

Almost all state-of-the-art methods for prediction accuracy of RNA secondary structure ignore the process of structure formation and focus on the final RNA structure. In this review, the existing evidence for cotranscriptional folding and the currently used strategies for RNA secondary-structure prediction are analyzed. Potential improvements to existing methods that would capture the process of cotranscriptional structure formation are suggested.
Keywords: RNA secondary-structure prediction, RNA structure formation in vivo, cotranscriptional RNA folding
Abstract
The expression of genes, both coding and noncoding, can be significantly influenced by RNA structural features of their corresponding transcripts. There is by now mounting experimental and some theoretical evidence that structure formation in vivo starts during transcription and that this cotranscriptional folding determines the functional RNA structural features that are being formed. Several decades of research in bioinformatics have resulted in a wide range of computational methods for predicting RNA secondary structures. Almost all state-of-the-art methods in terms of prediction accuracy, however, completely ignore the process of structure formation and focus exclusively on the final RNA structure. This review hopes to bridge this gap. We summarize the existing evidence for cotranscriptional folding and then review the different, currently used strategies for RNA secondary-structure prediction. Finally, we propose a range of ideas on how state-of-the-art methods could be potentially improved by explicitly capturing the process of cotranscriptional structure formation.
INTRODUCTION
The primary products of all DNA genomes are RNA transcripts consisting of linear sequences of four different types of ribonucleic acids (abbreviated A, C, G, and U and chemically different from the similarly abbreviated DNA building blocks, A, C, G, and T). When a gene of the genome is activated, a corresponding transcript is synthesized in a linear fashion with its 5′ end emerging first and its 3′ end emerging last. Primary transcripts vary greatly in length from a few nucleotides (nt) to 104 nt and longer. They may be processed in a number of ways, e.g., splicing and RNA editing, which may happen while the transcript is being made. The functional role of some transcripts is exerted by RNA structure that is formed when pairs of complementary nucleotides of the RNA sequence (C-G, A-U, G-U) form base pairs. In contrast to proteins, where we typically need to know its three-dimensional (3D) structure in order to study a protein's potential functional roles, it often suffices to only know the RNA secondary structure in order to investigate its potential functional role(s). This RNA secondary structure is defined by the pairs of base-paired sequence positions in the RNA. RNA structure can either be global, i.e., span most of the transcript, or more local, i.e., be confined to a subsequence of the transcript. During its life in the cell, a single transcript may assume more than one functionally relevant RNA structure, e.g., riboswitches, which can assume two mutually exclusive structures that are both functional.
Many computational methods for RNA structure prediction, in particular, earlier and noncomparative methods, implicitly focus on predicting global RNA structures only. They are typically applied to analyze the noncoding portion of a given transcriptome because this is where globally structured RNA genes are suspected. RNA structural features, however, are also known to play important functional roles in regulating protein-coding transcripts (e.g., splicing, localization, degradation, translation initiation), yet this typically involves only local RNA structures, which only some of the computational methods for RNA secondary-structure prediction can adequately model (Pedersen et al. 2004a,b).
Recent advances in nucleotide sequencing technologies have enabled the routine sequencing of entire transcriptomes, with methods such as strand-specific RNA-seq, enabling the discovery of novel transcripts en masse. Experimental methods for RNA structure determination such as X-ray crystallography and NMR can provide atomic-resolution 3D solutions, but remain relatively costly and comparatively slow. Computational methods for predicting RNA secondary structures based on RNA sequence information alone are therefore key to assigning potential functional roles to the transcriptome and identifying worthwhile targets for experimental validation.
When available, computational structure prediction can be aided by results from RNA footprinting experiments. Such experiments can estimate the pairing status of individual nucleotide positions in a single sequence with chemical probes, but cannot identify the pairing partner involved in a base pair. Such methods, when paired with next-generation sequencing technologies, in protocols such as Frag-seq, PARS, and SHAPE-seq, show great promise in generating high-throughput RNA secondary structure probe maps (Wan et al. 2011). Nonetheless, footprinting results still require algorithms to derive the overall most likely solution, again emphasizing the need for reliable and efficient computational methods.
There exists by now ample experimental evidence that RNA structure formation starts cotranscriptionally, i.e., while the RNA is transcribed from the genome. The process of cotranscriptional structure formation is key to determining the resulting functional RNA structure(s) in vivo and that this process can be influenced by a range of intrinsic as well as extrinsic factors. Yet, nearly all state-of-the-art methods for computational RNA secondary structure prediction ignore the structure formation process and focus exclusively on the end result, i.e., a single, final RNA structure. There already exist a few computational methods that aim to explicitly simulate the cotranscriptional folding pathway by capturing key features of the folding environment in vivo. Because their prediction accuracy has so far been evaluated on only a few select sequences of typically short length, however, they are currently viewed as folding-pathway prediction methods rather than RNA secondary-structure prediction methods.
We argue that ignoring the formation process often yields decent structure predictions, especially for short and globally structured transcripts (<200 nt), but that in order to increase the prediction accuracy for longer transcripts and to reach a conceptually better understanding, we ought to aim to take some effects of cotranscriptional folding into account.
In the following, we first review the variety of mechanisms that have been shown to influence cotranscriptional folding in vivo. This summarizes primarily experimental, but also some theoretical evidence for cotranscriptional folding. We then provide an overview of the currently existing methods for RNA secondary-structure prediction. This part of the review is not aimed at providing a detailed description of every existing method for RNA secondary-structure prediction, but rather at highlighting the different underlying concepts used by these methods. At this point, we also cover methods for predicting RNA folding pathways that already capture some effects of cotranscriptional folding. To conclude, we propose a range of ideas on how cotranscriptional folding could be captured in computational methods for RNA secondary-structure prediction in order to further improve their prediction accuracy.
EXPERIMENTAL AND THEORETICAL EVIDENCE FOR COTRANSCRIPTIONAL FOLDING
Directionality of transcription
One of the most obvious differences between the in vivo and the typical in vitro setting is that RNA transcripts in vivo emerge sequentially starting with the 5′ end, whereas in vitro experiments start with an already synthesized molecule. The directionality of the molecule's synthesis in vivo may thus lead to structural asymmetries during its cotranscriptional folding that may, in turn, influence the resulting functional RNA structure(s).
Transcription, transcription speed, and variations thereof
Whether or not folding can happen during synthesis depends, among other things, on how the timescale of RNA synthesis compares with that of RNA structure formation. The speed of transcription not only depends on the underlying organism, but also on the polymerase responsible for generating the transcript in question. It ranges from 200 nucleotides per second (nt/sec) in phages, to 20–80 nt/sec in bacteria and 10–20 nt/sec for human polymerase II (Pan and Sosnick 2006). On the other hand, RNA folding is known to occur on a wide range of time scales; some RNAs fold in 10–100 msec (Al-Hashimi and Walter 2008), whereas kinetically trapped conformations can persist for minutes or hours (Sosnick and Pan 2003; Thirumalai and Hyeon 2005; Al-Hashimi and Walter 2008). Experiments in the early 1980s have shown that RNA structure formation can happen during transcription (Boyle et al. 1980; Kramer and Mills 1981), i.e., cotranscriptionally, and that folding in vivo can happen on the same timescale as RNA synthesis (Brehm and Cech 1983). The latter was first shown for the cotranscriptional and structure-dependent self-splicing of the Tetrahymena group I intron (Brehm and Cech 1983).
Since then, several in vitro experiments have confirmed that RNA folding can happen cotranscriptionally and that the speed of transcription not only affects the overall folding rate, but also transient structures as well as the final structure (Pan et al. 1999; Heilmann-Miller and Woodson 2003a,b). Lewicki et al. (1993) and Chao et al. (1995) showed that altering the natural speed of transcription can yield misfolded and functionally inactive transcripts. Experimental studies of the Tetrahymena self-splicing intron are consistent with the view that a set of identical RNA molecules partitions into an active and an inactive pool, and that this partitioning is highly influenced by the cotranscriptional folding environment, including the RNA transcription rate (Koduvayur and Woodson 2004).
For a given transcript, the speed of transcription is not necessarily constant. Transcriptional pausing can serve as an additional mechanism for fine-tuning cotranscriptional folding (Toulme et al. 2005; Wickiser et al. 2005; Wong et al. 2007). This pausing happens at specific transcript positions and for well-defined time intervals (ranging from 10−6 sec to 10 sec). In bacteria, pausing can be due to interactions between the emerging RNA and the polymerase and/or polymerase-associated protein factors (Liu et al. 1996; Landick 1997; Mooney et al. 1998). The flavin mononucleotide (FMN)–dependent riboswitch in Bacillus subtilis (Wickiser et al. 2005) is a beautiful example of how these features can be combined into a cotranscriptional feedback loop in which the binding of a metabolite selects one of two possible cotranscriptional folding pathways whose resulting RNA structure determines whether transcription is terminated or not.
