

Abstract
Transforming growth factor betas (TGFβs) are pleiotropic cytokines involved in many biological processes. Genetic engineering and tissue explanation studies have revealed specific non-overlapping roles for TGFβ ligands and their signaling molecules in development and in normal function of the cardiovascular system in the adult. In the embryo, TGFβs appear to be involved in epithelial–mesenchymal transformations (EMT) during endocardial cushion formation, and in epicardial epithelial–mesenchymal transformations essential for coronary vasculature, ventricular myocardial development and compaction. In the adult, TGFβs are involved in cardiac hypertrophy, vascular remodeling and regulation of the renal renin–angiotensin system. The evidence for TGFβ activities during cardiovascular development and physiologic function will be given and areas which need further investigation will be discussed.
Keywords: TGFβ, Heart, Vasculature, Development, Physiology
1. Introduction
TGFβs are closely related members of a large family of structurally similar polypeptides referred to as the transforming growth factor beta (TGFβ) superfamily. Members of this superfamily include the activins, inhibins, bone morphogenetic proteins (BMPs), müllerian inhibiting substance, Drosophila decapentaplegic gene complex, and Xenopus Vg-1 gene [1]. The TGFβs 1, 2 & 3 exhibit a variety of proliferative, inductive and regulatory functions [2] and exhibit both overlapping and distinct spatial and temporal patterns of expression throughout development and in the adult, with pronounced embryonic expression in areas undergoing morphogenesis [3,4]. Knockout (KO) mice for these ligands display dozens of non-overlapping phenotypes in most major organ systems [5,6], indicating a high degree of functional specificity.
There is considerable evidence that TGFβs can signal through multiple pathways (Fig. 1). The three TGFβ cytokines are secreted as latent complexes that are activated by mechanisms only partially understood. Among the general mechanisms that can activate TGFβs are enzymatic mechanisms which include proteases such as plasmin, calpain and matrix metalloproteinases, conformation changing protein interactions with molecules such as thrombospondin and integrin αvβ6, physicochemical mechanisms such as pH, radiation and reactive oxygen species, and drugs such as antiestrogens, retinoids and glucocorticoids (reviewed in [7]). There are three classes of TGFβ receptors, the transmembrane serine–threonine kinase receptors, TGFβRI and TGFβRII [1,8] and TGFβRIII receptors which include an ubiquitous extracellular β-glycan [9] and the membrane glycoprotein endoglin (CD105) [10,11]. Since TGFβ2 has a lower affinity to TGFβRII than does TGFβ1 or 3, its stronger interaction with β-glycan facilitates its interaction with the TGFβRII/I complex [9], although a shed form of β-glycan may be inhibitory [12]. In addition, a splice variant of the TGFβRII allows TGFβ2 ligand–receptor interaction in the absence of the TGFβRIII [13]. In contrast, endoglin interacts with TGFβ1 and TGFβ3, but has low affinity for TGFβ2 [10]. In several cell types endoglin is inhibitory to TGFβ1 function [14,15]. The ligand–receptor complexes signal through SMAD proteins that exhibit regulatory (SMAD2 and 3), co-regulatory (SMAD4) and inhibitory (SMAD6 and 7) activities on TGFβ signaling [1,16].
Fig. 1.

TGFβ signaling can occur through multiple pathways. Many of these pathways are SMAD dependent, but there is evidence that pathways involving MAPK and FKBP12/Ca2+/calcineurin may be SMAD independent. The integrin pathway in platelets is independent of transcription.
As there is only partial ligand–receptor specificity, the functional specificity of TGFβs probably depends to a large degree upon extracellular ligand activation [7], differences in intracellular SMAD interactions [1,16], interactions with other signaling pathways such as the Ras/MAPK pathway [17], and interaction between SMADs and transcriptional co-activators and co-repressors [18]. TGFβ signaling can also occur through SMAD-independent pathways (e.g. MAPK and Ca2+) [19] and through a non-transcriptional pathway (e.g. platelet aggregation) [20].
Many members of the TGFβ superfamily that play important roles in the heart have been reviewed elsewhere [21–23]. This review will concentrate specifically on TGFβ1, TGFβ2 and TGFβ3 and their functions in the cardiovascular system where their importance has been evident for over a decade [24]. This review will subdivide the cardiovascular functions of the three TGFβs into two major categories: cardiovascular development and physiology. Because the TGFβ literature is so vast, we will limit our review for the most part to in vivo studies carried out primarily in chicken and genetically engineered mice.
2. Cardiovascular development
The propagation of the symmetrical embryonic cardiac tube into an asymmetrical four-chambered heart with proper alignment of the atrio-ventricular (AV) segments and the great vessels with their respective chambers requires considerable morphogenetic and remodeling processes. Cardiac looping, formation of endocardial cushions, and their remodeling, in which myocardialization plays a primary role, are all required for proper inflow and outflow tract (OT) alignment, cardiac septation, and valve formation. In addition, remodeling of the pharyngeal arch artery (PAA) system leads to an asymmetrical left-sided vascular system. Defects in any of these morphogenetic and remodeling steps will lead to congenital cardiovascular defects commonly found in humans [25].
2.1. Cardiovascular Tgfb1–3 expression
Tg fb1 is expressed in the endocardium of the developing mouse heart. In vessels, Tg fb1 is localized in the intima, whereas Tg fb2 and Tg fb3 are present in the media and adventitia of the great arteries (Molin et al, unpublished). Since only Tg fb2−/− mice have obvious congenital cardiovascular defects, it is first important to review its expression in the developing heart. The most thorough study on Tg fb2 expression during early mouse heart development has been carried out by Dickson et al. [26]. Tg fb2 message is found as early as embryonic day (E) 7.25 in the cardiogenic plate of the precardiac mesoderm, and it later becomes prominent in the myocardium of the aortic sac and OT regions. TGFβ2 protein is found in the entire myocardium of the heart at the time when looping occurs (E8.0–8.5).
From E8.5–9.5, when the morphogenetic process of cushion formation occurs, strong Tg fb2 expression becomes localized to the myocardium underlying the acellular cardiac jelly of the endocardial cushions of the OT region and AV canal where it remains strong during the epithelial-to-mesenchymal cell transformation (EMT) of endocardium to cushion mesenchyme and during invasion of the transforming cells into the cardiac jelly (Fig. 2A, B, D and E). At E9.5 the looped primitive heart consists of myocardial cells and endocardial cells facing the inner lumen of the heart (Figs. 2A and D and 4A). The endocardial cushions covering the AV canal (boxed section, Fig. 2A) and OT myocardium (boxed section, Fig. 2D) are free of mesenchymal cells at this stage. Tg fb2 is highly expressed in the myocardium of the AV canal (arrowheads, Fig. 2B and E; AVC, Fig. 2A and D) and in the OT (arrows, Fig. 2B and E; OT, Fig. 2A and D). There is no significant expression of Tg fb3 (Fig. 2C and F) with the exception of some scattered cells in the myocardium of the OT (Fig. 2C).
Fig. 2.

Expression of Tg fb2 and Tg fb3 in wildtype embryonic hearts. In situ hybridization (B, C, E, F, H, I, K–O) and cardiac α-actin antibody HHF35 (A, D, G) or α-smooth muscle actin antibody 1A4 (J). A: primitive atrium; Ao: ascending aorta; AoS: aortic sac; AoV: aortic valve; AVC: AV cushion; CM: condensed mesenchyme; DAo: dorsal aorta; LA: left atrium; OT: outflow tract; PT: pulmonary trunk; PV: pulmonary valve; RV: right ventricle; V: primitive ventricle. Bars: A–L, 100 nm; M–O, 200 nm.
After cushion formation and EMT and before myocardialization of the endocardial cushions begins (E10.5–11.5), Tg fb2 expression is strong in the OT myocardium and in the adjacent developing cushion mesenchyme. However, as myocardialization proceeds Tg fb2 expression is reduced in the myocardium so that from E12.5 onward it is predominantly expressed only in the mesenchyme of the cushion and OT septum, as well as in the prospective smooth muscle cells (SMC) of the great arteries [26].
During myocardialization, Tg fb2 expression remains high in the cushion mesenchyme of the OT septum (Fig. 2G and H). At the aortic valve and condensed mesenchyme levels of the OT septum the expression of Tg fb2 (Fig. 2H) and Tg fb3 (Fig. 2I) partly overlap, though Tg fb2 is more highly expressed in the area of myocardialization (arrowhead, Fig. 2H) and in the muscular ring surrounding the aortic valve (Fig. 2H). Tg fb2 is also highly expressed in the epicardium lining the atrium (arrows, Fig. 2G and H). Both Tg fb2 and Tg fb3 are only weakly expressed in the distal pulmonary valve (Fig. 2K and L). Relative to the aortic valve, the muscular ring surrounding the pulmonary valve, as well as the media of the ascending aorta and pulmonary trunk, show higher Tg fb2 than Tg fb3 expression (compare Fig. 2H and K to I and L). By E15.5 Tg fb1 is now the most highly expressed isoform in the endocardial cells of the myocardium, and Tg fb2 expression is low but predominantly myocardial in nature, whereas in the epicardium Tg fb1 and Tg fb3 expression is higher than that of Tg fb2 (Fig. 2M–O).
In general, the Tg fbs are not expressed in an overlapping fashion with the exceptions that from E12.5 onwards Tg fb2 and Tg fb3 are both expressed in the developing cushion mesenchyme of AV and OT cushions (Molin et al, unpublished; Fig. 2G–L), and all three are expressed in the epicardium (Fig. 2M–O). Interestingly, Tg fb2, and not Tg fb3, is expressed in the myocardium adjacent to the AV cushions and in the septal aspect which becomes myocardialized, suggesting that despite an overlap in expression they probably play non-redundant roles [27]. These observations are consistent with the fact that Tg fb2−/− mice have abnormal pharyngeal arch artery remodeling and defective myocardialization of the OT septum [28].
2.2. Specification of early cardiac precursor cells
The formation of the early tubular heart from splanchnic mesoderm along the anterior–posterior (AP) axis of the heart fields requires cross-talk between mesoderm and underlying endoderm. This cellular and molecular induction in the primary heart forming regions is important for the specification and differentiation of myocardial and endocardial precursor cells (reviewed in [29]). Many endoderm-derived growth factors such as BMP2, VEGF-2, FGF2 and TGFβs have been implicated in this process in the avian system [30]. TGFβ2 and TGFβ receptors are expressed in the precardiac mesoderm along with BMP2, and their receptors are also localized in the endoderm adjacent to the precardiac region [26,29,31]. Consequently, members of the TGFβ family can serve as putative molecular inductive signals at the heart forming fields for the formation of myocardial as well as endocardial precursor cells.
Numerous TGFβ family members, such as Activin, BMP, Nodal, Lefty and others, are essential for the establishment of embryonic asymmetry (reviewed in [22]). The establishment of this asymmetry is in turn critical for heart development [21]. Since this process has been covered elsewhere, this review will limit its focus to the functional activities of the three TGFβ ligands and their signaling molecules in cardiovascular development and homeostasis.
