

Abstract
Neural computations rely on ion channels that modify neuronal responses to synaptic inputs. While single cell recordings suggest diverse and neurone type-specific computational functions for HCN1 channels, their behavioural roles in any single neurone type are not clear. Using a battery of behavioural assays, including analysis of motor learning in vestibulo-ocular reflex and rotarod tests, we find that deletion of HCN1 channels from cerebellar Purkinje cells selectively impairs late stages of motor learning. Because deletion of HCN1 modifies only a subset of behaviours involving Purkinje cells, we asked whether the channel also has functional specificity at a cellular level. We find that HCN1 channels in cerebellar Purkinje cells reduce the duration of inhibitory synaptic responses but, in the absence of membrane hyperpolarization, do not affect responses to excitatory inputs. Our results indicate that manipulation of subthreshold computation in a single neurone type causes specific modifications to behaviour.
Key points
Purkinje cells in the cerebellum are important for motor learning and have electrical signalling properties determined by several different types of ion channel.
Using a restricted genetic deletion, we investigate the roles of HCN1 ion channels expressed by cerebellar Purkinje cells.
This deletion causes specific learning impairments in a subset of behaviours to which Purkinje cells contribute.
At a cellular level this specificity of function is mirrored by increases in the duration of responses to inhibitory synaptic input, without changes in responses to excitatory synaptic input activated in the absence of inhibition.
The results help us to understand how behaviours are influenced by ion channels important for aspects of computation in a defined neuronal cell type.
Introduction
Neuronal computation involves integration of inhibitory and excitatory synaptic inputs. Mechanisms of synaptic integration have been investigated in detail with single-cell electrophysiology techniques (Hausser et al. 2000; Magee, 2000; Branco et al. 2010). These studies suggest that different neurone types recruit distinct complements of voltage-gated ion channels to implement specific computational rules (London & Hausser, 2005; Nelson et al. 2006; O'Donnell & Nolan, 2011). Some ion channels are of particular interest for understanding the molecular basis for neural computations, as they primarily influence subthreshold integration of synaptic responses (Reyes, 2001). Among these channels, the HCN1 ion channel has important roles in motor and cognitive behaviours (Nolan et al. 2003; Nolan et al. 2004; Wang et al. 2007). A challenge to establishing the cellular mechanisms underlying these behavioural functions is that HCN1 channels, and other ion channels important for synaptic integration, can have distinct effects on subthreshold integration in different neurone types (London & Hausser, 2005; Nusser, 2009; Wahl-Schott & Biel, 2009). However, this functional diversity has been established primarily using in vitro electrophysiological recordings from identified neurones (Robinson & Siegelbaum, 2003; Wahl-Schott & Biel, 2009). In contrast, the behavioural roles of HCN1 ion channels in single neurone types have not previously been investigated.
Global deletion of HCN1 causes deficits in motor learning (Nolan et al. 2003) but, because HCN1 is expressed in many neurone types important for motor behaviour (Santoro et al. 2000; Notomi & Shigemoto, 2004), such a deletion provides no information about the specific roles played by HCN1 channels in particular neuronal populations. Here, we set out to address the roles in motor behaviours of HCN1 channels expressed by cerebellar Purkinje cells. The cerebellar circuit integrates diverse information to generate an output that is important for motor coordination (Apps & Garwicz, 2005; De Zeeuw et al. 2011). Each Purkinje cell integrates on the order of 100,000 glutamatergic inputs from cerebellar granule cells, with large numbers of inhibitory synaptic inputs from molecular layer interneurones and a single excitatory climbing fibre input (Harvey & Napper, 1991; Apps & Garwicz, 2005; Dean et al. 2010). Manipulations that modify the output from Purkinje cells typically have profound effects on motor behaviours (Karpova et al. 2005; Levin et al. 2006; Chen et al. 2010; Mark et al. 2011). HCN1 channels are required for a large hyperpolarization-activated current (Ih) found in cerebellar Purkinje cells (Nolan et al. 2003). While various studies have addressed roles for Ih in cerebellar Purkinje cells using single cell electrophysiology (e.g. Crepel & Penit-Soria, 1986; Williams et al. 2002; Nolan et al. 2003; Angelo et al. 2007; Oldfield et al. 2010), very little is known about the behavioural roles of Ih in cerebellar Purkinje cells. This is a difficult problem, as experiments using pharmacological blockers, or global deletion of HCN1, do not discriminate Ih in Purkinje cells and adjacent basket cells (Nolan et al. 2003; Maiz et al. 2012). Interpretation of the actions of pharmacological blockers of Ih is also confounded by off-target effects on Na+ and Ca2+ channels (Felix et al. 2003; Sanchez-Alonso et al. 2008; Wu et al. 2012). Here we address these issues by asking whether genetic deletion of HCN1 targeted to cerebellar Purkinje cells modifies motor behaviour.
To specifically evaluate the behavioural roles of HCN1 channels expressed by cerebellar Purkinje cells, we generated mice with deletion of HCN1 restricted to these neurones. We find that HCN1 channels in Purkinje cells are important specifically for the late stages of motor learning. Because HCN1 only appears to contribute to a subset of behaviours that involve Purkinje cells, we also asked whether the channel has functional selectivity at a cellular level. We find that HCN1 channels in Purkinje cells are specifically engaged by activation of inhibitory synaptic inputs. Our results suggest that molecularly definable components of synaptic integration by Purkinje cells have functions that differ between behaviours and stages of learning.
Methods
Ethical approval
Experimental studies conformed to the policies of the Journal of Physiology (described in Drummond, 2009), the UK Animals (Scientific Procedures) Act 1986 and European Directive 86/609/EEC on the protection of animals used for experimental purposes. Experiments were either carried out under a project licence granted by the UK Home Office and according to the guidelines laid down by the University of Edinburgh's Animal Welfare Committee, or following approval by the Centre National de Recherche Scientifique review board and the Direction Départementale des Services Vétérinaires.
Generation of mice
Mice with deletion of HCN1 restricted to cerebellar Purkinje cells were obtained by crossing mice expressing Cre-recombinase under the control of the L7 promoter (L7Cre, C57BL/6 background, obtained directly from Dr L. Reichardt and Dr B. Rico) (Rico et al. 2004), with mice that we developed previously and in which the exon of the HCN1 gene encoding the P region and the S6 transmembrane domain was flanked by LoxP sites (HCN1f/f, 129SVEV background) (Nolan et al. 2003). Their progeny were intercrossed for three further generations to produce HCN1f/f,L7Cre and HCN1f/f littermates. Mice with global deletion of HCN1 (HCN1−/−) and their wild-type littermates (HCN1+/+) were obtained as previously described (Nolan et al. 2003). For all experiments, the mice were on a mixed average 50:50% 129SVEV/C57 background. Genotype was determined by PCR analysis of DNA extracted from ear notch biopsies and confirmed after each experiment, using DNA from tail biopsies. HCN1f/f mice were used as controls for experiments with HCN1f/f,L7Cre mice. Separate control experiments to assess the possible effect of Cre expression in Purkinje cells compared L7Cre mice with wild-type animals. HCN1+/+ mice were used as controls for experiments with HCN1−/− mice. Male mutant and control littermates were used in all experiments. To confirm selective Cre expression, L7Cre mice were crossed with a Rosa26 β-gal reporter line. These mice demonstrated expression of the reporter protein exclusively in cerebellar Purkinje cells, but not in other brain areas (not shown).
Histology
Mice were deeply anaesthetized with sodium pentobarbitone [100 mg kg−1i.p.) and transcardially perfused with 0.9% saline followed by ice-cold 4% paraformaldehyde in phosphate-buffered saline (PBS). Images were acquired using a fluorescence microscope (Olympus) equipped with a CCD camera or a confocal microscope (Zeiss).
For Nissl staining, 24 μm thick cerebellar sections were cut with a freezing microtome, mounted, stained with 0.5% cresyl violet, dehydrated through an ethanol series, cleared in xylene and coverslipped with Eukitt (Sigma-Aldrich). At least three to six non-consecutive sagittal sections from the cerebellar vermis were stained, digitized and quantified for each animal. Quantification of the density of Purkinje cells was performed by counting the number of Purkinje cells along a 500 μm linear length in folia II, III, IV, VI, VIII, IX and averaging the counts for each animal. The thickness of the molecular layer was measured at three different points along the same length used to count Purkinje cells and the measurements averaged for each animal. Quantifications were carried out blind to the genotype, using ImageJ (http://rsb.info.nih.gov/ij/).
For immunofluorescence, 40 μm coronal or sagittal sections were obtained using a freezing microtome and stored in PBS containing 0.05% NaN2. Free-floating sections were washed for 3 × 10 min in PBS containing 0.1% Tween 20 (PBST) and incubated for 1 h in PBST containing 1% normal goat serum (PBST-NGS). The sections were then incubated overnight at 4°C in mouse monoclonal anti-HCN1 antibody (1:1000; Neuromab) and chicken polyclonal anti-MAP2 (1:5000; Abcam) diluted in PBST-NGS. The sections were washed three times in PBST and incubated in FITC-conjugated goat anti-mouse IgG (1:200; Abcam) and TRITC-conjugated goat anti-chicken (1:500; Abcam) in PBST-NGS for 1 h at RT. Some cerebellar sections were incubated with mouse anti-calbindin primary antibody (1:500; Sigma), followed by goat anti-mouse IgG (1:200; Abcam). To enhance staining, in some experiments, after post-fixation, the cerebellar tissue was transferred to 10 mm sodium citrate buffer (SCB; pH 6.0) overnight at 4°C and then incubated for 15 min in 10 mm SCB preheated at 95–98°C. This increased overall HCN1 labelling in the cerebellum and did not affect the staining in other brain regions.
Acute slice recordings
Electrophysiological recordings were made from Purkinje cells in cerebellar vermis slices prepared from 8–12 week-old male mice, as previously described (Nolan et al. 2003). Mice were euthanized by cervical dislocation and the brain carefully removed. Slices of thickness 200 μm were sectioned using a Vibratome 3000 (Intracel, UK). For sectioning, brains were submerged under cold (4–6°C) oxygenated modified artificial cerebrospinal fluid (ACSF) of the following composition (mm): NaCl 86, NaH2PO4 1.2, KCl 2.5, NaHCO3 25, CaCl2 0.5, MgCl2 7, glucose 25, sucrose 75. Slices were then maintained in oxygenated standard ACSF (mm): NaCl 124, NaH2PO4 1.2, KCl 2.5, NaHCO3 25, CaCl2 2, MgCl2 1, glucose 20. For recording, slices were maintained in oxygenated standard ACSF at 36 ± 1°C, unless stated otherwise. Recording electrodes were filled with intracellular solution (mm): potassium gluconate 130, KCl 10, Hepes 10, MgCl2 2, EGTA 0.1, Na2ATP 2, Na2GTP 0.3, sodium phosphocreatine 10 and biocytin 2.7. The electrode resistance in the bath containing standard ACSF was 3–5 MΩ. Cell-attached, voltage-clamp and current-clamp recordings were made with a Multiclamp 700A amplifier (Molecular Devices), sampled at 50 kHz and filtered at 10–20 kHz. Series resistance and capacitance neutralization were used as described previously (Purves, 1981; Nolan et al. 2003). Ih was recorded from Purkinje cells at room temperature in ACSF whose composition was designed to minimize the contribution of other voltage-gated currents (mm): NaCl 115, NaH2PO4 1.2, KCl 5, CaCl2 2, MgCl2 1, NaHCO3 25, glucose 20, BaCl2 1, CdCl2 0.1, NiCl2 1, tetraethylammonium 5, 4-aminopyridine 1, 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX) 0.005, d-(2R)-amino-5-phosphonovaleric acid (AP5) 0.005, picrotoxin 0.005 and tetrodotoxin 0.0005 (see also Nolan et al. 2003).
