

Abstract
The tumor microenvironment is characterized by of high levels of extracellular nucleotides that are metabolized through the dynamic and sequential action of cell surface enzymes (ectoenzymes). These ectoenzymes operate according to their spatial arrangement, as part of (1) continuous (molecules on the same cell) or (2) discontinuous (molecules on different cells) pathways, the latter being facilitated by restricted cellular microenvironment. The outcome of this catabolic activity is an increase in the local concentration of adenosine, a nucleoside involved in the control of inflammation and immune responses. The aim of the work presented here was to demonstrate that a previously unexplored enzymatic pathway may be an alternate route to produce extracellular adenosine. Our data show that this new axis is driven by the nucleotide-metabolizing ectoenzymes CD38 (an NAD+ nucleosidase), the ecto-nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1, also known as CD203a or PC-1) and the 5′ ectonucleotidase (5′-NT) CD73, while bypassing the canonical catabolic pathway mediated by the nucleoside tri- and diphosphohydrolase (NTPDase) CD39. To determine the relative contributions of these cell surface enzymes to the production of adenosine, we exploited a human T-cell model allowing for the modular expression of the individual components of this alternative pathway upon activation and transfection. The biochemical analysis of the products of these ectoenzymes by high-performance liquid chromatography (HPLC) fully substantiated our working hypothesis. This newly characterized pathway may facilitate the emergence of an adaptive immune response in selected cellular contexts. Considering the role for extracellular adenosine in the regulation of inflammation and immunogenicity, this pathway could constitute a novel strategy of tumor evasion, implying that these enzymes may represent ideal targets for antibody-mediated therapy.
Keywords: CD203a, CD38, NAD+, PC-1, adenosine, ectonucleotidases
Introduction
The immune system is endowed with multiple and diverse effector functions based upon the activity of cells or soluble factors, which are frequently shared by its innate and adaptive branches.1 Intuitively, one would expect immune defense mechanisms to work efficiently and with minimal collateral damage. Evolution has educated the immune system to achieve an acceptable balance between potent effector activities, on the one hand, and preservation of the host on the other. These intertwined biological systems are conventionally investigated by dissecting the different components and their roles in orchestrating events.2 Here, we analyze immunity-related extracellular nucleotides and the network of cell surface enzymes (ectoenzymes) involved in their metabolism, a set of proteins that are phylogenetically highly conserved.3,4
One component of the larger network integrating cellular energy status and immune signals is represented by nicotinamide adenine dinucleotide (NAD+).5,6 Results from a variety of studies consistently demonstrate that ectonucleotidases serve as bridges connecting cells with their microenvironment, including the immune system. The outcome of this interplay may be an alteration in the cellular energy status, as well as the induction of leukocyte activation or—counterintuitively—suppression. These effects appear to be mediated by the products of these ectoenzymes, including adenosine, a pleiotropic nucleoside involved in the modulation of immunity and inflammation.7,8 Indeed, adenosine acts through a negative feedback loop to inhibit the hyperactivation of immune cells, thereby increasing the gap between immune-mediated collateral tissue damage and disease.8,9
Extracellular ATP is considered as the primary substrate of the ectoenzymes that produce adenosine. At sites of immune activation, extracellular ATP is typically first hydrolyzed by the ecto-nucleoside triphosphate diphosphohydrolase (NTPDase) CD39. The AMP molecules produced by CD39 can be further hydrolyzed by the 5′-nucleotidase (5′-NT) CD73, thereby generating adenosine and inorganic phosphate, which complete the adenosinergic loop.10,11 The canonical pathway of adenosine production has various points of vulnerability. For instance, CD39 is responsible for the inefficient effector T cell responses in patients with chronic HIV-1 infection. However, blocking the enzymatic activity of CD39 is not sufficient to fully recover T-cell functions and overcome the immunosuppression associated with retroviral infections or cancer.12,13
Thus, we considered the hypothesis that CD39 is not the exclusive switch of the immune system to trigger immunosuppression, but an alternative adenosine-generating axis is operative. Our starting point was the notion that NAD+ and ATP share common structural characteristics, both possessing an adenine moiety. Moreover, NAD+ acts as an immunomodulator for human T lymphocytes.5,6 Finally, besides its vital functions in the generation of cellular energy, NAD+ has acquired new attention as a modulator of extracellular signaling pathways, mainly in cancer.14 NAD+ can be actively secreted across NAD+-impermeable plasma membranes, hence triggering a cascade of extracellular signals that leads to cell activation through purinergic P2 receptors.15,16 However, the concentration of NAD+ required for P2 activation is relatively high, suggesting the existence of alternative homeostatic mechanism(s). Indeed, the conversion of NAD+ into AMP in the immune microenvironment has been previously considered among the alternative mechanisms promoting the generation of extracellular adenosine.17,18
Important functions have been attributed to extracellular NAD+ including the regulation of inflammatory responses and regulatory T cells (Tregs).19 Such activities are finely tuned by interactions taking place between the NAD+ nucleosidase CD38 and a variety of other cell surface molecules.3,20 In light of these observations, we exploited a modular human T-cell model to demonstrate that the production of adenosine from NAD+ can mediated by a network of ectoenzymes involving CD38 as well as ecto-nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1, better known as CD203a or PC-1) and CD73. The main functional feature of this adenosinergic signaling axis that distinguishes it from canonical ectonucleotidase pathways is its independence from CD39. Indeed, NAD+ is hydrolyzed by CD38 generating adenosine diphosphate ribose (ADPR), either directly or through a cyclic ADPR (cADPR) intermediate.21 Our focus on CD203a derived from its broad substrate specificity, as this enzyme is capable of hydrolyzing both NAD+ and ADPR to produce AMP.22,23 Accordingly, NAD+ fluxes through an ectonucleotidase cascade involving CD73 culminate in the formation of immunosuppressive adenosine.24-26 In turn, adenosine can bind to specific purinergic P1 receptors and elicit immunosuppressive signals by increasing cAMP levels.27,28 Here, we test the assumption that the CD38/CD203a/CD73 pathway serves to balance the local concentrations of NAD+ and adenosine, hence determining the hierarchy of engagement of the P2 or P1 receptors to be preferentially activated.29,30
Results
Expression of nucleotide-hydrolyzing ectoenzymes by the human Jurkat T cell line in resting and activated conditions
Cytofluorometric analysis revealed that the NAD+-consuming CD38 ectoenzyme is expressed by the vast majority (95 ± 3% mean ± SEM) of Jurkat/VR cells, with minor variations in mean fluorescence intensity (MFI; mean, 152 ± 25) (Fig. 1A). Thus, the T-cell population is relatively homogeneous with regard to CD38 expression, and staining yielded a single peak. We next tested the expression of other ectoenzymes on the cell surface of either resting or activated Jurkat/VR cells, including CD203a, CD39, CD73, and CD26 (Fig. 1A-D). CD39, CD73 and CD26 were expressed neither by resting nor by activated Jurkat/VR (hereafter referred to as Jurkat/CD73-) cells. In contrast to CD38, CD203a was barely expressed by Jurkat/CD73− cells, unless these cells were activated by the mitogen phorbol 12-myristate 13-acetate (PMA) (Fig. 1B).

Figure 1. Expression of ectoenzymes on activated and resting Jurkat cell lines. (A-D) Comparative cytofluorometric analysis of Jurkat T cells stained with primary antibodies against various ectonucleotidases and detected with fluorescein (FITC)-conjugated secondary antibodies. Expression analysis of ectoenzymes (CD38, CD203a, CD73, CD39, and CD26) and T-cell activation marker (CD69) by Jurkat/CD73− (A and B) and Jurkat/CD73+ (C and D) cells either in resting conditions (A and C) or in response to phorbol 12-myristate 13-acetate (B and D). Representative histograms are shown. Grey peaks demarcate isotype control staining and black peaks depict the expression levels of the indicated markers.
The presence of CD38 and CD203a was confirmed both by the immunoblotting of plasma membrane-associated proteins from activated Jurkat/CD73−cells (Fig. 2A-B) and by enzymatic assays (see below). In addition to the constitutive expression of CD38, PMA-treated Jurkat/CD73− cells displayed a net increase of surface CD203a (Fig. 2B).
