclonal antibodies. In normal neurons, GFP–NF-H is well translocated into axons (a–c). However, in cortical neurons treated with OA for 2 h, NF proteins are hyperphosphorylated in cell bodies, and therefore NF translocation is impaired. NF proteins are not translocated until the distal end of the axons. The GFP–NF-H remains in the cell bodies and proximal axons but not the distal part of the axons (d–f). Similarly, in fostriecin-treated cortical neurons, GFP–NF-H is not translocated into axons (g–i). However, knockdown of Pin1 rescues the normal NF translocation into the axons in OA-treated neurons (j–l) and Fos-treated neurons (m–o). Scale bar, 10 μm. NT, Nontreated.
OA-induced phospho-NF accumulations in perikarya leads to apoptosis
Hyperphosphorylation of neurofilament and Tau may underlie the cytoskeletal abnormalities and neuronal death seen in several neurodegenerative diseases, including Alzheimer's disease (Ahlijanian et al., 2000, Kesavapany et al., 2007). Previous studies have described that hyperphosphorylation of NF in neuronal cell bodies leads to neuronal cell death (Ahlijanian et al., 2000; Rao and Nixon, 2003; Kesavapany et al., 2008). Furthermore, hyperphosphorylation of neurofilament in perikarya leads to inhibition of general anterograde transport of NF and eventually leads to cell death. To extend these findings, we asked whether hyperphosphorylation of NF in cell bodies in OA-treated neurons causes cell death. To test this possibility, we measured apoptosis by using TUNEL assay. Treatment of cortical neurons with 0.25 μm OA resulted in the increase in the TUNEL-positive neurons in a time-dependent manner (Fig. 10). The percentage apoptosis is gradually increased to 78% with 10 h OA treatment. Treatment of cortical neurons for longer times resulted in the detachment of neurons from the coverslip (data not shown).
Figure 10.

Inhibition of PP2A by OA increases apoptosis in a time-dependent manner. The presence of apoptotic neurons examined by TUNEL–tetramethylrhodamine red staining in 7 DIC cortical neurons. Nuclei were counterstained using DAPI. TUNEL-positive cells were increased during treatment of cortical neurons with OA in a time-dependent manner. A, The neuronal death is increased by nearly 28% (2.5 h), 43% (5 h), and 78% (10%) after treatment with 0.25 μm OA. B, The quantization in the bar graph represents TUNEL-positive counts from four separate experiments, in which 14 independent fields were counted. Ctrl, Control.
Inhibition of Pin1 rescues OA-induced apoptosis
We next examined whether Pin1 modulates OA-induced neuronal cell death. In normal neurons, ∼5% of untreated cortical neurons were TUNEL positive (basal level), and this increased to >46% during OA treatment for 5 h (Fig. 11 A,B). Inhibition of Pin1 by Pin1 siRNA reduced TUNEL-positive neurons to 10%, nearly comparable with nontreated neurons. Representative TUNEL images are shown, in which quantification in the bar graph represents TUNEL-positive counts from four separate experiments, in which 10 independent fields were counted (Fig. 11). Furthermore, treatment of cortical neurons with Fos increases the TUNEL-positive neurons by 38% (basal level TUNEL-positive neurons was 7%), and inhibition of Pin1 rescues the neuronal apoptosis in Fos-treated cells (Fig. 11 C). These results were further confirmed by transfecting cortical neurons with GFP–DN Pin1. Figure 11D shows that the GFP–DN Pin1-transfected neurons survive during OA-induced neuronal apoptosis.
Figure 11.
