

Abstract
Purpose
No mutations associated with Alström syndrome (AS), a rare autosomal recessive disease, have been reported in the Japanese population. The purpose of this study was to investigate the genetic and clinical features of two brothers with AS in a consanguineous Japanese family.
Methods
Whole-exome sequencing analysis was performed on two brothers with AS and their unaffected parents. We performed a complete ophthalmic examination, including decimal best-corrected visual acuity, slit-lamp and funduscopic examination, visual-field and color-vision testing, full-field electroretinography, and optical coherence tomography. Fasting blood tests and systemic examinations were also performed.
Results
A novel mutation (c.6151C>T in exon 8) in the Alström syndrome 1 (ALMS1) gene that causes a premature termination codon at amino acid 2051 (p.Q2051X), was identified in the homozygous state in the affected brothers and in the heterozygous state in the parents. The ophthalmologic findings for both brothers revealed infantile-onset severe retinal degeneration and visual impairment, marked macular thinning, and severe cataracts. Systemic findings showed hepatic dysfunction, hyperlipidemia, hypogonadism, short stature, and wide feet in both brothers, whereas hearing loss, renal failure, abnormal digits, history of developmental delay, scoliosis, hypertension, and alopecia were not observed in either brother. The older brother exhibited type 2 diabetic mellitus and obesity, whereas the younger brother had hyperinsulinemia and subclinical hypothyroidism.
Conclusions
A novel ALMS1 mutation was identified by using whole-exome sequencing analysis, which is useful not only to identify a disease causing mutation but also to exclude other gene mutations. Although characteristic ophthalmologic findings and most systemic findings were similar between the brothers, the brothers differed slightly in terms of glucose tolerance and thyroid function.
Introduction
Alström syndrome (AS; OMIM: 203800) is a rare and autosomal recessive hereditary disease with an estimated prevalence of less than 0.001% [1,2]. AS is caused by mutations in the ALMS1 gene, which is located on chromosome 2p13 [3,4]. ALMS1 is localized to centrosomes and ciliary basal bodies [5,6] and has been implicated in the function, formation, and maintenance of primary cilia [5,7–9]. Dysfunction of primary cilia caused by mutations in genes such as ALMS1 leads to a multitude of human monogenic disorders known as ciliopathies [10,11]; these include plural systemic diseases, such as AS, Usher syndrome, Bardet–Biedl syndrome (BBS), Senior–Løken syndrome, Joubert syndrome, Meckel–Gruber syndrome, and orofaciodigital syndrome 1 [11,12]. The majority of ALMS1 mutations are nonsense and frameshift variations (primarily clustered in exons 8, 10, and 16) that are predicted to cause truncated proteins [3,4,13]. In the photoreceptors, ALMS1 mutations lead to defective function of the connecting cilium.
AS is characterized by a wide spectrum of disorders, such as early onset severe retinal degeneration, obesity from childhood, hyperinsulinemia, type 2 diabetic mellitus (T2DM), hepatic dysfunction, heart failure, sensory hearing loss, and renal failure [14]. Other manifestations include acanthosis nigricans, alopecia, hypogonadism, hypothyroidism, hyperlipidemia, short stature, and scoliosis [15,16]. In most cases of AS, cone–rod degeneration in the first decade, normal intelligence, and no polydactyly serve as a differential diagnosis of BBS, which exhibits similar clinical findings to AS [17].
Almost all patients with AS show nystagmus and severe photophobia from infancy [14,18]. Visual impairment is usually seen at an age younger than 1 year [18]. Although the rate of progression of vision loss is variable, all patients show progressive retinal degeneration, with 90% becoming totally blind by the age of 16 years [19] and all becoming blind eventually [14,19]. Due to severe retinal degeneration and visual impairment during the first months of life, AS is often confused with congenital retinal degenerations, such as Leber congenital amaurosis (LCA) and congenital achromatopsia (ACHM) [20,21]. There are several reports of Japanese patients with AS [22–24]; however, there has been no report identifying any ALMS1 mutation associated with AS in the Japanese population.
Recently, the development of next-generation sequencing technology has facilitated biologic and biochemical research by enabling the broad analysis of genomes [25–28]. The whole genome of an individual can now be sequenced at great depth, and genomic capture technology can be used to isolate sequences of interest [29–32].
Here, we used whole-exome sequencing to identify a novel ALMS1 mutation in two Japanese brothers with AS. We also examined the clinical features of the two brothers in detail.
Methods
The protocol of this study was approved by the Institutional Review Board of the Jikei University School of Medicine and National Hospital Organization Tokyo Medical Center. The protocol adhered to the tenets of the Declaration of Helsinki, and written informed consent was obtained from all participants.
Clinical studies
The study was conducted in one consanguineous Japanese family (JU#0769–095JIKEI) with AS (Figure 1). The parents were second cousins. The clinical history was taken in detail, and the following ophthalmic examinations were performed: decimal best-corrected visual acuity (BCVA), slit-lamp and fundus examinations, and time-domain optical coherence tomography (TD-OCT; OCT3 Stratus; Carl Zeiss Meditec AG, Dublin, CA) or spectral-domain OCT (SD-OCT; Cirrus HD-OCT; Carl Zeiss Meditec AG). In color-vision tests, we used the Ishihara test (38-plate edition) and the Farnsworth Panel D-15 (Panel D-15). Visual-field testing by kinetic perimetry was conducted by using the Goldmann perimeter (GP; Haag Streit, Bern, Switzerland). Full-field electroretinography (ERG) was performed according to the protocols of the International Society for Clinical Electrophysiology of Vision. The procedure and conditions for ERG recording have been detailed previously [33].