Self-interactions including transient RNA structures
One of the key features of any RNA sequence is that it can interact with itself via base pairs between complementary nucleotides to form RNA structure. During cotranscriptional folding, already formed structures can unpair and yield to other structures, in which case, we refer to them as “transient structures.” In other cases, it is energetically unfavorable for an existing structure to yield to a new conformation, thereby forming a kinetic trap. Transient structural features thus have the potential to significantly influence the cotranscriptional folding pathway and the resulting functional RNA structure(s) (see Fig. 2, below). Most of our current knowledge of transient structures, which we also refer to as cis RNA–RNA interactions, stems from dedicated experimental studies of select folding pathways that explore how RNA structure changes as a function of time.
FIGURE 2.

Examples of cis and trans interactions during cotranscriptional folding. (A) Hypothetical RNA sequence, capable of forming helices h1–h4, at sites A–E. (B) Transcription of the sequence across time points t1–t5, with the sequential lengthening of the 3′ end. The transcription process limits the available sites for helix formation, imposing an order on helix formation. If an early-formed helix is stable, it can serve to block the formation of subsequent helices by occupying specific sites. (C) Sites may also be occupied due to interactions with other molecules; in this case, a protein-binding site (PBS) occupies site A, leading to a very different result. (D) If early helices are relatively unstable, they can be seen as transient helices that yield to new helices. This mechanism can aid the robust formation of desired structure features. Note that some of the conformations shown above correspond to the ones introduced and defined by Meyer and Miklós (2004). These are as follows: In B, h1 (iī) and h3 (ic) are 3′-trans, where h1 is stable, preventing the formation of h3, and h1 (īi) and h2 (ic) are 3′-cis, where h1 is stable, preventing the formation of h2; in D, h1 (ci) and h2 (iī) are 5′-cis, where h1 is an intermediate for h2, and h2 (ci) and h3 (iī) are 5′-cis, where h2 is an intermediate for h3.
Folding pathways of RNA transcripts in vitro have been the subject of intense study for a long time. Initial experiments primarily studied how already synthesized and fully denatured RNA molecules fold, whereas more recent studies examine cotranscriptional folding pathways in vitro and, most recently, also in vivo (Adilakshmi et al. 2009; Woodson 2010). Because any of these experiments are technically sophisticated, our current view derives from a few well-studied test cases such as the hairpin ribozyme (Donahue et al. 2000; Fedor 2002, 2009; Mahen et al. 2005, 2010) and the Tetrahymena intron (Koduvayur and Woodson 2004; Jackson et al. 2006). These ribozymes are comparatively easy to study in vivo because their cleavage relies on distinct structural features whose products are easier to detect than the corresponding functional structures.
Cotranscriptional folding—whether in vitro or in vivo—tends to happen sequentially (Mahen et al. 2005, 2010) because base pairs at the 5′ end of the RNA can form first, whereas base pairs involving the 3′ end can only form once transcription is complete. This folding often involves transient RNA structure elements, i.e., structural features that are only present for a specific time span (Kramer and Mills 1981; Repsilber et al. 1999). These can direct the structure formation via one or several folding pathways toward the desired structural configuration(s). These transient features may also play distinct functional roles. They may, for example, be required for template activity during (+)-strand synthesis in some viruses (Repsilber et al. 1999) or may serve as protein-binding sites during transcription (Ro-Choi and Choi 2003). These examples once again illustrate that any given RNA transcript may have more than a single functionally relevant RNA structure during its lifetime in the cell.
Cotranscriptional folding and other reaction rates in vivo typically differ from those in vitro with folding rates in vivo being typically (Mahen et al. 2005, 2010), but not always (Donahue et al. 2000), higher than in vitro. One example is the cotranscriptional folding of the Tetrahymena ribozyme in vitro, which is twice as fast as the refolding of the fully synthesized and denatured molecule, but slower than the cotranscriptional folding in vivo (Heilmann-Miller and Woodson 2003a). Cotranscriptional folding pathways in vivo need not be unique (Jackson et al. 2006), and tertiary interactions can determine which of several possible folding pathways is chosen (Chauhan and Woodson 2008). Factors such as transcription speed and flanking sequences can also influence which pathway dominates (Koduvayur and Woodson 2004). One of the few existing in vivo studies of cotranscriptional folding pathways (Sclavi et al. 1998) indirectly examined the structural folding intermediates of the Tetrahymena ribozyme at 10−5 sec time resolution using X-ray synchrotron radiation and chemical accessibility probing and found folding intermediates that are similar to those in vitro.
The tryptophan (trp) operon is a group of genes found in bacteria that act in the biosynthesis pathway of the amino acid tryptophan. The trp operon leader encodes a short peptide that is rich in tryptophan codons near the 5′ end of the RNA (Yanofsky 1981). Regulation of the trp operon is carried out in part by the trp operon leader through a mechanism that relies on the simultaneous transcription of a DNA gene and translation of the resulting RNA in bacteria. The trp operon leader assumes two mutually exclusive structural configurations that form cotranscriptionally: the attenuator, which prevents further transcription of the trp operon; and the anti-terminator, which permits transcription (Yanofsky 1981). When tryptophan levels are high, the ribosome proceeds rapidly through the operon leader and interferes with the anti-terminator hairpin. When tryptophan levels are low, the ribosome stalls while translating the leader peptide and allows the anti-terminator hairpin to form, and thus the trp operon is activated.
In addition to these experimental results, the bioinformatics community has conducted a range of computational studies to investigate cotranscriptional structure formation. Computational simulations of cotranscriptional folding pathways, e.g., Isambert and Siggia (2000), show that the basic features of cotranscriptional folding and their beneficial effects on RNA structure formation can be investigated in silico. Using a kinetic Monte Carlo Markov Chain (MCMC) to study the folding of the hepatitis delta virus ribozyme (87 nt in length), Isambert and Siggia (2000) show that cotranscriptional folding at the natural transcript speed of 50 nt/sec is significantly more efficient than when starting with a fully denatured sequence or when using the increased transcript speed of 1000 nt/sec that is typically used in in vitro experiments. By combining computational simulations of RNA folding pathways with phylogenetic structure analyses, Schoemaker and Gultyaev (2006) investigated the effect of sRNA binding on ribosomal RNA (rRNA) structure formation during cotranscriptional folding and find that it significantly facilitates structure formation.
A bioinformatics analysis of 361 structural RNA genes (Meyer and Miklós 2004) showed that these genes not only encode information on their known functional structure, but also on transient features of their respective cotranscriptional RNA folding pathways. For this, Meyer and Miklós (2004) examined helices (defined as contiguous stretches of adjacent base pairs) that could potentially out-compete helices of the known structure. They found statistically significant 5′-to-3′ asymmetries between these competing helices and the respective helices of the known structure. More specifically, they identified two different types of transient structures: those that can yield to the functional structure and help its cotranscriptional formation and those that are more likely to act as kinetic traps during cotranscriptional folding. They showed that the former are preferentially encoded in the underlying RNA sequences, whereas the latter are suppressed.
More recently, Zhu et al. (2013) conducted a computational study of six RNA families with known transient and alternative structures in order to test whether evolutionarily related sequences not only assume similar final structures, but also share common transient structures during their respective cotranscriptional folding pathways. They find that some transient structures have been evolutionarily conserved on a level that is similar to those of the final structure. Moreover, they find that evolutionarily related sequences encounter similar transient structure features during their respective, predicted cotranscriptional folding pathways and that these features often coincide with known transient features.
To conclude, naturally occurring transcripts not only encode their functional RNA structure, but also information on how to get there via transient features that help define the corresponding cotranscriptional folding pathway.
Interactions with other molecules
One key difference between the in vivo and in vitro settings is that the cellular environment typically contains a wealth of additional molecules. In vivo, these may interact with the RNA transcript and thereby influence its structure formation and the resulting RNA structure (see Fig. 2C, below). These molecules may comprise of proteins, RNA transcripts, metabolites, ligands, and different types of ions. Any intermolecular interaction between two distinct RNA molecules, i.e., any trans RNA–RNA interaction, has the potential to prevent the thus bound RNA nucleotides from engaging in other interactions including RNA structure (i.e., cis RNA–RNA interactions). This may either stabilize or destabilize existing RNA structure features, which may, in turn, influence the cotranscriptional folding pathway and the resulting RNA structures.
Due to the methodological challenges of investigating RNA folding in vivo and in real time, we currently have only limited insight into folding pathways in vivo (Sclavi et al. 1998; Heilmann-Miller and Woodson 2003a; Jackson et al. 2006; Chauhan and Woodson 2008). Numerous recent in vitro experiments that replicate specific aspects of the complex in vivo environment and rapid progress regarding in vivo methodologies (Adilakshmi et al. 2009; Alexander et al. 2011) are likely to change this.
So, which interactions between RNA transcripts and other molecules have been experimentally confirmed to be functionally important for RNA structure formation?
