

Abstract
Pathogenic mechanisms of Alzheimer’s disease (AD) are intensely investigated as it is the most common form of dementia and burdens society by its costs and social demands. While key molecules such as A-beta peptides and tau have been identified decades ago, it is still enigmatic what drives the disease in its sporadic manifestation. Synthesis of A-beta peptides as well as phosphorylation of tau proteins comprise normal cellular functions and occur in principle in the healthy as well as in dementia-affected persons. Dyshomeostasis of Amyloid Precursor Protein (APP) cleavage, energy metabolism or kinase/phosphatase activity due to stressors has been suggested as a trigger of the disease. One way for cells to escape stress based on dysfunction of ER is the unfolded protein response - the UPR. This pathway is composed out of three different routes that differ in proteins involved, targets and consequences for cell fate: activation of transmembrane ER resident kinases IRE1-alpha and PERK or monomerization of membrane-anchored activating transcription factor 6 (ATF6) induce activation of versatile transcription factors (XBP-1, eIF2-alpha/ATF4 and ATF6 P50). These bind to specific DNA sequences on target gene promoters and on one hand attenuate general ER-prone protein synthesis and on the other equip the cell with tools to de-stress. If cells fail in stress compensation, this signaling also is able to evoke apoptosis. In this review we summarized knowledge on how APP processing and phosphorylation of tau might be influenced by ER-stress signaling. In addition, we depicted the effects UPR itself seems to have on molecules closely related to AD and describe what is known about UPR in AD animal models as well as in human patients.
Keywords: Alzheimer’s disease, secretases, APP, tau, unfolded protein response, calcium homeostasis, autophagy, apoptosis
Introduction
The term amyloidosis describes a family of diseases that are characterized by abnormal protein deposition within the extracellular space. Examples are the cardiac amyloidosis or Alzheimer’s disease (AD). Pathological features of this type of disease are mediated on one hand by direct malfunction of the affected tissue or organ. Amyloid deposits in the ventricles and atria of the heart e.g. result in biventricular wall thickening with an ensuing elevation of pressure in the thin-walled part of the respective atrium [1,2]. On the other hand, interference of already deposited material or intermediate protein oligomers with cellular function has been described to lead to dysbalance and subsequent pathogenesis. For example, amyloidogenic light chains are able to evoke oxidative stress, cellular dysfunction, and apoptosis in primary cardiomyocyte cultures via MAPK signaling [3]. In case of Alzheimer’s disease the scientific landscape was dominated for a long time by the assumption that large aggregates of A-beta peptides (designated as senile plaques) are triggering neuronal degeneration. More recent investigations led to the insight that small oligomers (reviewed e.g. in [4]) or even intraneuronal A-beta peptides are culprit to pathogenic derailment [5,6]. Tau protein with its microtubule binding properties is another characteristic of the disease and has been suggested to act downstream of neurotoxic A-beta species (reviewed e.g. in [7]).
A-beta peptides derive from proteolytic processing of a large type I transmembrane protein - the amyloid precursor protein (APP). This protein matures within trafficking through ER and the Golgi-apparatus by being cleaved by signal peptidases and being modified in regard of carbohydrate-attachment. Despite having the knowledge about distinct function of each proteolysis product, the proteolytic degradation of this protein has been described in detail: subsequent cleavage by the beta-secretase BACE-1 (beta-site APP cleaving enzyme 1) and the gamma-secretase complex gives rise to the already mentioned A-beta peptides (e.g. [8,9]). Alternatively, alpha-secretase leads to prevention of A-beta peptide formation and to secretion of the so-called APPs-alpha fragment (for example [10-13]). The latter has been described to conduct neurotrophic and neuroprotective properties (reviewed in e.g. [14]). APP is a ubiquitously expressed protein and A-beta peptides are generated not only under pathological conditions but also in healthy human subjects. Nevertheless, under certain circumstances, synthesis and/or degradation of A-beta peptides are disturbed and oligomers and fibrils rise that are deposited in brain parenchyma or blood vessel walls. Some human beings seem to cope with these deposits very well and show no signs of cognitive decline despite having high plaque loads while others do not [15]. Understanding the underlying resilience factors might lead to development of new therapeutic approaches since directly inhibiting A-beta production or immunization strategies in the first line failed regarding curing this disease.
An adaptable access to gain resilience might be given by the cellular response to endogenous stressors. One cellular organelle that is related to stress mediation by its multifunctionality is the endoplasmic reticulum (ER, for example [16,17]): 1) the ER assists and closely monitors quality of nascent proteins. Protein disulfide isomerase (PDI) and ERp57 (thio-oxidoreductases) e.g. catalyze disulfide bond formation using the oxidative capacity provided by ER oxidoreduction 1 (ERO1). Glucose-regulated protein 78 (BiP or GRP78) or 94 (GRP94, calreticulin), stabilize as chaperones of the heat-shock protein family unfolded proteins. 2) Calcium-storage in the ER and regulated release protects the cell from cell death caused by calcium concentration dysbalance. This is driven by calcium import via SERCA (sarcoplasmic/endoplasmic reticulum calcium-ATPase) and ion release via Rhyanodine and IP3 receptors. Calcium binding proteins such as calreticulin and calnexin further buffer the calcium content of the cell.
Disturbance of ER homeostasis is not separated from general cellular function. The peripheral ER is in contact to other cellular organelles and for example forms physical interaction zones with mitochondria. These structures - designated as MAM in mammals (Mitochondrial-associated endoplasmic reticulum Membranes, [18], reviewed in [19]) - are enriched by certain proteins such as Mitofusin-2 or the autocrine motility factor receptor which allow attachment [20,21]. In case of perturbations regarding ER function these are not only sensed by the ER but also transduced to the cytoplasm and nucleus to evoke an appropriate response. This response includes enhanced expression of chaperones, transiently inhibited translation, increase in ER volume and enhanced degradation of misfolded proteins as well as enhanced autophagy [22,23]. If this attenuation of stressors and capacity compensation along the unfolded protein response (UPR) is not sufficient within a certain time frame, cells undergo apoptosis (reviewed in [24]).
APP as a central player in Alzheimer’s disease matures via bypassing the ER and beta- as well as gamma-secretase cleavage takes place in the Golgi apparatus [25,26]. In addition, mitochondrial dysfunction, disturbed autophagy and aberrant calcium signaling have been repeatedly connected to Alzheimer’s disease (reviewed e.g. in [27-29]). A general role of the UPR in neurodegenerative processes has been summarized previously [30]. Hence, a contribution of activated UPR signaling in the pathology of Parkinson’s or in particular Alzheimer’s disease has been discussed as an early event of disease progression [31]. Therefore, we focused on known correlations of APP, its processing products and related proteins with ER-stress events in this review.
ER-stress: three routes of UPR signal transduction
Three signal pathways operate in parallel to sense ER-stress and to react as the UPR (Figure 1). They have in common that signaling is transduced by a protein with an ER luminal domain, a transmembrane part and a cytoplasmic effector domain: IRE1-alpha (inositol requiring enzyme 1, [32-34]), PERK (double-stranded RNA-activated protein kinase (PKR)-like ER kinase, [35]), and ATF6 (activating transcription factor 6, [36-38]). All three proteins use a specific signal transduction mechanism but end in activation of b-Zip transcription factors that subsequently lead to altered transcription of target genes.
Figure 1.
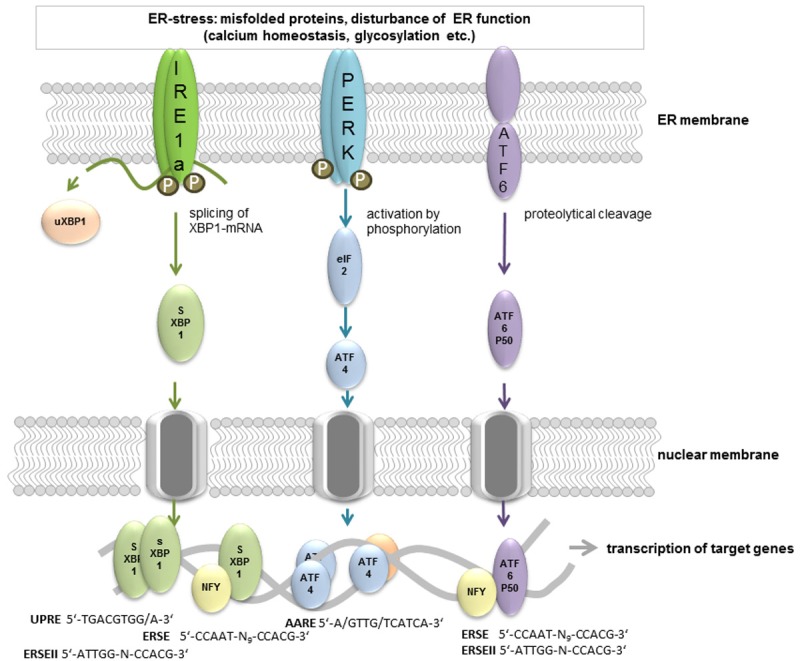
Three different signaling pathways conduct UPR upon ER-stress induction. ER-stress is sensed by the luminal proportion of three distinct transmembrane ER resident proteins: IRE1-alpha, PERK and ATF6. There are several mechanisms that have been proposed for this activation step such as direct binding of unfolded proteins or displacement of GRP78 binding (reviewed in [151]). Activation subsequently leads to changes in oligomerization status and the signal is transmitted via the C-terminus of all three proteins. PERK and IRE1-alpha homodimerize and perform autophosphorylation. PERK also phosphorylates the translation initiation factor eIF2-alpha resulting in a reversible attenuation of translation. Only single translational events such as ATF4 protein biosynthesis are enhanced under these conditions. Delayed reinitiation by reduced amount of eIF2-GTP allows ribosomes to scan through an inhibitory upstream open reading frame of ATF4 mRNA and instead reinitiate at the coding region [152]. ATF4 binds to a consensus motif of target gene promoters (e.g. AARE) as a homodimer or heterodimer with e.g. Fos, Jun [153] or other cofactors such as PCAF [154]. IRE1-alpha conducts its endonuclease function and splices XBP-1 mRNA as well as other mRNAs. This process is termed regulated IRE1-alpha-dependent decay (RIDD; [62]) and may further contribute to limitation of translation. Target genes of the active sXBP-1 (binding to UPRE, ERSE or ERSEII) produced by splicing are proteins involved in protein folding, maturation, secretion, and degradation. ATF6 translocates to the Golgi upon activation where it is cleaved by site-1-and 2 proteases. This generates the b-Zip transcription factor ATF6 P50 which initiates transcription of target genes in combination with NF-Y via ERSE motifs in its target genes.
