

Abstract
Aquatic organisms such as cichlids, coelacanths, seals, and cetaceans are active in UV-blue color environments, but many of them mysteriously lost their abilities to detect these colors. The loss of these functions is a consequence of the pseudogenization of their short wavelength-sensitive (SWS1) opsin genes without gene duplication. We show that the SWS1 gene (BdenS1ψ) of the deep-sea fish, pearleye (Benthalbella dentata), became a pseudogene in a similar fashion about 130 million years ago (Mya) yet it is still transcribed. The rates of nucleotide substitution (~1.4 × 10−9 /site/year) of the pseudogenes of these aquatic species as well as some prosimian and bat species are much smaller than the previous estimates for the globin and immunoglobulin pseudogenes.
Keywords: Aquatic animals, SWS1 pseudogenes, Molecular evolution
1. Introduction
The high level of DNA sequence variations found in nature has been explained by both neutral mutations (Nei 2005; Nei et al. 2010) and by adaptive mutations (Arbiza et al. 2006; Bakewell et al. 2007; Kosiol et al. 2008; Studer et al. 2008; Lindblad-Toh et al. 2011). The extremely high rates of nucleotide substitution (~5–13 × 10−9 /site/year) of pseudogenes have been used as strong supportive evidence for the neutral theory of molecular evolution (Li et al. 1981; Miyata and Yasunaga 1981; Nei 2005; Nei et al. 2010). However, recent molecular analyses of certain pseudogenes reveal that their presumed non-functionality is equivocal (Balakirev and Ayala 2003; Podlaha and Zhang 2010). The ENCODE team goes further by saying that more than two thirds of non-coding DNA sequences in the human genome are transcribed and have biochemical functions (The ENCODE Project Consortium 2012; Djebali et al. 2012). Yet, such pseudogenes are still subjected to much less selective constraint than protein-coding genes (Pei et al. 2012). Under these circumstances, it is of interest to re-evaluate the evolutionary rates of pseudogenes.
Many aquatic animals such as cichlids (e. g., Neolamprologus brichardi and N. mondabu) (O’Quin et al. 2010), coelacanths (Latimeria chalumnae and L. menadoensis) (Yokoyama et al. 1999; Yokoyama and Tada 2000), seals (Phoca groenlandica and P. vitulina), dolphin (Tursiops truncatus) (Newman and Robinson 2005), and whales (Globicephala melas, Mesoplodon densirostris, and Megaptera novaeangliae) (Newman and Robinson 2005; Koito et al. 2010) are active in UV-blue color environments, but they lost their abilities to make the UV/blue-sensitive or short wavelength-sensitive (SWS1) visual pigments. Their opsin genes have not been isolated, but many shark species have also lost the ability to make the SWS1 pigments (Hart et al. 2011).
Here, we isolated the SWS1 opsin gene from the deep-sea fish, pearleye (Benthalbella dentata). This gene (BdenS1ψ) contains premature stop codons, the typical characteristic of a pseudogene, but it is transcribed in the retina. Comparing the SWS1 pseudogenes of the pearleye, cichlids, coelacanths, cetaceans, seals, prosimians (Galago senegalensis and Nycticebus coucang) (Kawamura and Kubotera 2004), and bats (Rhinolophus affinis and Rhinolophus ferrumequinum) (Zhao et al. 2009) with opsin-coding SWS1 genes, we study the evolutionary rates of nucleotide substitution before and after pseudogenization.
2. Materials and Methods
2.1. Molecular cloning of the pearleye SWS1 gene
High molecular weight DNAs of the pearleye (B. dentate) was isolated from body tissues using a standard phenol-chloroform extraction procedure (e.g. Yokoyama et al. 1999). BdenS1ψ was cloned by polymerase chain reaction (PCR) first using a set of degenerate primers (F: 5′-GCNTCNACNCARAARGCNGA-3′ and R: 5′-ACRTANATNAYNGGRTTRTA-3′) and, then, by inverse PCR using another set of primers (F: 5′-GTGCACTTCTGAAGG-3′ and R: 5′-GGAGCCCACCGTCATCACG-3′). The PCR was performed by 30 cycles at 92°C for 45 sec, 55°C for 60 sec, and 72°C for 90 sec. At each cycle, the duration of the extension reaction was progressively extended by 3 sec.
The total retinal RNAs from the pearleye retina was also isolated as described previously (Yokoyama et al. 1995). To clone the SWS1 opsin cDNA of the pearleye, the internal sequence was cloned first by RT-PCR using the degenerate primers used for obtaining the first genomic sequence. To determine the rest of the cDNA sequences, we constructed additional gene specific primers (GSPs) and performed 5′ and 3′ rapid amplification of cDNA ends (RACE) analyses (e.g. Yokoyama et al. 1995). For the 3′ RACE, the first strand cDNA was made using the oligo (dT)-containing adaptor primer (AP), provided by the manufacturer (Gibco BRL, Gaitherburg, MD) and the original mRNA was degraded by RNase H. Then, two sequential PCR amplifications were performed applying two sets of GSPs and universal adapter primer (UAPs) to these cDNAs, first using (GSP1: 5′-CCGACGAGAACAAAGACTACCG-3′ and GSP2: 5′-CCATTCCAGCATTCTTCTCC-3′) with abridged UAPs supplied by the manufacturer. For the 5′ RACE, the cDNAs were first synthesized from total RNA using GSP1 (5′-CGGTAGTCTTTGTTCTCGTCGG-3′). The entire coding region was obtained by two sequential PCR amplifications, using two sets of nested GSPs (GSP2: 5′-AAGTACAACGCTGCGATGGC-3′ and GSP3: 5′-GGAGCCCACCGTCATCACG-3′) and abridged UAPs. Using these primers, cDNAs were reverse transcribed at 42°C for 1 hr, 95°C for 5 min and then PCR amplification was carried out for 30 cycles at 94°C for 45 sec, 55°C for 1.5 min, and 72°C for 2 min.
