

This paper describes the identification of an exonuclease in a kinetoplastid that is related to the XRN family of exonucleases present in higher eukaryotes. Functional studies demonstrate that knockdown of this exonuclease is not lethal but results in defects in rRNA processing.
Keywords: Ribosome biogenesis, ribonuclease, trypanosome, nucleolus, RNA processing
Abstract
Although biogenesis of ribosomes is a crucial process in all organisms and is thus well conserved, Trypanosoma brucei ribosome biogenesis, of which maturation of rRNAs is an early step, has multiple points of divergence. Our aim was to determine whether in the processing of the pre-rRNA precursor molecule, 5′→3′ exoribonuclease activity in addition to endonucleolytic cleavage is necessary in T. brucei as in other organisms. Our approach initiated with the bioinformatic identification of a putative 5′→3′ exoribonuclease, XRNE, which is highly diverged from the XRN2/Rat1 enzyme responsible for rRNA processing in other organisms. Tagging this protein in vivo allowed us to classify XRNE as nucleolar by indirect immunofluorescence and identify by copurification interacting proteins, many of which were ribosomal proteins, ribosome biogenesis proteins, and/or RNA processing proteins. To determine whether XRNE plays a role in ribosome biogenesis in procyclic form cells, we inducibly depleted the protein by RNA interference. This resulted in the generation of aberrant preprocessed 18S rRNA and 5′ extended 5.8S rRNA, implicating XRNE in rRNA processing. Polysome profiles of XRNE-depleted cells demonstrated abnormal features including an increase in ribosome small subunit abundance, a decrease in large subunit abundance, and defects in polysome assembly. Furthermore, the 5′ extended 5.8S rRNA in XRNE-depleted cells was observed in the large subunit, monosomes, and polysomes in this gradient. Therefore, the function of XRNE in rRNA processing, presumably due to exonucleolytic activity very early in ribosome biogenesis, has consequences that persist throughout all biogenesis stages.
INTRODUCTION
Most aspects of eukaryotic rRNA processing and ribosome biogenesis are conserved. They have been studied in Xenopus, human, and mouse cells, but are understood in greatest detail in yeast (Venema and Tollervey 1999; Fatica and Tollervey 2002; Gerbi et al. 2003). Generation of rRNAs occurs mainly in the nucleolus, where a polycistronic preribosomal RNA (pre-rRNA) transcribed by RNA Polymerase I is processed into mature small subunit (SSU) rRNA (here called 18S), 5.8S rRNA, and large subunit (LSU) rRNA (here called 25/28S), whereas 5S rRNA is transcribed separately in the nucleoplasm by RNA Polymerase III. The matured 18S rRNA is a component of the ribosome SSU, whereas 5.8S, 25/28S, and 5S rRNAs are components of the LSU. Pathways to maturation of rRNAs involve endonucleolytic cleavages, often directed by snoRNA complexes (Kiss 2001), and exonucleolytic trimming of the transcript ends. In yeast, four endonucleotytic cleavage events generate the mature 18S, whereas the processing of large subunit rRNA is more complex and involves both endonucleolytic cleavage and 5′ and 3′ exonucleolytic end trimming (Fatica and Tollervey 2002). Two pathways generate alternative forms of matured 5.8S rRNAs in yeast, metazoans, plants, and other eukaryotes except protozoa; the major short form (5.8SS) comprises 80% of the total 5.8S population and is ∼7 nucleotides (nt) shorter at the 5′ end than the long form (5.8SL) (Rubin 1974; Henry et al. 1994). The significance of this heterogeneity in 5.8S is unclear.
RNA processing often takes an unusual turn in the early diverging eukaryote Trypanosoma brucei, a parasitic protozoan responsible for human African sleeping sickness and Nagana in cattle. T. brucei is known for exotic and unique RNA processing events such as nuclear pre-mRNA trans-splicing and mitochondrial RNA editing (Liang et al. 2003; Simpson et al. 2003). The pre-rRNA of T. brucei itself is different from that in the previously mentioned well-studied eukaryotes, with the normally single 25/28S rRNA fragmented into six matured transcripts (LSUα, LSUβ, LSUγ, LSUδ, LSUϵ, and LSUζ). Additionally, T. brucei 18S rRNA is the largest known so far. Another striking difference between trypanosomatid rRNAs and those of other eukaryotes is that one rather than two forms of 5.8S rRNA are generated (White et al. 1986; Campbell et al. 1987; Hartshorne and Toyofuku 1999). Not surprisingly, along with these differences in rRNA species come differences in T. brucei pre-rRNA processing; and indeed, novel T. brucei factors have already been identified (Jensen et al. 2003, 2005; Hellman et al. 2007). For instance, the processing of pre-rRNA in yeast, humans, and mouse is initiated by cleavage events at the 5′ external transcribed spacer (5′ ETS) (Venema and Tollervey 1999; Fatica and Tollervey 2002; Gerbi et al. 2003); but in trypanosomatids, the initial cleavage event is usually that which separates 18S rRNA from the 5.8S and the 5.8S/LSU rRNAs (Hartshorne and Agabian 1993).
In eukaryotes a nuclear 5′→3′ exoribonuclease termed XRN2/Rat1 (henceforth called Rat1), and the exosome, a 3′→5′ exoribonuclease complex, are responsible for the bulk of trimming required in pre-rRNA maturation (Henry et al. 1994; Geerlings et al. 2000; Houseley et al. 2006). In yeast, Rat1’s roles include trimming the 5′ end of the major 5.8S species from an upstream cleavage site (Henry et al. 1994), trimming the 5′ end of 25/28S rRNA from its upstream cleavage site, and processing the 5′ end of intronic snoRNAs, which guide modification and cleavage events of pre-rRNA, notably upstream of 18S. In addition, it also degrades pre-rRNA spacer fragments, which is vital (Petfalski et al. 1998; Geerlings et al. 2000). We wanted to determine the extent to which the requirement for 5′→3′ exoribonuclease activity for pre-rRNA processing is conserved within the full breadth of eukaryotic evolution, given the known differences in T. brucei rRNAs and their maturation compared to those of well-studied eukaryotes.
5′→3′ exoribonucleases derive from the pfam XRN_N protein family (http://pfam.sanger.ac.uk/). Two major proteins delineate two functional classes of XRN family proteins in Saccharomyces cerevisiae; these are Rat1p and the cytosolic XRN1p. In yeast, conservation between these two well-studied XRNs occurs at the catalytic domain-containing N terminus comprising the first 765 amino acids of Rat1 and the first 671 amino acids of XRN1. Notably, there is no sequence similarity at the C termini among XRN family proteins (Larimer et al. 1992; Kenna et al. 1993).
Four T. brucei XRN family proteins (XRNA through D) were previously identified and partially characterized in Li et al. (2006). All four T. brucei XRNs contained a number of insertions and deletions within the conserved N termini compared to yeast and human XRNs. Further analysis demonstrated that XRNA is most similar to XRN1 in that it functions in mRNA decay in the cytosol, although a fraction of this enzyme is localized to the nucleus as well (Li et al. 2006; Manful et al. 2011). XRNB and XRNC appear cytosolic. Although XRND is nuclear and had the highest sequence similarity to yeast Rat1p, it does not appear to function in ribosomal RNA or snRNA processing (Li et al. 2006). So to date, no enzyme of the XRN family has been linked to pre-rRNA processing in T. brucei. Here, we report the identification of a T. brucei Rat1 functional homolog we term XRNE. XRNE is conserved in kinetoplastids, associates with a number of ribosome biogenesis and ribosomal proteins, localizes to the nucleolus, and is required for proper 5.8S rRNA maturation. Depletion of XRNE in procyclic form T. brucei results in a decreased growth rate, appearance of aberrant preprocessed 18S rRNA, and the generation of 5′ extended 5.8S rRNA that is able to incorporate into the LSU and ribosomes. Aberrant polysome profiles are also observed when XRNE is ablated. Thus, nuclear 5′→3′ exoribonuclease activity and its function in pre-rRNA processing are conserved in trypanosomes.
RESULTS
XRNE is a diverged XRN homolog
None of the four previously characterized T. brucei XRN family members are functional Rat1 homologs with respect to the role of this enzyme in rRNA processing (Li et al. 2006). Thus, we questioned whether 5′→3′ RNA decay activity was a part of normal rRNA precursor processing in trypanosomes. To answer this, we analyzed the T. brucei genome in an attempt to identify additional candidates for such a function. Using a BLAST search of TriTrypDB with the conserved N-terminal sequence of Rat1p (Fig. 1), we noted that, in addition to the four previously identified XRNs, which matched with strong E-values, two other candidates with much weaker E-values of 2.5 × 10−7 (Tb927.5.3850) and 3.2 × 10−8 (Tb11.55.0025) were evident. We termed these proteins, both of which are conserved in T. cruzi and Leishmania major, XRNE and XRNF, respectively. Comparisons of the sequence similarities between the N terminus of Rat1p and those of XRNE and XRNF are shown in Figure 1. Comparison of the conserved amino acids of the previously characterized trypanosome XRNA and XRND enzymes to XRNE and XRNF in Figure 1B emphasizes the highly diverged nature of the two newly identified XRN homologs. Neither XRNE nor XRNF have identifiable domains outside their N-terminal XRN_N domains, so very little function can be predicted based on primary sequence. However, XRNE, but not XRNF, possesses a C-terminal lysine rich region containing a predicted bipartite nuclear localization signal (see below); and thus, we considered this protein a potential player in rRNA processing. In further support of this hypothesis, the N terminus of XRNE displays higher homology to yeast Rat1p (44% identity/63% similarity over the first 655 amino acids of XRNE) than to XRN1 (24% identity/42% similarity over the first 486 amino acids of XRNE).