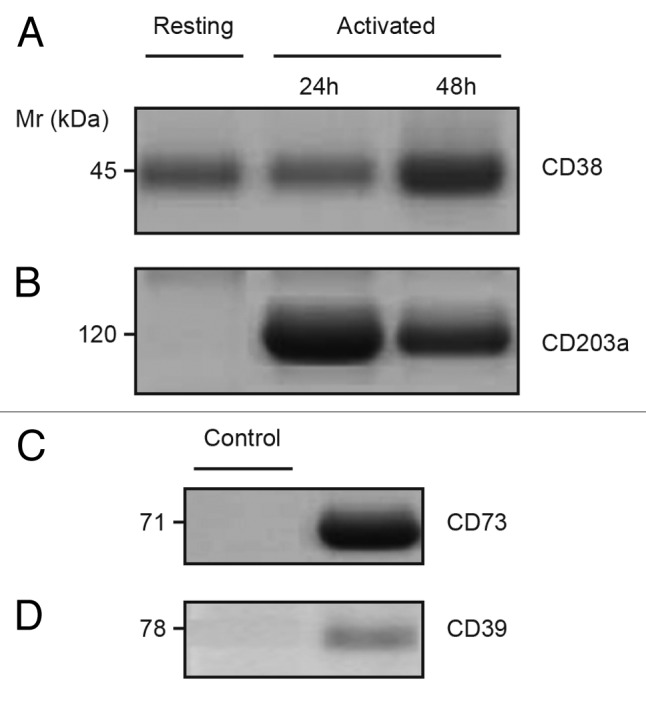
Figure 2. Constitutive expression of CD38 and elevated CD203a in activated Jurkat/CD73− cells. (A–B) Immunoblotting analysis of plasma membrane fractions from resting and phorbol 12-myristate 13-acetate (PMA)-activated Jurkat/CD73- cells probed with anti-CD38 (SUN-4B7, IgG1) mAbs or with anti-CD203a (3E8, IgG1) to detect the presence of 45-kDa CD38 (A) and 120-kDa CD203a (B) proteins. (C–D) Proteins with Mr of 71-kDa and 78-kDa, not expressed by Jurkat T cells (not shown), were immunodetected using lysates of epithelial cells from biopsied corneas used as positive controls and probed with anti-CD73 (CB73, IgG1) and anti-CD39 (IgG1) mAbs (right lanes). Isotype-matched irrelevant X63.Ag8 mAb was used as negative controls (left lanes). Mr = molecular weight”.
Metabolism of extracellular NAD+ in resting and activated Jurkat T cells: Extracellular conversion of NAD+ to ADPR via CD38
The HPLC-assisted analysis of the partial breakdown of exogenous NAD+ and the corresponding products in the supernatant of resting Jurkat/CD73− cells revealed the presence of non-consumed NAD+ together with the enzymatic products ADPR and nicotinamide (Nic) (Fig. 3A).

Figure 3. Analysis of NAD+ metabolites in the supernatant of Jurkat/CD73− cells. (A and B) The enzymatic conversion of NAD+ to adenosine diphosphate ribose (ADPR) (a CD38-dependent reaction) and to AMP (CD203a-dependent) was monitored by high-pressure liquid chromatography (HPLC). Representative resting (A) and phorbol 12-myristate 13-acetate (PMA)-activated-activated (B) Jurkat/CD73− cells upon incubation for 60 min at 37°C with 100 µM NAD+ are shown. The identity of peaks was confirmed by the co-migration and absorbance spectra of reference standards using a retention time (Rt) window of ± 5%. A representative plot of retention time of NAD+ metabolites in a single HPLC run is depicted on the right.
The same experiment performed 24–48 h after PMA activation revealed modifications on the output levels of NAD+ and ADPR (Fig. 3B), paralleling the changes in the pattern of expression of ectoenzymes. Besides the non-consumed NAD+, which could be identified by its unique retention time (Rt) of 2.80 min, the metabolic products of activated Jurkat/CD73− cells were ADPR, Nic and AMP, exhibiting corresponding Rt of 3.20, 6.87 and 2.15 min, respectively (Fig. 3B, right). The absence of transformation products with Rt ≥ 10 min rules out the presence of other catabolites in the supernatant from resting or PMA-activated Jurkat/CD73− cells (data not shown).
The NAD+ hydrolytic profile of PMA-treated Jurkat/CD73− cells, converting extracellular NAD+ into AMP, can be attributed to the nicotinamide adenine dinucleotidase (NADase) activity of CD38 and CD203a, which exhibits nucleotide pyrophosphatase/phosphodiesterase (NPP) activity. ADPR produced by CD38 upon the partial breakdown of NAD+ is generated outside the cell where it can subsequently be degraded to AMP by CD203a. In support of this notion, activated Jurkat/CD73− cells rapidly hydrolyzed extracellular NAD+, resulting in the accumulation of ADPR in the culture supernatant after 10–15 min of incubation (Fig. 3B). Since ADPR was the principal product of exogenously applied NAD+, we wondered whether it would be source of the AMP generated. Thus, we tested the functional activity of CD203a by directly applying ADPR, a known substrate for this ectoenzyme,23 to the Jurkat/CD73− cells. At variance with resting cells, PMA-activated Jurkat/CD73− cells displayed a marked increase in ADPR-hydrolyzing activity (data not shown). Another possible catabolite of this reaction, cyclic ADPR, could not be detected in the supernatant of resting or activated Jurkat/CD73− cells, either because it fell under the detection limit of the system (cADPR; Rt = 6.15 min) or was internalized.31
ATP is also a known substrate for CD203a.22 Time-course analysis of ATP consumption by activated Jurkat/CD73− cells allowed a comparison of the nucleotide transformation pathways. The incubation of activated Jurkat/CD73− cells with ATP led to a predominant accumulation of AMP, with no appreciable levels of ADP (Fig. 4). At variance with reports pointing to the CD39-mediated ATP hydrolysis to ADP and AMP by lymphoid cells,10 activated Jurkat/CD73− cells converted ATP directly to AMP, suggesting the presence of functional CD203a (Fig. 4). This finding was confirmed by the attenuation of this metabolic conversion in the presence of the CD203a inhibitor EDTA (data not shown). In contrast, sodium azide, a CD39 inhibitor, did not influence the production of AMP from NAD+, excluding a role for CD39 in the hydrolytic activity observed (data not shown). These findings support the view that the enzymatic activity of CD203a produce AMP by a secondary conversion of ADPR arising from the breakdown of NAD+ in a CD39-independent manner.
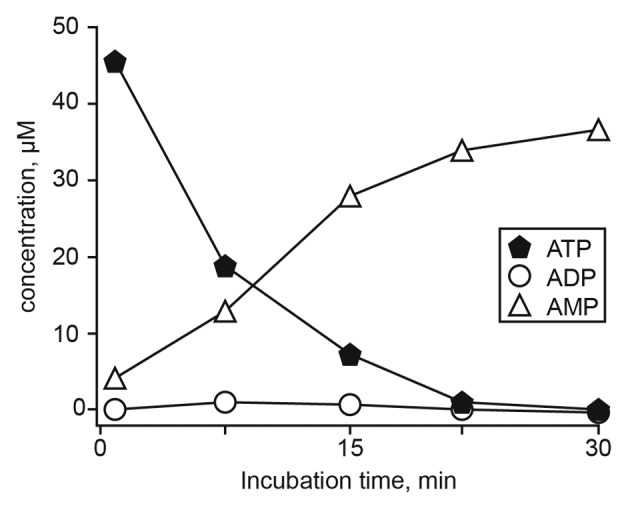
Figure 4. Time course of extracellular ATP metabolism in activated Jurkat/CD73− cells. ATP (100 μM) was added at time 0 and samples (500 µL) collected from the supernatants at the indicated times (abscissa). Sample were analyzed by HPLC to quantify ATP, ADP and AMP. The identity of peaks was confirmed by the co-migration and absorbance spectra of reference standards using a retention time (Rt) window of ± 5%. Vertical bars depicting the SEM do not exceed the size of symbols.
Metabolism of extracellular NAD+ in resting and activated Jurkat T cells: Extracellular conversion of NAD+ to nicotinamide mononucleotide and AMP via CD203a
Like nucleotides or nucleosides, NAD+ can be processed by more than one enzyme displaying the same or similar specificities, although the relative activity of each would depend upon the balance between the surface levels of the degradative machinery and modulations limiting the availability of various substrates.32 Indeed, activated Jurkat/CD73− cells generated AMP from NAD+ using either ADPR, as generated from CD38, or NAD+, as produced by CD203a.33 For this reason, we hypothesized that the observed kinetics of surface NADase activity in Jurkat/CD73− cells are driven in tandem with an NPP, which degrades NAD+ to nicotinamide mononucleotide (NMN) and AMP. Thus, we analyzed the products of NAD+ cleavage catalyzed by Jurkat/CD73− cells by HPLC to compare the relative ratios of ADPR (a NADase product) to NMN (a NPP product).