Nucleotide sequences of these cDNA clones were determined by cycle sequencing reactions using the Sequitherm Excel II long-read kits (Epicentre Technologies, Madison, WI) with dye-labeled M13 forward and reverse primers. Reactions were run on a LI-COR (Lincoln, NE) 4200LD automated DNA sequencer.
2.2. Inferences on phylogenetic trees and positive selection
In the analyses, we have aligned the nucleotide sequences of a total of 33 SWS1 opsin genes. The codons that are not shared by the functional SWS1 genes and pseudogenes were excluded (see Fig. S1). Then, the numbers of nucleotide substitutions per site (d) for pairwise comparisons were estimated by d = − (3/4) ln [1 − (4/3)p], where p is the proportion of different nucleotide per site (Jukes and Cantor 1969). The branch lengths of the composite phylogenetic tree of the 33 representative SWS1 opsin genes were inferred by PAML (Yang 2007) using the evolutionarily distantly-related RH1 gene of bovine (M21606) as well as RH2 (AB087805) and SWS2 (AB087809) genes of zebrafish as the outgroup.
A rooted phylogenetic tree of the SWS1 pseudogene (BdenS1ψ) and the RH1 (SanaRH1A and SanaRH1B) and RH2 (SanaRH2) genes of another pearleye species (Scopelarchus analis) (Table S1) was constructed applying the neighbor-joining (NJ) method (Saitou and Nei 1987) to their DNA sequences and those of the LWS opsin genes of zebrafish (AB087803) and goldfish (L11867) as the outgroup.
To search for positively selected amino acid sites, we studied the ratio of non-synonymous and synonymous nucleotide substitutions using the codon-based maximum likelihood (ML) based Bayesian method (Yang 2007). We considered the naive empirical Bayes (NEB) method without accounting sampling errors and Bayes empirical Bayes (BEB) method with accounting sampling errors (Yang et al. 2005). These Bayesian methods were applied to the SWS1 pseudogenes of 29 representative cetacean and 2 seal species (Table S1 and Fig. S2) using two initial ω values (0.4 and 3.4).
2.3. An evolutionary model for the SWS1 opsin genes
Using the two closely related SWS1 pseudogenes of coelacanths, seals, cetaceans, prosimians, and bats (sequences A and B) with known divergence times and two orthologous functional genes (sequences C and D), the times since pseudogenization (Tn) can be evaluated (Fig. 1A). In Fig. 1A, a and a′ describe the evolutionary rates of nucleotide substitution of historically younger and older groups of functional genes, respectively, while the parameter b denotes the evolutionary rate of the pseudogenes. For each data set of this four-sequence model, the numbers of nucleotide substitutions per site (dXY) for pairwise comparisons were estimated by dXY = − (3/4) ln [1 − (4/3) pXY], where pXY is again the proportion of different nucleotide per site (X, Y = A, B, C, D). Then, the relationships dAB = 2bT3, dAC = dBC = a(2T2 − Tn) + bTn, dAD = dBD = a′(2T1 − T2) + aT2 + (b − a)Tn, and dCD = a′(2T1 − T2) + aT2 hold. Hence, parameters a, a′, b, and Tn can be evaluated by
Fig. 1.
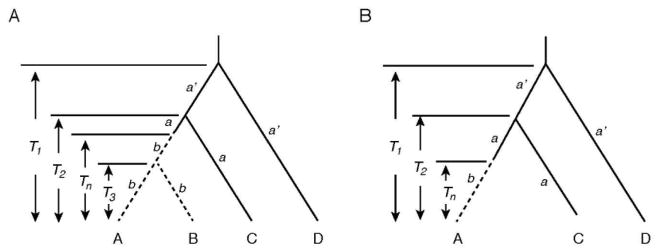
Plausible phylogenetic tree for pseudogenes and orthologous functional genes. (A) A tree consisting of two pseudogenes (sequences A and B) and two functional genes (sequences C and D). T1, T2, and T3 denote divergence times between sequence D and others, between sequence C and sequence A (or B), and between sequences A and B, respectively, and Tn the time of pseudogenization of sequences A and B. (B) A tree consisting of one pseudogene (sequence A) and two functional genes (sequences C and D).
| (1a) |
| (1b) |
| (1c) |
| (1d) |
When another closely related pseudogene is not known (pearleye) or the divergence time of the two known pseudogenes has not been determined (cichlids), Tn can be evaluated using the formula considering the three sequence model (Fig. 1B): Tn = [(y1 + y2)/2 − y3)/[a3 − (a1 + a2)/2], where yi = dADi − dCDi (or dBDi − dCDi) and ai is the rate of change at the ith position of a codon (i = 1, 2, 3) (Li et al. 1981).