FIGURE 1.

XRNE and XRNF are newly identified T. brucei homologs of yeast Rat1. (A) Comparison of the N terminus of yeast Rat1 (ScRAT1) with N termini of previously identified T. brucei pfam XRN_N domain-containing proteins exhibiting complete or partial nuclear localization (XRNA and XRND) and with the N termini of newly identified T. brucei XRN domain-containing proteins XRNE and XRNF, which have lower sequence similarity to Rat1. E-values indicated are between Rat1 amino acids 1–765 and each corresponding T. brucei XRN from amino acid 1 to the amino acid displayed at the end of the rectangle representing the aligned region. T. brucei XRNs are shown in order of decreasing sequence similarity. The total protein length in amino acids, which includes both the aligned N terminus and the unaligned C-terminal portion, is given at the far right of the protein schematic. The dotted region denotes the XRN_N domain. The entire XRNF protein aligns to the Rat1 N terminus. (B) Sequence alignment of the XRN domain of Rat1 with the T. brucei XRN domain-containing proteins shown in A. Amino acid conserved between three or more aligned sequences are indicated by black background; a gray background indicates conservative substitutions in three or more sequences. The lengths of the entire proteins are indicated by underlined terminal numbers. Asterisks are indicated below twelve of the thirteen amino acids determined to be crucial to catalysis in Johnson (1997), Page et al. (1998), Solinger et al. (1999), and Yang et al. (2006); the final conserved amino acid is located outside of this region.
Genome-wide analyses identified a single splice site in XRNE RNA 141 nt upstream of the start codon (Kolev et al. 2010; Siegel et al. 2010), thereby predicting an 891-residue, 98-kDa protein. Thirteen amino acids are essential for activity in the XRN1p exoribonuclease (Johnson 1997; Page et al. 1998; Solinger et al. 1999; Yang et al. 2006). All thirteen essential amino acids are conserved in XRNE except Cys201, which is Val243 in XRNE; these are indicated in Figure 1B. A Cys to Val substitution also occurs in T. brucei XRNB-D and in the enzymatically active mouse XRN enzyme Dhm1 (Bashkirov et al. 1997). Thus, although highly diverged, XRNE likely possesses the amino acids essential for exoribonuclease activity.
XRNE is localized to the nucleolus
The cleavage and processing steps required to produce mature rRNAs from the precursor rRNA transcript occur in the nucleolus. Consistent with a role in this process, XRNE is predicted by NucPred (http://www.sbc.su.se/~maccallr/nucpred/cgi-bin/single.cgi) to have 85% probability of nuclear localization and is predicted by motifscan (http://myhits.isb-sib.ch/cgi-bin/motif_scan) to contain a bipartite nuclear localization signal (NLS). However, T. brucei XRNA, B, C, and D all have moderate nuclear localization prediction despite the finding that only XRNA and XRND are nuclear (XRNA is both nuclear and cytosolic) (Li et al. 2006). To definitively determine whether XRNE is present in the nucleus and could therefore have a role in rRNA processing, we performed immunofluorescence microscopy. An exogenous copy of XRNE with Myc, His, and Tandem Affinity Purification (TAP) tags on its C terminus (XRNE-MHT) was inducibly expressed in the 29-13 parental cell line. Figure 2 shows the detection of XRNE-MHT with anti-myc primary and CY5-conjugated secondary antibodies as visualized by fluorescence microscopy. The signal is confined primarily to a region within the nucleus (visualized by DAPI staining) and colocalizes with the signal from the nucleolar marker NOG1 (Park et al. 2001; Jensen et al. 2003). Therefore, XRNE is a nucleolar protein and has the potential to be involved in rRNA processing.
FIGURE 2.
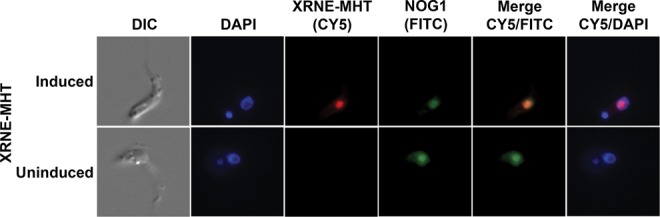
XRNE-MHT is nucleolar. Cells expressing XRNE-MHT in a tet-inducible manner were either induced or left uninduced as described. Expression and localization of XRNE-MHT was determined by immunofluorescence with α-Myc primary and Cy5-conjugated secondary antibody. Likewise, the nucleolar marker NOG1 was detected with α-NOG1 primary and a FITC-conjugated secondary antibody. DAPI staining indicates location of the nuclear DNA and kinetoplast. Appropriate images were merged.
Ribosomal and rRNA processing proteins copurify with XRNE
XRN family enzymes typically interact with other proteins to perform their functions. For example, Rat1p binds proteins such as Rai1 in the nucleus (Stevens and Poole 1995; Xue et al. 2000), and XRN1 is found in P bodies co-localizing with a number of proteins involved in mRNA turnover, notably mRNA decapping protein Dcp2 (Bashkirov et al. 1997). If XRNE acts in ribosome biogenesis, it may associate with other ribosome biogenesis proteins or with the ribosomal proteins themselves. We thus determined whether XRNE associates with other proteins, in part to obtain clues to its function, by performing copurification experiments. XRNE-MHT was tandem affinity (TAP) purified from procyclic form cells and associating proteins identified by LC-MS/MS. A number of additional proteins were identified in the XRNE-MHT preparation. We first excluded those proteins that are common contaminants in T. brucei TAP preparations (Supplemental Table 1; Hashimi et al. 2008; Ammerman et al. 2011; Ouna et al. 2012). Strikingly, associated with TAP-purified XRNE-MHT, we identified numerous ribosomal proteins, proteins reported or annotated as ribosome assembly proteins, and potential RNA processing factors (Supplemental Table 2). Ribosome biogenesis proteins and potential RNA processing proteins that copurified with XRNE are shown in Table 1. Four of the five ribosome biogenesis factors included in Table 1 are putative homologs of yeast ribosome biogenesis proteins, whereas one, P37 (or P34), has been characterized in and is unique to trypanosomes as a factor important for stability of 5S rRNA and ribosome biogenesis (Hellman et al. 2007). This protein is listed as P34/P37 in Table 1 because the recovered peptides are common to both of these almost identical paralogs.
TABLE 1.
Ribosome biogenesis and RNA processing proteins copurify with XRNE-MHT
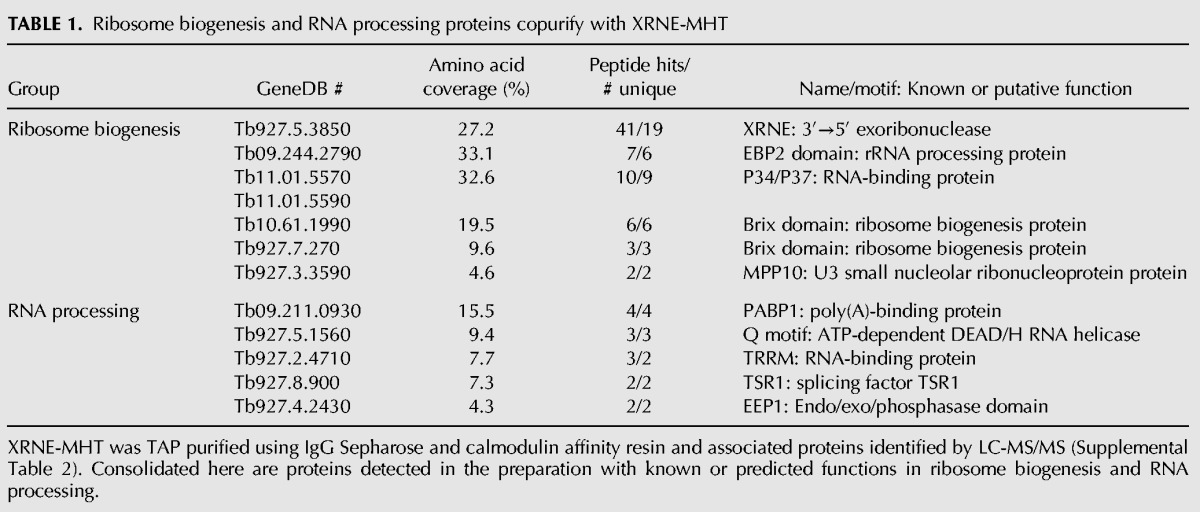
TABLE 2.