ATF6 and PERK represent evolutionary newer pathways that evolved in metazoans [39]. ATF6 exists partially oligomerized in the absence of stress through intermolecular disulfide bonds [40]. These are reduced upon accumulation of unfolded proteins, subsequently the monomeric form leaves the ER via transport vesicles and is cleaved within the Golgi apparatus by site-1 and site-2 proteases [41]. The cytoplasmic domain then is liberated from its transmembrane anchor and acts as a transcription factor on target genes such as GRP78 or GRP94 [42]. Both are part of the functional ER machinery and therefore might contribute to enhanced ER capacity.
PERK is a transmembrane kinase that oligomerizes during ER-stress and performs trans-autophosphorylation of its C-terminal cytoplasmic kinase domain at multiple residues [43,44] as well as phosphorylation of the general translation factor eIF2-alpha [45]. This inhibits eIF2-alpha which lowers translation rates regarding a wide variety of mRNAs [46,47] and consequently reduces protein burden within the ER. Only few mRNAs show increased translation upon eIF2-alpha phosphorylation such as ATF4 which by itself acts as a transcription factor on targets such as CHOP (transcription factor C/EBP homologous protein, [48]) or GADD34 (growth arrest and DNA damage-inducible 34, [49]). CHOP regulates expression of components of the apoptotic pathway (reviewed in [50]) while GADD34 encodes a regulatory subunit of PP1C - the protein phosphatase that dephosphorylates eIF2-alpha [51]. This on one hand indicates that the PERK branch contains both, alleviating strategies and death triggering ones, and that it is strictly regulated.
IRE1-alpha represents the third branch which also exists in lower eukaryotes [39]. By sensing ER-stress this bifunctional enzyme dimerizes and becomes activated [52]. During the dimer assembly trans-autophosphorylation takes place [53] which is discussed to be mandatory for the human enzyme activity or regulation [54,55]. Formation of the active dimer then evokes endonuclease function and the ER-associated mRNA of the transcription factor XBP-1 (X-box binding protein 1) is spliced to yield the active protein sXBP-1 (for the splicing mechanism see: [56]). In non-yeast organisms unspliced XBP-1-mRNA is translated to uXBP-1 which might act as an inhibitory counterbalance of sXBP1 action [57]. Downstream targets of activated XBP1 include ER chaperones as well as so-called ERAD (ER associated degradation) components such as HRD1 [58], PDI [59] or ER-localized DnaJ (Erdj4) [60]. Besides specifically activating XBP-1, IRE1-alpha is involved in activation of JNK [61] and degradation of other ER-associated mRNAs (e.g. its own mRNA: [53]; regulated IRE1-alpha-dependent decay (RIDD): [62]) further contributing to relief of ER machinery by decreasing translation into the ER lumen. In secretory cells such as B-cells IRE1-alpha also mediates UPR-independent enhancement of secretion [63,64].
The three signaling pathways are not fully isolated but also comprise cross-linkage. For example PERK facilitates synthesis and trafficking of ATF6 from ER to the Golgi [65] and IRE1-alpha activity also steers ATF6 activation [66].
Target genes of the UPR contain conserved binding sequences (Figure 1) designated as UPRE (UPR element, [67]) or ERSE (ER-stress element, [68]) or ERSEII [69]). ERSE sites consist of the conserved sequence CCAAT-N9-CCACG; the 9 nucleotide spacing between both half site motifs has been suggested to be quiet important for their functionality [70]. A recent report nevertheless described a functional XBP-1-responsive element with a 26 nt spacer [71]. ATF6 binds e.g. to the CCACG sequence of the ERSE motif, if the general transcription factor NF-Y has bound to the CCAAT part of the sequence. Active XBP-1 also binds to this sequence instead of ATF6 [70,72]. The ERSEII (ATTGG-N-CCACG) might be occupied by ATF6 NF-Y-dependent [69] or by XBP-1 without further binding of NF-Y [67]. To the UPRE (TGACGTGG/A) sXBP-1 binds as a homodimer [70].
A genome wide approach to identify targets of XBP-1 in myotubular, plasma and pancreatic cells by Acosta-Alvear and colleagues [73] revealed that XBP-1 is able to bind the promoters of a wide variety of target genes whereof 40% are not directly linked to ER-stress compensation. One of the unexpected GO categories contained disease-associated genes, including genes connected to Alzheimer’s disease. In cell culture studies UPR as indicated by eIF2-alpha phosphorylation is evoked by compounds such as Tunicamycin, Thapsigargin or DTT (e.g. [74]) which impair N-glycosylation of proteins, calcium homeostasis or formation of disulfide bonds. In addition, brefeldin A, 2-deoxy-glucose or eeyarestatin function as UPR inducers. These compounds seem to belong to two different clusters of drugs as shown by analysis of expression patterns of nine typical UPR target genes by [75]. Endogenous provokers of ER-stress are under intensive investigation and several molecules associated with Alzheimer’s disease are under suspicion.
Influence of APP-cleavage products on ER-stress
Shortly after cloning of the mammalian PERK and IRE1-alpha [43,32], first investigations indicated a potential link to Alzheimer dementia: Katayama et al. [76] described that mutations in Presenilin 1 (PS1), the catalytic component of the gamma-secretase complex, decreased GRP78 expression by interfering with IRE1-alpha function in cell culture experiments. Moreover, they already assessed reduced amount of GRP78 and GRP94 within temporal cortex tissue from sporadic and familiar Alzheimer patients. A potential role of PS1 in UPR was further confirmed by Niwa et al. [77], where a reduced nuclear translocation and attenuated UPR was reported for PS1 knockout fibroblasts. A direct cleavage of IRE1-alpha has been suggested but could not be confirmed for human fibroblasts due to technical problems [77]. In addition, cleavage of ATF6 has been analyzed but no influence of gamma-secretase inhibitor or dominant negative PS1 could be demonstrated [78]. Gamma-secretase is a very promiscuous endoproteolytic complex that cleaves a wide variety of substrates (e.g. reviewed in [79]) such as APP. Therefore, regulation of UPR by several proteolysis products could be assumed.
For APP as a substrate of gamma-secretase, there have been reports about correlation with ER-stress and subsequent UPR signaling: APP overexpression led for example to an enhanced upregulation of CHOP in PC12 cells upon Thapsigargin administration [80]. For other typical ER-stress associated chaperones such as GRP94 this was not confirmed in this cell line. However, colocalization of APP with ER chaperones calnexin, calreticulin, GRP94, GRP78, and associated with Alzheimer’s disease are under suspicion. ERp72 has been demonstrated in sporadic inclusion body myositis (s-IBM) muscle biopsies with APP containing aggregation products [81]. This was also confirmed for APP overexpressing human muscle fibers. While basal expression of UPR-related proteins in another report did not differ in APP overexpressing cells from wild type cells, UPR induction by ER-stress was augmented [82]. This was even higher in cells with overexpression of mutant APP. In contrast, elevated but comparable GRP78 mRNA levels were measured in B103 cells expressing either mutated or wild type APP after UPR induction with Tunicamycin [83]. In PC12 cells, APP has been reported to be protective against ER-stress evoked by brefeldin A application while GRP78 or CHOP expression remained unaffected. In sum, there is a rather inhomogeneous picture of how APP might interfere with ER-stress and subsequent signaling. This might be explained by different types of cells used in either investigation which might be characterized by differential APP processing activities since for proteolytic fragments of APP there is growing evidence for interfering with UPR and ER-stress (Figure 2). Mostly, reduction of A-beta is correlated with attenuated ER-stress and vice versa. For example, aged PS2 mutant mice revealed inhibited BACE-1 activity and C99 amount in brain tissue upon treadmill exercise [84]. This was accompanied by a down-regulation of GRP78 and PDI enzymes as well as an inhibited activation status of PERK, eIF2-alpha, ATF6 and sXBP-1. In GNE myopathy that is characterized by A-beta deposition, muscle biopsies showed enhanced expression of molecular chaperones such as GRP78 [85]. This might be interpreted as mere coincidence but direct application of A-beta also has been demonstrated to lead to ER-stress signaling: GRP78 and XBP-1 protein levels increased for example in neurons treated with A-beta [86] which could be further augmented by mitochondrial dysfunction. SK-S-SH cells displayed activation of the PERK pathway as well as increase in CHOP upon A-beta administration [87]. Another publication on the contrary found triggering of eIF2-alpha phosphorylation and calcium depletion from the ER, but no activation of UPR (as shown e.g. by splicing of XBP-1 mRNA, amount of XBP-1 mRNA and PERK phosphorylation) in primary cortical neurons [88]. The A-beta1-42 peptide used in this study comprised both, oligomeric and fibril forms and this might be the cause of observing no induction of UPR. Chafekar and colleagues demonstrated mild induction of the signaling pathway only by oligomers and not by fibrils [89]. A very recent report on human iPSC derived neurons from familial and sporadic Alzheimer cases confirmed that A-beta oligomer accumulation leads to ER as well as oxidative stress [90]. The mechanism behind this is still not fully understood: low molecular A-beta peptides have been described to interrupt mitochondria-ER anchoring and thereby to evoke ER collapse [91]. Yoon et al reported an AMPK-mediated translational block upon application of A-beta 42 oligomers [92]. This subsequently inhibited mTOR signaling and resulted in activated ER-stress as shown by eIF2-alpha phosphorylation in rat hippocampal neurons [92]. Interestingly, monomeric A-beta peptides as well as fibrils here failed to activate AMPK.
Figure 2.
