

Abstract
Purpose
gp96 (grp94) is a key downstream chaperone in the ER to mediate unfolded protein response (UPR) and the pathogenesis of multiple myeloma (MM) is closely linked to dysregulated UPR. In this study, we aimed to determine the roles of gp96 in the initiation and progression of MM in vivo and in vitro.
Experimental design
We generated a mouse model with over-expression of XBP1s and conditional deletion of gp96 in B cell compartment simultaneously to identify the roles of gp96 in the development of MM in vivo. Using a shRNA system, we silenced gp96 in multiple human MM cells and examined the effect of gp96 knockdown on MM cells by cell proliferation, cell cycle analysis, apoptosis assay, immunohistochemistry and human myeloma xenograft model. The anti-cancer activity of gp96 selective inhibitor, WS13 was evaluated by apoptosis assay and MTT assay.
Results
Genetic deletion of gp96 in XBP1s-Tg mice attenuates multiple myeloma. Silencing of gp96 causes severe compromise in human MM cell growth through inhibiting Wnt-LRP-survivin pathway. We also confirmed that knockdown of gp96 decreased human MM growth in a murine xenograft model. The targeted gp96 inhibitor induced apoptosis and blocked MM cell growth, but did not induce apoptosis in pre-B leukemic cells. We have demonstrated that myeloma growth is dependent on gp96 both genetically and pharmacologically.
Conclusions
gp96 is essential for MM cell proliferation and survival, suggesting that gp96 is a novel therapeutic target for multiple myeloma.
Keywords: Multiple myeloma, gp96, unfolded protein response, Wnt signaling, survivin
Introduction
As an ER chaperone, gp96 (1), also known as grp94 (2), endoplasmin (3), ERp99(4), and HSP90b1(5), is a paralogue of HSP90 and its expression can be induced by the accumulation of misfolded proteins (6). It binds to and hydrolizes ATP (7–9), and is the most abundant protein in the ER lumen. gp96 is a key downstream chaperone in the ER to mediate unfolded protein response (UPR) (10). UPR is an evolutionally conserved mechanism to maintain protein quality control in the secretory pathway. Accumulation of misfolded proteins in the ER triggers the activation of three well-known pathways including activating transcription factor 6 (ATF6), the double- stranded RNA-activated protein kinase-like ER kinase (PERK) and the spliced form of X box binding protein 1 (XBP1s) to induce the expression of several major ER HSPs including gp96, grp78 and calreticulin to enhance protein folding machinery (11). UPR plays critical roles for plasma cell differentiation as demonstrated by lack of plasma cells in the absence of XBP1 (12–14). Moreover, gp96 is induced more than 10 fold during B cell activation (4), and it has been shown to participate in the assembly of B cell receptor complexes through its association with Igα molecules (15). In addition to the critical roles of UPR in plasma cell differentiation, a picture has recently emerged of the roles of UPR, particularly XBP1s, in myeloma pathogenesis. XBP1s and downstream ER chaperones are consistently upregulated in myeloma cells (16). Most strikingly, transgenic expression of XBP1s in the B cell compartment of mice results in plasma cell dyscrasia with evidence of increased monoclonal antibodies (“M-spike”), lytic bone lesions, plasmacytosis and kidney damage (17). However, the roles of individual ER HSPs in myeloma have not been reported.
Using genetic strategies, we recently found that gp96 is an obligate master chaperone for multiple Toll-like receptors (TLRs) and integrins (18–21). However, under the steady state conditions, gp96 is not required for B cell activation, germinal center formation, plasma cell differentiation, and class-switching or affinity maturation (19). It is unclear if gp96 is required for plasma cell biology and for the development of myeloma during chronic ER stress conditions. In this study, we addressed the roles of gp96 in myeloma using XBP1s-transgenic mice as well as multiple human MM cell lines. We demonstrated that gp96 is required for myeloma progression both in vitro and in vivo. Mechanistically, we found that gp96 critically controls the Wnt-LRP6-survivin pathway. Our results indicated that gp96 is a novel therapeutic target for multiple myeloma.
Materials and Methods
Mice and cell lines
B cell specific gp96-deficient mice and wild type control littermates have been described (19). B cell specific XBP1s-transgenic and gp96 deficient mice were generated by crossing our B cell specific gp96-deficient mice with XBP1s transgenic mice. SCID mice were kindly provided by Jennifer Wu (Medical University of South Carolina, SC). Mice were bred and maintained according to the established guidelines and an approved protocol by Medical University of South Carolina Institutional Animal Care and Use Committee. Human multiple myeloma (MM) cell line RPMI 8226, U266B1, MM.1S, and MM.1R were purchased from ATCC. OPM1, JK-6L, and INA-6 were kindly provided by Yubin Kang (Medical University of South Carolina). MM cells were cultured in RPMI medium (Sigma-Aldrich) supplemented with 10% heat inactivated fetal calf serum (FCS, Atlas Biologicals), 55 μM 2- mercaptoethanol (2-ME, Gibco), 1 mM sodium pyruvate (Gibco), 10 mM HEPES (Gibco) and penicillin-streptomycin (Gibco). INA-6 cells were cultured with additional 2.5 ng/ml human IL-6. U266B1 cells were cultured in RPMI medium supplemented with 15% heat inactivated fetal calf serum, 1 mM sodium pyruvate (Gibco), 10 mM HEPES (Gibco) and penicillin-streptomycin (Gibco). All cells were cultured in 5% CO2 incubator. Gp96-mutant and WT pre-B cell lines were provided by Brian Seed (Harvard University).
