

Abstract
The centrality of phosphatidylinositol-3-kinase (PI3K) in cancer etiology is well established, but clinical translation of PI3K inhibitors has been limited by feedback signaling, suboptimal intra-tumoral concentration and an insulin resistance ‘class effect’. The current study was designed to explore the use of supramolecular nanochemistry for targeting PI3K to enhance antitumor efficacy and potentially overcome these limitations. PI3K inhibitor structures were rationally modified using a cholesterol-based derivative, facilitating supramolecular nanoassembly with L-α-phosphatidylcholine and DSPE-PEG. The supramolecular nanoparticles that were assembled were physicochemically characterized and functionally evaluated in vitro. Antitumor efficacy was quantified in vivo using 4T1 breast cancer and K-RasLSL/+/Ptenfl/fl ovarian cancer models, with effects on glucose homeostasis evaluated using an insulin sensitivity test. The use of PI103 and PI828 as surrogate molecules to engineer the supramolecular nanoparticles highlighted the need to keep design principles in perspective; specifically, potency of the active molecule and the linker chemistry were critical principles for efficacy, similar to antibody-drug conjugates. We found that the supramolecular nanoparticles exerted a temporally-sustained inhibition of phosphorylation of Akt, mTOR, S6K and 4EBP in vivo. These effects were associated with increased antitumor efficacy and survival as compared with PI103 and PI828. Efficacy was further increased by decorating the nanoparticle surface with tumor-homing peptides. Notably, the use of supramolecular nanoparticles abrogated the insulin resistance that has been associated widely with other PI3K inhibitors. This study provides a preclinical foundation for the use of supramolecular nanochemistry to overcome current challenges associated with PI3K inhibitors, offering a paradigm for extension to other molecularly targeted therapeutics being explored for cancer treatment.
Introduction
According to the World Health Organization, mortality due to cancer is expected to increase from 7.6 million in 2008 to 12 million deaths in 2030 (1). To address this growing problem, two emerging paradigms driving the evolution of newer treatment strategies are: 1. Better understanding of oncogenic drivers, leading to the development of molecularly ‘targeted’ therapeutics (2, 3); and 2. The use of nanotechnology to deliver cytotoxic drugs specifically to the tumor, resulting in improved therapeutic index (4, 5). However the interface between these two paradigms, which can offer unique opportunities for improving chemotherapeutic outcomes, currently remains largely underexplored. In this study, we demonstrate the potential advantages of bringing these two paradigms together through the rational design of supramolecular nanoparticles that target the PI3K pathway.
The centrality of the PI3K family of lipid kinases in the etiology of cancer in well established (6). Of the three classes of PI3K, Class IA PI3K is most implicated in driving human cancers (7). PIK3CA and PIK3R1, which encode the p110α catalytic subunit and the regulatory p85a subunit of PI3K respectively, are somatically mutated or amplified in multiple primary cancers, including of breast and ovarian origins (7). Similarly, the lipid phosphatase PTEN, an inhibitor of PI3K signaling, is a commonly inactivated tumor suppressor (8). Activation of this pathway can also occur upstream at the level of mutated or amplified tyrosine receptor kinase or downstream through mutations of AKT and RAS (7). Consequently, small molecule inhibitors that target PI3K pathway have emerged as an exciting area of research, and several molecules that either inhibit specific catalytic subunits (α, β, δ, γ) of p110 or act as pan-PI3K inhibitors are currently in development (9). However, recent studies have implicated p110α as also playing a predominant role in glucose homeostasis (10). Indeed, recent data from a Phase I clinical study with a pan-class I selective PI3K inhibitor (NVP-BKM120) showed dose-dependent hyperglycemia, possibly an example of a class effect consistent with PI3K inhibition (11). Furthermore, studies have reported that approximately 10-fold higher concentration of PI3K inhibitors might be required to block phosphorylation of downstream pathway proteins (such as ribosomal protein S6) than that needed for inhibiting more proximal AKT phosphorylation (12). We rationalized that a natural approach to overcome thesechallenges associated with targeting the PI3K pathway is through the use of nanotechnology.
Nanovectors capitalize on the unique leaky angiogenic tumor vasculature to preferentially home to tumors, which coupled with impaired lymphatic drainage results in increased intratumoral drug concentrations (termed as the enhanced permeation and retention or EPR effect) (13). However, traditional processes for nanoformulation are often incompatible with physicochemical properties of many chemotherapeutic agents, which can limit the entrapment efficiency or introduce sub-optimal release kinetics. Indeed, our early attempts in entrapping LY294002, one of the earliest PI3K inhibitors, resulted in sub-optimal loading efficiency that prevented translation to in vivo tumor efficacy studies (14). Similarly, in a recent study, wortmannin-encapsulated polymeric nanoparticles were shown to act as a radiosensitizer (15), but such formulations are limited by burst release, which complicate clinical translation. We rationalized that this can be addressed using supramolecular nanochemistry (16), i.e. evolution of complex nanostructures from molecular building blocks interacting via non-covalent intermolecular force (17, 18). Indeed, supramolecular nanochemistry is an emerging concept in cancer theranostics; for example, in a recent study, gandolinium (III)-encapsulated supramolecular nanoparticles were used in diagnosis of cancer metastasis (19). Here we report that rational modification of PI3K inhibitors facilitates supramolecular assembly in the nanoscale dimension. Such PI3K-targeting supramolecular nanoparticles (SNPs) exhibit the desired pharmacodynamic profile with enhanced antitumor efficacy, and can emerge as a new paradigm in targeted molecular therapeutics development.
Materials and Methods
Dichloromethane (DCM), anhydrous DCM, Methanol, Cholesterol, Dimethylamino Pyridine (DMAP), Succinic Anhydride, Sodium Sulfate, Pyridine, 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), L-α-phosphatidylcholine and Sephadex G-25 were purchased from Sigma-Aldrich (all analytical grades). PI103 and PI828 were obtained from Selleckchem and Tocris Biosciences respectively. 1,2-Distearoyl-sn-Glycero-3-Phosphoethanolamine-N-[Amino(Polythylene Glycol)2000], mini handheld Extruder kit was purchased from Avanti Polar Lipids Inc. 1H spectra were recorded on Bruker DPX 400MHz spectrometer. Chemical shifts are reported in δ (ppm) units using residual 1H signals from deuterated solvents as references. Spectra were analyzed with Mest-Re-C Lite (Mestrelab Research) and/or XWinPlot (Bruker Biospin) softwares. Electrospray ionization mass spectra were recorded on a Micromass Q Tof 2 (Waters) and data were analyzed with MassLynx 4.0 software (Waters). 4T1 and MDA-MB-231s cell lines were obtained ATCC and used within 6 months of resuscitation of frozen stock.