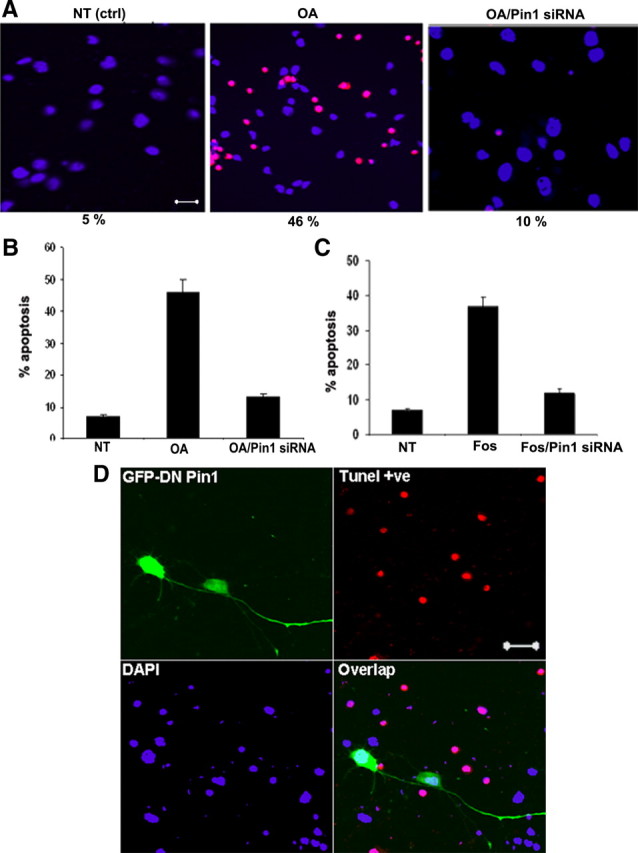
Inhibition of Pin1 reduces OA/Fos-induced neuronal death. A, Presence of apoptotic neurons examined by TUNEL–tetramethylrhodamine red staining in 7 DIC cortical neurons. Nuclei were counterstained using DAPI (blue). TUNEL-positive neurons were increased during OA treatment for 5 h and declined in OA-treated neurons transfected with Pin1 siRNA. Scale bar, 20 μm. The quantization in the bar graph (B) represents TUNEL-positive counts from four separate experiments, in which 12 independent fields were counted. NT (ctrl), Nontreated controls. B, The neuronal death is increased by 48% during exposure to OA (basal level, 5%), and this was reduced to 10% in neurons transfected with Pin1 siRNA and subjected to OA. C, The neuronal death is increased by 35% during exposure to Fos, and this was reduced to 12% in neurons transfected with Pin1 siRNA and subjected to Fos. D, OA-mediated neuronal apoptosis is reduced by overexpression of DN Pin1. Five-day-old cortical neurons were transfected with DN Pin1 and, after 24 h, were treated with 0.1 μm OA for 5 h. The GFP–DN Pin1-transfected neurons survive OA-induced cell death. Scale bar, 20 μm.
Downregulation of PP2A activity in ALS spinal cord and AD brain
Aberrant accumulation of extensively phosphorylated NF proteins are observed in ALS (Hirano et al., 1984; Hirano, 1991; Wharton and Ince, 2003; Green et al., 2005; Strong et al., 2005; Xiao et al., 2006). However, PP2A activity from ALS spinal cord lysate has not been studied so far. To determine whether PP2A activity is altered in ALS, we measured PP2A activity by a colorimetric assay. Homogenates were obtained from lumbar spinal cords of matched controls and ALS, immunoprecipitated with PP2A antibodies. The activities were determined in triplicate samples, by using phospho-peptide substrate that can be dephosphorylated by PP2A. Dephosphorylation of the peptide was determined by measuring the release of phosphate using a quantitative malachite-based colorimetric assay. The results show that PP2A activity is reduced in lumbar ALS spinal cord lysate (1520 ± 110 pmol/min) compared with matched controls (2611 ± 180 pmol/min). We measured the PP2A activity from adult AD brains and found that PP2A activity is downregulated in AD brain (997 ± 89 pmol/min) compared with matched controls (2016 ± 199 pmol/min).