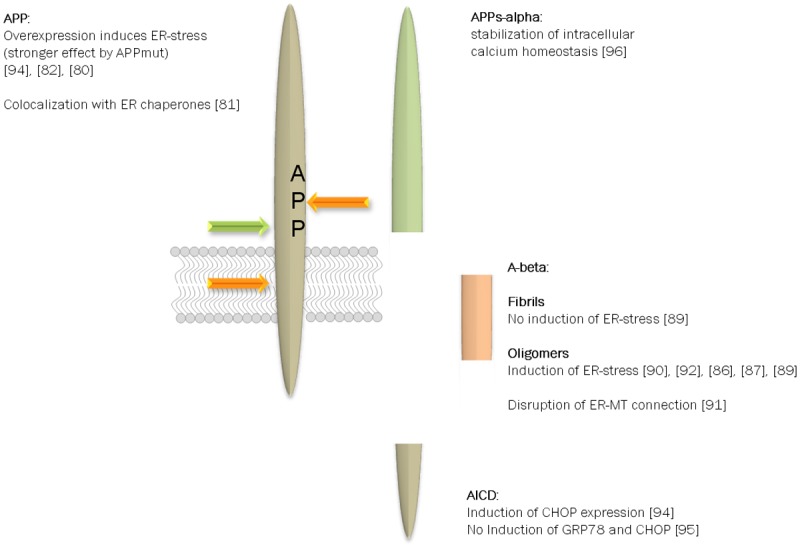
APP processing products and their potential role in UPR. APP is subject to a wide variety of posttranslational modifications such as phosphorylation and glycosylation (e.g. [62,155]). In addition, the type I transmembrane protein is cleaved by several proteases yielding protein fragments of different size and function [14]. In regard of ER-stress not all protein fragments have been analyzed but for the full length protein, A-beta peptides and the alpha-secretase derived soluble ectodomain correlation to subsequent signaling events have been reported. In this scheme only the most prominent facts are included but one has to consider that contradictory results have been published.
Not only A-beta peptides are thought to interfere with ER-stress but single reports also exist on other cleavage products - the APP intracellular domain (AICD) and the fragment produced by alpha-secretase-cleavage (APPs-alpha). The AICD is discussed to act as a transcription factor in analogy to the Notch C-terminal domain [93]. Regarding this, infliction in signal transduction to the nucleus by the UPR-prone transcription factors ATF6, 4 and sXBP-1 seems plausible. Takahashi and coworkers found increased CHOP mRNA and protein level in APP overexpressing cells. This could be attenuated by treatment with DAPT (a potent gamma-secretase inhibitor) and occurred also upon transfection with a tagged AICD variant [94]. CHIP assays in AICD transfected HEK293 cells additionally indicated a physical interaction of the AICD with the CHOP promoter region. Nevertheless, another group reported that AICD overexpression in SHEP neuroblastoma cells did not enhance GRP78 and CHOP expression but led to potentiation of ER-stress driven apoptosis [95]. This would rather indicate that AICD might act downstream or independently from UPR. APPs-alpha, which is released via alpha-secretase activity on APP, seems to provide protection against ER-stress: Guo et al. [96] described an NF-kappa B-dependent stabilization of intracellular calcium homeostasis in differentiated PC12 cells by APPs-alpha. In addition, enhancement of APPs-alpha secretion via DHA-treatment was observed to be protective against apoptosis induced by ER Ca(2+) store depletion via Thapsigargin in HEK293-APP cells [97]. This protective potential could be transferred to untransfected HEK293 or PC12 cells using the supernatant of DHA-treated cells.
For other cleavage products of APP such as APPs-beta, C99 and C83 as wells as p3 to our knowledge no literature exist so far that describes entangling in ER-stress and downstream signaling.
Influence of tau and APP-modifying molecules on ER-stress
A-beta peptides are just one component that triggers or induces AD pathology: a growing number of molecules has been correlated to this type of dementia, which makes it difficult to reflect a complete picture of interference with ER-stress. Therefore, we here focus on tau as the second central player and proteins, directly acting on APP.
An investigation by Unterberger [98] indicated the importance for tau proteins in ER-stress response by demonstrating that in human prion diseases activated PERK and eIF2-alpha only occurred concomitantly to neurofibrillary pathology. In contrast, phosphorylated PERK correlated with hyperphosphorylated forms of tau protein in AD. This connection of UPR activation and the microtubule-associated protein tau has been confirmed in a wide range of publications within the last years. Hoozemans and colleagues described that phospho-PERK was absent from neurofibrillary tangles but abundantly detectable in neurons with hyperphosphorylated tau [99]. The percentage of affected neurons thereby increased with the Braak stage for neurofibrillary changes. In frontotemporal lobar degeneration with Tau pathology (FTLD-tau) phospho-PERK and phospho-IRE1-alpha were increased in a similar way while FTLD without tau pathology or non-neurological control cases showed no signs of UPR activation in neurons and glia [100]. The implication of tau in UPR has also been shown in animal models of tau-pathology such as in [101] where high levels of activated PERK and eIF2-alpha were identified in the hippocampus of aged tau-transgenic mice (P301L). This is contradicted by a paper from Spatara and colleague [102]: they were not able to demonstrate activation of UPR in a mouse line where the P301S variant of tau was expressed (aged 6 and 11 months). They investigated XBP-1 splicing by splice variant specific PCR which might be difficult in tissue with low abundance of sXBP-1. No significant changes as compared to tissue of wild-type mice in calreticulin and GRP78 mRNA levels were found; unfortunately it is not clear whether those samples were from 6 month or 11 month old mice, which impairs a direct comparison with the data from Ho et al. [101]. In 9 month old rTg4510 mice (advanced stage of disease) phospho-PERK was significantly increased in comparison to non-transgenic control mice [103]. This was accompanied by an increase in GRP78 and occurred in the hippocampus and cortex of the mice, regions severely affected by tau pathology in this disease model. Suppression of the transgene by the TET-off-system for one month revised the phospho-PERK elevation as did the acute suppression for 4 days: decrease of phospho-PERK and GRP78 strongly suggests reversibility of ER-stress induction by tau protein [103].
Besides the colocalization of phospho-PERK and phospho-tau, Ho et al. [101] described a direct influence of ER-stress on tau metabolism: Thapsigargin stimulated phosphorylation of tau (Thr231, Ser262 and Ser396) as well as its cleavage [101]. This “vicious cycle” was also shown for neuronal and non-neuronal cell lines SH-SY5Y and HEK293 and rat brain preparations [104]. Recently, in addition an increase of total endogenous tau protein in cultured neurons and primary cultured neurons has been described due to a reduction in the degradation rate of tau under ER-stress [105].
Interestingly, UPR here directly links A-beta and tau pathology via ER-stress signaling: oligomeric A-beta peptides have repeatedly been linked to ER-stress (see Figure 2) and release of calcium from the ER leads to activation of GSK3-beta, a major tau-kinase, in primary rat embryonic cortical neurons [106]. Moreover, ventricular infusion of ER-stressors in rats resulted in GSK3-beta activation and tau hyperphosphorylation [107]. In addition, GRP78 was elevated and an enhanced binding of GSK3-beta and tau to this chaperone was demonstrated in brain tissue from Tunicamycin-treated animals. In HEK293 cells with overexpression of GRP78 and tau this increase in binding levels occurred in a similar manner. siRNA-mediated knock down of GRP78 in HEK/tau cells revised tau hyperphosphorylation upon Thapsigargin-application while GSK3-beta was still activated [107]. This in sum reflects the ambivalence of ER-stress driven signaling: once induced, it might even worsen degenerative processes, but activation of ER-stress induced UPR - e.g. via tau hyperphosphorylation - might also contribute to protection against apoptosis. HEK cells that overexpress tau for example revealed attenuated apoptosis in response to treatment with ER-stress inducers such as staurosporine or camptothecin [107].
For involvement of APP-modifying proteinases in ER-stress signaling, only scattered reports exist: for instance increased production of BACE-1 mRNA and non-coding antisense transcript has been reported in sporadic inclusion-body myositis muscle fibers [108] which have been correlated with increased ER-stress signaling [81]. Both transcripts were also elevated by experimentally evoked ER-stress via Tunicamycin or Thapsigargin in cultured human muscle cells [108]. In the APP/PS1 AD mouse model at 3, 6 and 12 months of age hippocampal neurons showed increased staining for PERK phosphorylation [109]. At 3 month also phospho-eI2Fa levels were elevated in brain tissue homogenates in comparison to wild type mice. Salubrinal, an inhibitor of eIF2-alpha phosphatase PP1c, increased BACE1 and subsequently A-beta production in primary neurons [110]. HSV1 transfection of SH-SY5Y cells which leads to activation of PRK - an alternative kinase that phosphorylates eI2F-alpha - strongly increased BACE-1 protein amounts [111]. Moreover, inhibition of eIF2-alpha phosphatase de-repressed the signal from a BACE1-5’UTR- luciferase reporter in HeLa cells, indicating that this is an important pathway regulating BACE1 translational repression [112]. If PKR or PERK both contribute in a similar manner to the regulation has to be investigated. However, CSF levels of PKR and phospho-PKR were found to be elevated in AD and amnestic mild cognitive impairment subjects [113] and the latter was also linked to cognitive decline measured by longitudinal MMSE changes [114].
Regarding gamma-secretase, very early on correlation to ER-stress has been suggested (see above) by the observation that PS1 knock-out fibroblasts revealed a reduction in nuclear translocation and lowered UPR signaling [77]. But again, the influence seems to be a mutual one: Tunicamycin application in vivo enhanced PS1 expression in mouse kidney [115]. Furthermore, it has been demonstrated that ATF4 binds to the amino acid response element (AARE) regulatory region of human PS1 gene. Quercetin, which induces XBP-1 splicing but suppresses ATF4 activation, prevented the Tunicamycin-evoked increase in PS1 and subsequently production of A-beta peptides in HEK293 cells [116].