Oligonucleotides used in this study

In an attempt to validate the mass spectrometry data, we performed reverse coimmunoprecipitations on two proteins that copurified with XRNE-MHT by TAP, PABP2 (Pitula et al. 1998), and P34/P37 (Zhang and Williams 1997) for which antibodies were readily available, keeping in mind that PABP2 is on our common contaminant list (Supplemental Table 1). However, XRNE-MHT did not reliably coprecipitate with PABP2 or P34/P37 (data not shown). This result suggests that either there are weak or no physical interactions or that the fraction of PABP2 and P34/37 associating with XRNE is very low. The fact that we did identify these proteins in the XRNE mass spectrometry analysis suggests that XRNE, P34/37, and PABP2 are at least sometimes in a similar cellular location.
XRNE is required for normal growth
We next determined whether XRNE is an essential enzyme in trypanosomes. To this end, we generated procyclic form T. brucei cells harboring tetracycline (tet)-induced expression of an RNAi construct targeting XRNE mRNA. We isolated RNA from 29-13 (parental) cells as well as from XRNE RNAi cells with and without 3 d of tet induction of RNAi. qRT-PCR analysis revealed that although XRNE mRNA levels in uninduced RNAi cells were equivalent to parental cells, levels in induced RNAi cells decreased to ∼30% of wild type (Fig. 3, inset). Growth of RNAi cells with and without RNAi induction was monitored over 10 d and compared to that of parental 29-13 cells. We observed a mild growth defect upon XRNE depletion starting at day 3 (compare RNAi induced to uninduced) (Fig. 3). This result is consistent with a genome-wide RNAi-based screen suggesting a mild loss of fitness upon XRNE depletion in procyclic cells. However, greater loss of fitness in bloodstream form cells and during differentiation would be expected based on the same screen (Alsford et al. 2011). We also note that the uninduced XRNE RNAi cell lines grew slower than the parental cells, potentially due to nonspecific effects of transformation. Thus, XRNE is required for normal growth rates in procyclic form T. brucei.
FIGURE 3.

XRNE depletion results in a modest growth phenotype in T. brucei. T. brucei cells harboring tet-inducible RNAi constructs were induced or left uninduced, and growth of these and tet-treated 29-13 parental cells was monitored for 10 d. (Inset) Quantitative RT-PCR demonstrating the relative abundance of XRNE mRNA from the tet-induced and uninduced RNAi cell lines compared to tet-treated 29-13 parental cells. A value of 1 indicates no change in transcript compared to 29-13 cells, whereas negative values indicate the level of depletion of XRNE mRNA. Error bars indicate standard deviation of triplicate determinations.
XRNE depletion leads to accumulation of aberrant 18S and 5.8S rRNAs
Rat1p is involved in the processing of a number of RNAs including rRNA, snRNAs, snoRNAs, and pre-mRNAs in the nucleus (Amberg et al. 1992; Bousquet-Antonelli et al. 2000; Lee et al. 2003; Fang et al. 2005). Of these RNA species, our focus regarding XRNE function was the polycistronic precursor rRNA transcript that is conserved, with some important differences, across the spectrum of life. Uniquely in trypanosomatids, the 25/28S rRNA normally residing as the third gene of the precursor is cleaved into two large and four small fragments (Fig. 4A; fragments beginning with RNA labeled “LSUα”). The 18S, 5.8S, and six LSU rRNAs are matured in a process with an acknowledged requirement for endonucleolytic cleavage and 3′→5′ exoribonucleolytic degradation (White et al. 1986; Campbell et al. 1987; Hartshorne and Toyofuku 1999). However, there has been no explicit evidence that the 5′→3′ processing activity of an XRN enzyme is essential for T. brucei rRNA maturation. Therefore, we asked whether XRNE-depleted cells display an rRNA processing defect. When total RNA was visualized with Ethidium bromide on 8% polyacrylamide or 1.2% agarose gels, we observed no changes in the abundance, sizes, or relative ratios of mature rRNAs at day 3 following induction of XRNE RNAi (Figs 4B,C). Consequently, we used RNA blots to detect potential increases in processing intermediates in XRNE RNAi cells. Transcripts that accumulate in XRNE-depleted cells likely represent those that are normally turned over by XRNE, and new RNAs that appear only in XRNE-depleted cells are presumably abnormal intermediates whose processing requires XRNE. Gray bars in Figure 4A show the locations of the five probes utilized for this study. A probe spanning the 5′ end of the LSUα rRNA and sequences immediately upstream revealed the absence of changes in size or abundance of the detected product upon XRNE depletion (data not shown). Similarly, probes to two intergenic regions within the LSU rRNAs (ITS3 and ITS7) (Table 2) did not reveal a buildup or change in the ratios of precursor rRNA molecules normally observed with these probes upon XRNE depletion (data not shown).
FIGURE 4.

Ablation of XRNE leads to accumulation of preprocessed rRNA species. RNA harvested from XRNE RNAi cells that were tet-induced or uninduced for 2, 3, or 4 d, and from 4-d tet-treated 29-13 cells, was fractionated by electrophoresis and stained with Ethidium bromide (EthBr) for direct visualization of mature rRNAs or subsequent Northern blot analysis. (A) Schematic representation of the T. brucei rRNA transcription unit, probes used for Northern blotting (gray bars below rRNA representations) labeled with italicized letters and identified in the accompanying legend, and processing products observed by Northern blotting in D, E, and F. Approximate sizes of each rRNA representation are indicated next to each schematic. Probes revealing no differences upon XRNE depletion are indicated with an asterisk. (B) Total RNA was electrophoresed on an 8% acrylamide/7M urea gel and stained with EthBr. (C) Total RNA was electrophoresed on a 1.2% formaldehyde agarose gel and stained with EthBr. (D) Northern blot utilizing the probe complementary to the region immediately upstream of 18S (pre-18S probe). Preprocessed products of 3.7, 2.7, and 9.6 kb accumulate in the tet-induced RNAi cell line. (E) RNA fractionated on a 1.2% formaldehyde agarose gel was analyzed with a probe spanning the 5′ end of the 5.8S RNA and upstream sequence (pre-5.8S probe). The 5.9-kb and ∼0.17-kb products that accumulate are noted. (F) RNA fractionated on 8% acrylamide/7M urea gel and analyzed with pre-5.8S probe. A 5′ extended product (ext. 5.8S) is noted. For D and E, EthBr-stained images of 18S and two largest LSU rRNAs from the gel used in that Northern blot are used as a loading control. For F, the loading control is the methylene blue-stained image of the small rRNA species on the membrane subsequently probed with pre-18S.
In contrast, a probe abutting the 5′ end of 18S yet located entirely within the 5′ ETS hybridized to two additional products upon XRNE silencing, as well as to the normal 3.7-kb processing intermediate consisting of 18S and additional upstream and downstream sequence (Fig. 4D). The longer additional fragment is likely the intact or nearly intact precursor rRNA molecule. The smaller additional product is of a size (2.7 kb) consistent with an 18S fragment possessing a cleaved or trimmed 5′ ETS. The T. brucei mature 18S rRNA is generated by a series of three U3 snoRNA-dependent cleavages within the 5′ ETS. Since, in other organisms, Rat1 is required for U3 snoRNA processing, one explanation for the presence of the aberrant SSU fragment is that XRNE normally processes U3 snoRNA. Consequently, we analyzed the abundance and size of the 144-nt U3 snoRNA (Tb927.8.2864) using an oligonucleotide probe that hybridizes to nt 1–30 (Pitula et al. 2002; results not shown). Surprisingly, no differences could be detected, suggesting that XRNE’s role generating the correctly processed 5′ end of 18S may be direct rather than indirect. The observation that a very long product also accumulates suggests that this inappropriate cleavage within the 5′ ETS can occur prior to the cleavage separating the SSU from the LSU rRNAs, normally thought to be the first step of the processing pathway.