Reagents
Antibodies used for flow cytometry were obtained from BD Biosciences (Mountain View, CA) and eBioscience (San Diego, CA). Antibodies against LRP6, β-catenin, survivin, c-myc and caspase 9 were purchased from Cell Signaling Technology (Danvers, MA), cyclin D1 was obtained from Abcam (Cambridge, MA) and gp96 Ab was bought from Enzo Life Sciences, Inc (Farmingdale, NY). Ig levels were determined by a sandwich ELISA kit from Southern Biotechnology Associates. All other chemicals were obtained from Sigma-Aldrich (St Louis, MO) and Fisher Scientific (Pittsburgh, PA). IL-4 and IL-21 were purchased from PeproTech (Rocky Hill, NJ). WS13, a gp96-specific Hsp90 inhibitor of the purine-scaffold class (22), was synthesized as previously described (23, 24) and the structure was recently reported (25).
Flow cytometry and in vitro plasma cell differentiation
Surface staining of cells and analysis on FACS Calibur and FACSVerse (Becton-Dickinson, Franklin Lakes, NJ) were done as described (18, 26). B cells were purified from spleens using CD19-magnetic beads according to the manufacturer’s protocol (Miltenyi Biotec, Auburn, CA). Purified murine splenic B cells were labeled with CFSE, and then stimulated with anti-μ antibody, agonistic CD40 antibody and IL-21 for 3 days. This was followed by flow cytometric analysis of expression levels of cell surface CD138 by dividing cells.
Viral vectors and transduction
Human gp96 and surivin shRNA lentiviral vectors as well as control vector were purchased from Open Biosystems (Huntsville, AL). Survivin retroviral vector and empty vector control were obtained from Origene Technologies (Rockville, MD). Cells were seeded in a 12-well plate and spin-infected with recombinant virus (3000 rpm, 32 °C, 90 min) in a desktop centrifuge as reported (26).
Protein extraction and Western blot
Protein extraction and immunoblot were performed as described previously (20). Briefly, cells were washed three times with ice-cold PBS and lysed in radioimmunoprecipitation assay (RIPA) lysis buffer (0.01 M sodium phosphate, pH 7.2, 150 mM NaCl, 2 mM EDTA, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 2 mM AEBSF, 130 mM bestatin, 14 mM E-64, 0.3 mM aprotinin, and 1 mM leupeptin). Total cell lysates was resolved on denaturing and reducing 10% to 12% SDS-PAGE, and the proteins were transferred from the gel onto Immobilon-P membranes. The membrane was blocked with 5% nonfat milk in PBS and then incubated with different Abs, followed by incubation with HRP-conjugated secondary Ab. Protein bands were visualized by using enhanced chemiluminescent substrate (Pierce, Rockford, IL) or clarity ECL substrate (Bio Rad, Hercules, CA).
Cell growth and proliferation assays
Cell growth was assessed through the trypan blue dye exclusion method and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) cell viability assay according to the manufacturer’s instruction. Cells were seeded at 1×105 cells/ml in 96-well plates. The cells were treated with 5 μM gp96 specific inhibitor WS13 or vehicle control and incubated in 5% CO2 incubator at 37°C for three time points (0, 24 and 72 hours). Plates were read at 570 nm by using iMark microplate absorbance reader (Bio Rad, Hercules, CA).
Cell cycle analyses
1×106 cells were washed twice in cold PBS and fixed with 4 mL of ice-cold 70% ethanol at 4°C overnight. Cells were washed once with PBS and incubated with 40 μg/mL Propidium iodide and 100 μg/mL RNAse for 30 minutes at 37°C in the dark. Cells were analyzed on a FACSCalibur. The percentage of cells in each phase of the cell cycle was quantitated using the FlowJo software (Tree Star, Ashland, OR).
Apoptosis assay
Apoptosis and cell death were determined by TUNEL staining according to manufacturer’s protocol (Trevigen, Gaithersburg, MD) and flow cytometry analysis of Annexin V and propidium iodide using Annexin V apoptosis detection Kit (eBioscience, San Diego, CA).
Human myeloma xenograft model
SCID mice (8–12 weeks old) were subcutaneously inoculated with 5×106 WT control and gp96 KD human myeloma RPMI 8226 cells respectively. Tumor growth was monitored every week using digital calipers to measure both the longitudinal (a, mm) and transverse (b, mm) diameters. Tumor area (a x b, mm2) was plotted. Mice were also monitored for the following general health indicators: overall behavior, feeding, neuromuscular tone, body weight, and appearance of fur, etc. At the end point (8 weeks after tumor inoculation), the primary tumor was also excised and weighed after the mice were sacrificed. Tumor tissues were fixed in 4% formalin or frozen in OCT medium.
Immunohistochemistry
Cryosections of fixed xenograft tumor tissues (5 μm thick) were treated with citrate antigen retrieval for 20 minutes at 95 degrees, allowed to cool, permeabilized with −20 degree methanol for 5 minutes, and exposed to 0.3% hydrogen peroxide for 5 minutes before blocking and incubation with primary antibody (Survivin, 1:800, Cell Signaling) for one hour at room temperature. Slides were washed and incubated with secondary antibody (Peroxidase rabbit IgG Vectastain ABC kit, Vector Lab, Burlingame, CA), and developed with DAB substrate kit (Vector Labs). Slides were counterstained with hematoxylin before visualization on Zeiss Axio microscope.