Takahashi et al. described in 2009 that Tunicamycin not only induces AICD production in HEK293 but also C83, the C-terminal fragment derived by cleavage of APP by alpha-secretase [94]. Treatment of retinal pigment epithelia cells (ARPE19) with Tunicamycin or Thapsigargin for 24hrs revealed enhanced amounts of TACE-mRNA [117]. TACE (tumour necrosis factor alpha converting enzyme, ADAM17) is widely accepted as the regulated alpha-secretase (for a comparison of ADAM10 and 17 physiological roles see [118]). The TACE-induction by ER-stress was also observed for several tumor cell lines such as HeLa or MCF7 [119]. By applying siRNA targeted against central players of the three arms of UPR - ATF4, XBP-1 and ATF6 - it was demonstrated that only ATF4 and 6 are able to act as regulators of TACE expression. ATF4 had the most prominent effect and was shown to bind at least to one of the three predicted binding sites of the TACE promoter by CHIP analysis [119]. This has been confirmed by overexpression of active XBP-1 in SH-SY5Y human neuroblastoma cells where TACE mRNA as well as protein was not elevated [120]. Interestingly, despite the failure of XBP-1 in enhancing TACE expression, secretion of APPs-alpha was induced. This has been shown to be based on upregulation of ADAM10 expression, the major alpha-secretase in neuronal tissue [11,121,122]. Mice with B-cells deleted in Adam10 contained normal numbers of plasma cells but were impaired in antibody responses [123]. This was accompanied by reduced amounts of transcription factors implicated in plasma cell function such as XBP-1.
In sum, different parts of ER-stress and UPR signaling are involved in a multimodal manner in APP processing as well as tau metabolism and players of AD pathogenesis by themselves affect ER-stress and adequate response of the cell due to stress.
ER-stress signaling in animal models of AD and human patients
Most transgenic models of AD are based on the amyloid hypothesis and therefore are predicated on genetic mutations of human AD-relevant genes such as APP or PS1, which have been identified in familial AD cases. These models mimic in part AD-like pathological features indicated e.g. by increased A-beta peptide generation, tau fibrillization or even cognitive impairment (reviewed in [124]). In this section we summarize the regulation of crucial ER-stress components belonging to one of the three branches (PERK, ATF6 or IRE1-alpha) in AD animal models and compare them to knowledge obtained by brain samples derived from human AD-patients (for an overview see Table 1).
Table 1.
Changes in ER-stress signaling in AD and AD animal models
| AD (human patients) | Severity of disease/tissue | Components of ER-stress pathway | Reference |
|
| |||
| Stage 5 to 6 | Accumulation of CD3-delta (ERAD substrate) | [103] | |
| Correlation of toxic turn A-beta with GRP78 | [133] | ||
| Reduced HRD1 protein levels, increased mRNA (function in ERAD) | [140] | ||
| Increase in p-eIF2-alpha, ATF4, CHOP, and PERK | [92] | ||
| Pro-apoptotic ER-stress pathway molecules increase with AD severity | [137] | ||
| Pro-homeostatic ER-stress molecules mainly upregulated in the intermediate stage of AD | |||
| Hippocampus and frontal lobe | PDI immuno-positive inclusions (NFTs) | [139] | |
| Stage 1 to 6 | Increased XBP-1 splicing and PDI expression | [87] | |
| Temporal cortex | CHOP activation | ||
| Temporal cortex | Reduction in GRP78 and 94 | [76] | |
| Frontal cortex | Phosphorylation of eIF2-alpha upregulated | [110] | |
| Temporal cortex | Phosphorylation of eIF2-alpha upregulated | [113] | |
| Hippocampus CA1 region | Phospho-PERK immunopositive neurons | [99] | |
| Hippocampus and temporal cortex | Phosphorylation of eIF2-alpha upregulated | [131] | |
| Stage 3 to 6 | sXBP-1 mRNA level downregulated | [120] | |
| Temporal and frontal cortex | |||
|
| |||
| AD models | Model | ||
|
| |||
| rTg4510 mice (tauopathy) | Activation of PERK | [103] | |
| 9 months | Accumulation of CD3-delta (ERAD substrate) | ||
| 3x Tg-AD mice | Increased GRP78 | [133] | |
| 2 months | |||
| APP(E693Δ) mice | up-regulation of GRP78 and HRD1 | [135] | |
| 18 months | |||
| A-beta transgenic flies | suppression of neurotoxicity by sXBP-1 | [138] | |
| Tg2576 mice | No activation of UPR or apoptosis | [87] | |
| 17 months | |||
| 5xFAD 6 months | Phosphorylation of eIF2-alpha upregulated | [110] | |
| APP/PS1 9 months | Phosphorylation of eIF2-alpha upregulated | [113] | |
| 5xFAD 1 and 9 month | sXBP-1 mRNA level increased (1 and 6 month) | [120] | |
| APP/PS1 6 and 9 month | sXBP-1 mRNA level decreased (9 month) | ||
Regarding the PERK pathway, in the rTg4510 tau-based Alzheimer mouse model a significant increase in activated phosphorylated PERK was observed at 9 month of age [103]. This is a rather aggressive model, where pathological features such as tangle formation, deficits in neuronal function or cognitive decline occur as early as 3 or 5.5 months of age [125,126]. Tissues severely affected by tau fibrillization such as hippocampus and cortex displayed a 2.4-fold increase in phospho-PERK [103]. These findings are consistent with analyses regarding AD brain samples: immunopositive phospho-PERK neurons were found in hippocampal CA1 region and the amount was significantly increased as compared to non-demented [99]. Furthermore, activation of PERK was positively correlated with advanced Braak scores and consequently with severity of disease. Longitudinal study of tau pathology and accumulation of phospho-PERK revealed also a correlation in the rTg4510 mouse model as early as 6 month of age (first time point investigated: 3 month) [103]. The influence of tau on ER-stress might be rather indirect because pathological tau did not colocalize with an ER-marker. Human cortical sections of advanced Braak stages (5-6) which also display robust accumulation of hyperphosphorylated tau confirmed that aberrant tau and the-ER marker calnexin are not colocalized. As tau was shown to co-immunoprecipitated with the ERAD protein complex members VCP and HRD1 this might explain triggering of ER-stress by tau.
In whole brain lysates of Tg2576 mice, which overexpress the Swedish mutant of human APP [127] activation/phosphorylation of the PERK downstream target eIF2-alpha was demonstrated and positively correlated with an increased expression of BACE-1 [110]. For this analysis 9 month old mice which already exhibit maximum plaque formation were further challenged by energy deprivation, therefore activation of PERK signaling might be due to the specific experimental paradigms. However, the induction of ER-stress in an APP-based mouse model was further substantiated by analysis using an aggressive amyloid deposition mouse model - 5xFAD, which display detectable A-beta peptide generation already at 1 month of age [128]. In 6 month old 5xFAD mice with a severe amyloid pathology the BACE-1 expression level was increased 1.7-fold as compared to non-transgenic littermates [110]. This was accompanied by an increased ratio of phospho-eIF2-alpha to total eIF2-alpha. The regulation of the BACE-1 expression by active eIF2-alpha could be mechanistically elucidated by Mouton-Liger and colleagues: in 9 month old APP/PS1 mice, which already show neuronal loss (detectable at 6 month of age [129]) the expression of BACE-1 was regulated by active eIF2-alpha under the control of PKR [113]. On the contrary, analysis of cortical samples of aged Tg2576 mice (17 month of age) with massive plaque formation revealed no significant change in CHOP expression, which is the most prominent downstream target of the PERK pathway [49]. These contradictory results might be explained by the different ages of animals used in the studies. ER-stress is known to be a transient process with the IRE1-alpha-branch being down-regulated with prolonged exposure to respective signals [130]. As CHOP induction was reported to maintain due to sustained stress at least in human cells, counter-regulation in old mice is to be debatable. Nevertheless, immunoblot analysis of human post-mortem frontal cortex tissue of AD-patients revealed a positive correlation of active eIF2-alpha with BACE-1 protein level and also with amyloid load of AD brain samples [110]. Increased levels of active eIF2-alpha have also been described in temporal cortex [113,131] and hippocampal tissue [131] of AD-patients. Moreover, CHOP has been reported to be increased in temporal [87] and frontal [92] cortex tissue as compared to age-matched controls.
In sum, literature is almost consistent with an overall upregulation of single PERK-pathway components e.g. PERK, eIF2-alpha and CHOP in AD or aggressive disease models. CHOP is responsible for the regulation of apoptosis under ER-stress conditions e.g. by ATF5 induction [132]. Therefore neuronal cell death in AD might be partially understood as a consequence of long lasting ER-stress in the brain.
For the most prominent ATF6-mediated ER-stress marker GRP78 the field of literature is more heterogeneous: for example it has been demonstrated that GRP78 is significantly upregulated in two different AD mouse models. In neurons of 3xTg AD mice aged 2 month an increase in GRP78 expression was detected [133]. Mice at this age are cognitively unimpaired und consequently are representative for a pre-pathological stage [134]. Umeda and colleagues described an increase of GRP78 expression in hippocampal neurons and cerebral cortex of APP (E693Δ) mice aged 18 month [135]. These mice display impaired hippocampal synaptic plasticity at an age of 8 month and even neuronal loss detectable at 24 month of age [136]. In addition, no expression changes of GRP78 were found in in the brain of 17 month old Tg2576 mice [87]. Comparably, analysis of human post mortem samples of AD-patients are characterized by contradictory results: a positive correlation of intraneuronal A-beta peptides with the expression of GRP78 has been reported by double-label immunostaining of AD brain sections as compared to those of healthy controls [133]. Consistent with this, the GRP78 protein level was significantly increased in cortical tissue derived from AD brains as compared to samples from non-demented [131]. On the contrary, Katayama and colleagues have shown that expression level of this protein was slightly reduced in temporal cortex samples derived from sporadic AD cases and even more reduced in the brain of patients with familial AD linked to PS1 mutations [76].
Existing literature does not allow a final appraisal of GRP78 expression in brain tissue of AD-patients due to missing characterization of samples regarding e.g. Braak stages. However, one possible attempt to explain these heterogeneous results might be the two-sited direction of ER-stress signaling dependent on the intensity and the duration of stress signals. Expression of molecules ensuring recovery of ER function and homeostasis were rather induced at intermediate disease stages while for progressed disease severity pro-apoptotic mediators were predominant in an investigation from de la Monte and colleagues [137].