We identified an additional function of XRNE using a probe designed to hybridize to the 9 nt immediately adjacent to the 5′ end of 5.8S rRNA and the first 15 nt of 5.8S rRNA. Using this probe, we detected an accumulation of an RNA of approximately the same size as 5.8S and a slight increase in the 5.9-kb precursor, which was not analyzed further (Fig. 4E). Since there was no observable change in the amount of mature 5.8S rRNA on EtBr gels, we hypothesized that the stronger signal represented accumulation of a precursor of similar size rather than an increase in mature transcript. To test this, we repeated the RNA blot using an 8% denaturing polyacrylamide gel with higher resolution (Fig. 4F). Although the mature 5.8S rRNA appears to be fairly equivalent in every sample, a second, higher band appears at 2, 3, and 4 d post-induction of XRNE RNAi. In these RNA blots, the extended 5.8S band is of equal or greater intensity to the normal despite it being far less abundant, in fact unobservable, on the EtBr stain; this is expected, considering the longer region of complementarity this probe shares to the extended product compared to the normal 5.8S rRNA. Based on its location relative to the location of LSUδ detected by methylene blue staining on this blot, the larger 5.8S transcript is not extended by >10 nt. Again, the appearance of this band suggests that endonucleolytic cleavage in the spacer between 18S and 5.8S occurs in a location upstream of the mature 5.8S and requires 5′→3′ exonucleolytic decay by XRNE to generate the proper 5.8S rRNA 5′ end. This is a pathway apparently common to other organisms (Henry et al. 1994; Mullineux and Lafontaine 2012). The fact that the 5.9-kb precursor also increases in abundance upon XRNE RNAi induction again suggests that the exonucleolytic trimming of the 5′ end can occur prior to additional endonucleolytic cleavages of LSU rRNAs but is not a prerequisite for these cleavages.
Both 5.8S rRNA and extended 5.8S can be incorporated into LSU, monosomes, and polysomes
Since aberrantly processed rRNA precursors accumulate upon XRNE depletion, we wanted to know whether these aberrant transcripts participate in later stages of ribosome biogenesis, including those outside of the nucleolus. We first determined whether the 2.7-kb or other preprocessed 18S rRNAs can be incorporated into ribosomes. XRNE RNAi cells induced with tet for 3 d and 29-13 parental cells were fractionated on 10%–40% sucrose polysome gradients. RNA was isolated from each fraction and subsequently analyzed by RNA blot with the pre-18S probe as in Figure 4D. Faint signals with sizes the same as those observed in the total RNA blots were detected in fractions corresponding to higher order complexes from both cell lines (Fig. 5A). That is, RNA from the induced XRNE RNAi cell line contained two preprocessed 18S rRNAs compared to one in 29-13 (Fig. 5A). If these RNAs were properly incorporated into SSU (40S), we would expect to observe them in fractions corresponding in the 40S region of the gradient. However, both the 2.7-kb RNA unique to XRNE RNAi cells and the 3.7-kb RNA present in both RNAi and parental cells occur in 60S fractions. Sedimentation at 60S does not imply incorporation into the LSU that also sediments at 60S but rather suggests the association of these RNAs with a complex of unknown composition. Because the aberrant 2.7- and 3.7-kb RNAs do not associate with complexes in the 40S region of the gradient, it is unlikely that the complexes containing these preprocessed 18S rRNAs are legitimate ribosomal subunits. Additionally, there is no indication that these aberrantly processed 18S rRNAs are incorporated into monosomes (80S) or polysomes.
FIGURE 5.

5′ extended 5.8S rRNA can integrate into ribosomes. Northern blot analysis of RNA isolated from fractions obtained from sucrose gradient fractionation (10%–40%) of induced XRNE RNAi and 29-13 parental cells, with fraction number shown across the top of each set of panels. Lanes indicated with a “C” contain total RNA from tet-induced XRNE RNAi cells and are used as a marker for sizes of aberrantly processed rRNAs. (A) Northern blot analysis of gradient fractions using pre-18S probe. (B) Northern blot analysis of select fractions using the pre-5.8S probe. Fractions corresponding to peaks containing 40S (SSU) and 60S (LSU) subunits, 80S monosomes, and polysomes are indicated. Bottom panels in both A and B are images of the methylene blue staining of the region of the membranes where SSU and two largest LSU rRNAs are located and demonstrate accuracy of fractionation.
In other eukaryotes, the two species of 5.8S rRNA, 5.8SS, and 5.8SL, incorporate into the LSU, monosomes, and polysomes. Only one type of 5.8S has been observed in kinetoplastid wild-type cells, but we demonstrate here (Fig. 4F) that two forms exist when XRNE is depleted. To ascertain whether abnormal, 5′ end-extended, 5.8S rRNAs can be incorporated into T. brucei ribosomes, polysome gradient fractions were analyzed by RNA blot with the pre-5.8S probe. As expected, in the polysome gradient from 29-13 cells, only the matured 5.8S form is present and incorporated into the LSU, monosomes, and polysomes. Interestingly, however, the extended 5.8S, despite being an abnormal rRNA, was incorporated along with the mature 5.8S into all of these complexes in XRNE-depleted cells (Fig. 5B). These data demonstrate that the effects of aberrant processing in the XRNE RNAi cells persist throughout ribosome biogenesis.
Large subunit biogenesis is impaired in XRNE depletion leading to polysome abnormalities
Because XRNE depletion leads to aberrant rRNA processing, we next wanted to ascertain whether the integrity or abundance of the 40S, 60S, 80S, or polysome complexes are affected upon XRNE RNAi. Parental 29-13 cells, as well as uninduced and 3-d induced XRNE RNAi cells, were fractionated on 10%–40% sucrose gradients and their polysome profiles compared (Fig. 6). The 29-13 cells exhibit a typical profile for T. brucei (Jensen et al. 2003, 2005; Hellman et al. 2007), and this profile was identical in the uninduced XRNE RNAi cell line. On the other hand, the profile of the XRNE RNAi cell line shows an increased amount of SSU and decreased amount of LSU compared to the controls. The monosomes for the different cell lines could not be compared because the signal exceeds the highest detection limit. Also, very distinct in the XRNE RNAi profile is the appearance of half-mers, or shoulders, which appear at the polysome peaks. Half-mers arise when small subunits stall at a start codon due to a lack of large subunit competent for subunit joining. This has been observed previously under conditions leading to large subunit defects in both T. brucei (Jensen et al. 2005) and yeast (Rotenberg et al. 1988; Zanchin et al. 1997). This result indicates that XRNE is required for proper LSU biogenesis, SSU-LSU ratios, and assembly of polysomes.
FIGURE 6.
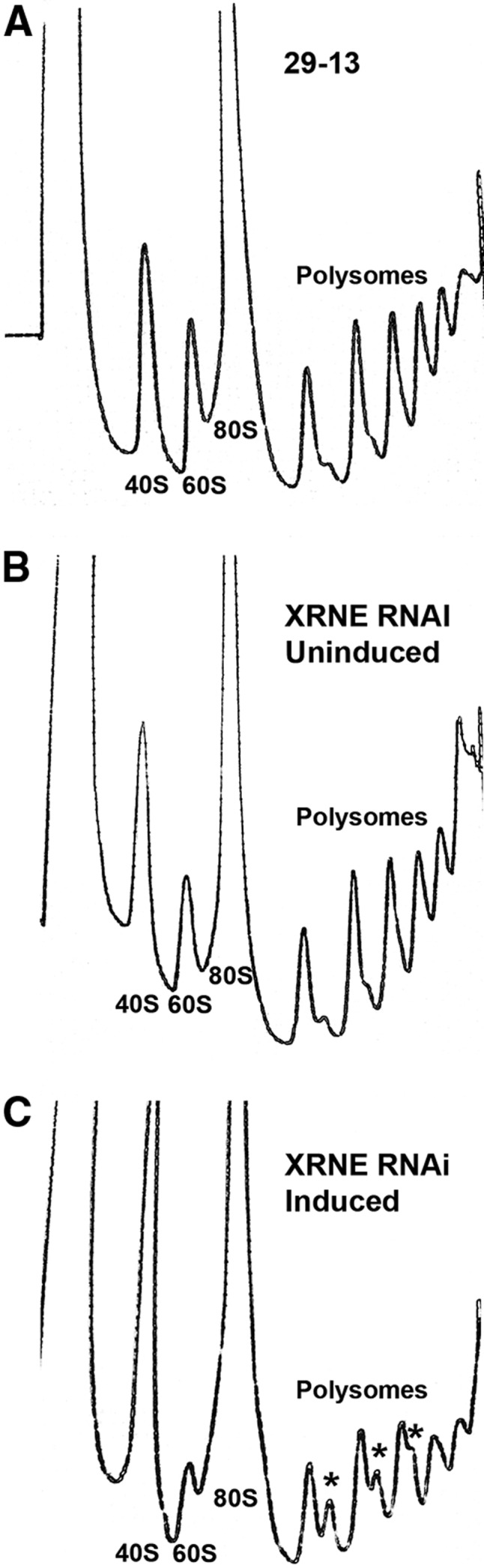
Ribosome biogenesis is impaired upon XRNE depletion. Total cell lysates from 29-13 parental cells (A), uninduced XRNE RNAi cells (B), and tet-induced XRNE RNAi cells (C) were fractionated on 10%–40% sucrose gradients. Absorbance readings were taken at 254 nm, and peaks representing 40S (SSU), 60S (LSU), 80S monosomes, and polysomes are indicated. Half-mer peaks are indicated with asterisks.