2.4. Method for estimating the variance of Tn
A resampling method based on using model generated population values was used to estimate the variance of Tn. Each resample used randomized branch lengths taken from the binomial distribution to calculate Tn values. For the three-sequence model there are three independent branch lengths (li, mi and ni in Li et al. 1981). In the four-sequence model there are five independent branches. Each of these lengths was used as the p parameter of a binomial distributed variate [k~B(N,p)] where N is the number of nucleotides compared and p (or d) is the proportion of changes in that branch. Replicate (1000) sets of randomized branch lengths (k/N) were used to generate distances (dAB, dAC, dAD, dBC, dBD, dCD) that were then used to calculate values of Tn according to equation (1d) in the four sequence model, or (6) in Li et al. (1981). The replicated values of Tn were used to estimate the variance of Tn. This procedure is analogous to the commonly used bootstrapping procedure where branch lengths are randomized by resampling the original sequence data (non-parametric bootstrapping), however, in the present case we used parametric bootstrapping by assuming the binomial is the appropriate distribution for the branch length observations.
3. Results
3.1. The genomic and cDNA sequences of the BdenS1ψ
The pearleye SWS1 gene (BdenS1ψ) is characterized by the deletions of two stretches of its DNA sequence: compared with a typical functional SWS1 gene (SleuS1) of the lampfish (Table S1), 1) the segments between codons 203 and 220 and the entire intron 4 are missing from BdenS1ψ and 2) the initiation codon ATG was replaced by ATA and a single nucleotide insertion and deletion can be found between codons 47 and 48 and at codon 224, respectively (Fig. 2A). These structural changes introduce several premature stop codons into BdenS1ψ (Fig. 2B).
Fig. 2.

The SWS1 opsin gene of the pearleye (BdenS1ψ). (A) The nucleotide sequences of the opsin cDNA (without introns) and genomic DNA (with introns). The deleted nucleotides in BdenS1ψ are shown by dashes (−). The codon sites follow those of the closely related functional SWS1 opsin gene of the lampfish (SleuS1). (B) The amino acid sequence deduced from the BdenS1ψ. Stars (*) indicate stop codons.
3.2. Branch lengths of the composite phylogenetic tree of the SWS1 opsin genes
For a total of 33 species, consisting of 15 species with the SWS1 pseudogenes and 18 representative species with the orthologous opsin-coding genes (Fig. 3A and Table S1), we first established a tree topology using “TimeTree of Life” web (www.timetree.org) server. This was done to avoid obtaining an erroneous tree topology that may be caused by the possibility of long branches of pseudogenes. This composite evolutionary tree shows that the SWS1 genes have become pseudogenes independently along seven separate lineages (pearleye, cichlids, coelacanths, bats, prosimians, cetaceans, and seals). Based on this tree topology and considering all positions of codons common to the 33 SWS1 genes, we evaluated the lengths of various branches (Fig. 3A).
Fig. 3.

Phylogenetic tree of SWS1 opsin genes. (A) The ML tree of 33 representative genes constructed using the program PAML. (For the names of specific species and the source of the sequence data, see table S1.) (B) The NJ tree of SWS1 BdenS1ψ, BdenRH1A, BdenRH1B, and BdenRH2 of the pearleye was constructed by using the NJ method, in which the time scale was based on the a value for the coelacanth data set.
Much to our surprise, we could not find extraordinarily long branches leading to the pseudogenes, which was unexpected from the previous results on the evolutionary rates of the globin and immunoglobulin pseudogenes (Li et al. 1981; Miyata and Hayashida 1981; Miyata and Yasunaga 1981). In particular, the branch leading to the coelacanth pseudogenes is the shortest among the 33 SWS1 genes. This ML tree has a striking resemblance to the phylogenetic tree based on the 251 concatenated protein-coding genes of various vertebrates (Amemiya et al. 2013), in which the coelacanth has the shortest branch length. In the coelacanth, therefore, both the SWS1 pseudogene and protein-coding genes have evolved very slowly. Similarly, the branch lengths of the pseudogenes of pearleye, cichlids, cetaceans, and seals are similar to those of the closely-related opsin-coding genes in other species. On the other hand, the pseudogenes of some bat species and prosimians (galago and loris) have much longer branch lengths than those of closely related opsin-coding genes; however, when they are compared to the orhtologous human gene, the branch length differences become less prominent.
3.3. Divergence times and evolutionary rates
Considering the seven sets of pseudogenes with orthologous opsin-coding SWS1 genes separately, we estimated the evolutionary rates of nucleotide substitution before (a) and after pseudogenizations (b) as well as the time of pseudogenization (Tn) by considering four sequence and three sequence models separately (see Section 2.3).
For the coelacanth, seal, cetacean, prosimian, and bat pseudogene data, divergence times (T1, T2, and T3) have been estimated by using the “TimeTree of Life” web server (www.timetree.org). Using these divergence times, Tn values vary from 16 Mya of the seal genes to 138 Mya of the coelacanth pseudogenes; for the cichlid and pearleye pseudogenes, Tn values are given by 16 and 134 Mya, respectively (Table 1).
Table 1.