In slices used for recordings, Purkinje cells continuously fired spontaneous action potentials, similar to reports of Purkinje cell firing in awake animals (Schonewille et al. 2006) and in previous cerebellar slice recordings (Hausser & Clark, 1997). In these recordings we did not observe spontaneous or evoked transitions between up and down states suggestive of bi-stability (Williams et al. 2002; Oldfield et al. 2010), or evidence of trimodal activity characterized previously in some slice preparations (Womack & Khodakhah, 2002). Synaptic responses of Purkinje cells were evoked using a glass stimulating electrode containing standard ACSF. Stimuli were delivered with a stimulus isolation unit (ISO-STIM 01D, NPI, Germany). In all experiments, at least three consecutive sweeps at 4 s intervals were recorded and analysed separately. Averaged data from each neurone were then used for statistical analysis of differences between groups. To reduce stimulation artifacts in voltage-clamp experiments, analysed synaptic responses were the differences between recordings from the same cell in the absence and presence of picrotoxin for inhibitory postsynaptic currents (IPSCs), or NBQX for excitatory postsynaptic currents (EPSCs). The baseline was also subtracted and residual stimulus artifacts were removed by interpolation. Series resistance and capacitance neutralization were used as described previously (Nolan et al. 2003), except during recording of synaptic responses, when to reduce capacitive transients they were not used. Synaptic inhibition was evoked by placing the stimulating electrode in the molecular layer 80–140 μm from the Purkinje cell layer and 100–150 μm along the long axis of the folia relative to the recorded Purkinje cell. The location of the electrode was adjusted to avoid climbing fibre stimulation and direct excitation of the recorded cell by parallel fibres. Recordings were made in the absence of drugs. Mediation of responses by GABAA receptors was confirmed using the blocker picrotoxin (50 μm). Parallel fibre responses were evoked with a stimulating electrode in the molecular layer, approximately 80–140 μm from the soma of the recorded Purkinje cell and within its dendritic arbor. Trains of 10 stimuli at 100 Hz were chosen to resemble sensory evoked burst firing patterns of granule cells recorded in vivo (Chadderton et al. 2004). Recordings were made in the presence of picrotoxin (50 μm) and the GABAB blocker CGP55845 (1 μm) to block inhibitory responses. The location of the electrode was adjusted to avoid climbing fibre stimulation. Mediation of responses by AMPA receptors was confirmed by blocking them with NBQX (5 μm). Climbing fibre responses were evoked by stimulation of the white matter or the granule cell layer with a stimulus of 0.02–0.05 mA amplitude and 1 ms duration. To ensure isolation of climbing fibre responses, the stimulus intensity and location of the stimulating electrode were adjusted for each cell until an all-or-nothing complex spike response was observed. For voltage-clamp recordings of IPSCs cells, they were held at −60 mV, whereas for recordings of EPSCs they were held at −75 mV. For examination of synaptic responses in current clamp there was no holding current and the Purkinje cells fired spontaneous action potentials. Data were analysed using custom-written routines in IGOR pro (Wavemetrics).
Behavioural testing
The mice were housed in groups in standard breeding cages at a constant temperature (22 ± 1°C) and with a 12:12 h light:dark cycle (light on 07.30–19.30 h). Food and water were available ad libitum, unless otherwise stated. All experiments were conducted during the light period. At the beginning of behavioural testing the mice were 8–12 weeks old. All animals were handled for at least 5 min/day for 2–3 days before starting the experiments. Behavioural parameters in the open field, parallel rod task and radial maze were analysed by automated recording software (ANY-maze). Experiments and analyses were performed blind to the animals’ genotypes.
Accelerating rotarod
The apparatus consisted of a rotating rod (diameter 3 cm) elevated 15 cm above a grid floor (TSE Systems). The rod was covered with grip tape to enhance grip for the mice. A rubber foam tube was used to adjust the diameter to 5 cm. The mice were tested with either the 3 cm or the 5 cm rod. They were trained for 4 days (3 cm rod) or 5 days (5 cm rod) with the rod accelerating from 4 to 40 rotations/min (rpm) in 300 s. Four trials were carried out each day with at least a 20 min intertrial interval (Nolan et al. 2003). Mice that completed a trial without falling were allowed to continue for an additional 30 s with the rod moving at 40 rpm. On the fifth day of testing with the 3 cm rod, the mice were tested for six trials with the rod moving for 180 s at constant speeds of 30, 25, 20, 15, 10 and 5 rpm (intertrial interval 20 min). The mice were allowed only one passive rotation/trial. At the second passive rotation they were gently pulled off the rod and the trial was considered complete. The latency to fall was recorded on each trial.
Vestibulo-ocular reflex (VOR)
Surgical preparation and postoperative care for head implant surgery, experimental set-up, apparatus, method of data acquisition used to record eye movements and methods for data analysis were performed as previously described (Beraneck & Cullen, 2007; Beraneck et al. 2012). Vestibulo-ocular reflex in the dark was recorded before and after each visuo-vestibular conflict training session by sinusoidal rotation of head-restrained mice en bloc at frequencies of 0.2, 0.5, 1.0, 1.5 and 2 Hz, with a fixed peak velocity of 50°/s. The training protocol was repeated for 5 consecutive days. The mice were left in their cages in normal lighting conditions between training sessions. Segments of data with saccades were removed from analysis. At least 10 cycles were analysed for each frequency at each time point. VOR gain and phase were determined by the least-squares optimization method. The variance accounted-for (VAF) of each fit was computed. VAF values were typically > 0.85, where VAF = 1 indicates a perfect fit to the data. Trials for which the VAF was < 0.5 were excluded from the analysis. During experiments on days 4 and 5, the amplitude of the eye movements occasionally decreased below the methodological threshold of gain < 0.05 and, thus, no gain or phase values were reported.
Open field
The apparatus consisted of an opaque Perspex floor (60 × 60 cm) surrounded by a 40 cm high wall. Mice were placed in the open field for two 5 min sessions (intersession interval 30 min), on 2 consecutive days (Bolivar, 2009).
Parallel Rod
The apparatus (Stoelting) consists of a clear acrylic box (21 × 21 cm, height 40 cm) whose floor is made of a series of parallel stainless steel rods placed 1 cm above a base plate. Each mouse was tested for 10 min. The distance moved in the apparatus and the number of paw slips through the parallel rods were recorded (Kamens & Crabbe, 2007).
Hanging wire test
The mice were allowed to grab a horizontal wire (diameter 2 mm, length 40 cm, suspended 50 cm above a cushioned table) with their front paws and the time spent hanging was measured (maximum time allowed 60 s). Behaviour was scored according to the following scale (Ogura et al. 2001): 1, hanging onto the bar with both forepaws; 2, in addition to 1, attempted to climb onto the bar; 3, hanging onto the bar with two forepaws and one or both hindpaws; 4, hung onto the bar with all four paws with tail wrapped around the bar; 5, escaped to one of the supports. Each mouse was tested twice, with an interval between sessions of 24 h.
Inverted grid test
The mice were placed in the centre of a wire grid (40 × 60 cm, suspended 50 cm above a cushioned table) and then the grid was inverted. The time spent hanging on to the grid was measured. Each mouse was trained for four trials, one trial/day (maximum time allowed 120 s) and then tested in a trial of 300 s maximum duration.
Grip strength test
The apparatus consisted of a grip strength meter (San Diego Instruments, CA, USA). The mouse was held at the base of the tail and allowed to grab the grid with either fore- or hind-limbs. The mouse was then pulled gently backwards until it released its grip. The peak force of each trial was taken as a measure of the grip strength.
Radial maze
The apparatus consisted of an eight-arm radial maze equipped with IR underlighting (Tracksys). It was located in a dimly illuminated room (2 × 2 m) and was surrounded by prominent extra-maze cues. Each arm was gated by a clear acrylic automated guillotine-door positioned by the central platform. Animals were food restricted to 85–90% of their body weight and trained in the radial maze for 25 days. First, the mice were trained with the fully baited procedure for 10 days, one trial/day. On each trial, all arms were baited with a small piece of standard pellet chow. A mouse was placed on the central platform and after 5 s all doors were simultaneously raised. The animal was allowed to explore the maze for 10 min or until all arms had been visited once. Between mice, the apparatus was thoroughly cleaned with 70% ethanol, followed by water. Several motor, spatial and procedural parameters were recorded following previously described protocols (Mandolesi et al. 2001). An arm entry was defined as the mouse having at least both forepaws in the second half of the arm. An error was defined as the mouse re-entering a previously visited arm. Upon completion of training with the fully baited procedure, the mice were trained with the delayed procedure for 15 days, one trial/day. Each trial consisted of two phases. In the first phase (sample phase) four randomly chosen arms were baited. The mouse was placed in the centre with all doors closed and after 5 s only the doors of the baited arms were raised. In the second phase (test phase), only the four previously closed arms were baited but the mouse was allowed to explore all arms. In each phase the mouse was allowed to explore the open arms for 10 min or until all baited arms had been visited. Between phases the mouse was placed in a holding cage for 1–2 min and the maze was cleaned. A different set of four randomly chosen arms was baited on each trial.
Acoustic startle response
Habituation and prepulse inhibition of the acoustic startle response were measured using the SR-LAB system (San Diego Instruments). The average reading of the system was calibrated to 1100 daily, using a vibrating standardization unit (San Diego Instruments). The sound levels were calibrated daily, using a digital sound level meter (Radio Shack). A continuous background noise of 70 dB was provided throughout habituation and testing to mask extraneous stimuli. The startle response was recorded for 65 ms (every 1 ms) from the onset of the startle stimulus and its amplitude was defined as the highest voltage peak produced by the acoustic stimulus. Short-term habituation of the startle response was measured in response to a 40 ms burst of white noise of 117 dB intensity, superimposed on the background noise. After 5 min of habituation to the startle chamber, the mouse was presented with 100 stimuli (interstimulus interval 10 s). Every 10 stimuli there was a trial in which no stimulus was presented, to measure baseline movements in the apparatus. Prepulse inhibition of the acoustic startle response was measured as previously described (Paylor & Crawley, 1997). The startle stimulus consisted of a 40 ms burst of white noise of 120 dB. The prepulse stimuli were 20 ms bursts of white noise of 74, 78, 82, 86 or 90 dB intensity, and were presented 100 ms before the onset of the startle stimulus.
Statistical analysis
Statistical analysis was performed using two-tailed Student's unpaired t test or two-way repeated-measures ANOVA followed by Fisher's PLSD post hoc test when appropriate. Level of significance was set at P < 0.05, unless otherwise stated. Asterisks in the figures denote significant differences assessed by t test or post hoc test, as appropriate.