As summarized in Table 1, the appearance of ADPR (under both resting and activated conditions, and irrespective of CD73 status) testifies the hydrolysis of NAD+ as operated by Jurkat/CD73− cells via CD38. Similar to ADPR, Nic was a major transformation product released by both resting and activated Jurkat/CD73− cells. ADPR and Nic were released by resting Jurkat/CD73− cells in nearly equimolar amounts, suggesting an equivalent extracellular processing of NAD+ by NADase activity. The amount of NMN originating from NAD+ via CD203a was low to nil in the supernatants from Jurkat/CD73− cells, regardless of activation state. Furthermore, the addition of EDTA to Jurkat/CD73− cells did not modify the production rate of ADPR (data not shown). This indicates that the direct hydrolysis of NAD+ by any pyrophosphatase does not interfere with the NADase activity of CD38, and thus is apparently not operative in the Jurkat model.
Table 1. Extracellular conversion of NAD+ to ADPR and nicotinamide via CD38 and to NMN and AMP via CD203a.
| Cell clone | consumed |
produced |
||||
|---|---|---|---|---|---|---|
| NAD+ |
ADPR |
Nic |
NMN |
AMP |
||
| (nmoles) | (nmoles) | (nmoles) | (nmoles) | (nmoles) | ||
|
Jurkat/VR |
Resting |
20.21 |
13.05 |
16.38 |
0 |
0 |
|
(CD73-) |
Activated |
19.91 |
17.69 |
14.27 |
0.33 |
4.91 |
|
Jurkat/B-NT5.1 |
Resting |
32.88 |
19.27 |
30.79 |
0 |
0 |
| (CD73+) | Activated | 34.81 | 15.81 | 30.73 | 0.14 | 0 |
The ectonucleotidase activity of CD38 and CD203a was measured by using Jurkat T cells (2 × 106/mL) incubated for 30 min at 37°C in AIM V medium supplemented with 100 µM NAD+. Abbreviations: ADPR, adenosine diphosphate ribose; AMP, adenosine 5’-monophosphate; Nic, nicotinamide; NAD+, nicotinamide adenine dinucleotide; NMN, nicotinamide mononucleotide.
Thus, we next sought to test the effect of CD203a activity on NAD+ degradation (which leads to direct generation of AMP rather than via an ADPR intermediate) using nicotinamide guanine dinucleotide (NGD+), a NAD+ analog used to track the NAD+-cyclase activity of CD38.34 The NGD+-consuming activity of CD203a+ activated Jurkat/CD73− cells was undetectable, as inferred from the absence of GMP in their supernatant. Rather, these supernatants contained cyclic guanosine diphosphoribose (cGDPR), a product of cyclase activity of CD38 (data not shown).
Taken together, these findings confirm that AMP in the supernatant of activated Jurkat/CD73− cells derives from the CD38-mediated conversion of NAD+ to ADPR, followed by the hydrolysis of ADPR by CD203a. This observation supports the hypothesis that the pyridine nucleotide-converting pathway leading to AMP is operative when cells concomitantly express CD38 and CD203a.
Metabolism of extracellular AMP to adenosine by CD73-expressing Jurkat T cells
The 5′-nucleotidase (5′-NT) CD73 is expressed on the surface of select lymphoid cells, while Jurkat are normally CD73-. A cell system displaying AMP-degrading activity was produced using a different clone of the Jurkat line, which was obtained by stably transfecting a construct for the constitutive expression of CD73 (Jurkat/B-NT5.4 cells, hereafter referred to as Jurkat/CD73+ cells).24 This cell line provided the proper background in which to dissect the effects of the expression of a true ecto-5′-NT with the capacity to degrade extracellular AMP to adenosine (see the complete cellular phenotype of resting and activated Jurkat/CD73+ cells in Figure 1C and D).
HPLC experiments confirmed that (1) the relative proportion of NAD+ products do not vary significantly between resting and activated Jurkat/CD73+ cells (Table 1), and that (2) Jurkat/CD73+ cells dephosphorylate extracellular AMP (Fig. 5A-C). A plot of the initial velocity at different substrate concentrations showed that AMP dephosphorylation to adenosine plateaus at 100 μM AMP (data not shown). AMP (Rt = 2.15 min) was metabolized (~80% within 30 min) by cultured Jurkat/CD73+ cells resulting in the production of adenosine (Rt = 5.56 min). The compounds detected in the supernatants were adenosine (187.50 ± 21.07 μmol/min/106cells) and (low levels of) hypoxanthine (Hxp, Rt = 3.26 min; 12.12 ± 25 μmol/min/106 cells) (Fig. 5A).

Figure 5. Metabolism of extracellular AMP consumption in Jurkat/CD73+ cells. (A-C) Jurkat/CD73+ cells were incubated with 100 µM AMP and the consequent (CD73-dependent) production of adenosine (ADO) was determined by HPLC. The identity of peaks was confirmed by the co-migration and absorbance spectra of reference standards using a retention time (Rt) window of ± 5%. (A) Experiments at t = 5 min and after 30 min incubation with 100 µM AMP. (B) AMP consumption in Jurkat/CD73+ cells exposed (solid symbols) or not exposed (open symbols) to 50 µM α,β-methylene-ADP (APCP). (C) Catabolism of AMP to ADO in the presence (solid symbols) or in the absence (open symbols) of 50 µM APCP. Representative Rt plots of metabolites in a single HPLC run are shown.
Inhibition experiments further evinced the role of CD73 in the conversion of AMP. Jurkat/CD73+ cells were incubated with AMP in the presence of increasing amounts of α,β-methylene-ADP (APCP, a 5′-NT inhibitor), followed by measurement of adenosine production. As expected for a reaction mediated by a 5′-NT, the catabolism of AMP and the formation of adenosine were strongly decreased by APCP (Fig. 5B-C). Even 10 μM APCP was sufficient to attenuate (~50%) the generation of adenosine, and a dose of 100 μM led to 75% inhibition levels. These results indicate that the ecto-5′NT CD73 is the main contributor to the AMP-hydrolytic activity observed and the predominant AMPase participating in adenosine generation by Jurkat/CD73+ cells. Nonspecific phosphatases were not involved in the conversion of AMP to adenosine since adenosine levels were unmodified by levamisole, an inhibitor of alkaline phosphatase (data not shown). These findings indirectly confirm the main role of CD73 in converting AMP to adenosine.
Extracellular adenosine may partially accumulate in the extracellular milieu (or culture medium) where it may bind specific P1 receptors or be internalized through nucleoside transporters. Alternatively, surface adenosine deaminase (ADA), complexed to CD26, may convert adenosine to Hxp via an inosine intermediate. Parental Jurkat cells do not express CD26,35 and we confirmed such a feature in Jurkat/CD73+ cells (Fig. 1B). Adenosine production increased upon the incubation of Jurkat/CD73+ cells with AMP in the presence of erytro-9 (2-hydroxy-3-nonyl) adenine (EHNA, an inhibitor of ADA). Conversely, the addition of dipyridamole (Dyp, an inhibitor of nucleoside transporters), produced no increase in adenosine, suggesting that other mechanisms contribute to adenosine homeostasis in this setting. One possibility is that ADA on the surface of Jurkat/CD73+ is anchored to a membrane receptor other than CD26.
Jurkat/CD73− and Jurkat/CD73+ cells offer a differential combination of ectoenzymes with NADase (CD38), NPP (CD203a), and 5′-NT (CD73) activities. Thus, the fate of NAD+ could be followed stepwise to validate the sequential contribution of these 3 ectoenzymes to the conversion of NAD+ to adenosine. The working hypothesis was that Jurkat/CD73− (CD38+) cells hydrolyze NAD+ to ADPR, which can then be converted into AMP by CD203a upregulated in the course of PMA-mediated activation. AMP-containing supernatants may then be transferred to Jurkat/CD73+ cell cultures, a setting in which AMP can be further hydrolyzed by CD73. This final reaction leads to the accumulation of adenosine in the extracellular milieu.
In the context of this dynamic model (Fig. 6), Jurkat/CD73− cells were incubated with NAD+ in an ADA-free medium. The resulting AMP-containing supernatants were then collected and either incubated with Jurkat/CD73+ cells or processed for HPLC analyses (Fig. 6A and B). In this setting, adenosine was produced by the Jurkat/CD73+ cells in high amounts (≥ 35 μmol/min/106 cells (Fig. 6B). In contrast, the Jurkat/CD73− cells did not produce detectable amounts of adenosine (Fig. 6A).