Synthesis of PI103-cholesterol conjugate
Cholesterol (500 mg, 1.29 mmol) was dissolved in 5 ml of anhydrous pyridine. Succinic anhydride (645 mg, 6.45 mmol) and catalytic amount of DMAP was added to the reaction mixture to form a clear solution. The reaction mixture was stirred under argon atmosphere for 12h. Pyridine was then removed under vacuum and the crude residue was diluted in 30 ml DCM. It was washed with 1N HCl (30 ml) and water (30 ml), and the organic layer was separated and dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. Completion of the reaction was confirmed by TLC in 1:99 Methanol:DCM solvent mixture. PI103 (25 mg, 0.072 mmol) was dissolved in 3 ml anhydrous DCM followed by addition of cholesterol-succinic acid (0.216 mmol, 105 mg), EDC (0.216 mmol, 41.4 mg) and DMAP (0.216mmol, 26 mg). The reaction mixture was stirred at room temperature for 12h under argon. Upon completion of reaction as monitored by TLC, the reaction mixture was diluted with 10 ml DCM and washed with dilute HCl and water. The organic layers were separated, combined and dried over anhydrous sodium sulfate. The solvent was evaporated under vacuum and the crude product was purified by using column chromatography, eluting with methanol:methylene chloride gradient, to give PI103 cholesterol conjugate as a light yellow solid (52 mg, 90 %). 1H NMR (CDCl3, 400MHz): δ 8.65 – 8.53 (m, 1H), 8.36 (d, J = 8.3 Hz, 1H), 8.19 (d, J = 1.7 Hz, 1H), 7.56 – 7.41 (m, 1H), 5.29 (s, 1H), 4.28 – 4.15 (m, 2H), 3.97 – 3.86 (m, 2H), 3.64 (s, 1H), 2.93 (d, J = 7.0 Hz, 1H), 2.76 (d, J = 7.0 Hz, 1H), 2.35 (s, 1H), 2.17 (s, 1H), 1.59 (s, 4H), 1.29 (d, J = 34.2 Hz, 3H), 1.25–1.23 (m, 6H), 1.13 –0.80 (m, 13H), 0.66 (s, 2H), 0.03 (m, 12H). HRMS Calculated for [C50H64N4O6+H]+:817.4899 Found: 817.4883.
Synthesis of PI828-cholesterol conjugate
PI-828 [28 mg (0.088 mmol) dissolved in 2.0 mL of dry DCM] was added to 20.0 mg (0.044 mmol) of cholesteryl chloroformate (dissolved in 2.0 mL dry DCM). Finally 15.5 μL (0.088 mmol) of dry DIPEA was added to it drop-wise at room temperature in an inert condition. Progress of the reaction was monitored by thin layer chromatography. After 24h, it was quenched with 100 mL 0.1(N) HCl and the compound was extracted in DCM. The desired product was separated by column chromatography using a solvent gradient of 0–5% MeOH in DCM. 1HNMR(300 MHz) δ(ppm) = 8.165–8.13(m); 7.59–7.40(m, aromatic); 6.72(s); 5.98–5.93(m); 5.42–5.40(m); 4.67–4.59(m); 3.75–3.74(m); 3.44–3.40(m); 2.43–2.34(m); 2.04–1.93(m); 1.86–1.77(m); 1.65–1.43(m); 1.35–1.43(m); 1.32–0.85(m).
Synthesis ansd characterization of SNPs
Drug-cholesterol conjugates, L-α-phosphatidylcholine, and DSPE-PEG2000 (at optimized weight ratios) were dissolved in 1.0 mL DCM. Resulting solutions were evaporated in a round-bottomed flask with the help of a rotary evaporator and thoroughly dried. The resulting thin films were hydrated with PBS with constant rotation at 55°C for 2h. Nanoparticles were eluted through a Sephadex column and extruded through 200 nm pores. The size was checked using DLS, and drug loading was determined by spectroscopy. For release kinetics studies, the drug-loaded nanoparticles (1mg drug/ml, 5ml) were suspended in PBS buffer (pH 7.4), 4T1 or 4306 cell lysates within a dialysis tube (MWCO= 3500 Dalton, Spectrum Lab). The dialysis tube was suspended in 1L PBS pH 7.4 with gentle stirring to simulate infinite sink tank condition. A 100 μL portion of the aliquot was collected from the sample at predetermined time intervals and replaced by equal volume of PBS buffer, and the released drug was quantified by spectrometry. More details are available in supplementary information.
In vitro Assays
4T1 and MDA MB 468 breast cancer cells were cultured in RPMI, while 4306 ovarian cancer cells were cultured in DMEM (supplemented with 10% FBS and 1% of antibiotic-antimycotic 100x solution). Cells (4×103) were seeded into 96-well flat-bottomed plates, and incubated with free drug or drug-loaded nanoparticles (normalized to equivalent amounts of free drug) for desired time periods. Cell viability was quantified using the CellTiter 96 Aqueous One Solution assay (Promega). To study drug internalization, 4T1 breast cancer cells were incubated with free PI103 or PI103-SNPs (with equivalent amount of PI103) for 4 hours, then washed and incubated in fresh media. After desired time of incubation, cells were lysed, and drug concentrations quantified using spectroscopy.
Murine 4T1 breast cancer model
4T1 breast cancer cells (1 × 105) were implanted subcutaneously in the flanks of 4-week-old BALB/c mice. The drug therapy was started on day 9. Animals were randomized into the following treatment groups: (i) Vehicle, (ii) free drug (5mg/kg), and (iii) SNPs (at dose equivalent to 5mg/kg of the PI3K inhibitor). In an separate experiment, we included an additional group treated with iRGD-PI103-SNPs to test for effect of active targeting on efficacy. Animals were dosed every 48 hours.
The tumors were measured regularly, and tumor volume (Vt) was calculated using the formula, L × B2/2, and tumor volume increments (as a function of growth rate) were calculated using the formula Vt/V0 (where V0 was tumor volume at the time of first injection). In a separate study, we monitored survival of 4T1 tumor-bearing mice were treated as described above. Animals (n=10 in each group) were sacrificed as soon as they reached moribund state, which is attained prior to reaching tumor volume cut-off in this model. All animal procedures were approved by the Harvard Institutional Use and Care of Animals Committee.