Concerning the IRE1-alpha pathway, a Drosophila model with human A-beta expression underlined a potential neuroprotective function of XBP-1, the main downstream target of IRE1-alpha [138]. Those flies show a strong A-beta-dependent phenotype in the eye regarding structure and size of the organ. These pathological changes were alleviated by introducing XBP-1. Such a protective effect of XBP-1 regarding neurodegenerative processes has been further substantiated by a sXBP-1-mediated induction of ADAM10 gene expression [120]. Within two different AD mouse models (APP/PS1 and 5xFAD) sXBP-1 mRNA level were significantly increased in whole brain samples as compared to non-transgenic littermates at early time points of pathological changes (APP/PS1: 6 month; 5xFAD: 1 month). This was accompanied by an increase of ADAM10 expression [120]. Analyzing more advanced stages (9 month of age for both models), XBP-1 returned to wild type levels or was even decreased [120]. However, mRNA level of spliced/active XBP-1 has been reported to be increased in temporal cortex tissue of AD-patients as compared to healthy controls (Braak scores ranged from 1 to 6 for AD samples, [87]). Our own study with a more defined stage of disease (Braak scores 3-6) demonstrated a significant decrease of sXBP-1 in temporal as well as frontal cortex tissue of AD-patients in a larger cohort of individuals [120]. Analyzing the same samples, a significant reduction of ADAM10 as a novel downstream target of XBP-1 in both tissue types was observed. Another downstream target of the transcription factor XBP-1 is the protein disulfide isomerase (PDI) for which Lee and colleagues revealed an unaltered expression level as demonstrated by immunoblot analysis in 17 month old Tg2576 mice (advanced stage of AD-like pathology [87]). However, in the same report this enzyme was found to be upregulated in cortical AD brain tissue and PDI immune-positive neurofibrillary tangles (NFTs) were described in slices from hippocampus and frontal lobe of AD-patients [139]. On the contrary, PDI protein level was unaffected in cerebral cortex samples derived from AD-patients as compared to healthy controls [140]. HRD-1, which is an ERAD-associated E3 ubiquitin ligase acting downstream of XBP-1 [58], was investigated immunohistochemically in 18 month old APP (E693Δ) mice with severe amyloid pathology [135]. Analysis revealed an intense immunoreactivity in hippocampal neurons and cerebral cortex tissue of the animals. However, Kaneko and colleagues showed that HRD1 is downregulated in the cerebral cortex of AD-patients [140]. This is in accordance with an increase of a subunit of the T-cell antigen receptor - CD3-delta, which is a well characterized substrate for ERAD driven proteolysis, in hippocampus of 9 month old rTg4510 mice [103]. Increased accumulation of CD3-delta was also described in temporal brain tissue from AD-patients (Braak scores: 5-6) as compared to non-demented controls (Braak scores: 1-2) and would rather support a defect in XBP-1 evoked ERAD in AD.
Conclusions
Literature predominantly hints at an early but transient induction of XBP-1 signaling in the AD context and an activation of the PERK-dominated pathway throughout to late stages of disease. This suggests ER-stress functioning as a scale in AD pathogenesis, balancing cells between coping with protein misfolding associated stress and undergoing cell death. Double-edged properties of ER-stress signaling comes clear e.g. in a recent investigation of a C. elegans model of Alzheimer’s disease: basal activity of the UPR was beneficial under normal conditions, but repression of the signaling delayed toxicity evoked by inducible A-beta peptide expression [141]. Age-onset loss of ER proteostasis could be reversed by neuronal expression of sXBP-1 in C. elegans and enhanced longevity [142], pointing at a potential role of UPR in age-related changes which will be interesting to investigate in humans.
All these observations lead to the rather alluring prospect that interfering with ER-stress might bear novel therapeutic approaches regarding neurodegenerative diseases such as AD and has been presented in previous reviews (e.g. [143-145]). For example, administration of CNB-001, a pyrazole-inhibitor of 5-lipoxygenase, was able to induce PERK signaling and improve memory in AD model mice [146]. Nevertheless, several uncertainties and open questions regarding the role of ER-stress in AD remain: 1) Animal models used for investigation of ER-stress signaling under pathological conditions are differing in time of onset of pathological features and no real scaling exists for comparison of “severity of disease” at the investigated age of animals. Only a minority of reports give a longitudinal picture on evolvement of stress which would allow estimating the role of single components in pathology. Investigations using human tissue partly miss characterization of disease stages and data concerning MCI patients and normal aging are missing - at least to our knowledge. 2) The actors of the three signaling branches are phosphorylated proteins and transcription factors and per se are rather unstable or regulated by further posttranslational processing, translocation events or protein interactions (e.g. interplay of ATF4 with p300 acetyltransferase, [147] or SUMO-conjugase UBC9 with sXBP-1, [148]). This might explain why a wide variety of investigations are based on mRNA quantitation which might distort the underlying scenario. 3) Crosstalk of the ER-stress signaling pathways among themselves and with other signal transduction pathways such as insulin signaling [149,150] further complicate entangling ER-stress involvement in AD. Nevertheless, elucidating ER-stress function or failure in AD might help turning the scale in therapeutic considerations or for evolvement of new highly diagnostic biomarkers.
Acknowledgements
The authors’ own work was supported by the Federal Ministry of Education and Research (BMBF) in the framework of the National Genome Research Network (NGFN, FKZ01GS08130) and by the Alfons Geib Stiftung of the University Medical Centre Mainz.
Disclosure of conflict of interest
The authors declare that they have no conflicts of interest.
References
- 1.Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation. 2005;112:2047–2060. doi: 10.1161/CIRCULATIONAHA.104.489187. [DOI] [PubMed] [Google Scholar]
- 2.Rapezzi C, Quarta CC, Riva L, Longhi S, Gallelli I, Lorenzini M, Ciliberti P, Biagini E, Salvi F, Branzi A. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol. 2010;7:398–408. doi: 10.1038/nrcardio.2010.67. [DOI] [PubMed] [Google Scholar]
- 3.Shi J, Guan J, Jiang B, Brenner DA, del Monte F, Ward JE, Connors LH, Sawyer DB, Semigran MJ, Macgillivray TE, Seldin DC, Falk R, Liao R. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway. Proc Natl Acad Sci U S A. 2010;107:4188–4193. doi: 10.1073/pnas.0912263107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kayed R, Lasagna-Reeves CA. Molecular mechanisms of amyloid oligomers toxicity. J Alzheimers Dis. 2013;33(Suppl 1):S67–S78. doi: 10.3233/JAD-2012-129001. [DOI] [PubMed] [Google Scholar]
- 5.Cuello AC, Allard S, Ferretti MT. Evidence for the accumulation of Abeta immunoreactive material in the human brain and in transgenic animal models. Life Sci. 2012;91:1141–1147. doi: 10.1016/j.lfs.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 6.Laferla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 7.Krstic D, Knuesel I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol. 2013;9:25–34. doi: 10.1038/nrneurol.2012.236. [DOI] [PubMed] [Google Scholar]
- 8.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 9.Anderson JP, Chen Y, Kim KS, Robakis NK. An alternative secretase cleavage produces soluble Alzheimer amyloid precursor protein containing a potentially amyloidogenic sequence. J Neurochem. 1992;59:2328–2331. doi: 10.1111/j.1471-4159.1992.tb10128.x. [DOI] [PubMed] [Google Scholar]
- 10.Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojro E, Prinzen C, Endres K, Hiemke C, Blessing M, Flamez P, Dequenne A, Godaux E, van Leuven F, Fahrenholz F. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J Clin Invest. 2004;113:1456–1464. doi: 10.1172/JCI20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 13.Skovronsky DM, Moore DB, Milla ME, Doms RW, Lee VM. Protein kinase C-dependent alpha-secretase competes with beta-secretase for cleavage of amyloid-beta precursor protein in the trans-golgi network. J Biol Chem. 2000;275:2568–2575. doi: 10.1074/jbc.275.4.2568. [DOI] [PubMed] [Google Scholar]
- 14.Chasseigneaux S, Allinquant B. Functions of Abeta, sAPPalpha and sAPPbeta : similarities and differences. J Neurochem. 2012;120(Suppl 1):99–108. doi: 10.1111/j.1471-4159.2011.07584.x. [DOI] [PubMed] [Google Scholar]
- 15.Rodrigue KM, Kennedy KM, Park DC. Beta-amyloid deposition and the aging brain. Neuropsychol Rev. 2009;19:436–450. doi: 10.1007/s11065-009-9118-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Araki K, Nagata K. Protein folding and quality control in the ER. Cold Spring Harb Perspect Biol. 2012;4:a015438. doi: 10.1101/cshperspect.a015438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benham AM. Protein secretion and the endoplasmic reticulum. Cold Spring Harb Perspect Biol. 2012;4:a012872. doi: 10.1101/cshperspect.a012872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem. 1990;265:7248–7256. [PubMed] [Google Scholar]
- 19.Raturi A, Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM) Biochim Biophys Acta. 2013;1833:213–224. doi: 10.1016/j.bbamcr.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 20.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 21.Registre M, Goetz JG, St Pierre P, Pang H, Lagacé M, Bouvier M, Le PU, Nabi IR. The gene product of the gp78/AMFR ubiquitin E3 ligase cDNA is selectively recognized by the 3F3A antibody within a subdomain of the endoplasmic reticulum. Biochem Biophys Res Commun. 2004;320:1316–1322. doi: 10.1016/j.bbrc.2004.06.089. [DOI] [PubMed] [Google Scholar]
