

Abstract
The mechanism for the biomimetic synthesis of flavonolignan diastereoisomers in milk thistle is proposed to proceed by single-electron oxidation of coniferyl alcohol, subsequent reaction with one of the oxygen atoms of taxifolin's catechol moiety, and finally, further oxidation to form four of the major components of silymarin: silybin A, silybin B, isosilybin A, and isosilybin B. This mechanism is significantly different from a previously proposed process that involves the coupling of two independently formed radicals.
Introduction
Milk thistle [Silybum marianum (L.) Gaertn. (Asteraceae)] has been used as a medicinal herb since antiquity. As outlined in several reviews, modern pharmacological studies typically focus on either the hepatoprotective properties,1-3 as it is the top herbal supplement for hepatitis C patients,2 and/or the prostate cancer chemopreventive properties,4-6 where promising results have been observed, especially for isosilybin B.7-10 The two most studied formulations are either silymarin, an extract of the seeds that contains at least seven major flavonolignans, or silibinin, a roughly equimolar mixture of silybin A and silybin B (Figure 1); a recent review delineates the somewhat confusing nomenclature surrounding the various permutations of milk thistle.11
Figure 1.

Flavonolignans from milk thistle; silibinin is a 1:1 mixture of 1 and 2, while isosilibinin is a 1:1 mixture of 3 and 4.11
The chemistry of milk thistle extract has been investigated since the 1960s, and impressive strides were made in the isolation and structure determination of the individual flavonolignans throughout the 60s, 70s and 80s, particularly by the competing groups of Hänsel and colleagues12-19 and Wagner and colleagues.20-25 However, likely due to improvements in chromatographic technology, the individual diastereoisomers were not isolated and characterized completely until 2003.26,27 Subsequently, gram-scale purifications of the individual flavonolignans were developed.28-30 Those materials likely facilitated several chemistry-driven investigations, including the generation of analogues,31-34 an X-ray crystallographic study to verify the structures of the four main isomers,35 and the development of tools to discern and quantify them by 1H NMR spectroscopy, despite near-identical spectra.36
Structurally, flavonolignans are characterized by the amalgamation of a flavonoid moiety (taxifolin) and a phenylpropane unit (coniferyl alcohol, Scheme 1).1,37,38 Within silibinin (silybin A and silybin B) and isosilibinin (isosilybin A and isosilybin B), each exists as a pair of trans-diasteroisomers with respect to the relative configuration at positions C-7″ and C-8″ in the 1,4-benzodioxane ring.26,27,35,36 Silychristin (5) and isosilychristin (6) each have coumaran ring systems, or dihydrobenzofuran bicycles, but the position of this bicycle differs by being formed at either the C-4′ and C-5′ positions in silychristin or the C-2′ and C-3′ positions in isosilychristin. Silydianin (7) is the most structurally complex of the flavonolignans in silymarin, since it contains a bicyclo[2.2.2]octenone with a transannular hemiketal.19,22,24
Scheme 1. Mechanistic Options for the Biomimetic Synthesis of Flavonolignans.

The first biomimetic synthesis of milk thistle flavonolignans was reported in 1977 by Schrall and Becker,39 who used horseradish peroxidase and a cell free extract of S. marianum suspension cultures to produce silibinin from taxifolin (8) and coniferyl alcohol (9, Scheme 1). Two years later, Merlini et al.17 reported an enzyme-free oxidative coupling of taxifolin and coniferyl alcohol using Ag2O to yield a mixture of silibinin and isosilibinin. Based on those results, multiple researchers have reported biomimetic syntheses of flavonolignans and related analogues, typically using a silver oxidant.18,31,40-42 The mechanism presented for these is based on the synthesis of lignans from coniferyl alcohol,43 termed Freudenberg's hypothesis, which itself has been the topic of controversy in recent years.44,45
The mechanism that has been proposed for the biomimetic synthesis of flavonolignans 1-4 involves single electron oxidation of both coniferyl alcohol and taxifolin individually, followed by combination of these two radicals to produce silibinin and isosilibinin (Scheme 1, Option 1). While there is a possibility that this type of pathway could occur within or near the active site of an enzyme,46,47 the probability of two radicals being formed independently in solution from Ag2O and then combining in a productive manner18 is unlikely based on first principles of reaction kinetics. Since the concentration of both radicals will be extremely low, the reaction rate will be essentially zero. Given the absence of an alternative mechanism for the biomimetic synthesis of flavonolignans in the current literature, we pursued a more thorough exploration of this process.