Discussion
Aberrant phosphorylation of NF-M/H causes the NF to form aggregates of phospho-NF-M/H in neuronal perikarya in AD and ALS. Our understanding of this phenomenon has improved over the past decade, although the mechanism underlying the hyperphosphorylation of NF is still unclear (Sternberger et al., 1985; Goldman and Yen, 1986; Lee et al., 1988; Manetto et al., 1988; Veeranna et al., 1998; Grant and Pant, 2000; Zheng et al., 2009). Proline-directed kinases, such as Cdk5, MAPKs, and glycogen synthase kinase 3, have been shown to be implicated in the aberrant phosphorylation of NF, but the role of phosphatases has not yet been explored. Compared with the hundreds of known kinases, there are only a handful of phosphatases, and each of them must dephosphorylate a wide range of protein substrates. Dephosphorylation of Tau has been extensively studied. PP1, PP2A, and PP2B have all been shown to dephosphorylate Tau in vitro. In transgenic mice with reduced PP2A activity, Tau becomes hyperphosphorylated and redistributed in CNS neurons (Kins et al., 2003). This is consistent with the reduced expression of the phosphatase in AD. Previous studies have suggested that Ser/Thr phosphatases are involved in neurodegeneration (Vogelsberg-Ragaglia et al., 2001). We found that the PP2A activity is reduced in ALS spinal cord and AD brain. In this study, we have used specific inhibitors to study the NF dephosphorylation in cortical neurons. The PP1/PP2A inhibitors, such as OA and microcystin LR, cause hyperphosphorylation of NF in primary cortical cultures. Treatment of cortical neurons with Fos, a more selective inhibitor of PP2A, results in the perikaryal hyperphosphorylation of NF. Conversely, the PP2B-specific inhibitor cyclosporine A did not affect the perikaryal hyperphosphorylation of NF. Previous studies performed by us and in other laboratories has shown that PP2A is the major NF phosphatase, and PP2B contributes less toward the dephosphorylation of NF. In addition, Strack et al. (1997) reported that PP2A and PP1 were associated with the spinal cord NF fraction, but PP2B was found exclusively in the low-molecular-weight fraction. They attributed 75% of NF dephosphorylation to PP2A and the rest to PP1. We performed dose-dependent analysis of cyclosporine A to verify the neurofilament phosphorylation and observed no effect on perikaryal phosphorylation of NF. In previous studies using the squid giant axon system, we have shown that PP2B is abundant in the axons compared with cell bodies (Krinks et al., 1988). PP2B levels and activity are shown to be increased in AD brain (Liu et al., 2005).
We show here that Pin1 affects the perikaryal phosphorylation of NF by PP2A in vitro and in primary neuronal cultures. Pin1 prevents the dephosphorylation of NF by PP2A in vitro.
Inhibition of Pin1 levels (Pin1 siRNA) as well as Pin1 activity (DN Pin1) inhibits the OA-induced perikaryal phosphorylation of NF. This study further shows that OA-mediated hyperphosphorylation of NF is not attributable to the activation of JNK, ERK, or Cdk5 but is a consequence of dephosphorylation of NF. In AD, the PP2A activity is downregulated (Sontag et al., 2004; Liu et al., 2005). Our study also shows that the KSP tail domain dephosphorylation of NF-M/H by PP2A is regulated by Pin1.
Previous studies by Mushynski and coworkers have reported an increase in phosphorylation of the N-terminal domain of NF-L and NF fragmentation in OA-treated DRG neurons. The punctuate appearance of axonal NF could be attributable to the fragmentation of hyperphosphorylation of these molecules as suggested previously (Sacher et al., 1992, 1994). OA has been noted to disrupt the NF network in DRG neurons and increase deposits of NF subunits in nb2a/d1 neuroblastoma cells (Shea et al., 1993).
NF phosphorylation has been shown to regulate the axonal transport of newly synthesized NFs. Shea et al. (2004) reported that the serine/threonine kinase Cdk5 regulates phosphorylation and axonal transport of NFs. They found that increasing Cdk5 activity in cultured chicken dorsal root ganglion neurons increases phosphorylation of the NFs, causes abnormal deposits of aggregated NFPs in the perikaryon, and reduces their axonal transport. Furthermore, they show that injection of OA into murine retina fostered the appearance of phospho-NF accumulations within the perikarya in situ and slowed the axonal transport (Jung and Shea, 2004). Previous studies performed with EGFP–NF-M have shown that NFs are transported rapidly with frequent pauses (Wang et al., 2000). Alterations of anterograde axonal transport and fragmentation have been observed in patients and animal models of ALS and other neurodegenerative diseases. Phosphorylated NF protein accumulations in the perikarya and proximal axons of motor neurons have been shown to block axonal transport of cytoskeletal proteins and mitochondria and eventually cause axonal degeneration (Hoffman and Lasek, 1975). Sporadic ALS patients have NF-H alleles with deletions in the KSP repeat domain, which suggests that altered phosphorylation may indeed be a cause for NF accumulations (Figlewicz et al., 1994., Julien, 1995). Our study reveals that inhibition of PP2A by OA affects axonal transport of transfected NF-H and induces accumulation of phospho-NF-M/H in these loci, whereas inhibition of Pin1 rescues this effect. These findings indicate that Pin1 regulates NF dynamics.