Insulin Tolerance Test using PI103-SNPs
Random fed mice (with 4T1 breast cancer) were injected with a single dose of empty nanoparticles (control), free PI103 or PI828 and PI828-SNP or PI103-SNPs (at doses equivalent to 5mg/kg of the parent inhibitor molecule) via the tail vein. The mice were injected with freshly prepared insulin solution (0.75 U/Kg) in 0.1 ml 0.9% NaCl at defined time points after drug administration. Blood glucose levels were measured before and 45 min after insulin injections using a glucometer.
Efficacy study of PI103-SNPs in Murine Ovarian Cancer Tumor Model
Ovarian adenocarcinomas were induced in genetically engineered K-rasLSL/+/Ptenfl/fl mice via intrabursal delivery of adenovirus-carrying Cre recombinase. Tumor cells were engineered to express luciferase activated by Adeno-Cre. Once mice developed medium to large tumors, they were placed into one of four groups, and treated with vehicle, free-PI103, PI103-SNP or iRGD-PI103-SNP at doses equivalent to 5mg/kg of PI103. Tumor imaging was performed using an IVIS Lumina II Imaging System. Quantification of bioluminescence was achieved by using Living Image Software 3.1 (Caliper Life Sciences). Images were taken a day prior to initial treatment (day 0, baseline image), and the day after 3 or 5 cycles of treatments. The 4306 cell line was established from these Cre-induced K-ras+/Pten- murine tumors in the Dinulescu laboratory, and genotyping is done periodically by PCR analysis of DNA for characterization.
Western Blot
For in vitro studies, 5 × 104 cells were seeded in each well of a 6 well plate and incubated with free drug or SNPs (with equivalent amount of drug) for 24 hours. For in vivo studies, tumor stored in −80°C were pulverized in a mortar and pestle using liquid nitrogen. Proteins were extracted with RIPA buffer. Protein lysates were fractionated by electrophoresis, transferred to membranes, which were incubated with antibodies against phosphorylated proteins, and probed with horseradish peroxidase–conjugated secondary antibody. Detection was done using a G-box (Syngene), and densitometric quantification was done by image J software. Expression was normalized to total expression of the specific protein or β-actin.
Tumor histocytochemistry
Tumors cryosections were directly imaged using a Nikon TE2000 epifluorescence microscope for studying the localization of FAM-labeled iRGD-SNP nanoparticles. Blood vessels were delineated using vonWillebrand factor (vWF) immunostaining. For studying apoptosis, formalin-fixed tumor sections were stained with a standard TMR red fluorescent terminal deoxynucleotidyl tranferase-mediated dUTP nick end labeling (TUNEL) kit following the manufacturer’s protocol (In Situ Cell Death Detection Kit, TMR-Red, Roche).
Statistics
The statistical analysis was done using two-tailed Student’s t-test or one-way ANOVA followed by Newman Keuls Post Hoc test, with P< 0.05 as the threshold for significance.
Results
Synthesis and characterization of PI3K-inhibiting SNPs
We used two different PI3K inhibitors, the pyridofuropyrimidine PI103, and PI828 (8-bromo-2-morpholin-4-yl-chromen-4-one) to engineer the supramolecular nanoparticles. PI828 is a derivative of the earlier generation and widely used PI3K inhibitor LY294002, where an amine linker has been inserted in 4-position hydrogen of the exocyclic phenyl substituent, enabling conjugation to cholesterol via a carbamate bond (Fig. 1A). Previous studies have demonstrated that conjugation via this linker maintains affinity for the catalytic site of PI3K class I isoforms (20). However, PI828, like LY294002, is a weak inhibitor (20). We therefore included, PI103, which has been reported to exhibit excellent potency in the low nanomolar range and selectivity for class IA PI3Ks as well as mTOR (12). However, PI103 was not found suitable for clinical development as the planar tricyclic structure resulted in limited aqueous solubility and the phenolic hydroxyl group is rapidly glucoronidated (12). These limitations, however, made PI103 a suitable molecule to engineer the supramolecular nanoparticles. As shown in Fig. 1B, the phenolic hydroxyl group was conjugated via an ester linkage to cholesterol-succinate complex. The intermediate and products were characterized by 1H NMR spectroscopy and mass spectrometry (Suppl. Fig. 1–3).
Figure 1. Synthesis and characterization of SNP.

Synthetic scheme showing conjugation of (A) PI-828 and (B) PI103 to cholesterol via carbamate and ester linkages respectively; (C) Schematic representation shows assembly of supramolecular nanoparticles (SNPs) from phosphatidylcholine (PC), PI103-/PI828-cholesterol conjugates and DSPE-PEG2000. Graph shows the distribution of hydrodynamic diameter of (D) PI828-SNPs and (E) PI103-SNPs measured using dynamic light scattering. (F) High resolution cryo-transmission electron microscopy image of PI103-SNPs (Scale Bar = 100 nm); (G) Graph shows the physical stability of PI103-SNPs during storage condition at 4°C as measured by changes in size and Zeta potential of nanoparticles; Graphs show (H) release kinetics of PI103 from SNPs in PBS, pH 7.4, and 4T1 breast cancer cell lysate, and (I) release kinetics of PI828 from SNPs in PBS, pH 7.4, 4T1 breast cancer cell lysate and PI3K-overexpressing 4306 ovarian cancer cell line. Data shown are mean ± SEM (at least triplicates at each condition).