Results and Discussion
The conversion of coniferyl alcohol and taxifolin to silibinin and isosilibinin necessarily requires an oxidation. This oxidation is presumably enzyme-catalyzed in nature,46,47 but it has been shown that silver salts can also efficiently effect this conversion.18,31,40-42 Since silver oxidations typically occur through a series of single electron transfers, various mechanistic options were explored and resolved (Scheme 1). Specifically, the reaction involves three steps: two single electron oxidations and the coupling of taxifolin to coniferyl alcohol (or an oxidized variant of either partner). The five options discussed below cover logical combinations of these three processes, although it is noted that alternative mechanisms could be taking place, and as such, a definitive mechanism cannot be absolutely determined.
As mentioned earlier, the mechanism proposed in the literature involves simultaneous single-electron oxidation of both coniferyl alcohol and taxifolin, followed by combination of the resultant radicals to form ether 14, which undergoes rapid addition of the phenol to the electrophilic p-quinone methide to yield silibinin (Option 1; Scheme 1). The same mechanism is possible using the other phenoxy radical of taxifolin to produce isosilibinin (omitted from Scheme 1 for clarity). Although this mechanism has been proposed based on Freudenberg's hypothesis for lignin biosynthesis,43 it was difficult to support due to the low concentration of each radical. Moreover, taxifolin and coniferyl alcohol would need to oxidize at nearly identical rates, otherwise dimerization would be the major pathway.
A second mechanistic option proceeds via two sequential oxidations of taxifolin to yield an o-quinone (12). This pathway seemed more plausible, given precedence for o-quinones to react with alkenes in the Diels-Alder reaction.48 Additionally, the stereospecific nature of the Diels-Alder reaction would conserve the relative configuration of the dienophile, i.e. the trans-configuration of the alkene in coniferyl alcohol would deliver the trans-configuration at C7″ and C8″, as observed in both silibinin and isosilibinin.35
Options 3 and 4 are similar to one another, since they both begin with initial oxidation of either taxifolin (Option 3) or coniferyl alcohol (Option 4), followed by coupling to either coniferyl alcohol or taxifolin, respectively. The resultant radicals (13 or 15) would be further oxidized to yield silibinin. Unlike Option 1, Options 3 and 4 both seemed plausible, since coniferyl alcohol and taxifolin are both electron-rich and, therefore, susceptible to oxidation or reaction with an electrophile.
The final option considered had two sequential oxidations of coniferyl alcohol and subsequent coupling to taxifolin (Option 5). It has been shown that coniferyl alcohol undergoes oxidation to yield coniferyl aldehyde (16),43 however, an unlikely redox reaction of this aldehyde and taxifolin would be required to yield silibinin. For the sake of this study, all five of these Options were considered while interrogating the mechanism. However, Option 1 seemed unlikely because it required a sufficient concentration of both reactive radical intermediates (10 and 11), while in contrast Options 2-5 were only dependent on the concentration of either of the singly-oxidized radicals.