Estimated times of pseudogenization (Tn).
| Group | Sequences | Divergence times (Mya)
|
|||
|---|---|---|---|---|---|
| T1a | T2a | T3a | Tn | ||
| coelacanths | A: LchaS1ψ, B: LmenS1ψ, C: XlaeS1; AcarS1; ClivS1; HsapS1, D: DrerS1; SsalS1 | 455 | 430 | 5.5 | 138 ± 119d |
| seals | A: PvitS1ψ, B: PgroS1ψ, C: CfamS1, D: BtauS1; SscrS1; HsapS1 | 89 | 44 | 7.8 | 16 ± 7d |
| cetaceans | A: MnovS1ψ, B: TtruS1ψ; GmelS1ψ; MdenS1ψ, C: BtauS1; SscrS1, D: HsapS1 | 91 | 61 | 32.3 | 61e |
| prosimians | A: GsenS1ψ NcouS1ψ, C: DmadS1; EfulS1, D: BtauS1; SscrS1; CfamS1 | 97 | 62 | 34.2 | 62e |
| bats | A: RferS1ψ RaffS1ψ, C: TausS1; MfulS1, D: CfamS1; BtauS1; SscrS1 | 83 | 62 | 17 | 50f |
| cichlids | A: NmonS1ψ; NbriS1ψ OnilS1, D: OlatS1; PretS1 | 104b | 36b | ? | 16g |
| pearleye | A: BdenS1ψ, C: SleuS1; OnilS1, D: DrerS1; SsalS1 | 284c | 160c | ? | 134 ± 30h |
The divergence times have been evaluated from TimeTree of Life (www.timetree.org).
The divergence times are taken from Genner et al. (2007).
The average divergence time between pearleye and lampfish (307 Mya) and between pearleye and tilapia (264 Mya).
The standard errors were estimated by a parametric Monte Carlo resampling (or the parametric bootstrap) method.
We were unable to compute proper standard errors because Tn was assumed to be equal to T2.
We were unable to compute proper standard errors because Tn was assumed to be equal to the divergence time (50 Mya) between the pseudogenes from Rhinolophus affinis (and Rhinolophus ferrumequinum) and the closely-related orthologous functional gene from Megaderma spasma (www.timetree.org)).
The data set used were dAC1 = 0.045, dAD1 = 0.125, dCD1 = 0.115, dAC2 = 0.022, dAD2 = 0.087, dCD2 = 0.067, dAC3 = 0.120, dAD3 = 0.505, and dCD3 = 0.481.
The data set used were dAC1 = 0.137, dAD1 = 0.221, dCD1 = 0.178, dAC2 = 0.090, dAD2 = 0.127, dCD2 = 0.094, dAC3 = 0.584, dAD3 = 0.677, and dCD3 = 0.795.
?: unknown.
The four-sequence analyses show that a values vary between 0.40 and 0.77 × 10−9 /site/year, while b varies between 0.84–1.65 × 10−9 /site/year, respectively (Table 2, the last column). The reliabilities of formulae (1a), (1c), and (1d) based on the four-sequence model can be tested by using the three-sequence model with the Tn values estimated and relationships ai = (dACi − dADi + dCDi)/(2T2) and bi = [dACi − ai (2T2 − Tn)]/Tn (i = 1, 2, and 3). The results show that the average values (a) of a1, a2, and a3 vary between 0.47 and 0.89 × 10−9 /site/year with the overall average of 0.65 × 10−9 /site/year; similarly, the average values (b) of b1, b2, and b3 vary between 0.80 and 2.0 × 10−9 /site/year with the overall average of 1.37 × 10−9 /site/year (Table 2). These a and b values are very similar to the corresponding a and b values estimated using formulae (1a) and (1c), respectively, justifying the use of these formulae. In addition, despite significant differences between T3 and Tn (Table 1), the evolutionary rates of the pseudogenes estimated using the three- and four-sequence models (b vs b) are similar, again justifying the use of formulae (1c) and (1d) in estimating b and Tn, respectively.
Table 2.
Evolutionary rates of nucleotide substitution of SWS1 opsin genes.