We engineered the SNPs from the cholesterol-PI828 or cholesterol-PI103 conjugates, phosphatidylcholine (PC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (DSPEPEG2000) at optimized weight ratios using a lipid-film hydration self-assembly method (21) (Fig. 1C). The incorporation efficiency for the cholesterol-PI828 SNPs was 43%, and 60±5% for PI103-cholesterol conjugate SNPs. As shown in Fig. 1D, cholesterol-PI828 conjugates resulted in the formation of SNPs with hydrodynamic diameter of 108 ± 8.9 nm as determined by dynamic light scattering (Fig. 1D). PI103-SNPs showed a mean particle diameter of 172 ± 1.8 nm (Fig. 1E). Ultrastructure analysis using cryo-transmission electron microscopy (cryo-TEM) (Fig. 1F) revealed the formation of predominantly unilamellar structures 100 nm or less in diameter. The size difference between TEM and DLS measurements can be attributed to the hydration sphere arising from the PEG coating, which can facilitate the masking from the reticuloendothelial system (22). Additionally, aliquots of the PI103-SNPs stored for a period of over a month exhibited no changes in size and zeta potential, indicating that the formulations were stable (Fig. 1G). Temporal release kinetics revealed a sustained release of active drug in cell lysate (Fig. 1H, I), consistent with the cleavage of the linkers in acidic and enzymatic (esterase) conditions. Interestingly, the rate of release of PI828 was significantly lower, consistent with the more stable carbamate linker (cleaved by carboxyesterases). As a control experiment, we tried engineering nanoparticles using traditional approaches of nanoformulation, where we entrapped PI103 in the lipid bilayer (Suppl. Fig. 4A). Using the lipid ratio employed with the SNPs resulted in minimal incorporation efficiency of 2% PI103, which could be optimized by changing the compositional ratio (Suppl Fig. 4B). A sustained release of PI103 was observed from the formulation (Suppl. Fig. 4C), resulting in similar effects on cell viability (Suppl. Fig. 4D) and inhibition of Akt phosphorylation (Suppl. Fig. 4E). However, light scattering studies revealed a temporal increase in the size of the nanoparticles (Suppl. Fig. 4F), leading to precipitation (instability) (Suppl. Fig. 4G), consistent with the fact that currently available strategies for nanoformulation are not be compatible with many molecules, which has limited the repertoire of nanomedicines.
In vitro efficacy of supramolecular nanoparticles
We evaluated the efficacy of the SNPs in vitro using 4T1 (murine breast cancer), MDA-MB-468 (human breast cancer) and a PI3K-overexpressing 4306 (ovarian cancer) cell lines. Temporal effect on cell viability and IC50 values are shown in Fig. 2 and Table 1. While we observed a decrease in the potency following conjugation of the active molecules to cholesterol, this is consistent with a prodrug approach, where the construct requires activation to the parent molecule for efficacy. Western blot analysis showed that continuous incubation with both the free drug as well the SNP (at equimolar concentrations of PI103) induced a sustained inhibition of basal phosphorylation of Akt (Fig 3A). Interestingly, on the other hand, a transient exposure over 4 hours resulted in a rebound increase in phosphorylation of AKT in the case of free PI103, while SNP-PI103 inhibited Akt phosphorylation in a more sustained manner (Fig. 3B–D). Indeed, the transient treatment resulted in an initial higher intracellular concentration of PI103 in the cells treated with the free drug compared with SNP-PI103. However, while the concentration remained elevated in the PI103-SNP-treated cells, only traces of the drug were detected in the cells treated with the free drug by 18 hours (Fig. 3E) in line with the need for conversion to active drug in the case of SNPs. Consistent with the above observations, PI828-SNPs and free PI828 exhibited similar cytotoxic effect on the 4T1 cells (Fig. 2G) and 4306 cells (Fig. 2H). The cells treated with PI103SNPs and PI828-SNPs exhibited similar inhibition of Akt phosphorylation after 36 hours of treatment (Fig. 3F).
Figure 2. Cell viability assays of PI3K-inhibiting SNPs.
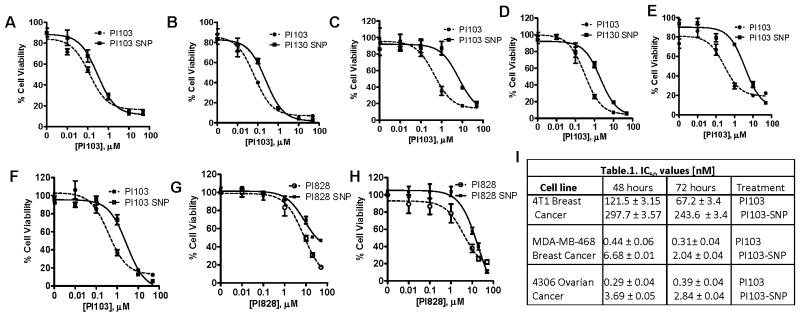
MTS assay showing the effect of free or PI103 SNPs at different concentrations on 4T1 cells at (A) 48h and (B) 72h; MDA-MB-468 cells at (C) 48h and (D) 72h; and 4306 cells at (E) 48h and (F) 72h. Graphs show the effect of treatment with PI828 or PI828SNPs on viability of (G) 4T1 breast cancer cells or (H) 4306 cells. (I) Table shows IC50 of PI103 and PI103 SNP in different cell lines at 48h and 72h. Data shown are mean ± SEM (n=3, with at least triplicates in each independent experiment).
Table 1.
IC50 values [nM]
| Cell line | 48 hours | 72 hours | Treatment |
|---|---|---|---|
| 4T1 Breast Cancer | 121.5 ± 3.15 297.7 ±3.57 |
67.2 ±3.4 243.6 ±3.4 |
PI103 PI103-SNP |
| MDA-MB-468 Breast Cancer | 0.44 ± 0.06 6.68 ±0.01 |
0.31±0.04 2.04 ± 0.04 |
PI103 PI103-SNP |
| 4306 Ovarian Cancer | 0.29 ± 0.04 3.69 ±0.05 |
0.39 ±0.04 2.84 ± 0.04 |
PI103 PI103-SNP |
Figure 3. In vitro characterization of PI3K-inhibiting SNPs. Representative Western blots show.
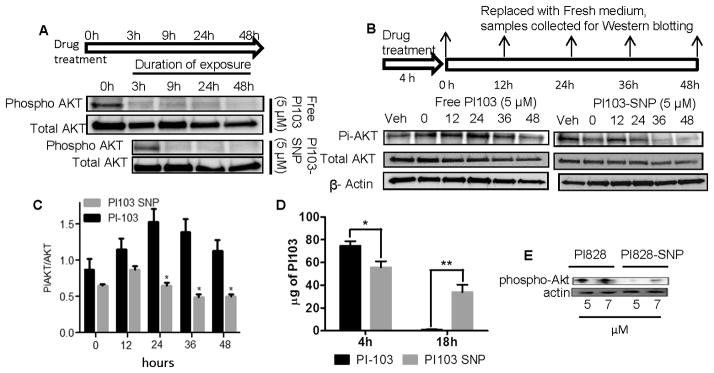
(A) Expression of phospho-AKT and Total AKT in 4T1 cells at 3,9,24 and 48 hours after treatment with either 5 μM of Free PI103 or PI103-SNP. Effect of acute treatment (4 hours incubation) with (B, D) PI103 or (C, D) PI103-SNP on PI3K activity over time. After 4 hours of exposure to drug, the cells were washed thrice with cold PBS to remove additional drug outside cells and then incubated with fresh media with 1% FBS. Cells were collected at 0, 12, 24, 36 and 48 hours. PI103-SNPs induced sustained inhibition of phosphorylation of Akt. (F) Graph showing internalization of free PI103 and PI103 nanoparticles at 4h and 18h. The amount of drug internalized was quantified by UV-vis spectroscopy. Data shown are mean ± SEM from at least 3 replicates; *p < 0.05; **p < 0.01 (Student t-test). (G) Effect of treatment with PI828 or PI828-SNPs (5.0 and 7.0 μM) on phospho-Akt levels at 36 hours post-treatment.