Figure 1.
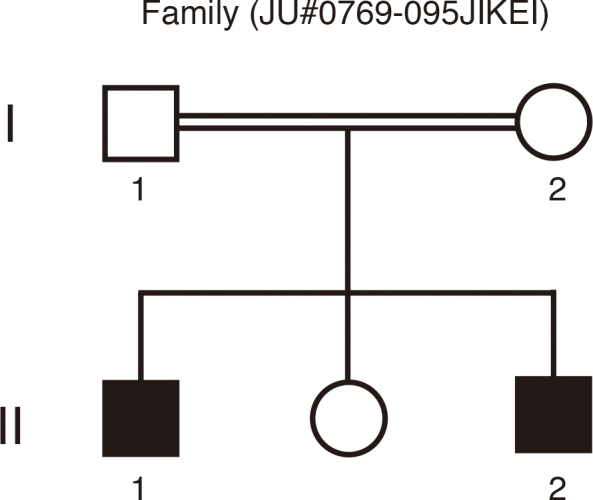
A consanguineous family (JU#0769–095JIKEI) with Alström syndrome. Two affected brothers (II-1 and II-2) with Alström syndrome and their unaffected parents are depicted.
Fasting venous blood samples were analyzed for glucose, lipid, lipoprotein, and hemogram levels and renal, liver, and thyroid function tests. In addition, hemoglobin A1c, insulin, anti-thyroid peroxidase, anti-thyroglobulin antibodies, cortisol, luteinizing hormone, follicle stimulating hormone, testosterone, estradiol, prolactin, parathyroid hormone, and thyroid receptor antibody levels were examined. Chest X-rays and electrocardiograms were also performed.
DNA preparation and exome sequencing analysis
We obtained venous blood samples from the affected brothers and their unaffected parents. Genomic DNA was extracted from the blood samples by using a Gentra Puregene Blood kit (Qiagen, Tokyo, Japan) and sheared with a Covaris Ultrasonicator (Covaris, Woburn, MA). Construction of paired-end sequence libraries and exome capture were performed by using the Agilent Bravo automated liquid-handling platform with SureSelect XT Human All Exon kit V4 + UTRs kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s instructions. Enriched libraries were sequenced by using in Illumina HiSeq2000 sequencer (San Diego, CA), according to the manufacturer’s instructions for 100-bp paired-end sequencing. Reads were mapped to the reference human genome (1000 genomes phase 2 reference, hs37d5) with Burrows–Wheeler Aligner software version 0.6.2 [34]. Duplicated reads were then removed by Picard MarkDuplicates module version 1.62, and mapped reads around insertion–deletion polymorphisms (INDELs) were realigned by using the Genome Analysis Toolkit (GATK) version 2.1–13 [35]. Base-quality scores were recalibrated by using GATK. Calling of mutations was performed by using the GATK UnifiedGenotyper module, and called single-nucleotide variants and INDELs were annotated by using snpEff software version 3.0 [36]. The mutations were annotated with the snpEff score (“HIGH,” “MODERATE,” or “LOW”) and with the allele frequency in the 1000 genomes database. The mutations were then filtered so that only those with “HIGH” or “MODERATE” snpEff scores (indicating that the amino acid sequence would be functionally affected) and a frequency of less than 1% in the 1000 genome database were analyzed further. We also used new variations, which were not found in the in-house database of seven people exome data with control individuals without ocular diseases. Mutations were classified by hereditary information into homozygous recessive, heterozygous recessive, and de novo mutations in the family members. Filtered mutations were scored with PolyPhen software version 2.2.2 [37], which predicts the effect on the structure and function of the protein. The above exome analysis pipeline is available at Cell Innovation.
Results
Ophthalmologic findings for patient II-1
Patient II-1, the elder of the two brothers, was referred to our hospital at the age of 7 years and 4 months for the assessment of poor visual acuity from infancy. His BCVA was 0.04 (with +2.00 diopter [dpt], cylinder [cyl] –1.00 dpt axis [Ax] 180°) in the right eye and 0.06 (with +2.00 dpt, cyl –1.00 dpt Ax 180°) in the left eye. Fundus examination showed slight retinal degeneration in both eyes. At the age of 9 years, the patient recognized only the first plate in the Ishihara test for color vision, the panel D-15 test for color vision showed irregular arrangements along no particular axis, and the ERG showed no standard combined, photopic, or 30-Hz flicker responses in either eye (Figure 2). GP analysis at the age of 11 years showed markedly constricted visual fields in V-4e and I-4e isopters of both eyes (Figure 3A). The fundus photographs at the age of 14 years showed retinal degeneration with attenuated vessels from the arcade to the periphery in both eyes (Figure 4A). GP analysis at the age of 16 years showed more marked constricted visual fields of V-4e and I-4e isopters in both eyes than those observed at the age of 11 years (Figure 3B); a similar analysis at the age of 22 years showed a small visual field of V-4e isopter in the right eye and no visual field in the left eye (Figure 3C). TD-OCT at the age of 22 years showed total macular thinning in both eyes (Figure 5A). At the age of 29 years, his BCVA was light perception (LP) in the right eye and no light perception in the left eye. Intraocular pressure in each eye was within the normal range. He had severe cortical and subcapsular cataracts in the right (Figure 6) and left eyes, and the fundi were not visible due to these cataracts.