Immunofluorescence
Cryosections of xenograft tumor tissues (5 μm thick) were fixed with 4% paraformaldehyde, permeabilized with cold methanol, blocked and stained with gp96 Ab (9G10), and costained with AlexaFluor488-conjugated mouse anti-HLA-ABC (BD biosciences). Images of sections were taken under a fluorescent microscope (Zeiss, Chester, VA) and analyzed by AxioVision 4.4 software (Carl Zeiss Micro Imaging, Thornwood, NY).
Microarray analysis
Raw cDNA microarray data of myeloma samples and their corresponding clinical parameters were downloaded from Oncomine database (27). The most specific probe for gp96 was used to represent gene expression levels. Unpaired, 2-tailed student t-test was used for comparison of gp96 expression.
Statistical analysis
Error bars represent the standard deviation (SD) or the standard error of the arithmetic mean (SEM). Student t test and ANOVA were used for statistical analysis. Values of P less than 0.05 were considered to represent statistically significant differences.
Results
Genetic deletion of gp96 in XBP1s-Tg mice attenuates myeloma
Using CD19cre-mediated deletion of gp96, we previously found that gp96 is dispensable for immunoglobulin (Ig) synthesis, Ig assembly, B cell differentiation, plasma cell development and Ig class-switching.(19) It is unclear, however, if gp96 plays important roles in antibody production and myeloma progression during chronic ER stress. To answer this question, we generated B cell-specific XBP1s-Tg and gp96 KO mice (named TKO mice) by crossing B cell specific gp96 KO mice with XBP1s-Tg mice; the latter spontaneously develop multiple myeloma after reaching 40 weeks of age (17). The constitutive expression of XBP1s created a scenario of chronic unresolving ER stress. As expected, without transgenic expression of XBP1s, gp96 deletion does not negatively affect plasma cell number or the level of serum antibodies (Figure 1) (19). Strikingly, we found that plasma cells (B220−/lowCD138+) were significantly reduced in number in both the bone marrow and the spleen of the TKO mice when gp96 was deleted in XBP1s transgenic mouse (Figure 1A and 1B). To directly examine the plasma cell differentiation in TKO mice, we stimulated carboxyfluorescein succinimidyl ester (CFSE)–labeled splenic B cells from WT, KO, Tg and TKO mice with IL-4 alone or the combination of IL-4, anti-μ antibody, agonistic CD40 Ab and IL-21 for 3 days (28). We found that TKO B cells differentiated to plasma cells as efficiently as WT, KO or XBP1s Tg B cells based on CD138 expression (Figure 1C). This data indicated that the reduction of plasma cells in TKO mice is not due to inefficent plasma cell differentiation. Consistent with the reduced plasma cell number, we also found significant reduction of Ig level (both IgG1 and IgM) in TKO mice (Figure 2A and 2B), along with significant reduction of γ globulin on serum protein electrophoresis (Figure 2C and 2D) and decreased Ig deposition in the glomeruli (Figure 2E). These results suggest that gp96 is required for myeloma progression during chronic ER stress.
Figure 1. Gp96 is required for the maintenance but not the differentiation of plasma cells in XBP1s-Tg mice.

(A) Representative FACS plots of B220−CD138+ plasma cells in the bone marrow of four types of age/gender-matched mice (WT-wild type mice; KO-CD19cregp96flox/flox mice; Tg-B cell specific XBP1s-Tg mice; TKO-B cell specific gp96 KO and XBP1s-Tg mice). Number represents % of boxed cells over total number of cells analyzed. (B) Quantification of plasma cells in both spleen and bone marrow of four types of mice. The reduction of plasma cells in TKO mice is statistically significant (n=5 per group, *p < 0.05, T-test). Error bars indicate standard error of mean. (C) In vitro differentiation of plasma cells from TKO B cells is not impaired. Splenic B cells were purified with CD19 microbeads, labeled with CFSE, followed by stimulation with either IL4 alone (top row) or IL4 in combination with anti-μ antibody, agonistic CD40 antibody and IL21 (bottom row). Three days later, plasma cells were defined by CD138 expression and CFSE dilution. Number represents percentage of cells in each quandrant.
Figure 2. Development of myeloma in XBP1-Tg mice was blocked in the absence of gp96.

(A, B) Sera from 5–16 weeks (n=9) (A) and 20–29 weeks (n=10) (B) Tg and TKO mice were collected. Total levels of IgG1 and IgM were measured by ELISA. (C) Serum protein electrophoresis was performed. Shown is a piece of representative data demonstrating the loss of γ globulin fraction in TKO mice. (D) Quantification of γ globulin fraction over the total protein in the serum. (E) Frozen kidney sections from 37-week-old Tg and TKO mice were analyzed for glomerular immunoglobulin deposition by immunofluorescence using anti-mouse IgG, IgM, IgA, κ and λ Abs. Error bars indicate standard error of mean. *p<0.05; **p<0.01; ***p<0.001 (T-test).
gp96 is essential for multiple myeloma (MM) cell proliferation and survival
We next mined the Oncomine database (27) to gain a possible clue for the clinical significance of gp96 expression in myeloma. We found that gp96 expression is significantly elevated in MM samples from relapsed patients compared to the ones at the initial diagnosis (Figure 3A, p<0.05). To further study the mechanism of gp96 dependency by MM cells, we next silenced gp96 in human MM cell lines using a gp96 short hairpin RNA (shRNA) lentiviral vector. We found that silencing of gp96 causes significantly compromised MM cell growth. By comparison, a pre-B leukemic cell line however does not depend on gp96 for growth (Figure 3B and 3C). Cell cycle analysis of MM cells revealed that there were significantly fewer cells in G2 phase and more cells in over-G2 phase upon gp96 knockdown (KD) (Figure 3D). PI stain confirmed that more gp96 KD MM cells undergo constitutive death (PI+ cells: 60.4% ± 6.62%) than their wild type counterparts (PI+ cells: 35.3% ± 2.05%) (Figure 3E, p<0.001). To further determine if gp96 is required for human MM cell growth in vivo, we performed xenograft model by inoculating WT and gp96KD RPMI8226 MM cells to SCID mice followed by monitoring tumor growth. Indeed, knockdown of gp96 significantly inhibited MM tumor growth (Figure 3F and 3G). TUNEL analysis of the xenografts tumor tissues confirmed that the gp96 KD MM cells undergo massive apoptosis when compared to its wild type counterpart (Figure 3H and 3I).