Efficacy of SNP in an in vivo 4T1 breast cancer model
We next investigated the anti-tumor efficacy of PI103-SNPs in the 4T1 cell line, which is negative for ER and PR, and expresses a low level of the mouse Her2/neu equivalent (23). Transplanted into syngeneic mice, the 4T1s form aggressive, highly metastatic breast cancers. Mutations in genes that constitute the PI3K pathway occur in >70% of breast cancers (24). We have previously demonstrated that the 4T1 cells mount a survival response to standard chemotherapy via an upregulation of PI3K signaling (25). As shown in Fig. 4A, treatment with PI103 resulted in tumor growth inhibition relative to PBS-treated controls, but a tumor rebound was observed after the treatment was stopped. In contrast, treatment with PI103-SNP resulted in a sustained tumor growth inhibition over the study period. To test whether targeting the nanoparticles to the tumor using ‘homing’ peptides increases antitumor efficacy, a separate group of tumor-bearing mice were treated with PI103-SNPs that were surface-decorated with iRGD peptide. As shown in Fig. 4A, such a treatment resulted in greater tumor inhibition compared with SNPs that accumulated via passive uptake. Indeed, previous observations have shown that iRGD-coated nanostructures exhibit increased extravasation and tissue penetration in a tumor-specific and neuropilin-1-dependent manner (26). To elucidate the mechanism underlying the increased in vivo efficacy, the tumors were excised post-treatment, and processed for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) as a marker for apoptosis. As shown in Fig. 4B, C, treatment with PI103-SNPs resulted in greater apoptosis than treatment with free PI103. While we did observe an enhanced antitumor efficacy with iRGD-coated PI103-SNPs, stereological analysis of multiple tumor sections revealed a statistically insignificant increase in the level of apoptosis compared with PI103-SNP. Epifluorescence imaging of tumor cross-sections did reveal intra-tumoral localization of FAM-labeled iRGD-coated PI103-SNPs (Fig. 4D). We next studied the effect of treatment on survival of 4T1-bearing mice. The 4T1 model is an aggressive form of breast tumor, and the animals become moribund and have to be sacrificed prior to the tumor reaching maximum tumor cut-offs. Hence the 4T1 syngeneic implants serves as an excellent model for generating Kaplan Mier survival curves. As shown in Fig. 4E, three cycles of PI103-SNP significantly increased the median survival by two days as compared with free PI103 treatment (P<0.05). Pharmacodynamic monitoring revealed that phosphorylation of Akt and downstream signaling molecules mTOR and 4EBP were significantly inhibited in the PI103-SNP-treated tumors than in the PI103-treated tumors (P<0.05, t test). Treatment with PI828-SNPs (5mg/kg PI828 equivalent, 3 doses) also exerted a inhibitory effect on Akt phosphorylation in vivo translating into superior tumor growth inhibition as compared to free PI828 (Suppl. Fig 5). However, consistent with its low potency, the antitumor efficacy of PI828 or PI828-SNP was significantly lower than seen with PI103-SNPs. However, given that the both PI103-SNPs and PI828-SNPs did inhibit PI3K signaling, it is possible that the release kinetics (rate of release) of the active agent plays a critical role in efficacy and needs to be considered in the design of supramolecular nanoparticles.
Figure 4. In Vivo efficacy of PI103 or PI103-SNP in a syngenic 4T1 breast cancer BALB/c mice model.

(A) Growth curves show effect of different multi-dose treatments on tumor volume. Each animal were injected with three doses of either PBS (for control group), 5 mg/kg of free PI013, 5mg/kg of PI103SNP or iRGD tagged PI103-SNPs (at dose equivalent to PI103) on every alternate day. First day of treatment was considered as Day 1. End point for each animal was tumor size>2000cm3 or tumor ulceration or necrosis or animal death. Treatment with PI103-SNPs or iRGD tagged PI103-SNP was statistically more effective than treatment with free PI103. Data shown are mean ± SEM, P<0.01, ANOVA). Top panel shows representative tumors from each group; (B) Representative epifluorescent images of tumor sections from animals treated as above were labeled for apoptosis using TUNEL (red) and counterstained with DAPI (blue). (C) Graph shows the quantification of apoptosis from the labeled tumor sections as a percentage of TUNEL+ve cells as a function of total nuclei. (D) Immunoflurescence image of the frozen section of tumor from either control or iRGD tagged PI103-SNP, showing effective targeting with iRGD (iRGD peptide was attached with FAM)-coated PI103-SNP; the section was counterlabeled for vonWillebrand factor (Texas-Red), which delineates the vasculature; (E) Kaplan Mier survival curves show that treatment with PI103-SNP increases survival (P<0.05) as compared with free PI103 (n=10 in each treatment group). (F) Representative Western blots show the expression of phospho and total forms of mTOR, Akt, 4EBP (normalized to actin) in tumors 72 hours after a single dose injection of either free PI103 or PI103-SNP (at 5mg/kg PI103 dose equivalent) in a syngenic 4T1 breast model. (G) Graph shows densitometric quantification from the above study. Data shown are mean ± SEM from n=3, *P<0.05, **P<0.01, ***P<0.01.