Figure 2.

Full-field electroretinogram. The electroretinograms (ERGs; patient II-1) at the age of 9 years, showing no standard combined, photopic, or 30-Hz flicker responses in either eye. The ERGs (patient II-2) at the age of 7 years, showing no rod, standard combined, photopic, or 30-Hz flicker responses in either eye.
Figure 3.

Visual fields assayed by Goldmann perimetry in patient II-1. A–C: Visual fields at the age of 11 years (A), at the age of 16 years (B), and at the age of 22 years (C). Markedly constricted visual fields (V-4e and I-4e isopters) are observed in both eyes, and the visual fields become constricted as the patient ages.
Figure 4.

Fundus photographs of patients II-1 and II-2. A and B: Fundus photographs of patient II-1 at the age of 14 years (A) and patient II-2 at the age of 8 years (B) show retinal degeneration with attenuated vessels in the posterior poles of both eyes.
Figure 5.

Optic coherence tomography findings. A and B: Time domain optic coherence tomography (OCT; retinal mapping) of patient II-1 at the age of 22 years (A) and II-2 at the age of 16 years (B) show total macular thinning in both eyes. C: Spectral-domain OCT (HD-5-line raster) of patient II-2 at the age of 23 years, showing marked macular thinning with indistinguishable retinal layers in the macular areas of both eyes.
Figure 6.
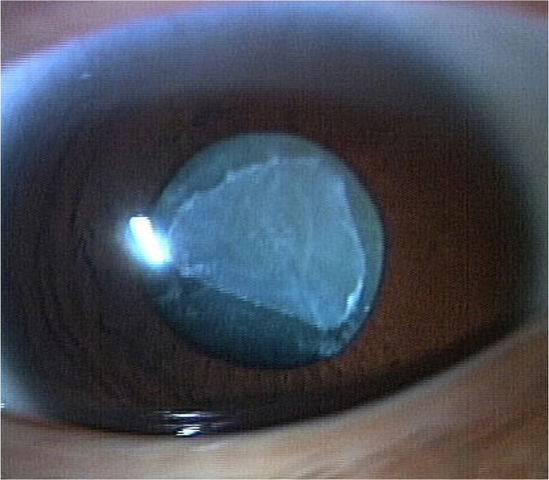
Anterior segment of the right eye in patient II-1. A severe cortical and anterior subcapsular cataract is present at the age of 29 years.
Ophthalmologic findings for patient II-2
Patient II-2, the younger of the two brothers, visited our hospital at the age of 2 years and 6 months with the main complaint of poor visual acuity and photophobia. At the age of 3 years, his BCVA was 0.01 (+1.50 dpt) in the right eye and 0.01 (+1.50 dpt) in the left eye. Fundus examination showed retinal degeneration with slight attenuation of peripheral vessels. At the age of 4 years, he failed the Ishihara test. At the age of 6 years, the panel D-15 test showed irregular arrangements along no particular axis, and the GP could not be measured well because of low visual acuity and nystagmus. The ERG at the age of 7 years showed no rod, standard combined, photopic, or 30-Hz flicker responses in either eye (Figure 2). The fundus examination at the age of 8 years showed retinal degeneration with attenuated vessels from the arcade artery to the periphery in both eyes (Figure 4B). TD-OCT at the age of 16 years showed total macular thinning in both eyes (Figure 5B). At the age of 23 years, his BCVA was LP in both eyes, the intraocular pressure was within the normal range in both eyes, posterior subcapsular cataracts were present in both eyes, and SD-OCT showed total macular thinning with indistinguishable retinal layers in both eyes (Figure 5C).
Systemic features except ocular findings
Systemic examinations were performed for patient II-1 at the age of 29 and patient II-2 at the age of 23. Both patients had hepatic dysfunction, hyperlipidemia, hypogonadism, short stature, and flat feet, and neither patient had hearing loss, renal failure, abnormal digits, history of developmental delay, mental retardation, scoliosis, hypertension, or alopecia. Obesity was present in patient II-1 only. Patient II-1 had T2DM, whereas patient II-2 showed hyperinsulinemia. Subclinical hypothyroidism was diagnosed in patient II-2 only. Recurrent pulmonary infections were not observed, and chest X-rays showed neither fibrotic infiltrations nor cardiac dilation in either patient. Infantile asthma was experienced by both patients. Electrocardiogram analysis showed no arrhythmia in either patient. Summaries of the clinical features, bio-information, and detailed laboratory data are presented in Table 1 and Table 2. Collectively, the phenotypes of the brothers were consistent with those described for AS.
Table 1. Clinical characteristics.
| Clinical findings | Patient II-1 | Patient II-2 | Percentage (%) in 182 casesa |
|---|---|---|---|
| Low vision |
+ |
+ |
100 |
| Subcapsular cataracts |
+ |
+ |
32 |
| Hearing loss |
– |
– |
88 |
| Cardiomyopathy |
– |
– |
62 |
| Type 2 diabetes mellitus |
+ |
– |
68 |
| Childhood obesity |
+ |
– |
98 |
| Hyperinsulinemia |
– |
+ |
92 |
| Short stature |
+ |
+ |
98 |
| Hypertriglyceridemia |
+ |
+ |
52 |
| Hypothyroidism |
– |
+ (subclinical) |
17 |
| Hypogonadism |
+ |
+ |
78 |
| Elevated hepatic enzyme levels |
+ |
+ |
92 |
| Renal insufficiency |
– |
– |
50 |
| Renal hypertension |
– |
– |
30 |
| Pulmonary symptoms |
– |
– |
52 |
| Asthma |
+ (childhood) |
+ (childhood) |
19 |
| Muscle weakness |
– |
– |
29 |
| Global development delay |
– |
– |
49 |
| Wide feet |
+ |
+ |
ND |
| Abnormal digits |
– |
– |
ND |
| Scoliosis |
– |
– |
ND |
| Alopecia | – | – | ND |
ND, not described. aThe cases are cited [13].