- 22.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 24.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 25.Dyrks T, Dyrks E, Mönning U, Urmoneit B, Turner J, Beyreuther K. Generation of beta A4 from the amyloid protein precursor and fragments thereof. FEBS Lett. 1993;335:89–93. doi: 10.1016/0014-5793(93)80446-2. [DOI] [PubMed] [Google Scholar]
- 26.Sambamurti K, Shioi J, Anderson JP, Pappolla MA, Robakis NK. Evidence for intracellular cleavage of the Alzheimer’s amyloid precursor in PC12 cells. J Neurosci Res. 1992;33:319–329. doi: 10.1002/jnr.490330216. [DOI] [PubMed] [Google Scholar]
- 27.Selfridge JE, E L, Lu J, Swerdlow RH. Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol Dis. 2013;51:3–12. doi: 10.1016/j.nbd.2011.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barnett A, Brewer GJ. Autophagy in aging and Alzheimer’s disease: pathologic or protective? J Alzheimers Dis. 2011;25:385–394. doi: 10.3233/JAD-2011-101989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stutzmann GE, Mattson MP. Endoplasmic reticulum Ca(2+) handling in excitable cells in health and disease. Pharmacol Rev. 2011;63:700–727. doi: 10.1124/pr.110.003814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bernales S, Soto MM, McCullagh E. Unfolded protein stress in the endoplasmic reticulum and mitochondria: a role in neurodegeneration. Front Aging Neurosci. 2012;4:5. doi: 10.3389/fnagi.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Scheper W. Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener Dis. 2012;10:212–215. doi: 10.1159/000334536. [DOI] [PubMed] [Google Scholar]
- 32.Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998;17:5708–5717. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73:1197–1206. doi: 10.1016/0092-8674(93)90648-a. [DOI] [PubMed] [Google Scholar]
- 34.Nikawa J, Yamashita S. IRE1 encodes a putative protein kinase containing a membrane-spanning domain and is required for inositol phototrophy in Saccharomyces cerevisiae. Mol Microbiol. 1992;6:1441–1446. doi: 10.1111/j.1365-2958.1992.tb00864.x. [DOI] [PubMed] [Google Scholar]
- 35.Meurs E, Chong K, Galabru J, Thomas NS, Kerr IM, Williams BR, Hovanessian AG. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell. 1990;62:379–390. doi: 10.1016/0092-8674(90)90374-n. [DOI] [PubMed] [Google Scholar]
- 36.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu C, Johansen FE, Prywes R. Interaction of ATF6 and serum response factor. Mol Cell Biol. 1997;17:4957–4966. doi: 10.1128/mcb.17.9.4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hai TW, Liu F, Coukos WJ, Green MR. Transcription factor ATF cDNA clones: an extensive family of leucine zipper proteins able to selectively form DNA-binding heterodimers. Genes Dev. 1989;3:2083–2090. doi: 10.1101/gad.3.12b.2083. [DOI] [PubMed] [Google Scholar]
- 39.Hollien J. Evolution of the unfolded protein response. Biochim Biophys Acta. 2013;1833:2458–2463. doi: 10.1016/j.bbamcr.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 40.Nadanaka S, Okada T, Yoshida H, Mori K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol Cell Biol. 2007;27:1027–1043. doi: 10.1128/MCB.00408-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ye J, Rawson RB, Komuro R, Chen X, Davé UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 42.Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct. 2008;33:75–89. doi: 10.1247/csf.07044. [DOI] [PubMed] [Google Scholar]
- 43.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic- reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 44.Marciniak SJ, Garcia-Bonilla L, Hu J, Harding HP, Ron D. Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J Cell Biol. 2006;172:201–209. doi: 10.1083/jcb.200508099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sood R, Porter AC, Ma K, Quilliam LA, Wek RC. Pancreatic eukaryotic initiation factor-2alpha kinase (PEK) homologues in humans, Drosophila melanogaster and Caenorhabditis elegans that mediate translational control in response to endoplasmic reticulum stress. Biochem J. 2000;346:281–293. [PMC free article] [PubMed] [Google Scholar]
- 46.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 47.Ernst V, Levin DH, London IM. Inhibition of protein synthesis initiation by oxidized glutathione: activation of a protein kinase that phosphorylates the alpha subunit of eukaryotic initiation factor 2. Proc Natl Acad Sci U S A. 1978;75:4110–4114. doi: 10.1073/pnas.75.9.4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–1365. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- 49.Lee YY, Cevallos RC, Jan E. An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2alpha phosphorylation. J Biol Chem. 2009;284:6661–6673. doi: 10.1074/jbc.M806735200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu CY, Schröder M, Kaufman RJ. Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J Biol Chem. 2000;275:24881–24885. doi: 10.1074/jbc.M004454200. [DOI] [PubMed] [Google Scholar]
- 53.Tirasophon W, Lee K, Callaghan B, Welihinda A, Kaufman RJ. The endoribonuclease activity of mammalian IRE1 autoregulates its mRNA and is required for the unfolded protein response. Genes Dev. 2000;14:2725–2736. doi: 10.1101/gad.839400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ali MM, Bagratuni T, Davenport EL, Nowak PR, Silva-Santisteban MC, Hardcastle A, McAndrews C, Rowlands MG, Morgan GJ, Aherne W, Collins I, Davies FE, Pearl LH. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 2011;30:894–905. doi: 10.1038/emboj.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mannan MA, Shadrick WR, Biener G, Shin BS, Anshu A, Raicu V, Frick DN, Dey M. An ire1-phk1 chimera reveals a dispensable role of autokinase activity in endoplasmic reticulum stress response. J Mol Biol. 2013;425:2083–2099. doi: 10.1016/j.jmb.2013.02.036. [DOI] [PubMed] [Google Scholar]
- 56.Korennykh AV, Korostelev AA, Egea PF, Finer-Moore J, Stroud RM, Zhang C, Shokat KM, Walter P. Structural and functional basis for RNA cleavage by Ire1. BMC Biol. 2011;9:47. doi: 10.1186/1741-7007-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoshida H, Nadanaka S, Sato R, Mori K. XBP1 is critical to protect cells from endoplasmic reticulum stress: evidence from Site-2 protease-deficient Chinese hamster ovary cells. Cell Struct Funct. 2006;31:117–125. doi: 10.1247/csf.06016. [DOI] [PubMed] [Google Scholar]
- 58.Yamamoto K, Suzuki N, Wada T, Okada T, Yoshida H, Kaufman RJ, Mori K. Human HRD1 promoter carries a functional unfolded protein response element to which XBP1 but not ATF6 directly binds. J Biochem. 2008;144:477–486. doi: 10.1093/jb/mvn091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang S, Chen Z, Lam V, Han J, Hassler J, Finck BN, Davidson NO, Kaufman RJ. IRE1alpha-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab. 2012;16:473–486. doi: 10.1016/j.cmet.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kanemoto S, Kondo S, Ogata M, Murakami T, Urano F, Imaizumi K. XBP1 activates the transcription of its target genes via an ACGT core sequence under ER stress. Biochem Biophys Res Commun. 2005;331:1146–1153. doi: 10.1016/j.bbrc.2005.04.039. [DOI] [PubMed] [Google Scholar]
- 61.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 62.Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 64.Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 65.Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG, Wek RC. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell. 2011;22:4390–4405. doi: 10.1091/mbc.E11-06-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem. 2000;275:27013–27020. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
- 67.Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J Biochem. 2004;136:343–350. doi: 10.1093/jb/mvh122. [DOI] [PubMed] [Google Scholar]
- 68.Roy B, Lee AS. The mammalian endoplasmic reticulum stress response element consists of an evolutionarily conserved tripartite structure and interacts with a novel stress-inducible complex. Nucleic Acids Res. 1999;27:1437–1443. doi: 10.1093/nar/27.6.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kokame K, Kato H, Miyata T. Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J Biol Chem. 2001;276:9199–9205. doi: 10.1074/jbc.M010486200. [DOI] [PubMed] [Google Scholar]
- 70.Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, Mori K. Endoplasmic reticulum stress-induced formation of transcription factor complex ERSF including NF-Y (CBF) and activating transcription factors 6alpha and 6beta that activates the mammalian unfolded protein response. Mol Cell Biol. 2001;21:1239–1248. doi: 10.1128/MCB.21.4.1239-1248.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Misiewicz M, Déry MA, Foveau B, Jodoin J, Ruths D, le Blanc AC. Identification of a Novel Endoplasmic Reticulum Stress Response Element Regulated by XBP1. J Biol Chem. 2013;288:20378–20391. doi: 10.1074/jbc.M113.457242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem. 1998;273:33741–33749. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- 73.Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, Lennon CJ, Kluger Y, Dynlacht BD. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell. 2007;27:53–66. doi: 10.1016/j.molcel.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 74.Prostko CR, Brostrom MA, Malara EM, Brostrom CO. Phosphorylation of eukaryotic initiation factor (eIF) 2 alpha and inhibition of eIF-2B in GH3 pituitary cells by perturbants of early protein processing that induce GRP78. J Biol Chem. 1992;267:16751–16754. [PubMed] [Google Scholar]
- 75.Shinjo S, Mizotani Y, Tashiro E, Imoto M. Comparative analysis of the expression patterns of UPR-target genes caused by UPR-inducing compounds. Biosci Biotechnol Biochem. 2013;77:729–735. doi: 10.1271/bbb.120812. [DOI] [PubMed] [Google Scholar]
- 76.Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, Nakano Y, Takeda J, Tsuda T, Itoyama Y, Murayama O, Takashima A, St George-Hyslop P, Takeda M, Tohyama M. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol. 1999;1:479–485. doi: 10.1038/70265. [DOI] [PubMed] [Google Scholar]