Figure 6. Jurkat T cells generates adenosine from NAD+. (A) The enzymatic conversion of NAD+ to AMP (a CD38/CD203a-dependent reaction) and to adenosine (ADO) (a CD73-dependent reaction) was evaluated. Activated Jurkat/CD73− cells were incubated for 60 min in buffer supplemented with 100 µM NAD+ and the resulting supernatant transferred to Jurkat/CD73+ cells, followed by further incubation for 30 min. (B) Supernatants from each step depicted in (A) were analyzed by HPLC for the presence of NAD+ catabolites. The identity of peaks was confirmed by the co-migration and absorbance spectra of reference standards using a retention time (Rt) window of ± 5%. Representative Rt plots of metabolites in a single HPLC run are shown.
Therefore, HPLC assays confirmed that exogenous NAD+ can be sequentially converted into ADPR, AMP, and adenosine, with this latter transformation being mediated by Jurkat/CD73+ T cells only. In conclusion, the CD38/CD203a/CD73 axis represents a functional enzymatic cascade leading to adenosine synthesis in this T-cell model system.
NAD+ conversion and intracellular cyclic AMP production by Jurkat T cells
The CD38/CD203a/CD73 axis generates extracellular adenosine, which can bind P1 receptors.36 Jurkat/CD73+ express high levels of purinergic A2A receptors (A2AR) of the P1 type, a feature that we exploited to evaluate the production of intracellular cyclic AMP (cAMP) elicited by adenosine signaling. Since A2AR is functional in the Jurkat model, A2AR engagement by CGS21680, a specific agonist, induced a significant increase in the levels of cytoplasmic cAMP. Exogenous adenosine was bound by A2AR, resulting in the activation of the associated stimulatory G protein and the expected increase of intracellular cAMP (from a basal value of 14.04 ± 2.13 fmol/well to 71.00 ± 0.67 fmol/well). Moreover, when Jurkat/CD73+ cells were treated with AMP-containing supernatants as generated by Jurkat/CD73− cells exposed to extracellular NAD+, the intracellular concentration of cAMP increased to 23.79 ± 0.97 fmol/well. Such a synthesis of cAMP is mediated by the adenosinergic loop specifically in CD73-proficient Jurkat/CD73+ cells. Indeed, Jurkat/CD73+ cells pre-treated with the CD73 inhibitor APCP displayed a marked attenuation in cAMP synthesis as stimulated by AMP-containing supernatants obtained from Jurkat/CD73− cells provided with NAD+. Thus, in the presence of exogenous adenosine or (in a CD73-dependent manner) AMP, the intracellular levels of cAMP increase. Conversely, the addition of extracellular NAD+ to resting Jurkat/CD73+ (CD38+/CD203a-/CD73+) cells did not provoke an elevation of cAMP levels, as expected when the substrate AMP is not produced and NAD+ is not processed by 5′-NT (Table 1).37
Taken together, these results indicate that the CD38/CD203a/CD73 pathway can mediate the sequential conversion of NAD+ to adenosine in a Jurkat T-cell model and that adenosine, the final product of the reaction, can elicit cAMP-dependent signaling in A2AR+ cells.
Discussion
Inferences derived from ectoenzymes38 and from NAD+ metabolomics14 have completely reshaped our understanding of the key steps involved in the processing and activities of extracellular nucleotides.9 The canonical substrate for ectonucleotidases is ATP, hence the nucleotide is a primary source of adenosine in the extracellular milieu. Another possibility is that pericellular adenosine can arise from the degradation of the pyridine nucleotide NAD+, but experimental confirmation in support of this hypothesis was lacking.
Our results obtained here indicate that the NADase CD38, the NPP CD203a, and the 5′NT CD73 catalytic cascade forms an unexplored but robust pathway for the production of adenosine from NAD+. These ectoenzymes may function according to their spatial arrangement as part of (1) continuous (molecules on the same cell) or (2) discontinuous (molecules on different cells) pathways. A peculiar feature of the CD38/CD203a/CD73 axis is that it can exhibit bimodal operation. When the individual components are located on different cells, their interactions are facilitated in spatially restricted systems (or niches). Irrespective of this point, the biochemical complexity of the axis in terms of substrates, ectoenzymes, products and receptors has made it difficult to validate its operational status in a single cell type.
We provide conclusive evidence of the existence of this pathway by means of a T lymphocyte model constructed ad hoc in vitro. T lymphocytes exert key roles in the generation of tolerance in inflammation, cancer and transplantation.39,40 The human model adopted for this study was the human Jurkat T-cell leukemia cell lines (mostly composed of CD38+/CD203a-/CD39-/CD73− cells), which we selected because of the capacity for clonal derivation of cellular subsets that recapitulate discrete steps of normal and pathological T-cell differentiation in vitro. Environmental pressures were mimicked in vitro by adding the differentiating agent PMA, which also fostered the expression of the NPP CD203a. However, neither resting nor activated parental Jurkat/VR cells expressed CD73. This was circumvented by stably transfecting Jurkat cells with a CD73-coding construct.24 By means of these two different cell lines (expressing or not CD73), under either resting or activated conditions (CD203a- and CD203a+ cells, respectively), the exogenously applied substrates (i.e., NAD+ and ATP) and products (i.e., ADPR, NMN, AMP, and adenosine) that accumulated in the culture supernatant could be quantified without derivatization by means of dedicated HPLC assays.
The first player in the network found to be responsible for the conversion of extracellular nucleotides was CD38, as expected due to its topological features (i.e., accessibility to exogenous NAD+) and known enzymatic function as the primary regulator of extracellular NAD+ levels.41,42 The role of the paralogue CD157 was not considered here, mainly due to reduced expression levels and limited enzymatic activity in T lymphocytes.43
The second player in this novel ectonucleotidase cascade was identified as PC-1, an NPP ectoenzyme clustered as CD203a.44,45 In this enzymatic context, NAD+, ADPR (a product of the enzymatic activity of CD38) and ATP are all prospective substrates for the NPP activity of CD203a. When NAD+ is a substrate of CD203a,46 the product of the reaction is AMP along with NMN, a product that mainly proceeds intracellularly.32 Conversely, when CD203a metabolizes ADPR, the main reaction products are AMP and PPi. ATP, a potent modulator of immune cell responses,47 shares structural characteristics with NAD+. As a consequence of the activity of CD203a, ATP is degraded in the extracellular space into AMP and PPi.22 The finding that CD203a hydrolyzes these purine analogs in our Jurkat T-cell model validates the appropriateness of our system to address our working hypothesis.
Our findings also indicate that the interactions between extracellular NAD+ and CD38/CD203a can be exploited by T lymphocytes to generate AMP. Indeed, the addition of NAD+ to Jurkat/CD73− cells, in resting conditions (CD38+/CD203a- cells) and after PMA-mediated activation (CD38+/CD203a+ cells), caused a remarkable variation in the levels of ADPR, AMP and Nic, in the culture supernatants (NMN was only seen in trace amounts). These results indicate that the outer plasma membrane of a subset of Jurkat cells is equipped with distinct hydrolytic activities that determine the fate of extracellular NAD+. The first (the NADase CD38) cleaves a glycosidic link in NAD+ to produce ADPR and Nic. The second (the NPP CD203a) operates on the pyrophosphate bond in NAD+ to yield NMN and AMP.32 It is therefore possible that the activity of CD203a might reduce the availability of substrates for CD38 in activated Jurkat/CD73− cells, modifying the balance of NAD+ consumption. A comparison between ADPR (a NADase product) and NMN (a NPP product) generated by activated Jurkat/CD73− cells revealed that the NAD+-consuming role of the NADase-CD38 dominates over that of the NPP CD203a. Indeed, the amounts of ADPR generated from NAD+ hydrolysis by activated (CD38+/CD203a+/CD73-) Jurkat cells remained unchanged in the presence of EDTA, a calcium chelator and CD203a inhibitor. Conversely, AMP production was reduced when the assay was performed in the absence of Ca2+. Therefore, we assert that the Ca2+-dependent activity of CD203a ensures the conversion of ADPR (generated by CD38) into AMP in Jurkat T cells. These findings sustain the view that extracellular NAD+ is efficiently metabolized to AMP when both surface CD38 and CD203a are present (Fig. 7).

Figure 7. CD38/CD203a ectoenzyme tandem catalytic cascade to degrade NAD+ in activated Jurkat/CD73− cells. Extracellular NAD+ is metabolized by the nicotinamide adenine dinucleotidase (NADase) CD38 expressed by resting Jurkat cells, generating nicotinamide (Nic), cyclic adenosine diphosphate ribose (cADPR) and ADPR. The latter product is transformed to AMP by the nucleotide pyrophosphatase/phosphodiesterase (NPP) CD203a expressed by phorbol 12-myristate 13-acetate (PMA)-activated Jurkat cells.
