

Summary
The pyrimidine biosynthesis pathway in the protozoan pathogen Toxoplasma gondii is essential for parasite growth during infection. To investigate the properties of dihydroorotate dehydrogenase (TgDHOD), the fourth enzyme in the T. gondii pyrimidine pathway, we expressed and purified recombinant TgDHOD. TgDHOD exhibited a specific activity of 84 U/mg, a kcat of 89 sec−1, a Km = 60 μM for L-dihydroorotate, and a Km = 29 μM for decylubiquinone (QD). Quinones lacking or having short isoprenoid side chains yielded lower kcats than QD. As expected, fumarate was a poor electron acceptor for this family 2 DHOD. The The IC50s determined for A77-1726, the active derivative of the human DHOD inhibitor leflunomide, and related compounds MD249 and MD209 were, 91 μM, 96 μM, and 60 μM, respectively. The enzyme was not significantly affected by brequinar or TTFA, known inhibitors of human DHOD, or by atovaquone. DSM190, a known inhibitor of Plasmodium falciparum DHOD, was a poor inhibitor of TgDHOD. TgDHOD exhibits a lengthy 157-residue N-terminal extension, consistent with a potential organellar targeting signal. We constructed C-terminally c-myc tagged TgDHODs to examine subcellular localization of TgDHOD in transgenic parasites expressing the tagged protein. Using both exogenous and endogenous expression strategies, anti-myc fluorescence signal colocalized with antibodies against the mitochondrial marker ATPase. These findings demonstrate that TgDHOD is associated with the parasite’s mitochondrion, revealing this organelle as the site of orotate production in T gondii. The TgDHOD gene appears to be essential because while gene tagging was successful at the TgDHOD gene locus, attempts to delete the TgDHOD gene were not successful in the KU80 background. Collectively, our study suggests that TgDHOD is an excellent target for the development of anti-Toxoplasma drugs.
Keywords: Dihydroorotate dehydrogenase, pyrimidine biosynthesis, Toxoplasma gondii, mitochondria, oxidative phosphorylation
1. Introduction
Toxoplasma gondii is an obligate intracellular parasite from the phylum Apicomplexa with worldwide distribution. Infections are usually asymptomatic; however, life-threatening illness occurs in immunocompromised patients and in the fetus [1]. Current prophylactic treatments with sulfadiazine and pyrimethamine are effective, but cannot be used for pregnant women [2], and cause severe side effects in some HIV/AIDS patients [3, 4, 5]. Thus, there is a need to identify new targets for design of less toxic and more effective drugs.
Recent work has shown that enzymes of the de novo pyrimidine biosynthetic pathway in T. gondii are potential drug targets [6, 7, 8]. Products of the pathway are required for the synthesis of DNA, RNA, and other metabolically important molecules. T. gondii mutants lacking the first enzyme or mutants lacking the last enzyme in the de novo pyrimidine biosynthetic pathway are avirulent in mice, and are unable to replicate in cell culture in the absence of added uracil [6, 7]. An alternate route for obtaining pyrimidines is to recycle host or parasite pyrimidines via salvage pathways. In T. gondii, salvage enzymes do not appear to contribute significantly to the pyrimidine pools; the growth of parasite mutants lacking the principle salvage enzyme, uracil phosphoribosyltransferase (UPRT), is similar to that of wild type parasites in cell culture and in mice [9, 10].
The fourth enzyme of the de novo pathway, dihydroorotate dehydrogenase (DHOD, E.C. 1.3.5.2), catalyzing dihydroorotate oxidation to orotate, appears to be a promising therapeutic target. This enzyme is the target of drugs used for the treatment of rheumatoid arthritis and other autoimmune diseases [11], and is being intensively studied as an antimalarial therapeutic target [12 – 18]. Studies on the Plasmodium falciparum DHOD (PfDHOD) show that potent human DHOD inhibitors have no significant effect on the parasite enzyme, and PfDHOD inhibitors are not cytotoxic to kidney tissue [19]. Recently, a series of triazolopyrimidine compounds that inhibit PfDHOD at nanomolar concentrations were shown to have high bioavailability, long half-life, and low clearance in rodents [20].
DHODs are classified into two families. Family 1 DHODs are soluble enzymes found in gram-positive bacteria, archaea, and lower eukaryotes. These are further subdivided into family 1A, FMN-containing homodimeric enzymes that use fumarate as the electron acceptor [21], and family 1B heterotetrameric enzymes that use FMN, FAD and iron/sulfur clusters as redox centers, and NAD+ as the electron acceptor [22, 23]. Family 2 DHODs are membrane-associated and found in gram-negative bacteria and eukaryotes. They are flavoproteins, usually anchored on the periplasmic side of the inner cytoplasmic membrane in bacteria or the outer surface of the inner mitochondrial membrane in eukaryotes, where they transfer electrons via FMN to quinones and are thus linked to the respiratory chain. Similarities are observed among DHODs in the mechanisms of the first half of the reaction catalyzed involving the oxidation of dihydroorotate and subsequent reduction of a FMN. However, because different electron acceptors are used by the different DHODs [24], mechanisms diverge in the second half of the reaction involving the oxidation of the FMN.
The T. gondii DHOD (TgDHOD) is most similar to family 2 enzymes [25]. An important difference between family 1 and family 2 enzymes is that the latter contain extended N-termini that play roles in targeting and membrane association [26, 24, 27]. The N-terminal extension of TgDHOD is comprised of ~157 residues on the N-terminal side of a predicted transmembrane segment, and this enzyme possesses the longest extension found to date in any reported family 2 DHOD enzyme [25] (Fig. 1). A slightly shorter N-terminal extension (~143 residues) is found in PfDHOD, while a relatively short extension is present (~13 residues) in the human enzyme (HsDHOD). DHOD crystal structures of family 2 enzymes are available for Escherichia coli [24, 28], human [29, 30], rat [27] and P. falciparum [31, 18]. The structures of the eukaryotic enzymes are of recombinant proteins truncated at the N-termini, thereby eliminating targeting signals and a transmembrane segment thought to serve as a membrane anchor (Fig. 1). These structural studies reveal a large domain, consisting of an α/β barrel structure containing the active site with a catalytic serine (S175 in the E. coli enzyme, S215 in HsDHOD) that is highly conserved among family 2 enzymes (Fig. 1). The serine is in close contact with the substrate, dihydroorotate, and has been proposed to remove a proton from C5 during oxidation [29, 24, 27]. A second, smaller, domain near the N-terminus is composed of two α-helices forming the entrance of a tunnel to the dihydroorotate oxidation site. This domain is the binding site for leflunomide, brequinar, atovaquone and their derivatives, and triazolopyrimidine derivatives, and is the predicted binding site for quinone [32]. Differences in longitudes and orientations of the first α-helix in the different DHODs [24] result in differences in quinone- and inhibitor-binding that could account for the observed differences in sensitivity to inhibitors [32, 18]. Here we report the purification, kinetic characterization, and initial inhibition studies of an active, recombinant DHOD from T. gondii, and show that this enzyme is located in the parasite mitochondrion. Our biochemical and genetic studies indicate that TgDHOD is an excellent target for new anti-Toxoplasma drugs.