Ligand–RNA interactions
One of the most-obvious examples in which RNA structure formation is influenced by trans interactions is so-called riboswitches. The change of one distinct RNA structure to another one is usually triggered by the binding of a metabolite or ion, but can also be induced by a temperature change, at least in bacteria (thermoswitches) (Johansson et al. 2002; Giuliodori et al. 2010; Narberhaus 2010). The two distinct structural conformations of a riboswitch are typically located in the 5′ UTRs of messenger RNAs (mRNAs) and are mutually exclusive because they engage two overlapping subsequences. The structural change triggers a change of the gene's expression by altering either its transcription, translation, or splicing (Serganov 2009; Roth and Breaker 2010). Nechooshtan et al. (2009) identified a pH-responsive riboregulator upstream of the alx open reading frame (ORF). For a high pH, the translationally active RNA structure is formed during transcription, which involves two well-defined transcriptional pausing sites. Frieda and Bock (2012) succeeded in directly observing the cotranscriptional folding of the pbuE adenine riboswitch. Using an optical assay that allowed them to monitor folding transitions in individual transcripts in real time, they showed that the transcriptional outcome of the riboswitch is kinetically controlled. Perdrizet et al. (2012) present strong evidence that the btuB riboswitch in Escherichia coli depends on the precise transcriptional pausing of its polymerase to guide its folding into its native structure (Hopkins et al. 2011).
Protein–RNA interactions
In order for many large RNAs to fold in vitro into their functional structure without any other trans-acting molecules (apart from water), it is necessary to raise the concentration of metal ions (e.g., of Mg2+) significantly above normal levels in vivo (Gregan et al. 2001; Fedorova et al. 2002). Several in vitro experiments have shown that the ion concentration can be lowered if specific proteins are added that stabilize the RNA structure (Gampel and Cech 1991; Caprara et al. 1996; Matsuura et al. 1997; Weeks 1997; Ostersetzer et al. 2005) and that can bind folding intermediates (Caprara et al. 1996). This has also been confirmed by several in vivo experiments (Mohr et al. 1992; Waldsich et al. 2002a,b).
RNA-binding proteins often play different functional roles depending on the binding interface they use to interact with different partners. One example is Cyt-18 in Neurospora crassa, which not only aids RNA folding, but also acts as a splicing factor and an aminoacyl-tRNA synthetase (Mohr et al. 1992, 1994). Most of these proteins bind an RNA in a sequence- or structure-specific way (Caprara et al. 1996; Weeks and Cech 1996; Matsuura et al. 1997; Webb and Weeks 2001; Bassi et al. 2002; Paukstelis et al. 2005, 2008; Talkington et al. 2005; Adilakshmi et al. 2008; Dai et al. 2008). There are also proteins, however, that interact with RNAs in a less specific way such as RNA helicases, which help anneal and unwind RNAs while requiring ATP (Hickman and Dyda 2005; Bleichert and Baserga 2007; Halls et al. 2007; Pyle 2008; Fairman-Williams et al. 2010), and hnRNP proteins, which bind single-stranded stretches of pre-mRNAs and thereby aid splicing (Farina and Singer 2002). Some protein–RNA interactions are required to happen at very specific times. One key example is ribosomal RNAs, which are modified and processed with the corresponding ribosomes pre-assembled cotranscriptionally in a tightly coregulated way as shown in several in vivo experiments (Udem and Warner 1973; Oakes et al. 1993; Granneman and Baserga 2005; Kos and Tollervey 2010). There is also recent experimental evidence that cotranscriptional splicing is coupled to transcriptional pausing in yeast (Alexander et al. 2010) and that, interestingly, cotranscriptional splicing can also be coupled to translation as shown in vivo for the thymidylate synthase intron of the T4-phage (Semrad and Schroeder 1998). Therefore, RNA-binding proteins involved in splicing may thus act cotranscriptionally.
Chaperone–RNA interactions
Chaperones are molecules, usually proteins, that assist a molecule's correct folding by refolding misfolded structure features. Based on this definition, the trans-interaction partners of a given RNA transcript described above are not chaperones because they guide the correct cofolding pathway rather than help already misfolded RNA transcripts refold correctly. Many detailed experiments have shown that RNA transcripts can misfold in vitro and that it takes these molecules minutes to many hours or longer to escape these structural traps (Turner et al. 1990; Treiber and Williamson 2001; Baird et al. 2007; Shcherbakova et al. 2008). This may be attributed to several alternative folding pathways of the in vitro folding landscape, which tends to be more rugged than the cotranscriptional folding landscape in vivo (Nikolcheva and Woodson 1999; Schroeder et al. 2002; Zemora and Waldsich 2010), but can also be due to individual RNA structure elements that keep the structure trapped.
There is some evidence that RNA structures can also misfold in vivo (Semrad and Schroeder 1998; Jackson et al. 2006) and that there exist dedicated cellular mechanisms for dealing with misfolded RNA structures, e.g., by sequestering and degrading them as shown for the Tetrahymena intron (Jackson et al. 2006). Most RNA chaperones identified so far are proteins that resolve misfolded RNA structures by binding stretches of double-stranded RNA with low affinity and in a sequence-unspecific way. Other RNA chaperones bind single-stranded RNA and facilitate the transition from the incorrect to the correct structural conformation by lowering specific kinetic barriers (Herschlag 1995).
Chaperone-assisted folding has been extensively studied for proteins, whereas comparatively little is known about the extent and mechanisms underlying chaperone-assisted RNA folding. What we know is that most of these proteins play a wide range of other functional roles in addition to being RNA chaperones and that they share no obvious similarities in terms of sequence and structure motifs (Woodson 2010). Additionally, unlike protein chaperones, RNA chaperones typically do not require any ATP to encourage refolding (Herschlag 1995; Weeks 1997; Schroeder et al. 2004; Rajkowitsch et al. 2007).
Trans RNA–RNA interactions, i.e., interactions with other transcripts
Trans RNA–RNA interactions, i.e., interactions with other transcripts, involve the same elementary building blocks as RNA structure or cis RNA–RNA interactions, namely, base pairs between pairs of complementary nucleotides. This implies that trans RNA–RNA interactions involve two single-stranded stretches of RNAs. They differ in that regard from protein–RNA interactions, which may involve single-stranded or double-stranded RNA (and may happen in a sequence-specific or unspecific way).
If a single-stranded stretch of RNA sequence is to be bound in a sequence-specific way, it should be much easier in terms of evolution to come up with a corresponding, near-complementary RNA sequence than to devise an RNA-binding protein that would bind in an equally sequence-specific way. One would therefore expect trans RNA–RNA interactions to be much more abundant than sequence-specific protein–RNA interactions with single-stranded RNAs (Smit et al. 2007; Meyer 2008).
Functionally important trans RNA–RNA interactions include the well-known class of microRNA–mRNA interactions, which alter gene expression on the mRNA level (Lagos-Quintana et al. 2001), interactions between snoRNAs and ribosomal RNAs, which edit rRNAs before ribosome assembly (Bachellerie et al. 2002); and snRNA–mRNA interactions, which are key during mRNA splicing (Horowitz 2012). Both mRNA splicing and ribosome assembly can occur cotranscriptionally.
Large-scale transcriptome studies of higher organisms such as mouse and human show that a large fraction of the transcriptome does not encode any proteins, e.g., Carninci (2010). These noncoding transcripts are diverse with regard to length, expression patterns and levels, and functional roles, if known. This has given rise to a wealth of different names for these transcripts, which we shall simply call noncoding RNAs (ncRNAs) in the following.
One well-studied example is the short DsrA ncRNA in E. coli, which alters the structure of the rpoS mRNA upon binding, thereby enabling its translation. In order for this trans RNA–RNA interaction to happen, the structure of the ncRNA DsrA first needs to be destabilized by binding the Sm-like protein Hfq (Mikulecky et al. 2004; Soper and Woodson 2008; Soper et al. 2010; Hopkins et al. 2011). Several other examples of structure-mediated translation regulation via trans RNA–RNA interactions between a short ncRNA and an mRNA have been found, primarily in bacteria (Geissmann et al. 2010; Lioliou et al. 2010). The short ncRNA is often an anti-sense transcript of the corresponding mRNA, the trans RNA–RNA interaction typically involves a short stretch of near-complementarity, and a protein is often required as a third ingredient for the regulatory mechanism to be functional. Yet another example of a functionally relevant trans RNA–RNA interaction is the formation of the 30S ribosomal subunit in bacteria, which requires the transient interaction with the leader sequence of the rRNA operons (Balzer and Wagner 1998).
Another well-studied example is the hok/sok toxin–antitoxin system in E. coli, which provides a mechanism for preservation of the R1 plasmid after cell division (see Fig. 1; Steif and Meyer 2012). This system consists of three overlapping genes. The host-killing hok gene induces cell death upon translation of its protein. The mok (modulation of killing) gene overlaps hok on the same mRNA transcript, and translation of the mok reading frame must occur in order for translation of hok to occur. The sok (suppression of killing) gene encodes a short anti-sense RNA that binds and prevents translation of mok and thus, indirectly, also the translation of hok. In cells that possess the R1 plasmid, the unstable sok RNA is produced in high quantities and prevents cell death caused by the longer-lived hok RNAs. Following mitosis, the sok RNA is rapidly degraded in any daughter cells that lack the R1 plasmid, allowing the hok gene to induce cell death. The mechanism of the hok/sok system depends on several structural features of the hok mRNA. Alternative structural configurations reduce the degradation rate of the hok mRNA, and several transient hairpins at the 5′ end prevent binding of sok RNA during transcription (Steif and Meyer 2012).
FIGURE 1.