Results
Deletion of HCN1 from mature cerebellar Purkinje cells
To selectively delete HCN1 channels from cerebellar Purkinje cells, we generated mice with deletion of HCN1 restricted by expression of Cre under the control of the L7 promoter (HCN1f/f,L7Cre mice) (see Methods and Fig. 1 and 2). We confirmed deletion of HCN1 by the absence of Ih in Purkinje cells from adult HCN1f/f,L7Cre mice (P= 0.02; two-way ANOVA) (Fig. 1A). In contrast, Purkinje cells from control (HCN1f/f) mice showed a large Ih (Fig. 1A) that was abolished by the blocker ZD7288 (P < 0.0001; two-way ANOVA) (Supplementary Fig. 1). HCN1 currents recorded from HCN1f/f,L7Cre Purkinje cells were indistinguishable from controls at 1 week after birth (P= 0.57; two-way ANOVA) but became progressively smaller from week 2 (P < 0.0001) to week 5 (P < 0.0001) after birth (Fig. 1B). From 5 weeks onwards, Ih was reduced in all Purkinje cells recorded from HCN1f/f,L7Cre mice (P < 0.0001) (Fig. 1C). We found no evidence for changes in HCN1 expression in cell types other than Purkinje cells, including cells in the cerebellum (Fig. 2A), neocortex and hippocampus (Fig. 2B). There was no obvious reduction in HCN1 labelling in sections of cerebellar cortex, suggesting that either HCN1 channels in Purkinje cells are not accessible to labelling by immunohistochemistry (Lorincz & Nusser, 2008) or that any labelling of Purkinje cells in wild-type animals is saturated by signal from nearby interneurones, which strongly express HCN1 (Santoro et al. 1997; Notomi & Shigemoto, 2004; Lujan et al. 2005). Importantly, HCN1 channel expression in basket cell terminals is maintained in HCN1f/f,L7Cre mice (Fig. 2A), enabling HCN1f/f,L7Cre mice to be used to test the influence of Purkinje cell HCN1 channels on responses to inhibitory synaptic input without interfering with the presynaptic Ih found in basket cell terminals (Southan et al. 2000). In contrast, experiments that rely on extracellular blockers of HCN channels, or global deletion of HCN1, are unable to clearly distinguish roles of HCN1 channels expressed by Purkinje cells from functions of the channels in nearby interneurones.
Figure 1. Postnatal reduction of Ih in Purkinje cells from HCN1f/f,L7Cre mice.
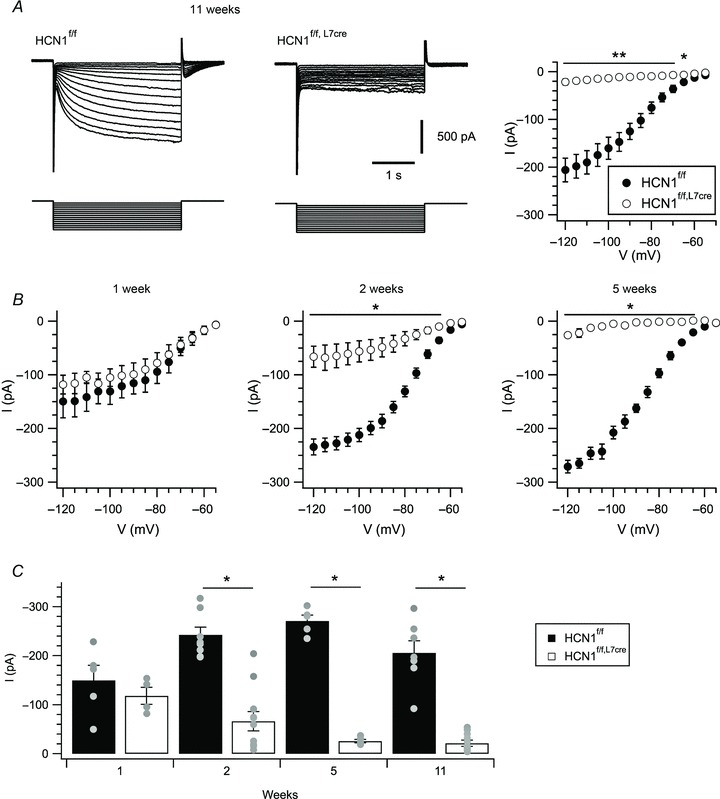
A, hyperpolarization to voltages between −55 mV and −120 mV in 5 mV increments from a holding potential of −50 mV (lower traces) activates a prominent Ih in HCN1f/f mice (upper left), while in HCN1f/f,L7Cre mice Ih is absent (upper middle). Tail currents measured upon return to −50 mV, plotted as a function of the preceding test potential (right), quantify reduction of Ih in HCN1f/f,L7Cre (n= 11) compared to HCN1f/f (n= 7) mice (genotype X test potential F13,208= 66.9, P= 0.02, two-way ANOVA). B, mean tail current amplitude plotted as a function of test potential for Purkinje cells from 1 (genotype X test potential F1,13= 0.89, P= 0.57, n= 4–5/group, two-way ANOVA), 2 (F13,23= 35.99, P < 0.0001, n= 9–11/group) and 5 week-old HCN1f/f and HCN1f/f,L7Cre mice (F13,104= 166.81, P < 0.0001, n= 5/group). C, tail currents measured following a test step to −120 mV are plotted as a function of age of the mouse (genotype X age F3,50= 8.06, P < 0.0001, two-way ANOVA). Grey circles represent individual cells. Data are presented as mean ± SEM; *P < 0.05, **P < 0.001 HCN1f/fversus HCN1f/f,L7Cre, Fisher's PLSD.
Figure 2. Deletion of HCN1 is restricted to cerebellar Purkinje cells and does not affect cerebellar morphology.
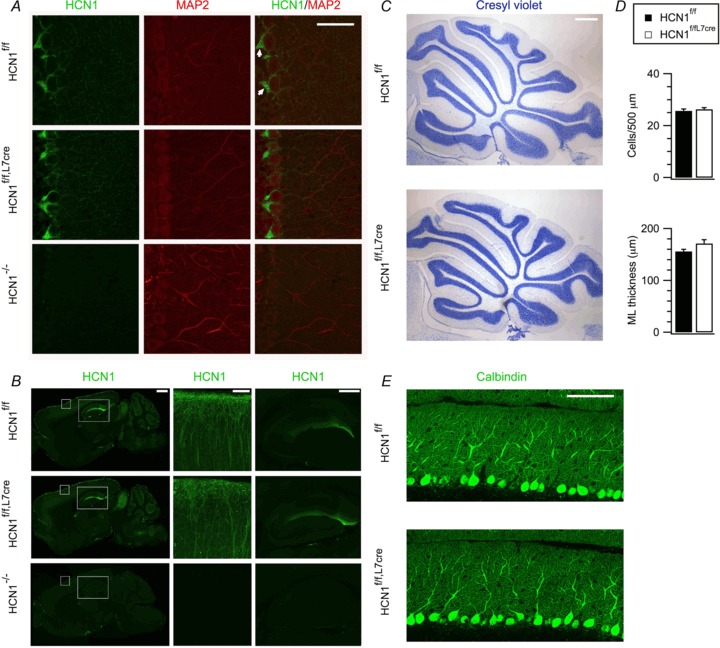
A, labelling of sagittal cerebellar sections from HCN1f/f, HCN1f/f,L7Cre and HCN1−/– mice with anti-HCN1 and anti-MAP2 (a dendritic marker) antibodies. Anti-HCN1 antibodies clearly labelled basket cell terminals (arrows) and MAP2-negative processes; scale bar = 50 μm. B, anti-HCN1-labelled sagittal brain sections from HCN1f/f, HCN1f/f,L7Cre and HCN1−/− mice (left). Higher magnification images of the boxes are shown in the right panels, focusing on the neocortex (middle) and the hippocampus (right); scale bars = 1000, 100 and 500 μm. C, Nissl-stained sections of the cerebellar vermis from HCN1f/f and HCN1f/f,L7Cre, mice demonstrating similar foliar architecture and laminar organization. D, quantification of mean Purkinje cell density and molecular layer (ML) thickness (right) did not reveal any significant differences (t10= 0.55, P= 0.6 and t10= 1.78, P= 0.11, respectively, n= 6 mice/group, Student's unpaired t test); scale bar = 500 μm. E, cerebellar cortex labelled with antibodies against calbindin, a Purkinje cell marker, show comparable dendritic arborization of Purkinje cells in HCN1f/f and HCN1f/f,L7Cre mice; scale bar = 75 μm. Data are presented as mean ± SEM.
To further assess the specificity of HCN1 deletion, we investigated cerebellar morphology and the firing properties of Purkinje cells. In adult HCN1f/f,L7Cre mice, the morphology of the cerebellar cortex appeared indistinguishable from control HCN1f/f mice: foliar architecture and laminar organization were similar (Fig. 2C); there were no significant differences in the density of Purkinje cells (P= 0.6, Student's unpaired t test) or the thickness of the molecular layer (P= 0.11, Student's unpaired t test) (Fig. 2D); and the Purkinje cell dendritic tree showed a similar organization (Fig. 2E). The frequency and waveform of spontaneous action potentials fired by Purkinje cells were not affected by HCN1 deletion (Supplementary Fig. 2A, B) and bistability, previously reported following pharmacological block of Ih (Williams et al. 2002; but see Maiz et al. 2012) was not observed (Supplementary Fig. 2A). Responses of Purkinje cells from adult HCN1f/f,L7Cre mice to injected current were consistent with previous results from global HCN1 knockout mice (Nolan et al. 2003) and did not show any evidence for secondary changes in intrinsic properties following loss of HCN1 (Supplementary Figs 2 and 3). Together, these results demonstrate that in HCN1f/f,L7Cre mice, deletion of HCN1 occurs relatively late in postnatal development, is restricted to cerebellar Purkinje cells, does not interfere directly with action potential firing and does not lead to changes in cerebellar morphology.
HCN1 channels in cerebellar Purkinje cells influence late stages of motor learning
We reasoned that if HCN1 channels influence all aspects of motor behaviour that involve Purkinje cells, then behavioural deficits in HCN1f/f,L7Cre mice would resemble deficits caused by manipulations that target other aspects of Purkinje cell function, such as action potential firing (Levin et al. 2006; Chen et al. 2010; Mark et al. 2011) or synaptic plasticity (De Zeeuw et al. 1998; Koekkoek et al. 2005; Schonewille et al. 2010; Gao et al. 2012). Alternatively, HCN1 channels in Purkinje cells may be engaged only during specific behaviours or stages of learning.