2.3. Cardiac looping
Cardiac looping is one of the important early events of cardiac morphogenesis. It is a dynamic process and can be categorized into two phases [32]. The first phase involves rightward bending of the linear heart tube at the interface between the primitive right and left ventricles into a S-shaped heart (E8.0–8.5 in mouse). In the second phase, the inner curvature tightens and the OT moves over the atrio-ventricular canal. Simultaneously, septal connections are made between the primary ring or fold of the ventricle and the atrial septation, resulting in a four-chambered heart. Although Tg fb2 expression data would be consistent with a function in cardiac looping, analysis of heart looping in Tg fb2−/− mice shows no gross morphological abnormalities in the initial bending of the linear heart tubes. Similarly, Tg fb3−/− mice exhibit normal bending of early developing hearts (M. Azhar and T. Doetschman, data not shown). Therefore, the cardiovascular malformations in Tg fb2 null mice might arise from defects in the second phase of cardiac looping which involves septation and remodeling of the developing heart [33].
2.4. Endocardial epithelial–mesenchymal transformation
As the ventricular-infundibular fold deepens during looping, the endocardial cushions begin to form at the future AV and OT regions of the heart. This involves an AV- and OT-myocyte-induced EMT of endocardial cells that break away from the endocardium, migrate into the cardiac jelly, and transform into cushion mesenchyme cells [34] (Fig. 3A). These cushions expand and fuse to eventually form aspects of the atrial, ventricular, AV and OT septa and valves. From endocardial cushion explant studies [35] there is considerable evidence that this transformation involves signals from the adjacent myocardium [36], the extracellular matrix (ECM) of the cardiac jelly [37], and TGFβs [38]. In chicken, TGFβ2 and TGFβ3 are thought to decrease cell adhesion of the transforming endocardial cells [39] and to enhance their cell migration into the cardiac jelly [40,41], though in mouse TGFβ2 is thought to carry out both of these functions [27,39]. Also, in chicken the β-glycan TGFβRIII, has been localized to the endocardium overlying both the AV and OT cushions [41]. In addition, β-glycan, but not TGFβRII, has been shown to mediate endothelial activation by triggering distinct cellular and molecular responses [42]. Together, these data imply a prominent role for TGFβ2 in the EMT of mammals.
Fig. 3.

Endocardial epithelial–mesenchymal transformation (A) to form endocardial cushions (E9.5) and myocardialization (B) to form muscularized OT/AV septum (E12.5–16.5). OTC: outflow tract cushion; AVC: atrio-ventricular cushion; IC: inner curvature; M: myocardium; IVS: interventricular septum, RV: right ventricle; LV: left ventricle.
Expression studies have shown that Tg fb2 is localized to the cushion myocardium as well as to the invading mesenchymal cells during mouse EMT. These data also indicate that Tg fb3 is not expressed in early cardiac endocardial cushions of the mouse (Fig. 2C and F) but only in the established mesenchymal cells at later stages [27]. TGFβ2, and not TGFβ3, has been found to induce mouse EMT in collagen gel explant cultures [27]. The role of TGFβ1, if any, is not clear, although it is expressed in the endocardial cells of the AV canal during mouse EMT [43], where TGFβ3 is found to be expressed in avian models [39]. No cardiac developmental defects have been detected in TGFβ1-deficient mice [44,45].
In the mammalian system only Tg fb2−/− mice have clear congenital heart defects [5]. Analysis of Tg fb3−/− mice has revealed minor differences in position and curvature of the aortic arches and in ventricular wall thickness, though the significance of these differences from wildtype animals is not clear since they do not affect survival (compare Fig. 4G with I, and Fig. 4A and B with E and F). Interestingly, several Tg fb2−/− fetuses exhibit early post-transformation embryonic lethality between E9.5 and E13.5, suggesting that heart malformations due to defects in EMT may occur. Also, Tg fb2 Tgfb3 double knockout (DKO) mice do not survive beyond embryonic stage E15.0, suggesting either a redundant or an additional role for TGFβ3 in EMT or post-transformation expansion of cushion mesenchyme during valvular development (M. Azhar and T. Doetschman, unpublished data). This is consistent with the overlap in expression of Tg fb2 and Tg fb3 in the OT cushion mesenchyme of E13.5 mice (Fig. 2H and I; Molin et al. unpublished data). The combined functions of the three TGFβs in cardiovascular development are yet to be determined.
Fig. 4.
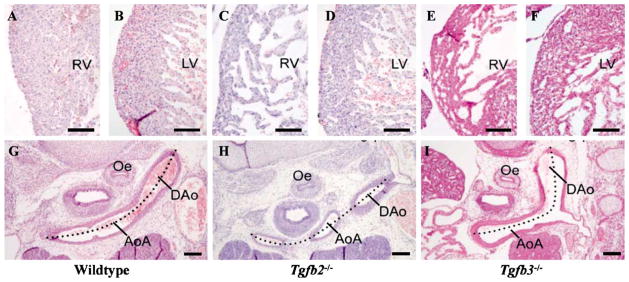
Histochemical analysis of E15.5 Tg fb2 and Tg fb3 KO hearts at the ventricular wall (G–I) and aortic arch artery (A–F). AoA: aortic arch; DAo: dorsal aorta; LV: left ventricle, Oe: oesophagus; RV: right ventricle. Bars: A–F, 100 nm; G–I, 200 nm.
Gene expression profiling of TGFβ-induced EMT in vitro in human keratinocytes [46] and in chick hearts [42] have revealed several common candidate genes involved in TGFβ-mediated EMT. Among these are BMPs and their cognate receptors which are known to be expressed in the myocardium adjacent to the developing cushions and to regulate EMT and post-transformation cushion remodeling (reviewed in [23]). BMP2 and TGFβ3 are thought to act synergistically in avian cushion development [47]. In the mammalian system, Bmp6 Bmp7 DKO mice display abnormal cushion development [48]. Recently Schneider and associates [49] have demonstrated that conditional ablation (CKO) of Tg fbr1a (gene for BMP receptor ALK3) in the myocardium can result in failure of AV cushion formation, and abnormal trabeculae and ventricular myocardial compaction. Notably, no defects were found in the OT of Tg fbr1a CKO mice. A downregulation of TGFβ2 was found, suggesting that a severe reduction in TGFβ2 in the myocardium could contribute to the defects seen in Tg fbr1a CKO hearts. It is noteworthy that Tg fb2−/− mice have hyperplastic AV cushions along with major defects in the OT segment, as well as spongy or less compact ventricular myocardium (Fig. 3B). In contrast, despite reduced levels of TGFβ2, Tg fbr1a CKO hearts show reduced AV cushions and no OT defects, indicating that BMP6 and BMP7 also function independently of and antagonistically to TGFβ2 in AV cushion formation. Knockout of the inhibitory Smad6 gene results in hyperplastic cardiac valves [50], a phenotype similar to that of Tg fb2−/− mice and opposite to that of the Bmp6 Bmp7 DKO mice, suggesting that during AV cushion formation the inhibitory activity of SMAD6 affects BMP and not TGFβ2 signaling.
2.5. Epicardial epithelial–mesenchymal transformation
Most studies on epicardial EMT have been done in the chicken heart. Epicardium, a third cell layer which develops over the myocardium and covers the entire mouse heart by E11–12, originates from the mesothelial cells which in turn are derived from cells migrating from the proepicardial organ (PEO) at E9.0–9.5 [51]. During heart development the epicardium transforms to mesenchyme giving rise to coronary pericytes and vascular smooth muscle cells (VSMC), invasive mesenchymal cells of the AV cushions and valves, and intermyocardial fibroblasts [52,53]. Using tissue explants it has recently been shown that treatment with FGF1, 2 and 7 promotes epicardial EMT, whereas treatment with TGFβ1, 2 and 3 inhibits epicardial EMT [54]. This is in contrast to the known functions of TGFβ in avian endocardial EMT discussed above in which TGFβs play an inductive role. The expression of TGFβ2 in the PEO and the early migrating epicardial cell, and the expression of all three isoforms in later stages of the epicardium suggest a TGFβ function in epicardial cardiogenesis (Fig. 2M–O; Molin et al., unpublished data).
It is possible that the cardiac phenotypes of Tg fb2−/−mice might result, at least in part, from an epicardial EMT defect resulting in hyperplastic AV/OT cushions [33], segmentally thick epicardium (Azhar & Doetschman, unpublished data) and underdeveloped ventricular myocardium relative to lumen volume (Figs. 3B and 5). Defects at the level of the myocardium are evident in both Tg fb2−/− and Tg fb3−/− mice. Cross-sections of the right and left ventricle wall show a different wall thickness and compactness between wildtype (Fig. 4A and B), Tg fb2−/− (Fig. 4C and D) and Tg fb3−/− (Fig. 4E and F) mice, being reduced and spongier in the two knockouts. Tg fb1−/− mice have no congenital heart defects related to epicardial EMT. However, as shown above we have preliminary indications that Tg fb3−/− mice have minor differences in myocardial architecture (Fig. 4E and F). It remains to be determined whether multiple TGFβs synergistically affect epicardial EMT in vivo.
Fig. 5.

Morphometric analysis of ventricular myocardial and lumen volumes of E14.5 & E15.5 Tg fb2+/+ and Tg fb2−/− embryos. Paired t-test comparison between Tg fb2+/+ and Tg fb2−/− embryos at E14.5 and E15.5.
2.6. Myocardialization
Myocardialization is the process by which myocardial cells invade the endocardial cushions. This process is most prominent in the proximal part of the OT cushions (Fig. 3B). After fusion these cushions will form the myocardial infundibulum that lines the OT of the right ventricle. Myocardialization in the OT has been described in human embryos [55,56], chicken embryos [32,57–59] and mouse embryos [60]. The left ventral OT cushion merges with the superior AV cushion and the latter fuses with the inferior AV cushion. Also, during the formation of the atrial and ventricular septum in the AV cushion region myocardialization has been described. Notably, most cardiac malformations associated with defective myocardialization involve OT segments. Defective myocardialization can result in a thickening of the OT septum as is seen in some cases of tetralogy of Fallot, in OT obstruction in neonates with subpulmonary stenosis [55], and in diabetic rat embryos [61]. It is also possible that a lack of myocardialization could lead to a fibrotic and diminished OT septum. In neonates this leads to a double committed ventricular septal defect. It is also encountered in the trisomy 16 mouse model which lacks myocardialization of proximal cushions with the resultant failure to form a muscular OT septum [62]. A similar observation is seen in chicken embryos in which hemodynamics are altered by a venous clip procedure [63]. The cellular and molecular mechanisms underlying defective myocardialization remain to be solved, but it has been postulated that in chicken embryos TGFβ2 plays a role [58,59].
The hearts of E18.5 Tg fb2 null fetuses have, with varying penetrance, double outlet right ventricle, double inlet left ventricle, atrial and ventral septal defects, hyperplastic inlet valves, and occasional thickened semilunar valves and persistent truncus arteriosus [5,33]. The most notable defect in the embryonic hearts of these mice is a lack of myocardialization of the OT cushions and in the portion of the AV cushions that are involved in septation (Fig. 3B). The intracardiac malformations named above result from deficient secondary looping of the heart. Whether TGFβs play a primary role in this process is unknown. It is reasonable to assume that the process of myocardialization would involve decreased myocardial cell adhesion, a more receptive (differentiated) cushion mesenchyme, or a myocardium more responsive to signals from the adjacent cushion mesenchyme. It has been noted that cushion mesenchyme close to the myocardium is more condensed and differentiated, whereas the cushion mesenchyme more luminal to the myocardium is less condensed and more highly proliferating. It is hypothesized that this gradient of proliferation to differentiation could regulate cushion mesenchyme differentiation and induce myocardialization (reviewed in [34]).