Fig. 1.
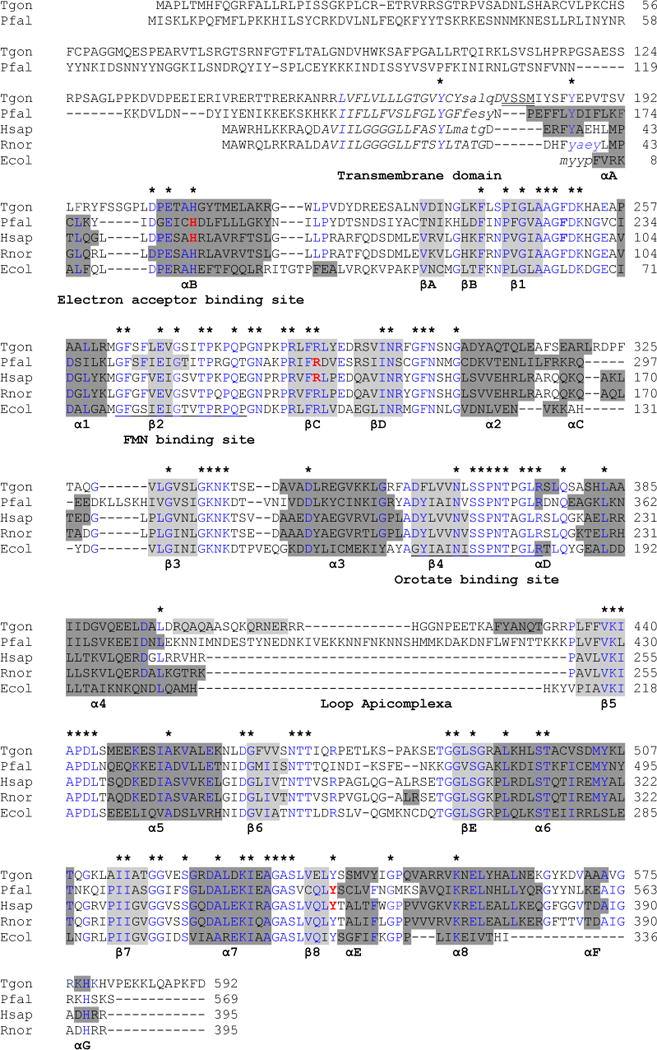
Alignment of the T gondii DHOD predicted amino acid sequence with other family 2 DHODs. Alignment of the T gondii DHOD predicted amino acid sequence with other family 2 DHODs based on [27]. Secondary structural representation of the DHOD motifs from the PDB-viewer; alpha helices show in dark gray and beta sheets in light gray. The N-termini of the predicted 3D structure for TgDHOD and for the crystallographic structures (1TV5, 1D3G, 1UUO and 1F76) are shown in lowercase. In the central barrel α-helices are named α1–α8, and β-sheets are named β1–β8. Alpha-helices and β-sheets outside the barrel are named αA–αG and βA–βE as in [27]. Partially conserved amino acids within the sequences are shown in blue and completely conserved residues are indicated above the alignment with asterisks. The N-terminal transmembrane domains predicted by HMMTOP are shown in italics. The first four residues of the recombinant protein described in the present report are indicated with double underlining. FMN and orotate binding site described in [76] are underlined. Amino acids interacting with A77-1726 are in red and bold (PfDHOD: H185, R265, Y528 [31], HsDHOD: H55, R135, Y355 [29]).
2. Materials and Methods
2.1 Materials
Reagents were from Sigma-Aldrich, unless otherwise specified. Inhibitors used were: 6-fluoro-2-(2′-fluoro-1,1-biphenyl-4-yl)-3-methyl-4-quinoline carboxylic acid (brequinar sodium salt, NSC 368390; DuPont Pharma GmbH, Bad Homburg); 2-hydroxyethylidene-cyano acetic acid 4-trifluoromethyl anilide (A77-1726; Sanofi Aventis, Germany); trans-2-[4-(chlorophenyl)cyclohexyl]-3-hydroxy-1,4-naphthoquinone, (atovaquone, 566C80; Wellcome Foundation, Dartford, UK); (2,2′-[3,3′-dimethoxy[1,1′-biphenyl]-4,4′-diyl]diimino)bis-benzoic acid (redoxal, NSC-73735); 2-hydroxy-3-(3,3-dichloroallyl)-1,4-naphthoquinone, (DCL, NSC-126771; NIH, Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment Bethesda, USA); 1-(3-methyl-4(4′-trifluoromethylthiophenoxy)phenyl)-3-methyl-1,3,5-triazine-2,4,6-(1H,3H,5H)-trione (toltrazuril; Bayer AG, Leverkusen, Germany). 4,4,4-trifluoro-1-(thienyl)-1,3-butanedione (2-thenoyltrifluoroacetone, TTFA); 2,3-dimethoxy-5-methyl-6-decyl-1,4-benzoquinone (decylubiquinone, QD); 2,3-dimethoxy-5-methyl-1,4-benzoquinone (Q0); ubiquinone-30 (Q6); and 2-methyl-1,4-naphthoquinone (menadione or vitamin K3) were from Sigma. Ubiquinone-50 (Q10) was from Kaneka, Japan; and 2,5-dimethyl-p-benzoquinone (PQ0) from Acros, Belgium. MD249 (compound 20) 2-cyano-3-hydroxy-N-/2′,3,3′-trichlorobiphenyl-4-yl)but-2-enamide, MD209 (compound 19) N-(3-chloro-2′-methoxybiphenyl-4-yl)-2-cyano-3-hydroxybut-2-enamide, were gifts from Colin W. G. Fishwick and A. Peter Johnson (University of Leeds) [17]. DSM190, N-(3,5-difluoro-4-(trifluoromethyl)phenyl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine, was the gift of Margaret A. Phillips (University of Texas Southwestern Medical Center) [16]. Additional compounds tested are listed in Table S2.