Table 2. Bio-information and biochemical assessment.
| Bio-information and blood test results | Normal range | Patient II-1 | Patient II-2 |
|---|---|---|---|
| Bio-information |
|||
| Weight (kg) |
60 |
52 |
|
| Height (m) |
1.52 |
1.55 |
|
| Body mass index (kg/m2) |
18.5–25 |
25.96 |
21.6 |
| Biochemical assessment |
|||
| Fasting blood glucose (mg/dl) |
65–109 |
247 |
77 |
| Hemoglobin A1c (%) |
4.6–6.2 |
12.5 |
6.0 |
| Urea (mg/dl) |
8–20 |
16 |
10 |
| Creatinine (mg/dl) |
0.50–1.10 |
0.99 |
0.63 |
| Uric acid (mg/dl) |
3.1–6.9 |
4.2 |
3.6 |
| Sodium (mmol/l) |
136–146 |
140 |
141 |
| Potassium (mmol/l) |
3.6–4.8 |
4.1 |
4.3 |
| Chloride (mmol/l) |
98–109 |
100 |
104 |
| Calcium (mg/dl) |
8.6–10.2 |
10.3 |
10.2 |
| Aspartate aminotransferase (U/l) |
10–33 |
76 |
81 |
| Alanine aminotransferase (U/l) |
6–35 |
99 |
241 |
| Gamma glutamyl transpeptidase (U/l) |
12–65 |
134 |
135 |
| Alkaline phosphatase (U/l) |
96–300 |
285 |
319 |
| Low density lipoprotein-cholesterol (mg/dl) |
70–139 |
120 |
282 |
| Total cholesterol (mg/dl) |
120–219 |
211 |
441 |
| Triglycerides (mg/dl) |
30–149 |
309 |
761 |
| Albumin/globulin (g/dl) |
3.5–5.2 |
5.0 |
5.1 |
| Hemogram |
|||
| White blood cells (103/ml) |
3.3–8.6 |
4.7 |
6.4 |
| Red blood cells (106/ml) |
4.10–5.50 |
5.00 |
4.88 |
| Hemoglobin (g/dl) |
13.5–16.5 |
14.0 |
14.3 |
| Hematocrit (%) |
40.0–50.0 |
42.3 |
42.7 |
| Mean corpuscular volume (fl) |
83.0–101.0 |
84.6 |
87.5 |
| Platelets (103/ml) |
150–350 |
166 |
251 |
| Erythrocyte sedimentation rate (mm/h) |
2–10 |
21 |
19 |
| Hormones and autoantibodies |
|||
| Free T3 (pg/ml) |
2.36–5.00 |
2.34 |
2.47 |
| Free T4 (pmol/l) |
0.88–1.67 |
1.33 |
0.79 |
| Thyroid stimulating hormone (mIU/l) |
0.34–4.04 |
3.06 |
4.67 |
| Anti-thyroid peroxidase antibody (U/ml) |
0.0–15 |
10 |
10 |
| Anti-thyroglobulin antibody (U/ml) |
0.0–27 |
11 |
<10 |
| Thyroglobulin (ng/ml) |
0.0–32.7 |
6.1 |
29.4 |
| Thyroid stimulating hormone receptor antibody (IU/ml) |
<1.0 |
0.9 |
<1.0 |
| Parathyroid hormone (pg/ml) |
10–65 |
26 |
20 |
| Luteinizing hormone (mIU/ml) |
1.7–8.6 |
13.2 |
9.1 |
| Follicle stimulating hormone (mIU/ml) |
1.5–12.4 |
30.7 |
20.3 |
| Estradiol (pg/ml) |
14.0–43.9 |
18.7 |
10.6 |
| Total testosterone (ng/ml) |
250.0–1100.0 |
90.7 |
53.5 |
| Prolactin (ng/ml) |
4.3–13.7 |
3.7 |
2.5 |
| Cortisol (mg/dl) |
4.0–18.3 |
8.1 |
10.6 |
| Insulin (µU/ml) |
0.0–13.0 |
9.2 |
63.6 |
| Growth hormone (ng/ml) | 0–2.47 | 0.3 | 0.27 |
Bold type indicates values outside the normal range
Exome sequencing analysis and identification of a gene mutation
We performed whole-exome sequencing of the two affected brothers and their parents by using the Agilent SureSelect Human All Exon kit followed by Illumina HiSeq 2000 platforms. Sequences of average length 11.8 Gb were generated from 101-bp paired-end sequences. After eliminating reads from PCR duplicates by discarding reads with duplicated start sites, we achieved 58-fold depth and 87% coverage in Refseq annotated regions (Table 3). When the sequences were compared with the reference human genome (hs37d5), 3,506,741 mutations were found in the two brothers and their parents (Table 4). To distinguish potentially causal mutations from other mutations, we focused only on mutations that could change the amino acid sequence (19,574 mutations), such as nonsynonymous mutations, splice acceptor and donor site mutations, and INDELs. We also assumed the frequency of the mutations responsible for AS is likely to be under 1%. After filtering with snpEff score and frequency criteria, we filtered the remaining 3,685 mutations by using the pattern of inheritance and identified 17 gene mutations as causal candidates. Among these mutations, nine mutations were found homozygous in the HECT domain containing E3 ubiquitin protein ligase 3 (HECTD3), the vitrin (VIT), the protein kinase domain containing, cytoplasmic (PKDCC), the ATP-binding cassette, sub-family G (WHITE), member 8 (ABCG8), the leucine-rich pentatricopeptide repeat containing (LRPPRC), the G protein-coupled receptor 75 (GPR75), the notochord homeobox (NOTO), the matrix-remodelling associated 5 (MXRA5), and the ALMS1 genes. Eight mutations were found as compound heterozygous mutations within the PERP, TP53 apoptosis effector (PERP), the transforming, acidic coiled-coil containing