Based on our findings, we propose a model for topographic phosphorylation of NF (Fig. 12). In normal neurons, PP2A activity is high in cell bodies, and phosphorylation of NF occurs selectively in axonal compartments. Glial–axonal interactions may activate the proline-directed Ser/Thr kinases; phosphorylation of S/T-P residues of the tail domain of NF-M/H occurs in axons (Dashiell et al., 2002). The phosphorylation of initial S/T-P residues in the tail domain may induce spontaneous transitions to cis conformation for some of the peptidyl-prolyl bonds. Pin1 binding to phospho-S/T-P sites converts cis-peptidyl bonds to the trans configuration. Thus, Pin1 stabilizes the NF tail domain phosphorylation of S/T-P motifs in the axonal compartment. Pin1 facilitates NF phosphorylation in the axonal compartment of almost all S/T-P residues. However, when PP2A activity is inhibited, NF-M/H are phosphorylated on S/T-P sites within the cell body compartment. This is consistent with the known requirement of Pin1 for S/T phosphorylation before its binding and isomerization of such sites. Aggregated phospho-NF proteins accumulate in the cell bodies and proximal axons and impair NF axonal transport. Accordingly, inhibition of Pin1 inhibits the OA-induced hyperphosphorylation of NF and rescues the axonal transport of NF.
Figure 12.

Role of Pin1 in the progression of compartment-specific NF-M/H phosphorylation. Top, In normal neurons, the NF phosphorylation is selective in the axonal compartment in normal neurons. After NF protein synthesis, higher activity of PP2A in cell bodies inhibits the NF tail domain phosphorylation. Although Pin1 is localized in cell bodies, it cannot act on the non-S/T-P phosphorylated proteins. During assembly of NF in axonal hillock region, NFs are transported by slow axonal transport. In the axonal compartment, the axonal–glial interaction activates the Ser/Thr kinases and phosphorylates some of pSer/Thr-Pro residues. The phosphorylation of these residues renders the phospho-Ser/Thr-Pro to remain in cis configuration and blocks additional phosphorylation. However, Pin1 induces cis-trans isomerization, increases the accessibility of the Ser/Thr kinases, and stabilizes the NF phosphorylation. Because of the high PP2A activity, there is no NF phosphorylation in the cell bodies. Bottom, Inhibition of PP2A activity leads to aberrant perikaryal hyperphosphorylation of NF proteins in cell bodies. Pin1 stabilizes the NF phosphorylation in the cell bodies, phospho-NF proteins are accumulated in the cell bodies and proximal axons, and NF transport is impaired. This is consistent with the observation that PP2A activity is reduced in AD brain and ALS spinal cord with higher accumulations of phospho-NF in the perikarya.
Pin1 action on its target proteins depends on their structure. Pin1 facilitates the dephosphorylation of Tau by PP2A at a specific single residue, Thr 231 (Lu et al., 1999). In contrast, Pin1 inhibits the dephosphorylation of multiple SP repeats of RNA polymerase C-terminal domain (CTD) by FCP1 phosphatase (Xu et al., 2003). Similar to CTD, rat NF-H has 52 SP repeats in the tail domain. The mechanism of Pin1 action on NF appears to be similar to RNA polymerase II. This raises an intriguing possibility that proteins with multiple SP repeats have similar mechanisms of Pin1 action.
Protein–protein interactions have been reported to sterically inhibit protein phosphatase activity (Palancade et al., 2004). For example, Pin1 binding to multiphosphorylated transcription factor NFAT (nuclear cofactor of activated T cells) inhibits its additional dephosphorylation by calcineurin. The binding of Pin1 to its phosphorylated substrate can inhibit its subsequent dephosphorylation by specific protein phosphatases. This inhibition might be attributable to either a direct masking of the phosphorylated site(s) targeted by the phosphatase or a failure to recruit the phosphatase by masking the docking site within the substrate.
Juglone has been used previously as a pharmacological Pin1 inhibitor. Accumulating evidence suggests that juglone is a nonspecific inhibitor of Pin1, because it also blocks transcription (Chao et al., 2001). In addition, juglone inhibits numerous parvulins as well as other enzymes with Cys residues in their catalytic domains (Fila et al., 2008). A specific Pin1 inhibitor has not been identified so far. To inhibit cellular Pin1 function, we used DN Pin1 (Ryo et al., 2002; Lam et al., 2008). We also used Pin1 siRNA to specifically inhibit Pin1 levels.