- 77.Niwa M, Sidrauski C, Kaufman RJ, Walter P. A role for presenilin-1 in nuclear accumulation of Ire1 fragments and induction of the mammalian unfolded protein response. Cell. 1999;99:691–702. doi: 10.1016/s0092-8674(00)81667-0. [DOI] [PubMed] [Google Scholar]
- 78.Steiner H, Winkler E, Shearman MS, Prywes R, Haass C. Endoproteolysis of the ER stress transducer ATF6 in the presence of functionally inactive presenilins. Neurobiol Dis. 2001;8:717–722. doi: 10.1006/nbdi.2001.0405. [DOI] [PubMed] [Google Scholar]
- 79.Haapasalo A, Kovacs DM. The many substrates of presenilin/gamma-secretase. J Alzheimers Dis. 2011;25:3–28. doi: 10.3233/JAD-2011-101065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Copanaki E, Schürmann T, Eckert A, Leuner K, Müller WE, Prehn JH, Kögel D. The amyloid precursor protein potentiates CHOP induction and cell death in response to ER Ca2+ depletion. Biochim Biophys Acta. 2007;1773:157–165. doi: 10.1016/j.bbamcr.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 81.Vattemi G, Engel WK, McFerrin J, Askanas V. Endoplasmic reticulum stress and unfolded protein response in inclusion body myositis muscle. Am J Pathol. 2004;164:1–7. doi: 10.1016/S0002-9440(10)63089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chafekar SM, Zwart R, Veerhuis R, Vanderstichele H, Baas F, Scheper W. Increased Abeta1-42 production sensitizes neuroblastoma cells for ER stress toxicity. Curr Alzheimer Res. 2008;5:469–474. doi: 10.2174/156720508785908883. [DOI] [PubMed] [Google Scholar]
- 83.Esposito L, Gan L, Yu GQ, Essrich C, Mucke L. Intracellularly generated amyloid-beta peptide counteracts the antiapoptotic function of its precursor protein and primes proapoptotic pathways for activation by other insults in neuroblastoma cells. J Neurochem. 2004;91:1260–1274. doi: 10.1111/j.1471-4159.2004.02816.x. [DOI] [PubMed] [Google Scholar]
- 84.Kang EB, Kwon IS, Koo JH, Kim EJ, Kim CH, Lee J, Yang CH, Lee YI, Cho IH, Cho JY. Treadmill exercise represses neuronal cell death and inflammation during Abeta-induced ER stress by regulating unfolded protein response in aged presenilin 2 mutant mice. Apoptosis. 2013;18:1332–1347. doi: 10.1007/s10495-013-0884-9. [DOI] [PubMed] [Google Scholar]
- 85.Li H, Chen Q, Liu F, Zhang X, Li W, Liu S, Zhao Y, Gong Y, Yan C. Unfolded protein response and activated degradative pathways regulation in GNE myopathy. PLoS One. 2013;8:e58116. doi: 10.1371/journal.pone.0058116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Costa RO, Ferreiro E, Oliveira CR, Pereira CM. Inhibition of mitochondrial cytochrome c oxidase potentiates Abeta-induced ER stress and cell death in cortical neurons. Mol Cell Neurosci. 2013;52:1–8. doi: 10.1016/j.mcn.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 87.Lee JH, Won SM, Suh J, Son SJ, Moon GJ, Park UJ, Gwag BJ. Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp Mol Med. 2010;42:386–394. doi: 10.3858/emm.2010.42.5.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yu MS, Suen KC, Kwok NS, So KF, Hugon J, Chang RC. Beta-amyloid peptides induces neuronal apoptosis via a mechanism independent of unfolded protein responses. Apoptosis. 2006;11:687–700. doi: 10.1007/s10495-006-5540-1. [DOI] [PubMed] [Google Scholar]
- 89.Chafekar SM, Hoozemans JJ, Zwart R, Baas F, Scheper W. Abeta 1-42 induces mild endoplasmic reticulum stress in an aggregation state-dependent manner. Antioxid Redox Signal. 2007;9:2245–2254. doi: 10.1089/ars.2007.1797. [DOI] [PubMed] [Google Scholar]
- 90.Kondo T, Asai M, Tsukita K, Kutoku Y, Ohsawa Y, Sunada Y, Imamura K, Egawa N, Yahata N, Okita K, Takahashi K, Asaka I, Aoi T, Watanabe A, Watanabe K, Kadoya C, Nakano R, Watanabe D, Maruyama K, Hori O, Hibino S, Choshi T, Nakahata T, Hioki H, Kaneko T, Naitoh M, Yoshikawa K, Yamawaki S, Suzuki S, Hata R, Ueno S, Seki T, Kobayashi K, Toda T, Murakami K, Irie K, Klein WL, Mori H, Asada T, Takahashi R, Iwata N, Yamanaka S, Inoue H. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell. 2013;12:487–496. doi: 10.1016/j.stem.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 91.Lai CS, Preisler J, Baum L, Lee DH, Ng HK, Hugon J, So KF, Chang RC. Low molecular weight Abeta induces collapse of endoplasmic reticulum. Mol Cell Neurosci. 2009;41:32–43. doi: 10.1016/j.mcn.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 92.Yoon SO, Park DJ, Ryu JC, Ozer HG, Tep C, Shin YJ, Lim TH, Pastorino L, Kunwar AJ, Walton JC, Nagahara AH, Lu KP, Nelson RJ, Tuszynski MH, Huang K. JNK3 perpetuates metabolic stress induced by Abeta peptides. Neuron. 2012;75:824–837. doi: 10.1016/j.neuron.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pardossi-Piquard R, Checler F. The physiology of the beta-amyloid precursor protein intracellular domain AICD. J Neurochem. 2012;120(Suppl 1):109–124. doi: 10.1111/j.1471-4159.2011.07475.x. [DOI] [PubMed] [Google Scholar]
- 94.Takahashi K, Niidome T, Akaike A, Kihara T, Sugimoto H. Amyloid precursor protein promotes endoplasmic reticulum stress-induced cell death via C/EBP homologous protein-mediated pathway. J Neurochem. 2009;109:1324–1337. doi: 10.1111/j.1471-4159.2009.06067.x. [DOI] [PubMed] [Google Scholar]
- 95.Kögel D, Concannon CG, Müller T, König H, Bonner C, Poeschel S, Chang S, Egensperger R, Prehn JH. The APP intracellular domain (AICD) potentiates ER stress-induced apoptosis. Neurobiol Aging. 2012;33:2200–2209. doi: 10.1016/j.neurobiolaging.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 96.Guo Q, Robinson N, Mattson MP. Secreted beta-amyloid precursor protein counteracts the proapoptotic action of mutant presenilin-1 by activation of NF-kappaB and stabilization of calcium homeostasis. J Biol Chem. 1998;273:12341–12351. doi: 10.1074/jbc.273.20.12341. [DOI] [PubMed] [Google Scholar]
- 97.Eckert GP, Chang S, Eckmann J, Copanaki E, Hagl S, Hener U, Müller WE, Kögel D. Liposome- incorporated DHA increases neuronal survival by enhancing non-amyloidogenic APP processing. Biochim Biophys Acta. 2011;1808:236–243. doi: 10.1016/j.bbamem.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 98.Unterberger U, Höftberger R, Gelpi E, Flicker H, Budka H, Voigtländer T. Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. J Neuropathol Exp Neurol. 2006;65:348–357. doi: 10.1097/01.jnen.0000218445.30535.6f. [DOI] [PubMed] [Google Scholar]
- 99.Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol. 2009;174:1241–1251. doi: 10.2353/ajpath.2009.080814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nijholt DA, van Haastert ES, Rozemuller AJ, Scheper W, Hoozemans JJ. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J Pathol. 2012;226:693–702. doi: 10.1002/path.3969. [DOI] [PubMed] [Google Scholar]
- 101.Ho YS, Yang X, Lau JC, Hung CH, Wuwongse S, Zhang Q, Wang J, Baum L, So KF, Chang RC. Endoplasmic reticulum stress induces tau pathology and forms a vicious cycle: implication in Alzheimer’s disease pathogenesis. J Alzheimers Dis. 2012;28:839–854. doi: 10.3233/JAD-2011-111037. [DOI] [PubMed] [Google Scholar]
- 102.Spatara ML, Robinson AS. Transgenic mouse and cell culture models demonstrate a lack of mechanistic connection between endoplasmic reticulum stress and tau dysfunction. J Neurosci Res. 2010;88:1951–1961. doi: 10.1002/jnr.22359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Abisambra JF, Jinwal UK, Blair LJ, O’Leary JC 3rd, Li Q, Brady S, Wang L, Guidi CE, Zhang B, Nordhues BA, Cockman M, Suntharalingham A, Li P, Jin Y, Atkins CA, Dickey CA. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J Neurosci. 2013;33:9498–9507. doi: 10.1523/JNEUROSCI.5397-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fu ZQ, Yang Y, Song J, Jiang Q, Lin ZC, Wang Q, Zhu LQ, Wang JZ, Tian Q. LiCl attenuates thapsigargin-induced tau hyperphosphorylation by inhibiting GSK-3beta in vivo and in vitro. J Alzheimers Dis. 2010;21:1107–1117. doi: 10.3233/jad-2010-100687. [DOI] [PubMed] [Google Scholar]
- 105.Sakagami Y, Kudo T, Tanimukai H, Kanayama D, Omi T, Horiguchi K, Okochi M, Imaizumi K, Takeda M. Involvement of endoplasmic reticulum stress in tauopathy. Biochem Biophys Res Commun. 2013;430:500–504. doi: 10.1016/j.bbrc.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 106.Resende R, Ferreiro E, Pereira C, Oliveira CR. ER stress is involved in Abeta-induced GSK-3beta activation and tau phosphorylation. J Neurosci Res. 2008;86:2091–2099. doi: 10.1002/jnr.21648. [DOI] [PubMed] [Google Scholar]
- 107.Liu ZC, Fu ZQ, Song J, Zhang JY, Wei YP, Chu J, Han L, Qu N, Wang JZ, Tian Q. Bip enhanced the association of GSK-3beta with tau during ER stress both in vivo and in vitro. J Alzheimers Dis. 2012;29:727–740. doi: 10.3233/JAD-2012-111898. [DOI] [PubMed] [Google Scholar]
- 108.Nogalska A, Engel WK, Askanas V. Increased BACE1 mRNA and noncoding BACE1-antisense transcript in sporadic inclusion-body myositis muscle fibers--possibly caused by endoplasmic reticulum stress. Neurosci Lett. 2010;474:140–143. doi: 10.1016/j.neulet.2010.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Page G, Rioux Bilan A, Ingrand S, Lafay-Chebassier C, Pain S, Perault Pochat MC, Bouras C, Bayer T, Hugon J. Activated double-stranded RNA-dependent protein kinase and neuronal death in models of Alzheimer’s disease. Neuroscience. 2006;139:1343–1354. doi: 10.1016/j.neuroscience.2006.01.047. [DOI] [PubMed] [Google Scholar]