protein 2 (TACC2), the zinc finger protein, FOG family member 1 (ZFPM1), and the lipoxygenase homology domains 1 (LOXHD1) genes. No de novo mutations were found. To determine the causative gene, we investigated SAGE (EyeSAGE) database to determine if the candidate genes are expressed in the retina. Nine candidate mutations were identified within VIT, LRPPRC, PERP, TACC2, ZFPM1, and ALMS1 genes. These nine candidate genes were further reduced by the BIOBASE Biologic Database and RetNet to determine which of the candidate genes would be likely to progress known phenotype with syndromic disorders. Finally, ALMS1 was speculated to be the disease-causing gene. The ALMS1 sequence was compared with the NCBI reference sequence of the ALMS1 transcript (GenBank NM_015120.4).
Table 3. DNA sequence statistics.
| Family members | Read length (bp) | Number of reads | Mapping rate (%) | Mean depth (fold) | Coverage (%) |
|---|---|---|---|---|---|
| II-2 (younger brother) |
101 |
47,724,724 |
99.4 |
46.6 |
88.5 |
| II-1 (elder brother) |
101 |
68,584,852 |
99.4 |
59.6 |
86.1 |
| I-1 (father) |
101 |
57,103,807 |
99.3 |
69.3 |
88.5 |
| I-2 (mother) |
101 |
59,776,264 |
99.3 |
56.8 |
86 |
| Average | 101 | 58,297,412 | 99.3 | 58.1 | 87.3 |
Table 4. Number of mutations after each filtering step.
| Filtering step | Number of mutations |
|---|---|
| 1. Raw single-nucleotide variants plus insertion–deletion polymorphisms |
3,506,741 |
| 2. Mutations capable of changing amino acid sequence |
19,574 |
| 3. Mutations filtering by the snpEff score and existing at a frequency of less than 1% in 1000 genomes |
3,685 |
| 4. Mutations filtering by the pattern of inheritance |
17 |
| 5. Mutations expressed in retina, confirmed by SAGE databasea |
9 |
| 6. Mutations narrowed down using BIOBASE Biologic Databaseb and RetNet databasec | 1 |
As a result, in the two affected brothers we identified a novel single-nucleotide substitution at position 6151 (c.6151C>T in exon 8) that causes a premature termination codon at amino acid 2051 (p.Q2051X) of the ALMS1 gene resulting in a truncated protein. Both brothers were homozygous for the mutant allele, whereas the unaffected parents were heterozygous carriers of the allele, also reconfirmed by Sanger sequencing. The novel ALMS1 mutation (p.Q2051X) was not found in any of 100 Japanese individuals without ocular disease in the Single Nucleotide Polymorphism Database or in the Human Gene Mutation Database.
Discussion
To date, no patient with ALMS1-associated AS has been reported in the Japanese population. Here, we identified a novel ALMS1 mutation (p.Q2051X) in two Japanese brothers with AS.
Marshall et al. advocate criteria for the diagnosis of AS [19]. In patients over the age of 15, it is necessary to fulfill “two major and two minor criteria” or “one major and four minor criteria” [19]. Our brother patients exhibited two major (ALMIS1 mutation and loss of vision, such as legal blindness) and more than four minor criteria (obesity, insulin resistance, and/or T2DM, hepatic dysfunction, short stature, and hypogonadism). Also, the phenotypic expression of AS is differentiated from BBS characterized by later onset retinal dystrophy, polydactyly, central obesity, learning disabilities, hypogonadism, and renal anomalies [38].
In patients with AS, phenotypic variability in disease severity and retinal function assessed by electroretinographic and visual-field testing [39] and variability in pathological or anatomic changes of the retina [14,18,21,40] have been reported. For instance, a study of the pathology of the retina of a 2-year-old girl with AS showed hypocellularity of the ganglionic cell layer, the inner nuclear layer, and the outer nuclear layer (ONL) in addition to an absence of rod and cone outer segments and disruption of retinal pigment epithelium [18,21]; a study of a 42-year-old female with AS revealed severe reduction of all retinal layers containing a complete lack of photoreceptors and deposits of melanin pigments in the inner nuclear layer [14]; and OCT findings of a 5-year-old boy with AS showed only a slight thinning of the central retina [40]. In our patients, OCT findings showed marked retinal thinning (Figure 5A,B). The retinal layers of patient II-2 could not be distinguished because of marked retinal thinning (Figure 5C).