Our finding that Pin1 regulates dephosphorylation of NF mediated by PP2A indicates that Pin1 has a direct role in phospho-NFP aggregation in AD, ALS, and related neurodegenerative disorders. Pin1 also increases the phosphorylation of NF by proline-directed kinases, such as MAPKs, Cdk5, and JNK3 (Rudrabhatla et al., 2008). This study suggests a novel mechanism whereby Pin1 not only inhibits dephosphorylation but induces sequential phosphorylation of S/T-P residues in a protein. Exactly how this occurs is unknown, but it is consistent with previous findings that Pin1 regulates the function of its substrate by different mechanisms (Lu and Zhou, 2007; Rudrabhatla and Pant, 2009). Thus, phosphorylation-specific Pin1 could be an attractive therapeutic target to inhibit the perikaryal hyperphosphorylation of NF caused by downregulation of PP2A activity in AD, PD, ALS, and/or related pathology.
Footnotes
This work was supported by the National Institutes of Health intramural research programs of National Institute of Neurological Disorders and Stroke. We thank Christine Winters for providing rat cortices. We thank Dr. Veeranna for helpful discussions.
References
- Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci U S A. 2000;97:2910–2915. doi: 10.1073/pnas.040577797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao SH, Greenleaf AL, Price DH. Juglone, an inhibitor of the peptidyl-prolyl isomerase Pin1, also directly blocks transcription. Nucleic Acids Res. 2001;29:767–773. doi: 10.1093/nar/29.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin SS, Liem RK. Transfected rat high-molecular-weight neurofilament (NF-H) coassembles with vimentin in a predominantly nonphosphorylated form. J Neurosci. 1990;10:3714–3726. doi: 10.1523/JNEUROSCI.10-11-03714.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashiell SM, Tanner SL, Pant HC, Quarles RH. Myelin-associated glycoprotein modulates expression and phosphorylation of neuronal cytoskeletal elements and their associated kinases. J Neurochem. 2002;81:1263–1272. doi: 10.1046/j.1471-4159.2002.00927.x. [DOI] [PubMed] [Google Scholar]
- de Waegh SM, Lee VM, Brady ST. Local modulation of neurofilament phosphorylation, axonal caliber and slow axonal transport by mylenating Schwann cells. Cell. 1992;68:451–463. doi: 10.1016/0092-8674(92)90183-d. [DOI] [PubMed] [Google Scholar]
- Eagles PAM, Pant HC, Gainer H. Neurofilaments. In: Goldman RD, Steinert PM, editors. Cellular and molecular biology of intermediate filaments. New York: Plenum; 1990. pp. 951–958. [Google Scholar]
- Figlewicz DA, Krizus A, Martinoli MG, Meininger V, Dib M, Rouleau GA, Julien JP. Familial amyotrophic lateral sclerosis: a review. Hum Mol Genet. 1994;3:1757–1761. doi: 10.1093/hmg/3.10.1757. [DOI] [PubMed] [Google Scholar]
- Fila C, Metz C, van der Sluijs P. Juglone inactivates cysteine-rich proteins required for progression through mitosis. J Biol Chem. 2008;283:21714–21724. doi: 10.1074/jbc.M710264200. [DOI] [PubMed] [Google Scholar]
- Goldman JE, Yen SH. Cytoskeletal protein abnormalities in neurodegenerative diseases. Ann Neurol. 1986;19:209–223. doi: 10.1002/ana.410190302. [DOI] [PubMed] [Google Scholar]
- Grant P, Pant HC. Neurofilament protein synthesis and phosphorylation. J Neurocytol. 2000;29:843–872. doi: 10.1023/a:1010999509251. [DOI] [PubMed] [Google Scholar]
- Green SL, Westendorf JM, Jaffe H, Pant HC, Cork LC, Ostrander EA, Vignaux F, Ferrell JE., Jr Allelic variants of the canine heavy neurofilament (NFH) subunit and extensive phosphorylation in dogs with motor neuron disease. J Comp Pathol. 2005;132:33–50. doi: 10.1016/j.jcpa.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Guan L, Song K, Pysz MA, Curry KJ, Hizli AA, Danielpour D, Black AR, Black JD. Protein kinase C-mediated down-regulation of cyclin D1 involves activation of the translational repressor 4E-BP1 via a phosphoinositide 3-kinase/Akt-independent, protein phosphatase 2A-dependent mechanism in intestinal epithelial cells. J Biol Chem. 2007;282:14213–14225. doi: 10.1074/jbc.M610513200. [DOI] [PubMed] [Google Scholar]