Figure 3. Gp96 is essential for multiple myeloma cell proliferation and survival.

(A) Comparison of gp96 expression level in MM patients by cDNA microarray analysis. (B) Immunoblot analysis of gp96 and β-actin in WT and gp96 KO pre-B leukemic cells as well as empty vector (EV) control and gp96 knockdown (KD) U266B1, RPMI8226, OPM1, and INA-6 MM cells. (C) The growth of EV and gp96 KD MM cells was assessed over 72h- 96h; WT and gp96 KO pre-B leukemic cells were assessed over 72 h. (D) Cell cycle analysis of EV and gp96 KD RPMI8226 cells by propidium iodide staining and flow cytometry. The fraction of cells in subG1, G1, S, G2 and over G2 was quantified by FlowJo and data was plotted. Error bars indicate standard deviation. *p<0.05; **p<0.01 (T-test). (E) Representative plots of EV control and gp96 KD RPMI8226 cells were analyzed by flow cytometry for necrotic cells (PI+). (F) SCID mice were challenged subcutaneously with 5×106 WT and gp96 KD RPMI8226 cells. The kinetics of tumor growth was measured using a digital caliper. (G) Weight of excised tumor at the end of the experiment on week 8 (left panel) and the rate of tumor formation (right panel). Error bars indicate standard error of mean. ***p<0.001 (ANOVA). (H) Xenograft tumors were stained for gp96 and class I molecule of human leukocyte antigen (HLA) by immunofluorescence. (Scale bar: 25 μm). (I) The apoptotic cells were detected by TUNEL staining in xenografted WT and gp96 KD MM. (Scale bar: 25 μm).
gp96 is required for canonical Wnt signaling by controlling LRP6 expression
gp96 has recently been implicated in controlling cell surface expression of LRP6 (29). We also demonstrated that there is a strong interaction between LRP6 and gp96 (30). LRP6 is a co-receptor for the cell surface Wnt receptor Frizzled, and is required for canonical Wnt signaling. The Wnt/β-catenin signaling pathway is well known to modulate cell differentiation, proliferation and apoptosis. The roles of the Wnt pathway in the pathogenesis of MM have received increasing attention recently (31–33). To determine whether Wnt/β-catenin signaling defect is responsible for decreasing multiple myeloma cell growth in the absence of gp96, we compared the canonical Wnt signaling between empty vector (EV) control and gp96 KD RPMI 8226 cells by western blot. We found that the expression of LRP6 is compromised in gp96 KD MM cells, consistent with the post-translational chaperoning roles of LRP6 by gp96. Moreover, β-catenin, as well as the Wnt downstream targets, cyclin D1 and c-myc were significantly decreased in gp96 KD RPMI8226 MM cells (Figure 4A). Furthermore, the growth defect of gp96 knockdown RPMI8226 cells was significantly rescued by GSK3β inhibitors (TWS119 and Lithium Chloride) (Figure 4B), demonstrating that gp96 promotes myeloma growth via Wnt signaling.
Figure 4. gp96 knockdown in MM cells decreases canonical Wnt signaling, reduced expression of survivin, and causes mitotic catastrophe.

(A) Immunoblot analysis of gp96, LRP6, β-catenin, cyclin D1, C-myc, grp78 and β-actin from EV control and gp96 KD RPMI 8226 cell lysates. (B) EV control and gp96 KD RPMI 8226 cells were treated with TWS119 and Lithium Chloride (LiCl) and the cell growth was assessed at 72 hours. Cell number without inhibitors was set as 1. Error bars indicate standard deviation. *p<0.05 (T-test). (C) Immunoblot analysis of gp96, survivin, caspase 9 and β-actin from EV control and gp96 KD RPMI 8226 cell lysates. (D) Xenograft MM as in Figure 3 was stained for survivin by immunohistochemistry. (E) Representative images of Hema 3 staining on EV, gp96 KD, gp96 KD and survivin overexpression, and survivin KD RPMI 8226 cells. The arrows show multi-nucleation in gp96 KD and survivin KD cells. (F, G, H) Quantification of multi-nucleated cells from EV and gp96 KD RPMI 8226 cells (F), gp96 KD and overexpression of survivin on gp96 KD RPMI 8226 cells (G), and EV and survivin KD RPMI 8226 cells (H). The increase of multi-nucleated cells in gp96 KD and survivin KD cells (F, H) and the reduction of multi-nucleated cells in gp96 KD cells by overexpression of survivin (G) are statistically significant. Error bars indicate standard deviation. *p < 0.05, **p<0.01, ***p <0.001 (T-test).
gp96 knockdown compromises expression of survivin and induces mitotic catastrophe in human multiple myeloma cells
We next studied the candidate molecules downstream of Wnt in controlling cell survival. We focused on examining the expression of survivin and caspases in gp96 KD MM cells. We found that survivin levels were significantly reduced in the gp96 KD RPMI 8226 cells which was associated with an increased level of cleaved caspase 9 (Figure 4C). The reduction of nuclear survivin in gp96 KD cells was further confirmed by immunohistochemistry (Figure 4D). Survivin is known to play important roles in spindle separation during mitosis (34). As expected, morphological analysis of the gp96 KD MM cells revealed a marked tendency toward multi-nucleation (Figure 4E and 4F), a sign of mitotic catastrophe (35), which was phenocopied by survivin silencing (Figure 4E and 4H). To further address that survivin is the key downstream target of gp96 that is responsible for our observed phenotype, we over-expressed survivin in gp96 KD cells. The mitotic arrest was significantly rescued (Figure 4E and 4G).