DISCUSSION
In this study, we demonstrate that the highly diverged T. brucei XRN family protein XRNE functions in 5′ end processing of rRNAs. That XRNE is, at least in part, a functional homolog of the Rat1/XRN2 enzyme involved in precursor rRNA processing in other organisms is surprising, considering that of the six conserved kinetoplastid XRN family members, two are nuclear and have a much higher sequence similarity to Rat1p than does XRNE (Li et al. 2006). Database searches suggest yeast and most metazoans have only two XRN genes (Chang et al. 2011) with isoforms generated through alternative splicing (Li et al. 2005). Plants and algae have three XRN genes (Kastenmayer and Green 2000; Souret et al. 2004; Zimmer et al. 2008). Thus, of the organisms studied to date, kinetoplastids may have the highest number of XRN family protein coding genes. What roles might proteins of this expanded gene family play in trypanosomatids besides the roles of XRNA and XRNE as the closest functional homologs of XRN1 and XRN2/Rat1? XRNB is apparently not essential in either life cycle stage (Li et al. 2006; Manful et al. 2011), and XRNC likewise has no effect on PF growth (Li et al. 2006). Thus, there could be functional redundancy between at least some members of this family to complicate analysis. A clue to the essentiality of XRNF is an RNAi library screen suggesting that loss of XRNF results in a loss of fitness for procyclic form cells (Alsford et al. 2011), a result that requires validation. Gross differences in mRNA abundance between life cycle stages for XRN-containing genes are not obvious (Siegel et al. 2010; Archer et al. 2011). However, regulation of protein expression for the other XRN family proteins may occur at the level of protein stability or translation, as it does for the mitochondrial ribonuclease TbRND (Zimmer et al. 2011), so we should not rule out stage-specific roles for XRN-containing proteins. Consistent with this possibility, global proteomic analyses identified XRNA, XRND, and XRNF as more abundant in procyclic compared to bloodstream form cells, although the number of peptides found and analyzed was as low in some instances; and XRNF was only observed in one of the studies (Urbaniak et al. 2012; Butter et al. 2013). Finally, there are potential roles for post-translational modifications in altering the function of XRN-containing protein between life cycle stages. Interestingly, phosphorylation of T108 in XRNF is >10-fold more abundant in the procyclic than the bloodstream life cycle stage (Urbaniak et al. 2012), and phosphorylation of XRNA Y343 is >10-fold more abundant in bloodstream form cells.
XRNE-MHT copurifies with a number of ribosomal, ribosome biogenesis, and RNA processing proteins. Although we were not able to validate the XRNE interaction with P34/P37, trypanosome-specific RNA binding protein paralogs involved in the biogenesis of 80S monosomes and interacting with and affecting the abundance of 5S rRNA (Hellman et al. 2007), we note that P34 and P37 are nucleolar, nucleoplasmic, and cytosolic (Prohaska and Williams 2009), whereas we show here that XRNE-MHT appears exclusively nucleolar. Therefore, any XRNE interactions would only occur with a subset of P34 and P37. The interactions may also be mediated by other factors and thus not be direct. Importantly, we note that other nucleolar proteins involved in ribosome biogenesis in trypanosomes did not copurify with XRNE. For example, NOG1 was absent from the XRNE TAP purification and NOPP44/46 was identified with only one peptide and was thus below our stringency cut-off (Jensen et al. 2003, 2005). Thus, at least some of the XRNE interactions we observe are likely due to more than simple proximity or binding on common RNAs. Also of note, XRNE-MHT copurified with two currently uncharacterized proteins of the Brix/Imp4 superfamily, which are known to be involved in the processing of ribosomal RNA (Eisenhaber et al. 2001). Based on sequence similarity, one of these, Tb10.61.1990, is a member of family V, represented by yeast Brx1p. The other is a member of family VI, represented by yeast Rpf2p. Conditional mutations of the essential nucleolar Brx1p and Rpf2p proteins result in large subunit assembly blockages (Kaser et al. 2001; Bogengruber et al. 2003) similar to what is observed with XRNE depletion. Finally, a homolog of the MPP10 subunit of the U3 snoRNP complex that functions in cleavage events resulting in mature 18S rRNAs (Hughes and Ares 1991) also copurified with XRN-MHT. In summary, XRNE associates with other proteins that likely play roles in both small and large ribosome subunit biogenesis.
Our inability to detect XRNE-MHT outside the nucleolus suggests that this protein acts in a transient fashion and does not form a stable complex with ribosomal subunits as they continue maturation outside the nucleus. Other transient players in ribosome biogenesis, such as T. brucei NOPP44/46 and the yeast Rpf2p, migrate in low molecular weight regions on sucrose gradients (Bogengruber et al. 2003; Jensen et al. 2005); their lack of colocalization with ribosome subunits in a sucrose gradient demonstrates a lack of participation in the subunit complexes. XRNE-MHT migrates in the low molecular weight fractions as well (data not shown). This is an additional indication that like NOPP44/46 and Rpf2p, XRNE is not part of a stable structure of a ribosome subunit or monomer but rather functions transiently in early stages of ribosome assembly.
This current study is the first evidence that 5′→3′ exoribonuclease activity of an XRN family protein is required for normal processing of pre-rRNA in T. brucei. One result of XRNE depletion is the accumulation of preprocessed 18S rRNA products. In T. brucei as well as in other organisms, a number of cleavage events at the 5′ ETS generates the mature 5′ end of 18S rRNA, and these cleavages are guided by the U3 snoRNP complex (Hughes and Ares 1991). U3 snoRNA binding sites on 5′ ETS have been mapped in T. brucei (Hartshorne 1998; Hartshorne and Toyofuku 1999) and are required for 5′ ETS cleavage events (Hartshorne and Agabian 1994; Hartshorne 1998; Hartshorne and Toyofuku 1999). Interestingly, although the U3 snoRNA component of the RNP is processed by Rat1p in yeast (Petfalski et al. 1998), we did not observe a role for XRNE in trypanosome U3 processing; rather, XRNE may be directly responsible for removing 5′ leader sequences on the small rRNA fragment. Yet another explanation is that XRNE is required for appropriate recruitment of endonucleolytic activities. It must be emphasized that despite the accumulation of preprocessed 18S rRNA, the integrity of the 40S subunit is not impacted significantly based on the polysome profile.
Depletion of XRNE also led to the appearance of two 5.8S rRNA species—the normal mature form (5.8S rRNA) and another possessing a short 5′ extension (5′ ext 5.8S). Likely, the 5′ extended 5.8S rRNA is an XRNE substrate, and XRNE efficiently processes it to obtain the matured 5.8S in T. brucei. In eukaryotes other than protozoa, two matured 5.8 species (5.8SL and 5.8SS) are generated by two separate pathways and incorporated into polysomes (Fatica and Tollervey 2002). The 5′ end of metazoan 5.8SL is generated by a cleavage event in the preceeding ITS1, whereas the 5′ end of 5.8SS, ∼7 nt shorter than 5.8SL, requires a different cleavage event followed by XRN2/Rat1 trimming at the 5′ end (Henry et al. 1994; Zakrzewska-Placzek et al. 2010). The 5.8S rRNA of trypanosomes is unique in that a single species is produced (Campbell et al. 1987). Based on this work, the parasite utilizes a pathway similar to the 5.8SS rather than the 5.8SL pathway to generate its single 5.8S species. Currently, it is not clear whether the 5.8SS and 5.8SL rRNAs in other organisms have distinct functions (Mullineux and Lafontaine 2012), and perhaps more extensive investigation into the effects of multiple 5.8S species in T. brucei may shed some light on potential effects that a long vs. short 5.8S rRNA could have in ribosomes of those organisms where two are normally present.
A remaining question is the link between the observed growth and ribosome biogenesis defects and XRNE activity. At least some preprocessed 18S rRNA resulting from XRNE depletion is incorporated into a high molecular weight complex, but these complexes are too large to be consistent with SSU that have incorporated preprocessed transcript. The identity of this complex is unknown; therefore, it is not possible to speculate what its impact might be on growth or ribosome assembly. On the other hand, incorporation of the 5′ extended 5.8S rRNA into 60S and 80S complexes and polysomes is potentially detrimental to cells, as only one 5.8S rRNA is normally generated and incorporated into trypanosome 60S subunits. Although the 5.8S rRNA 5′ end is buried within the LSU by visual analysis of the cryo-electron microscopy structure (Hashem et al. 2013), there appears to be space to accommodate an extended 5′ end. A 5′ extension of 5.8S rRNA could result in 5.8S interactions with ribosomal proteins L13A and L3 that are unlikely to exist with the normal form of 5.8S rRNA. Alternately or additionally, an extended 5′ end could change the 5.8S rRNA secondary structure on the 5′ terminus; and since a portion of the L17 protein runs alongside the 5.8S terminus, this could affect interactions with L17 indirectly. Therefore, incorporation of a longer version of 5.8S rRNA into large subunits may result in changes to L3, L13A, and L17 interactions with RNA in the LSU in such a way that large subunit loading on polysomes or translation is decreased. In support of this hypothesis, the polysome profile of XRNE-depleted cells revealed the appearance of half-mers, suggesting a problem with subunit joining on polysomes. Half-mers have previously been observed when ribosome biogenesis proteins such as NOPP44/46 and NOG1 have been depleted, leading to growth defects (Jensen et al. 2003, 2005). Whether it is aberrantly extended 5.8S or 18S rRNAs or an unidentified function of XRNE that contributes to the accumulation of half-mers and/or the growth phenotype of XRNE-depleted cells will be an interesting topic for further study.