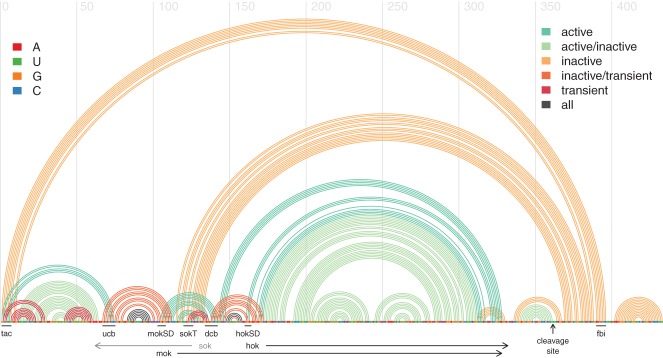
RNA structure features for the reference sequence from E. coli plasmid R1 encoding the hok and mok proteins. The horizontal line depicts the plasmid's sequence with its nucleotides color-coded according to the legend on the top left. Underneath the sequence line, black arrows indicate the protein-coding regions of the hok and mok proteins. The gray arrow shows the sequence region that is complementary to the sok anti-sense RNA, which is part of a different transcript. Each arc above the horizontal line represents a base pair between the two corresponding positions along the sequence and is color-coded according to the structure conformation to which it belongs (active, inactive, or transient; see the legend on the top right). Below the horizontal sequence line, black lines indicate the location of known sequence motifs: (tac) translational activator element; (ucb) upstream complementary box; (dcb) downstream complementary box; (mokSD) mok Shine-Dalgarno sequence; (hokSD) hok Shine-Dalgarno sequence; (fbi) fold-back inhibitory element. This arc-diagram was first published by Steif and Meyer (2012) and generated using the R-chie web server (Lai et al. 2012).
Summary
The overall view that emerges is that the cotranscriptional folding pathways are determined both by intrinsic features encoded in the RNA sequence itself such as transient and final structural features, and by extrinsic features such as the speed of the transcribing polymerase, and trans-interaction partners (e.g., proteins, ligands, RNA transcripts, and other trans-interaction partners). In vivo, both types of features are combined in the appropriate cellular context and determine the functional RNA structure(s) being formed.
A range of experimental evidence supports the notion of fairly well-defined cofolding pathways in vivo. These pathways are, on the one hand, robust enough to guide the formation of the correct functional RNA structure under typical cellular conditions, but, on the other hand, are—if required—flexible enough to yield different structural and functional outcomes, if the cellular environment significantly changes (Wickiser et al. 2005).
CAPTURING COTRANSCRIPTIONAL FOLDING IN METHODS FOR RNA SECONDARY-STRUCTURE PREDICTION
Existing methods for RNA secondary-structure prediction
A wide variety of computational methods already exist for predicting RNA structural features. Most RNA structure prediction methods that can technically handle long, naturally occurring transcripts such as rRNAs only aim to capture the RNA secondary structure rather than its tertiary structure. Fortunately, many functional features can already be studied on this level of abstraction. In the following, we therefore focus on methods for RNA secondary-structure prediction (rather than also covering methods for predicting tertiary RNA structure, which are currently limited to sequences of ∼100 nt length).
Existing methods for predicting RNA secondary structure can be broadly grouped into two categories: those that take a single RNA sequence as input and those that work in a comparative way by taking a set of homologous RNA sequences as input. There also exists a different class of prediction methods that explicitly predict cotranscriptional folding pathways in terms of RNA secondary-structure changes over time. They aim to capture the structure formation process in vivo and are typically limited to analyzing transcripts of a few hundred nucleotides in length. These methods are currently viewed as folding-pathway prediction methods rather than RNA secondary-structure prediction methods.
Comparative methods for RNA secondary-structure prediction currently provide the state-of-art in terms of prediction accuracy, in particular, for long RNA sequences. Apart from one recently introduced new method, CoFold (Proctor and Meyer 2013), none of the currently existing noncomparative or comparative methods for predicting RNA secondary structures, however, explicitly capture cotranscriptional folding or its overall effects.
In the following, we review the existing methods and propose ways of capturing some effects of cotranscriptional folding explicitly in order to further improve their prediction accuracy.
Noncomparative, MFE methods for RNA secondary-structure prediction
Historically, noncomparative methods that take a single RNA sequence as input came first. These use the so-called minimum-free energy (MFE) approach, which aims to identify the (usually pseudoknot-free) RNA secondary structure that minimizes the overall free Gibbs energy of the transcript. They include well-known methods such as MFold, RNAfold, and related programs (Zuker and Stiegler 1981; Hofacker et al. 1994; Mathews et al. 1999; Zuker 2003). These methods mirror the in vitro setting, where a fully synthesized RNA has infinite time to settle into its thermodynamically most favorable configuration. They implicitly assume that the functionally relevant secondary structure is the thermodynamically most stable one. Predictions are generated by efficiently searching the search space of all possible (usually, pseudoknot-free) RNA secondary structure for the structure with the lowest overall MFE. This is typically done using a dynamic programming algorithm.
Several methods based on the suboptimal folding algorithm introduced by Wuchty et al. (1999) have been developed that explicitly consider an ensemble of RNA secondary structures close to the minimum free energy. RNAsubopt, a program included in the ViennaRNA package (Hofacker et al. 1994; Hofacker 2003), provides a list of low-energy secondary structures above a user-defined energy cutoff above the minimum free energy. Sfold (Ding and Lawrence 2003; Ding et al. 2004; Chan et al. 2005) uses a statistical approach to sample RNA secondary structures from the ensemble of RNA secondary structures at thermodynamic equilibrium, where the probability that the algorithm picks a particular structure is proportional to the structure's probability in the structural ensemble. While these methods consider structures that differ from the MFE configuration, they still assume that the RNAs are in thermodynamic equilibrium. Moreover, they ignore the kinetic nature of cotranscriptional formation and the effect it may have on the resulting structure or ensemble of structures.
In 1996, Morgan and Higgs (1996) investigated a set of long RNAs (comprising 16S rRNAs, 23S rRNAs, and RNase P) and found significant discrepancies between the evolutionarily conserved RNA structure features and the respective predicted MFE structures. They concluded that these differences “cannot simply be put down to errors in the free energy parameters used in the model” (Morgan and Higgs 1996) and hypothesized that these may be due to effects of kinetic folding in vivo.
To test this hypothesis, Proctor and Meyer (2013) recently introduced the new RNA secondary-structure prediction method called CoFold, which is the first to combine thermodynamic with kinetic considerations. They incorporate one overall effect of kinetic folding into a minimum free-energy prediction method: the reachability of potential pairing partners during cotranscriptional folding. CoFold demonstrates a significant performance improvement over minimum free-energy methods alone, particularly for longer RNA sequences of >1000 nt for which one usually observes a marked decrease in prediction accuracy. Capturing this overall effect of cotranscriptional folding yields RNA secondary structures with similar, but slightly higher free energies compared with the MFE structure. These results promise that there may be great value in accounting for other effects of cotranscriptional folding to improve noncomparative methods for RNA secondary-structure prediction.
Comparative methods for RNA secondary-structure prediction
Rapidly increasing amounts of genome sequencing data for a variety of organisms have given rise to a conceptually new approach to RNA secondary-structure prediction that takes as input a set of homologous RNA sequences rather than a single RNA sequence of interest (e.g., Knudsen and Hein 1999, 2003; Hofacker et al. 2002; Mathews and Turner 2002; Perriquet et al. 2003; Ji et al. 2004; Pedersen et al. 2004a,b; Ruan et al. 2004; Touzet and Perriquet 2004; Witwer et al. 2004; Havgaard et al. 2005; Holmes 2005; Mathews 2005; Dowell and Eddy 2006; Meyer and Miklós 2007). Even though these comparative methods differ considerably regarding their underlying algorithms, they all aim to identify the consensus RNA secondary structure that has been conserved during evolution. The underlying working hypothesis is that RNA structures that are functionally relevant should also be conserved. This assumption usually holds because RNA structures tend to be more conserved than the underlying primary sequences. Depending on the evolutionary distances among the input sequences, however, this approach may fail to detect species-specific structure features that have only developed recently.
Overall, comparative methods for RNA secondary-structure prediction currently provide the state-of-art in terms of prediction accuracy. They tend to significantly outperform noncomparative methods (Gardner and Giegerich 2004), but typically require a high-quality input alignment provided by the user to reach their optimal performance (see, e.g., Perriquet et al. 2003; Ji et al. 2004; Touzet and Perriquet 2004; Holmes 2005; Meyer and Miklós 2007 for methods that do not require a fixed input alignment).
All of these methods generate predictions by first identifying pairs of covarying alignment columns to detect conserved base pairs and then combining these into a single (and, usually, global) consensus RNA secondary structure. For this, they use (1) a modified MFE framework that also accounts for conservation of base pairs and aims for overall energy minimization; (2) a probabilistic framework such as stochastic context-free grammars (SCFGs) combined with likelihood maximization; (3) a nondeterministic, yet probabilistic approach such as Bayesian Markov Chain Monte Carlos (MCMCs) that samples from a posterior distribution that is subsequently combined with a post-processing step to extract a consensus structure; or (4) a combination of heuristic, ad hoc procedures.