Targeted gp96 inhibitor WS13 inhibits growth and induces death of myeloma cells
HSP90 inhibitors have been shown to be potent and promising anti-cancer agents (36, 37). Recent studies showed that pan-HSP90 inhibitors such as 17-AAG, Radicicol and PU-H71 are potential therapeutics for treatment of multiple myeloma (38, 39). However, none of these studies specifically looked into the contribution by different members of HSP90 (cytosolic HSP90, gp96 and mitochondrial TRAP-1) in myeloma (40). We next determined if inhibiting gp96 could be therapeutically beneficial to myeloma without inhibiting cytosolic HSP90. WS13, a gp96-specific HSP90 inhibitor of the purine-scaffold class (22), was synthesized as previously described (23, 24). We treated multiple MM cells and pre-B leukemic cells with it. We observed that WS13 induced both apoptosis and necrosis, and inhibited growth of multiple MM cells, but did not induce death in pre-B leukemic cells (Figure 5A and 5B). Thus we have demonstrated that myeloma growth is dependent on gp96 both genetically and pharmacologically, strongly indicating that gp96 is a promising therapeutic target in multiple myeloma.
Figure 5. Selective gp96 inhibitor WS13 inhibits growth of human MM cells and induces apoptosis.

(A) Pre-B leukemic cells, RMPI 8226, MM.1S, MM.1R, JK-6L, INA-6, OPM1 and U266B1 MM cells, were treated with 5 μM WS13 or vehicle control for 24 hours followed by flow cytometry analysis for necrotic cells (PI+ Annexin V+) or apoptotic cells (PI- Annexin V+). (B) Pre-B leukemic cells RPMI and multiple human MM cells were treated with 5 μM WS13 or vehicle control for 24, 72 and 120 hours followed by quantification of live cells by MTT assay. Error bars indicate standard deviation. *p<0.05; **p<0.01; ***p<0.001 (T-test).
Discussion
UPR plays critical roles in plasma cell differentiation as well as in the pathogenesis of multiple myeloma (MM). XBP1s and downstream ER chaperones are consistently up regulated in myeloma cells in patients (16), inspiring increasing efforts to develop UPR-targeted anti-MM therapy (41–43). However, no studies have looked specifically into the roles of individual ER-resident chaperones in MM. gp96 is a key downstream chaperone in the ER and mediates UPR, and yet its potential roles in MM was unexplored (10). In this study, we selectively deleted gp96 in B cells in XBP1s transgenic mice which overexpress XBP1s in B cells and plasma cells and spontaneously develop multiple myeloma after reaching advanced age (17). We found that gp96 is required for maintenance of plasma cells in this model and it is a key driver for development of MM. Moreover, we have demonstrated both genetically and pharmacologically that targeted inhibition of gp96 resulted in significant compromise in MM cell growth and death that was mediated at least in part by the loss of canonical Wnt pathway (Figure 1–3). It would be interesting to test the roles of gp96 in other myeloma models such as a transgenic mouse model that expresses myc transgene in a activation-induced deaminase (AID)-dependent manner (44). This is important given the limitation of XBP1s-transgenic model in faithfully representing human myeloma (45).
Our study has added significantly to the emerging role of Wnt signaling in MM (31–33). Human MM cells appear to have hallmarks of active Wnt signaling by overexpressing β-catenin which promotes proliferation of multiple myeloma (46). Furthermore, blocking β-catenin with small-molecule inhibitors, AV-65 (33) or PKF115-584 (31, 32) specifically inhibits MM cell proliferation. Paradoxically, higher expression of Wnt signaling inhibitor, Dickkopf1 (Dkk1) in bone marrow stromal cells can promote myeloma growth (47). Still other studies demonstrated that increasing Wnt signaling did not significantly affect the proliferation of MM cells (48, 49). These studies suggest that Wnt signaling can play both positive and negative roles in the progression of MM, likely due to the difference in the underlying pathogenesis. Our study has firmly linked the roles of gp96 with its ability to chaperone Wnt co-receptor LRP6. We showed that gp96 is required for MM development in XBP1s transgenic mouse model whose underlying pathogenesis is dysregulated UPR. To illustrate the importance of cell type-selective roles of gp96, we found that silencing of gp96 causes significant compromise in the growth of MM cells but not pre-B leukemic cells (Figure 3). Thus, it is possible that MM cells, with hallmarks of ER stress, are uniquely dependent on gp96. The explanation could be that MM cells have a particularly stressed ER due to heightened production of immunoglobulin. When rate of protein synthesis is increased, UPR leads to increased level of HSPs including gp96 to accelerate protein folding. In the absence of gp96, the folding capacity of the ER in MM cells is exceeded by chronic accumulation of misfolded proteins. In addition, loss of gp96 causes collapse of canonical Wnt signaling. Therefore, gp96 regulates multiple myeloma via both UPR and Wnt pathways. Our work thus suggests that Wnt pathway is under the regulation of UPR. The interplay between Wnt pathway and UPR in MM deserves further study. Although speculative, it might also be fruitful to look at the combination of targeted therapy against both Wnt and UPR pathway for the treatment of MM in the future.