We first investigated the influence of Purkinje cell HCN1 channels on basal coordination and learning in an accelerating rotarod task. Ion channels that contribute to action potential firing by Purkinje cells are important for initial and maintained performance in rotarod tasks (Levin et al. 2006; Chen et al. 2010; Mark et al. 2011). In contrast, we find that HCN1f/f,L7Cre mice are indistinguishable from controls on the first day of training in a rotarod task (Fig. 3A), but their performance is impaired on the second and subsequent training days (genotype P= 0.02, genotype X trial P= 0.0008, two-way ANOVA) (Fig. 3A). Since rotarod configuration can affect performance, and as in the initial experiment the latencies on day 1 were already close to their maximal value, with the result that the observed learning was relatively modest, we wondered whether the strong initial performance was masking an early learning deficit. We therefore examined a second cohort of mice using a modified, more challenging version of the task, in which the diameter of the rod was reduced. In this experiment, control mice showed large performance improvements with training (Fig 3B). Again, HCN1f/f,L7Cre mice did not differ from controls on the first day of training, but demonstrated profound deficits during the second and subsequent days (genotype P= 0.01, genotype X trial P < 0.0001, two-way ANOVA) (Fig. 3B). When tested with the rod moving at constant speeds, following 4 days of training with the accelerating rod, this deficit was apparent as an inability to balance at speeds > 10 rpm (P= 0.002, two-way ANOVA) (Fig. 3C). The differences between groups did not appear to be a result of altered consolidation of memories, as we found no significant effect of genotype on the difference between the latency to fall in the first trial of the day and latency to fall in the last trial of the preceding day (5 cm rod, genotype P= 0.373, genotype X day P= 0.218; 3 cm rod, genotype P= 0.354, genotype X day P= 0.806, two-way ANOVA). These deficits are also not explained by Cre expression in Purkinje cells, as expressing Cre alone did not affect rotarod performance (genotype P= 0.23, genotype X trial P= 0.35, two-way ANOVA) (Supplementary Fig. 4). Together, these data indicate that HCN1 channels in Purkinje cells are required for late phases of rotarod learning.
Figure 3. Late stages of motor learning are selectively impaired by deletion of HCN1 from Purkinje cells.
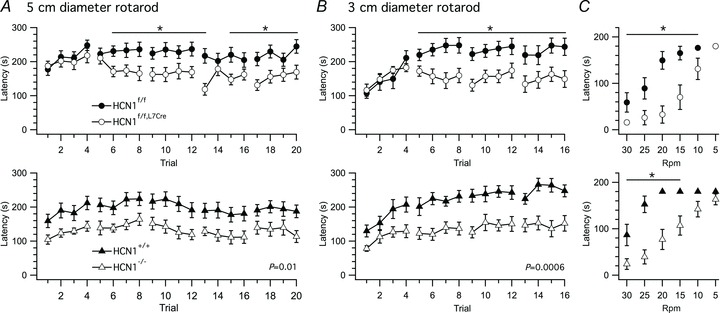
A and B, time spent on a 5 cm wide (A) and a 3 cm wide (B) accelerating rotarod is plotted as a function of training session for Purkinje cell restricted HCN1 deletion (upper) and global HCN1 deletion (lower). In (A) HCN1f/f,L7Cre mice (n= 15) are progressively impaired compared to HCN1f/f mice (n= 17) (genotype F1,30= 6.36, P= 0.02; genotype X trial F19,570= 2.4, P= 0.0008, two-way ANOVA), while HCN1−/− (n= 11) compared to HCN1+/+ mice (n= 13) are impaired at all times (genotype F1,22= 8.59, P= 0.01; genotype X trial F19,418= 0.33, P= 1). In B, HCN1f/f,L7Cre mice are progressively impaired compared to HCN1f/f mice (genotype F1,20= 7.66, P= 0.01; genotype X trial F15,300= 3.85, P < 0.0001; n= 11/group), whereas HCN1−/− (n= 14) compared to HCN1+/+ mice (n= 10) are impaired at all time points (genotype F1,22= 16.27, P= 0.0006; genotype X trial F15,330= 1.46, P= 0.12). C, performance in the rotarod tested at constant speeds following training in the accelerating version of the task (HCN1f/f,L7Creversus HCN1f/f mice: genotype F1,20= 16.71, P= 0.0006; genotype X rpm F5,100= 4.15, P= 0.002, HCN1−/−versus HCN1+/+: genotype F1,22= 19.05, P= 0.0002; genotype X rpm F5,110= 4.552, P= 0.0008, two-way ANOVA). Data are presented as mean ± SEM; *P < 0.05, control (HCN1f/f or HCN1+/+) versus knock-out (HCN1f/f,Cre or HCN1−/−), Fisher's PLSD.
How does the phenotype obtained following deletion of HCN1 restricted to cerebellar Purkinje cells compare to global functions of HCN1 channels? Because, in addition to Purkinje cells, HCN1 is expressed in other neurone types involved in motor learning (Notomi & Shigemoto, 2004), and because previous experiments with global knockout mice suggest that HCN1 channels are important on the first day of training on a rotarod (Nolan et al. 2003), we compared results from HCN1f/f,L7Cre mice with experiments under similar conditions using global HCN1 knock-out mice (HCN1−/−). We find that, in contrast to HCN1f/f,L7Cre mice, HCN1−/− mice were impaired from the very first day of training in both versions of the rotarod task (5 cm rod, genotype P= 0.01, genotype X trial P= 1; 3 cm rod, genotype P= 0.0006, genotype X trial P= 0.12, two-way ANOVA) (Fig. 3A and B). After 4 days of training with the accelerating rotarod protocol, HCN1f/f,L7Cre and HCN1−/− mice showed similar impairments when tested with the rod moving at constant speeds (P= 0.0008, two-way ANOVA) (Fig. 3C). The additional HCN1-expressing cell types that affect motor learning are therefore likely to be found in series with Purkinje cells, for example, HCN1-expressing neurones in the inferior olive or cerebellar nuclei (Notomi & Shigemoto, 2004).
Does this selective role of HCN1 extend to other motor behaviours? To address this, we focused on the vestibulo-ocular reflex (VOR) and its adaptation (Fig. 4A), as this is a behaviour for which the underlying circuit mechanisms have been investigated in some detail and in which cerebellar Purkinje cells play an instrumental role (Boyden et al. 2004). To measure VOR performance, we tracked eye movement in the dark during sinusoidal rotation of the mouse around a vertical axis, with varying frequencies and constant peak velocity of 50/s (Fig. 4A). We quantified the gain and phase of the eye velocity relative to the head rotation velocity (Fig. 4A–C). Performance of naive HCN1f/f and HCN1f/f,L7Cre mice was similar (Fig. 4D and Supplementary data). Introduction of a visuo-vestibular conflict (VVC) by rotation at 0.5 Hz in the light for 1 h caused adaptation of the VOR, with a similar reduction in gain in both groups of mice (Fig. 4D–G, upper panels). However, as adaptation progressed, the absence of HCN1 from Purkinje cells caused the phase to increase relative to the optimal value for accurate eye movement. This became apparent as a significant increase in phase after 40 min of VVC on day 1 (P= 0.01, two-way ANOVA) (Fig. 4E, lower panel) and was maintained during VVC on all 5 days (P= 0.04, two-way ANOVA) (Fig. 4F, lower panel). The effects of HCN1 deletion on the phase of VVC-induced modifications extended to VOR in the dark at lower frequencies (0.2 Hz, P= 0.009; 0.5 Hz, P= 0.03) (Fig. 4D and G and Supplementary Data), but not higher frequencies (data not shown). These results are in contrast to those from manipulations that target intracellular signalling pathways important for synaptic plasticity within Purkinje cells, and which cause substantial deficits in gain adaptation during the initial stages of learning (De Zeeuw et al. 1998; Schonewille et al. 2010). Thus, while Purkinje cells profoundly influence gain and phase at all stages of VOR adaptation (De Zeeuw et al. 1998; Boyden et al. 2004; Schonewille et al. 2010), the influence of HCN1 channels in cerebellar Purkinje cells is relatively small, depends on the stage of learning and appears to be manifested primarily through significant differences in phase.
Figure 4. HCN1 channels in Purkinje cells are important for integration of visual-vestibular inputs during later stages of learning.
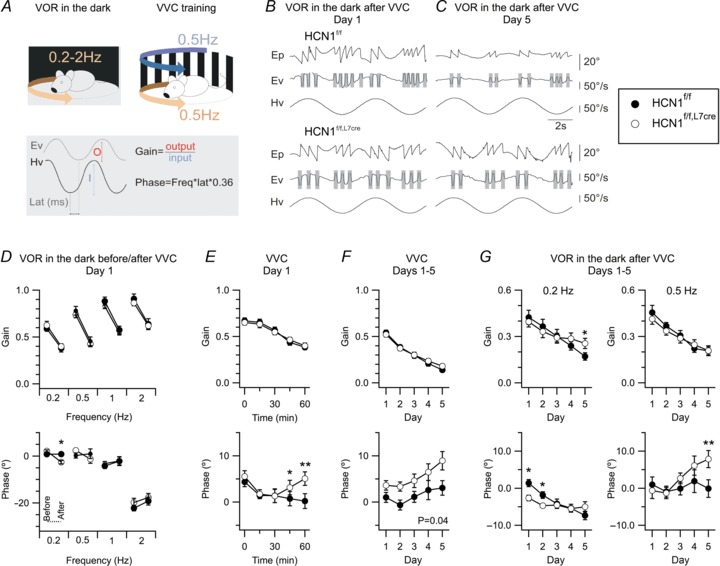
A, illustration of the training paradigm (top) and gain and phase calculations based on head (Hv) and eye (Ev) velocity measurements. B and C, example traces of 0.2 Hz VOR in the dark, with amplitude of rotation ∼80°, recorded from HCN1f/f (top) and HCN1f/f,L7Cre (bottom) mice after the first (B) and last (C) VVC training session. Each panel shows eye position (Ep), Ev and corresponding Hv. Shaded areas represent segments of data removed from the analysis because they contained saccades. D, gain (upper) and phase (lower) of the VOR in the dark, plotted as a function of rotation frequency. VOR phase at 0.2 Hz following VVC was reduced in HCN1f/f,L7Cre mice compared to HCN1f/f (genotype X trial F1,23= 13.89, P= 0.001, n= 10–15, two-way ANOVA). There was no other significant difference (P > 0.1; see Supplementary Data). E and F, mean gain (upper) and phase (lower) during VVC are plotted as a function of time during the first day (E) and as a function of day (F). HCN1f/f (n= 11) and HCN1f/f,L7Cre (n= 13) mice showed a comparable gain decrease on day 1 (genotype X time F4,88= 0.86, P= 0.49, two-way ANOVA), but a significant difference developed in the phase (F4,88= 3.33, P= 0.01). Gain was similar in HCN1f/f (n= 10) and HCN1f/f,L7Cre mice (n= 15) (genotype X day F4,92= 1.78, P= 0.14, two-way ANOVA), while the phase difference was maintained across consecutive training days (genotype F1,23= 4.66, P= 0.04). G, VOR gain adaptation was reduced by HCN1 deletion at the lowest frequency tested (genotype X day F4,72= 3.05, P= 0.02, two-way ANOVA) but not at other frequencies (P > 0.1; see Supplementary Data), while phase adaptation was modified at 0.2 Hz (genotype X day F4,72= 3.67, P= 0.009) and 0.5 Hz (F4,84= 3, P= 0.03). Data are presented as mean ± SEM; *P < 0.05, **P < 0.001 HCN1f/fversus HCN1f/f,L7Cre, Fisher's PLSD.