MATERIALS AND METHODS
Cloning of XRNE RNAi construct and XRNE-MHT
Oligonucleotides used for cloning are shown in Table 2. The gene encoding the XRNE open reading frame (ORF; Tb927.5.3850) was inserted in pLEW-79MHT plasmid containing a C-terminal myc-6xhis-TAP (MHT) tag (Jensen et al. 2007) between the HindIII and BamHI sites to generate pXRNE-MHT. This allows for tet-regulated expression of XRNE with a C-terminal myc-6x histidine-Tandem Affinity Purification tag (XRNE-MHT) in T. brucei. To generate a tet-inducible XRNE RNAi plasmid (p2T7–177XRNE), a 524 nucleotide fragment (nucleotides 1–524) of the XRNE gene was cloned into p2T7–177 plasmid (Wickstead et al. 2002) between XbaI and HindIII restriction sites internal to opposing T7 promoters.
T. brucei cell culture, transfection, and induction
Procyclic form T. brucei strain 29-13 was grown in SM media supplemented with 10% fetal bovine serum. Not1 linearized pXRNE-MHT was transfected into 29-13 cells using an Amaxa Nucleofector II device with the Basic Parasite I kit, and positive transfectants were selected by phleomycin resistance. Tet-induced cells at 1 × 106 cells/mL starting concentration were harvested at 2 d post-induction for all experiments involving expression of XRNE-MHT.
Similarly, a tet-inducible clonal XRNE RNAi cell line was generated by transfection of 29-13 cells with Not1 linearized p2T7–177XRNE, and positive transformants were selected by addition of puromycin, resulting in three puromycin-resistant polyclonal cultures. A single clone was obtained from one of these polyclonal cultures by limiting dilution. Tet-induced XRNE RNAi cells were harvested at day 3 except where noted otherwise. In all cases, cells were induced with 2.5 µg/mL tet, and for growth curves, cells were induced at a concentration of 5 × 105 cells/mL and diluted as necessary every 24–48 h. Values from three independent growth experiments were averaged to generate growth curves with experimental error bars depicting standard deviation.
TAP-purification of XRNE-MHT
Cells (4.5 × 1010) containing tet-induced XRNE-MHT were lysed on ice for 30 min in 20 mL of IPP150 buffer (Puig et al. 2001) containing 1% Triton X-100 and two tablets of EDTA-free protease inhibitors (Roche). The supernatant after centrifugation at 14,000g for 10 min was considered crude lysate. The crude lysate was treated with 177 units of DNAse1 for 60 min before incubation with 200 μL of the IgG Sepharose Fast Flow beads (Pharmacia) for 4 h at 4°C. After washing with IPP150, beads were equilibrated with TEVCB (Invitrogen) and incubated for 15 h at 4°C with 10 units of TEV protease (Invitrogen) in 1 mL of TEVCB buffer. The TEV eluate was combined with a 3× volume of CBB buffer (Puig et al. 2001) and incubated for 4 h at 4°C with 200 μL calmodulin affinity resin equilibrated with CBB. The tagged XRNE and its interacting partners were eluted with 1 mL of the CEB buffer (Puig et al. 2001) after washing. One hundred microliters of the eluate was concentrated and separated by 10% SDS-PAGE and visualized by silver staining (data not shown). The remaining eluate was sent for LC-MS/MS analysis.
LC-MS/MS analysis
The TAP-purified XRNE-MHT sample was digested overnight with 5 ng/µL trypsin (Promega Corporation) in 50 mM ammonium bicarbonate at 37°C. LC-MS/MS analysis was performed using LTQ Orbitrap mass spectrometer (Thermo Scientific). The LC system configured in a vented format (Licklider et al. 2002) consisted of a fused-silica nanospray needle packed in-house with Magic C18 AQ 100A reverse-phase media (Michrom Bioresources Inc.) (25 cm) and a trap (2 cm) containing Magic C18 AQ 200A reverse-phase media. The peptide samples were loaded onto the column, and chromatographic separation was performed using a two-mobile-phase solvent system consisting of 0.1% formic acid in water and 0.1% acetic acid in acetonitrile. The mass spectrometer operated in a data-dependent MS/MS mode over the m/z range of 400–1800. For each cycle, the five most abundant ions from each MS scan were selected for MS/MS analysis using 35% normalized collision energy. Selected ions were dynamically excluded for 45 sec.
Data analysis
Raw MS/MS data were submitted to the Computational Proteomics Analysis System (CPAS), a web-based system built on the LabKey Server v11.2 (Rauch et al. 2006) and searched using the X!Tandem search engine (Craig and Beavis 2004) against T. brucei protein database v. 4.0 (ftp://ftp.sanger.ac.uk/pub/databases/T.brucei_sequences/T.brucei_genome_v4/), which included additional common contaminants such as human keratin. The search output files were analyzed and validated by ProteinProphet (Nesvizhskii et al. 2003). First, peptide hits were filtered with PeptideProphet (Keller et al. 2002) error rate less than 0.05, and proteins with probability scores of greater than 0.9 were accepted.
Coimmunoprecipitation studies
Crude lysate from 1.4 × 1010 cells expressing XRNE-MHT was prepared as described above and treated with 177 units of DNAse1. The lysate was then incubated with 200 µg Protein A-Sepharose beads crosslinked with either T. brucei anti-P34/P37 rabbit polyclonal antibodies or preimmune serum from the same rabbit (Zhang and Williams 1997). Likewise, beads were prepared with either anti-PABP2 rabbit polyclonal antibodies (α-PABP2) or preimmune serum from the corresponding rabbit (Pitula et al. 2002; called PABP1 in this reference), for 3 h at 4°C. Beads were washed with 50 bed volumes of phosphate-buffered saline (PBS pH 7.5), and bound proteins were eluted with 100 mM triethanolamine (pH 12.5) and dialysed into PBS. Crude lysate (input) and an amount of P34/P37 eluate corresponding to 33% of loaded input or PABP2 eluate corresponding to 16% of total input was separated by 10% SDS-PAGE. Protein was transferred to PVDF membranes and used in immunoblotting experiments. Reaction with Peroxidase-Anti-Peroxidase soluble complex (PAP; Sigma-Aldrich) was used to detect the presence of XRNE-MHT.
qRT-PCR
RNA was isolated from 29-13 cells and uninduced and tet-induced XRNE RNAi cells using RiboZol RNA extraction reagent (Amresco) according to manufacturer’s instructions. Forty to fifty micrograms of isolated RNA was DNAse treated with DNAse 1 (Ambion DNA free kit) and cDNA was prepared from 4 μg RNA using random hexamers and Taqman reverse transcription kit using conditions recommended by manufacturer (Applied Biosystem). qRT-PCR was performed and analyzed as previously (Carnes et al. 2005) using primers shown in Table 2 and iQ5 software (Bio-Rad). cDNA starting quantities were normalized with telomerase reverse transcriptase (TERT) (Brenndorfer and Boshart 2010).
Indirect fluorescence microscopy
After harvest, uninduced and induced XRNE-MHT-expressing cells were treated using a modification of the protocol in Engstler and Boshart (2004). Briefly, cells were fixed in suspension on ice for 30 min with 4% paraformaldehyde and permeabilized as described. Fixed cells were incubated for 1 h with Anti-c-Myc (9E10) mouse monoclonal IgG (Santa Cruz Biotechnology) at a dilution of 1:50 and the nucleolar marker Anti-NOG1 (Park et al. 2001) at a 1:500 dilution. Anti-mouse Cy5 conjugated antibody (Chemicon) at a 1:200 dilution and Anti-rabbit FITC (Millipore) at a 1:100 dilution were used as secondary antibodies in a 30-min incubation, and cells were mounted in ProLong Gold antifade reagent with DAPI (Molecular Probes) as described. A Zeiss Axioimager Z1 Fluorescence microscope and AxioVision software was used to visualize trypanosomes. Cell preparations were also made and visualized lacking either of the primary antibodies to verify that the FITC and Cy5 signals did not overlap (data not shown).