Cancer cell metabolism is necessarily abnormal to accommodate the internal and external pressures which are secondary to excessive growth: nutrient insufficiency, free radical generation, and cellular machinery overloaded with accelerated DNA replication and protein production. gp96 stands at the intersection of causative and responsive in regard to such changes. Increased protein production upregulates gp96, making it an important compensatory response to malignant growth, and acting to protect the cell from ER stress. However, upregulated gp96 may also actively support enhanced growth signaling in the form of the canonical Wnt pathway, a commonly upregulated signaling cascade in cancer. Interestingly, survivin has been reported to be a target of Wnt signaling (33). Survivin is normally absent in healthy cells, appearing only during regulated passage through G2/M. Overexpression of survivin is correlated with multidrug-resistance in multiple myeloma (50). Here we have demonstrated that gp96 knockdown induces mitotic catastrophe and apoptosis in human MM cells, which is characterized by loss of survivin and appearance of multi-nucleated cells (Figure 4). Our data allow us to propose a model of the roles of gp96 in multiple myeloma cell survival (Figure 6). We believe that gp96 is an especially important mediator of malignant growth due to its dual roles of absorbing the ER stress of cancer-driven protein production and also instigating further Wnt-survivin growth signaling.
Figure 6. A model for the roles of gp96 in myeloma.
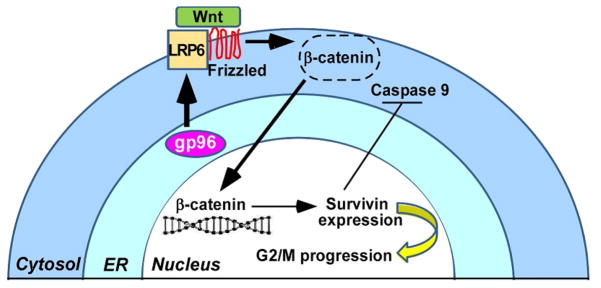
gp96 is a critical chaperone for LRP6 and is therefore required for canonical Wnt signaling, leading to release of β-catenin from its destruction complex (dotted circle), accumulation of nuclear β-catenin and upregulation of Wnt targets such as survivin Deletion of gp96 compromises survivin expression, leading to its failure to safeguard mitotic spindles, and therefore initiation of apoptosis. The mechanism of XBP1s or other UPR sensors to induce gp96 expression in MM cells is not depicted.
In summary, we have uncovered that both murine and human myeloma cells are exquisitely dependent on gp96 for growth in vivo and in vitro. Mechanistically, we have linked the roles of gp96 in promoting multiple myeloma to its function in chaperoning Wnt-LRP-survivin pathway. Thus, gp96-targeted inhibitors such as WS13 may prove to be a novel therapeutic against multiple myeloma in the future.
Translational relevance.
Multiple myeloma (MM) is an incurable plasma cell neoplasm whose pathogenesis is closely linked to dysregulated unfolded protein response (UPR) in the endoplasmic reticulum (ER). In this study, we demonstrated that the persistence of plasma cells as well as the development of myeloma is critically dependent on gp96, a major molecular chaperone in the ER in vivo. Furthermore, our results demonstrated that gp96 knockdown causes severe compromise in human multiple myeloma (MM) cell growth through attenuating Wnt-LRP-survivin pathway. Targeting gp96 in MM cells by a gp96 specific inhibitor also blocks proliferation and induces apoptosis. This is the first study to uncover the critical roles of gp96 in the initiation and progression of multiple myeloma, suggesting that blockade of gp96 is a novel therapeutic strategy against this disease.
Acknowledgments
Grant Support: This work was supported by the South Carolina Clinical & Translational Research Institute at the Medical University of South Carolina KL2RR029880 and UL1RR029882 (to B.L.), the American Cancer Society Institutional Research Grant from the American Cancer Society IRG-97-219-14 (to B.L.), NIH grants AI070603 and AI077283 (to Z.L.), NIH grants CA155226 and AG032969 (to G.C.).
We would like to thank Dr. Yusheng Zhu for assistance with the serum protein electrophoresis. B.L. is an NIH KL2 scholar and is supported by the South Carolina Clinical & Translational Research Institute (SCTR) at the Medical University of South Carolina (KL2RR029880 and UL1RR029882). B.L. is also supported by the American Cancer Society Institutional Research Grant from the American Cancer Society (IRG-97-219-14). G.C. and Z.L. are supported by NIH grants. Z.L. is the Abney Chair Remembering Sally Abney Rose in Stem Cell Biology & Therapy.
Footnotes
Disclosure of Conflicts of Interest: The authors declare no competing financial interests.
Authorship Contributions
B.L. conceived the idea and designed the research. Y.H., S.W., J.K., S.R., B.L. performed research. Y.H., S.W., J.K., S.R., S.Z.U., Z.L., B.L. analyzed data. G.C. provided reagent. Y.H., S.W. and B.L. wrote the manuscript which was also critically read by J.K., S.R., S.Z.U., G.C., R.D. and Z.L.