ECM is a major component of myocardial and cushion tissues, and TGFβs are known to strongly affect ECM composition. Consequently, it is likely that ECM components such as collagens, chondroitin sulfate proteoglycan (CSPG2) and fibronectin are involved in the process of myocardialization, and they are also expressed in the cushion mesenchyme [34]. Since fibronectin can either inhibit or enhance invasion of a tissue depending on the cellularity of the invaded tissue [64], it is likely to significantly affect myocardialization because of the cell density gradient of the cushion mesenchyme cells. In addition, fibulin-2 is associated with the early, more proliferative stages of endocardial cushion development [65], whereas fibrillin-2 [34] is more highly expressed in the differentiated mesenchyme. Finally, for myocardial cells to leave myocardium they must presumably alter their differentiation state in order to weaken adhesive interactions with other myocardial cells before invading the cushion mesenchyme. Determination of alterations in these ECM components in Tg fb2−/− mice should shed light on the mechanisms by which TGFβ2 facilitates the process of myocardialization.
2.7. Roles of neural crest cells in myocardialization
Neural crest cells (NCC) migrate into the OT cushions in the chicken embryo prior to myocardialization and cushion fusion, and are thought to be involved in the differentiation of cushion mesenchyme and myocardialization of the developing OT septum [62]. Experiments showing that quail NCC migrate into the developing chick cardiovascular system [66] and that NCC ablation leads to outlet and inlet defects [67] demonstrated that NCC migration into the aorti-copulmonary region is required for proper alignment of the OT and ventricles (reviewed in [68]). Since Pexieder’s discovery that cushion mesenchymal cells of the proximal OT undergo apoptosis [69], several lines of evidence have suggested that NCC and/or their apoptosis could play a critical role in cardiovascular remodeling (reviewed in [60]). At the site of myocardialization the NCC population goes into apoptosis [59].
TGFβs are thought to be important for NCC-mediated morphogenetic processes because NCC produce TGFβ2 [70,71]. Tg fb2 is highly expressed in the OT cushions, especially in the NCC-populated condensed mesenchyme of the OT [72] (Fig. 2G and H, and J and K). At the period of myocardialization the expression of Tg fb2 and Tg fb3 is restricted to the mesenchyme. The underlying myocardium, which at previous stages was the main source of their expression, ceases to express them (Fig. 2G–L). Recent unpublished data (Molin & Gittenberger-de Groot) reveal high expression levels only of Tg fb2 and only in the septal portion that becomes myocardialized. In Tg fb2−/−mice defective myocardialization was found to be associated with a 1-day delay in the peak of apoptosis and an additional delay in its regression. There was no subsequent decrease in the volume of total endocardial cushion, and a fibrous OT septum developed [33]. From recent data it has become clear that NCC also populate the OT septum in both Tg fb2+/+ and Tg fb2−/− mouse embryos (Fig. 3B; Gittenberger-de Groot, unpublished observations; [72]). Hence, TGFβ2 is not required for NCC migration to the OT region, but may function in regulating NCC apoptosis. One possibility is that TGFβ2 is activated by proteolysis due to NCC apoptosis [59]. Since TGFβs are cytokines known for their involvement in apoptosis and chemotactic signaling (reviewed in [6]), it is possible that a positive feedback loop of TGFβ2 activation and apoptosis, coupled with chemotaxis, leads to myocardial cell invasion and the replacement of dying cushion mesenchyme cells.
The potential roles of NCC in pharyngeal arch remodeling will be discussed at the end of the Section 2.8.
2.8. Pharyngeal arch artery remodeling
The early embryonic mammalian pharyngeal arch artery system consists of five paired arch arteries, numbered I–VI in order of appearance from rostral to caudal, the fifth artery normally considered to be absent [73]. The remodeling of the arch artery system into the adult left-sided vascular system requires the regression of the right dorsal aorta (α-segment), the right VIth arch artery and both carotid ducts (γ-segments). When remodeling of the arch system is complete (E15.5), defects at the aortic arch level are evident in both Tg fb2−/− and Tg fb3−/− mice. The normally curved (dotted line) left-sided AoA in the wildtype (Fig. 4G) is regression in the Tg fb2−/− mouse (Fig. 4H), and Tg fb3−/− mice have minor defects in position and curvature of the aortic arches (Fig. 4I), though these do not affect embryo survival [74,75]. Molin et al. [28] have found that in normal regression, the highest degree of apoptosis shifts from the mesenchyme at the onset of remodeling towards the media and SMC at the latest stages of regression. A parallel between lumen diameter, wall volume reduction and apoptosis was found. The presence of a relatively high incidence of apoptosis at the earliest point when left/right differences become apparent underlines the relation between the early onset of remodeling and apoptosis. This increase in apoptosis might imply a relation between a reduction in lumen diameter and associated flow [76,77].
Tg fb2−/− embryos exhibit decreased vascular regression in the α-segment of the right dorsal aorta [28]. This malformation is characterized by lack of a reduction in vessel lumen diameter, wall thickness and apoptosis, leading to persistence of the right dorsal aorta α-segment. On the other hand, the normally non-regressing segments of the IVth arch artery regress in Tg fb2−/− embryos and exhibit vascular hypoplasia and interruption of the IV arch artery. Occasionally, an aberrant right subclavian artery is found due to combined interruption of the right IVth arch artery segment and concomitant persistence of the right dorsal aorta α-segment. Consequently, TGFβ2 appears to regulate apoptosis, both positively and negatively, depending upon the arch segment, during normal pharyngeal arch artery remodeling.
There is considerable in vitro evidence for TGFβ effects on VSMC differentiation [78,79]. With respect to pharyngeal arch artery remodeling a difference in cellular origin of the VSMC population between segments could account for the remodeling differences which underlie formation of the asymmetric vascular system. SMC are known to originate either from ectodermally-derived neural crest or from local mesenchyme [80]. If SMC originate from ectodermally-derived cardiac neural crest, their in vitro growth response is enhanced by TGFβ. However, if they are derived from local mesenchyme, TGFβ inhibits their in vitro growth response [78]. Consequently, it is possible that varying compositions of SMC with different growth and apoptosis responses to TGFβ2 may play an important role in determining the persistence or regression of different arch arteries.
2.9. Calcium signaling in heart development
Morphometric analysis of the developing total ventricular myocardial volume and total ventricular luminal volume show higher total ventricular luminal volume in Tg fb2−/−hearts (Fig. 5), such that the ventricular myocardial architecture appears to be more spongy and dilated and less compact (also see Fig. 3B) [5,33]. FKBP12 is postulated to interact with TGFβR1 [1] as well as with cardiac ryanodine and inositol 1,4,5-triphosphate receptors which regulate intracellular Ca2+ levels [81]. FKBP12 deficiency in mice leads to high intracellular Ca2+ and non-compaction of ventricular myocardium, cardiac dysfunction and dilated cardiomyopathy [82]. Intracellular Ca2+ is involved in controlling gene expression by activating the nuclear factor of activated T cells (NF-AT) transcription factors through dephosphorylation by calcineurin. Since both Fkbp12−/− and Tg fb2−/−mice have non-compaction of ventricular myocardium, and Nfat−/− mice have reduced cardiac cushions [83,84], it is reasonable to postulate that a TGFβ2–FKBP12–calcineurin pathway is involved in cardiovascular development. In this regard, it is of interest that a defect in such a pathway may be the cause of autoimmune disease in Tg fb1−/− mice [85].
2.10. Congenital heart diseases associated with chromosome 22q11
Genomic disorders involving amplification (tetrasomy & trisomy) and hemizygous deletion of regions/genes present on human chromosome 22 (22q11) are often associated with human cat-eye syndrome (CES), deletion (22) syndrome and velocardiofacial/DiGeorge syndrome (VCFS)/DGS), respectively. Of these, VCFS/DGS is the most frequently found congenital disorder (1/4000 live births; reviewed in [25]). Genetically engineered mice with alterations in genes or chromosomal regions syntenic to human 22q11 (mouse 16) show promise for unraveling the complexity of these congenital disorders. Some of the features typical of disorders associated with human 22q11 or mouse 16 include abnormalities of cardiac outflow and pharyngeal arch artery system, craniofacial defects, hypoplasia of thymus and parathyroid, anomalies of the eye and inner ear, and cleft palate (reviewed in [86]). Genetic pathways involving proper NCC development and function are considered to be the major pathways leading to DGS [87,88], and trisomy 16 in mice is involved in NCC-mediated abnormalities in myocardialization of the OT septum [62]. Tg fb2−/− mice have multiple cardiovascular defects associated with NCC [28,33] along with other multiple congenital malformations often found associated with 22q11 disorders such as cranial defects leading to cleft palate [5]. Hence, since the human and mouse genes for TGFβ2 are not linked to human 22q11 or mouse 16, it is reasonable to hypothesize that TGFβ2 modifies, or is modified by, genetic pathways on those chromosomes.
3. Cardiovascular physiology
Cell culture studies have indicated that TGFβ1 inhibits mitotic growth of cardiomyocytes [89], and stimulates hypertrophic growth [90], fibrosis [90,91] and re-expression of the fetal isoforms of myofibrillar protein genes [92]. TGFβ1 is secreted from cultured cardiomyocytes and fibroblasts during cyclic stretch [93]. In the vascular system TGFβ1 switches the response of angiotensin II (Ang II)-stimulated cultured VSMC from mitotic to hypertrophic growth [94], TGFβ1 treatment affects Ca2+ mobilization in VSMC from spontaneously hypertensive rats [95], vascular remodeling of coronary and aortic arteries in response to hypertension is associated with increases in TGFβ1 and TGFβ3 levels [96], and intimal thickening following shear stress is associated with increases in TGFβ1 [97]. The renin–angiotensin system plays an important role in cardiovascular homeostasis through regulation of blood pressure and extracellular fluid volume [98]. Physiological stress from salt and water imbalance can lead to hypertrophy of the juxtaglomerular apparatus and induce both TGFβ2 and renin [99], suggesting that TGFβ2 may regulate pathophysiological renal responses involving blood pressure control. Altogether, these data suggest important roles for TGFβs in several aspects of cardiovascular physiology ranging from the cardiomyocyte to vascular smooth muscle and renal control of blood pressure.