The biomimetic synthesis was performed with natural taxifolin, isolated from milk thistle extract in >90% purity (data not shown), and commercially available coniferyl alcohol. The reactions were explored initially on a small scale (∼1 mg taxifolin) and monitored by HPLC; they were later increased to the >100 mg scale. The reaction conditions were based upon the procedure described by Merlini et al.,17 with moderate optimizations. Several solvents, oxidizing agents, and temperatures were tested, and the best results were obtained when one equiv of taxifolin was reacted with two equiv of coniferyl alcohol in ethyl acetate containing four equiv of Ag2O at 75 °C for 96 h (Scheme 2). These conditions afforded a mixture of silybin A (1), silybin B (2), isosilybin A (3), and isosilybin B (4; Scheme 2) in a combined 52% yield with nearly equimolar amounts of each flavonolignan (Figure 2 and Supporting Information).
Scheme 2. Biomimetic Synthesis of Silibinin, Isosilibinin, and Other Byproducts.
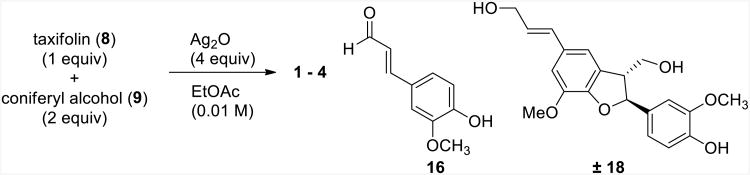
Figure 2.

UPLC chromatogram of biomimetic reaction using conditions in Scheme 2. UPLC was conducted using a CH3OH/H2O (0.1% formic acid) gradient that initiated at 5:95 and increased to 50:50 over 10 min and held at that ratio for 2 min at a flow rate of 0.6 mL/min (50°C) using an HSST3 column monitored at 288 nm.
In addition to flavonolignans 1-4, coniferyl aldehyde (16) and lignin 18 were produced in moderate amounts in the biomimetic reaction (Figure 2). Trace amounts of flavonolignans 5-7 also formed during some of the reactions, as determined by UPLC-MS, but the quantities were never sufficient to confirm by isolation and NMR analysis. It has been determined previously that silymarin consists of silybin A (16.0%), silybin B (23.8%), isosilybin A (6.4%), isosilybin B (4.4%), silychristin (11.6%), isosilychristin (2.2%), silydianin (16.7%), and taxifolin (1.6%; see Supporting Information).10 The biomimetic conditions described herein increased the yield of isosilybin B relative to other flavonolignans, which was noteworthy given studies that demonstrate its potential in prostate cancer chemoprevention7-10 and the challenges it presents when isolating it on a multigram scale.30 Flavonolignans 1-4 were isolated by HPLC in greater than 99% purity, and their structures were confirmed by NMR spectroscopy (see Supporting Information).
To explore the mechanistic possibilities involved in this biomimetic process, the oxidative coupling of cis-coniferyl alcohol (19) and taxifolin using Ag2O was examined (Scheme 3). If Option 2 (Scheme 1) occurred, the product would maintain a cis-relationship at C7″ and C8″, since the Diels-Alder reaction of an o-quinone has been shown to be stereospecific with respect to the relative configuration of the dienophile.48 With all of the other options, the stereochemical information of coniferyl alcohol would be lost when either radical 11 or 13 was formed, and a trans-relationship at C7″ and C8″ would be produced as the major product since this is thermodynamically more favorable. By running the oxidative coupling of cis-coniferyl alcohol with taxifolin it was determined that the identical products (1-4) as trans-coniferyl alcohol were generated and that the cis-related products (19-22) were not observed. Although it was determined that cis-coniferyl alcohol isomerized to trans-coniferyl alcohol under the reaction conditions, it occurred much slower than the rate of formation of silibinin and isosilibinin. Unless either the Diels-Alder reaction is not concerted, which is inconsistent with prior results,48 or the cis-related products (21-24) rapidly isomerize to the trans-related products (1-4), Option 2 is not viable. Based on the results of the reaction of cis-coniferyl alcohol (Scheme 3), Option 2 was considered unlikely, but as it could not be definitively excluded, additional reactions were performed.
Scheme 3. Mechanistic Investigation for the o-Quinone Diels-Alder Option.