Finally, to demonstrate that extracellular AMP is further catabolized to produce adenosine, we used a Jurkat cell line genetically modified to express active CD73. Jurkat/CD73+ cells metabolize AMP, which is converted by dephosphorylation into adenosine and small amounts of Hxp. Intermediate products of AMP catabolism were detected at any time point, suggesting that AMP degradation is led by a cell surface mechanism. The direct processing of extracellular NAD+ by the 5′-NT CD73, as observed in other systems,48 was excluded in our Jurkat/CD73+ cells, as well as in fresh leukocytes and Epstein-Barr virus (EBV)-derived lymphoblastoid lines obtained from a CD73−/− patient (ref. 49 and A.L. Horenstein and F. Malavasi, unpublished observations, 2013).
Adenosine generated by AMP hydrolysis may either (1) accumulate in the extracellular medium and hence bind to specific P1 receptors; (2) be inactivated at the cell surface by an ADA/CD26 complex that converts it into Hxp via inosine; or (3) internalized by nucleoside transporters.19 Our results indicate that adenosine levels increase in the extracellular medium when Jurkat/CD73+ cells are incubated with AMP in the presence of EHNA (an ADA inhibitor). One possible interpretation is that adenosine homeostasis is influenced by ADA due to deamidation of adenosine. However, like other Jurkat clones, Jurkat/CD73+ cells do not express CD26, suggesting that ADA may have a surface anchor different from CD26, at least in this system. A possible alternative ADA-anchoring candidate is the purinergic A2AR.50 In this complex, A2AR may exhibit and altered affinity for its ligand, thereby finely tuning the biological effects of adenosine, as it occurs in vivo. Other mechanisms involved in adenosine homeostasis, such as the internalization through nucleoside transport, were not highly operative in our system. Indeed, no increase in adenosine was detected following the addition of an inhibitor of nucleoside transporters, confirming previous observations obtained in parental Jurkat T cells.24
Cells can simultaneously express a variety of related ectonucleotidases that are functionally competent to metabolize different nucleotides, such as the NPP CD203a and NTPDase CD39, either on the surface of the same cells or on that of different, but adjacent cells.45 Such complexities obfuscate the assignment of specific functions unless the kinetic properties of each contributing enzyme are analyzed in distinct physiological conditions. Like NAD+, ATP is released from inflammatory cells into the extracellular space. The conversion of extracellular ATP to adenosine by the NTPDase CD39 is kinetically complex, with the upstream metabolite ADP acting as a crucial feed-forward inhibitor of the 5′NT CD73,51 and resulting in a tendency to AMP accumulation (A.L. Horenstein, unpublished observations, 2013).
Physiologically, such an ADP-dependent feed-forward inhibition does not appear to significantly modulate purinergic signaling, as human cell surfaces are exposed to low levels of ATP (< 1 μM).52 However, high ATP levels in the context of CD203a might induce the NPP to blunt signals mediated by P2 receptors through an ATP conversion step that bypasses the formation of ADP. The lower affinity displayed by ATP for CD203a as compared with CD3953 offers indirect support for such a view. Alternatively, the ectoenzymatic CD38/CD203a tandem may become relevant when ATP is released after injury or inflammation. The extracellular microenvironment likely tends to compensate for a lack of adenosine that could result from an ADP feed-forward inhibition by activating the NAD+-dependent CD38/CD203a/CD73 adenosinergic loop.
This ectoenzymatic pathway hydrolyzes NAD+ and AMP in sequence to produce functional adenosine that, upon binding to P1 receptors, increases intracellular cAMP concentrations.27 Thus, these cellular processes generate a biologically active form of adenosine. In fact, the addition of NAD+ to CD73+CD203a- Jurkat T cells did not stimulate cAMP production. Conversely, Jurkat/CD73+ cells exposed to AMP-containing supernatants from activated Jurkat/CD73− cultures synthesize cAMP, an effects that can be blocked by the CD73 inhibitor APCP. Our conclusion is that, in our Jurkat T-cell system, the NADase CD38, the NPP CD203a and the 5′NT CD73 form a functionally interrelated pathway that acts independently of the NTPDase CD39 (Fig. 8).
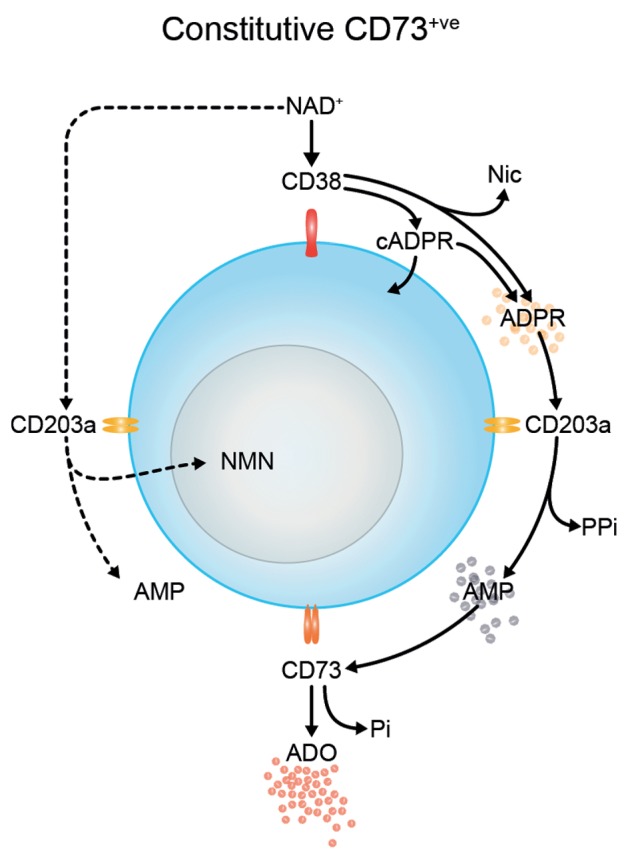
Figure 8. Biochemical activity of the CD38/CD203a/CD73 pathway in Jurkat T cells upon activation or transgene-driven CD73 expression. Extracellular NAD+ is metabolized by the nicotinamide adenine dinucleotidase (NADase) CD38, generating nicotinamide (Nic), cyclic adenosine diphosphate ribose (cADPR) and ADPR. The latter compound is transformed to AMP by nucleotide pyrophosphatase/phosphodiesterase (NPP) CD203a and then converted to adenosine (ADO) and inorganic pyrophosphate (PPi) by 5′-nucleotidase (5′NT) CD73. NMN, nicotinamide mononucleotide.
This ectonucleotidase model is centered around the NAD+/adenosine axis, a checkpoint that determines whether the extracellular environment is pro-inflammatory (nucleotide-mediated responses) or anti-inflammatory (nucleoside-mediated responses). On one side, the NAD+ substrate triggering the CD38/CD203a/CD73 pathway may influence the immune system by switching the balance from activatory (P2-mediated) to suppressive (P1-mediated) signals. This task may be altered in specific lymphocyte subsets in the pathogenesis of autoimmune diseases.54 On the other side, the interplay between adenosine and purinergic receptors generates local cellular anergy, which is a relevant mechanism of peripheral tolerance in both normal tissues and in the course of oncogenesis and tumor progression.52 Therefore, the NAD+-catalytic network that we delineated here is now under evaluation in normal cells (natural killer cells, mesenchymal stem cells, Tregs, regulatory B cells, and myeloid-derived suppressor cells) as well as in the human cornea, which was selected because of its immunologically privileged status and proven expression of CD38 (and CD157).55 The results of these studies will indicate whether this adenosinergic ectoenzyme network contributes to the generation of local tolerance or rather facilitates the elicitation of adaptive immune responses. Another physiological model of relevance in this setting is the tolerogenic environment regulating the interactions between mother and fetus.56
Immunohistochemical studies showed that CD38, CD203a and CD73 are co-expressed in lymph node sections, and in particular that CD203a is markedly expressed by stromal cells.20 It seems reasonable to postulate that a discontinuous ectoenzymatic pathway provided by the proximity of different cell types can generate not only AMP but also adenosine. Thus, the CD38/CD203a/CD73 pathway may represent one of multiple strategies adopted by tumor cells to exploit the normal machinery for peripheral tolerance to elude the antitumor activity of the host in spatially restricted systems.57-59 Interventions targeting this pathway may be instrumental for reinstating immunosurveillance.60 The biochemical observations derived from our T-lymphocyte model are undergoing further analysis using myeloma and melanoma patient samples, a disease-relevant context in which this ectoenzymatic axis may be properly evaluated for the potential development of therapeutic targeting strategies.