Do HCN1 channels in Purkinje cells contribute to other behaviours in which the cerebellum is implicated? Whereas HCN1−/− mice showed a significant reduction of distance moved (P= 0.04, two-way ANOVA) and mean speed (P= 0.001, two-way ANOVA) in the open field compared to wild-type littermates, these measures were unaffected by deletion of HCN1 from Purkinje cells (P > 0.1, two-way ANOVA) (Fig. 5A). Deletion of HCN1 from Purkinje cells also does not appear to be associated with changes in parameters that could indirectly influence motor behaviour, including body size, weight and behaviour, in the home cage (data not shown); limb control and muscle fatigue assessed with the parallel rod floor (Fig. 5B), suspended wire (Fig. 5C) inverted grid and grip strength tests (Fig. 5D and E). There is evidence for roles of the cerebellum in spatial learning in the radial maze (Mandolesi et al. 2001) and habituation of the acoustic startle response (Leaton & Supple, 1986). However, HCN1f/f,L7Cre mice did not show any difference in their radial maze performance compared to HCN1f/f mice (Supplementary Figs 5, 6), in short- and long-term habituation of the acoustic startle response (Supplementary Fig. 7) or in the prepulse inhibition of the startle response (Supplementary Fig. 7). Therefore, as well as being limited to specific stages of learning, the roles of HCN1 may be selective for a subset of the behaviours in which the cerebellar cortex plays a role.
Figure 5. General neurological tests do not reveal effects of HCN1 deletion from Purkinje cells.
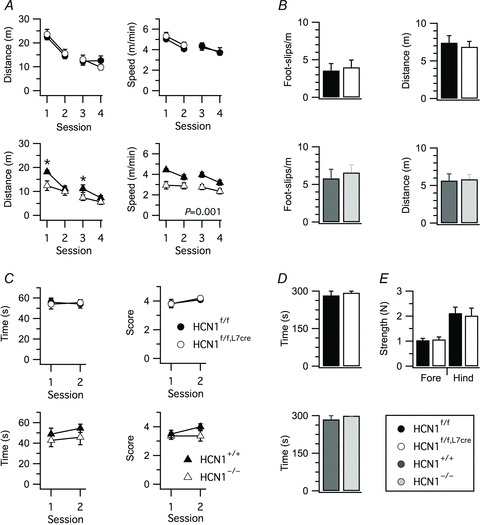
A, distance travelled and movement speed in an open field, plotted as a function of test session. Locomotor activity was not affected by HCN1 deletion in HCN1f/f,L7Cre mice (distance, genotype X session F3,90= 1.87, P= 0.14; speed, genotype X session F3,90= 0.47, P= 0.71, n= 15–17/group, two-way ANOVA), whereas locomotor activity of HCN1−/− mice was reduced compared to HCN1+/+ mice (distance, genotype F1,22= 3.88, P= 0.06; genotype X session F3,66= 2.91, P= 0.04; mean speed, genotype F1,22= 13.59, P= 0.001; genotype X session F3,66= 1.84, P= 0.15, n= 11–13/group). B, foot slips and distance travelled in a parallel rod floor test did not differ significantly between HCN1f/f and HCN1f/f,L7Cre mice (t30= 0.95, P= 0.35 and t30= 0.66, P= 0.51, respectively, n= 15–17, Student's unpaired t test) or between HCN1−/− and HCN1+/+ mice (t22= 0.48, P= 0.63 and t22= 0.63, P= 0.53, n= 11–13). C, time spent hanging on to a suspended wire and score plotted as a function of session. Performance was not significantly affected by deletion of HCN1 from Purkinje cells (time, genotype X session F1,18= 0.12, P= 0.73; score, genotype X session F1,18= 0.06, P= 0.81, n= 10, two-way ANOVA) or by global deletion of HCN1 (time, genotype X session F1,21= 0.06, P= 0.82; score, genotype X session F1,21= 1.2, P= 0.8228, n= 11–12). D, time spent hanging on to a grid in the inverted grid test. No differences were observed between HCN1f/f and HCN1f/f,L7Cre mice (t18= 1, P= 0.33, Student's unpaired t test) and between HCN1+/+ and HCN1−/− mice (t21= 0.59, P= 0.56). E, Purkinje cell-specific HCN1 deletion did not affect grip strength (forelimbs, t28= 0.27, P= 0.79; hindlimbs, t28= 0.26, P= 0.8, n= 15, Student's unpaired t test). Data are presented as mean ± SEM; *P < 0.05 control (HCN1f/f or HCN1+/+) versus knock-out (HCN1f/f,Cre or HCN1−/−), Fisher's PLSD.
HCN1 channels in Purkinje cells selectively influence integration of inhibitory synaptic inputs
Because deletion of HCN1 channels from Purkinje cells affected only a subset of the behaviours to which these neurones are known to contribute, we wondered whether a similar selectivity of function for HCN1 channels might be manifested at a single-cell level. To address this possibility, we first investigated responses of Purkinje cells to excitatory synaptic input isolated by pharmacological block of GABAA and GABAB receptors. To evoke parallel fibre responses, we positioned stimulating electrodes above or below the dendritic arbor of the recorded Purkinje cell at a distance of 80–140 μm from its cell body. Activation of excitatory postsynaptic potentials following stimulation caused a similar increase in the firing frequency of Purkinje cells from HCN1f/f and HCN1f/f,L7Cre mice (P= 0.94, two-way ANOVA) (Fig. 6A, B). Consistent with a parallel fibre origin, this increase in firing was completely abolished by the GluA antagonist NBQX (Fig. 6A, B). We also found no difference in the amplitude of pharmacologically isolated GluA-mediated parallel fibre-evoked EPSCs (P= 0.95) (Fig. 6C, D). To evoke climbing fibre responses, we moved stimulating electrodes around the white matter or granule cell layer until we evoked all-or-nothing complex spike responses. Deletion of HCN1 in Purkinje neurones also did not affect the properties of the complex spikes evoked by stimulating climbing fibre inputs (P > 0.2, Student's unpaired t test) (Fig. 6E, F). This is in contrast to pharmacological block of Ih, which increases the duration of the pause following a complex spike (Maiz et al. 2012). This difference could be because pharmacological block might also affect HCN1 channels expressed by molecular layer interneurones (Santoro et al. 2000), which are known to contribute to the complex spike pause (Mathews et al. 2012), whereas in HCN1f/f,L7Cre mice HCN1 channels in interneurones are intact (Fig. 2A). Together, these data indicate that HCN1 channels in Purkinje cells do not influence the integration of excitatory inputs in isolation when Purkinje cells are in their physiologically normal spontaneously spiking state. We note that HCN1 channels may still influence responses of Purkinje cells to excitatory inputs, but this will only occur when the membrane potential is hyperpolarized (Angelo et al. 2007).
Figure 6. Excitatory synaptic input is not modified by deletion of HCN1 from Purkinje cells.
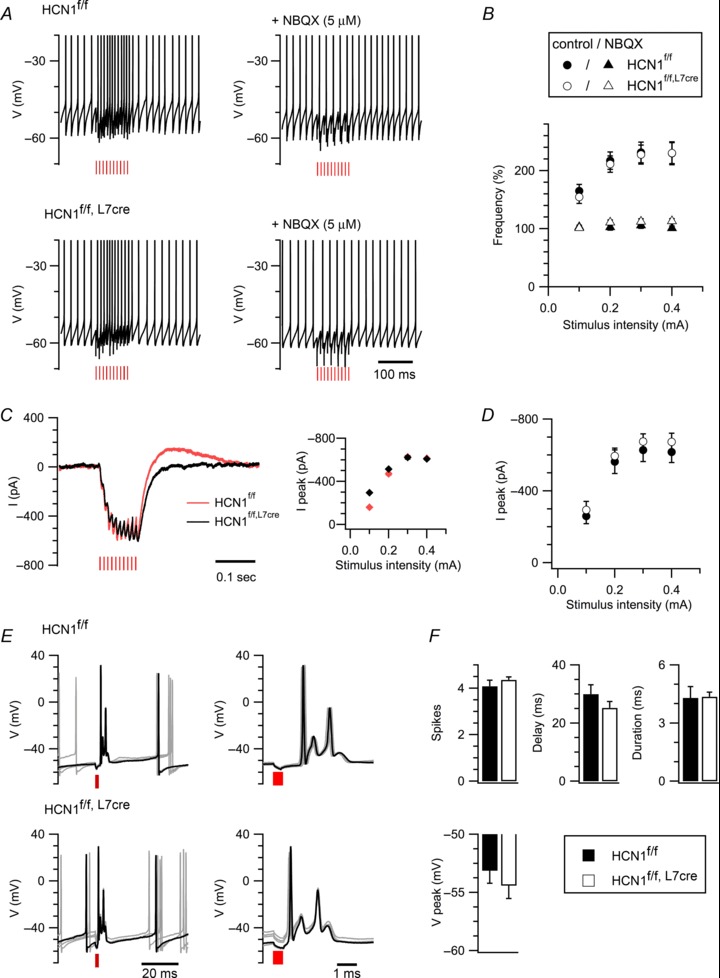
A, responses (left) of Purkinje cells from HCN1f/f (upper) and HCN1f/f,L7Cre mice (lower) to activation of parallel fibres with 10 stimuli at 100 Hz (0.3 mA) are abolished by the GluA antagonist NBQX (right). Action potentials are truncated. B, action potential frequency during parallel fibre stimulation is plotted as a function of stimulus intensity (genotype F1,22= 0.06, P= 0.8; genotype X stimulus intensity F3,66= 0.13, P= 0.94, n= 10–14/group; NBQX F1,14= 85.68, P= 0.0001; genotype X stimulus intensity X NBQX F3,42= 0.09, P= 0.97, n= 7–9, two-way ANOVA). C, examples of membrane currents recorded in voltage-clamp from HCN1f/f and HCN1f/f,Cre Purkinje cells in response to parallel fibre stimulation. Each trace is the pharmacologically isolated GluA current, measured as the difference between the recordings from the same cell in the absence and in the presence of NBQX. The graph plots the peak current as a function of stimulus intensity. Cells were voltage-clamped at –75 mV. The rebound outward current at the end of the train reflects activation of Ih during the train, most likely because of imperfect voltage-clamp. D, the peak current in response to parallel fibre stimulation was similar in HCN1f/f and HCN1f/f,L7Cre mice (genotype F1,16= 0.36, P= 0.56; genotype X stimulus intensity F3,48= 0.11, P= 0.95, n= 8–10, two-way ANOVA). E, examples of complex spike responses of Purkinje cells from HCN1f/f and HCN1f/f,L7Cre mice following stimulation of climbing fibres. On the right the complex spikes are plotted on an extended time scale. F, the number of spikes within the complex spike (t11= 0.28, P= 0.31, Student's unpaired t test), the duration of the pause in spontaneous firing following a complex spike (t11= 4.72, P= 0.23), the duration of the complex spike (t11= 0.12, P= 0.91) and the peak hyperpolarization after a complex spike (t11= 0.78, P= 0.45) were similar in HCN1f/f and HCN1f/f,L7Cre mice (n= 5–8). Red vertical lines denote stimulation. Data are presented as mean ± SEM.
In contrast to the integration of excitatory synaptic inputs, we found that HCN1 channels affect the responses of spontaneously spiking Purkinje cells to inhibitory synaptic input. To evoke inhibitory responses, we positioned stimulating electrodes in the molecular layer, distal to the recorded Purkinje cell (Cohen & Yarom, 1998; Gao et al. 2006; Dizon & Khodakhah, 2011). This induced a transient GABAA receptor-mediated suppression of spontaneous spiking (Fig. 7A–D). Inhibitory suppression of firing lasted significantly longer (P= 0.017, two-way ANOVA) and the membrane hyperpolarization was larger (P < 0.0001, two-way ANOVA) in Purkinje cells from HCN1f/f,L7Cre mice compared with HCN1f/f mice (Fig. 7A, C, E). The duration of the pause in firing was also more variable in HCN1f/f,L7Cre mice (P= 0.04, Student's unpaired t test) (Fig. 7F). These differences are not due to changes in the chloride reversal potential during whole-cell recording, as we found similar results with cell-attached recordings (Fig. 7G). These effects of HCN1 deletion are also not accounted for by changes in synaptic transmission, as the amplitude of IPSCs was not different between HCN1f/f and HCN1f/f,L7Cre mice (P= 0.35, two-way ANOVA) (Fig. 7H). Thus, in spontaneously spiking Purkinje cells, HCN1 channels are engaged during the integration of inhibitory synaptic input. They then act to reduce the duration of pauses in spontaneous action potential firing and limit variability in the timing of the first spike following a pause.