A study using retinal sections of Alms1 knockout (Alms1−/−) mice showed loss of the cell bodies in the ONL, shortening of the inner and outer segments, and incorrect localization of rhodopsin to the ONL [7]. The mislocalization of rhodopsin in the Alms1−/− mice indicates a defective rhodopsin transport system through the photoreceptor-connecting cilium [7]. The connecting cilium, damaged by loss of function of ALMS1, modifies the outer segments of the photoreceptors. Therefore, it has been suggested that defective protein transport across the connecting cilium is the probable cause of early onset severe retinal degeneration in AS patients [10]. We consider that the marked retinal thinning (Figure 5) and loss of retinal function (Figure 2) observed in our patients are due to a defective transport system across the photoreceptor-connecting cilium, resulting from the homozygous truncated mutation (p.Q2051X).
Variability in the phenotypic expression of AS is observed within sets of affected siblings [14,41–43]. Most patients with AS eventually develop T2DM, although there is wide variability in the age of onset [14]. Here, patient II-1 showed T2DM, but patient II-2 exhibited hyperinsulinemia, a predictor of T2DM (Table 2), suggesting that he might develop T2DM in the future. In addition, patient II-2 showed subclinical hypothyroidism, whereas patient II-1 did not exhibit hypothyroidism (Table 2). Hypothyroidism or subclinical hypothyroidism is reported to exist in approximately 20% of AS patients [14,19]. Most clinical features, such as retinal degeneration, hepatic dysfunction, hyperlipidemia, hypogonadism, short stature, and wide feet, were common features of the affected brothers (Table 1, Table 2); however, slight phenotypic differences in terms of glucose tolerance and thyroid function were observed between them.
ALMS1 protein has several notable sequence features, including an extensive tandem repeat domain (34×47 amino acid approximate tandem repeat, residues 538–2,199), a putative leucine-zipper motif (residues 2,480–2,501), and an ALMS motif (residues 4,035–4,167). Although the precise roles of the above domain and motifs are unknown, it is suggested that two regions of ALMS1—a relatively small internal region (residues 2,261–2,602) and a larger C-terminal region (residues 3,176–4,169)—play important roles in targeting ALMS1 to the centrosomes and ciliary basal bodies [44]. In our patients, if the truncated protein caused by the mutation (p.Q2051X) is expressed in the retina, the protein would not contain the two regions important for targeting (residues 2,261–2,602 and residues 3,176–4,169) or the putative leucine-zipper and ALMS motifs. Therefore, this truncated mutation would cause loss of function of ALMS1, resulting in the AS phenotype. Although genotype–phenotype correlations are not clear among AS patients with ALMS1 mutations [45,46], patients with mutations in exon 8 are reported to have delayed and milder renal complications compared with those with mutations in exons 10 and 16 [13]. In our patients, the p.Q2051X mutation was present in exon 8, explaining normal renal function.
The syndromic disorder AS is often misdiagnosed as LCA, ACHM, or other ciliopathies [11,20,21], so the identification of diagnostic mutations is important. Also, early diagnosis may improve longevity and long-term quality of life. By the whole-exome sequencing analysis technique, we were able to comprehensively determine the disease-causing gene mutation by using the fewest samples possible from the pedigree and analyzing all exon sequences in a relatively short time. Because of the autosomal recessive inheritance pattern, the parents and two affected brothers were enough to narrow down the candidate genes. Consequently, we identified a single causative gene mutation (p.Q2051X of ALMS1). Whole-exome sequence analysis should play an important role in future diagnostics for AS.
In conclusion, there has been no report of any AS patient with an ALMS1 mutation in the Japanese population, probably because AS is an extremely rare inherited disease. We identified a novel ALMS1 mutation in two brothers of a consanguineous family and examined their clinical features in detail. Our results indicate the presence of different mutations in AS between Japanese and other populations.
ACKNOWLEGMENTS
We thank the patients and their families for participation in this study. This study was supported by grants to T.I. from the Ministry of Health, Labor and Welfare of Japan (13,803,661), to M.A. and T.H. from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grant-in-Aid for Scientific Research C, 25,462,744 and 25,462,738), to T.H. from the Vehicle Racing Commemorative Foundation, and to M.F. from the Research Grant for RIKEN Omics Science Center MEXT. The authors wish to acknowledge RIKEN GeNAS for the sequencing of the Exome enriched libraries using the Illumina HiSeq2000.