- 110.O’Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hébert SS, de Strooper B, Haass C, Bennett DA, Vassar R. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ill-Raga G, Palomer E, Wozniak MA, Ramos-Fernández E, Bosch-Morató M, Tajes M, Guix FX, Galán JJ, Clarimón J, Antúnez C, Real LM, Boada M, Itzhaki RF, Fandos C, Muñoz FJ. Activation of PKR causes amyloid ss-peptide accumulation via de-repression of BACE1 expression. PLoS One. 2011;6:e21456. doi: 10.1371/journal.pone.0021456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lammich S, Schöbel S, Zimmer AK, Lichtenthaler SF, Haass C. Expression of the Alzheimer protease BACE1 is suppressed via its 5’-untranslated region. EMBO Rep. 2004;5:620–625. doi: 10.1038/sj.embor.7400166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mouton-Liger F, Paquet C, Dumurgier J, Bouras C, Pradier L, Gray F, Hugon J. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2alpha pathway. Biochim Biophys Acta. 2012;1822:885–896. doi: 10.1016/j.bbadis.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 114.Dumurgier J, Mouton-Liger F, Lapalus P, Prevot M, Laplanche JL, Hugon J, Paquet C. Cerebrospinal fluid PKR level predicts cognitive decline in Alzheimer’s disease. PLoS One. 2013;8:e53587. doi: 10.1371/journal.pone.0053587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mitsuda T, Hayakawa Y, Itoh M, Ohta K, Nakagawa T. ATF4 regulates gamma-secretase activity during amino acid imbalance. Biochem Biophys Res Commun. 2007;352:722–727. doi: 10.1016/j.bbrc.2006.11.075. [DOI] [PubMed] [Google Scholar]
- 116.Ohta K, Mizuno A, Li S, Itoh M, Ueda M, Ohta E, Hida Y, Wang MX, Furoi M, Tsuzuki Y, Sobajima M, Bohmoto Y, Fukushima T, Kobori M, Inuzuka T, Nakagawa T. Endoplasmic reticulum stress enhances gamma-secretase activity. Biochem Biophys Res Commun. 2011;416:362–366. doi: 10.1016/j.bbrc.2011.11.042. [DOI] [PubMed] [Google Scholar]
- 117.Koyama Y, Matsuzaki S, Gomi F, Yamada K, Katayama T, Sato K, Kumada T, Fukuda A, Matsuda S, Tano Y, Tohyama M. Induction of amyloid beta accumulation by ER calcium disruption and resultant upregulation of angiogenic factors in ARPE19 cells. Invest Ophthalmol Vis Sci. 2008;49:2376–2383. doi: 10.1167/iovs.07-1067. [DOI] [PubMed] [Google Scholar]
- 118.Saftig P, Reiss K. The “A Disintegrin And Metalloproteases” ADAM10 and ADAM17: novel drug targets with therapeutic potential? Eur J Cell Biol. 2011;90:527–535. doi: 10.1016/j.ejcb.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 119.Rzymski T, Petry A, Kračun D, Riess F, Pike L, Harris AL, Görlach A. The unfolded protein response controls induction and activation of ADAM17/TACE by severe hypoxia and ER stress. Oncogene. 2012;31:3621–3634. doi: 10.1038/onc.2011.522. [DOI] [PubMed] [Google Scholar]
- 120.Reinhardt S, Schuck F, Grösgen S, Riemenschneider M, Hartmann T, Postina R, Grimm M, Endres K. Unfolded protein response signaling by transcription factor XBP-1 regulates ADAM10 and is affected in Alzheimer’1(1):s disease. FASEB J. 2013 doi: 10.1096/fj.13-234864. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 121.Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010;29:3020–3032. doi: 10.1038/emboj.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, Snellinx A, Craessaerts K, Thathiah A, Tesseur I, Bartsch U, Weskamp G, Blobel CP, Glatzel M, de Strooper B, Saftig P. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci. 2010;30:4833–4844. doi: 10.1523/JNEUROSCI.5221-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chaimowitz NS, Kang DJ, Dean LM, Conrad DH. ADAM10 regulates transcription factor expression required for plasma cell function. PLoS One. 2012;7:e42694. doi: 10.1371/journal.pone.0042694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Elder GA, Gama Sosa MA, de Gasperi R. Transgenic mouse models of Alzheimer’s disease. Mt Sinai J Med. 2010;77:69–81. doi: 10.1002/msj.20159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Abisambra JF, Blair LJ, Hill SE, Jones JR, Kraft C, Rogers J, Koren J 3rd, Jinwal UK, Lawson L, Johnson AG, Wilcock D, O’Leary JC, Jansen-West K, Muschol M, Golde TE, Weeber EJ, Banko J, Dickey CA. Phosphorylation dynamics regulate Hsp27-mediated rescue of neuronal plasticity deficits in tau transgenic mice. J Neurosci. 2010;30:15374–15382. doi: 10.1523/JNEUROSCI.3155-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 128.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004;165:1289–1300. doi: 10.1016/s0002-9440(10)63388-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hoozemans JJ, Veerhuis R, van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, Scheper W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005;110:165–172. doi: 10.1007/s00401-005-1038-0. [DOI] [PubMed] [Google Scholar]
- 132.Teske BF, Fusakio ME, Zhou D, Shan J, McClintick JN, Kilberg MS, Wek RC. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol Biol Cell. 2013;24:2477–2490. doi: 10.1091/mbc.E13-01-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Soejima N, Ohyagi Y, Nakamura N, Himeno E, Iinuma KM, Sakae N, Yamasaki R, Tabira T, Murakami K, Irie K, Kinoshita N, Laferla FM, Kiyohara Y, Iwaki T, Kira J. Intracellular accumulation of toxic turn amyloid-beta is associated with endoplasmic reticulum stress in Alzheimer’s disease. Curr Alzheimer Res. 2013;10:11–20. [PubMed] [Google Scholar]
- 134.Oddo S, Caccamo A, Cheng D, Jouleh B, Torp R, Laferla FM. Genetically augmenting tau levels does not modulate the onset or progression of Abeta pathology in transgenic mice. J Neurochem. 2007;102:1053–1063. doi: 10.1111/j.1471-4159.2007.04607.x. [DOI] [PubMed] [Google Scholar]
- 135.Umeda T, Tomiyama T, Sakama N, Tanaka S, Lambert MP, Klein WL, Mori H. Intraneuronal amyloid beta oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J Neurosci Res. 2011;89:1031–1042. doi: 10.1002/jnr.22640. [DOI] [PubMed] [Google Scholar]
- 136.Tomiyama T, Matsuyama S, Iso H, Umeda T, Takuma H, Ohnishi K, Ishibashi K, Teraoka R, Sakama N, Yamashita T, Nishitsuji K, Ito K, Shimada H, Lambert MP, Klein WL, Mori H. A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J Neurosci. 2010;30:4845–4856. doi: 10.1523/JNEUROSCI.5825-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.de La Monte SM, Re E, Longato L, Tong M. Dysfunctional pro-ceramide, ER stress and insulin/ IGF signaling networks with progression of Alzheimer’s disease. J Alzheimers Dis. 2012;30(Suppl 2):S217–S229. doi: 10.3233/JAD-2012-111728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Casas-Tinto S, Zhang Y, Sanchez-Garcia J, Gomez- Velazquez M, Rincon-Limas DE, Fernandez- Funez P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum Mol Genet. 2011;20:2144–2160. doi: 10.1093/hmg/ddr100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Honjo Y, Ito H, Horibe T, Takahashi R, Kawakami K. Protein disulfide isomerase-immunopositive inclusions in patients with Alzheimer disease. Brain Res. 2010;1349:90–96. doi: 10.1016/j.brainres.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 140.Kaneko M, Koike H, Saito R, Kitamura Y, Okuma Y, Nomura Y. Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-beta generation. J Neurosci. 2010;30:3924–3932. doi: 10.1523/JNEUROSCI.2422-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Safra M, Ben-Hamo S, Kenyon C, Henis-Korenblit S. The ire-1 ER stress-response pathway is required for normal secretory-protein metabolism in C. elegans. J Cell Sci. 2013;126:4136–4146. doi: 10.1242/jcs.123000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Taylor RC, Dillin A. XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell. 2013;153:1435–1447. doi: 10.1016/j.cell.2013.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Halliday M, Mallucci GR. Targeting the unfolded protein response in neurodegeneration: A new approach to therapy. Neuropharmacology. 2014;76:169–174. doi: 10.1016/j.neuropharm.2013.08.034. [DOI] [PubMed] [Google Scholar]
- 144.Chadwick W, Mitchell N, Martin B, Maudsley S. Therapeutic targeting of the endoplasmic reticulum in Alzheimer’s disease. Curr Alzheimer Res. 2012;9:110–119. doi: 10.2174/156720512799015055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rochet JC. Novel therapeutic strategies for the treatment of protein-misfolding diseases. Expert Rev Mol Med. 2007;9:1–34. doi: 10.1017/S1462399407000385. [DOI] [PubMed] [Google Scholar]
- 146.Valera E, Dargusch R, Maher PA, Schubert D. Modulation of 5-lipoxygenase in proteotoxicity and Alzheimer’s disease. J Neurosci. 2013;33:10512–10525. doi: 10.1523/JNEUROSCI.5183-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lassot I, Estrabaud E, Emiliani S, Benkirane M, Benarous R, Margottin-Goguet F. p300 modulates ATF4 stability and transcriptional activity independently of its acetyltransferase domain. J Biol Chem. 2005;280:41537–41545. doi: 10.1074/jbc.M505294200. [DOI] [PubMed] [Google Scholar]
- 148.Uemura A, Taniguchi M, Matsuo Y, Oku M, Wakabayashi S, Yoshida H. UBC9 regulates the stability of XBP1, a key transcription factor controlling the ER stress response. Cell Struct Funct. 2013;38:67–79. doi: 10.1247/csf.12026. [DOI] [PubMed] [Google Scholar]
- 149.Park SW, Zhou Y, Lee J, Lu A, Sun C, Chung J, Ueki K, Ozcan U. The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP-1 and increase its nuclear translocation. Nat Med. 2010;16:429–437. doi: 10.1038/nm.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Winnay JN, Boucher J, Mori MA, Ueki K, Kahn CR. A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat Med. 2010;16:438–445. doi: 10.1038/nm.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Carrara M, Prischi F, Ali MM. UPR Signal Activation by Luminal Sensor Domains. Int J Mol Sci. 2013;14:6454–6466. doi: 10.3390/ijms14036454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hai T, Curran T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci U S A. 1991;88:3720–3724. doi: 10.1073/pnas.88.9.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chérasse Y, Maurin AC, Chaveroux C, Jousse C, Carraro V, Parry L, Deval C, Chambon C, Fafournoux P, Bruhat A. The p300/CBP-associated factor (PCAF) is a cofactor of ATF4 for amino acid-regulated transcription of CHOP. Nucleic Acids Res. 2007;35:5954–5965. doi: 10.1093/nar/gkm642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lyckman AW, Confaloni AM, Thinakaran G, Sisodia SS, Moya KL. Post-translational processing and turnover kinetics of presynaptically targeted amyloid precursor superfamily proteins in the central nervous system. J Biol Chem. 1998;273:11100–11106. doi: 10.1074/jbc.273.18.11100. [DOI] [PubMed] [Google Scholar]