Existing methods for predicting RNA folding pathways
In parallel to the development of the RNA secondary-structure prediction methods, several methods have been developed that aim to explicitly simulate cotranscriptional structure formation as a function of time. All of these methods—e.g., RNAkinetics (Mironov et al. 1985; Mironov and Lebedev 1993; Danilova et al. 2006), Kinfold (Flamm et al. 2000), Kinefold (Isambert and Siggia 2000; Xayaphoummine et al. 2003, 2005), and Kinwalker (Geis et al. 2008)—take as input a single RNA sequence and use a range of different statistical models, approximations, and heuristics to arrive at their predictions. Typically, they use stochastic simulation that extends the input RNA sequence at regular intervals, and simulates helix formation and disruption events over a simulated timescale. The probability that each event occurs is proportional to its theoretical chemical rate of change. They have, however, conceptual difficulties dealing with long sequences (over a few hundred nucleotides), and their performance has until recently (Zhu et al. 2013) been only benchmarked for a few select sequences. They are thus currently viewed as folding-pathway prediction methods rather than RNA secondary-structure prediction methods.
The recent study by Zhu et al. (2013) uses three of these existing methods to show that evolutionarily related RNA sequences share common transient structural features during their predicted folding pathways, and that these features often coincide with known transient structures. The investigators propose an analysis pipeline that applies several folding-pathway prediction methods in a comparative manner by combining folding predictions across evolutionarily related RNA sequences. Moreover, this study provides solid evidence that some transient helices have been conserved during evolution.
Ideas for capturing cotranscriptional folding in methods for RNA secondary-structure prediction
The key effect of cotranscriptional folding is to make the formation of the final structure depend on its wider context, both along the sequence and in terms of time.
The key feature common to all existing noncomparative and comparative methods for RNA secondary-structure prediction is that they search the space of all possible (typically pseudoknot-free) RNA secondary structure for the optimal structure without having any notion of a folding pathway or a timewise ordering of events (see Fig. 2). The recently introduced method CoFold (Proctor and Meyer 2013) is an exception, yet it currently only models a single overall effect, namely, the reachability of base-pairing partners during cotranscriptional folding, which effectively amounts to a reweighing of different regions of the structure search space. The search of the structure space usually involves a scoring function whose overall value is being optimized during the search. The overall score for any candidate RNA structure is typically expressed as the sum or product of scores for individual structural building blocks that, taken together, cover the entire sequence. These elementary scores and the way in which they are combined by the scoring function during optimization, however, only depends on the local building blocks of the subsequence under consideration, but neither on their location within the sequence nor the RNA structure context of the surrounding sequence (see Fig. 2). Most optimization algorithms are dynamic programming algorithms that combine optimal structures for adjacent subsequences into one optimal structure for the resulting merged subsequence. The order of these steps, however, does not replicate the events during cotranscriptional folding. In particular, no region of the theoretical structural search space is marked as unlikely, if the corresponding structure feature could not readily form cotranscriptionally in vivo (see Fig. 2).
One of the intrinsic features that are known to influence the formation of RNA structure in vivo are transient structures as discussed above. Because these features are encoded in the RNA sequence itself, they could, in principle, be detected by any method for RNA secondary-structure prediction and subsequently used to bias the optimization process yielding the final RNA structure. Their detection could be implemented via a straightforward dynamic programming procedure that swiftly identifies all candidate helices (of some minimum length or stability) in the given input RNA sequence (Meyer and Miklós 2004). The conceptual problem is that these helices would naturally comprise both candidate transient helices as well as candidate helices of the final RNA secondary structure. These helices could be used in the optimization procedure in order to influence the local decision making (how to combine optimal structures for two subsequences into a single optimal structure for the merged subsequence). This would be one conceptual way of taking the wider structure context into account during the optimization procedure yielding the predicted final RNA structure. In the spirit of Meyer and Miklós (2004), these modifications could, for example, penalize any candidate structure that has strong competing transient helices upstream that could jeopardize its cotranscriptional formation.
Whereas the identification of candidate helices and relevant competing helices for a single sequence may be complicated due to the relatively large search space, comparative methods may generate a more accurate and smaller set of evolutionarily conserved competing helices to consider, such as those output by the conservation-based helix-finding algorithm Transat (Wiebe and Meyer 2010). If transient RNA structural features turn out to be evolutionarily conserved on a similar level to those of the final RNA structure, which is what recent results by Zhu et al. (2013) indicate, however, this may actually lower the prediction accuracy of comparative RNA secondary-structure prediction methods because they may erroneously incorporate these conserved transient helices into the predicted final RNA secondary structure. Whether or not this is the case and a cause for concern remains to be shown.
In addition to the ideas used by CoFold (Proctor and Meyer 2013) discussed above, the directionality of transcription could also be captured by rendering the scores assigned to the structural building blocks dependent on their position within the transcript, whether they are nearer to the 5′ end or the 3′ end.
It is less obvious how one should account for the speed of transcription, let alone variations of transcription speed and transcriptional pausing. At least for now, there is too little experimental information to hope to identify transcriptional pausing sites computationally. A change in overall transcription speed alters the ratio between the speed of transcript synthesis and the rate of structure formation. This has been experimentally shown to influence cotranscriptional folding pathways and their structural outcome. On the structure prediction side, the speed of transcription could be captured by altering the effective distances between structural features. This is exactly what the free parameter in CoFold (Proctor and Meyer 2013) is for. By changing its value, one can effectively account for different (yet constant) transcription rates and thereby optimize the program's performance for different species. If the transcription speed is high with respect to the rate of structure formation, the emerging transcript has less time and hence fewer opportunities to explore the surrounding structure space. This has the overall effect of enlarging effective distances, whereas a low transcription speed should have the overall effect of reducing effective distances.
A biologically diverse set of molecules can form trans interactions with transcripts in vivo. All of the existing methods for predicting RNA secondary structure including methods for folding pathway prediction assume an isolated RNA sequence as input and ignore any potential trans-interaction partners (the bulk effects of water and some ions is taken into account by most folding-pathway prediction methods). If and how these trans interactions influence the cotranscriptional structure formation not only depends on the type of interaction (RNA–RNA, RNA–protein, etc.), but also very much on the timing of the interaction with respect to the structure formation. For example, a protein that binds the emerging transcript early on and for a short time has a very different influence on structure formation from that of a protein that binds the final RNA structure only.
Early and persistent types of trans interactions could be captured in RNA secondary-structure prediction methods by preventing the bound (and either single-stranded RNA [ssRNA] or double-stranded RNA [dsRNA]) subsequence from engaging in other interactions, in particular, other RNA structural features. Technically, this is fairly easy to achieve via a slight modification of the default optimization procedure by assigning a large penalty to all structure solutions that do not keep the bound subsequence single or double stranded. This feature is already implemented by all RNA secondary-structure prediction methods that allow known RNA structural features to be taken into account (e.g., Zuker and Stiegler 1981; Knudsen and Hein 2003; Pedersen et al. 2004b). This assumes, however, that details about the interaction site (subsequence, ssRNA vs. dsRNA) are known up-front, which is often not the case.
Any trans interactions of a more transient nature, however, are hard to capture computationally by any of the existing methods for RNA secondary-structure prediction because this would require them to have some notion of time-ordered steps, which they currently do not have.
Suggestions for further improving methods for folding-pathway prediction
The existing folding-pathway prediction methods already mimic the in vivo folding as they fold the RNA sequence cotranscriptionally at a constant transcription speed (which needs to be specified by the user). This is, however, only a first approximation of the complex in vivo situation. Because these methods explicitly predict folding pathways, they already model cis RNA–RNA interactions and, in particular, transient RNA structural features. At least for now, these methods do not predict variations of transcription speed and do not capture potential trans interactions with other molecules from the in vivo environment.
If details about trans interactions are known up-front (timing, binding site, ssRNA vs. dsRNA), these could be fairly easily captured by preventing the known binding site from engaging in other interactions. This has already been done for select examples and allowed us to computationally investigate the effect of trans interactions on cotranscriptional RNA structure formation (Schoemaker and Gultyaev 2006).
SUMMARY
With 75% of the human genome being transcribed (Djebali et al. 2012), the investigation of transcriptomes and how they are regulated has never been more important. RNA structure is one important feature by which transcripts can influence their fate in the cell. There is by now ample experimental and solid theoretical evidence that RNA structure formation already starts during transcription and that events during the cotranscriptional folding determine which functional RNA structure(s) are being formed. Yet, as of now, the process of structure formation is completely ignored by almost all state-of-the-art methods for RNA secondary-structure prediction. We argue that capturing some aspects of the structure formation process in predictive models could significantly improve these methods and provide evidence for this in form of a new method (Proctor and Meyer 2013). These initial results are very encouraging because they show that a significant improvement in prediction accuracy can already be gained by modeling a single overall effect of cotranscriptional folding and without making the underlying prediction algorithm much more complex. Beyond this, we propose detailed ideas of how different aspects of cotranscriptional folding in vivo could also be captured in silico.