3.1. Blood pressure and cardiac contractility
Blood pressure regulation is a multiorgan-regulated process involving many signaling pathways. TGFβ1 has been implicated in several of the chronic pathologies developed from poorly controlled blood pressure, including left ventricular cardiac hypertrophy, vascular remodeling, and renal disease. There is clinical and genetic evidence demonstrating a correlation between TGFβ1 polymorphisms and hypertension [100]. One clinical study revealed that TGFβ1 mRNA and protein levels are significantly higher in hypertensive patients compared with normotensives [101]. This overexpression of TGFβ1 is more frequent in hypertensive African-Americans compared with hypertensive Caucasians and correlates with increased frequency of the proline allele at codon 10, a signal peptide region of the TGFβ1 gene. Tg fb1 mRNA is elevated in aortae of hypertensive rats [102], and recent evidence demonstrates that mice deficient in SMAD6, an inhibitory protein in TGFβ signaling, has a significant elevation in mean arterial blood pressure compared to wildtype mice [50]. However, mice deficient in TGFβ1 display no difference in mean arterial blood pressure [103], and no increase in blood pressure was reported in transgenic mice with cardiac overexpression of Tg fb1 [104]. Nonetheless, diastolic dysfunction has been found in work-performing heart preparations [105] from Tg fb1−/−mice (Fig. 6A). These results are consistent with a TGFβ1 deficiency leading to impairment of inotropic responsiveness to β-adrenergic stimulation, but do not demonstrate causality.
Fig. 6.

Diastolic and β-adrenergic deficiency in Tg fb1−/− hearts. Isolated, work-performing hearts from adult Tg fb1−/− Scid mice (A). β-Adrenergic-stimulated contractility (+dP/dt) and relaxation (−dP/dt) rates in LFA-1 antibody-treated adult Tg fb1−/− mice. Immunodeficiency (Scid) or immunosuppression (LFA-1 antibody treatment) is required to circumvent weaning age lethality (see Section 3.2 for explanation). IVP: interventricular pressure.
The adrenergic receptor system is critical to the regulation of blood pressure and cardiac contractility/relaxation. There is evidence that the β-adrenergic system regulates Tg fb1 and Tg fb3 expression in cultured cardiac cells [106,107]. Similarly, in vitro studies have demonstrated that TGFβ1 can modulate α- and β-adrenergic signaling in various cell types by altering adrenergic receptor density and function [108,109]. Recent Tg fb1 transgenic and KO mouse studies have supported these in vitro observations. In work-performing heart preparations from Tg fb1−/− animals there is a decrease in both contraction and relaxation rates in response to the β-adrenergic agonist, isoproterenol (Fig. 6B and C). This correlates with a reduction in β-adrenergic receptor density (not shown). In addition, consistent with the diastolic dysfunction results mentioned above, only the relaxation rate is depressed in unstimulated hearts (Fig. 6C). These alterations in β-adrenergic signaling result in an enhancement of the inotropic responsiveness to β-receptor stimulation [104]. Consequently, TGFβ1 is required for full Ca2+ uptake in the unstimulated heart, and is required for full positive contractile and relaxation responses to β-adrenergic stimulation.
3.2. Cardiac hypertrophy
There is clinical evidence that patients with idiopathic hypertrophic obstructive cardiomyopathy have elevated cardiac Tg fb1 mRNA and protein levels and increased TGFβ1 receptor levels on both cardiomyocytes and fibroblasts [110]. In addition, human atrial tissue stimulated with Ang II results in a significant increase in Tg fb1 expression [111], which correlates with cardiac hypertrophy [112], fibrosis [113] and recapitulation of fetal isoforms of cardiac myofibrillar proteins [114]. However, in a survey for candidate genes related to increased susceptibility to hypertrophic cardiomyopathy, Tg fb1 polymorphisms were not associated with the risk or severity of the disease [115]. From these data, it was unclear whether there is a causal relationship between increased levels of Tg fb1 and hypertrophy, isoform switching and fibrosis.
The role of TGFβ1 in cardiac hypertrophy has been assessed by applying non-pressor doses of the hypertrophic stimulant Ang II to Tg fb1−/− Rag1−/− mice [103]. Ablation of the Rag1 immunodeficiency gene is required [116] to circumvent the weaning-age lethality caused by a multifocal autoimmune disease [44,45]. Non-pressor doses were used to avoid compounding the Ang II stimulus with hemodynamic stimulation. Echocardiographic, gravimetric and morphometric analysis indicated that TGFβ1 is required for Ang II-induced cardiac hypertrophy since only the control (Tg fb1+/+ Rag1−/−) mice developed hypertrophy. No difference was found in levels of fibrosis, mitotic growth or cytokine infiltration between wildtype and TGFβ1-deficient mice. TGFβ and fibrosis in the cardiovascular system will be discussed in a later section. Finally, recapitulation of the fetal myosin heavy chain (β-MHC) protein was not observed in Ang II-treated Tg fb1+/+ Rag1−/− or Tg fb1−/− Rag1−/−mice, indicating that isoform switching is not obligatorily coupled with hypertrophy or TGFβ1.
Re-expression of fetal structural protein isoforms has often been used as an indicator of cardiac hypertrophy [117,118]. However, cardiac hypertrophy has been dissociated from the re-expression of fetal isoforms under specific types of stimulation [119], suggesting that the isoform switch is dependent upon pressure overload [120,121] rather than on growth factors such as FGF2 and TGFβ1 [103,121].
3.3. Vascular smooth muscle and vessel remodeling
Several studies have implicated TGFβ1 in arterial remodeling following hypertrophic stress [94,96,122] or in hypertensive rats [95,123]. The vasoconstrictor thromboxane A2 causes hypertrophy in cultured VSMC [124], and thromboxane A2 and Ang II induce hypertrophy in cultured VSMC through the effects of the endogenously produced growth modifiers FGF2 and TGFβ1 [125]. However, overexpression of the human FGF2, especially the high molecular weight isoforms, in mouse aortic VSMC from transgenic mice enhances serum-stimulated mitotic growth [126]. Thus, it has been hypothesized that after stimulation with hypertrophic agents FGF2 functions to promote mitotic growth, while TGFβ1 prevents full cell cycle progression such that their combined activity would result in hypertrophy.
To test this hypothesis cultured Tg fb1+/+ and Tg fb1−/−aortic VSMC were stimulated with the hypertrophic thromboxane mimetic U46619. Fig. 7 shows that in Tg fb1−/−cells mitotic growth is more than doubled relative to vehicle alone, whereas the increase in protein synthesis over vehicle is independent of the presence or absence of TGFβ1. Addition of TGFβ1 to the U44619-treated Tg fb1−/− cells drops mitotic growth to vehicle levels, but increases protein synthesis by nearly 50% (not shown). From this and previous studies we postulate that TGFβs may function as a general response to vasoconstricting hypertrophic agonists in several cardiovascular compartments.
Fig. 7.

Thromboxane induces mitotic growth of Tg fb1−/− VSMC. Quiescent VSMC derived from Tg fb1+/+ and Tg fb1−/− mice were stimulated with vehicle or 1 μM U44619. [3H]thymidine (A) and [3H]leucine incorporation (B) after 24 and 48 h exposure, respectively. n = 7, (*) P < 0.05 vs. Tg fb1+/+ mice).
Several studies have found an association of TGFβ activity with SMC differentiation as it relates to intimal thickening following both vascular injury [127,128] and fluid shear stress [97], as well as in plaque development during atherogenesis in hypercholesterolemic animals [129]. These studies suggest important inhibitory roles for TGFβ in all of these responses, and they are consistent with in vitro studies that suggest growth inhibitory and differentiation stimulatory roles for TGFβ in VSMC [78,79,130]. One must caution, however, that an association only suggests causality. In addition, treatments that inhibit TGFβ activity with antibodies or antisense RNA can result in inconsistencies, as is the case with intimal thickening following vascular injury (compare [131] (no effect) with [128] (intimal thickening)).
Since TGFβ1 is known to be a potent regulator of the immune system [85,132], immune sources of TGFβ1 may also affect the intimal thickening that occurs during cardiac transplant arteriosclerosis. Using a heterotopic mouse transplant model cardiac grafts from CBA mice were placed into Tg fb1+/+ and Tg fb1+/− C57BL/6 recipients [133]. Arteriosclerosis was reduced in TGFβ1-deficient mice, indicating an important role for immune-cell-derived TGFβ1 in regulating intimal thickening.
From these studies it is clear that TGFβ signaling plays significant roles in vascular remodeling, but the mechanisms are unclear. The application of vascular stress and injury models to genetically engineered mice will help delineate what aspect of each response requires TGFβ signaling. The specific TGFβ signaling mechanisms underlying these responses can then be determined.
3.4. Renal pathophysiology
When the renin–angiotensin system is stimulated by physiologic or pathologic stimuli, renin is released from intracellular vesicles of the modified SMC of the medial layer of the afferent portion of glomerular arterioles in the juxtaglomerular apparatus [134]. Hypertension [135], myocardial infarction [136] and diabetic nephropathy [137] can all result from a hyperactive renin–angiotensin system. Both co-localization and co-induction of renin and TGFβ2, as well as hypertrophy, have been observed in the juxtaglomerular apparatus after water deprivation, potassium depletion or inhibition of angiotensin I converting enzyme [99,138]. This suggests a potential role for TGFβ2 in the control of renin synthesis and/or release. However, the activities of TGFβ2 and perhaps other TGFβs would be complex, as low doses (10−12 M) of TGFβ1 and TGFβ2 stimulate renin secretion in rat renal cortical slices, whereas higher doses (10−10 M) inhibit renin levels [139].
The role of TGFβ2 in renal renin regulation under dehydration conditions has been investigated in Tg fb2+/− mice [140]. Since Tg fb2−/− mice die around the time of birth due to multiple developmental defects [5], the renal studies were done on Tg fb2+/− mice whose TGFβ2 message and protein levels were approximately half those of wildtype animals. Dehydration-induced increase in plasma renin levels was not different between Tg fb2+/+ and Tg fb2+/− mice. However, in the kidneys, in the presence of a full dose of TGFβ2, dehydration led to a large increase in renin mRNA and little increase in renin activity, whereas with a half dose of TGFβ2 there was a slight increase in renin mRNA and more than a doubling of renin activity (Fig. 8). Hence, TGFβ2 acts primarily at the transcriptional level in hydrated conditions and at the translational level in dehydrating conditions where it may act as a check on renin activity, thereby protecting the juxtaglomerular apparatus from potential pathophysiological effects of excessive renin. It is not known whether other TGFβs may have independent functions in the homeostasis and pathophysiology of the kidney. This is not unreasonable given the association of higher levels of circulating TGFβ1 in African-Americans with end-stage renal disease and a coding polymorphism in TGFβ1 that associates with hypertension in African-Americans [101].
Fig. 8.

Determination of renal renin mRNA and protein levels in the kidneys of hydrated or dehydrated Tg fb2+/+ and Tg fb2+/− mice. The relative intensity of the renin signal was estimated as the sum of intensities for each glomerulus, divided by the number of glomeruli observed [140].