References
- 1.Paisey RB, Carey CM, Bower L, Marshall J, Taylor P, Maffei P, Mansell P. Hypertriglyceridaemia in Alström's syndrome: causes and associations in 37 cases. Clin Endocrinol (Oxf) 2004;60:228–31. doi: 10.1111/j.1365-2265.2004.01952.x. [DOI] [PubMed] [Google Scholar]
- 2.Joy T, Cao H, Black G, Malik R, Charlton-Menys V, Hegele RA, Durrington PN. Alström syndrome (OMIM 203800): a case report and literature review. Orphanet J Rare Dis. 2007;2:49. doi: 10.1186/1750-1172-2-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, Beck S, Boerkoel CF, Sicolo N, Martin M, Nishina PM, Naggert JK. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat Genet. 2002;31:74–8. doi: 10.1038/ng867. [DOI] [PubMed] [Google Scholar]
- 4.Hearn T, Renforth GL, Spalluto C, Hanley NA, Piper K, Brickwood S, White C, Connolly V, Taylor JF, Russell-Eggitt I, Bonneau D, Walker M, Wilson DI. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alström syndrome. Nat Genet. 2002;31:79–83. doi: 10.1038/ng874. [DOI] [PubMed] [Google Scholar]
- 5.Hearn T, Spalluto C, Phillips VJ, Renforth GL, Copin N, Hanley NA, Wilson DI. Subcellular localization of ALMS1 supports involvement of centrosome and basal body dysfunction in the pathogenesis of obesity, insulin resistance, and type 2 diabetes. Diabetes. 2005;54:1581–7. doi: 10.2337/diabetes.54.5.1581. [DOI] [PubMed] [Google Scholar]
- 6.Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426:570–4. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- 7.Collin GB, Cyr E, Bronson R, Marshall JD, Gifford EJ, Hicks W, Murray SA, Zheng QY, Smith RS, Nishina PM, Naggert JK. Alms1-disrupted mice recapitulate human Alström syndrome. Hum Mol Genet. 2005;14:2323–33. doi: 10.1093/hmg/ddi235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graser S, Stierhof YD, Lavoie SB, Gassner OS, Lamla S, Le Clech M, Nigg EA. Cep164, a novel centriole appendage protein required for primary cilium formation. J Cell Biol. 2007;179:321–30. doi: 10.1083/jcb.200707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li G, Vega R, Nelms K, Gekakis N, Goodnow C, McNamara P, Wu H, Hong NA, Glynne R. A role for Alström syndrome protein, alms1, in kidney ciliogenesis and cellular quiescence. PLoS Genet. 2007;3:e8. doi: 10.1371/journal.pgen.0030008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adams M, Smith UM, Logan CV, Johnson CA. Recent advances in the molecular pathology, cell biology and genetics of ciliopathies. J Med Genet. 2008;45:257–67. doi: 10.1136/jmg.2007.054999. [DOI] [PubMed] [Google Scholar]
- 11.Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–48. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 12.Adams NA, Awadein A, Toma HS. The retinal ciliopathies. Ophthalmic Genet. 2007;28:113–25. doi: 10.1080/13816810701537424. [DOI] [PubMed] [Google Scholar]
- 13.Marshall JD, Hinman EG, Collin GB, Beck S, Cerqueira R, Maffei P, Milan G, Zhang W, Wilson DI, Hearn T, Tavares P, Vettor R, Veronese C, Martin M, So WV, Nishina PM, Naggert JK. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alström syndrome. Hum Mutat. 2007;28:1114–23. doi: 10.1002/humu.20577. [DOI] [PubMed] [Google Scholar]
- 14.Marshall JD, Bronson RT, Collin GB, Nordstrom AD, Maffei P, Paisey RB, Carey C, Macdermott S, Russell-Eggitt I, Shea SE, Davis J, Beck S, Shatirishvili G, Mihai CM, Hoeltzenbein M, Pozzan GB, Hopkinson I, Sicolo N, Naggert JK, Nishina PM. New Alström syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med. 2005;165:675–83. doi: 10.1001/archinte.165.6.675. [DOI] [PubMed] [Google Scholar]
- 15.Koç E, Bayrak G, Suher M, Ensari C, Aktas D, Ensari A. Rare case of Alström syndrome without obesity and with short stature, diagnosed in adulthood. Nephrology (Carlton) 2006;11:81–4. doi: 10.1111/j.1440-1797.2006.00443.x. [DOI] [PubMed] [Google Scholar]
- 16.Akdeniz N, Bilgili SG, Aktar S, Yuca S, Calka O, Kilic A, Kosem M. Alström syndrome with acanthosis nigricans: a case report and literature review. Genet Couns. 2011;22:393–400. [PubMed] [Google Scholar]
- 17.Dyer DS, Wilson ME, Small KW, Pai GS. Alström syndrome: a case misdiagnosed as Bardet-Biedl syndrome. J Pediatr Ophthalmol Strabismus. 1994;31:272–4. doi: 10.3928/0191-3913-19940701-19. [DOI] [PubMed] [Google Scholar]
- 18.Russell-Eggitt IM, Clayton PT, Coffey R, Kriss A, Taylor DS, Taylor JF. Alström syndrome. Report of 22 cases and literature review. Ophthalmology. 1998;105:1274–80. doi: 10.1016/S0161-6420(98)97033-6. [DOI] [PubMed] [Google Scholar]
- 19.Marshall JD, Beck S, Maffei P, Naggert JK. Alström syndrome. Eur J Hum Genet. 2007;15:1193–202. doi: 10.1038/sj.ejhg.5201933. [DOI] [PubMed] [Google Scholar]
- 20.Lambert SR, Kriss A, Taylor D, Coffey R, Pembrey M. Follow-up and diagnostic reappraisal of 75 patients with Leber's congenital amaurosis. Am J Ophthalmol. 1989;107:624–31. doi: 10.1016/0002-9394(89)90259-6. [DOI] [PubMed] [Google Scholar]
- 21.Russell-Eggitt IM, Taylor DS, Clayton PT, Garner A, Kriss A, Taylor JF. Leber's congenital amaurosis–a new syndrome with a cardiomyopathy. Br J Ophthalmol. 1989;73:250–4. doi: 10.1136/bjo.73.4.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ikeda Y, Morita Y, Matsuo Y, Akanuma Y, Itakura H. A case of Alström syndrome associated with situs inversus totalis and characteristic liver cirrhosis. Nippon Naika Gakkai Zasshi. 1974;63:1303–11. [PubMed] [Google Scholar]