One of the most simple and encouraging messages from the mounting (and sometimes dauntingly complex) experimental results is certainly the realization that the transcript in the cell does not explore all of the structure search space.
ACKNOWLEDGMENTS
This project was supported by grants to I.M.M. from the Natural Sciences and Engineering Research Council (NSERC) of Canada and from the Canada Foundation for Innovation (CFI). D.L. is funded by an NSERC Postgraduate Scholarship. J.R.P. holds an Alexander Graham Bell Canada Graduate Scholarship from NSERC, with additional funding from the CIHR/MSFHR Bioinformatics Training Program at the University of British Columbia. CIHR is the Canadian Institutes of Health Research, and MSFHR is the Michael Smith Foundation for Health Research in Canada.
Footnotes
Freely available online through the RNA Open Access option.
REFERENCES
- Adilakshmi T, Bellur D, Woodson S 2008. Concurrent nucleation of 16S folding and induced fit in 30S ribosome assembly. Nature 455: 1268–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adilakshmi T, Soper S, Woodson S 2009. Structural analysis of RNA in living cells by in vivo synchrotron X-ray footprinting. Methods Enzymol 468: 239–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander R, Innocente S, Barrass J, Beggs J 2010. Splicing-dependent RNA polymerase pausing in yeast. Mol Cell 40: 582–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander R, Barrass J, Dichtl B, Kos M, Obtulowicz T, Robert M, Koper M, Karkusiewicz I, Mariconti L, Tollervey D, et al. 2011. RiboSys, a high-resolution, quantitative approach to measure the in vivo kinetics of pre-mRNA splicing and 3′-end processing in Saccharomyces cerevisiae. RNA 16: 2570–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hashimi HM, Walter NG 2008. RNA dynamics: It is about time. Curr Opin Struct Biol 18: 321–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachellerie J, Cavaille J, Hiittenhofer A 2002. The expanding snoRNA world. Biochimie 84: 775–790 [DOI] [PubMed] [Google Scholar]
- Baird N, Fang X, Srividya N, Pan T, Sosnick T 2007. Folding of a universal ribozyme: The ribonuclease P RNA. Q Rev Biophys 40: 113–161 [DOI] [PubMed] [Google Scholar]
- Balzer M, Wagner R 1998. Mutations in the leader region of ribosomal RNA operons cause structurally defective 30 S ribosomes as revealed by in vivo structural probing. J Mol Biol 276: 547–557 [DOI] [PubMed] [Google Scholar]
- Bassi G, de Oliveira D, White M, Weeks K 2002. Recruitment of intron-encoded and co-opted proteins in splicing of the bI3 group I intron RNA. Proc Natl Acad Sci 99: 128–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleichert F, Baserga S 2007. The long unwinding road of RNA helicases. Mol Cell 27: 339–352 [DOI] [PubMed] [Google Scholar]
- Boyle J, Robillard G, Kim S 1980. Sequential folding of transfer RNA. A nuclear magnetic resonance study of successively longer tRNA fragments with a common 5′ end. J Mol Biol 139: 601–625 [DOI] [PubMed] [Google Scholar]
- Brehm SL, Cech TR 1983. Fate of an intervening sequence ribonucleic acid: Excision and cyclization of the Tetrahymena ribosomal ribonucleic acid intervening sequence in vivo. Biochemistry 22: 2390–2397 [DOI] [PubMed] [Google Scholar]
- Caprara M, Mohr G, Lambowitz A 1996. A tyrosyl-tRNA synthetase protein induces tertiary folding of the group I intron catalytic core. J Mol Biol 257: 512–531 [DOI] [PubMed] [Google Scholar]
- Carninci P 2010. RNA dust: Where are the genes? DNA Res 17: 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C, Lawrence C, Ding Y 2005. Structure clustering features on the Sfold Web server. Bioinformatics 21: 3926–3928 [DOI] [PubMed] [Google Scholar]
- Chao MY, Kan M, Lin-Chao S 1995. RNAII transcribed by IPTG-induced T7 RNA polymerase is nonfunctional as a replication primer for ColE1-type plasmids in Escherichia coli. Nucleic Acids Res 23: 1691–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan S, Woodson S 2008. Tertiary interactions determine the accuracy of RNA folding. J Am Chem Soc 130: 1296–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L, Chai D, Gu S, Gabel J, Noskov S, Blocker FJH, Lambowitz A, Zimmerly S 2008. A three-dimensional model of a group II intron RNA and its interaction with the intron-encoded reverse transcriptase. Mol Cell 30: 472–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilova L, Pervouchine D, Favorov A, Mironov A 2006. RNAkinetics: A web server that models secondary structure kinetics of an elongating RNA. J Bioinform Comput Biol 4: 589–596 [DOI] [PubMed] [Google Scholar]
- Ding Y, Lawrence C 2003. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res 31: 7280–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Chan C, Lawrence C 2004. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res 32: W135–W141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue C, Yadava R, Nesbitt S, Fedor M 2000. The kinetic mechanism of the hairpin ribozyme in vivo: Influence of RNA helix stability on intracellular cleavage kinetics. J Mol Biol 295: 693–707 [DOI] [PubMed] [Google Scholar]
- Dowell RD, Eddy SR 2006. Efficient pairwise RNA structure prediction and alignment using sequence alignment constraints. BMC Bioinformatics 7: 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman-Williams M, Guenther U, Jankowsky E 2010. SF1 and SF2 helicases: Family matters. Curr Opin Struct Biol 20: 313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina K, Singer R 2002. The nuclear connection in RNA transport and localization. Trends Cell Biol 12: 466–472 [DOI] [PubMed] [Google Scholar]
- Fedor M 2002. The catalytic mechanism of the hairpin ribozyme. Biochem Soc Trans 30: 1109–1115 [DOI] [PubMed] [Google Scholar]
- Fedor M 2009. Comparative enzymology and structural biology of RNA self-cleavage. Annu Rev Biophys 38: 271–299 [DOI] [PubMed] [Google Scholar]
- Fedorova O, Su L, Pyle A 2002. Group II introns: Highly specific endonucleases with modular structures and diverse catalytic functions. Methods 28: 323–335 [DOI] [PubMed] [Google Scholar]
- Flamm C, Fontana W, Hofacker I, Schuster P 2000. RNA folding at elementary step resolution. RNA 6: 325–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieda KL, Bock SM 2012. Direct observation of cotranscriptional folding in an adenine riboswitch. Science 338: 397–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gampel A, Cech T 1991. Binding of the CBP2 protein to a yeast mitochondrial group-I intron requires the catalytic core of the RNA. Genes Dev 5: 1870–1880 [DOI] [PubMed] [Google Scholar]
- Gardner PP, Giegerich R 2004. A comprehensive comparison of comparative RNA structure prediction approaches. BMC Bioinformatics 5: 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geis M, Flamm C, Wolfinger MT, Tanzer A, Hofacker IL, Middendorf M, Mandl C, Stadler PF, Thurner C 2008. Folding kinetics of large RNAs. J Mol Biol 379: 160–173 [DOI] [PubMed] [Google Scholar]
- Geissmann T, Marzi S, Romby P 2010. The role of mRNA structure in translational control in bacteria. RNA Biol 6: 153–160 [DOI] [PubMed] [Google Scholar]
- Giuliodori A, Pietro FD, Marzi S, Masquida B, Wagner R, Romby P, Gualerzi C, Pon C 2010. The cspA mRNA is a thermosensor that modulates translation of the cold-shock protein CspA. Mol Cell 37: 21–33 [DOI] [PubMed] [Google Scholar]
- Granneman S, Baserga S 2005. Crosstalk in gene expression: Coupling and co-regulation of rDNA transcription, pre-ribosome assembly and pre-rRNA processing. Curr Opin Cell Biol 17: 281–286 [DOI] [PubMed] [Google Scholar]
- Gregan J, Kolisek M, Schweyen R 2001. Mitochondrial Mg2+ homeostasis is critical for group II intron splicing in vivo. Genes Dev 15: 2229–2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halls C, Mohr S, Campo MD, Yang Q, Jankowsky E, Lambowitz A 2007. Involvement of DEAD-box proteins in group I and group II intron splicing. Biochemical characterization of Mss116p, ATP hydrolysis-dependent and -independent mechanisms, and general RNA chaperone activity. J Mol Biol 365: 835–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havgaard JH, Lyngsø RB, Stormo GD, Gorodkin J 2005. Pairwise local structural alignment of RNA sequences with sequence similarity less than 40%. Bioinformatics 21: 1815–1824 [DOI] [PubMed] [Google Scholar]
- Heilmann-Miller SL, Woodson SA 2003a. Effect of transcription on folding of the Tetrahymena ribozyme. RNA 9: 722–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilmann-Miller SL, Woodson SA 2003b. Perturbed folding kinetics of circularly permuted RNAs with altered topology. J Mol Biol 328: 385–394 [DOI] [PubMed] [Google Scholar]
- Herschlag D 1995. RNA chaperones and the RNA folding problem. J Biol Chem 270: 20871–20874 [DOI] [PubMed] [Google Scholar]