Northern blot analysis
RNA was collected as described for qRT-PCR, with an additional Phenol:Chloroform:Isoamyl Alcohol (25:24:1) pH 5.2 extraction. RNA was then precipitated and resuspended in RNase-free water at a concentration >1 mg/mL. Two micrograms RNA (5 μg in the case of pre-5.8S agarose Northern) was electrophoresed on 1.2% formaldehyde agarose gels or 8% polyacrylamide/7M urea gels and transferred to neutral nylon membranes by capillary transfer (agarose) or positively charged nylon membranes by semidry transfer in 0.5X TBE (polyacrylamide) and subsequently crosslinked with UV illumination. 5′ radiolabeled oligonucleotide probes described in Table 2 were used to probe membranes in ULTRAhyb-Oligo buffer (Ambion) according to the manufacturer’s instructions.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We are grateful to Dr. Marilyn Parsons (Seattle BioMed) for the anti-NOG antibody and Dr. Yuko Ogata (Fred Hutchinson Cancer Research Center) for LC-MS/MS analysis. This work was supported by NIH grants RO1 AI077520 to L.K.R.; RO1 GM92719 to N.W.; and F32 AI092902 to S.L.Z.
REFERENCES
- Alsford S, Turner DJ, Obado SO, Sanchez-Flores A, Glover L, Berriman M, Hertz-Fowler C, Horn D 2011. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res 21: 915–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg DC, Goldstein AL, Cole CN 1992. Isolation and characterization of RAT1: An essential gene of Saccharomyces cerevisiae required for the efficient nucleocytoplasmic trafficking of mRNA. Genes Dev 6: 1173–1189 [DOI] [PubMed] [Google Scholar]
- Ammerman ML, Hashimi H, Novotna L, Cicova Z, McEvoy SM, Lukes J, Read LK 2011. MRB3010 is a core component of the MRB1 complex that facilitates an early step of the kinetoplastid RNA editing process. RNA 17: 865–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer SK, Inchaustegui D, Queiroz R, Clayton C 2011. The cell cycle regulated transcriptome of Trypanosoma brucei. PloS One 6: e18425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashkirov VI, Scherthan H, Solinger JA, Buerstedde JM, Heyer WD 1997. A mouse cytoplasmic exoribonuclease (mXRN1p) with preference for G4 tetraplex substrates. J Cell Biol 136: 761–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogengruber E, Briza P, Doppler E, Wimmer H, Koller L, Fasiolo F, Senger B, Hegemann JH, Breitenbach M 2003. Functional analysis in yeast of the Brix protein superfamily involved in the biogenesis of ribosomes. FEMS Yeast Res 3: 35–43 [DOI] [PubMed] [Google Scholar]
- Bousquet-Antonelli C, Presutti C, Tollervey D 2000. Identification of a regulated pathway for nuclear pre-mRNA turnover. Cell 102: 765–775 [DOI] [PubMed] [Google Scholar]
- Brenndorfer M, Boshart M 2010. Selection of reference genes for mRNA quantification in Trypanosoma brucei. Molecular Biochem Parasitol 172: 52–55 [DOI] [PubMed] [Google Scholar]
- Butter F, Bucerius F, Michel M, Cicova Z, Mann M, Janzen CJ 2013. Comparative proteomics of two life cycle stages of stable isotope-labeled Trypanosoma brucei reveals novel components of the parasite’s host adaptation machinery. Mol Cell Proteomics 12: 172–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DA, Kubo K, Clark CG, Boothroyd JC 1987. Precise identification of cleavage sites involved in the unusual processing of trypanosome ribosomal RNA. J Mol Biol 196: 113–124 [DOI] [PubMed] [Google Scholar]
- Carnes J, Trotter JR, Ernst NL, Steinberg A, Stuart K 2005. An essential RNase III insertion editing endonuclease in Trypanosoma brucei. Proc Natl Acad Sci 102: 16614–16619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HJ, Xiang S, Tong L 2011. 5′-3′ exoribonucleases. In Nucleic acids and molecular biology 26 (ed. AW Nicholson), Vol. 26, pp. 167–192 Springer, Heidelberg [Google Scholar]
- Craig R, Beavis RC 2004. TANDEM: Matching proteins with tandem mass spectra. Bioinformatics 20: 1466–1467 [DOI] [PubMed] [Google Scholar]
- Eisenhaber F, Wechselberger C, Kreil G 2001. The Brix domain protein family—a key to the ribosomal biogenesis pathway? Trends Biochem Sci 26: 345–347 [DOI] [PubMed] [Google Scholar]
- Engstler M, Boshart M 2004. Cold shock and regulation of surface protein trafficking convey sensitization to inducers of stage differentiation in Trypanosoma brucei. Genes Dev 18: 2798–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang F, Phillips S, Butler JS 2005. Rat1p and Rai1p function with the nuclear exosome in the processing and degradation of rRNA precursors. RNA 11: 1571–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A, Tollervey D 2002. Making ribosomes. Curr Opin Cell Biol 14: 313–318 [DOI] [PubMed] [Google Scholar]
- Geerlings TH, Vos JC, Raué HA 2000. The final step in the formation of 25S rRNA in Saccharomyces cerevisiae is performed by 5′→3′ exonucleases. RNA 6: 1698–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbi SA, Borovjagin AV, Lange TS 2003. The nucleolus: A site of ribonucleoprotein maturation. Curr Opin Cell Biol 15: 318–325 [DOI] [PubMed] [Google Scholar]
- Hartshorne T 1998. Distinct regions of U3 snoRNA interact at two sites within the 5′ external transcribed spacer of pre-rRNAs in Trypanosoma brucei cells. Nucleic Acids Res 26: 2541–2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorne T, Agabian N 1993. RNA B is the major nucleolar trimethylguanosine-capped small nuclear RNA associated with fibrillarin and pre-rRNAs in Trypanosoma brucei. Mol Cell Biol 13: 144–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorne T, Agabian N 1994. A common core structure for U3 small nucleolar RNAs. Nucleic Acids Res 22: 3354–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorne T, Toyofuku W 1999. Two 5′-ETS regions implicated in interactions with U3 snoRNA are required for small subunit rRNA maturation in Trypanosoma brucei. Nucleic Acids Res 27: 3300–3309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashem Y, des Georges A, Fu J, Buss SN, Jossinet F, Jobe A, Zhang Q, Liao HY, Grassucci RA, Bajaj C, et al. 2013. High-resolution cryo-electron microscopy structure of the Trypanosoma brucei ribosome. Nature 494: 385–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimi H, Zikova A, Panigrahi AK, Stuart KD, Lukes J 2008. TbRGG1, an essential protein involved in kinetoplastid RNA metabolism that is associated with a novel multiprotein complex. RNA 14: 970–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman KM, Ciganda M, Brown SV, Li J, Ruyechan W, Williams N 2007. Two trypanosome-specific proteins are essential factors for 5S rRNA abundance and ribosomal assembly in Trypanosoma brucei. Eukaryot Cell 6: 1766–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry Y, Wood H, Morrissey JP, Petfalski E, Kearsey S, Tollervey D 1994. The 5′ end of yeast 5.8S rRNA is generated by exonucleases from an upstream cleavage site. EMBO J 13: 2452–2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseley J, LaCava J, Tollervey D 2006. RNA-quality control by the exosome. Nat Rev Mol Cell Biol 7: 529–539 [DOI] [PubMed] [Google Scholar]
- Hughes JM, Ares M Jr 1991. Depletion of U3 small nucleolar RNA inhibits cleavage in the 5′ external transcribed spacer of yeast pre-ribosomal RNA and impairs formation of 18S ribosomal RNA. EMBO J 10: 4231–4239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen BC, Wang Q, Kifer CT, Parsons M 2003. The NOG1 GTP-binding protein is required for biogenesis of the 60 S ribosomal subunit. J Biol Chem 278: 32204–32211 [DOI] [PubMed] [Google Scholar]
- Jensen BC, Brekken DL, Randall AC, Kifer CT, Parsons M 2005. Species specificity in ribosome biogenesis: A nonconserved phosphoprotein is required for formation of the large ribosomal subunit in Trypanosoma brucei. Eukaryot Cell 4: 30–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen BC, Kifer CT, Brekken DL, Randall AC, Wang Q, Drees BL, Parsons M 2007. Characterization of protein kinase CK2 from Trypanosoma brucei. Mol Biochem Parasitol 151: 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AW 1997. Rat1p and Xrn1p are functionally interchangeable exoribonucleases that are restricted to and required in the nucleus and cytoplasm, respectively. Mol Cell Biol 17: 6122–6130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A, Bogengruber E, Hallegger M, Doppler E, Lepperdinger G, Jantsch M, Breitenbach M, Kreil G 2001. Brix from Xenopus laevis and Brx1p from yeast define a new family of proteins involved in the biogenesis of large ribosomal subunits. Biol Chem 382: 1637–1647 [DOI] [PubMed] [Google Scholar]