Materials and Methods
Cells and treatments
Human T-cell leukemia Jurkat/VR cells were propagated in a humidified atmosphere (95% air, 5% CO2) at 37°C in RPMI-1640 medium (Sigma) supplemented with 5% heat-inactivated fetal calf serum (FCS; Euroclone), 50 μg/mL kanamycin, 100 U/mL penicillin and 100 μg/mL streptomycin (all from Sigma) (hereafter referred to as complete medium). For all experiments, cells were plated at a density of 0.25 × 106 cells/mL and used after 24 h adaptation time. Activation was induced with 10 ng/mL phorbol 12-myristate 13-acetate (PMA; Sigma) and cellular responses monitored by increased expression of CD69 on the cell surface. B-NT5.1 cells, Jurkat derivatives stably transfected with the human CD73 gene (kindly provided by Dr. L. Thompson,61) were grown in complete medium supplemented with 400 µg/mL G418 (Gibco).24
Chemicals
Nicotinamide adenine dinucleotide (NAD+), nicotinamide guanine dinucleotide (NGD+), adenosine diphosphate ribose (ADPR), adenosine, adenosine 5′-monophosphate (AMP), adenosine 5′-diphosphate (ADP), adenosine 5′-triphosphate (ATP), potassium dihydrogen phosphate (KH2PO4) and acetonitrile (HPLC-grade reagent) were all from Sigma as well as α,β-methylene ADP (APCP, CD73 inhibitor), erythro-9-(2-hydroxy-3-nonyl)-adenine (EHNA, adenosine deaminase inhibitor), deoxycoformycin (DCF, adenosine deaminase inhibitor), CGS21680 (A2A receptor agonist), levamisole (alkaline phosphatase inhibitor), EDTA (ethylene-diamine-tetraacetic acid; CD39 inhibitor) and dipyridamole (DYP, nucleoside transporter inhibitor). Sodium azide (NaN3), was from Merck (Merck, Darmstadt, Germany). Sample buffer and markers for sodium dodecyl sulfate PAGE (SDS-PAGE) were from Bio-Rad (Milan, Italy). Milli-Q water was employed throughout the study (Millipore, Bedford, MA). Before use, high-performance liquid chromatography (HPLC) solutions were filtered through 0.45 µm membranes (Millipore). All other chemical reagents used were of analytical quality.
Antibodies
The antibodies used in this study include two anti-CD38 monoclonal antibodies (IB4, IgG2a; and SUN-4B7, IgG1),62 and one anti-CD203a antibody (PC-1, clone 3E8, kindly provided by J. Goding).23 Anti-CD73, -CD26, and -CD69 were IgG1 monoclonal antibodies produced in our Lab and purified in-house by affinity chromatography on protein A-Sepharose and HPLC on hydroxylapatite.63 Anti-CD39 monoclonal antibodies were obtained from eBioscience. Isotype-matched irrelevant mAb used was X63.Ag8, an IgG1κ secreted by a parental mouse myeloma.64 The fluorescein (FITC)-conjugated F(ab’)2 fragment goat anti-mouse IgG+IgM (GAMIg) was from Jackson Immuno-Research Laboratories (West Grove, PA).
Flow cytometry
Indirect immunofluorescence tests were done on cells washed in PBS containing 1% bovine serum albumin (BSA) + NaN3 and incubated with primary monoclonal antibodies for 1 h at 4°C. Cells were then washed twice and incubated with FITC-conjugated anti-mouse antibody for 30 min at 4°C. The samples were washed, resuspended in PBS and acquired on a FACSort flow cytometer (Becton-Dickinson, San Jose, CA), using CellQuest Software (Becton-Dickinson,). Data were analyzed using FlowJo Software (TreeStar Inc., Ashland, OR).
Immunoblotting analysis
The expression of CD38, CD203a, CD39, and CD73 was determined by conventional immunoblotting techniques. To this aim, resting and PMA-activated Jurkat T cells as well as epithelial cells obtained from human corneal tissues (Piedmont Cornea Bank, “Città della Salute e della Scienza” Hospital, Torino, Italy), which were used as CD39/CD73 positive controls, were washed with PBS and lysed as previously reported.65 Whole cell lysates were resolved by SDS-PAGE and transferred to nitrocellulose membrane (Hybond ECL; GE Healthcare, Milan, Italy), using a tank blotting apparatus (GE Healthcare). After incubation with the indicated primary and secondary horseradish peroxidase-conjugated goat anti-mouse antibodies, blots were developed using enhanced chemiluminescence (Hyperfilm ECL) and an ECL kit (Amersham Biosciences).
HPLC analysis
Chromatographic analysis was performed with an HPLC System (Beckman Gold 126/166NM, Beckman Coulter) equipped with a reverse-phase column (Hamilton C18, 5 µm; 250 × 4.5 mm). Separation of nucleotides and nucleosides was performed using a mobile-phase buffer (0.125 M citric acid and 0.025 M KH2PO4), pH 5.1 with 8% acetonitrile (Sigma) over 10 min at a flow rate of 0.8 mL/min. UV absorption spectra were measured at 254 nm. HPLC-grade standards used to calibrate the signals were dissolved in AIM V serum-free medium (Invitrogen, Paisley, UK), pH 7.4, 0.2 μm sterile-filtered and injected in a buffer volume of 20 μL. The retention times (Rt, in min) of standards were: AMP, 2.15; NAD+, 2.8; ADPR, 3.2; cADPR, 6.15; Nic, 6.87; and adenosine; 5.56. Peak integration was performed using a Karat software (Beckman Coulter).
The ectonucleotidase activity of CD38 and CD203a was measured using 2 × 106/mL Jurkat/CD73-cells maintained for 30 min at 37°C in AIM V medium (Invitrogen) containing NAD+, ADPR, or ATP (100 μM). Following collection (at the indicated time points) and centrifugation (700 g; 5 min at 4°C) supernatants were transferred to Jurkat/CD73+ cells (2 × 106cells/mL) to metabolize AMP. Adenosine production (CD73 activity) was evaluated using supernatants collected in tubes containing 1 mL of acetonitrile (Sigma) at 4°C, to stabilize adenosine. Supernatants were evaporated by speed-vac, reconstituted in mobile-phase buffer, and assayed by HPLC. In some experiments, cells were incubated with or without the selective 5′-NT inhibitor APCP (50 μM, 15 min) before addition of 100 μM AMP, and supernatants collected in microcentrifuge tubes containing 100 μM EHNA, 10 μM dipyridamole, 10 μM α,β-methylene ADP and 10 μM 5′-deoxycoformycin (in 0.1 mL AIM V medium), and processed for HPLC analysis.
The qualitative identity of HPLC peaks was confirmed by co-migration and absorbance spectra of known reference standards using an Rt window of ± 5%. The presence of adenosine was also confirmed by spiking standard (100 μM adenosine), followed by chromatography. Quantitative measurements were inferred by comparing the peak area of samples with calibration curves for peak areas of each standard compound. Product concentrations were expressed as micromoles/min/106 cells.
cAMP production in response to adenosine
Twenty thousand Jurkat/CD73+ cells per well were incubated for 1 h at 37°C with or without 50 µM adenosine, the supernatants collected from activated Jurkat/CD73− cells, or 10 µM CGS21680, a selective agonist of the A2A purinergic receptor. After incubation, cells were lysed on ice and intracellular cAMP levels determined using an enzymatic assay (GE Healthcare).
Statistical analyses
Data are expressed as means ± SEM. The GraphPad software package was used for the construction of concentration–response curves.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. L. Thompson for the Jurkat B-NT5.1/CD73+ cell line and Professor J. Goding for the anti-human PC-1 (3E8 clone). This work was supported by research grants from PRIN (Ministry of Education, University, and Research), FIRB (Fondo per gli Investimenti della Ricerca di Base), the ex 60% Project (University of Torino) and partially from AIRC (5 × 1000). The Fondazione Ricerca Molinette provided valuable assistance.