2.2 Expression and purification of N-terminally truncated recombinant TgDHOD
The TgDHOD coding region was amplified with reagents of the Expand Long Template PCR system (Roche) using a full-length clone (MAPL-PKFD) [25] as a template. Three sense primers were used to produce sequences for constructs encoding three different N-terminally truncated proteins: VSSMs, MIYSs, and FYEPs, where the four underlined letters indicate the N-terminal amino acid sequence corresponding to the 5′-end of the T. gondii DHOD (Table S1). The same antisense primer was used for all three constructs, TgDHODas (Table S1). Sense and antisense primers incorporated NdeI restriction sites to facilitate cloning. Plasmids TgDHODpET19b-MIYS, TgDHODpET19b-FYEP, and TgDHODpET19b-VSSM were constructed by cloning the amplified sequences into the NdeI site of pET19b (Novagen). The N-termini of the recombinant proteins included the tagging sequence of the expression vector, MGH10SSGHIDDDDKHM, followed by the TgDHOD sequence indicated.
One hundred μL starter cultures of BL21-CodonPlus(DE3)-RP cells (Stratagene) transformed with TgDHODpET19b-MIYS, TgDHODpET19b-FYEP, or TgDHODpET19b-VSSM were inoculated into 100 mL of LB media [33] containing 100 μg mL−1 ampicillin and were grown at 37°C. When cultures reached OD600 = 0.5, 1 mM isopropylthio-β-galactoside (IPTG) and 0.1 mM riboflavin 5′-phosphate sodium salt (FMN) were added and the temperature was lowered to 25°C. Cells were harvested 20 hrs after induction. The purification procedure was based on that developed for recombinant PfDHOD [12] with modifications. Cells were resuspended in 5 mL of buffer A (2 mM beta-mercaptoethanol, 2% Triton X-100, 10% glycerol, 0.5 mM FMN, 50 mM Tris-HCl, pH 8.5) in the presence of 1 mM phenylmethanesulfonyl fluoride (PMSF) and 1 mM benzamidine. The solution was frozen at −80°C, thawed, and submitted to sonication on ice for a total of 12 min (model W-220F sonicater, Heat Systems Ultrasonics), followed by centrifugation at 10,000 × g and 4°C for 30 min to remove cell debris. An ultracentrifugation step was found to be unnecessary. Recombinant proteins were purified from the supernatant using Ni-NTA technology (Qiagen) on resin equilibrated with buffer B (buffer A containing 300 mM NaCl, 20 mM imidazole). Fractions containing recombinant protein were eluted with buffer B containing 300 mM imidazole. Prior to measuring activity, recombinant protein-containing fractions were prepared by using PD-10 desalting columns (Amersham) equilibrated with 300 mM NaCl, 10% glycerol, 0.1% Triton, 50 mM Tris-HCl, pH 8.5, at 4°C or by dialyzing against the same buffer at 4°C with three buffer changes.
2.3 Enzyme and protein assays
Protein concentration was measured by the bicinchoninic acid (BCA) protein assay (Pierce) with bovine serum albumin (BSA) as the standard, using the microplate procedure performed according to the manufacturer’s instructions.
Activities of purified recombinant proteins were measured by monitoring 2,6 dichlorophenol-indophenol (DCIP) reduction, a reaction coupled to dihydroorotate substrate via quinone cosubstrate oxidation [34]. DCIP reduction assays were performed at 30°C in a reaction buffer containing 50 mM Tris-HCl pH 8.0, 150 mM KCl, 0.1% Triton X-100, 10% glycerol, 1 mM dihydroorotate, 0.1 mM DCIP, 0.1 mM ubiquinone, with a concentration of 6.2 nM of recombinant TgDHOD. [34]. DCIP reduction was measured at 600 nm (ɛ = 18,800 M−1cm−1) [34]. Since DCIP can accept electrons in the absence of ubiquinone, this background activity was measured (kcat = 28.1 ± 0.7 sec−1) and was subtracted from activities in the presence of the ubiquinones shown in Table 1. The activities of quinones at 0.1 mM, such as PQ0, Q10, and menadione [35, 36], were measured and compared to the activity in the presence of QD.
Table 1.
Use of alternative electron acceptors by TgDHOD-VSSM
| Electron acceptor | Enzyme Activity %(a) |
|---|---|
| QD | 100 |
| Q0 | 44 ± 5 |
| Q1 | 89 ± 5 |
| Q6 | 34 ± 2 |
| Q10 | 21 ± 3 |
| PQ0 | 35 ± 1 |
| Menadione | 24 ± 1 |
| Ferricyanide | 12 ± 1 |
| Fumarate | 0.3 ± 0.1 |
| 1,4-Naphthoquinone | 35 ± 1 |
| 2,5-Dimethyl-p-benzoquinone | 27 ± 1 |
Activity was measured with the DCIP reduction assay using 1 mM dihydroorotate, 0.1 mM electron acceptor and 0.1 mM DCIP. The activity measured with QD was taken as 100%. Values are followed by standard deviations.
Quinones were dissolved in absolute ethanol to make a stock solution of 10 mM, resulting in a final ethanol concentration of 1% in the assay. Q10 presented solubility problems; to prevent precipitation the ethanol concentration in the reaction assay was raised to 10%, a concentration that did not affect TgDHOD activity, as is the case for other DHODs [36].
The kinetic constants of both substrates dihydroorotate and decylubiquinone (QD) were determined by varying dihydroorotate concentration (5 μM – 1.0 mM) while keeping QD constant at 100 μM, or by varying QD (0.1 μM – 100 μM) at a fixed dihydroorotate concentration of 1mM. Saturation curves were also performed using 1 mM dihydroorotate, 0.1 mM DCIP, while varying concentrations (0.1 μM – 100 μM) of ubiquinone-0 (Q0), ubiquinone-6 (Q6), PQ0, 1,4-naphthoquinone, and 2,5-dimethyl-p-benzoquinone. The Michaelis-Menten equation v = Vmax x [S] / (Km+ [S]) was used to calculate Km (SigmaPlot 8.0). The kcat was calculated from kcat= Vmax/[ET], where [ET] is total enzyme concentration, based on one active site monomer.