3.5. Fibrosis
It has long been known that TGFβ can induce fibrosis [141], (reviewed in [142]). With respect to cardiac fibrosis, however, it should be noted that in the autoimmune disease of Tg fb1−/− mice there is extensive fibrosis in the heart and liver, though not in the lung, in the absence of TGFβ1 [44,143]. Furthermore, cardiac-restricted Tg fb1 transgenic animals have atrial but not ventricular fibrosis [144]. Finally, alterations in levels of Tg fb1 mRNA do not correlate with the increased mRNA expression of fibronectin and collagens I and III during low-dose isoprenaline treatment, again suggesting that cardiac fibrosis is not significantly mediated by TGFβ1 [145]. On the other hand, Langendorff preparations of hearts from 2-year-old Tg fb1+/+ and Tg fb1+/− mice found greater diastolic dysfunction in the wildtype than in heterozygous mice [146]. Histological analysis of the hearts indicates that there is more fibrosis in Tg fb1+/+ hearts, suggesting that more fibrosis occurs in the presence of a full dose of TGFβ1, which in turn results in decreased left ventricle compliance. Consequently, it is the context of the heart and its environment which may determine whether and to what extent TGFβ1 induces cardiac fibrosis. Whether other TGFβs are involved has not been tested but can not be ruled out since decreased inhibitory SMAD7 in cardiac fibroblasts has been implicated in the pathogenesis of cardiac fibrosis in post-myocardial infarct rat hearts [147].
It is thought that the association between increased renin–angiotensin activity and renal fibrosis lies in the co-induction and co-localization of renin and TGFβ2 [148]. In experimental models, controlled infusion of TGFβ2 into adult mice leads to kidney fibrosis in the cortical tubular interstitium and vasculature, particularly at the cortical–medullary junction and medullary vasa recta, but not in glomeruli [149]. In a diabetic rat model TGFβ2 antibody treatment decreases glomerular and tubular fibrosis in streptozotocin-treated rats with diabetic nephropathy [150]. The extent to which Ang II-induced vasoconstriction and tissue hypoxia and/or increased TGFβ are responsible for the fibrosis is not clear, and a possible role for other TGFβs has not been ruled out. Since these results seem at odds with the renal renin study in Tg fb2+/− mice in which less, rather than more, TGFβ2 leads to increased renin levels, we must consider the possibility that TGFβ2 affects the development of fibrosis by a renin-independent mechanism. More highly controlled dosing of TGFβ2 along with tissue and temporal specificity in its expression will be required to delineate the complex roles of TGFβ in blood pressure homeostasis and renal pathophysiology.
4. Perspectives
Analysis of genetically engineered mouse models of TGFβ deficiencies and overexpression have revealed numerous in vivo functions for TGFβs in cardiovascular development and physiology. Nonetheless, there remain many aspects of cardiovascular development for which the roles of TGFβ have yet to be determined. It is not clear how TGFβs are involved in molecular specification and formation of early endocardial and myocardial precursors from the primary heart forming fields or in the recruitment of cells from the secondary heart-field to the outflow myocardium. And it is likely that TGFβs have complementary as well as redundant roles in endocardial and epicardial EMT and in NCC–SMC differentiation. Likewise, functional studies on TGFβ activities in response to most forms of stress and injury to the heart, kidney and vasculature have hardly begun. Finally, since TGFβ signaling can bring a multitude of pathways into play, it will be necessary to decipher what pathway networks are utilized to carry out these activities. From the practical standpoint, elucidating these cellular and molecular mechanisms will provide a greater understanding of cardiovascular diseases.
Acknowledgments
Grant support for the studies presented here: NIH grants HD26471, HL70174, HL58511, ES06096 to TD; INSERM and Association Claude Bernard to PM; VA Merit Review to GWD; NHS: ECCARD to ACG.
References
- 1.Massague J, Chen YG. Controlling TGF-beta signaling. Genes Dev. 2000;14:627–44. [PubMed] [Google Scholar]
- 2.Sporn MB, Roberts AB. Transforming growth factor-beta: recent progress and new challenges. J Cell Biol. 1992;119:1017–21. doi: 10.1083/jcb.119.5.1017. review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heine U, Munoz EF, Flanders KC, Ellingsworth LR, Lam HY, Thompson NL, et al. Role of transforming growth factor-beta in the development of the mouse embryo. J Cell Biol. 1987;105:2861–76. doi: 10.1083/jcb.105.6.2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pelton RW, Saxena B, Jones M, Moses HL, Gold LI. Immunohistochemical localization of TGF beta 1–3 in the mouse embryo: expression patterns suggest multiple roles during embryonic development. J Cell Biol. 1991;115:1091–105. doi: 10.1083/jcb.115.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanford LP, Ormsby I, Gittenberger-de GA, Sariola H, Friedman R, Boivin GP, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–70. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunker N, Krieglstein K. Targeted mutations of transforming growth factor-beta genes reveal important roles in mouse development and adult homeostasis. Eur J Biochem. 2000;267:6982–8. doi: 10.1046/j.1432-1327.2000.01825.x. [DOI] [PubMed] [Google Scholar]
- 7.Saharinen J, Hyytiainen M, Taipale J, Keski-Oja J. Latent transforming growth factor-beta binding proteins (LTBPs): structural extracellular matrix proteins for targeting TGF-beta action. Cytokine Growth Fact Rev. 1999;10:99–117. doi: 10.1016/s1359-6101(99)00010-6. [DOI] [PubMed] [Google Scholar]
- 8.Wrana JL. TGF-beta receptors and signalling mechanisms. Miner Electrolyte Metab. 1998;24:120–30. doi: 10.1159/000057359. [DOI] [PubMed] [Google Scholar]
- 9.Wang XF, Lin HY, Ng-Eaton E, Downward J, Lodish HF, Weinberg RA. Expression cloning and characterization of the TGF-beta type III receptor. Cell. 1991;67:797–805. doi: 10.1016/0092-8674(91)90074-9. [DOI] [PubMed] [Google Scholar]
- 10.Cheifetz S, Bellon T, Cales C, Vera S, Bernabeu C, Massague J, et al. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J Biol Chem. 1992;267:19027–30. [PubMed] [Google Scholar]
- 11.Lopez-Casillas F, Cheifetz S, Doody J, Andres JL, Lane WS, Massague J. Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-beta receptor system. Cell. 1991;67:785–95. doi: 10.1016/0092-8674(91)90073-8. [DOI] [PubMed] [Google Scholar]
- 12.Lopez-Casillas F, Payne HM, Andres JL, Massague J. Betaglycan can act as a dual modulator of TGF-beta access to signaling receptors: mapping of ligand binding and GAG attachment sites. J Cell Biol. 1994;124:557–68. doi: 10.1083/jcb.124.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rotzer D, Roth M, Lutz M, Lindemann D, Sebald W, Knaus P. Type III TGF-beta receptor-independent signalling of TGF-beta2 via TbetaRII-B, an alternatively spliced TGF-beta type II receptor. EMBO J. 2001;20:480–90. doi: 10.1093/emboj/20.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lastres P, Letamendia A, Zhang H, Rius C, Almendro N, Raab U, et al. Endoglin modulates cellular responses to TGF-beta 1. J Cell Biol. 1996;133:1109–21. doi: 10.1083/jcb.133.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li C, Guo B, Bernabeu C, Kumar S. Angiogenesis in breast cancer: the role of transforming growth factor beta and CD105. Microsc Res Technol. 2001;52:437–49. doi: 10.1002/1097-0029(20010215)52:4<437::AID-JEMT1029>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 16.Moustakas A, Heldin CH. From mono- to oligo-Smads: the heart of the matter in TGF-β signal transduction. Genes Dev. 2002;16:1867–71. doi: 10.1101/gad.1016802. [DOI] [PubMed] [Google Scholar]
- 17.Mulder KM. Role of Ras and Mapks in TGFbeta signaling. Cytokine Growth Fact Rev. 2000;11:23–35. doi: 10.1016/s1359-6101(99)00026-x. [DOI] [PubMed] [Google Scholar]
- 18.Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-beta responses. Cell. 1998;95:737–40. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- 19.Nesti LJ, Caterson EJ, Wang M, Chang R, Chapovsky F, Hoek JB, et al. TGF-beta1-stimulated osteoblasts require intracellular calcium signaling for enhanced alpha5 integrin expression. Ann NY Acad Sci. 2002;961:178–82. doi: 10.1111/j.1749-6632.2002.tb03078.x. [DOI] [PubMed] [Google Scholar]
- 20.Hoying JB, Yin M, Diebold R, Ormsby I, Becker A, Doetschman T. Transforming growth factor beta1 enhances platelet aggregation through a non-transcriptional effect on the fibrinogen receptor. J Biol Chem. 1999;274:31008–13. doi: 10.1074/jbc.274.43.31008. [DOI] [PubMed] [Google Scholar]
- 21.Whitman M, Mercola M. TGF-beta superfamily signaling and left–right asymmetry. Science STKE. 2001 doi: 10.1126/stke.2001.64.re1. http://www.stke.org/cgi/content/full/OCsigtrans2001;/64/re1. [DOI] [PubMed]
- 22.Hamada H, Meno C, Watanabe D, Saijoh Y. Establishment of vertebrate left–right asymmetry. Nat Rev Genet. 2002;3:103–13. doi: 10.1038/nrg732. [DOI] [PubMed] [Google Scholar]
- 23.Schneider MD, Gaussin V, Lyons KM. Tempting fate: BMP signals for cardiac morphogenesis. Cytokine Growth Fact Rev. 2003;14:1–4. doi: 10.1016/s1359-6101(02)00053-9. [DOI] [PubMed] [Google Scholar]
- 24.MacLellan WR, Brand T, Schneider MD. Transforming growth factor-beta in cardiac ontogeny and adaptation. Circ Res. 1993;73:783–91. doi: 10.1161/01.res.73.5.783. [DOI] [PubMed] [Google Scholar]
- 25.McDermid HE, Morrow BE. Genomic disorders on 22q11. Am J Hum Genet. 2002;70:1077–88. doi: 10.1086/340363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dickson MC, Slager HG, Duffie E, Mummery CL, Akhurst RJ. RNA and protein localisations of TGF beta 2 in the early mouse embryo suggest an involvement in cardiac development. Development. 1993;117:625–39. doi: 10.1242/dev.117.2.625. [DOI] [PubMed] [Google Scholar]