References
- 1.Srivastava PK, DeLeo AB, Old LJ. Tumor rejection antigens of chemically induced sarcomas of inbred mice. Proc Natl Acad Sci U S A. 1986;83:3407–11. doi: 10.1073/pnas.83.10.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee AS, Delegeane A, Scharff D. Highly conserved glucose-regulated protein in hamster and chicken cells: preliminary characterization of its cDNA clone. Proc Natl Acad Sci U S A. 1981;78:4922–5. doi: 10.1073/pnas.78.8.4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koch G, Smith M, Macer D, Webster P, Mortara R. Endoplasmic reticulum contains a common, abundant calcium-binding glycoprotein, endoplasmin. J Cell Sci. 1986;86:217–32. doi: 10.1242/jcs.86.1.217. [DOI] [PubMed] [Google Scholar]
- 4.Lewis MJ, Mazzarella RA, Green M. Structure and assembly of the endoplasmic reticulum. The synthesis of three major endoplasmic reticulum proteins during lipopolysaccharide-induced differentiation of murine lymphocytes. J Biol Chem. 1985;260:3050–7. [PubMed] [Google Scholar]
- 5.Chen B, Piel WH, Gui L, Bruford E, Monteiro A. The HSP90 family of genes in the human genome: insights into their divergence and evolution. Genomics. 2005;86:627–37. doi: 10.1016/j.ygeno.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–4. doi: 10.1038/332462a0. [DOI] [PubMed] [Google Scholar]
- 7.Li Z, Srivastava PK. Tumor rejection antigen gp96/grp94 is an ATPase: implications for protein folding and antigen presentation. Embo J. 1993;12:3143–51. doi: 10.1002/j.1460-2075.1993.tb05983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol Cell. 2007;28:41–56. doi: 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frey S, Leskovar A, Reinstein J, Buchner J. The ATPase cycle of the endoplasmic chaperone Grp94. J Biol Chem. 2007;282:35612–20. doi: 10.1074/jbc.M704647200. [DOI] [PubMed] [Google Scholar]
- 10.Yang Y, Li Z. Roles of heat shock protein gp96 in the ER quality control: redundant or unique function? Mol Cells. 2005;20:173–82. [PubMed] [Google Scholar]
- 11.Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–31. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–7. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 13.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–9. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 14.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 15.Foy SP, Matsuuchi L. Association of B lymphocyte antigen receptor polypeptides with multiple chaperone proteins. Immunol Lett. 2001;78:149–60. doi: 10.1016/s0165-2478(01)00256-5. [DOI] [PubMed] [Google Scholar]
- 16.Bagratuni T, Wu P, Gonzalez de Castro D, Davenport EL, Dickens NJ, Walker BA, et al. XBP1s levels are implicated in the biology and outcome of myeloma mediating different clinical outcomes to thalidomide-based treatments. Blood. 2010;116:250–3. doi: 10.1182/blood-2010-01-263236. [DOI] [PubMed] [Google Scholar]
- 17.Carrasco DR, Sukhdeo K, Protopopova M, Sinha R, Enos M, Carrasco DE, et al. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell. 2007;11:349–60. doi: 10.1016/j.ccr.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y, Liu B, Dai J, Srivastava PK, Zammit DJ, Lefrancois L, et al. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity. 2007;26:215–26. doi: 10.1016/j.immuni.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu B, Li Z. Endoplasmic reticulum HSP90b1 (gp96, grp94) optimizes B-cell function via chaperoning integrin and TLR but not immunoglobulin. Blood. 2008;112:1223–30. doi: 10.1182/blood-2008-03-143107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu B, Yang Y, Qiu Z, Staron M, Hong F, Li Y, et al. Folding of Toll-like receptors by the HSP90 paralogue gp96 requires a substrate-specific cochaperone. Nat Commun. 2010;1:79. doi: 10.1038/ncomms1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Staron M, Yang Y, Liu B, Li J, Shen Y, Zuniga-Pflucker JC, et al. gp96, an endoplasmic reticulum master chaperone for integrins and Toll-like receptors, selectively regulates early T and B lymphopoiesis. Blood. 2010;115:2380–90. doi: 10.1182/blood-2009-07-233031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taldone T, Chiosis G. Purine-scaffold Hsp90 inhibitors. Curr Top Med Chem. 2009;9:1436–46. doi: 10.2174/156802609789895737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He H, Zatorska D, Kim J, Aguirre J, Llauger L, She Y, et al. Identification of potent water soluble purine-scaffold inhibitors of the heat shock protein 90. J Med Chem. 2006;49:381–90. doi: 10.1021/jm0508078. [DOI] [PubMed] [Google Scholar]
- 24.Llauger L, He H, Kim J, Aguirre J, Rosen N, Peters U, et al. Evaluation of 8-arylsulfanyl, 8-arylsulfoxyl, and 8-arylsulfonyl adenine derivatives as inhibitors of the heat shock protein 90. J Med Chem. 2005;48:2892–905. doi: 10.1021/jm049012b. [DOI] [PubMed] [Google Scholar]
- 25.Patel PD, Yan P, Seidler PM, Patel HJ, Sun W, Yang C, et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat Chem Biol. 2013 doi: 10.1038/nchembio.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu B, Staron M, Li Z. Murine but not human basophil undergoes cell-specific proteolysis of a major endoplasmic reticulum chaperone. PLoS One. 2012;7:e39442. doi: 10.1371/journal.pone.0039442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burington B, Barlogie B, Zhan F, Crowley J, Shaughnessy JD., Jr Tumor cell gene expression changes following short-term in vivo exposure to single agent chemotherapeutics are related to survival in multiple myeloma. Clin Cancer Res. 2008;14:4821–9. doi: 10.1158/1078-0432.CCR-07-4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ozaki K, Spolski R, Ettinger R, Kim HP, Wang G, Qi CF, et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J Immunol. 2004;173:5361–71. doi: 10.4049/jimmunol.173.9.5361. [DOI] [PubMed] [Google Scholar]