Efficacy of PI103-SNPs in an in vivo K-RasLSL/+/Ptenfl/fl ovarian cancer model
We further evaluated the effect of PI103-SNP in a K-RasLSL/+/Ptenfl/fl ovarian cancer model (27). We selected this model because tumors that lack Pten have been reported to be addicted to PI3K signaling (7). On the other hand, tumors that present a mutated or activated Ras have been reported to be less responsive to PI3K inhibitors (7). As shown in Fig. 5A–C, bioluminescence quantification of tumor luciferase signal indicates that free PI103, PI103SNP and iRGD-PI103-SNP resulted in significant tumor regression as compared to vehicle control. The anti-tumor response to iRGD-PI103-SNP was statistically significantly superior to free PI103 after 3 cycles of treatments as quantified by the bioluminescence signal. This distinction between free drug and PI103-SNPs was attained only after 5 cycles of treatment, consistent with previous observations that iRGD facilitates intratumoral penetrance and accumulation. No change in body weight was observed in any treatment group (Fig. 5D). The expression levels of PI3K/mTOR pathway markers, as assessed by Western blot analysis of tumor samples from different groups, showed a significant decrease in the expression of phospho-mTOR, phospho-AKT, phospho-S6 and phospho-4EBP1 in the PI103-SNP and the iRGD-PI103-SNP-treated groups as compared to the free PI103-treated tumors (Fig. 5E).
Figure 5. PI103-SNP inhibits tumor growth in a K-rasLSL/+/Ptenfl/fl ovarian cancer model.

(A) Representative pictures from the 4 treatment groups before and after 3 and 5 treatments (n = 3). Tumor images was obtained using a IVIS Lumina II Imaging System. Quantification of bioluminescence was achieved by using the Living Image Software 3.1. Mice received 150 mg/kg of D-luciferin firefly potassium salt via intraperitoneal injection prior to imaging; (B) Bioluminescence quantification indicates a significantly decreased tumor luciferase signal in mice treated with Free-PI103, PI103-SNP, and iRGD-PI103-SNP compared to vehicle (p<0.05, one-way ANOVA analysis) after 3 treatments. Following 5 treatments, bioluminescence quantification indicates response to PI103-SNP was significantly higher compared to free PI103 (p<0.01, one-way ANOVA analysis); (C) Representative K-rasLSL/+/Ptenfl/fl tumors excised from animals treated with free-PI103, PI103-SNP, and iRGD-PI103-SNP; (D) Graph shows drug toxicity assessed by measurements in overall body weight. Daily recordings indicate no difference in body weight after 5 treatments of all treatment groups; (E) PI3K/mTOR pathway markers were assessed in K-rasLSL/+/Ptenfl/fl tumors treated with vehicle, 5 mg/kg Free-PI103, 5 mg/kg PI103-SNP, and 5 mg/kg iRGDPI103-SNP by western blotting as described in Materials and Methods. Inhibition of mTOR substrates by PI103-SNP and iRGD-PI103-SNP treatment was much greater in comparison to free drug in K-rasLSL/+/Ptenfl/fl tumors.
Effect of PI103-SNP on glucose homeostasis
PI3K plays a central role in mediating insulin signaling that is conserved throughout eukaryotic evolution. We therefore investigated the effect of PI103-SNPs on insulin tolerance in a 4T1 breast cancer model. Consistent with previous studies (28), mice injected with free PI103 exhibited a transient tolerance to insulin compared with the significant insulin-induced decrease in blood glucose level observed in mice pre-treated with PI103-SNP or with empty nanoparticles as a control (Fig. 6). The insulin response was restored at later time points, consistent with previous studies with PI103 (28), and can arise from the short half-life of PI103. Interestingly, we did not observe any change in glucose levels following PI103-SNP treatment compared with vehicle-treated controls over the study period, suggesting that the sustained release of PI103 from the nanoparticles and the increased efficacy did not manifest in a delayed onset of insulin resistance. Treatment with free PI828 or PI828-SNP did not affect glucose response to insulin at the doses used (Fig. 6B), consistent with the low potency of the active agent as compared with PI103. Furthermore, we analyzed tissue sections from liver, kidney and spleen, three organs that are implicated in clearance of nanoparticles. As shown in Fig. 6C, TUNEL study revealed no distinctions between the different treatments. This was validated using Western blotting for monitoring the expression of phospho-mTOR as well as cleaved caspase 3 and PARP (as markers of induction of apoptosis), which revealed no differences between free PI103- or PI103-SNP-treated animals (Fig. 6D).
Figure 6. Testing off-tumor effects of PI3K inhibition.
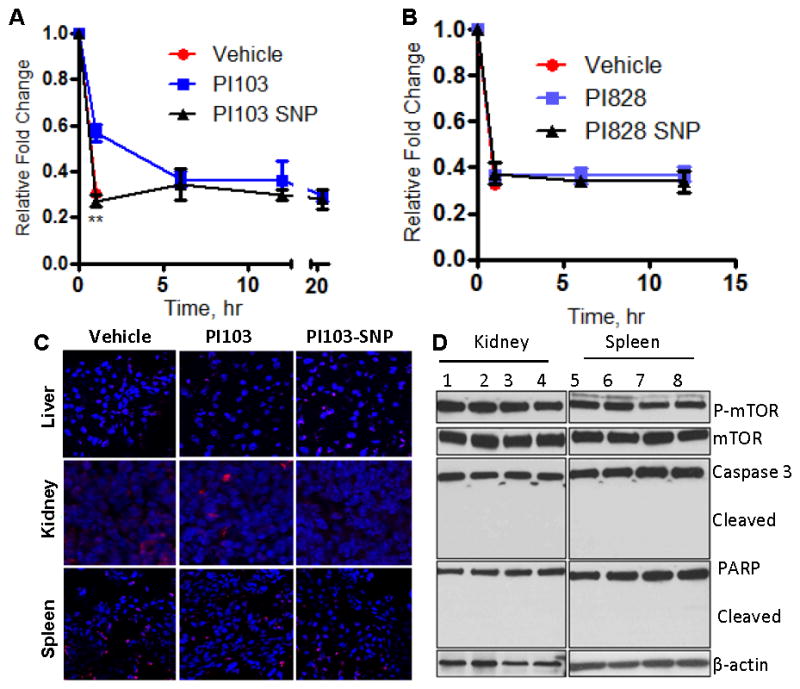
Graphs show the effect of (A) PI103-SNP and (B) PI828-SNP on insulin tolerance. Each animal was injected with a single dose of empty nanoparticles, free PI3K inhibitor (5mg/kg) or SNP (equivalent to 5 mg/kg of corresponding PI3K inhibitor). At defined time points animals were injected with insulin (0.75 units/kg). Glucose levels in blood were measured in blood samples before insulin injections and 45 min after insulin injection. Results are mean ± SEM (n = 5) **p < 0.01. (C) Representative epifluorescence images of cross sections of different organs from animals treated with PI103 or PI103-SNP. (D) Western blot of kidney and spleen cells isolated from animals treated with blank SNPs (1, 5), free PI103 (2, 6), PI103-SNP (3,7), and iRGD-coated PI103-SNP (4,8) shows expression levels of phospho- and total mTOR (downstream of Akt) and state of apoptosis markers, Caspase 3 and PARP (cleaved and total).