Glossary
Abbreviations:
- ADA
adenosine deaminase
- ADO
adenosine
- ADP
adenosine 5′-diphosphate
- ADPR
adenosine diphosphate ribose
- AMP
adenosine 5′-monophosphate
- APCP
α,β-methylene-ADP
- ATP
adenosine 5′-triphosphate
- cADPR
cyclic ADPR
- cGDPR
cyclic guanosine diphosphoribose
- DCF
deoxycoformycin
- Dyp
dipyridamole
- EHNA
erythro-9-(2-hydroxy-3-nonyl)-adenine
- Hxp
hypoxanthine
- HPLC
high-performance liquid chromatography
- NAD+
nicotinamide adenine dinucleotide
- NADase
nicotinamide adenine dinucleotidase
- NGD+
nicotinamide guanine dinucleotide
- Nic
nicotinamide
- NMN
nicotinamide mononucleotide
- NPP
nucleotide pyrophosphatase/phosphodiesterase
- 5′-NT
5′-nucleotidase
- NTPDase
ectonucleoside triphosphate diphosphohydrolase
- PMA
phorbol 12-myristate 13-acetate
- PPi
inorganic pyrophosphate
- Rt
retention time
- Treg
regulatory T cell
Citation: Horenstein AL, Chillemi A, Zaccarello G, Bruzzone S, Quarona V, Zito A, Serra S, Malavasi F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. OncoImmunology 2013; 2:e26246; 10.4161/onci.26246
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/26246
References
- 1.Lanier LL. Shades of grey--the blurring view of innate and adaptive immunity. Nat Rev Immunol. 2013;13:73–4. doi: 10.1038/nri3389. [DOI] [PubMed] [Google Scholar]
- 2.Kirschner MW. The meaning of systems biology. Cell. 2005;121:503–4. doi: 10.1016/j.cell.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, Vaisitti T, Aydin S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. 2008;88:841–86. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- 4.Zimmermann H, Zebisch M, Sträter N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012;8:437–502. doi: 10.1007/s11302-012-9309-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grahnert A, Grahnert A, Klein C, Schilling E, Wehrhahn J, Hauschildt S. Review: NAD+: a modulator of immune functions. Innate Immun. 2011;17:212–33. doi: 10.1177/1753425910361989. [DOI] [PubMed] [Google Scholar]
- 6.Ziegler M. New functions of a long-known molecule. Emerging roles of NAD in cellular signaling. Eur J Biochem. 2000;267:1550–64. doi: 10.1046/j.1432-1327.2000.01187.x. [DOI] [PubMed] [Google Scholar]
- 7.Drury AN, Szent-Györgyi A. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J Physiol. 1929;68:213–37. doi: 10.1113/jphysiol.1929.sp002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramakers BP, Wever KE, Kox M, van den Broek PH, Mbuyi F, Rongen G, Masereeuw R, van der Hoeven JG, Smits P, Riksen NP, et al. How systemic inflammation modulates adenosine metabolism and adenosine receptor expression in humans in vivo. Crit Care Med. 2012;40:2609–16. doi: 10.1097/CCM.0b013e318259205b. [DOI] [PubMed] [Google Scholar]
- 9.Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2012;367:2322–33. doi: 10.1056/NEJMra1205750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serra S, Horenstein AL, Vaisitti T, Brusa D, Rossi D, Laurenti L, D’Arena G, Coscia M, Tripodo C, Inghirami G, et al. CD73-generated extracellular adenosine in chronic lymphocytic leukemia creates local conditions counteracting drug-induced cell death. Blood. 2011;118:6141–52. doi: 10.1182/blood-2011-08-374728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sträter N. Ecto-5′-nucleotidase: Structure function relationships. Purinergic Signal. 2006;2:343–50. doi: 10.1007/s11302-006-9000-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mandapathil M, Lang S, Gorelik E, Whiteside TL. Isolation of functional human regulatory T cells (Treg) from the peripheral blood based on the CD39 expression. J Immunol Methods. 2009;346:55–63. doi: 10.1016/j.jim.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nikolova M, Carriere M, Jenabian MA, Limou S, Younas M, Kök A, Huë S, Seddiki N, Hulin A, Delaneau O, et al. CD39/adenosine pathway is involved in AIDS progression. PLoS Pathog. 2011;7:e1002110. doi: 10.1371/journal.ppat.1002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiarugi A, Dölle C, Felici R, Ziegler M. The NAD metabolome--a key determinant of cancer cell biology. Nat Rev Cancer. 2012;12:741–52. doi: 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- 15.Fruscione F, Scarfì S, Ferraris C, Bruzzone S, Benvenuto F, Guida L, Uccelli A, Salis A, Usai C, Jacchetti E, et al. Regulation of human mesenchymal stem cell functions by an autocrine loop involving NAD+ release and P2Y11-mediated signaling. Stem Cells Dev. 2011;20:1183–98. doi: 10.1089/scd.2010.0295. [DOI] [PubMed] [Google Scholar]
- 16.Penuela S, Gehi R, Laird DW. The biochemistry and function of pannexin channels. Biochim Biophys Acta. 2013;1828:15–22. doi: 10.1016/j.bbamem.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 17.Deterre P, Gelman L, Gary-Gouy H, Arrieumerlou C, Berthelier V, Tixier JM, Ktorza S, Goding J, Schmitt C, Bismuth G. Coordinated regulation in human T cells of nucleotide-hydrolyzing ecto-enzymatic activities, including CD38 and PC-1. Possible role in the recycling of nicotinamide adenine dinucleotide metabolites. J Immunol. 1996;157:1381–8. [PubMed] [Google Scholar]
- 18.Massaia M, Perrin L, Bianchi A, Ruedi J, Attisano C, Altieri D, Rijkers GT, Thompson LF. Human T cell activation. Synergy between CD73 (ecto-5′-nucleotidase) and signals delivered through CD3 and CD2 molecules. J Immunol. 1990;145:1664–74. [PubMed] [Google Scholar]
- 19.Hubert S, Rissiek B, Klages K, Huehn J, Sparwasser T, Haag F, Koch-Nolte F, Boyer O, Seman M, Adriouch S. Extracellular NAD+ shapes the Foxp3+ regulatory T cell compartment through the ART2-P2X7 pathway. J Exp Med. 2010;207:2561–8. doi: 10.1084/jem.20091154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quarona V, Zaccarello G, Chillemi A, Brunetti E, Singh VK, Ferrero E, Funaro A, Horenstein AL, Malavasi F. CD38 and CD157: a long journey from activation markers to multifunctional molecules. Cytometry B Clin Cytom. 2013;84:207–17. doi: 10.1002/cyto.b.21092. [DOI] [PubMed] [Google Scholar]
- 21.Malavasi F, Funaro A, Roggero S, Horenstein A, Calosso L, Mehta K. Human CD38: a glycoprotein in search of a function. Immunol Today. 1994;15:95–7. doi: 10.1016/0167-5699(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 22.Goding JW. Ecto-enzymes: physiology meets pathology. J Leukoc Biol. 2000;67:285–311. doi: 10.1002/jlb.67.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goding JW, Terkeltaub R, Maurice M, Deterre P, Sali A, Belli SI. Ecto-phosphodiesterase/pyrophosphatase of lymphocytes and non-lymphoid cells: structure and function of the PC-1 family. Immunol Rev. 1998;161:11–26. doi: 10.1111/j.1600-065X.1998.tb01568.x. [DOI] [PubMed] [Google Scholar]
- 24.Resta R, Yamashita Y, Thompson LF. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol Rev. 1998;161:95–109. doi: 10.1111/j.1600-065X.1998.tb01574.x. [DOI] [PubMed] [Google Scholar]
- 25.Whiteside TL, Jackson EK. Adenosine and prostaglandin e2 production by human inducible regulatory T cells in health and disease. Front Immunol. 2013;4:212. doi: 10.3389/fimmu.2013.00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sorrentino R, Pinto A, Morello S. The adenosinergic system in cancer: Key therapeutic target. Oncoimmunology. 2013;2:e22448. doi: 10.4161/onci.22448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haskó G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–70. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. 2011;11:201–12. doi: 10.1038/nri2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kukulski F, Lévesque SA, Sévigny J. Impact of ectoenzymes on p2 and p1 receptor signaling. Adv Pharmacol. 2011;61:263–99. doi: 10.1016/B978-0-12-385526-8.00009-6. [DOI] [PubMed] [Google Scholar]
- 30.Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim Biophys Acta. 2008;1783:673–94. doi: 10.1016/j.bbamcr.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 31.Podestà M, Benvenuto F, Pitto A, Figari O, Bacigalupo A, Bruzzone S, Guida L, Franco L, Paleari L, Bodrato N, et al. Concentrative uptake of cyclic ADP-ribose generated by BST-1+ stroma stimulates proliferation of human hematopoietic progenitors. J Biol Chem. 2005;280:5343–9. doi: 10.1074/jbc.M408085200. [DOI] [PubMed] [Google Scholar]