- 29.Weekes MP, Antrobus R, Talbot S, Hor S, Simecek N, Smith DL, et al. Proteomic plasma membrane profiling reveals an essential role for gp96 in the cell surface expression of LDLR family members, including the LDL receptor and LRP6. J Proteome Res. 2012;11:1475–84. doi: 10.1021/pr201135e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu B, Staron M, Hong F, Wu BX, Sun S, Morales C, et al. Essential roles of grp94 in gut homeostasis via chaperoning canonical Wnt pathway. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1302933110. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sukhdeo K, Mani M, Zhang Y, Dutta J, Yasui H, Rooney MD, et al. Targeting the beta-catenin/TCF transcriptional complex in the treatment of multiple myeloma. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:7516–21. doi: 10.1073/pnas.0610299104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ashihara E, Kawata E, Nakagawa Y, Shimazaski C, Kuroda J, Taniguchi K, et al. beta-catenin small interfering RNA successfully suppressed progression of multiple myeloma in a mouse model. Clin Cancer Res. 2009;15:2731–8. doi: 10.1158/1078-0432.CCR-08-1350. [DOI] [PubMed] [Google Scholar]
- 33.Yao H, Ashihara E, Strovel JW, Nakagawa Y, Kuroda J, Nagao R, et al. AV-65, a novel Wnt/beta-catenin signal inhibitor, successfully suppresses progression of multiple myeloma in a mouse model. Blood Cancer J. 2011;1:e43. doi: 10.1038/bcj.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamers F, van der Ploeg I, Schild L, Ebus ME, Koster J, Hansen BR, et al. Knockdown of survivin (BIRC5) causes apoptosis in neuroblastoma via mitotic catastrophe. Endocr Relat Cancer. 2011;18:657–68. doi: 10.1530/ERC-11-0207. [DOI] [PubMed] [Google Scholar]
- 35.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerchietti LC, Lopes EC, Yang SN, Hatzi K, Bunting KL, Tsikitas LA, et al. A purine scaffold Hsp90 inhibitor destabilizes BCL-6 and has specific antitumor activity in BCL-6-dependent B cell lymphomas. Nat Med. 2009;15:1369–76. doi: 10.1038/nm.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caldas-Lopes E, Cerchietti L, Ahn JH, Clement CC, Robles AI, Rodina A, et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:8368–73. doi: 10.1073/pnas.0903392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishii T, Seike T, Nakashima T, Juliger S, Maharaj L, Soga S, et al. Anti-tumor activity against multiple myeloma by combination of KW-2478, an Hsp90 inhibitor, with bortezomib. Blood Cancer J. 2012;2:e68. doi: 10.1038/bcj.2012.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stuhmer T, Iskandarov K, Gao Z, Bumm T, Grella E, Jensen MR, et al. Preclinical activity of the novel orally bioavailable HSP90 inhibitor NVP-HSP990 against multiple myeloma cells. Anticancer Res. 2012;32:453–62. [PubMed] [Google Scholar]
- 40.Usmani SZ, Bona RD, Chiosis G, Li Z. The anti-myeloma activity of a novel purine scaffold HSP90 inhibitor PU-H71 is via inhibition of both HSP90A and HSP90B1. J Hematol Oncol. 2010;3:40. doi: 10.1186/1756-8722-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117:1311–4. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mimura N, Fulciniti M, Gorgun G, Tai YT, Cirstea D, Santo L, et al. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood. 2012;119:5772–81. doi: 10.1182/blood-2011-07-366633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ri M, Tashiro E, Oikawa D, Shinjo S, Tokuda M, Yokouchi Y, et al. Identification of Toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress-induced XBP1 mRNA splicing. Blood Cancer J. 2012;2:e79. doi: 10.1038/bcj.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell. 2008;13:167–80. doi: 10.1016/j.ccr.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuehl WM. Modeling multiple myeloma by AID-dependent conditional activation of MYC. Cancer Cell. 2008;13:85–7. doi: 10.1016/j.ccr.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 46.Derksen PW, Tjin E, Meijer HP, Klok MD, MacGillavry HD, van Oers MH, et al. Illegitimate WNT signaling promotes proliferation of multiple myeloma cells. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:6122–7. doi: 10.1073/pnas.0305855101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fowler JA, Mundy GR, Lwin ST, Edwards CM. Bone marrow stromal cells create a permissive microenvironment for myeloma development: a new stromal role for Wnt inhibitor Dkk1. Cancer Res. 2012;72:2183–9. doi: 10.1158/0008-5472.CAN-11-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qiang YW, Endo Y, Rubin JS, Rudikoff S. Wnt signaling in B-cell neoplasia. Oncogene. 2003;22:1536–45. doi: 10.1038/sj.onc.1206239. [DOI] [PubMed] [Google Scholar]
- 49.Qiang YW, Walsh K, Yao L, Kedei N, Blumberg PM, Rubin JS, et al. Wnts induce migration and invasion of myeloma plasma cells. Blood. 2005;106:1786–93. doi: 10.1182/blood-2005-01-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsubaki M, Satou T, Itoh T, Imano M, Komai M, Nishinobo M, et al. Overexpression of MDR1 and survivin, and decreased Bim expression mediate multidrug-resistance in multiple myeloma cells. Leuk Res. 2012;36:1315–22. doi: 10.1016/j.leukres.2012.07.003. [DOI] [PubMed] [Google Scholar]