| Group | Evolutionary rates (× 10−9) | All positions (× 10−9) | ||||
|---|---|---|---|---|---|---|
| a1 | a2 | a3 | a1+2 | a | a | |
| coelacanths | 0.27 ± 0.05 | 0.15 ± 0.04** | 0.99 ± 0.13**,†† | 0.21 ± 0.03†† | 0.47 ± 0.04 | 0.40 |
| seals | 0.05 ± 0.06** | 0.24 ± 0.13 | 1.46 ± 0.33**,†† | 0.15 ± 0.07†† | 0.58 ± 0.12 | 0.56 |
| cetaceans | 0.31 ± 0.13 | 0.16 ± 0.09** | 1.41 ± 0.28**,†† | 0.24 ± 0.08†† | 0.63 ± 0.11 | 0.60 |
| prosimians | 0.44 ± 0.15 | 0.24 ± 0.11** | 0.93 ± 0.22**,† | 0.34 ± 0.09† | 0.54 ± 0.10 | 0.52 |
| bats | 0.53 ± 0.20 | 0.25 ± 0.13** | 1.61 ± 0.35**,†† | 0.39 ± 0.12†† | 0.80 ± 0.24 | 0.77 |
| cichlids | 0.48 ± 0.20 | 0.02 ± 0.04** | 1.34 ± 0.34**,†† | 0.25 ± 0.10†† | 0.61 ± 0.13 | ND |
| pearleye | 0.29 ± 0.08 | 0.18 ± 0.06** | 2.19 ± 0.26**,†† | 0.23 ± 0.05†† | 0.89 ± 0.08 | ND |
|
|
|
|||||
| Average | 0.34 ± 0.04 | 0.18 ± 0.03** | 1.42 ± 0.19**,†† | 0.25 ± 0.02†† | 0.65 ± 0.07 | 0.57 |
| b1 | b2 | b3 | b1+2 | b | b | |
| coelacanths | 1.13 ± 0.20 | 0.52 ± 0.13** | 1.25 ± 0.22** | 0.83 ± 0.12 | 0.97 ± 0.11 | 0.94 |
| seals | 2.57 ± 0.71* | 1.06 ± 0.45* | 1.20 ± 0.47 | 1.81 ± 0.42 | 1.61 ± 0.32 | 1.65 |
| cetaceans | 0.65 ± 0.19 | 0.75 ± 0.21 | 1.30 ± 0.28† | 0.68 ± 0.14† | 0.90 ± 0.13 | 0.84 |
| prosimians | 1.88 ± 0.33 | 1.12 ± 0.25 | 1.65 ± 0.30 | 1.50 ± 0.20 | 1.55 ± 0.17 | 1.26 |
| bats | 1.22 ± 0.34* | 2.76 ± 0.53* | 2.03 ± 0.44 | 1.99 ± 0.31 | 2.00 ± 0.44 | 1.61 |
| cichlids | 1.12 ± 0.47 | 1.29 ± 0.50 | 2.82 ± 0.75 | 1.22 ± 0.34 | 1.74 ± 0.34 | ND |
| pearleye | 0.56 ± 0.11 | 0.39 ± 0.09** | 1.46 ± 0.20**,† | 0.52 ± 0.08† | 0.80 ± 0.08 | ND |
|
|
|
|||||
| Average | 1.31 ± 0.16 | 1.13 ± 0.13 | 1.65 ± 0.25 | 1.22 ± 0.10 | 1.37 ± 0.17 | 1.26 |
The largest difference among ai or bi (i = 1, 2, 3) is significantly different at the 5% (*) and 1% level (**). The difference between a1+2 and a3 or between b1+2 and b3 is significantly different at the 5% (†) and 1% level (††). ND: not determined.
In these analyses, the numbers of nucleotide substitutions per site (d) were estimated using the Jukes and Cantor (JC) method (Section 2.2), which underestimates the d value as the divergence time of a pair of sequences increases. However, the d values for the SWS1 gene pairs are usually much smaller than 1.0 and the JC method gives reasonably accurate Tn, a, and b values. For example, Tamura and Nei’s (1993) method corrects the underestimation very effectively (see Fig. 3.1 in Nei and Kumar 2000). For the coelacanth data, for example, Tn (138 Mya), a (0.40 × 10−9) and b (0.94 × 10−9) values obtained using Tamura and Nei method are virtually identical to the corresponding estimates of 138 Mya, 0.40 × 10−9, and 0.94 × 10−9 under the JC model.
For the opsin-coding genes, a3 is generally the largest, followed by a1 and a2, in that order, showing the functional constraint on the nucleotide changes expected for the protein-coding genes (Kimura 1983). As may be expected from the relationship a3 > a1 > a2 for the opsin-coding genes, the evolutionary rate at the first and second positions of a codon (a1+2) is always much smaller than a3 (Table 2).
For the pseudogenes, the b1, b2, and b3 values cannot be ordered in a uniform fashion and any one of them can be largest. The overall average values of b1, b2, and b3 for the seven sets of pseudogenes are given by 1.31 × 10−9, 1.13 × 10−9, and 1.65 × 10−9 /site/year, respectively. Compared with those of opsin-coding SWS1 genes, the corresponding b1+2 and b3 values do not differ significantly, for five out of the seven sets of pseudogenes, revealing dramatically different patterns of nucleotide substitution before and after pseudogenization (Table 2). This supports the basic assumption that the rates of nucleotide substitution at the three positions of codons are uniform for the pseudogenes (Li et al. 1981; Miyata and Hayashida 1981; Miyata and Yasunaga 1981). The conservative nature of the SWS1 pseudogene evolution is suspected (Fig. 3A), but it is still surprising to see that this evolutionary rate is 4–10 times smaller than those of the globin and immunoglobulin pseudogenes.
When all positions of a codon are considered, the opsin-coding SWS1 genes and SWS1 pseudogenes have evolved at rates of ~0.6 × 10−9 and ~1.4 × 10−9 /site/year, respectively. Since, the evolutionary rates of the opsin-coding rhodopsin (RH1) and middle and long wavelength-sensitive (M/LWS) genes are 0.3–0.6 /site/year (Yokoyama and Yokoyama 1990a, b), the evolutionary rates of the opsin-coding SWS1 genes are similar to those of the other opsin genes and the SWS1 pseudogenes have evolved 2–3 times faster than the opsin-coding genes.