An alternative assay measuring the appearance of orotate at 30°C at 280 nm (ɛ = 7,500 M−1cm−1) [36] was performed using 1 mM fumarate as the electron acceptor [35]. To examine ferricyanide (K3Fe(CN)6) as an alternate electron acceptor (1 mM), the change in absorbance of this acceptor was measured at 420 nm (ɛ = 1, 020 M−1cm−1) [35].
To determine the effect of inhibitors, DHOD activity was measured by the DCIP reduction assay with QD as the quinone in the presence of different inhibitors at 1 mM concentration, except for toltrazuril and atovoquone, which were used at 0.5 mM and 0.1 mM, respectively. Stock solutions of A77-1726, TTFA, and brequinar, were prepared in the assay buffer solution. In the case of brequinar, Triton X-100 was not included in the stock solution because it decreased that compound’s solubility. Stock solutions and serial dilutions of redoxal, toltrazuril, atovoquone, DCL, MD209, MD241, MD249, and DSM190 were prepared in dimethyl sulfoxide (DMSO). To achieve the desired inhibitor concentration in the assay solution for these compounds, 100 μL of an appropriate dilution of the inhibitor in DMSO were added to a final assay volume of 1 mL. The final concentration of DMSO used in all assay solutions (10%) had no effect on activity.
A total of 34 compounds were tested for inhibition (Table S2), and the best inhibitors were further characterized (Table 3). IC50 values were determined using fixed saturating concentrations of the substrates dihydroorotate (1 mM) and QD (0.1 mM), with varying inhibitor concentrations. Kis were determined with QD as the variable substrate, and saturating concentrations of dihydroorotate using SigmaPlot12 to fit the data (Table 3). The best fits were obtained for partial mixed inhibition, with goodness of fit R2 values of 0.991 (A77-1726), 0.988 (MD249), and 0.993 (MD209). The next best fits were to full mixed inhibition, with decreases in Akaika criteria (AICc) of 17 units for A77-1726, and 4 units for MD249, where a decrease of 2 units is considered significant. The difference in AICc for full mixed and partial mixed for MD209 was only 0.62, with the third best fit to full noncompetitive, with a decrease in AICc of 5.8. Partial mixed inhibition was confirmed for A77-1726 and MD249 by plotting v/(v0−v) vs. 1/[I], where v0 is the velocity at a given concentration of substrate in absence of inhibitor [37].
Table 3.
Inhibition of DHODs
| Compound | TgDHOD | PfDHOD | HsDHOD | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| IC50 μM | Ki μM | IC50 μM |
|
IC50 μM |
|
||
| A77-1726 | 91 ± 2 | 45 | 190 ± 10 [14] | 22 [17] | 1.08 ± 0.1 [34] 0.26 ± 0.10 [13] |
0.032 [17] | |
| MD209 | 60 ± 8 | 18 | 8.6 [17] | 1.0 [17] | 0.20 [17] | 0.025 [17] | |
| MD249 | 96 ± 18 | 16 | 9.9 [17] | 1.2 [17] | 0.33 [17] | 0.040 [17] | |
| DSM190 | >100 | – | 0.19 [16] | – | >30 [16] | – | |
(13) Baldwin et al., 2005; (14) Heikkila et al., 2007; (16) Gujjar et al., 2011; (17) Davies et al., 2009; (34) Knecht et al., 1998. IC50 values for TgDHOD are followed by standard deviations.
2.4 Antibody production and affinity purification
Antibodies were purified prior to use by chromatography on a column containing purified TgDHOD-VSSM that had been previously immobilized using the AminoLink Immobilization Kit (Pierce, #44890), followed by elution with 150 mM glycine, pH 2.5, and neutralization with 1M TrisHCl, pH 9.0. New polyclonal antibodies were raised against active, truncated TgDHOD-VSSM in mice. Although the mouse and rabbit antibodies described above recognized TgDHOD on immunoblots, both antisera appeared to react non-specifically when employed in immunolocalization experiments with tachyzoites on slides.
2.5 Protein electrophoresis and Immunoblots
SDS-PAGE was carried out on 1.0 mm, 7.5 – 15% gradient, or 12% non-gradient running gels with 5% stacking gels, using the buffer system described by Laemmli [38]. Electrophoresis was performed in BioRad Mini-PROTEAN or PROTEAN II xi electrophoresis cells. Immunoblot samples were prepared by resuspending tachyzoites in 100°C denaturing gel loading buffer. Protein samples were electrotransferred from gels to PVDF (Millipore) membranes using a Trans-blot® SD Semi-Dry Transfer Cell (BioRad). Western blotting was performed as described previously [39] using enhanced chemiluminescence. Rabbit anti-DHOD was used at a 1:10,000 dilution, mouse anti-DHOD at 1:2000, and mouse anti-myc 9E10 at 1:5000.
2.6 Gene knockout and gene replacements at the DHOD locus
The type I DHOD gene locus is defined by TGGT1_124080 in the current T. gondii genome database www.Toxodb.org (version 6.4). Gene knockout targeting plasmid pΔDHOD was constructed by fusing through yeast recombinational cloning [40] in the following order, a ~1.1 kb 5′ DHOD target flank amplified from RH genomic DNA, the hypoxanthine guanine phosphoribosyl transferase (HXGPRT) minigene cassette [41] and a ~1.2 kb 3′ DHOD target flank amplified from RH genomic DNA in the above order into the yeast-shuttle plasmid pRS416. The deletion was engineered to remove a small portion of the 5′ UTR and essentially all the coding region of DHOD. Using previously described methods [42, 43], pΔDHODCD was engineered by incorporating a functional cytosine deaminase gene downstream of the pΔDHOD 3′ target flank. Plasmid pDHOD/HA was assembled using recombinational cloning to C-terminal hemagglutinin (HA) tag the DHOD gene and inserting the HXGPRT selectable marker between the coding region and the 3′UTR of DHOD. The oligonucleotide primers used in gene knockout and tagging plasmid construction are shown in Table S1. Targeting plasmids were validated by restriction digest and by DNA sequencing to verify 100% homology in gene targeting flanks.