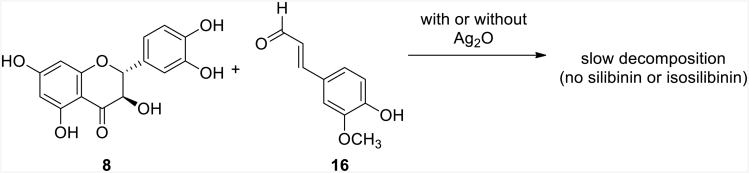
The next reactions that were examined were the oxidation of either coniferyl alcohol or taxifolin individually with Ag2O in the absence of the other compound (Scheme 4). Interestingly, coniferyl alcohol was rapidly oxidized by Ag2O, whereas taxifolin was practically inert to those conditions (Scheme 4 and Supporting Information). The lack of reactivity towards oxidation of taxifolin implies that Options 1-3 in Scheme 1 were all not viable mechanisms and simplifies the possibilities to only Options 4 or 5. Importantly, further scrutiny of the oxidation of coniferyl alcohol in the absence of taxifolin revealed the production of two major products, coniferyl aldehyde and lignan 18. This verified that two oxidations of coniferyl alcohol yielded coniferyl aldehyde, as expected, and that the initial radical from single-electron oxidation was prone to reaction with an electron-rich phenol of a different molecule of coniferyl alcohol. Hypothetically, if taxifolin was in solution with the oxidized radical of coniferyl alcohol, the nucleophilic catechol moiety could similarly react to give silibinin and isosilibinin (Option 4).
Scheme 4. Individual Oxidations of Taxifolin and Coniferyl Alcohol.

As mentioned earlier, it was not anticipated that coniferyl aldehyde (16) would react with taxifolin to undergo a redox reaction and yield silibinin and isosilibinin. To test this, the reaction of coniferyl aldehyde with taxifolin was examined in both the presence and absence of Ag2O (Scheme 5). As anticipated, silibinin and isosilibinin were not observed with this reaction. Thus, the only remaining viable mechanism of those considered was Option 4, where coniferyl alcohol was oxidized, this radical (11) reacted with taxifolin, and finally, the compound oxidized further to yield silibinin and isosilibinin (Scheme 6). This pathway accounts for the formation of flavonolignans 1-4, coniferyl aldehyde (16), and lignin 18. Although not specifically tested, it is plausible that the silver salts are involved in the process, beyond acting as the single-electron oxidants, and coordinate the phenols to position and stabilize the various charges and radicals.
Scheme 5. Attempted Coupling of Coniferyl Aldehyde to Taxifolin.
Scheme 6. Mechanistically Supported Biomimetic Synthesis of Flavonolignans.

Conclusions
In summary, a biomimetic synthesis of four major flavonolignans present in silymarin is reported, and the analyses of related reactions were used to support or refute possible mechanisms. From this analysis it is proposed that the mechanism for the biomimetic synthesis of flavonolignans proceeds by single electron oxidation of coniferyl alcohol, addition of taxifolin, and finally, oxidation to yield silibinin and isosilibinin. This is contrary to the mechanism proposed previously for this process,18 which involved the coupling of two independently formed radicals. While the study presented herein has exclusively examined oxidative couplings using Ag2O instead of enzymes to form flavonolignans, it is proposed that similar reactivity should be considered for the biosynthesis of related compounds such as lignans.