Discussion
The challenges faced in clinical translation of PI3K inhibitors have highlighted the need to overcome feedback signaling and insulin resistance ‘class effect’, and to achieve high intratumoral concentration of the drug (29). Here we demonstrate that the supramolecular nanoparticles can induce a sustained inhibition of the PI3K resulting in potentially overcoming feedback signaling. Furthermore, this sustained inhibition of PI3K signaling results in increased antitumor efficacy without insulin resistance, indicating that the use of supramolecular nanochemistry can emerge as a powerful strategy for overcoming the challenges faced during clinical translation of PI3K inhibitors.
Indeed, our study demonstrates that an acute exposure to the free drug (PI103) results in an increase in the phospho-Akt levels at later time-points. Such a rebound activation of the pathway is consistent with previous reports (29, 30), arising from a homeostatic feedback loop via the up-regulation of receptor tyrosine kinases (31). Interestingly, treatment with the PI103-SNPs could potentially overcome this feedback loop as evident from a sustained inhibition of the phospho-Akt signal. This could arise from an increase in the intracellular concentration of PI103 with time, achieved with SNPs. Indeed, Western blot analysis of the in vivo tumor samples (extracted 72 hours of administration of dose) revealed a robust PI3K signaling in tumors from animals that were treated with free PI103. In contrast, treatment with PI103-SNP resulted in complete shutdown of the pathway, as evident from decreased levels of phosphorylated forms of Akt, S6K, 4E-BP1 or mTOR in both tumor models (indeed PI828-NPs exerted a similar sustained inhibition on phosphorylation of Akt in vitro and in vivo as compared with free PI828, but it is unlikely that PI828-SNP will emerge as a potential drug candidate for reasons described later). Indeed, in a recent commentary, Engleman et al had posed the question whether lack of efficacy of PI3K inhibitors is due to inadequate inhibition of the target or because complete inhibition of the target is not sufficient to produce antitumor activity (31). The current results indicate that in addition to the level of inhibition of the pathway, the temporality or kinetics of inhibition may be a critical element in determining antitumor outcome. Interestingly, a similar observation was made during the evolution of a current clinical candidate GDC-0941 from PI103, where ~90% inhibition of Akt phosphorylation for several hours was seen as a requirement for antitumor activity, establishing a link between pharmacokinetic exposure and pharmacodynamic biomarker changes (9). Indeed, this is validated by the fact that free inhibitors were found to be more potent that the SNPs in vitro under conditions of sustained exposure (as the latter has to be activated into the active moiety), while SNPs were more effective in vivo, potentially arising from sustained intratumoral concentrations as compared with the free drug. An additional point to note was that we did see a disconnect between the expression levels of phosphorylated Akt and downstream phosphorylated proteins, such as S6K, 4E-BP1, where although p-Akt was observed in some SNP-treated tumor tissues the downstream proteins were still inhibited. This captures the kinetics of flow of information through a signaling cascade, potentially dissected due to the temporal inhibition attained with the SNPs as opposed to an ‘all or none’ inhibition seen with free inhibitors. This also highlights the necessity to include the distinct levels of PI3K signal transduction pathway, as opposed to monitoring only phosphorylation of Akt, as biomarkers for efficacy (32).
Clearly, the transient resistance to insulin observed with PI103, together with its short half-life and its ability to inhibit tumor growth, suggests that insulin resistance could possibly be a Cmax-driven effect, while the anti-tumor efficacy is a function of sustained exposure (area under curve or AUC-driven effect). This is supported by the results with PI828, which showed no resistance to insulin at the dose used but did exert an inhibitory effect on tumor growth. This could also explain why PI103-SNP exerted a significant intratumoral PI3K inhibition resulting in increased antitumor efficacy (arising from a sustained release of active drug) but did not induce insulin tolerance, indicating that the supramolecular nanoparticle approach an indeed overcome the current challenges associated with PI3K inhibitors.
Indeed, the integration of supramolecular nanochemistry with targeted therapeutics can open up new opportunities to harness the full potential of targeted therapeutics by enhancing the therapeutic index. As seen in the case of PI103, it can potentially overcome limitations that had prevented potent molecules from advancing further, and can enable the rescue of ‘failed’ drugs (12). Furthermore, the current study reveals that the supramolecular nanochemistry-based approach can potentially enable the fabrication of nanomedicines from drugs that are not compatible with currently used techniques of nanoformulation. Indeed, in a recent study, we had demonstrated that such supramolecular nanoparticles of cisplatin resulted in stable formulations, increased drug loading efficiency and enhanced delivery to the tumors as opposed to free cisplatin (16). However, the contrasts in the outcomes with PI103- and PI828-SNPs also highlight the need for keeping design principles in perspective when engineering the supramolecular nanoparticles. In some manner, this is similar to the design principles for a successful antibody-drug conjugate (33), where selection of a potent active agent and the optimal linker chemistry is critical to efficacy, with the nanoparticle enabling a preferential delivery to the tumor.
In summary, several components of the current approach can facilitate future therapy in humans. First, the ability of nanoparticles to accumulate in tumors can lead to increased efficacy. Secondly, the sustained release resulting in prolonged inhibition of the PI3K pathway and absence of the ‘feedback loop’ could be a critical determinant in clinical success. Third, the absence of insulin resistance with the PI103-SNP indicates that supramolecular nanochemistry can significantly impact the therapeutic index. We also demonstrate that the efficacy can be further improved by placing active targeting moieties such as iRGD peptides on the nanoparticle surface. Indeed, the fact that the nanoparticle assembles from unit molecules offers an exquisite control over the stoichiometry in terms of active agents as well as the valency of targeting agents. This stoichiometric control also means that supramolecular nanochemistry platform can potentially be extended to additional cytotoxics and molecularly targeted therapeutics including enabling combination therapy from a single nanoparticle, thereby facilitating an integrative approach towards cancer chemotherapy.
Supplementary Material
Acknowledgments
Funding: This work was supported by US Department of Defense (DOD) Breast Cancer Research Program (BCRP) Era of Hope Scholar Award W81XWH-07-1-0482, a DOD Collaborative Innovator Grant W81XWH-09-0698/700, National Institutes of Health Grant R01 1R01CA135242-01A2 and an American Lung Association Discovery Award ( LCD-259932-N) (to SS), Department of Defense Ovarian Cancer Research Program Award W81XWH-10-1-0263, a V Foundation Scholar Award, an Ovarian Cancer Research Fund Liz Tilberis Award, by the Burroughs Wellcome, Mary Kay Ash and Rivkin Foundations, and by a generous contribution from the Mildred Moorman Ovarian Cancer Research Fund (to DMD).