- Guru SC, Shetty KT, Shankar SK. Effect of chronic ethanol ingestion on phosphate content of NF proteins and NF associated protein phosphatase in rat spinal cord. Neurochem Res. 1991;16:1193–1197. doi: 10.1007/BF00966695. [DOI] [PubMed] [Google Scholar]
- Hirano A. Cytopathology of amyotrophic lateral sclerosis. Adv Neurol. 1991;56:91–101. [PubMed] [Google Scholar]
- Hirano A, Donnenfeld H, Sasaki S, Nakano I. Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1984;43:461–470. doi: 10.1097/00005072-198409000-00001. [DOI] [PubMed] [Google Scholar]
- Hoffman PN, Lasek RJ. The slow component of axonal transport. Identification of major structural polypeptides of the axon and their generality among mammalian neurons. J Cell Biol. 1975;66:351–366. doi: 10.1083/jcb.66.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe H, Veeranna, Pant HC. Characterization of serine and threonine phosphorylation sites in beta-elimination/ethanethiol addition-modified proteins by electrospray tandem mass spectrometry and database searching. Biochemistry. 1998;37:16211–16224. doi: 10.1021/bi981264p. [DOI] [PubMed] [Google Scholar]
- Julien JP. A role of neurofilaments in the pathogenesis of amyotrophic lateral sclerosis. Biochem Cell Biol. 1995;73:593–597. doi: 10.1139/o95-064. [DOI] [PubMed] [Google Scholar]
- Jung C, Shea TB. Neurofilament subunits undergo more rapid translocation within retinas than in optic axons. Brain Res Mol Brain Res. 2004;122:188–192. doi: 10.1016/j.molbrainres.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Kesavapany S, Patel V, Zheng YL, Pareek TK, Bjelogrlic M, Albers W, Amin N, Jaffe H, Gutkind JS, Strong MJ, Grant P, Pant HC. Inhibition of Pin1 reduces glutamate-induced perikaryal accumulation of phosphorylated neurofilament-H in neurons. Mol Biol Cell. 2007;18:3645–3655. doi: 10.1091/mbc.E07-03-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kins S, Kurosinski P, Nitsch RM, Götz J. Activation of the ERK and JNK signaling pathways caused by neuron-specific inhibition of PP2A in transgenic mice. Am J Pathol. 2003;163:833–843. doi: 10.1016/S0002-9440(10)63444-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krinks MH, Klee CB, Pant HC, Gainer H. Identification and quantification of calcium-binding proteins in squid axoplasm. J Neurosci. 1988;8:2172–2182. doi: 10.1523/JNEUROSCI.08-06-02172.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam PB, Burga LN, Wu BP, Hofstatter EW, Lu KP, Wulf GM. Prolyl isomerase Pin1 is highly expressed in Her2-positive breast cancer and regulates erbB2 protein stability. Mol Cancer. 2008;7:91. doi: 10.1186/1476-4598-7-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Otvos L, Jr, Carden MJ, Hollosi M, Dietzschold B, Lazzarini RA. Identification of the major multiphosphorylation site in mammalian neurofilaments. Proc Natl Acad Sci U S A. 1988;85:1998–2002. doi: 10.1073/pnas.85.6.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22:1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signaling and disease. Nat Rev Mol Cell Biol. 2007;8:904–916. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- Manetto V, Perry G, Tabaton M, Mulvihill P, Fried VA, Smith HT, Gambetti P, Autilio-Gambetti L. Ubiquitin is associated with abnormal cytoplasmic filaments characteristic of neurodegenerative diseases. Proc Natl Acad Sci U S A. 1988;85:4501–4505. doi: 10.1073/pnas.85.12.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata M, Kupina N, Fink DJ. Phosphorylation dependent neurofilament epitopes are reduced at the node of Ranvier. J Neurocytol. 