Experimental Section
General information
All reactions were carried out under a N2 atmosphere with anhydrous conditions. All reagents and solvents were purchased and used without further purification. NMR experiments were conducted using a spectrometer operating at 500 MHz for 1H and 125 MHz for 13C. Accurate mass measurements were acquired using an Orbitrap mass analyzer and an electrospray ionization (ESI) source for compounds 1-4 in negative ionization mode via a liquid chromatographic/autosampler system that consisted of a UPLC system. Accurate mass measurements of 4-(3-hydroxyprop-1-yn-1-yl)-2-methoxyphenol were accomplished using an atmospheric pressure chemical ionization (APCI) source under direct infusion flow conditions in positive mode ionization. HPLC and UPLC were analyzed using a photodiode array (PDA) detector. For preparative HPLC, a YMC ODS-A (5 μm, 250 × 20 mm) column was used at a 7 mL/min flow rate and a pentafluorophenyl propyl (PFP; 5 μm, 250 × 21 mm) column was used at a 21.2 mL/min flow rate. For analytical HPLC, a YMC ODS-A (5 μm, 250 × 4.6 mm) column and a PFP (5 μm, 150 × 4.6 mm) column were used, both a 1 mL/min flow rate. For UPLC, an HSST3 (1.8 μm, 2.1 × 100 mm) column was used at 50 °C at a 0.6 mL/min flow rate and monitored at 288 nm.
Procedure of Biomimetic Synthesis
Taxifolin was isolated in >90% purity from milk thistle extract (silymarin) via two successive reverse phase HPLC methods. The first method utilized a gradient of 15:85 to 50:50 MeOH/H2O over 60 min using the YMC ODS-A (5 μm, 250 × 20 mm) column and detected at 288 nm. The second method utilized a gradient of 5:90 to 70:30 CH3CN/H2O (0.1% formic acid) over 30 min using a PFP (5 μm, 250 × 21 mm) column.
To a 100 mL round-bottom flask with a stirred solution of taxifolin (106 mg, 0.348 mmol) and trans-coniferyl alcohol (125 mg, 0.696 mmol) in ethyl acetate (30 mL, 0.01 M) under a nitrogen atmosphere at ambient temperature was added Ag2O (323 mg, 1.39 mmol). The flask was covered with foil and equipped with a reflux condenser. The solution was stirred and heated to 75 °C for 96 h under an atmosphere of nitrogen gas. The reaction mixture was cooled to rt, filtered through Celite, and washed with ethyl acetate. A yellow filtrate was concentrated under reduced pressure, dissolved in ethyl acetate (2 mL), and centrifuged through a polypropylene Eppendorf tube filter (0.22 μm) to remove any residual silver salts. The crude product (225 mg) was purified by reverse-phase HPLC as described below to afford flavonolignans with a total yield of 82.6 mg, 52% (21.4 mg, 21.1 mg, 20.7 mg, and 19.5 mg for 1, 2, 3, and 4, respectively). In addition to the four major compounds, coniferyl aldehyde (16, 2.3 mg) and lignan 18 (13.6 mg) were isolated. In addition to having 1H and 13C NMR spectra that were identical to prior reports,36 co-injection of coniferyl aldehyde or the individual natural flavonolignans by UPLC was used to confirm their identity (see Supporting Information). 1H and 13C NMR data were used to confirm the structure of known lignin 18.49
To purify the reaction mixtures, two different reverse-phase columns were utilized, ODS-A (5 μm, 250 × 20 mm) and PFP (5 μm, 250 × 21 mm). The reaction mixture was first purified using a gradient of 20:80 to 50:50 CH3OH/H2O over 90 min and then held for 20 min. Partially purified fractions were chromatographed using a similar procedure. Then, for the final purification, the PFP column was used with a gradient of 20:80 to 40:60 CH3CN/H2O (0.1% formic acid) over 30 min. Each synthetic flavonolignan was purified until >99% pure, as measured by analytical UPLC (see Supporting Information).