- Hickman A, Dyda F 2005. Binding and unwinding: SF3 viral helicases. Curr Opin Struct Biol 15: 77–85 [DOI] [PubMed] [Google Scholar]
- Hofacker I 2003. The Vienna RNA secondary structure server. Nucleic Acids Res 31: 3429–3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker I, Fontana W, Stadler P, Bonhoeffer S, Tacker M, Schuster P 1994. Fast folding and comparison of RNA secondary structures. Monatshefte fur Chemie 125: 167–188 [Google Scholar]
- Hofacker I, Fekete M, Stadler P 2002. Secondary structure prediction for aligned RNA sequences. J Mol Biol 319: 1059–1066 [DOI] [PubMed] [Google Scholar]
- Holmes I 2005. Accelerated probabilistic inference of RNA structure evolution. BMC Bioinformatics 6: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins J, Panja S, Woodson S 2011. Rapid binding and release of Hfq from ternary complexes during RNA annealing. Nucleic Acids Res 39: 5193–5202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz DS 2012. The mechanism of the second step of pre-mRNA splicing. Wiley Interdiscip Rev RNA 3: 331–350 [DOI] [PubMed] [Google Scholar]
- Isambert H, Siggia E 2000. Modeling RNA folding paths with pseudoknots: Application to hepatitis δ virus ribozyme. Proc Natl Acad Sci 97: 6515–6520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S, Koduvayur S, Woodson S 2006. Self-splicing of a group I intron reveals partitioning of native and misfolded RNA populations in yeast. RNA 12: 2149–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Xu X, Stormo G 2004. A graph theoretical approach for predicting common RNA secondary structure motifs including pseudoknots in unaligned sequences. Bioinformatics 20: 1591–1602 [DOI] [PubMed] [Google Scholar]
- Johansson J, Mandin P, Renzoni A, Chiaruttini C, Springer M, Cossart P 2002. An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell 110: 551–561 [DOI] [PubMed] [Google Scholar]
- Knudsen B, Hein J 1999. RNA secondary structure prediction using stochastic context-free grammars and evolutionary history. Bioinformatics 15: 446–454 [DOI] [PubMed] [Google Scholar]
- Knudsen B, Hein J 2003. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res 31: 3423–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koduvayur S, Woodson S 2004. Intracellular folding of the Tetrahymena group I intron depends on exon sequence and promoter choice. RNA 10: 1526–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kos M, Tollervey D 2010. Yeast pre-rRNA processing and modification occur cotranscriptionally. Mol Cell 37: 809–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer F, Mills D 1981. Secondary structure formation during RNA-synthesis. Nucleic Acids Res 9: 5109–5124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T 2001. Identification of novel genes coding for small expressed RNAs. Science 294: 853–858 [DOI] [PubMed] [Google Scholar]
- Lai D, Proctor JR, Zhu JY, Meyer IM 2012. R-Chie: A web server and R package for visualizing RNA secondary structures. Nucleic Acids Res 40: e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landick R 1997. RNA polymerase slides home: Pause and termination site recognition. Cell 88: 741–744 [DOI] [PubMed] [Google Scholar]
- Lewicki B, Margus T, Remme J, Nierhaus K 1993. Coupling of rRNA transcription and ribosomal assembly in vivo: Formation of active ribosomal-subunits in Escherichia coli requires transcription of RNA genes by host RNA polymerase which cannot be replaced by T7 RNA polymerase. J Mol Biol 231: 581–593 [DOI] [PubMed] [Google Scholar]
- Lioliou E, Romilly C, Romby P, Fechter P 2010. RNA-mediated regulation in bacteria: From natural to artificial systems. N Biotechnol 27: 222–235 [DOI] [PubMed] [Google Scholar]
- Liu K, Zhang Y, Severinov K, Das A, Hanna M 1996. Role of Escherichia coli RNA polymerase α subunit in modulation of pausing, termination and anti-termination by the transcription elongation factor NusA. EMBO J 15: 150–161 [PMC free article] [PubMed] [Google Scholar]
- Mahen E, Harger J, Calderon E, Fedor M 2005. Kinetics and thermodynamics make different contributions to RNA folding in vitro and in yeast. Mol Cell 19: 27–37 [DOI] [PubMed] [Google Scholar]
- Mahen E, Watson P, Cottrell J, Fedor M 2010. mRNA secondary structures fold sequentially but exchange rapidly in vivo. PLoS Biol 8: e1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DH 2005. Predicting a set of minimal free energy RNA secondary structures common to two sequences. Bioinformatics 21: 2246–2253 [DOI] [PubMed] [Google Scholar]
- Mathews DH, Turner DH 2002. Dynalign: An algorithm for finding the secondary structure common to two RNA sequences. J Mol Biol 317: 191–203 [DOI] [PubMed] [Google Scholar]
- Mathews D, Burkard M, Freier S, Wyatt J, Turner D 1999. Predicting oligonucleotide affinity to nucleic acid targets. RNA 5: 1458–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura M, Saldanha R, Ma H, Wank H, Yang J, Mohr G, Cavanagh S, Dunny G, Belfort M, Lambowitz A 1997. A bacterial group II intron encoding reverse transcriptase, maturase, DNA endonuclease activities: Biochemical demonstration of maturase activity and insertion of new genetic information within the intron. Genes Dev 11: 2910–2924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer I 2008. Predicting novel RNA–RNA interactions. Curr Opin Struct Biol 18: 387–393 [DOI] [PubMed] [Google Scholar]
- Meyer IM, Miklós I 2004. Co-transcriptional folding is encoded within RNA genes. BMC Mol Biol 10: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer IM, Miklós I 2007. SimulFold: Simultaneously inferring an RNA structure including pseudo-knots, a multiple sequence alignment and an evolutionary tree using a Bayesian Markov Chain Monte Carlo framework. PLoS Comput Biol 3: e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikulecky P, Kaw M, Brescia C, Takach J, Sledjeski D, Feig A 2004. Escherichia coli Hfq has distinct interaction surfaces for DsrA, rpoS and poly(A) RNAs. Nat Struct Mol Biol 11: 1206–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov A, Lebedev V 1993. A kinetic model of RNA folding. Biosystems 30: 49–56 [DOI] [PubMed] [Google Scholar]
- Mironov A, Dyakonova L, Kister A 1985. A kinetic approach to the prediction of RNA secondary structures. J Biomol Struct Dyn 2: 953–962 [DOI] [PubMed] [Google Scholar]
- Mohr G, Zhang A, Gianelos J, Belfort M, Lambowitz A 1992. The Neurospora CYT-18 protein suppresses defects in the phage T4 td intron by stabilizing the catalytically active structure of the intron core. Cell 69: 483–494 [DOI] [PubMed] [Google Scholar]
- Mohr G, Caprara M, Guo Q, Lambowitz A 1994. A tyrosyl-transfer-RNA synthetase can function similarly to an RNA structure in the Tetrahymena ribozyme. Nature 370: 147–150 [DOI] [PubMed] [Google Scholar]
- Mooney R, Artsimovitch I, Landick R 1998. Information processing by RNA polymerase: Recognition of regulatory signals during RNA chain elongation. J Bacteriol 180: 3265–3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan S, Higgs P 1996. Evidence for kinetic effects in the folding of large RNA molecules. J Chem Phys 105: 7152–7157 [Google Scholar]
- Narberhaus F 2010. Translational control of bacterial heat shock and virulence genes by temperature-sensing mRNAs. RNA Biol 7: 84–89 [DOI] [PubMed] [Google Scholar]
- Nechooshtan G, Egrably-Weiss M, Sheaffer A, Westhof E, Altuvia S 2009. A pH-responsive riboregulator. Genes Dev 23: 2650–2662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolcheva T, Woodson S 1999. Facilitation of group 1 splicing in vivo: Misfolding of the Tetrahymena IVS and the role of ribosomal RNA exons. J Mol Biol 292: 557–567 [DOI] [PubMed] [Google Scholar]
- Oakes M, Nogi I, Clark M, Nomura M 1993. Structural alterations of the nucleolus in mutants of Saccharomyces cerevisiae defective in RNA polymerase I. Mol Cell Biol 13: 2441–2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostersetzer O, Cooke A, Watkins K, Barkan A 2005. CRS1, a chloroplast group II intron splicing factor, promotes intron folding through specific interactions with two intron domains. Plant Cell 17: 241–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan T, Sosnick T 2006. RNA folding during transcription. Annu Rev Biophys Biomol Struct 35: 161–175 [DOI] [PubMed] [Google Scholar]
- Pan T, Fang X, Sosnick T 1999. Pathway modulation, circular permutation and rapid RNA folding under kinetic control. J Mol Biol 286: 721–731 [DOI] [PubMed] [Google Scholar]
- Paukstelis P, Coon R, Madabusi L, Nowakowski J, Monzingo A, Robertus J, Lambowitz A 2005. A tyrosyl-tRNA synthetase adapted to function in group I intron splicing by acquiring a new RNA binding surface. Mol Cell 17: 417–428 [DOI] [PubMed] [Google Scholar]
- Paukstelis P, Chen J, Chase E, Lambowitz A, Golden B 2008. Structure of a tyrosyl-tRNA synthetase splicing factor bound to a group I intron RNA. Nature 451: 94–97 [DOI] [PubMed] [Google Scholar]