4. Discussion
The SWS1 pseudogene of pearleye (B. dentata) lost its opsin-coding ability about 130 Mya (Table 1). From another genus of pearleye (Scopelarchus analis), three functional opsin genes have been sequenced: two RH1 (SanaRH1A and SanaRH1B) genes and one RH2 gene (SanaRH2) (Pointer et al. 2007). The NJ tree shows that SanaRH1A and SanaRH1B are most closely related, their ancestor diverged from the ancestor of SanaRH2, and their common ancestor diverged from the ancestor of BdenS1ψ before that (Fig. 3B). This was expected from the phylogenetic relationship of the five paralogous opsin genes (Yokoyama 2000). If we take the a value as 0.5 × 10−9 /site/year, then the divergence time between the RH1 and RH2 genes is 540 Mya (Fig. 3B), which is also consistent with the previous observation that the vertebrate ancestor already possessed all five groups of evolutionarily distant visual pigments (Yokoyama and Yokoyama 1996). However, the totally unexpected feature of the NJ tree is that despite the old pseudogenization event of BdenS1ψ, the pseudogene and the paralogous opsin-coding genes have maintained similar evolutionary rates for the last 800 My.
Recent ENCODE analyses suggest that a significant portion of non-coding DNA sequences, including over a 17,000 DNA stretches of pseudogenes (www.pseudogene.org), is transcribed and is used for gene regulation (Djebali et al. 2012; however see Graur et al. 2013; Doolittle 2013). From this survey, the expression of BdenS1ψ may not be surprising. In animals, short antisense RNAs are used to inhibit translation or to degrade cytoplasmic mRNA post-transcriptionally, which not only protect against viral infection, prevent transposon mobilization, and regulate the expression of endogenous genes but also maintain genome integrity by preventing transposon mobilization and double-stranded break repair (Castel and Martienssen 2013; Leslie 2013). One way to determine whether or not these SWS1 pseudogenes have such biochemical functions may be to try to isolate double-stranded RNAs using methods applied to the functional analyses of pseudogenes in mice (Tam et al. 2008; Watanabe et al. 2008). However, since the pearleye does not have any evolutionarily closely related opsin-coding SWS1 gene, the isolation of double stranded RNAs may not be a fruitful approach. The problem is further complicated by the fact that no mRNAs of the SWS1 pseudogenes of the cichlid, coelacanth, prosimian, bat, seal, and cetacean species has been identified, strongly suggesting that they are not transcribed. For these reasons, we cannot offer any plausible explanation for the slow evolution of the SWS1 pseudogenes.
In analysing various DNA sequence data, molecular evolutionists and molecular biologists often claim the existence of adaptive evolution by showing that the number of nonsynonymous substitutions per nonsynonymous site (dn) is greater than that of synonymous substitutions per synonymous site (ds) (Hughes and Nei 1988; Nei and Kumar 2000). The condition of “dn > ds (or ω = dn/ds > 1),” however, is an untested assumption and these statistical results contain significant proportions of false positives and false negatives (Yokoyama et al. 2008; Nozawa et al. 2009a). Recently, theoretical basis and reliabilities of these statistical methods have been debated intensely among statistical evolutionary geneticists (Nozawa et al. 2009a; Nozawa et al. 2009b; Yang et al. 2009; Yang and dos Reis 2011; Nei 2013). These authors, however, seem to agree that the final answer of adaptive evolution can be obtained only by subjecting the statistical results to some form of experimental test.
SWS1 pseudogenes do not encode functional opsins and the concept of codon is irrelevant. Consequently, the positively selected sites inferred by any statistical methods based on the condition of “dn > ds” are false positives and, therefore, the pseudogenes can be used as a negative control in evaluating the reliabilities of such statistical methods. Hence, a total of 88 codons that are common to the 29 representative cetacean and two seal SWS1 pseudogenes were analysed by the NEB and BEB approaches of Bayesian method. We found false-positives at 3 codon sites (69, 107, and 118) and 2 sites (107 and 118) using the NEB and BEB models, respectively (Table S2). If these sequences could encode amino acids, then 4, 8, and 3 amino acid changes would have occurred at sites 69, 107, and 118, respectively (Fig. 4). At site 107, a total of 8 nucleotide substitutions occurred only at the first and second positions of codons: A → C (once), G → A (twice), G → T (once), C → A (once), and C → T (three times). These nucleotide changes agree well with the mutation profiles of pseudogenes observed, where the mutations C → T (66%) and G → A (62%) are particularly high (Li et al. 1984). It is expected that 5 and 3 changes should occur at the first two positions and at the third position of a codon, respectively, but the chance that all 8 mutations occur at the first two positions is still 0.1. Similarly, the four hypothetical nonsynonymous substitutions each at sites 69 and 118 exhibit the pseudogene-characteristic nucleotide substitutions. Therefore, we do not have to invoke any positive selection for the biased nucleotide substitutions in the cetacean pseudogenes. These observations again warn the danger of the blind use of the untested assumption of dn > ds (or ω > 1) in inferring positive selection and show the necessity of experimental tests of such statistical predictions.
Fig. 4.

Positively selected codon sites with hypothetical amino acid replacements predicted by the NEB and BEB approaches of the Bayesian method. The composite phylogenetic tree is based on the three sources (Price et al. 2005; Koito et al. 2010; McGowen 2011) and the sources of their nucleotide sequences are given in Table S1. *significant at the 5% level. ** significant at the 1% level.
Supplementary Material
Highlights.