4-(3-Hydroxyprop-1-yn-1-yl)-2-methoxyphenol
To a stirred solution of 4-bromo-2-methoxyphenol (1.00 g, 4.93 mmol), CuI (282 mg, 0.148 mmol, 3 mol %), and bis(triphenylphosphine)palladium(II) dichloride (104 mg, 0.148 mmol, 3 mol %) in triethylamine (10 mL) under a nitrogen atmosphere at ambient temperature was added propargyl alcohol (440 mg, 7.9 mmol). The reaction was heated to 95 °C for 4 h, cooled to rt, filtered through Celite, and concentrated under reduced pressure. The crude extract was purified by silica gel column chromatography (85:15, hexanes:ethyl acetate) to yield 80 mg of 4-(3-hydroxyprop-1-yn-1-yl)-2-methoxyphenol (9% yield) as a white solid. 1H NMR (500 MHz, CDCl3) δ = 3.89 (s, 3H), 4.48 (s, 2H), 5.74 (s, 1H), 6.86 (d, J = 8.1 Hz, 1H), 6.95 (d, J = 1.5 Hz, 1H), 7.0 ppm (dd, J = 8.1, 1.7 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ = 51.9, 56.1, 85.5, 86.1, 114.1, 114.2, 114.7, 125.8, 146.3, 146.6. HRMS (APCI) m/z 179.0698 [M+H]+ (calcd for C10H11O3 179.0703).
(Z)-4-(3-Hydroxyprop-1-en-1-yl)-2-methoxyphenol or cis-coniferyl alcohol (19)
To a stirred solution of 4-(3-hydroxyprop-1-yn-1-yl)-2-methoxyphenol (72 mg, 0.40 mmol) and Lindlar's catalyst (0.016 g, 37 mol %) in 10 mL of methanol was added an atmosphere of hydrogen gas at ambient temperature. The reaction mixture was stirred for 1 h, filtered through Celite, concentrated under reduced pressure, and purified by silica gel column chromatography (80:20 hexanes/ethyl acetate) to yield 37 mg of cis-coniferyl alcohol (51% yield) as a white solid. The 1H NMR spectrum was consistent with previously reported data (Supporting Information).50
Supplementary Material
Acknowledgments
This research was supported in part by the National Institutes of Health/National Center for Complementary and Alternative Medicine via grant R01 AT006842. H.S.A was supported by the King Abdullah Scholarship Program from the Ministry of Higher Education of Saudi Arabia. The authors thank Mr. T.N. Graf (UNCG) for helpful discussions regarding milk thistle and Dr. I.F.D. Hyatt (UNCG) for assistance with this mechanistic study. The authors thank Dr. Brandie M. Ehrmann (UNCG) for assistance with the high resolution mass spectrometry data at the Triad Mass Spectrometry Laboratory at the University of North Carolina at Greensboro.
Footnotes
Supporting Information: UPLC chromatograms, 1H and 13C NMR spectra, and tabulated comparisons between isolated and synthesized compounds. This material is available free of charge via the Internet at http://pubs.acs.org
Notes: The authors declare no competing financial interest
References
- 1.Polyak SJ, Oberlies NH, Pecheur EI, Dahari H, Ferenci P, Pawlotsky JM. Antivir Ther. 2013;18:141. doi: 10.3851/IMP2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polyak SJ, Ferenci P, Pawlotsky JM. Hepatology. 2013;57:1262. doi: 10.1002/hep.26179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abenavoli L, Capasso R, Milic N, Capasso F. Phytother Res. 2010;24:1423. doi: 10.1002/ptr.3207. [DOI] [PubMed] [Google Scholar]