Approximately 15 μg of PmeI linearized pΔDHOD, pΔDHODCD, or pDHOD/HA targeting plasmid was individually transfected into T. gondii strain RHΔku80Δhxgprt that exhibits highly enhanced homologous recombination [39, 43]. Knockouts were then selected in mycophenolic acid (MPA) or in MPA and 5-fluorocytosine (5FC) in the presence of uracil supplementation (250 μM) using previously described methods [7, 39]. Stable MPA resistant clones were isolated and the genotype of the clones was evaluated by PCR as previously described [43]. Validation primers used to measure genotype of parasites selected following transfection with pDHOD/HA are shown in Table S1. Briefly, primers DHODHACXF and 5′DHFRCXR were used in PCR 1 (1,219 bp product) and primers 3′DHFRCXF and DHODCXR were used in PCR 2 (1,259 bp product) to verify C-terminal HA tagging and functional deletion of the DHOD 3′UTR by targeted insertion of the HXGPRT selectable marker following the translation termination codon of DHOD.
2.7 Construction of tagged TgDHODs for immunolocalization
T. gondii RH strain genomic DNA was used to PCR amplify a 1 kb fragment of the DHOD 3′-end containing ligation independent cloning (LIC) sequences (underlined) using forward primer DHOD.LIC.9385.F and reverse primer DHOD.LIC.YFP.R (Table S1). This fragment was TA-subcloned into pGEM-T-Easy (Promega). To introduce a unique restriction enzyme site for endogenous tagging, primers were used to change one nucleotide (in bold) in the sequence to be recognized by the restriction enzyme MfeI (underlined) using forward primer DHOD.MfeI.QC.F and reverse primer DHOD.MfeI.QC.R (Table S1). This was generated using the QuikChange mutagenesis strategy (Stratagene). Following sequence verification, this plasmid was used as a template to amplify the mutated DHOD fragment with the above primers DHOD.LIC.9385.F and DHOD.LIC.YFP.R. LIC cloning into endogenous tagging vectors, transfection, and selection was performed as described previously [39].
For exogenous expression of DHOD, the DHOD cDNA was amplified from a T. gondii cDNA library (V. Carruthers, unpublished) with the primers: rorward primer TgDHOD.BglII.F and reverse primer TgDHOD.myc.AscI.R (Table S1). A c-myc tag was incorporated into the reverse primer for identification by fluorescence and immunoblotting. The cDNA was subcloned into the BglII and AscI restriction sites of the pTubYFPYFP vector [43], thus replacing YFPYFP with TgDHOD. This construct was transfected into RH parasites and selected with chloramphenicol following transfection [45].
2.8 Immunofluorescence microscopy
Immunofluorescence staining of intracellular parasites was performed as described previously [46]. Briefly, slides of parasites replicating in HFF cells for 24 hrs were fixed, permeabilized with 0.1% Triton X-100, blocked with 10% FBS, and stained with primary antibodies (Rbamyc 1:250, MsαATPase 1:5,000, MsαATrx1 1:5,000) and 4′,6-diamino-2-phenylindole (DAPI) at 5 μg mL−1. MsαATPase (MAb 5F4 [47]) and MsαATrx1 (MAb 11G8 [48]) were kindly provided by Dr. Peter J. Bradley (Department of Microbiology, Immunology and Molecular Genetics, University of California Los Angeles, USA). Secondary Abs goat-anti-mouse Alexa-Fluor594 and goat-anti-rabbit Alexa-Fluor488 (Molecular Probes/Invitrogen) were used at 1:1,000. Slides were mounted with Mowiol and examined on a Zeiss Axio imager inverted microscope at 1,000× total magnification. Z-stack images were deconvolved using the Zeiss Axio Deconvolution software.
3. Results
3.1 T. gondii DHOD appears to be an essential gene
Previous genetic studies have shown that disruption of the first and last enzymes in the de novo pyrimidine biosynthetic pathway of T. gondii establish uracil auxotroph mutants that are avirulent in mice, and are unable to replicate in cell culture in the absence of added uracil [6,7] Consequently, assuming a singular role for TgDHOD in pyrimidine biosynthesis, genetic disruption of the parasite DHOD gene was expected to establish a uracil auxotroph. However, using the KU80 background [39, 43], repeated attempts to target deletion of the DHOD coding region were unsuccessful following transfection of plasmid pDHOD. Targeted disruption of DHOD was not obtained even after employing a strategy using plasmid pΔDHODCD that included a negative selection with a downstream cytosine deaminase gene and 5FC selection [43] (Fig. 2A). Targeted DHOD disruption experiments using plasmids pΔDHOD and pΔDHODCD did produce replicating parasites that were selected in MPA and uracil and grew for 10 to 14 days, then parasites simply stopped growing. This observation suggested parasite growth was initially supported by an episomal HXGPRT selectable marker and that after ~ 14 days of selection the episomes were lost and any targeted DHOD deletions, if present, were not viable. Next, to verify that gene targeting was feasible at the DHOD locus we targeted the C-terminal HA tagged DHOD using the strategy depicted in Fig. 2B. The HA tag was added to the C-terminus of DHOD and the (2 kb) HXGPRT selectable marker was inserted immediately following the termination codon of DHOD, functionally deleting the 3′ UTR of DHOD by moving this genetic element 2 kb 3′ of the coding region. Parasites were transfected with plasmid pDHOD/HA and were selected in the presence of uracil supplementation and MPA (Fig. 2B). MPA resistant clones (lanes 1 – 11) showed the expected targeted genotype with the integration of the HXGPRT selectable marker immediately following the termination codon of the C-terminal HA-tagged DHOD (Fig. 2C). These parasites exhibited identical growth rates in the presence or absence of uracil supplementation (data not shown), demonstrating that expression of a functional DHOD did not require the 3′ UTR element. While the DHOD gene can be tagged at its endogenous locus by gene targeting in the KU80 background, we were unable to select a knockout in the presence of uracil. The TgDHOD gene appears to be essential.
Fig. 2.