- 32.Nikiforov A, Dölle C, Niere M, Ziegler M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J Biol Chem. 2011;286:21767–78. doi: 10.1074/jbc.M110.213298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di Lisa F, Ziegler M. Pathophysiological relevance of mitochondria in NAD(+) metabolism. FEBS Lett. 2001;492:4–8. doi: 10.1016/S0014-5793(01)02198-6. [DOI] [PubMed] [Google Scholar]
- 34.Graeff RM, Walseth TF, Fryxell K, Branton WD, Lee HC. Enzymatic synthesis and characterizations of cyclic GDP-ribose. A procedure for distinguishing enzymes with ADP-ribosyl cyclase activity. J Biol Chem. 1994;269:30260–7. [PubMed] [Google Scholar]
- 35.Dong RP, Tachibana K, Hegen M, Munakata Y, Cho D, Schlossman SF, Morimoto C. Determination of adenosine deaminase binding domain on CD26 and its immunoregulatory effect on T cell activation. J Immunol. 1997;159:6070–6. [PubMed] [Google Scholar]
- 36.Burnstock G. Discovery of purinergic signalling, the initial resistance and current explosion of interest. Br J Pharmacol. 2012;167:238–55. doi: 10.1111/j.1476-5381.2012.02008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garavaglia S, Bruzzone S, Cassani C, Canella L, Allegrone G, Sturla L, Mannino E, Millo E, De Flora A, Rizzi M. The high-resolution crystal structure of periplasmic Haemophilus influenzae NAD nucleotidase reveals a novel enzymatic function of human CD73 related to NAD metabolism. Biochem J. 2012;441:131–41. doi: 10.1042/BJ20111263. [DOI] [PubMed] [Google Scholar]
- 38.Zola H, Swart B, Banham A, Barry S, Beare A, Bensussan A, Boumsell L, D Buckley C, Bühring HJ, Clark G, et al. CD molecules 2006--human cell differentiation molecules. J Immunol Methods. 2007;319:1–5. doi: 10.1016/j.jim.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 39.Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;19:355–67. doi: 10.1016/j.molmed.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T. Regulatory T cells: how do they suppress immune responses? Int Immunol. 2009;21:1105–11. doi: 10.1093/intimm/dxp095. [DOI] [PubMed] [Google Scholar]
- 41.Iqbal J, Zaidi M. Extracellular NAD+ metabolism modulates osteoclastogenesis. Biochem Biophys Res Commun. 2006;349:533–9. doi: 10.1016/j.bbrc.2006.08.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Q, Kriksunov IA, Graeff R, Munshi C, Lee HC, Hao Q. Crystal structure of human CD38 extracellular domain. Structure. 2005;13:1331–9. doi: 10.1016/j.str.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 43.Ortolan E, Vacca P, Capobianco A, Armando E, Crivellin F, Horenstein A, Malavasi F. CD157, the Janus of CD38 but with a unique personality. Cell Biochem Funct. 2002;20:309–22. doi: 10.1002/cbf.978. [DOI] [PubMed] [Google Scholar]
- 44.Goding JW, Grobben B, Slegers H. Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim Biophys Acta. 2003;1638:1–19. doi: 10.1016/S0925-4439(03)00058-9. [DOI] [PubMed] [Google Scholar]
- 45.Stefan C, Jansen S, Bollen M. NPP-type ectophosphodiesterases: unity in diversity. Trends Biochem Sci. 2005;30:542–50. doi: 10.1016/j.tibs.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 46.Katada T, Kontani K, Wada T, Hosoda N, Hoshino S, Nishina H. Enzymic and signal transduction properties of CD38/NADase and PC-1/phosphodiesterase. Chem Immunol. 2000;75:60–78. doi: 10.1159/000058762. [DOI] [PubMed] [Google Scholar]
- 47.Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther. 2006;112:358–404. doi: 10.1016/j.pharmthera.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 48.Grozio A, Sociali G, Sturla L, Caffa I, Soncini D, Salis A, Raffaelli N, De Flora A, Nencioni A, Bruzzone S. CD73 as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J Biol Chem. 2013;288:25938–49. doi: 10.1074/jbc.M113.470435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.St Hilaire C, Ziegler SG, Markello TC, Brusco A, Groden C, Gill F, Carlson-Donohoe H, Lederman RJ, Chen MY, Yang D, et al. NT5E mutations and arterial calcifications. N Engl J Med. 2011;364:432–42. doi: 10.1056/NEJMoa0912923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gracia E, Pérez-Capote K, Moreno E, Barkešová J, Mallol J, Lluís C, Franco R, Cortés A, Casadó V, Canela EI. A2A adenosine receptor ligand binding and signalling is allosterically modulated by adenosine deaminase. Biochem J. 2011;435:701–9. doi: 10.1042/BJ20101749. [DOI] [PubMed] [Google Scholar]
- 51.Gordon EL, Pearson JD, Slakey LL. The hydrolysis of extracellular adenine nucleotides by cultured endothelial cells from pig aorta. Feed-forward inhibition of adenosine production at the cell surface. J Biol Chem. 1986;261:15496–507. [PubMed] [Google Scholar]
- 52.Di Virgilio F. Purines, purinergic receptors, and cancer. Cancer Res. 2012;72:5441–7. doi: 10.1158/0008-5472.CAN-12-1600. [DOI] [PubMed] [Google Scholar]
- 53.Grobben B, Anciaux K, Roymans D, Stefan C, Bollen M, Esmans EL, Slegers H. An ecto-nucleotide pyrophosphatase is one of the main enzymes involved in the extracellular metabolism of ATP in rat C6 glioma. J Neurochem. 1999;72:826–34. doi: 10.1046/j.1471-4159.1999.0720826.x. [DOI] [PubMed] [Google Scholar]
- 54.Pavón EJ, Zumaquero E, Rosal-Vela A, Khoo KM, Cerezo-Wallis D, García-Rodríguez S, Carrascal M, Abian J, Graeff R, Callejas-Rubio JL, et al. Increased CD38 expression in T cells and circulating anti-CD38 IgG autoantibodies differentially correlate with distinct cytokine profiles and disease activity in systemic lupus erythematosus patients. Cytokine. 2013;62:232–43. doi: 10.1016/j.cyto.2013.02.023. [DOI] [PubMed] [Google Scholar]
- 55.Horenstein AL, Sizzano F, Lusso R, Besso FG, Ferrero E, Deaglio S, Corno F, Malavasi F. CD38 and CD157 ectoenzymes mark cell subsets in the human corneal limbus. Mol Med. 2009;15:76–84. doi: 10.2119/molmed.2008.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cecati M, Emanuelli M, Giannubilo SR, Quarona V, Senetta R, Malavasi F, Tranquilli AL, Saccucci F. Contribution of adenosine-producing ectoenzymes to the mechanisms underlying the mitigation of maternal-fetal conflicts. J Biol Regul Homeost Agents. 2013;27:519–29. [PubMed] [Google Scholar]
- 57.Jun JE, Goodnow CC. Scaffolding of antigen receptors for immunogenic versus tolerogenic signaling. Nat Immunol. 2003;4:1057–64. doi: 10.1038/ni1001. [DOI] [PubMed] [Google Scholar]
- 58.Zhang B. CD73 promotes tumor growth and metastasis. Oncoimmunology. 2012;1:67–70. doi: 10.4161/onci.1.1.18068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma Y, Adjemian S, Yang H, Catani JP, Hannani D, Martins I, Michaud M, Kepp O, Sukkurwala AQ, Vacchelli E, et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology. 2013;2:e24568. doi: 10.4161/onci.24568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. 2013;39:74–88. doi: 10.1016/j.immuni.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 61.Gutensohn W, Resta R, Misumi Y, Ikehara Y, Thompson LF. Ecto-5′-nucleotidase activity is not required for T cell activation through CD73. Cell Immunol. 1995;161:213–7. doi: 10.1006/cimm.1995.1029. [DOI] [PubMed] [Google Scholar]
- 62.Ausiello CM, Urbani F, Lande R, la Sala A, Di Carlo B, Baj G, Surico N, Hilgers J, Deaglio S, Funaro A, et al. Functional topography of discrete domains of human CD38. Tissue Antigens. 2000;56:539–47. doi: 10.1034/j.1399-0039.2000.560608.x. [DOI] [PubMed] [Google Scholar]
- 63.Horenstein AL, Durelli I, Malavasi F. Purification of clinical-grade monoclonal antibodies by chromatographic methods. Methods Mol Biol. 2005;308:191–208. doi: 10.1385/1-59259-922-2:191. [DOI] [PubMed] [Google Scholar]
- 64.Alessio M, Roggero S, Funaro A, De Monte LB, Peruzzi L, Geuna M, Malavasi F. CD38 molecule: structural and biochemical analysis on human T lymphocytes, thymocytes, and plasma cells. J Immunol. 1990;145:878–84. [PubMed] [Google Scholar]
- 65.Horenstein AL, Stockinger H, Imhof BA, Malavasi F. CD38 binding to human myeloid cells is mediated by mouse and human CD31. Biochem J. 1998;330:1129–35. doi: 10.1042/bj3301129. [DOI] [PMC free article] [PubMed] [Google Scholar]