- 4.Deep G, Agarwal R. Cancer Metastasis Rev. 2010;29:447. doi: 10.1007/s10555-010-9237-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gazak R, Walterova D, Kren V. Curr Med Chem. 2007;14:315. doi: 10.2174/092986707779941159. [DOI] [PubMed] [Google Scholar]
- 6.Ramasamy K, Agarwal R. Cancer Lett. 2008;269:352. doi: 10.1016/j.canlet.2008.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deep G, Oberlies NH, Kroll DJ, Agarwal R. Carcinogenesis. 2007;28:1533. doi: 10.1093/carcin/bgm069. [DOI] [PubMed] [Google Scholar]
- 8.Deep G, Oberlies NH, Kroll DJ, Agarwal R. Oncogene. 2008;27:3986. doi: 10.1038/onc.2008.45. [DOI] [PubMed] [Google Scholar]
- 9.Deep G, Oberlies NH, Kroll DJ, Agarwal R. Int J Cancer. 2008;123:41. doi: 10.1002/ijc.23485. [DOI] [PubMed] [Google Scholar]
- 10.Davis-Searles PR, Nakanishi Y, Kim NC, Graf TN, Oberlies NH, Wani MC, Wall ME, Agarwal R, Kroll DJ. Cancer Res. 2005;65:4448. doi: 10.1158/0008-5472.CAN-04-4662. [DOI] [PubMed] [Google Scholar]
- 11.Kroll DJ, Shaw HS, Oberlies NH. Integr Cancer Ther. 2007;6:110. doi: 10.1177/1534735407301825. [DOI] [PubMed] [Google Scholar]
- 12.Janiak B, Hänsel R. Planta Med. 1960;8:71. [Google Scholar]
- 13.Pelter A, Hänsel R. Tetrahedron Lett. 1968:2911. [Google Scholar]
- 14.Hänsel R, Schulz J, Pelter A, Rimpler H, Rizk AF. Tetrahedron Lett. 1969:4417. [Google Scholar]
- 15.Hänsel R, Schulz J, Pelter A. J Chem Soc Chem Commun. 1972:195. [Google Scholar]
- 16.Pelter A, Hänsel R. Chem Ber. 1975;108:790. [Google Scholar]
- 17.Merlini L, Zanarotti A, Pelter A, Rochefort MP, Hänsel R. J Chem Soc Chem Commun. 1979:695. [Google Scholar]
- 18.Merlini L, Zanarotti A, Pelter A, Rochefort MP, Hänsel R. J Chem Soc Perkin Trans 1. 1980:775. [Google Scholar]
- 19.Hänsel R, Kaloga M, Pelter A. Tetrahedron Lett. 1976;2241 [Google Scholar]
- 20.Wagner H, Hörhammer L, Münster R. Naturwissenschaften. 1965;52:305. [Google Scholar]
- 21.Wagner H, Hörhammer L, Munster R. Arzneim Forsch. 1968;18:688. [PubMed] [Google Scholar]
- 22.Wagner H, Seligmann O, Hörhammer L, Seitz M, Sonnenbichler J. Tetrahedron Lett. 1971;22:1895. [Google Scholar]
- 23.Wagner H, Diesel P, Seitz M. Arzneim Forsch. 1974;24:466. [PubMed] [Google Scholar]
- 24.Wagner H, Seligmann O, Seitz M, Abraham D, Sonnenbichler J. Z Naturforsch. 1976;31:876. [Google Scholar]
- 25.Lotter H, Wagner H. Z Naturforsch C. 1983;38:339. [Google Scholar]
- 26.Kim NC, Graf TN, Sparacino CM, Wani MC, Wall ME. Org Biomol Chem. 2003;1:1684. doi: 10.1039/b300099k. [DOI] [PubMed] [Google Scholar]
- 27.Lee DYW, Liu YZ. J Nat Prod. 2003;66:1171. doi: 10.1021/np030163b. [DOI] [PubMed] [Google Scholar]
- 28.Kren V, Gazak R, Purchartova K, Marhol P, Biedermann D, Sedmera P. J Mol Catal B Enzym. 2009;61:247. [Google Scholar]
- 29.Monti D, Gazak R, Marhol P, Biedermann D, Purchartova K, Fedrigo M, Riva S, Kren V. J Nat Prod. 2010;73:613. doi: 10.1021/np900758d. [DOI] [PubMed] [Google Scholar]