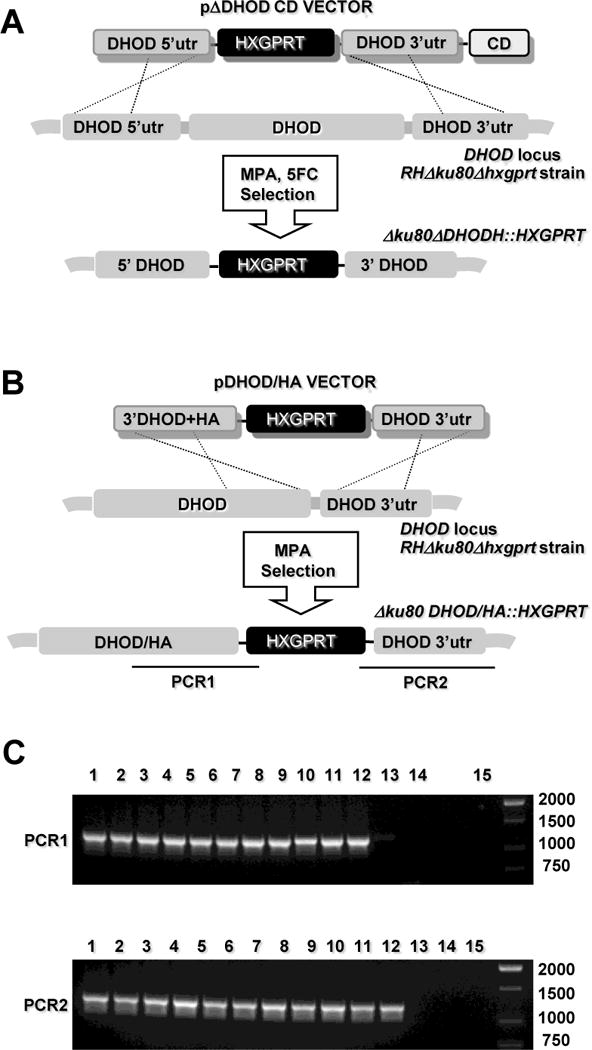
Targeted deletion or insertion at the DHOD gene. (A) Strategy for disrupting the DHOD gene via integration of the HXGPRT marker. Targeting plasmid pΔDHOD and pΔDHODCD targets a ~ 10 kb deletion of the DHOD coding region (see Materials and Methods). Parasites were first selected by positive selection for HXGPRT in MPA + xanthine, then later by negative selection against the downstream cytosine deaminase (CD) marker in MPA + xanthine + 5-fluorocytosine (5FC) to enrich for clones lacking inserting of the CD gene. (B) Strategy for HA tagging the C-terminus of DHOD and inserting the (2 kb) HXGPRT selectable marker immediately following the termination codon of DHOD. (C) Genotype validation of MPA resistant clones selected following transfection with plasmid pDHOD/HA. Top gel panel (PCR 1): selected MPA resistant clones (lanes 1 – 12); parental RHΔku80Δhxgprt (lane 13), RH (lane 14), DNA size ladder (lane 15). PCR 1 produces a 1,219 bp product from a correctly targeted clone (lanes 1 – 11), but not from the parent strains. Bottom gel panel (PCR 2): selected MPA resistant clones (lanes 1 – 12); parental RHΔku80Δhxgprt (lane 13), RH (lane 14), DNA size ladder (lane 15). PCR 2 produces a 1,259 bp product from a correctly targeted clone (lanes 1 – 11), but not from the parent strains.
3.2 Expression and purification of active TgDHOD recombinant protein
In previous work, we showed that an N-terminally truncated form of recombinant TgDHOD termed TgDHOD-MIYS (MIYS indicates the first four amino acids of the truncated N-terminus, see Fig. 1) functionally complemented a DHOD-deficient E. coli cell strain [25]. No complementation was observed for full-length recombinant protein, or for another N-terminally truncated recombinant protein, TgDHOD-ALQD, that included an additional 7 residues of DHOD coding sequence at the N-terminus of TgDHOD-MIYS; both of these appeared in the pellets of detergent-extracted E. coli cells. Although we observed TgDHOD-MIYS recombinant protein in detergent extracts [25], these supernatants had no measurable DHOD activity. In the current study, we constructed new N-terminally truncated clones using a different bacterial expression vector (pET19b, Novagen) to produce the active recombinant proteins TgDHOD-pET19bVSSM, TgDHOD-pET19bMIYS, and TgDHOD-pET19bFYEP. The TgDHOD-VSSM recombinant protein was chosen for further characterization. The expressed recombinant protein was purified employing a ten-histidine tag at its N-terminus (Fig. 3). The yield of purified recombinant TgDHOD-VSSM was approximately 2.8 mg L−1 of bacterial culture. The specific activity measured for TgDHOD-VSSM using QD as electron acceptor was 83.8 U/mg, where one unit is defined as the amount of enzyme required to catalyze the reduction of 1 μmol of substrate in 1 min.
Fig. 3.
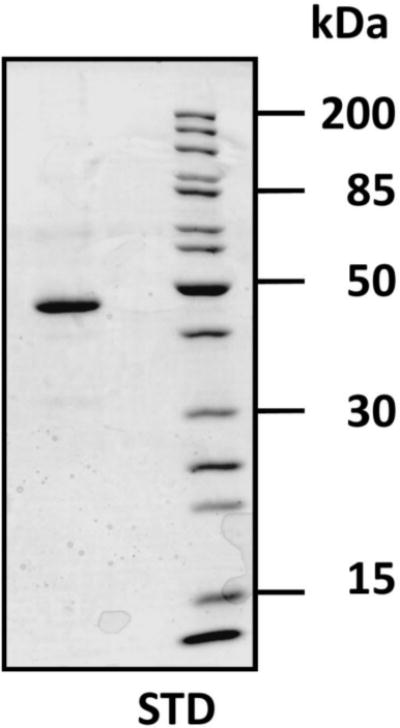
Denaturing gel electrophoresis of T. gondii DHOD truncated recombinant protein purified from bacterial extract. The pET19b expression system was used to produce a recombinant T. gondii DHOD protein missing the first 177 residues on the N-terminus, TgDHOD-VSSM (predicted molecular mass, 48.3 kDa). The N-terminally histidine-tagged recombinant protein was purified using Ni-NTA resin.
3.3 Activities and effect of inhibitors on purified T. gondii DHOD-VSSM recombinant protein
The TgDHOD-catalyzed oxidation of dihydroorotate was measured in the presence of different electron acceptors using the DCIP reduction assay. The activities were expressed as a percentage, taking the activity measured with QD as 100% (Table 1). Low activity was observed using ferricyanide as an artificial electron acceptor. As expected, negligible activity was observed with fumarate, the electron acceptor of the family 1 DHODs. Kinetic parameters were determined for the acceptors exhibiting the highest activities (Table 2).
Table 2.