Figure 7. HCN1 channels in cerebellar Purkinje cells control integration of inhibitory synaptic inputs.
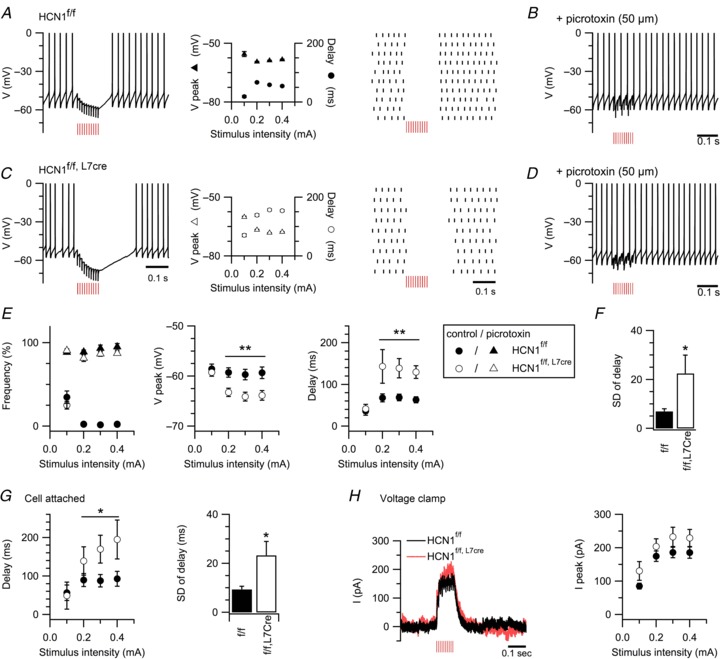
A and C, synaptic inhibition of Purkinje cells from HCN1f/f and HCN1f/f,L7Cre mice in response to 10 stimuli at 100 Hz (0.3 mA). Action potentials are truncated. Duration of firing suppression and peak hyperpolarization are plotted as a function of stimulus intensity (centre). Raster plots (right) show timing of action potentials in 10 consecutive trials repeated at 0.25 Hz. B and D, inhibitory responses are abolished by the GABAA blocker picrotoxin (50 μm). E, firing frequency (left), peak hyperpolarization (middle, genotype F1,29= 7.46, P= 0.01; genotype X stimulus intensity F3,87= 8.47, P < 0.0001) and duration of suppression of spontaneous firing (right, genotype F1,29= 6.6, P= 0.016; genotype X stimulus intensity F3,87= 3.59, P= 0.017), each plotted as a function of stimulus intensity (HCN1f/f,L7Cren= 15, HCN1f/fn= 16, two-way ANOVA). F, variability in the duration of firing inhibition following 0.3 mA stimulation (t29= 2.16, P= 0.04, Student's unpaired t test). G, duration of suppression of spontaneous firing (genotype F1,11= 2.39, P= 0.14; genotype X stimulus intensity F3,33= 9.93, P < 0.0001, two-way ANOVA) and variability in the duration of firing inhibition (t11= 2.98, P= 0.01, Student's unpaired t test) measured with cell-attached recordings of Purkinje cells from HCN1f/f (n= 8) and HCN1f/f,L7Cre mice (n= 5). H, examples of voltage-clamp recordings in response to stimulation of Purkinje cells as in A–D. The graph plots the average peak current as a function of stimulus intensity (genotype F1,12= 1.96, P= 0.18; genotype X stimulus intensity F3,36= 1.13, P= 0.35, n= 7/group, two-way ANOVA). Red vertical lines denote stimulation time. Data are presented as mean ± SEM; *P < 0.05, **P < 0.001 HCN1f/fversus HCN1f/f,L7Cre, Fisher's PLSD or Student's t test.
Discussion
To understand neural computations at a molecular level requires that specific behaviours are mapped onto molecules expressed by single neurone types. We show that when deletion of HCN1 channels is restricted to cerebellar Purkinje cells, late stages of motor learning are impaired but other behaviours are intact. This behavioural phenotype reflects only a subset of behaviours to which Purkinje cells contribute, and is distinct from the phenotype caused by global deletion of HCN1. We find a similar selectivity at a cellular level, where deletion of HCN1 from cerebellar Purkinje cells modifies their responses to inhibitory synaptic input, but not to excitatory synaptic inputs evoked in the absence of hyperpolarization due to synaptic inhibition. Based on this selectivity of HCN1 channel function, we suggest that integrative mechanisms engaged by Purkinje cells may vary between behaviours and at different stages of learning.
While the importance of HCN1 channels for motor learning and other behaviours has previously been established (Nolan et al. 2003; Nolan et al. 2004; Wang et al. 2007), this study is the first to address the role of HCN1 specifically in a genetically defined neurone type – cerebellar Purkinje cells. In contrast to manipulations that abolish the spike output from Purkinje cells (Levin et al. 2006; Chen et al. 2010; Mark et al. 2011), deletion of HCN1 affects a much smaller set of behaviours (Figs 3–5 and Supplementary Figs 5–7). Several observations suggest that the phenotypes we observe are a result of direct loss of HCN1 from Purkinje cells. First, a developmental role of HCN1 is unlikely to explain our results, as in HCN1f/f,L7Cre mice Ih is only reduced from 2 weeks after birth (Fig. 1). Second, cerebellar architecture and the morphology of Purkinje cells in HCN1f/f,L7Cre mice are similar to control mice, also arguing against developmental changes (Fig. 2). Third, deletion of HCN1 from Purkinje cells does not appear to affect their synaptic inputs, as both inhibitory and excitatory synaptic currents were similar in Purkinje cells from HCN1f/f,L7Cre and control mice (Figs 6, 7). Finally, when Purkinje cell function is impaired from early in development, compensatory changes do not prevent much broader deficits than those we observe here (Zuo et al. 1997; Levin et al. 2006; Chen et al. 2010; Mark et al. 2011). Therefore, even if adaptation occurs in HCN1f/f,L7Cre mice, the principle that HCN1 channels in Purkinje cells selectively influence a subset of behaviours that involve these neurones nevertheless holds.
How do behavioural roles of HCN1 channels relate to functions at a cellular level? The roles attributed to any ion channel depend in part on the nature of the spontaneous membrane potential activity of the cell in which they are expressed. We focused our in vitro electrophysiological analysis on functions of HCN1 channels during ongoing simple spike firing by Purkinje cells, as this activity pattern is consistently recorded from Purkinje cells, both in cerebellar slices (Hausser & Clark, 1997; Williams et al. 2002; Nolan et al. 2003; Zonta et al. 2011) and in vivo in the absence of anaesthesia (Schonewille et al. 2006). Under certain conditions, recordings from Purkinje cells in cerebellar slices and in anaesthetized animals have also revealed bistable and multimodal activity patterns involving large membrane potential hyperpolarizations that are likely to engage HCN1 channels (Womack & Khodakhah, 2002; Loewenstein et al. 2005; Oldfield et al. 2010). However, because bistable and multimodal patterns of activity are either very rare or completely absent from Purkinje cells in vivo in the absence of anaesthesia (e.g. Schonewille et al. 2006; Jelitai & Duguid, 2012), their functional relevance is unclear. We therefore consider the relationship between our biophysical and behavioural analysis of the roles of HCN1 channels primarily in the context of ongoing simple spike firing. In this context, a possible function for HCN channels revealed by blocking with ZD7288 is to prevent the emergence of bistability (Williams et al. 2002). However, this observation is not consistent across slice preparations (Maiz et al. 2012), while spontaneous spiking is clearly maintained following genetic deletion of HCN1 (Supplementary Fig. 2 and Nolan et al. 2003). Indeed, while induction of bistability in Purkinje cells would be expected to impact many aspects of cerebellar function, the maintenance of spontaneous spiking in Purkinje cells is consistent with the relatively restricted behavioural phenotypes that we find for HCN1f/f,L7Cre mice.
Our results instead point towards a more specific cellular function of HCN1 in Purkinje cells. HCN1 channels activated by inhibition oppose further hyperpolarization (Fig. 7). In contrast, when excitatory synaptic responses are evoked in the absence of inhibition, and without artificial hyperpolarization of the membrane potential, HCN1 channels are not engaged because the membrane potential is depolarized relative to the voltages at which HCN1 channels activate (Fig. 6). This is consistent with the idea that HCN1 channels ensure that the relationship between the input to and output from Purkinje cells is not modified by hyperpolarization (Nolan et al. 2003). In contrast to this selective engagement of HCN1 channels by synaptic inhibition, when firing of spontaneous action potentials by Purkinje cells is prevented by hyperpolarization of their membrane potential, HCN1 channels suppress summation of excitatory postsynaptic potentials (Angelo et al. 2007). The latter role is similar to the function of HCN channels in many cell types with hyperpolarized resting potentials, in which HCN channels are tonically open and therefore oppose responses to excitatory and inhibitory synaptic inputs (Magee, 1999; Williams & Stuart, 2003). The situation is different for Purkinje cells responding to excitatory input during spontaneous action potential firing (Hausser & Clark, 1997; Nolan et al. 2003; Schonewille et al. 2006; Mittmann & Hausser, 2007), as HCN1 channels are closed and so their influence on excitatory synaptic responses is minimal (Fig. 6). Distinguishing further between these scenarios will require experiments that identify Purkinje cells contributing to specific behaviours and that record their membrane potential before, during and after learning.
The acquisition and performance of learned behaviours requires interaction between neuronal electrical activity and mechanisms that control plasticity of synaptic connections and intrinsic excitability (Paulsen & Moser, 1998; Martin et al. 2000; Gao et al. 2012). Thus, modification of learned behaviours can be brought about by altering either the patterns of activity that lead to induction of plasticity, the plasticity mechanisms that control synaptic efficacy and intrinsic excitability, or the patterns of activity that trigger readout of plasticity. Since HCN1 channels influence electrical signalling, the simplest explanation for the learning deficits caused by deletion of HCN1 from Purkinje cells is that activity patterns required for induction or readout of plasticity are modified by deletion of the channel. A related possibility is that HCN1 channels play direct roles in activity-induced plasticity of intrinsic excitability, which contributes to control of the gain of Purkinje cell responses to parallel fibre input (Belmeguenai et al. 2010). However, whereas this plasticity is manifest as a change in the frequency of spontaneous action potentials, our results suggest that HCN1 channels do not influence spontaneous firing of Purkinje cells. Nevertheless, our data do not rule out the possibility that HCN1 channels in Purkinje cells play a direct role in plasticity mechanisms contributing to cerebellar-dependent learning. Because of the diversity of candidate plasticity mechanisms and uncertainty about their relationship to behaviour (Schonewille et al. 2011; Gao et al. 2012), we have not attempted to address this experimentally.