- Pedersen J, Forsberg R, Meyer I, Hein J 2004a. An evolutionary model for protein-coding regions with conserved RNA structure. Mol Biol Evol 21: 1913–1922 [DOI] [PubMed] [Google Scholar]
- Pedersen J, Meyer I, Forsberg R, Simmonds P, Hein J 2004b. A comparative method for finding and folding RNA secondary structures within protein-coding regions. Nucleic Acids Res 32: 4925–4936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdrizet G, Artsimovitch I, Furman R, Sosnick TR, Pan T 2012. Transcriptional pausing coordinates folding of the aptamer domain and the expression platform of a riboswitch. Proc Natl Acad Sci 109: 3323–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perriquet O, Touzet H, Dauchet M 2003. Finding the common structure shared by two homologous RNAs. Bioinformatics 19: 108–116 [DOI] [PubMed] [Google Scholar]
- Proctor JR, Meyer IM 2013. CoFold: An RNA secondary structure prediction method that takes co-transcriptional folding into account. Nucleic Acids Res 41: e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle A 2008. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu Rev Biophys 37: 317–336 [DOI] [PubMed] [Google Scholar]
- Rajkowitsch L, Chen D, Starnpfl S, Semrad K, Waldsich C, Mayer O, Jantsch M, Konrat R, Blaesi U, Schroeder R 2007. RNA chaperones, RNA annealers and RNA helicases. RNA Biol 4: 118–130 [DOI] [PubMed] [Google Scholar]
- Repsilber D, Wiese S, Rachen M, Schroder A, Riesner D, Steger G 1999. Formation of metastable RNA structures by sequential folding during transcription: Time-resolved structural analysis of potato spindle tuber viroid (−)-stranded RNA by temperature-gradient gel electrophoresis. RNA 5: 574–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ro-Choi T, Choi Y 2003. Structural elements of dynamic RNA strings. Mol Cells 16: 201–210 [PubMed] [Google Scholar]
- Roth A, Breaker R 2010. The structural and functional diversity of metabolite-binding riboswitches. Annu Rev Biochem 78: 305–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan J, Stormo G, Zhang W 2004. An iterated loop matching approach to the prediction of RNA secondary structures with pseudoknots. Bioinformatics 20: 58–66 [DOI] [PubMed] [Google Scholar]
- Schoemaker RJW, Gultyaev AP 2006. Computer simulation of chaperone effects of Archael C/D box sRNA binding on rRNA folding. Nucleic Acids Res 34: 2015–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder R, Grossberger R, Pichler A, Waldsich C 2002. RNA folding in vivo. Curr Opin Struct Biol 12: 296–300 [DOI] [PubMed] [Google Scholar]
- Schroeder R, Barta A, Semrad K 2004. Strategies for RNA folding and assembly. Nat Rev Mol Cell Biol 5: 908–919 [DOI] [PubMed] [Google Scholar]
- Sclavi B, Sullivan M, Chance M, Brenowitz M, Woodson S 1998. RNA folding at millisecond intervals by synchrotron hydroxyl radical footprinting. Science 279: 1940–1943 [DOI] [PubMed] [Google Scholar]
- Semrad K, Schroeder R 1998. Ribosomal function is necessary for efficient splicing of the T4 phage thymidylate synthase intron in vivo. Genes Dev 12: 1327–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serganov A 2009. The long and the short of riboswitches. Curr Opin Struct Biol 19: 251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova I, Mitra S, Laederach A, Brenowitz M 2008. Energy barriers, pathways, and dynamics during folding of large, multidomain RNAs. Curr Opin Chem Biol 12: 655–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit S, Widmann J, Knight R 2007. Evolutionary rates vary among rRNA structural elements. Nucleic Acids Res 35: 3339–3354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soper T, Woodson S 2008. The rpoS mRNA leader recruits Hfq to facilitate annealing with DsrA sRNA. RNA 14: 1907–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soper T, Mandin P, Majdalani N, Gottesman S, Woodson S 2010. Positive regulation by small RNAs and the role of Hfq. Proc Natl Acad Sci 107: 9602–9607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosnick TR, Pan T 2003. RNA folding: Models and perspectives. Curr Opin Struct Biol 13: 309–316 [DOI] [PubMed] [Google Scholar]
- Steif A, Meyer IM 2012. The hok mRNA family. RNA Biol 9: 1399–1404 [DOI] [PubMed] [Google Scholar]
- Talkington M, Siuzdak G, Williamson J 2005. An assembly landscape for the 30S ribosomal subunit. Nature 438: 628–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirumalai D, Hyeon C 2005. RNA and protein folding: Common themes and variations. Biochemistry 44: 4957–4970 [DOI] [PubMed] [Google Scholar]
- Toulme F, Mosrin-Huaman C, Artsimovitch I, Rahmouni A 2005. Transcriptional pausing in vivo: A nascent RNA hairpin restricts lateral movements of RNA polymerase in both forward and reverse directions. J Mol Biol 351: 39–51 [DOI] [PubMed] [Google Scholar]
- Touzet H, Perriquet O 2004. CARNAC: Folding families of related RNAs. Nucleic Acids Res 32: W142–W145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiber D, Williamson J 2001. Beyond kinetic traps in RNA folding. Curr Opin Struct Biol 11: 309–314 [DOI] [PubMed] [Google Scholar]
- Turner D, Sugimoto N, Freier S 1990. Thermodynamics and kinetics of base-pairing and of DNA and RNA self-assembly and helix coil transition. In Nucleic acids (ed. Saenger W), Vol. 1C of Landolt–Bornstein Series, pp. 201–227 Springer-Verlag, Berlin [Google Scholar]
- Udem S, Warner J 1973. Cytoplasmic maturation of a ribosomal precursor ribonucleic-acid in yeast. J Biol Chem 248: 1412–1416 [PubMed] [Google Scholar]
- Waldsich C, Grossberger R, Schroeder R 2002a. RNA chaperone StpA loosens interactions of the tertiary structure in the td group I intron in vivo. Genes Dev 16: 2300–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldsich C, Masquida B, Westhof E, Schroeder R 2002b. Monitoring intermediate folding states of the td group I intron in vivo. EMBO J 21: 5281–5291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Kertesz M, Spitale RC, Segal E, Chang HY 2011. Understanding the transcriptome through RNA structure. Nat Rev Genet 12: 641–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb A, Weeks K 2001. A collapsed state functions to self-chaperone RNA folding into a native ribonucleoprotein complex. Nat Struct Biol 8: 135–140 [DOI] [PubMed] [Google Scholar]
- Weeks KM 1997. Protein-facilitated RNA folding. Curr Opin Struct Biol 7: 336–342 [DOI] [PubMed] [Google Scholar]
- Weeks K, Cech T 1996. Assembly of a ribonucleoprotein catalyst by tertiary structure capture. Science 271: 345–348 [DOI] [PubMed] [Google Scholar]
- Wickiser J, Winkler W, Breaker R, Crothers D 2005. The speed of RNA transcription and metabolite binding kinetics operate an FMN riboswitch. Mol Cell 18: 49–60 [DOI] [PubMed] [Google Scholar]
- Wiebe NJP, Meyer IM 2010. Transat—a method for detecting the conserved helices of functional RNA structures, including transient, pseudo-knotted and alternative structures. PLoS Comput Biol 6: e1000823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witwer C, Hofacker IL, Stadler PF 2004. Prediction of consensus RNA structures including pseudoknots. IEEE/ACM Trans Comput Biol Bioinform 1: 66–77 [DOI] [PubMed] [Google Scholar]
- Wong T, Sosnick T, Pan T 2007. Folding of noncoding RNAs during transcription facilitated by pausing-induced nonnative structures. Proc Natl Acad Sci 104: 17995–18000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodson S 2010. Taming free energy landscapes with RNA chaperones. RNA Biol 7: 677–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuchty S, Fontana W, Hofacker I, Schuster P 1999. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers 49: 145–165 [DOI] [PubMed] [Google Scholar]
- Xayaphoummine A, Bucher T, Thalmann F, Isambert H 2003. Prediction and statistics of pseudoknots in RNA structures using exactly clustered stochastic simulations. Proc Natl Acad Sci 100: 15310–15315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xayaphoummine A, Bucher T, Isambert H 2005. Kinefold web server for RNA/DNA folding path and structure prediction including pseudoknots and knots. Nucleic Acids Res 33: W605–W610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanofsky C 1981. Attenuation in the control of expression of bacterial operons. Nature 289: 751–758 [DOI] [PubMed] [Google Scholar]
- Zemora G, Waldsich C 2010. RNA folding in living cells. RNA Biol 7: 634–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JY, Steif A, Proctor JR, Meyer IM 2013. Transient RNA structure features are evolutionarily conserved and can be computationally predicted. Nucleic Acids Res 41: 6273–6285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M, Stiegler P 1981. Optimal computer folding of large RNA sequences using thermodynamic and auxiliary information. Nucleic Acids Res 9: 133–148 [DOI] [PMC free article] [PubMed] [Google Scholar]