We cloned the SWS1 opsin pseudogene of the deep-sea fish pearleye
SWS1 genes in several vertebrate lineages have lost their abilities to make opsins
The pseudogenization events took place separately 15 - 140 × 106 years ago
The evolutionary rates of these pseudogenes ranged between 0.9–2.0 × 10−9/site/year
Acknowledgments
We thank K. Carleton, T. Gojobori, P. Robinson, and R. Yokoyama for their comments. This work was supported by the National Eye Institute at the National Institutes of Health (EY016400) and Emory University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amemiya CT, Alfoldi J, Lee AP, et al. The African coelacanth genome provides insights into tetrapod evolution. Nature. 2013;496:311–316. doi: 10.1038/nature12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbiza L, Dopazo J, Dopazo H. Positive selection, relaxation, and acceleration in the evolution of the human and chimp genome. PLoS Comput Biol. 2006;2:e38. doi: 10.1371/journal.pcbi.0020038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakewell MA, Shi P, Zhang J. More genes underwent positive selection in chimpanzee evolution than in human evolution. Proc Natl Acad Sci USA. 2007;104:7489–7494. doi: 10.1073/pnas.0701705104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakirev ES, Ayala FJ. Pseudogenes: are they “junk” or functional DNA? Annu Rev Genet. 2003;37:123–151. doi: 10.1146/annurev.genet.37.040103.103949. [DOI] [PubMed] [Google Scholar]
- Castel SE, Martienssen RA. RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nature Rev Genet. 2013;14:100–112. doi: 10.1038/nrg3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF. Is junk DNA bunk? A critique of ENCODE. Proc Natl Acad Sci USA. 2013;110:5294–5300. doi: 10.1073/pnas.1221376110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genner MJ, Seehausen O, Lunt DH, Joyce DA, Shaw PW, Carvalho GR, Turner GF. Age of cichlids: new dates for ancient lake fish radiations. Mol Biol Evol. 2007;24:1269–1282. doi: 10.1093/molbev/msm050. [DOI] [PubMed] [Google Scholar]
- Graur D, Zheng Y, Price N, Azevedo RB, Zufall RA, Elhaik E. On the immortality of television sets: “function” in the human genome according to the evolution-free gospel of ENCODE. Genome Biol Evol. 2013;5:578–590. doi: 10.1093/gbe/evt028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart NS, Theiss SM, Harahush BK, Collin SP. Microspectrophotometric evidence for cone monochromacy in sharks. Naturwissenschaften. 2011;98:193–201. doi: 10.1007/s00114-010-0758-8. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Nei M. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature. 1988;335:167–170. doi: 10.1038/335167a0. [DOI] [PubMed] [Google Scholar]
- Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro HN, editor. Mammalian protein metabolism. New York: Academic Press; 1969. pp. 21–132. [Google Scholar]
- Kawamura S, Kubotera N. Ancestral loss of short wave-sensitive cone visual pigment in lorisiform prosimians, contrasting with its strict conservation in other prosimians. J Mol Evol. 2004;58:314–321. doi: 10.1007/s00239-003-2553-z. [DOI] [PubMed] [Google Scholar]
- Kimura M. The neutral theory of molecular evolution. Cambrige: Cambridge University Press; 1983. [Google Scholar]
- Koito T, Kubotera K, Tanabe S, Miyazaki N. Phylogenetic analyses in cetacean species of the family Delphinidae using a showrt wavelength sensitive opsin gene sequence. Fish Sci. 2010;76:571–576. [Google Scholar]
- Kosiol C, Vinar T, da Fonseca RR, Hubisz MJ, Bustamante CD, Nielsen R, Siepel A. Patterns of positive selection in six Mammalian genomes. PLoS Genet. 2008;4:e1000144. doi: 10.1371/journal.pgen.1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie M. Cell biology. The immune system’s compact genomic counterpart. Science. 2013;339:25–27. doi: 10.1126/science.339.6115.25. [DOI] [PubMed] [Google Scholar]
- Li WH, Gojobori T, Nei M. Pseudogenes as a paradigm of neutral evolution. Nature. 1981;292:237–239. doi: 10.1038/292237a0. [DOI] [PubMed] [Google Scholar]
- Li WH, Wu CI, Luo CC. Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J Mol Evol. 1984;21:58–71. doi: 10.1007/BF02100628. [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, Garber M, Zuk O, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478:476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowen MR. Toward the resolution of an explosive radiation--a multilocus phylogeny of oceanic dolphins (Delphinidae) Mol Phylogenet Evol. 2011;60:345–357. doi: 10.1016/j.ympev.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Miyata T, Hayashida H. Extraordinarily high evolutionary rate of pseudogenes: evidence for the presence of selective pressure against changes between synonymous codons. Proc Natl Acad Sci USA. 1981;78:5739–5743. doi: 10.1073/pnas.78.9.5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T, Yasunaga T. Rapidly evolving mouse alpha-globin-related pseudo gene and its evolutionary history. Proc Natl Acad Sci USA. 1981;78:450–453. doi: 10.1073/pnas.78.1.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Selectionism and neutralism in molecular evolution. Mol Biol Evol. 2005;22:2318–2342. doi: 10.1093/molbev/msi242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Mutation-driven evolution. Oxford: Oxford University Press; 2013. [Google Scholar]