- 27.Camenisch TD, Molin DG, Person A, Runyan RB, Gittenberger-de Groot AC, McDonald JA, et al. Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev Biol. 2002;248:170–81. doi: 10.1006/dbio.2002.0731. [DOI] [PubMed] [Google Scholar]
- 28.Molin DG, DeRuiter MC, Wisse LJ, Azhar M, Doetschman T, Poelmann RE, et al. Altered apoptosis pattern during pharyngeal arch artery remodelling is associated with aortic arch malformations in Tgfbeta2 knock-out mice. Cardiovasc Res. 2002;56:312–22. doi: 10.1016/s0008-6363(02)00542-4. [DOI] [PubMed] [Google Scholar]
- 29.Lough J, Sugi Y. Endoderm and heart development. Dev Dyn. 2000;217:327–42. doi: 10.1002/(SICI)1097-0177(200004)217:4<327::AID-DVDY1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 30.Sugi Y, Markwald RR. Early endocardial formation originates from precardiac mesoderm as revealed by QH-1 antibody staining. Ital J Anat Embryol. 1995;100(Suppl 1):263–72. [PubMed] [Google Scholar]
- 31.Brown CB, Drake CJ, Barnett JV. Antibodies directed against the chicken type II TGFbeta receptor identify endothelial cells in the developing chicken and quail. Dev Dyn. 1999;215:79–85. doi: 10.1002/(SICI)1097-0177(199905)215:1<79::AID-DVDY9>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 32.Bouman HG, Broekhuizen ML, Baasten AM, Gittenberger-de Groot AC, Wenink AC. Spectrum of looping disturbances in stage 34 chicken hearts after retinoic acid treatment. Anat Rec. 1995;243:101–8. doi: 10.1002/ar.1092430112. [DOI] [PubMed] [Google Scholar]
- 33.Bartram U, Molin DG, Wisse LJ, Mohamad A, Sanford LP, Doetschman T, et al. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in Tg fb2 knockout mice. Circulation. 2001;103:2745–52. doi: 10.1161/01.cir.103.22.2745. [DOI] [PubMed] [Google Scholar]
- 34.Mjaatvedt CH, Yamamura H, Wessels A, Ramsdell A, Turner D, Markwald RR. Mechanisms of segmentation, septation, and remodeling of the tubular heart: endocardial cushion fate and cardiac looping. In: Harvey RP, Rosenthal N, editors. Heart development. New York: Academic Press; 1999. pp. 159–77. [Google Scholar]
- 35.Runyan RB, Markwald RR. Invasion of mesenchyme into three-dimensional collagen gels: a regional and temporal analysis of interaction in embryonic heart tissue. Dev Biol. 1983;95:108–14. doi: 10.1016/0012-1606(83)90010-6. [DOI] [PubMed] [Google Scholar]
- 36.Potts JD, Runyan RB. Epithelial–mesenchymal cell transformation in the embryonic heart can be mediated, in part, by transforming growth factor beta. Dev Biol. 1989;134:392–401. doi: 10.1016/0012-1606(89)90111-5. [DOI] [PubMed] [Google Scholar]
- 37.Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res. 1995;77:1–6. doi: 10.1161/01.res.77.1.1. [DOI] [PubMed] [Google Scholar]
- 38.Huang JX, Potts JD, Vincent EB, Weeks DL, Runyan RB. Mechanisms of cell transformation in the embryonic heart. Ann NY Acad Sci. 1995;752:317–30. doi: 10.1111/j.1749-6632.1995.tb17441.x. [DOI] [PubMed] [Google Scholar]
- 39.Boyer AS, Ayerinskas II, Vincent EB, McKinney LA, Weeks DL, Runyan RB. TGFbeta2 and TGFbeta3 have separate and sequential activities during epithelial–mesenchymal cell transformation in the embryonic heart. Dev Biol. 1999;208:530–45. doi: 10.1006/dbio.1999.9211. [DOI] [PubMed] [Google Scholar]
- 40.Boyer AS, Erickson CP, Runyan RB. Epithelial–mesenchymal transformation in the embryonic heart is mediated through distinct pertussis toxin-sensitive and TGFbeta signal transduction mechanisms. Dev Dyn. 1999;214:81–91. doi: 10.1002/(SICI)1097-0177(199901)214:1<81::AID-DVDY8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 41.Brown CB, Boyer AS, Runyan RB, Barnett JV. Requirement of type III TGF-beta receptor for endocardial cell transformation in the heart. Science. 1999;283:2080–2. doi: 10.1126/science.283.5410.2080. [DOI] [PubMed] [Google Scholar]
- 42.Boyer AS, Runyan RB. TGFbeta Type III and TGFbeta Type II receptors have distinct activities during epithelial–mesenchymal cell transformation in the embryonic heart. TA, Dev Dyn. 2001;221:454–9. doi: 10.1002/dvdy.1154. [DOI] [PubMed] [Google Scholar]
- 43.Akhurst RJ, Lehnert SA, Faissner A, Duffie E. TGF beta in murine morphogenetic processes: the early embryo and cardiogenesis. Development. 1990;108:645–56. doi: 10.1242/dev.108.4.645. [DOI] [PubMed] [Google Scholar]
- 44.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–9. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. 1993;90:770–4. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zavadil J, Bitzer M, Liang D, Yang YC, Massimi A, Kneitz S, et al. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc Natl Acad Sci USA. 2001;98:6686–91. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakajima Y, Yamagishi T, Hokari S, Nakamura H. Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: roles of transforming growth factor (TGF)-beta and bone morphogenetic protein (BMP) Anat Rec. 2000;258:119–27. doi: 10.1002/(SICI)1097-0185(20000201)258:2<119::AID-AR1>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 48.Kim RY, Robertson EJ, Solloway MJ. Bmp6 and Bmp7 are required for cushion formation and septation in the developing mouse heart. Dev Biol. 2001;235:449–66. doi: 10.1006/dbio.2001.0284. [DOI] [PubMed] [Google Scholar]
- 49.Gaussin V, Van de PT, Mishina Y, Hanks MC, Zwijsen A, Huylebroeck D, et al. Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor ALK3. Proc Natl Acad Sci USA. 2002;99:2878–83. doi: 10.1073/pnas.042390499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galvin KM, Donovan MJ, Lynch CA, Meyer RI, Paul RJ, Lorenz JN, et al. A role for smad6 in development and homeostasis of the cardiovascular system. Nat Genet. 2000;24:171–4. doi: 10.1038/72835. [DOI] [PubMed] [Google Scholar]
- 51.Viragh S, Challice CE. The origin of the epicardium and the embryonic myocardial circulation in the mouse. Anat Rec. 1981;201:157–68. doi: 10.1002/ar.1092010117. [DOI] [PubMed] [Google Scholar]
- 52.Dettman RW, Denetclaw W, Jr, Ordahl CP, Bristow J. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev Biol. 1998;193:169–81. doi: 10.1006/dbio.1997.8801. [DOI] [PubMed] [Google Scholar]
- 53.Gittenberger-de Groot AC, Vrancken Peeters MP, Mentink MM, Gourdie RG, Poelmann RE. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ Res. 1998;82:1043–52. doi: 10.1161/01.res.82.10.1043. [DOI] [PubMed] [Google Scholar]
- 54.Morabito CJ, Dettman RW, Kattan J, Collier JM, Bristow J. Positive and negative regulation of epicardial mesenchymal transformation during avian heart development. Dev Biol. 2001;234:204–15. doi: 10.1006/dbio.2001.0254. [DOI] [PubMed] [Google Scholar]
- 55.Bartelings MM, Gittenberger-de Groot AC. The outflow tract of the heart: embryologic and morphologic correlations. Int J Cardiol. 1989;22:289–300. doi: 10.1016/0167-5273(89)90270-2. [DOI] [PubMed] [Google Scholar]
- 56.Lamers WH, Viragh S, Wessels A, Moorman AF, Anderson RH. Formation of the tricuspid valve in the human heart. Circulation. 1995;91:111–21. doi: 10.1161/01.cir.91.1.111. [DOI] [PubMed] [Google Scholar]
- 57.Rosenquist TH, Fray-Gavalas C, Waldo K, Beall AC. Development of the musculoelastic septation complex in the avian truncus arteriosus. Am J Anat. 1990;189:339–56. doi: 10.1002/aja.1001890406. [DOI] [PubMed] [Google Scholar]
- 58.van den Hoff MJ, Moorman AF, Ruijter JM, Lamers WH, Bennington RW, Markwald RR, et al. Myocardialization of the cardiac outflow tract. Dev Biol. 1999;212:477–90. doi: 10.1006/dbio.1999.9366. [DOI] [PubMed] [Google Scholar]
- 59.Poelmann RE, Mikawa T, Gittenberger-de Groot AC. Neural crest cells in outflow tract septation of the embryonic chicken heart: differentiation and apoptosis. Dev Dyn. 1998;212:373–84. doi: 10.1002/(SICI)1097-0177(199807)212:3<373::AID-AJA5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 60.Poelmann RE, Molin D, Wisse LJ, Gittenberger-de GA. Apoptosis in cardiac development. Cell Tissue Res. 2000;301:43–52. doi: 10.1007/s004410000227. [DOI] [PubMed] [Google Scholar]
- 61.Siman CM, Gittenberger-de Groot AC, Wisse B, Eriksson UJ. Malformations in offspring of diabetic rats: morphometric analysis of neural crest-derived organs and effects of maternal Vitamin E treatment. Teratology. 2000;61:355–67. doi: 10.1002/(SICI)1096-9926(200005)61:5<355::AID-TERA7>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 62.Waller BR, III, McQuinn T, Phelps AL, Markwald RR, Lo CW, Thompson RP, et al. Conotruncal anomalies in the trisomy 16 mouse: an immunohistochemical analysis with emphasis on the involvement of the neural crest. Anat Rec. 2000;260:279–93. doi: 10.1002/1097-0185(20001101)260:3<279::AID-AR65>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 63.Hogers B, DeRuiter MC, Gittenberger-de Groot AC, Poelmann RE. Extraembryonic venous obstructions lead to cardiovascular malformations and can be embryolethal. Cardiovasc Res. 1999;41:87–99. doi: 10.1016/s0008-6363(98)00218-1. [DOI] [PubMed] [Google Scholar]
- 64.Armstrong PB, Armstrong MT. Intercellular invasion and the organizational stability of tissues: a role for fibronectin. Biochim Biophys Acta. 2000;1470:9–20. doi: 10.1016/s0304-419x(00)00003-2. [DOI] [PubMed] [Google Scholar]
- 65.Zhang HY, Chu ML, Pan TC, Sasaki T, Timpl R, Ekblom P. Extracellular matrix protein fibulin-2 is expressed in the embryonic endocardial cushion tissue and is a prominent component of valves in adult heart. Dev Biol. 1995;167:18–26. doi: 10.1006/dbio.1995.1003. [DOI] [PubMed] [Google Scholar]
- 66.Le Lievre CS, Le Douarin NM. Mesenchymal derivatives of the neural crest: analysis of chimeric quail and chick embryos. J Embryol Exp Morphol. 1975;34:125–54. [PubMed] [Google Scholar]
- 67.Kirby ML, Waldo KL. Role of neural crest in congenital heart disease. Circulation. 1990;82:332–40. doi: 10.1161/01.cir.82.2.332. [DOI] [PubMed] [Google Scholar]
- 68.Maschhoff KL, Baldwin HS. Molecular determinants of neural crest migration. Am J Med Genet. 2000;97:280–8. doi: 10.1002/1096-8628(200024)97:4<280::aid-ajmg1278>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 69.Pexieder T. Cell death in the morphogenesis and teratogenesis of the heart. Adv Anat Embr Cell Biol. 1975;51:1–100. doi: 10.1007/978-3-642-66142-6. [DOI] [PubMed] [Google Scholar]