1992;21:199–210. doi: 10.1007/BF01194978. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Lewis SE, Marotta CA. Posttranslational modification of neurofilament proteins by phosphate during axoplasmic transport in retinal ganglion cell neurons. J Neurosci. 1987;7:1145–1158. doi: 10.1523/JNEUROSCI.07-04-01145.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palancade B, Marshall NF, Tremeau-Bravard A, Bensaude O, Dahmus ME, Dubois MF. Dephosphorylation of RNA polymerase II by CTD-phosphatase FCP1 is inhibited by phospho-CTD associating proteins. J Mol Biol. 2004;335:415–424. doi: 10.1016/j.jmb.2003.10.036. [DOI] [PubMed] [Google Scholar]
- Rao MV, Nixon RA. Defective neurofilament transport in mouse models of amyotrophic lateral sclerosis: a review. Neurochem Res. 2003;28:1041–1047. doi: 10.1023/a:1023259207015. [DOI] [PubMed] [Google Scholar]
- Rudrabhatla P, Pant HC. Phosphorylation-specific peptidyl-prolyl isomerization of neuronal cytoskeletal proteins by Pin1: implications for therapeutics in neurodegeneration. J Alzheimers Dis. 2009 doi: 10.3233/JAD-2010-1243. Advance online publication. Retrieved November 12, 2009. doi: 10.3233/JAD-2009-1243. [DOI] [PubMed] [Google Scholar]
- Rudrabhatla P, Zheng YL, Amin ND, Kesavapany S, Albers W, Pant HC. Pin1-dependent prolyl isomerization modulates the stress-induced phosphorylation of high molecular weight neurofilament protein. J Biol Chem. 2008;283:26737–26747. doi: 10.1074/jbc.M801633200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryo A, Liou YC, Wulf G, Nakamura M, Lee SW, Lu KP. PIN1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Mol Cell Biol. 2002;22:5281–5295. doi: 10.1128/MCB.22.15.5281-5295.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacher MG, Athlan ES, Mushynski WE. Okadaic acid induces the rapid and reversible disruption of the neurofilament network in rat dorsal root ganglion neurons. Biochem Biophys Res Commun. 1992;186:524–530. doi: 10.1016/s0006-291x(05)80839-3. [DOI] [PubMed] [Google Scholar]
- Sacher MG, Athlan ES, Mushynski WE. Increased phosphorylation of the amino-terminal domain of the low molecular weight neurofilament subunit in okadaic acid-treated neurons. J Biol Chem. 1994;269:18480–18484. [PubMed] [Google Scholar]
- Schilling K, Duvernoy C, Keck S, Pilgrim C. Detection and partial characterization of a developmentally regulated nuclear antigen in neural cells in vitro and in vivo . J Histochem Cytochem. 1989;37:241–247. doi: 10.1177/37.2.2492046. [DOI] [PubMed] [Google Scholar]
- Shea TB, Paskevich PA, Beermann ML. The protein phosphatase inhibitor okadaic acid increases axonal neurofilaments and neurite caliber, and decreases axonal microtubules in NB2a/d1 cells. J Neurosci Res. 1993;35:507–521. doi: 10.1002/jnr.490350507. [DOI] [PubMed] [Google Scholar]
- Shea TB, Zheng YL, Ortiz D, Pant HC. Cyclin-dependent kinase 5 increases perikaryal neurofilament phosphorylation and inhibits neurofilament axonal transport. J Neurosci Res. 2004;76:795–800. doi: 10.1002/jnr.20099. [DOI] [PubMed] [Google Scholar]
- Shetty KT, Veeranna, Guru SC. Effect of chronic ethanol ingestion on phosphate content of neurofilament proteins and neurofilament associated protein phosphatase in rat spinal cord. Neurosci Lett. 1992;137:83–86. [Google Scholar]
- Sim ATR. The regulation and function of protein phosphatases in the brain. Mol Neurobiol. 1991;5:229–246. doi: 10.1007/BF02935548. [DOI] [PubMed] [Google Scholar]
- Sontag E, Luangpirom A, Hladik C, Mudrak I, Ogris E, Speciale S, White CL., Jr Altered expression levels of the protein phosphatase 2A ABalphaC enzyme are associated with Alzheimer disease pathology. J Neuropathol Exp Neurol. 2004;63:287–301. doi: 10.1093/jnen/63.4.287. [DOI] [PubMed] [Google Scholar]