- 23.Awazu M, Tanaka T, Sato S, Anzo M, Higuchi M, Yamazaki K, Matsuo N. Hepatic dysfunction in two sibs with Alström syndrome: case report and review of the literature. Am J Med Genet. 1997;69:13–6. [PubMed] [Google Scholar]
- 24.Awazu M, Tanaka T, Yamazaki K, Kato S, Higuchi M, Matsuo N. A 27-year-old woman with Alström syndrome who had liver cirrhosis. Keio J Med. 1995;44:67–73. [PubMed] [Google Scholar]
- 25.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–45. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 26.Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–41. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 27.Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 28.Ansorge WJ. Next-generation DNA sequencing techniques. New Biotechnol. 2009;25:195–203. doi: 10.1016/j.nbt.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 29.Teer JK, Mullikin JC. Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet. 2010;19(R2):R145–51. doi: 10.1093/hmg/ddq333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H, Albrechtsen A, Andersen G, Cao H, Korneliussen T, Grarup N, Guo Y, Hellman I, Jin X, Li Q, Liu J, Liu X, Sparso T, Tang M, Wu H, Wu R, Yu C, Zheng H, Astrup A, Bolund L, Holmkvist J, Jorgensen T, Kristiansen K, Schmitz O, Schwartz TW, Zhang X, Li R, Yang H, Wang J, Hansen T, Pedersen O, Nielsen R, Wang J. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet. 2010;42:969–72. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- 31.Kim DW, Nam SH, Kim RN, Choi SH, Park HS. Whole human exome capture for high-throughput sequencing. Genome. 2010;53:568–74. doi: 10.1139/g10-025. [DOI] [PubMed] [Google Scholar]
- 32.Hodges E, Rooks M, Xuan Z, Bhattacharjee A, Benjamin Gordon D, Brizuela L, Richard McCombie W, Hannon GJ. Hybrid selection of discrete genomic intervals on custom-designed microarrays for massively parallel sequencing. Nat Protoc. 2009;4:960–74. doi: 10.1038/nprot.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeuchi T, Hayashi T, Bedell M, Zhang K, Yamada H, Tsuneoka H. A novel haplotype with the R345W mutation in the EFEMP1 gene associated with autosomal dominant drusen in a Japanese family. Invest Ophthalmol Vis Sci. 2010;51:1643–50. doi: 10.1167/iovs.09-4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cingolani P, Platts A. Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 2013; Chapter 7:Unit7 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, Heath O, McManamon PJ, O'Leary E, Pryse-Phillips W. The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med. 1989;321:1002–9. doi: 10.1056/NEJM198910123211503. [DOI] [PubMed] [Google Scholar]
- 39.Malm E, Ponjavic V, Nishina PM, Naggert JK, Hinman EG, Andreasson S, Marshall JD, Moller C. Full-field electroretinography and marked variability in clinical phenotype of Alström syndrome. Arch Ophthalmol. 2008;126:51–7. doi: 10.1001/archophthalmol.2007.28. [DOI] [PubMed] [Google Scholar]
- 40.Vingolo EM, Salvatore S, Grenga PL, Maffei P, Milan G, Marshall J. High-resolution spectral domain optical coherence tomography images of Alström syndrome. J Pediatr Ophthalmol Strabismus. 2010;47 Online:e1–3. doi: 10.3928/01913913-20100507-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marshall JD, Ludman MD, Shea SE, Salisbury SR, Willi SM, LaRoche RG, Nishina PM. Genealogy, natural history, and phenotype of Alström syndrome in a large Acadian kindred and three additional families. Am J Med Genet. 1997;73:150–61. doi: 10.1002/(sici)1096-8628(19971212)73:2<150::aid-ajmg9>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 42.Hoffman JD, Jacobson Z, Young TL, Marshall JD, Kaplan P. Familial variable expression of dilated cardiomyopathy in Alström syndrome: a report of four sibs. Am J Med Genet A. 2005;135:96–8. doi: 10.1002/ajmg.a.30688. [DOI] [PubMed] [Google Scholar]
- 43.Hung YJ, Jeng C, Pei D, Chou PI, Wu DA. Alström syndrome in two siblings. J Formos Med Assoc. 2001;100:45–9. [PubMed] [Google Scholar]
- 44.Knorz VJ, Spalluto C, Lessard M, Purvis TL, Adigun FF, Collin GB, Hanley NA, Wilson DI, Hearn T. Centriolar association of ALMS1 and likely centrosomal functions of the ALMS motif-containing proteins C10orf90 and KIAA1731. Mol Biol Cell. 2010;21:3617–29. doi: 10.1091/mbc.E10-03-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bond J, Flintoff K, Higgins J, Scott S, Bennet C, Parsons J, Mannon J, Jafri H, Rashid Y, Barrow M, Trembath R, Woodruff G, Rossa E, Lynch S, Sheilds J, Newbury-Ecob R, Falconer A, Holland P, Cockburn D, Karbani G, Malik S, Ahmed M, Roberts E, Taylor G, Woods CG. The importance of seeking ALMS1 mutations in infants with dilated cardiomyopathy. J Med Genet. 2005;42:e10. doi: 10.1136/jmg.2004.026617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Minton JA, Owen KR, Ricketts CJ, Crabtree N, Shaikh G, Ehtisham S, Porter JR, Carey C, Hodge D, Paisey R, Walker M, Barrett TG. Syndromic obesity and diabetes: changes in body composition with age and mutation analysis of ALMS1 in 12 United Kingdom kindreds with Alström syndrome. J Clin Endocrinol Metab. 2006;91:3110–6. doi: 10.1210/jc.2005-2633. [DOI] [PubMed] [Google Scholar]