- 70.Garcia-Castro M, Bronner-Fraser M. Induction and differentiation of the neural crest. Curr Opin Cell Biol. 1999;11:695–8. doi: 10.1016/s0955-0674(99)00038-1. [DOI] [PubMed] [Google Scholar]
- 71.Brauer PR, Yee JA. Cranial neural crest cells synthesize and secrete a latent form of transforming growth factor beta that can be activated by neural crest cell proteolysis. Dev Biol. 1993;155:281–5. doi: 10.1006/dbio.1993.1026. [DOI] [PubMed] [Google Scholar]
- 72.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–16. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- 73.de Ruiter MC, Gittenberger-de Groot AC, Rammos S, Poelmann RE. The special status of the pulmonary arch artery in the branchial arch system of the rat. Anat Embryol (Berl) 1989;179:319–25. doi: 10.1007/BF00305058. [DOI] [PubMed] [Google Scholar]
- 74.Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, et al. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–14. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, et al. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial–mesenchymal interaction. Nat Genet. 1995;11:415–21. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- 76.Moene RJ, Gittenberger-de Groot AC, Oppenheimer-Dekker A, Bartelings MM. Anatomic characteristics of ventricular septal defect associated with coarctation of the aorta. Am J Cardiol. 1987;59:952–5. doi: 10.1016/0002-9149(87)91132-5. [DOI] [PubMed] [Google Scholar]
- 77.Meeson A, Palmer M, Calfon M, Lang R. A relationship between apoptosis and flow during programmed capillary regression is revealed by vital analysis. Development. 1996;122:3929–38. doi: 10.1242/dev.122.12.3929. [DOI] [PubMed] [Google Scholar]
- 78.Topouzis S, Majesky MW. Smooth muscle lineage diversity in the chick embryo: two types of aortic smooth muscle cell differ in growth and receptor-mediated transcriptional responses to transforming growth factor-beta. Dev Biol. 1996;178:430–45. doi: 10.1006/dbio.1996.0229. [DOI] [PubMed] [Google Scholar]
- 79.Owens GK. Molecular control of vascular smooth muscle cell differentiation. Acta Physiol Scand. 1998;164:623–35. doi: 10.1111/j.1365-201x.1998.tb10706.x. [DOI] [PubMed] [Google Scholar]
- 80.Waldo KL, Kumiski D, Kirby ML. Cardiac neural crest is essential for the persistence rather than the formation of an arch artery. Dev Dyn. 1996;205:281–92. doi: 10.1002/(SICI)1097-0177(199603)205:3<281::AID-AJA8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 81.Cameron AM, Nucifora FCJ, Fung ET, Livingston DJ, Aldape RA, Ross CA, et al. FKBP12 binds the inositol 1,4,5-trisphosphate receptor at leucine–proline (1400–1401) and anchors calcineurin to this FK506-like domain. J Biol Chem. 1997;272:27582–8. doi: 10.1074/jbc.272.44.27582. [DOI] [PubMed] [Google Scholar]
- 82.Shou W, Aghdasi B, Armstrong DL, Guo Q, Bao S, Charng MJ, et al. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391:489–92. doi: 10.1038/35146. [DOI] [PubMed] [Google Scholar]
- 83.de la Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, et al. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature. 1998;392:182–6. doi: 10.1038/32419. [DOI] [PubMed] [Google Scholar]
- 84.Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, de la Brousse FC, Hoey T, et al. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 1998;392:186–90. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- 85.Bommireddy R, Ormsby I, Yin M, Boivin GP, Babcock GF, Doetschman T. Transforming growth factor beta1 inhibits Ca2+–calcineurin-mediated activation in thymocytes. J Immunol. 2003;170:3645–52. doi: 10.4049/jimmunol.170.7.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goldmuntz E, Emanuel BS. Genetic disorders of cardiac morphogenesis: the DiGeorge and velocardiofacial syndromes. Circ Res. 1997;80:437–43. doi: 10.1161/01.res.80.4.437. [DOI] [PubMed] [Google Scholar]
- 87.Farrell MJ, Stadt H, Wallis KT, Scambler P, Hixon RL, Wolfe R, et al. HIRA: a DiGeorge syndrome candidate gene, is required for cardiac outflow tract septation. Circ Res. 1999;84:127–35. doi: 10.1161/01.res.84.2.127. [DOI] [PubMed] [Google Scholar]
- 88.Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- 89.Kardami E. Stimulation and inhibition of cardiac myocyte proliferation in vitro. Mol Cell Biochem. 1990;92:129–35. doi: 10.1007/BF00218130. [DOI] [PubMed] [Google Scholar]
- 90.Villarreal FJ, Dillmann WH. Cardiac hypertrophy-induced changes in mRNA levels for TGF-beta 1, fibronectin, and collagen. Am J Physiol. 1992;262:H1861–6. doi: 10.1152/ajpheart.1992.262.6.H1861. [DOI] [PubMed] [Google Scholar]
- 91.Eghbali M, Tomek R, Sukhatme VP, Woods C, Bhambi B. Differential effects of transforming growth factor-beta 1 and phorbol myristate acetate on cardiac fibroblasts: regulation of fibrillar collagen mRNAs and expression of early transcription factors. Circ Res. 1991;69:483–90. doi: 10.1161/01.res.69.2.483. [DOI] [PubMed] [Google Scholar]
- 92.Parker TG, Packer SE, Schneider MD. Peptide growth factors can provoke “fetal” contractile protein gene expression in rat cardiac myocytes. J Clin Invest. 1990;85:507–14. doi: 10.1172/JCI114466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ruwhof C, van Wamel AE, Egas JM, van der LA. Cyclic stretch induces the release of growth promoting factors from cultured neonatal cardiomyocytes and cardiac fibroblasts. Mol Cell Biochem. 2000;208:89–98. doi: 10.1023/a:1007046105745. [DOI] [PubMed] [Google Scholar]
- 94.Gibbons GH, Pratt RE, Dzau VJ. Vascular smooth muscle cell hypertrophy vs. hyperplasia: autocrine transforming growth factor-beta 1 expression determines growth response to angiotensin II. J Clin Invest. 1992;90:456–61. doi: 10.1172/JCI115881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhu Z, Tepel M, Neusser M, Zidek W. Transforming growth factor beta 1 modulates angiotensin II-induced calcium influx in vascular smooth muscle. Eur J Clin Invest. 1995;25:317–21. doi: 10.1111/j.1365-2362.1995.tb01708.x. [DOI] [PubMed] [Google Scholar]
- 96.Xu C, Lee S, Singh TM, Sho E, Li X, Sho M, et al. Molecular mechanisms of aortic wall remodeling in response to hypertension. J Vasc Surg. 2001;33:570–8. doi: 10.1067/mva.2001.112231. [DOI] [PubMed] [Google Scholar]
- 97.Song RH, Kocharyan HK, Fortunato JE, Glagov S, Bassiouny HS. Increased flow and shear stress enhance in vivo transforming growth factor-beta1 after experimental arterial injury. Arterioscler Thromb Vasc Biol. 2000;20:923–30. doi: 10.1161/01.atv.20.4.923. [DOI] [PubMed] [Google Scholar]
- 98.Nicholls MG, Robertson JI. The renin–angiotensin system in the year 2000. J Hum Hypertens. 2000;14:649–66. doi: 10.1038/sj.jhh.1001056. [DOI] [PubMed] [Google Scholar]
- 99.Ray PE, McCune BK, Gomez RA, Horikoshi S, Kopp JB, Klotman PE. Renal vascular induction of TGF-beta 2 and renin by potassium depletion. Kidney Int. 1993;44:1006–13. doi: 10.1038/ki.1993.342. [DOI] [PubMed] [Google Scholar]
- 100.Cambien F, Ricard S, Troesch A, Mallet C, Generenaz L, Evans A, et al. Polymorphisms of the transforming growth factor-beta 1 gene in relation to myocardial infarction and blood pressure: the Etude Cas-Temoin de l’Infarctus du Myocarde (ECTIM) Study. Hypertension. 1996;28:881–7. doi: 10.1161/01.hyp.28.5.881. [DOI] [PubMed] [Google Scholar]
- 101.Suthanthiran M, Li B, Song JO, Ding R, Sharma VK, Schwartz JE, et al. Transforming growth factor-beta 1 hyperexpression in African-American hypertensives: a novel mediator of hypertension and/or target organ damage. Proc Natl Acad Sci USA. 2000;97:3479–84. doi: 10.1073/pnas.050420897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ohta K, Kim S, Hamaguchi A, Yukimura T, Miura K, Takaori K, et al. Role of angiotensin II in extracellular matrix and transforming growth factor-beta 1 expression in hypertensive rats. Eur J Pharmacol. 1994;269:115–9. doi: 10.1016/0922-4106(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 103.Schultz JJ, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, et al. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002;109:787–96. doi: 10.1172/JCI14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rosenkranz S, Flesch M, Amann K, Haeuseler C, Kilter H, Seeland U, et al. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1) Am J Physiol Heart Circ Physiol. 2002;283:H1253–62. doi: 10.1152/ajpheart.00578.2001. [DOI] [PubMed] [Google Scholar]
- 105.Grupp IL, Subramaniam A, Hewett TE, Robbins J, Grupp G. Comparison of normal, hypodynamic, and hyperdynamic mouse hearts using isolated work-performing heart preparations. Am J Physiol. 1993;265:H1401–1410. doi: 10.1152/ajpheart.1993.265.4.H1401. [DOI] [PubMed] [Google Scholar]
- 106.Long CS, Hartogensis WE, Simpson PC. Beta-adrenergic stimulation of cardiac non-myocytes augments the growth-promoting activity of non-myocyte conditioned medium. J Mol Cell Cardiol. 1993;25:915–25. doi: 10.1006/jmcc.1993.1104. [DOI] [PubMed] [Google Scholar]
- 107.Taimor G, Uter KD, Frischkopf K, Flesch M, Rosenkranz S, Piper HM. Autocrine regulation of TGF beta expression in adult cardiomyocytes. J Mol Cell Cardiol. 1999;31:2127–36. doi: 10.1006/jmcc.1999.1055. [DOI] [PubMed] [Google Scholar]
- 108.Nogami M, Romberger DJ, Rennard SI, Toews ML. TGF-beta 1 modulates beta-adrenergic receptor number and function in cultured human tracheal smooth muscle cells. Am J Physiol. 1994;266:L187–91. doi: 10.1152/ajplung.1994.266.2.L187. [DOI] [PubMed] [Google Scholar]
- 109.Miki N, Hamamori Y, Hirata K, Suematsu M, Kawashima S, Akita H, et al. Transforming growth factor-beta 1 potentiated alpha 1-adrenergic and stretch-induced c-fos mRNA expression in rat myocardial cells. Circ Res. 1994;75:8–14. doi: 10.1161/01.res.75.1.8. [DOI] [PubMed] [Google Scholar]
- 110.Li RK, Li G, Mickle DA, Weisel RD, Merante F, Luss H, et al. Overexpression of transforming growth factor-beta1 and insulin-like growth factor-I in patients with idiopathic hypertrophic cardiomyopathy. Circulation. 1997;96:874–81. doi: 10.1161/01.cir.96.3.874. [DOI] [PubMed] [Google Scholar]