- Sternberger NH, Sternberger LA, Ulrich J. Aberrant neurofilament phosphorylation in Alzheimer disease. Proc Natl Acad Sci U S A. 1985;82:4274–4276. doi: 10.1073/pnas.82.12.4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, Westphal RS, Colbran RJ, Ebner FF, Wadzinski BE. Protein serine/threonine phosphatase 1 and 2A associate with and dephosphorylate neurofilaments. Brain Res Mol Brain Res. 1997;49:15–28. doi: 10.1016/s0169-328x(97)00117-4. [DOI] [PubMed] [Google Scholar]
- Strong MJ, Kesavapany S, Pant HC. The pathobiology of amyotrophic lateral sclerosis: a proteinopathy? J Neuropathol Exp Neurol. 2005;64:649–664. doi: 10.1097/01.jnen.0000173889.71434.ea. [DOI] [PubMed] [Google Scholar]
- Tokutake S, Hutchison SB, Pachter JS, Liem RK. A batchwise purification of neurofilament proteins. Anal Biochem. 1983;135:102–105. doi: 10.1016/0003-2697(83)90736-4. [DOI] [PubMed] [Google Scholar]
- Veeranna, Shetty KT, Link WT, Jaffe H, Wang J, Pant HC. Neuronal cyclin-dependent kinase-5 phosphorylation sites in neurofilament protein (NF-H) are dephosphorylated by protein phosphatase 2A. J Neurochem. 1995;64:2681–2690. doi: 10.1046/j.1471-4159.1995.64062681.x. [DOI] [PubMed] [Google Scholar]
- Veeranna, Amin ND, Ahn NG, Jaffe H, Winters CA, Grant P, Pant HC. Mitogen-activated protein kinases (Erk1,2) phosphorylate Lys-Ser-Pro (KSP) repeats in neurofilament proteins NF-H and NF-M. J Neurosci. 1998;18:4008–4021. doi: 10.1523/JNEUROSCI.18-11-04008.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VM. PP2A mRNA expression is quantitatively decreased in Alzheimer's disease hippocampus. Exp Neurol. 2001;168:402–412. doi: 10.1006/exnr.2001.7630. [DOI] [PubMed] [Google Scholar]
- Wang L, Ho CL, Sun D, Liem RKH, Brown A. Rapid movement of axonal neurofilaments interrupted by prolonged pauses. Nat Cell Biol. 2000;2:137–141. doi: 10.1038/35004008. [DOI] [PubMed] [Google Scholar]
- Weigum SE, García DM, Raabe TD, Christodoulides N, Koke JR. Discrete nuclear structures in actively growing neuroblastoma cells are revealed by antibodies raised against phosphorylated neurofilament proteins. BMC Neurosci. 2003;4:6. doi: 10.1186/1471-2202-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton S, Ince PG. Pathology of motor neuron disorders. In: Shaw PJ, Strong MJ, editors. Motor neuron disorders blue books of practical neurology. Philadelphia: Butterworth Heinemann; 2003. pp. 17–49. [Google Scholar]
- Xiao S, McLean J, Robertson J. Neuronal intermediate filaments and ALS: a new look at an old question. Biochim Biophys Acta. 2006;1762:1001–1012. doi: 10.1016/j.bbadis.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Xu YX, Hirose Y, Zhou XZ, Lu KP, Manley JL. Pin1 modulates the structure and function of human RNA polymerase II. Genes Dev. 2003;17:2765–2776. doi: 10.1101/gad.1135503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabe JT, Pimenta A, Shea TB. Kinesin-mediated transport of neurofilament protein oligomers in growing axons. J Cell Sci. 1999;112:3799–3814. doi: 10.1242/jcs.112.21.3799. [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld JU, Xu J, Kuang J, Kirschner MW, Fischer G, Cantley LC, Lu KP. Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science. 1997;278:1957–1960. doi: 10.1126/science.278.5345.1957. [DOI] [PubMed] [Google Scholar]
- Zheng YL, Amin ND, Rudrabhatla P, Kesavapany S, Pant HC. Neuronal cytoskeleton regulation and neurodegeneration. In: Maccioni RB, Perry G, editors. Current hypotheses and research milestones in Alzheimer's disease. New York: Springer; 2009. pp. 1–16. [Google Scholar]
- Zhou XZ, Kops O, Werner A, Lu PJ, Shen M, Stoller G, K̈ullertz G, Stark M, Fischer G, Lu KP. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol Cell. 2000;6:873–883. doi: 10.1016/s1097-2765(05)00083-3. [DOI] [PubMed] [Google Scholar]