- Kastenmayer JP, Green PJ 2000. Novel features of the XRN-family in Arabidopsis: Evidence that AtXRN4, one of several orthologs of nuclear Xrn2p/Rat1p, functions in the cytoplasm. Proc Natl Acad Sci 97: 13985–13990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74: 5383–5392 [DOI] [PubMed] [Google Scholar]
- Kenna M, Stevens A, McCammon M, Douglas MG 1993. An essential yeast gene with homology to the exonuclease-encoding XRN1/KEM1 gene also encodes a protein with exoribonuclease activity. Mol Cell Biol 13: 341–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T 2001. Small nucleolar RNA-guided post-transcriptional modification of cellular RNAs. EMBO J 20: 3617–3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolev NG, Franklin JB, Carmi S, Shi H, Michaeli S, Tschudi C 2010. The transcriptome of the human pathogen Trypanosoma brucei at single-nucleotide resolution. PLoS Pathog 6: e1001090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larimer FW, Hsu CL, Maupin MK, Stevens A 1992. Characterization of the XRN1 gene encoding a 5′→3′ exoribonuclease: Sequence data and analysis of disparate protein and mRNA levels of gene-disrupted yeast cells. Gene 120: 51–57 [DOI] [PubMed] [Google Scholar]
- Lee CY, Lee A, Chanfreau G 2003. The roles of endonucleolytic cleavage and exonucleolytic digestion in the 5′-end processing of S. cerevisiae box C/D snoRNAs. RNA 9: 1362–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zheng H, Ji C, Fei X, Zheng M, Gao Y, Ren Y, Gu S, Xie Y, Mao Y 2005. A novel splice variant of human XRN2 gene is mainly expressed in blood leukocyte. DNA Seq 16: 143–146 [DOI] [PubMed] [Google Scholar]
- Li CH, Irmer H, Gudjonsdottir-Planck D, Freese S, Salm H, Haile S, Estévez AM, Clayton C 2006. Roles of a Trypanosoma brucei 5′→3′ exoribonuclease homolog in mRNA degradation. RNA 12: 2171–2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Haritan A, Uliel S, Michaeli S 2003. trans and cis splicing in trypanosomatids: Mechanism, factors, and regulation. Eukaryot Cell 2: 830–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licklider LJ, Thoreen CC, Peng J, Gygi SP 2002. Automation of nanoscale microcapillary liquid chromatography-tandem mass spectrometry with a vented column. Anal Chem 74: 3076–3083 [DOI] [PubMed] [Google Scholar]
- Manful T, Fadda A, Clayton C 2011. The role of the 5′–3′ exoribonuclease XRNA in transcriptome-wide mRNA degradation. RNA 17: 2039–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullineux ST, Lafontaine DL 2012. Mapping the cleavage sites on mammalian pre-rRNAs: Where do we stand? Biochimie 94: 1521–1532 [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R 2003. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75: 4646–4658 [DOI] [PubMed] [Google Scholar]
- Ouna BA, Stewart M, Helbig C, Clayton C 2012. The Trypanosoma brucei CCCH zinc finger proteins ZC3H12 and ZC3H13. Mol Biochem Parasitol 183: 184–188 [DOI] [PubMed] [Google Scholar]
- Page AM, Davis K, Molineux C, Kolodner RD, Johnson AW 1998. Mutational analysis of exoribonuclease I from Saccharomyces cerevisiae. Nucleic Acids Res 26: 3707–3716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Jensen BC, Kifer CT, Parsons M 2001. A novel nucleolar G-protein conserved in eukaryotes. J Cell Sci 114: 173–185 [DOI] [PubMed] [Google Scholar]
- Petfalski E, Dandekar T, Henry Y, Tollervey D 1998. Processing of the precursors to small nucleolar RNAs and rRNAs requires common components. Mol Cell Biol 18: 1181–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitula J, Ruyechan WT, Williams N 1998. Trypanosoma brucei: Identification and purification of a poly(A)-binding protein. Exp Parasitol 88: 157–160 [DOI] [PubMed] [Google Scholar]
- Pitula J, Ruyechan WT, Williams N 2002. Two novel RNA binding proteins from Trypanosoma brucei are associated with 5S rRNA. Biochem Biophys Res Commun 290: 569–576 [DOI] [PubMed] [Google Scholar]
- Prohaska K, Williams N 2009. Assembly of the Trypanosoma brucei 60S ribosomal subunit nuclear export complex requires trypanosome-specific proteins P34 and P37. Eukaryot Cell 8: 77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Séraphin B 2001. The tandem affinity purification (TAP) method: A general procedure of protein complex purification. Methods 24: 218–229 [DOI] [PubMed] [Google Scholar]
- Rauch A, Bellew M, Eng J, Fitzgibbon M, Holzman T, Hussey P, Igra M, Maclean B, Lin CW, Detter A, et al. 2006. Computational Proteomics Analysis System (CPAS): An extensible, open-source analytic system for evaluating and publishing proteomic data and high throughput biological experiments. J Proteome Res 5: 112–121 [DOI] [PubMed] [Google Scholar]
- Rotenberg MO, Moritz M, Woolford JL Jr 1988. Depletion of Saccharomyces cerevisiae ribosomal protein L16 causes a decrease in 60S ribosomal subunits and formation of half-mer polyribosomes. Genes Dev 2: 160–172 [DOI] [PubMed] [Google Scholar]
- Rubin GM 1974. Three forms of the 5.8-S ribosomal RNA species in Saccharomyces cerevisiae. Eur J Biochem 41: 197–202 [DOI] [PubMed] [Google Scholar]
- Siegel TN, Hekstra DR, Wang X, Dewell S, Cross GA 2010. Genome-wide analysis of mRNA abundance in two life-cycle stages of Trypanosoma brucei and identification of splicing and polyadenylation sites. Nucleic Acids Res 38: 4946–4957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson L, Sbicego S, Aphasizhev R 2003. Uridine insertion/deletion RNA editing in trypanosome mitochondria: A complex business. RNA 9: 265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinger JA, Pascolini D, Heyer WD 1999. Active-site mutations in the Xrn1p exoribonuclease of Saccharomyces cerevisiae reveal a specific role in meiosis. Mol Cell Biol 19: 5930–5942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souret FF, Kastenmayer JP, Green PJ 2004. AtXRN4 degrades mRNA in Arabidopsis and its substrates include selected miRNA targets. Mol Cell 15: 173–183 [DOI] [PubMed] [Google Scholar]
- Stevens A, Poole TL 1995. 5′-exonuclease-2 of Saccharomyces cerevisiae. Purification and features of ribonuclease activity with comparison to 5′-exonuclease-1. J Biol Chem 270: 16063–16069 [DOI] [PubMed] [Google Scholar]
- Urbaniak MD, Guther ML, Ferguson MA 2012. Comparative SILAC proteomic analysis of Trypanosoma brucei bloodstream and procyclic lifecycle stages. PloS One 7: e36619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venema J, Tollervey D 1999. Ribosome synthesis in Saccharomyces cerevisiae. Annu Rev Genet 33: 261–311 [DOI] [PubMed] [Google Scholar]
- White TC, Rudenko G, Borst P 1986. Three small RNAs within the 10 kb trypanosome rRNA transcription unit are analogous to Domain VII of other eukaryotic 28S rRNAs. Nucleic Acids Res 14: 9471–9489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickstead B, Ersfeld K, Gull K 2002. Targeting of a tetracycline-inducible expression system to the transcriptionally silent minichromosomes of Trypanosoma brucei. Mol Biochem Parasitol 125: 211–216 [DOI] [PubMed] [Google Scholar]
- Xue Y, Bai X, Lee I, Kallstrom G, Ho J, Brown J, Stevens A, Johnson AW 2000. Saccharomyces cerevisiae RAI1 (YGL246c) is homologous to human DOM3Z and encodes a protein that binds the nuclear exoribonuclease Rat1p. Mol Cell Biol 20: 4006–4015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Lee JY, Nowotny M 2006. Making and breaking nucleic acids: Two-Mg2+-ion catalysis and substrate specificity. Mol Cell 22: 5–13 [DOI] [PubMed] [Google Scholar]
- Zakrzewska-Placzek M, Souret FF, Sobczyk GJ, Green PJ, Kufel J 2010. Arabidopsis thaliana XRN2 is required for primary cleavage in the pre-ribosomal RNA. Nucleic Acids Res 38: 4487–4502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanchin NI, Roberts P, DeSilva A, Sherman F, Goldfarb DS 1997. Saccharomyces cerevisiae Nip7p is required for efficient 60S ribosome subunit biogenesis. Mol Cell Biol 17: 5001–5015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Williams N 1997. Purification, cloning, and expression of two closely related Trypanosoma brucei nucleic acid binding proteins. Mol Biochem Parasitol 87: 145–158 [DOI] [PubMed] [Google Scholar]
- Zimmer SL, Fei Z, Stern DB 2008. Genome-based analysis of Chlamydomonas reinhardtii exoribonucleases and poly(A) polymerases predicts unexpected organellar and exosomal features. Genetics 179: 125–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer SL, McEvoy SM, Li J, Qu J, Read LK 2011. A novel member of the RNase D exoribonuclease family functions in mitochondrial guide RNA metabolism in Trypanosoma brucei. J Biol Chem 286: 10329–10340 [DOI] [PMC free article] [PubMed] [Google Scholar]