How do the behavioural consequences of deleting HCN1 relate to the effects of other manipulations targeted to Purkinje cells? Increased efficacy of inhibitory synaptic input to cerebellar Purkinje cells may be particularly important for long-term learning (Dean et al. 2010; Gao et al. 2012). Consistent with this idea, genetic ablation of inputs from inhibitory interneurones to Purkinje cells by deletion of the GABAA receptor γ2 subunit causes specific memory consolidation impairments (Wulff et al. 2009). However, these deficits in plasticity of gain and phase of the VOR are more general (Wulff et al. 2009) compared with those following HCN1 deletion, suggesting that HCN1 channels are not critical for all stages of learning in which inhibition is engaged. These observations suggest a model whereby initial learning involves changes to synaptic input that do not lead to hyperpolarization of Purkinje cells sufficient to activate HCN1, whereas with increased efficacy of inhibitory inputs during iterated long-term learning (Dean et al. 2010; Gao et al. 2012), hyperpolarization of Purkinje cells engages HCN1 channels, which then return the membrane potential towards the spike threshold. A complementary possibility is that Purkinje cells implement different mechanisms for pattern recognition at different stages of learning, for example, using linear integration without pauses in spike firing (Walter & Khodakhah, 2009) or non-linear integration requiring spike pauses (Steuber et al. 2007). In this scenario, HCN1 channels in Purkinje cells might exert their greatest influence on behaviour during membrane potential hyperpolarizations that cause spike pauses, while having less impact on linear integration during tonic action potential firing. Distinguishing these and other possible mechanisms will require a better understanding of the codes used by populations of Purkinje cells to represent information.
Neural computations that account for behaviour are often assumed to be primarily determined by connectivity and synaptic weights, with integrative properties of circuit components simply setting the spike threshold and controlling the gain of synaptic responses. Recent molecular genetic experiments suggest that, instead, ion channels that influence subthreshold integration of synaptic input may enable computations important for behaviour (Nolan et al. 2003; Nolan et al. 2004; Hammond et al. 2006; Wang et al. 2007). The data described here delineate specific behavioural and cellular roles for HCN1 channels in a genetically identified neurone type. While further investigation of the mechanistic relationship between these cellular and behavioural phenotypes is required, our data suggest that the influence of HCN1 channels in Purkinje cells on behaviour is not fixed, but depends on the stage of motor learning. Therefore, as behavioural demands vary, cerebellar Purkinje cells may engage different mechanisms of synaptic integration that can be defined at a molecular level by the contribution of particular ion channels.
Acknowledgments
We thank Dr L. Reichardt and Dr B. Rico for L7Cre mice; Ruth Milne for assistance with radial maze experiments; C. Levenes, C. Meunier, Ian Duguid, Mike Shipston and members of the Nolan laboratory for discussions and comments on the manuscript; and Trudi Gillespie and the IMPACT facility for assistance with imaging.
Glossary
- AP5
D-(2R)-amino-5-phosphonovaleric acid
- HCN
hyperpolarization-activated cyclic nucleotide gated
- Ih
hyperpolarization-activated current
- NBQX
2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione
- PBS
phosphate-buffered saline
- PBST
PBS containing 0.1% Tween 20
- PBST-NGS
PBST containing 1% normal goat serum
- SCB
sodium citrate buffer
- VAF
variance accounted for
- VOR
vestibulo-ocular reflex
- VVC
visuo-vestibular conflict
Additional information
Competing interests
None.
Author contributions
A.R. designed, performed, analysed and interpreted behavioural, electrophysiological and histological experiments; A.M. and M.B. designed, performed and interpreted VOR experiments; C.D. performed and analysed behavioural experiments; D.L.F.G. performed and analysed electrophysiological experiments; M.F.N. conceived the study and designed and interpreted experiments; M.F.N. and A.R. wrote the paper; and M.B. contributed to writing the paper.
Funding
This study was supported by funding from the Medical Research Council (to M.F.N.), Marie Curie (to M.F.N.), NARSAD (to M.F.N.) the Wellcome Trust (to M.F.N.) and Centre National d’Études Spatiales (to M.B.).
Supplementary material
Supporting Information
References
- Angelo K, London M, Christensen SR, Hausser M. Local and global effects of Ih distribution in dendrites of mammalian neurons. J Neurosci. 2007;27:8643–8653. doi: 10.1523/JNEUROSCI.5284-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apps R, Garwicz M. Anatomical and physiological foundations of cerebellar information processing. Nat Rev Neurosci. 2005;6:297–311. doi: 10.1038/nrn1646. [DOI] [PubMed] [Google Scholar]
- Belmeguenai A, Hosy E, Bengtsson F, Pedroarena CM, Piochon C, Teuling E, He Q, Ohtsuki G, De Jeu MT, Elgersma Y, De Zeeuw CI, Jorntell H, Hansel C. Intrinsic plasticity complements long-term potentiation in parallel fibre input gain control in cerebellar Purkinje cells. J Neurosci. 2010;30:13630–13643. doi: 10.1523/JNEUROSCI.3226-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraneck M, Bojados M, Le Seac’h A, Jamon M, Vidal PP. Ontogeny of mouse vestibulo-ocular reflex following genetic or environmental alteration of gravity sensing. PLoS One. 2012;7:e40414. doi: 10.1371/journal.pone.0040414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraneck M, Cullen KE. Activity of vestibular nuclei neurons during vestibular and optokinetic stimulation in the alert mouse. J Neurophysiol. 2007;98:1549–1565. doi: 10.1152/jn.00590.2007. [DOI] [PubMed] [Google Scholar]
- Bolivar VJ. Intrasession and intersession habituation in mice: from inbred strain variability to linkage analysis. Neurobiol Learn Mem. 2009;92:206–214. doi: 10.1016/j.nlm.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, Katoh A, Raymond JL. Cerebellum-dependent learning: the role of multiple plasticity mechanisms. Annu Rev Neurosci. 2004;27:581–609. doi: 10.1146/annurev.neuro.27.070203.144238. [DOI] [PubMed] [Google Scholar]
- Branco T, Clark BA, Hausser M. Dendritic discrimination of temporal input sequences in cortical neurons. Science. 2010;329:1671–1675. doi: 10.1126/science.1189664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Hausser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428:856–860. doi: 10.1038/nature02442. [DOI] [PubMed] [Google Scholar]
- Chen X, Kovalchuk Y, Adelsberger H, Henning HA, Sausbier M, Wietzorrek G, Ruth P, Yarom Y, Konnerth A. Disruption of the olivo-cerebellar circuit by Purkinje neuron-specific ablation of BK channels. Proc Natl Acad Sci USA. 2010;107:12323–12328. doi: 10.1073/pnas.1001745107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D, Yarom Y. Patches of synchronized activity in the cerebellar cortex evoked by mossy-fibre stimulation: questioning the role of parallel fibers. Proc Natl Acad Sci USA. 1998;95:15032–15036. doi: 10.1073/pnas.95.25.15032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepel F, Penit-Soria J. Inward rectification and low threshold calcium conductance in rat cerebellar Purkinje cells. An in vitro study. J Physiol. 1986;372:1–23. doi: 10.1113/jphysiol.1986.sp015993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Zeeuw CI, Hansel C, Bian F, Koekkoek SK, van Alphen AM, Linden DJ, Oberdick J. Expression of a protein kinase C inhibitor in Purkinje cells blocks cerebellar LTD and adaptation of the vestibulo-ocular reflex. Neuron. 1998;20:495–508. doi: 10.1016/s0896-6273(00)80990-3. [DOI] [PubMed] [Google Scholar]
- De Zeeuw CI, Hoebeek FE, Bosman LW, Schonewille M, Witter L, Koekkoek SK. Spatiotemporal firing patterns in the cerebellum. Nat Rev Neurosci. 2011;12:327–344. doi: 10.1038/nrn3011. [DOI] [PubMed] [Google Scholar]
- Dean P, Porrill J, Ekerot CF, Jorntell H. The cerebellar microcircuit as an adaptive filter: experimental and computational evidence. Nat Rev Neurosci. 2010;11:30–43. doi: 10.1038/nrn2756. [DOI] [PubMed] [Google Scholar]
- Dizon MJ, Khodakhah K. The role of interneurons in shaping Purkinje cell responses in the cerebellar cortex. J Neurosci. 2011;31:10463–10473. doi: 10.1523/JNEUROSCI.1350-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in the Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix R, Sandoval A, Sanchez D, Gomora JC, De la Vega-Beltran JL, Trevino CL, Darszon A. ZD7288 inhibits low-threshold Ca2+ channel activity and regulates sperm function. Biochem Biophys Res Commun. 2003;311:187–192. doi: 10.1016/j.bbrc.2003.09.197. [DOI] [PubMed] [Google Scholar]
- Gao W, Chen G, Reinert KC, Ebner TJ. Cerebellar cortical molecular layer inhibition is organized in parasagittal zones. J Neurosci. 2006;26:8377–8387. doi: 10.1523/JNEUROSCI.2434-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, van Beugen BJ, De Zeeuw CI. Distributed synergistic plasticity and cerebellar learning. Nat Rev Neurosci. 2012;13:619–635. doi: 10.1038/nrn3312. [DOI] [PubMed] [Google Scholar]
- Hammond RS, Bond CT, Strassmaier T, Ngo-Anh TJ, Adelman JP, Maylie J, Stackman RW. Small-conductance Ca2+-activated K+ channel type 2 (SK2) modulates hippocampal learning, memory, and synaptic plasticity. J Neurosci. 2006;26:1844–1853. doi: 10.1523/JNEUROSCI.4106-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey RJ, Napper RM. Quantitative studies on the mammalian cerebellum. Prog Neurobiol. 1991;36:437–463. doi: 10.1016/0301-0082(91)90012-p. [DOI] [PubMed] [Google Scholar]
- Hausser M, Clark BA. Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron. 1997;19:665–678. doi: 10.1016/s0896-6273(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Hausser M, Spruston N, Stuart GJ. Diversity and dynamics of dendritic signalling. Science. 2000;290:739–744. doi: 10.1126/science.290.5492.739. [DOI] [PubMed] [Google Scholar]
- Jelitai M, Duguid IC. In Society for Neuroscience. New Orleans, LA: 2012. Membrane potential dynamics of cerebellar Purkinje cells and interneurons during voluntary locomotion. 2012; 477.411. [Google Scholar]
- Kamens HM, Crabbe JC. The parallel rod floor test: a measure of ataxia in mice. Nat Protoc. 2007;2:277–281. doi: 10.1038/nprot.2007.19. [DOI] [PubMed] [Google Scholar]