- Nei M, Kumar S. Molecular evolution and phylogenetics. Oxford: Oxford University Press; 2000. [Google Scholar]
- Nei M, Suzuki Y, Nozawa M. The neutral theory of molecular evolution in the genomic era. Annu Rev Genomics Hum Genet. 2010;11:265–289. doi: 10.1146/annurev-genom-082908-150129. [DOI] [PubMed] [Google Scholar]
- Newman LA, Robinson PR. Cone visual pigments of aquatic mammals. Vis Neurosci. 2005;22:873–879. doi: 10.1017/S0952523805226159. [DOI] [PubMed] [Google Scholar]
- Nozawa M, Suzuki Y, Nei M. Reliabilities of identifying positive selection by the branch-site and the site-prediction methods. Proc Natl Acad Sci USA. 2009a;106:6700–6705. doi: 10.1073/pnas.0901855106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa M, Suzuki Y, Nei M. Response to Yang et al. : Problems with Bayesian methods of detecting positive selection at the DNA sequence level. Proc Natl Acad Sci USA. 2009b;106:10. 1073/pnas.0906089106. [Google Scholar]
- O’Quin KE, Hofmann CM, Hofmann HA, Carleton KL. Parallel evolution of opsin gene expression in African cichlid fishes. Mol Biol Evol. 2010;27:2839–2854. doi: 10.1093/molbev/msq171. [DOI] [PubMed] [Google Scholar]
- Pei B, Sisu C, Frankish A, et al. The GENCODE pseudogene resource. Genome Biol. 2012;13:R51. doi: 10.1186/gb-2012-13-9-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlaha O, Zhang J. Encyclopedia of Life Sciences. Chichester: John Wiley & Sons, Ltd; 2010. Pseudogenes and their evolution. [Google Scholar]
- Pointer MA, Carvalho LS, Cowing JA, Bowmaker JK, Hunt DM. The visual pigments of a deep-sea teleost, the pearl eye Scopelarchus analis. J Exp Biol. 2007;210:2829–2835. doi: 10.1242/jeb.006064. [DOI] [PubMed] [Google Scholar]
- Price SA, Bininda-Emonds OR, Gittleman JL. A complete phylogeny of the whales, dolphins and even-toed hoofed mammals (Cetartiodactyla) Biol Rev. 2005;80:445–473. doi: 10.1017/s1464793105006743. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Studer RA, Penel S, Duret L, Robinson-Rechavi M. Pervasive positive selection on duplicated and nonduplicated vertebrate protein coding genes. Genome Res. 2008;18:1393–1402. doi: 10.1101/gr.076992.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam OH, Aravin AA, Stein P, et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008;453:534–538. doi: 10.1038/nature06904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Totoki Y, Toyoda A, et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008;453:539–543. doi: 10.1038/nature06908. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang Z, dos Reis M. Statistical properties of the branch-site test of positive selection. Mol Biol Evol. 2011;28:1217–1228. doi: 10.1093/molbev/msq303. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Goldman N. In defense of statistical methods for detecting positive selection. Proc Natl Acad Sci USA. 2009;106:10. doi: 10.1073/pnas.0904550106. 1073/pnas.0904550106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Wong WS, Nielsen R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- Yokoyama R, Knox BE, Yokoyama S. Rhodopsin from the fish, Astyanax: role of tyrosine 261 in the red shift. Invest Ophthalmol Visual Sci. 1995;36:939–945. [PubMed] [Google Scholar]
- Yokoyama R, Yokoyama S. Convergent evolution of the red- and green-like visual pigment genes in fish, Astyanax fasciatus, and human. Proc Natl Acad Sci USA. 1990a;87:9315–9318. doi: 10.1073/pnas.87.23.9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama R, Yokoyama S. Isolation, DNA sequence and evolution of a color visual pigment gene of the blind cave fish Astyanax fasciatus. Vision Res. 1990b;30:807–816. doi: 10.1016/0042-6989(90)90049-q. [DOI] [PubMed] [Google Scholar]
- Yokoyama S. Molecular evolution of vertebrate visual pigments. Prog Retin Eye Res. 2000;19:385–419. doi: 10.1016/s1350-9462(00)00002-1. [DOI] [PubMed] [Google Scholar]
- Yokoyama S, Tada T. Adaptive evolution of the African and Indonesian coelacanths to deep-sea environments. Gene. 2000;261:35–42. doi: 10.1016/s0378-1119(00)00474-1. [DOI] [PubMed] [Google Scholar]
- Yokoyama S, Tada T, Zhang H, Britt L. Elucidation of phenotypic adaptations: Molecular analyses of dim-light vision proteins in vertebrates. Proc Natl Acad Sci USA. 2008;105:13480–13485. doi: 10.1073/pnas.0802426105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama S, Yokoyama R. Adaptive evolution of photoreceptors and visual pigments in vertebrates. Annu Rev Ecol Syst. 1996;27:543–567. [Google Scholar]
- Yokoyama S, Zhang H, Radlwimmer FB, Blow NS. Adaptive evolution of color vision of the Comoran coelacanth (Latimeria chalumnae) Proc Natl Acad Sci USA. 1999;96:6279–6284. doi: 10.1073/pnas.96.11.6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Rossiter SJ, Teeling EC, Li C, Cotton JA, Zhang S. The evolution of color vision in nocturnal mammals. Proc Natl Acad Sci USA. 2009;106:8980–8985. doi: 10.1073/pnas.0813201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.