Kinetic constants of purified recombinant TgDHOD-VSSM
| Varied co-substrate | Fixed Substrate | kcat(S−1) | Km(μM) |
|---|---|---|---|
| L-DHO | QD | 82 ± 1 | 60 ± 0.003 |
| QD | L-DHO | 89 ± 2 | 29 ± 2 |
| Q0 | L-DHO | 32 ± 1 | 99 ± 8 |
| Q1 | L-DHO | 61 ± 1 | 27 ± 2 |
| Q6 | L-DHO | 21 ± 1 | 51 ± 7 |
| PQ0 | L-DHO | 44 ± 5 | 132 ± 25 |
| 1,4-Naphthoquinone | L-DHO | 29 ± 1 | 28 ± 3 |
| 2,5-Dimethyl-p-benzoquinone | L-DHO | 42 ± 7 | 149 ± 39 |
Values are followed by standard deviations.
Compounds known to inhibit human or Plasmodium DHODs were tested to evaluate their effect on TgDHOD-VSSM (Table 3, Table S2). Little inhibition was observed in the presence of atovaquone (0.1 mM, 80.6% activity compared to the control) or toltrazuril (0.5 mM, 82.9% activity compared to the control). No significant inhibition of TgDHOD was observed in the presence of brequinar (1 mM) or TTFA (1 mM) (Table S2). The PfDHOD inhibitor DSM190 was a poor inhibitor of TgDHOD. While none of the inhibitors were very potent, the best were redoxal, and A77-1726, the active metabolite of leflunomide (Arava™) a drug used to treat rheumatoid arthritis, and derivatives of A77-1726 (MD209 and MD249 [14]). The IC50s determined for redoxal, A77-1726, MD249, MD209, and DSM190 were 253.3 μM ± 13.3, 91.2 μM ± 2.2, 95.6 μM ± 17.8, 60.4 μM ± 7.6, and > 100 μM, respectively. The Kis determined for A77-1726, MD249, and MD209 are shown in Table 3.
3.4 Dihydroorotate dehydrogenase localizes to mitochondria in T. gondii
Bioinformatic analysis did not reveal a consensus subcellular destination of the TgDHOD, although mitochondrial targeting scored the highest. Initial immunolocalization experiments with antibodies raised in rabbits against the inactive recombinant TgDHOD-ALQD suggested a possible association of DHOD with the apicoplast (data not shown), which is a remnant chloroplast organelle. However, purification of these antibodies, and new antibodies raised in mice against the active TgDHOD-VSSM failed to intensely stain tachyzoites despite specifically recognizing TgDHOD by immunoblotting. To further explore the localization of this enzyme, epitope tagging experiments were undertaken. Parasites were transfected with two different constructs: TgDHOD with a C-terminal three myc epitope tag for homologous targeting to the DHOD locus and expression from the endogenous TgDHOD promoter, and TgDHOD with a C-terminal single myc epitope tag under the control of the tubulin promoter for observing exogenous expression. TgDHOD was found to colocalize with the mitochondrial F1 ATPase in parasites with both endogenously and exogenously expressed enzyme (Fig. 4A, B). Interestingly, TgDHOD seems to localize in particular subregions of the mitochondrion, a pattern that is distinct from the more uniform localization of the F1 ATPase throughout the organelle. Tagged TgDHOD showed little or no colocalization with the apicoplast marker Atrx1. These findings are consistent with TgDHOD being a member of the mitochondrially-associated family 2 DHODs.
Fig. 4.
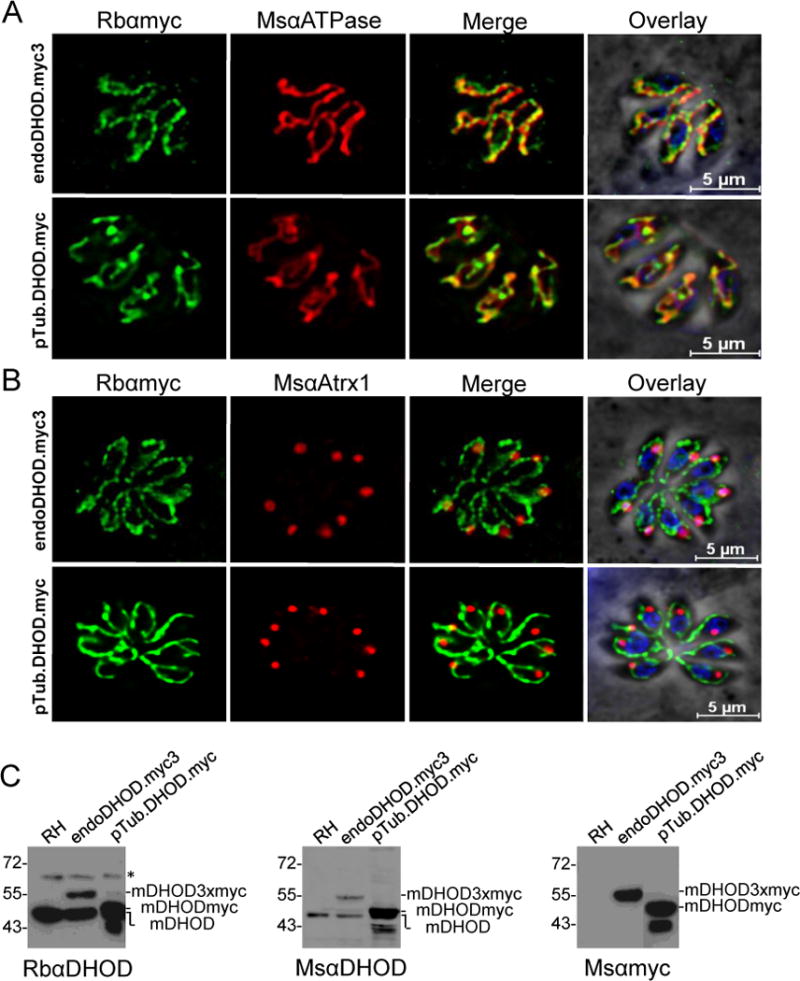
Immunolocalization of TgDHOD in intracellular tachyzoites. A. Parasites expressing endogenous TgDHOD tagged with three copies of c-myc or parasites exogenously expressing TgDHOD tagged with one copy of c-myc under the control of the tubulin promoter were visualized by deconvolution immunofluorescence with antibodies against myc (green) and the mitochondrial marker ATPase (red); nuclei were stained with DAPI. B. Dual staining of DHOD and the apicoplast marker Atrx (red); nuclei were stained with DAPI. C. Immunoblots of tachyzoite extracts from wild-type RH, endogenously tagged TgDHOD, and exogenously expressed TgDHOD were probed with affinity purified rabbit antibodies raised against TgDHOD-ALQD (left panel), antibodies raised against the myc-tag (middle panel), or mouse antibodies raised against TgDHOD-VSSM (right panel). An unknown antigen recognized by the rabbit antibodies is marked with an asterisk.