- Karpova AY, Tervo DG, Gray NW, Svoboda K. Rapid and reversible chemical inactivation of synaptic transmission in genetically targeted neurons. Neuron. 2005;27:727–735. doi: 10.1016/j.neuron.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Koekkoek SK, Yamaguchi K, Milojkovic BA, Dortland BR, Ruigrok TJ, Maex R, De Graaf W, Smit AE, VanderWerf F, Bakker CE, Willemsen R, Ikeda T, Kakizawa S, Onodera K, Nelson DL, Mientjes E, Joosten M, De Schutter E, Oostra BA, Ito M, De Zeeuw CI. Deletion of FMR1 in Purkinje cells enhances parallel fibre LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron. 2005;27:339–352. doi: 10.1016/j.neuron.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Leaton RN, Supple WF., Jr Cerebellar vermis: essential for long-term habituation of the acoustic startle response. Science. 1986;232:513–515. doi: 10.1126/science.3961494. [DOI] [PubMed] [Google Scholar]
- Levin SI, Khaliq ZM, Aman TK, Grieco TM, Kearney JA, Raman IM, Meisler MH. Impaired motor function in mice with cell-specific knockout of sodium channel Scn8a (NaV1.6) in cerebellar Purkinje neurons and granule cells. J Neurophysiol. 2006;27:785–793. doi: 10.1152/jn.01193.2005. [DOI] [PubMed] [Google Scholar]
- Loewenstein Y, Mahon S, Chadderton P, Kitamura K, Sompolinsky H, Yarom Y, Hausser M. Bistability of cerebellar Purkinje cells modulated by sensory stimulation. Nat Neurosci. 2005;8:202–211. doi: 10.1038/nn1393. [DOI] [PubMed] [Google Scholar]
- London M, Hausser M. Dendritic computation. Annu Rev Neurosci. 2005;28:503–532. doi: 10.1146/annurev.neuro.28.061604.135703. [DOI] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Specificity of immunoreactions: the importance of testing specificity in each method. J Neurosci. 2008;28:9083–9086. doi: 10.1523/JNEUROSCI.2494-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan R, Albasanz JL, Shigemoto R, Juiz JM. Preferential localization of the hyperpolarization-activated cyclic nucleotide-gated cation channel subunit HCN1 in basket cell terminals of the rat cerebellum. Eur J Neurosci. 2005;21:2073–2082. doi: 10.1111/j.1460-9568.2005.04043.x. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci. 1999;2:508–514. doi: 10.1038/12229. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic integration of excitatory synaptic input. Nat Rev Neurosci. 2000;1:181–190. doi: 10.1038/35044552. [DOI] [PubMed] [Google Scholar]
- Maiz J, Karakossian MH, Pakaprot N, Robleto K, Thompson RF, Otis TS. Prolonging the postcomplex spike pause speeds eyeblink conditioning. Proc Natl Acad Sci USA. 2012;109:16726–16730. doi: 10.1073/pnas.1214274109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandolesi L, Leggio MG, Graziano A, Neri P, Petrosini L. Cerebellar contribution to spatial event processing: involvement in procedural and working memory components. Eur J Neurosci. 2001;14:2011–2022. doi: 10.1046/j.0953-816x.2001.01819.x. [DOI] [PubMed] [Google Scholar]
- Mark MD, Maejima T, Kuckelsberg D, Yoo JW, Hyde RA, Shah V, Gutierrez D, Moreno RL, Kruse W, Noebels JL, Herlitze S. Delayed postnatal loss of P/Q-type calcium channels recapitulates the absence epilepsy, dyskinesia, and ataxia phenotypes of genomic Cacna1a mutations. J Neurosci. 2011;31:4311–4326. doi: 10.1523/JNEUROSCI.5342-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Mathews PJ, Lee KH, Peng Z, Houser CR, Otis TS. Effects of climbing fibre driven inhibition on Purkinje neuron spiking. J Neurosci. 2012;32:17988–17997. doi: 10.1523/JNEUROSCI.3916-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittmann W, Hausser M. Linking synaptic plasticity and spike output at excitatory and inhibitory synapses onto cerebellar Purkinje cells. J Neurosci. 2007;27:5559–5570. doi: 10.1523/JNEUROSCI.5117-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson SB, Sugino K, Hempel CM. The problem of neuronal cell types: a physiological genomics approach. Trends Neurosci. 2006;29:339–345. doi: 10.1016/j.tins.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsaki G, Siegelbaum SA, Kandel ER, Morozov A. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119:719–732. doi: 10.1016/j.cell.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Lee KH, Gibbs E, Dudman JT, Santoro B, Yin D, Thompson RF, Siegelbaum SA, Kandel ER, Morozov A. The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar Purkinje cells. Cell. 2003;115:551–564. doi: 10.1016/s0092-8674(03)00884-5. [DOI] [PubMed] [Google Scholar]
- Notomi T, Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1–4, in the rat brain. J Comp Neurol. 2004;471:241–276. doi: 10.1002/cne.11039. [DOI] [PubMed] [Google Scholar]
- Nusser Z. Variability in the subcellular distribution of ion channels increases neuronal diversity. Trends Neurosci. 2009;32:267–274. doi: 10.1016/j.tins.2009.01.003. [DOI] [PubMed] [Google Scholar]
- O’Donnell C, Nolan MF. Tuning of synaptic responses: an organizing principle for optimization of neural circuits. Trends Neurosci. 2011;34:51–60. doi: 10.1016/j.tins.2010.10.003. [DOI] [PubMed] [Google Scholar]
- Ogura H, Matsumoto M, Mikoshiba K. Motor discoordination in mutant mice heterozygous for the type 1 inositol 1,4,5-trisphosphate receptor. Behav Brain Res. 2001;122:215–219. doi: 10.1016/s0166-4328(01)00187-5. [DOI] [PubMed] [Google Scholar]
- Oldfield CS, Marty A, Stell BM. Interneurons of the cerebellar cortex toggle Purkinje cells between up and down states. Proc Natl Acad Sci USA. 2010;107:13153–13158. doi: 10.1073/pnas.1002082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen O, Moser EI. A model of hippocampal memory encoding and retrieval: GABAergic control of synaptic plasticity. Trends Neurosci. 1998;21:273–278. doi: 10.1016/s0166-2236(97)01205-8. [DOI] [PubMed] [Google Scholar]
- Paylor R, Crawley JN. Inbred strain differences in prepulse inhibition of the mouse startle response. Psychopharmacology (Berl) 1997;132:169–180. doi: 10.1007/s002130050333. [DOI] [PubMed] [Google Scholar]
- Purves RD. London, New York: Academic Press; 1981. Microelectrode Methods for Intracellular Recording and Ionophoresis. [Google Scholar]
- Reyes A. Influence of dendritic conductances on the input–output properties of neurons. Annu Rev Neurosci. 2001;24:653–675. doi: 10.1146/annurev.neuro.24.1.653. [DOI] [PubMed] [Google Scholar]
- Rico B, Beggs HE, Schahin-Reed D, Kimes N, Schmidt A, Reichardt LF. Control of axonal branching and synapse formation by focal adhesion kinase. Nat Neurosci. 2004;7:1059–1069. doi: 10.1038/nn1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- Sanchez-Alonso JL, Halliwell JV, Colino A. ZD 7288 inhibits T-type calcium current in rat hippocampal pyramidal cells. Neurosci Lett. 2008;439:275–280. doi: 10.1016/j.neulet.2008.05.016. [DOI] [PubMed] [Google Scholar]
- Santoro B, Chen S, Luthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, Siegelbaum SA. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci. 2000;20:5264–5275. doi: 10.1523/JNEUROSCI.20-14-05264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Grant SG, Bartsch D, Kandel ER. Interactive cloning with the SH3 domain of N-src identifies a new brain specific ion channel protein, with homology to eag and cyclic nucleotide-gated channels. Proc Natl Acad Sci USA. 1997;94:14815–14820. doi: 10.1073/pnas.94.26.14815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonewille M, Belmeguenai A, Koekkoek SK, Houtman SH, Boele HJ, van Beugen BJ, Gao Z, Badura A, Ohtsuki G, Amerika WE, Hosy E, Hoebeek FE, Elgersma Y, Hansel C, De Zeeuw CI. Purkinje cell-specific knockout of the protein phosphatase PP2B impairs potentiation and cerebellar motor learning. Neuron. 2010;67:618–628. doi: 10.1016/j.neuron.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonewille M, Gao Z, Boele HJ, Veloz MF, Amerika WE, Simek AA, De Jeu MT, Steinberg JP, Takamiya K, Hoebeek FE, Linden DJ, Huganir RL, De Zeeuw CI. Reevaluating the role of LTD in cerebellar motor learning. Neuron. 2011;70:43–50. doi: 10.1016/j.neuron.2011.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonewille M, Khosrovani S, Winkelman BH, Hoebeek FE, De Jeu MT, Larsen IM, Van der Burg J, Schmolesky MT, Frens MA, De Zeeuw CI. Purkinje cells in awake behaving animals operate at the upstate membrane potential. Nat Neurosci. 2006;9:459–461. doi: 10.1038/nn0406-459. author reply 461. [DOI] [PubMed] [Google Scholar]
- Southan AP, Morris NP, Stephens GJ, Robertson B. Hyperpolarization-activated currents in presynaptic terminals of mouse cerebellar basket cells. J Physiol. 2000;526 (pt 1):91–97. doi: 10.1111/j.1469-7793.2000.t01-1-00091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steuber V, Mittmann W, Hoebeek FE, Silver RA, De Zeeuw CI, Hausser M, De Schutter E. Cerebellar LTD and pattern recognition by Purkinje cells. Neuron. 2007;54:121–136. doi: 10.1016/j.neuron.2007.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl-Schott C, Biel M. HCN channels: structure, cellular regulation and physiological function. Cell Mol Life Sci. 2009;66:470–494. doi: 10.1007/s00018-008-8525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter JT, Khodakhah K. The advantages of linear information processing for cerebellar computation. Proc Natl Acad Sci USA. 2009;106:4471–4476. doi: 10.1073/pnas.0812348106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley A, Nou E, Mazer JA, McCormick DA, Arnsten AF. α2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signalling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Williams SR, Christensen SR, Stuart GJ, Hausser M. Membrane potential bistability is controlled by the hyperpolarization-activated current IH in rat cerebellar Purkinje neurons in vitro. J Physiol. 2002;539:469–483. doi: 10.1113/jphysiol.2001.013136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Stuart GJ. Voltage- and site-dependent control of the somatic impact of dendritic IPSPs. J Neurosci. 2003;23:7358–7367. doi: 10.1523/JNEUROSCI.23-19-07358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack M, Khodakhah K. Active contribution of dendrites to the tonic and trimodal patterns of activity in cerebellar Purkinje neurons. J Neurosci. 2002;22:10603–10612. doi: 10.1523/JNEUROSCI.22-24-10603.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Liao L, Liu X, Luo F, Yang T, Li C. Is ZD7288 a selective blocker of hyperpolarization-activated cyclic nucleotide-gated channel currents. Channels (Austin) 2012;6:438–442. doi: 10.4161/chan.22209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff P, Schonewille M, Renzi M, Viltono L, Sassoe-Pognetto M, Badura A, Gao Z, Hoebeek FE, van Dorp S, Wisden W, Farrant M, De Zeeuw CI. Synaptic inhibition of Purkinje cells mediates consolidation of vestibulo-cerebellar motor learning. Nat Neurosci. 2009;12:1042–1049. doi: 10.1038/nn.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonta B, Desmazieres A, Rinaldi A, Tait S, Sherman DL, Nolan MF, Brophy PJ. A critical role for neurofascin in regulating action potential initiation through maintenance of the axon initial segment. Neuron. 2011;69:945–956. doi: 10.1016/j.neuron.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J, De Jager PL, Takahashi KA, Jiang W, Linden DJ, Heintz N. Neurodegeneration in Lurcher mice caused by mutation inδ2 glutamate receptor gene. Nature. 1997;388:769–773. doi: 10.1038/42009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.