- 30.Graf TN, Wani MC, Agarwal R, Kroll DJ, Oberlies NH. Planta Med. 2007;73:1495. doi: 10.1055/s-2007-990239. [DOI] [PubMed] [Google Scholar]
- 31.Zhao HP, Brandt GE, Galam L, Matts RL, Blagg BSJ. Bioorg Med Chem Lett. 2011;21:2659. doi: 10.1016/j.bmcl.2010.12.088. [DOI] [PubMed] [Google Scholar]
- 32.Gazak R, Purchartova K, Marhol P, Zivna L, Sedmera P, Valentova K, Kato N, Matsumura H, Kaihatsu K, Kren V. Eur J Med Chem. 2010;45:1059. doi: 10.1016/j.ejmech.2009.11.056. [DOI] [PubMed] [Google Scholar]
- 33.Gazak R, Valentova K, Fuksova K, Marhol P, Kuzma M, Medina MA, Oborna I, Ulrichova J, Kren V. J Med Chem. 2011;54:7397. doi: 10.1021/jm201034h. [DOI] [PubMed] [Google Scholar]
- 34.Sy-Cordero AA, Graf TN, Runyon SP, Wani MC, Kroll DJ, Agarwal R, Brantley SJ, Paine MF, Polyak SJ, Oberlies NH. Bioorg Med Chem. 2013;21:742. doi: 10.1016/j.bmc.2012.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sy-Cordero AA, Day CS, Oberlies NH. J Nat Prod. 2012;75:1879. doi: 10.1021/np3005369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Napolitano JG, Lankin DC, Graf TN, Friesen JB, Chen SN, McAlpine JB, Oberlies NH, Pauli GF. J Org Chem. 2013;78:2827. doi: 10.1021/jo302720h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kurkin VA. Chemistry of Natural Compounds. 2003;39:123. [Google Scholar]
- 38.Elwekeel A, Elfishway A, AbouZid S. Phytochemistry Lett. 2012;5:393. [Google Scholar]
- 39.Schrall R, Becker H. Planta Med. 1977;32:27. doi: 10.1055/s-0028-1097604. [DOI] [PubMed] [Google Scholar]
- 40.Guz NR, Stermitz FR. J Nat Prod. 2000;63:1140. doi: 10.1021/np000166d. [DOI] [PubMed] [Google Scholar]
- 41.Guz NR, Stermitz FR, Johnson JB, Beeson TD, Willen S, Hsiang J, Lewis K. J Med Chem. 2001;44:261. doi: 10.1021/jm0004190. [DOI] [PubMed] [Google Scholar]
- 42.Begum SA, Sahai M, Ray AB. Progress in the Chemistry of Organic Natural Products. Vol. 93. Springer-Verlag Wien; Vienna: 2010. p. 1. [DOI] [PubMed] [Google Scholar]
- 43.Freudenberg K. In: Constitution and Biosynthesis of Lignin. Freudenberg K, Neish AC, editors. Springer-Verlag; Berlin-Heidelberg: 1968. p. 47. [Google Scholar]
- 44.Davin LB, Lewis NG. Curr Opin Biotechnol. 2005;16:407. doi: 10.1016/j.copbio.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 45.Ralph J, Brunow G, Harris PJ, Dixon RA, Schatz PF, Boerjan W. In: Recent Advances in Polyphenol Research. Daayf F, Lattanzio V, editors. Blackwell Publishing, Ltd; West Sussex: 2008. p. 36. [Google Scholar]
- 46.Nyiredy S, Samu Z, Szucs Z, Gulacsi K, Kurtan T, Antus S. J Chromatogr Sci. 2008;46:93. doi: 10.1093/chromsci/46.2.93. [DOI] [PubMed] [Google Scholar]
- 47.Samu Z, Nyiredy S, Baitz-Gacs E, Varga Z, Kurtan T, Dinya Z, Antus S. Chem Biodivers. 2004;1:1668. doi: 10.1002/cbdv.200490125. [DOI] [PubMed] [Google Scholar]
- 48.Nair V, Menon RS, Biju AT, Abhilash KG. Chem Soc Rev. 2012;41:1050. doi: 10.1039/c1cs15186j. [DOI] [PubMed] [Google Scholar]
- 49.Quideau S, Ralph J. Holzforschung. 1994;48:12. [Google Scholar]
- 50.Ralph J, Zhang YS. Tetrahedron. 1998;54:1349. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.