Footnotes
Supplementary information: Detailed Materials and Methods + Supplementary figures (5).
Conflict of Interest: E.R. and V.R.K. are shareholders in CendR Therapeutics Inc., which has rights to the peptide technology described in this paper. AAK, AG, SS, are listed on a patent filed by BWH on this technology. SS is a cofounder of Cerulean Pharmaceuticals and Invictus Oncology, which focus on nanomedicine.
References
- 1.World Health Organization. WHO Cancer Report. 2008. [Google Scholar]
- 2.Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nature Rev Cancer. 2004;4(5):361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 3.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nature Rev Cancer. 2009;9(1):28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 4.Ferrari M. Cancer nanotechnology: opportunities and challenges. Nature Rev Cancer. 2005;5(3):161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 5.Sengupta S, Sasisekharan R. Exploiting nanotechnology to target cancer. Br J Cancer. 2007;96(9):1315–1319. doi: 10.1038/sj.bjc.6603707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nature Rev Genetics. 2006;7(8):606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 7.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nature Rev Cancer. 2009;9(8):550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 8.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96(8):4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Workman P, Clarke PA, Raynaud FI, van Montfort RL. Drugging the PI3 kinome: from chemical tools to drugs in the clinic. Cancer Res. 2010;70(6):2146–2157. doi: 10.1158/0008-5472.CAN-09-4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knight ZA, Gonzalez B, Feldman ME. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006;125(4):733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bendell JC, Rodon J, Burris HA. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30(3):282–290. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 12.Raynaud FI, Eccles SA, Patel S. Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: from PI103 through PI-540, PI-620 to the oral agent GDC0941. Mol Cancer Ther. 2009;8(7):1725–1738. doi: 10.1158/1535-7163.MCT-08-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan F, Leunig M, Huang SK. Microvascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res. 1994;54(13):3352–3356. [PubMed] [Google Scholar]
- 14.Harfouche R, Basu S, Soni S. Nanoparticle-mediated targeting of phosphatidylinositol-3-kinase signaling inhibits angiogenesis. Angiogenesis. 2009;12(4):325–338. doi: 10.1007/s10456-009-9154-4. [DOI] [PubMed] [Google Scholar]
- 15.Karve S, Werner ME, Sukumar R. Revival of the abandoned therapeutic wortmannin by nanoparticle drug delivery. Proc Natl Acad Sci U S A. 2012;109(21):8230–8235. doi: 10.1073/pnas.1120508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sengupta P, Basu S, Soni S. Cholesterol-tethered platinum II-based supramolecular nanoparticle increases antitumor efficacy and reduces nephrotoxicity. Proc Natl Acad Sci U S A. 2012;109(28):11294–11299. doi: 10.1073/pnas.1203129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lehn JM. Supramolecular Chemistry: Concepts and Perspectives. VCH Newyork; 1995. [Google Scholar]
- 18.Lehn JM. Toward complex matter: supramolecular chemistry and self-organization. Proc Natl Acad Sci U S A. 2002;99(8):4763–4768. doi: 10.1073/pnas.072065599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen KJ, Wolahan SM, Wang H. A small MRI contrast agent library of gadolinium(III)encapsulated supramolecular nanoparticles for improved relaxivity and sensitivity. Biomaterials. 2011;32(8):2160–2165. doi: 10.1016/j.biomaterials.2010.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gharbi SI, Zvelebil MJ, Shuttleworth SJ. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem J. 2007;404(1):15–21. doi: 10.1042/BJ20061489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sengupta S, Eavarone D, Capila I. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature. 2005;436(7050):568–572. doi: 10.1038/nature03794. [DOI] [PubMed] [Google Scholar]
- 22.Schipper ML, Iyer G, Koh AL. Particle size, surface coating, and PEGylation influence the biodistribution of quantum dots in living mice. Small. 2009;5(1):126–134. doi: 10.1002/smll.200800003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seavey MM, Pan ZK, Maciag PC. A novel human Her-2/neu chimeric molecule expressed by Listeria monocytogenes can elicit potent HLA-A2 restricted CD8-positive T cell responses and impact the growth and spread of Her-2/neu-positive breast tumors. Clin Cancer Res. 2009;15(3):924–932. doi: 10.1158/1078-0432.CCR-08-2283. [DOI] [PubMed] [Google Scholar]
- 24.Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13(6):224. doi: 10.1186/bcr3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldman AJ, RB, Ravi S, Rivera F, Kulkarni AA, Sengupta S. CD44 mediates chemotherapy tolerance through Akt and Ezrin/Radixin/Moesin in an EGFR-dependent manner. Cancer Research. 2012;72:27. [Google Scholar]
- 26.Sugahara KN, Teesalu T, Karmali PP. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer cell. 2009;16(6):510–520. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dinulescu DM, Ince TA, Quade BJ. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med. 2005;11(1):63–70. doi: 10.1038/nm1173. [DOI] [PubMed] [Google Scholar]
- 28.Smith GC, Ong WK, Rewcastle GW, Kendall JD, Han W, Shepherd PR. Effects of acutely inhibiting PI3K isoforms and mTOR on regulation of glucose metabolism in vivo. Biochem J. 2012;442(1):161–9. doi: 10.1042/BJ20111913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turke AB, Engelman JA. PIKing the right patient. Clin Cancer Res. 2010;16(14):3523–3525. doi: 10.1158/1078-0432.CCR-10-1201. [DOI] [PubMed] [Google Scholar]
- 30.Muranen T, Selfors LM, Worster DT. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer cell. 2012;21(2):227–239. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28(6):1075–1083. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clarke PA, Workman P. Phosphatidylinositide-3-kinase inhibitors: addressing questions of isoform selectivity and pharmacodynamic/predictive biomarkers in early clinical trials. J Clin Oncol. 2012;30(3):331–333. doi: 10.1200/JCO.2011.38.7167. [DOI] [PubMed] [Google Scholar]
- 33.Sengupta S, Kulkarni A. Design principles for clinical efficacy of cancer nanomedicine: a look into the basics. ACS Nano. 2013;7(4):2878–82. doi: 10.1021/nn4015399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.