- 111.Kupfahl C, Pink D, Friedrich K, Zurbrugg HR, Neuss M, Warnecke C, et al. Angiotensin II directly increases transforming growth factor beta1 and osteopontin and indirectly affects collagen mRNA expression in the human heart. Cardiovasc Res. 2000;46:463–75. doi: 10.1016/s0008-6363(00)00037-7. [DOI] [PubMed] [Google Scholar]
- 112.Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc Res. 1998;40:352–63. doi: 10.1016/s0008-6363(98)00121-7. [DOI] [PubMed] [Google Scholar]
- 113.Lee AA, Dillmann WH, McCulloch AD, Villarreal FJ. Angiotensin II stimulates the autocrine production of transforming growth factor-beta 1 in adult rat cardiac fibroblasts. J Mol Cell Cardiol. 1995;27:2347–57. doi: 10.1016/s0022-2828(95)91983-x. [DOI] [PubMed] [Google Scholar]
- 114.Sadoshima J, Izumo S. Molecular characterization of angiotensin II: induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts, critical role of the AT1 receptor subtype. Circ Res. 1993;73:413–23. doi: 10.1161/01.res.73.3.413. [DOI] [PubMed] [Google Scholar]
- 115.Patel R, Lim DS, Reddy D, Nagueh SF, Lutucuta S, Sole MJ, et al. Variants of trophic factors and expression of cardiac hypertrophy in patients with hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2000;32:2369–77. doi: 10.1006/jmcc.2000.1267. [DOI] [PubMed] [Google Scholar]
- 116.Diebold RJ, Eis MJ, Yin M, Ormsby I, Boivin GP, Darrow BJ, et al. Early-onset multifocal inflammation in the transforming growth factor beta 1-null mouse is lymphocyte mediated. Proc Natl Acad Sci USA. 1995;92:12215–9. doi: 10.1073/pnas.92.26.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tsuchimochi H, Sugi M, Kuro-o M, Ueda S, Takaku F, Furuta S, et al. Isozymic changes in myosin of human atrial myocardium induced by overload. Immunohistochemical study using monoclonal antibodies. J Clin Invest. 1984;74:662–5. doi: 10.1172/JCI111466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lompre AM, Mercadier JJ, Schwartz K. Changes in gene expression during cardiac growth. Int Rev Cytol. 1991;124:137–86. doi: 10.1016/s0074-7696(08)61526-0. [DOI] [PubMed] [Google Scholar]
- 119.Izumo S, Lompre AM, Matsuoka R, Koren G, Schwartz K, Nadal-Ginard B, et al. Myosin heavy chain messenger RNA and protein isoform transitions during cardiac hypertrophy: interaction between hemodynamic and thyroid hormone-induced signals. J Clin Invest. 1987;79:970–7. doi: 10.1172/JCI112908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Susic D, Nunez E, Frohlich ED, Prakash O. Angiotensin II increases left ventricular mass without affecting myosin isoform mRNAs. Hypertension. 1996;28:265–8. doi: 10.1161/01.hyp.28.2.265. [DOI] [PubMed] [Google Scholar]
- 121.Schultz JJ, Witt SA, Nieman ML, Reiser PJ, Engle SJ, Zhou M, et al. Fibroblast growth factor-2 mediates pressure-induced hypertrophic response. J Clin Invest. 1999;104:709–19. doi: 10.1172/JCI7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schmidt A, Gopfert C, Vlodavsky I, Volker W, Buddecke E. Induction of a hypertrophic growth status of coronary smooth muscle cells is associated with an overexpression of TGF-beta. Eur J Cell Biol. 2002;81:138–44. doi: 10.1078/0171-9335-00234. [DOI] [PubMed] [Google Scholar]
- 123.Bouillier H, Samain E, Miserey S, Perret C, Renaud JF, Safar M, et al. Transforming growth factor-beta1 modulates angiotensin II-induced calcium release in vascular smooth muscle cells from spontaneously hypertensive rats. J Hypertens. 2000;18:733–42. doi: 10.1097/00004872-200018060-00011. [DOI] [PubMed] [Google Scholar]
- 124.Ali S, Davis MG, Becker MW, Dorn GW., 2 Thromboxane A2 stimulates vascular smooth muscle hypertrophy by up-regulating the synthesis and release of endogenous basic fibroblast growth factor. J Biol Chem. 1993;268:17397–403. [PubMed] [Google Scholar]
- 125.Itoh H, Mukoyama M, Pratt RE, Gibbons GH, Dzau VJ. Multiple autocrine growth factors modulate vascular smooth muscle cell growth response to angiotensin II. J Clin Invest. 1993;91:2268–74. doi: 10.1172/JCI116454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Davis MG, Zhou M, Ali S, Coffin JD, Doetschman T, Dorn GW. Intracrine and autocrine effects of basic fibroblast growth factor in vascular smooth muscle cells. J Mol Cell Cardiol. 1997;29:1061–72. doi: 10.1006/jmcc.1997.0383. [DOI] [PubMed] [Google Scholar]
- 127.Kanzaki T, Tamura K, Takahashi K, Saito Y, Akikusa B, Oohashi H, et al. In vivo effect of TGF-beta 1: enhanced intimal thickening by administration of TGF-beta 1 in rabbit arteries injured with a balloon catheter. Arterioscler Thromb Vasc Biol. 1995;15:1951–7. doi: 10.1161/01.atv.15.11.1951. [DOI] [PubMed] [Google Scholar]
- 128.Merrilees M, Beaumont B, Scott L, Hermanutz V, Fennessy P. Effect of TGF-beta(1) antisense S-oligonucleotide on synthesis and accumulation of matrix proteoglycans in balloon catheter-injured neointima of rabbit carotid arteries. J Vasc Res. 2000;37:50–60. doi: 10.1159/000025713. [DOI] [PubMed] [Google Scholar]
- 129.Chen YL, Wu HW, Jiang MJ. Transforming growth factor-beta1 gene and protein expression associated with atherogenesis of cholesterol-fed rabbits. Histol Histopathol. 2000;15:421–8. doi: 10.14670/HH-15.421. [DOI] [PubMed] [Google Scholar]
- 130.Flaumenhaft R, Abe M, Sato Y, Miyazono K, Harpel J, Heldin CH, et al. Role of the latent TGF-beta binding protein in the activation of latent TGF-beta by co-cultures of endothelial and smooth muscle cells. J Cell Biol. 1993;120:995–1002. doi: 10.1083/jcb.120.4.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Paiement P, Bertrand O, Scortichini D, Meerkin D, Cloutier MJ, Methot D, et al. Local delivery of TGF-b antibodies to prevent neointima formation after balloon injury in a pig coronary artery model. J Invasive Cardiol. 1998;10:470–6. [PubMed] [Google Scholar]
- 132.Wahl SM, Orenstein JM, Chen W. TGF-beta influences the life and death decisions of T lymphocytes. Cytokine Growth Fact Rev. 2000;11:71–9. doi: 10.1016/s1359-6101(99)00030-1. [DOI] [PubMed] [Google Scholar]
- 133.Koglin J, Glysing-Jensen T, Raisanen-Sokolowski A, Russell ME. Immune sources of transforming growth factor-beta1 reduce transplant arteriosclerosis: insight derived from a knockout mouse model. Circ Res. 1998;83:652–60. doi: 10.1161/01.res.83.6.652. [DOI] [PubMed] [Google Scholar]
- 134.Kurtz A, Wagner C. Cellular control of renin secretion. J Exp Biol. 1999;202:219–25. doi: 10.1242/jeb.202.3.219. [DOI] [PubMed] [Google Scholar]
- 135.Allikmets K, Parik T, Viigimaa M. The renin–angiotensin system in essential hypertension: associations with cardiovascular risk. Blood Press. 1999;8:70–8. doi: 10.1080/080370599438239. [DOI] [PubMed] [Google Scholar]
- 136.Farmer JA. Renin–angiotensin system and ASCVD. Curr Opin Cardiol. 2000;15:141–50. doi: 10.1097/00001573-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 137.Rabelink TJ, Bakris GL. The renin–angiotensin system in diabetic nephropathy: the endothelial connection. Miner Electrolyte Metab. 1998;24:381–8. doi: 10.1159/000057399. [DOI] [PubMed] [Google Scholar]
- 138.Horikoshi S, McCune BK, Ray PE, Kopp JB, Sporn MB, Klotman PE. Water deprivation stimulates transforming growth factor-beta 2 accumulation in the juxtaglomerular apparatus of mouse kidney. J Clin Invest. 1991;88:2117–22. doi: 10.1172/JCI115541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Antonipillai I, Horton R. Paracrine regulation of the renin–aldosterone system. J Steroid Biochem Mol Biol. 1993;45:27–31. doi: 10.1016/0960-0760(93)90118-g. [DOI] [PubMed] [Google Scholar]
- 140.Pietri L, Bloch-Faure M, Belair MF, Sanford LP, Doetschman T, Menard J, et al. Altered renin synthesis and secretion in the kidneys of heterozygous mice with a null mutation in the TGF-beta(2) gene. Exp Nephrol. 2002;10:374–82. doi: 10.1159/000065302. [DOI] [PubMed] [Google Scholar]
- 141.Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Nat Acad Sci USA. 1986;83:4167–71. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331:1286–92. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 143.Boivin GP, O’Toole BA, Orsmby IE, Diebold RJ, Eis MJ, Doetschman T, et al. Onset and progression of pathological lesions in transforming growth factor-beta 1-deficient mice. Am J Pathol. 1995;146:276–88. [PMC free article] [PubMed] [Google Scholar]
- 144.Nakajima H, Nakajima HO, Salcher O, Dittie AS, Dembowsky K, Jing S, et al. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ Res. 2000;86:571–9. doi: 10.1161/01.res.86.5.571. [DOI] [PubMed] [Google Scholar]
- 145.Omura T, Kim S, Takeuchi K, Iwao H, Takeda T. Transforming growth factor beta 1 and extracellular matrix gene expression in isoprenaline induced cardiac hypertrophy: effects of inhibition of the renin–angiotensin system. Cardiovasc Res. 1994;28:1835–42. doi: 10.1093/cvr/28.12.1835. [DOI] [PubMed] [Google Scholar]
- 146.Brooks WW, Conrad CH. Myocardial fibrosis in transforming growth factor beta(1) heterozygous mice. J Mol Cell Cardiol. 2000;32:187–95. doi: 10.1006/jmcc.1999.1065. [DOI] [PubMed] [Google Scholar]
- 147.Wang B, Hao J, Jones SC, Yee MS, Roth JC, Dixon IM. Decreased Smad7 expression contributes to cardiac fibrosis in the infarcted rat heart. Am J Physiol Heart Circ Physiol. 2002;282:H1685–96. doi: 10.1152/ajpheart.00266.2001. [DOI] [PubMed] [Google Scholar]
- 148.Border WA, Noble NA. Interactions of transforming growth factor-beta and angiotensin II in renal fibrosis. Hypertension. 1998;31:181–8. doi: 10.1161/01.hyp.31.1.181. [DOI] [PubMed] [Google Scholar]
- 149.Ledbetter S, Kurtzberg L, Doyle S, Pratt BM. Renal fibrosis in mice treated with human recombinant transforming growth factor-beta2. Kidney Int. 2000;58:2367–76. doi: 10.1046/j.1523-1755.2000.00420.x. [DOI] [PubMed] [Google Scholar]
- 150.Hill C, Flyvbjerg A, Rasch R, Bak M, Logan A. Transforming growth factor-beta2 antibody attenuates fibrosis in the experimental diabetic rat kidney. J Endocrinol. 2001;170:647–51. doi: 10.1677/joe.0.1700647. [DOI] [PubMed] [Google Scholar]