3.5 The TgDHOD mitochondrial targeting sequence is proteolytically removed
Human DHOD displays a short (~13 aa) N-terminal mitochondrial targeting sequence that is not removed upon import whereas TgDHOD has a much longer (~157 aa) N-terminal putative mitochondrial targeting sequence. We performed immunoblotting to assess whether this sequence is retained or removed during mitochondrial import. A band migrating near the truncated recombinant TgDHOD-VSSM (48 kDa) in tachyzoite lysates (Fig. 4C) was recognized by both mouse and rabbit antibodies, likely reflects in vivo processing coincident with mitochondrial import and is probably the mature enzyme (mDHOD). Both anti-TgDHOD and anti-myc antibodies detected bands corresponding to processed TgDHOD tagged with three copies of myc (~55 kDa, mDHOD3xmyc) or one copy of myc (~50 kDa, mDHODmyc) (Fig. 4C). The exogenously and endogenously tagged TgDHODs appeared to be susceptible to further proteolysis, removing the C-terminal epitope tag to produce a species that comigrates with mDHOD (Fig. 4C). A precursor species (expected size 65 kDa) was not detected even after long exposure, implying that TgDHOD is rapidly imported and processed. Collectively, the above findings suggest that unlike many other family 2 DHODs [26], the N-terminal mitochondrial targeting sequence of TgDHOD is proteolytically removed during mitochondrial import.
4. Discussion
Pyrimidine biosynthesis has been proposed as a drug target for apicomplexan parasites [49, 50, 6, 13, 8]. Plasmodium depends completely on de novo pyrimidine biosynthesis for pyrimidine nucleotides because it lacks salvage enzymes. Although T. gondii is able to salvage uracil using uracil phosphoribosyltransferase [9, 51], this enzyme is nonessential [10]. In the present work we showed that although tagging the C-terminus or functionally deleting the 3′ UTR of DHOD was possible, the coding region of DHOD could not be deleted even in the presence of uracil supplementation that is known to bypass the requirement for a functional de novo pyrimidine synthesis pathway [6, 7]. Other enzymes of the pyrimidine biosynthetic pathway such as carbamoyl phosphate synthetase II (CPSII) [6] and orotidine 5′-monophosphate decarboxylase (ODC) [7] have been demonstrated to be essential enzymes but these enzymes can be deleted and the parasite growth rate can be easily rescued in vitro with uracil supplementation. The inability to delete DHOD was not because of inefficient gene targeting at this locus; we functionally deleted the 3′ UTR of DHOD and tagged the C-terminus of the enzyme. Similarly, while it has been possible to chemically inactivate P. falciparum DHOD enzyme function and rescue pyrimidine biosynthesis and parasite growth using a cytosolic fumarate-dependent yeast DHOD, a knockout of the P. falciparum DHOD has not been reported [52, 53]. The TgDHOD gene appears to be essential. Future studies will further investigate the essentiality of the pyrimidine-dependent and, possibly, the pyrimidine-independent roles of TgDHOD.
DHOD is being intensively studied as a potential target in Plasmodium [18]. The production of an active, purified, recombinant TgDHOD has allowed us to perform a kinetic characterization of the enzyme, a fundamental step for defining the effects of potential inhibitors. The kinetic parameters determined for the TgDHOD-VSSM recombinant protein are found in Table 2. TgDHOD exhibited the lowest Kms for QD, Q1 and Q6, and a higher Km for Q0. Baldwin and co-workers [12] observed similar trends for recombinant DHOD from PfDHOD, with lower Kms for Q4, Q6 and QD (~ 15 – 20 μM), and higher Kms for quinones lacking isoprenoid chains, such as Q0 or menadione (~ 55 – 115 μM). The Kms for quinones synthesized by P. falciparum, Q7, Q8 and Q9[54, 55], have not been determined but are probably in the former range. The quinones present in T. gondii have not yet been identified. Q10 is the most common quinone present in humans and higher animals [56]. HsDHOD exhibits a Km for QD of 14 μM, similar to the values measured for the parasite enzymes [57]. The Km for dihydroorotate for TgDHOD with QD as the co-substrate was in the same range as that reported for PfDHOD, 30 – 90 μM [14, 15], but higher than that measured for HsDHOD, 9.5 μM [58].
The kcat measured for TgDHOD was similar to that of HsDHOD, 107 s−1 [58], and 6 to 11-fold higher than those measured for recombinant PfDHOD proteins [13, 14]. The lower activity for the Plasmodium enzyme may be accounted for by differences in assay conditions, including a lower temperature.
We tested the effect of compounds known to inhibit human and rodent DHOD on our purified recombinant enzyme (Table 3, Table S2). We found that the nanomolar inhibitors of mammalian DHODs, A77-1726, redoxal, and brequinar, were not efficient inhibitors of TgDHOD, as is also observed for PfDHOD. TTFA, shown to bind to ubiquinone sites in complex II of the respiratory chain [59], had no effect on TgDHOD activity. Atovaquone, an inhibitor that binds to the ubiquinol oxidation pocket of complex III (IC50 = 0.04 μM for Saccharomyces cerevisiae complex III, IC50 = 0.4 μM for human complex III [60], IC50 = 14.5 μM for HsDHOD), and a drug used in the treatment of toxoplasmosis, malaria, and pneumocystis pneumonia, had little effect on the TgDHOD activity. Toltrazuril, an anticoccidial drug and an inhibitor of rat DHOD [61], also had little effect on TgDHOD activity. It is evident from many studies [12 – 18] that there are significant differences between the binding sites of PfDHOD and HsDHOD that can be exploited for producing Plasmodium-specific inhibitors. Our data suggest that this will also be the case for TgDHOD.
The IC50s for the A77-1726 derivatives MD209 and MD249 were an order of magnitude higher in concentration for TgDHOD than the IC50s reported for PfDHOD [17] (Table 3). DSM190, a representative of the triazolopyrimidine compounds that have been developed for inhibition of PfDHOD [16], exhibited an IC50 for TgDHOD three orders of magnitude higher than that reported for the Plasmodium enzyme (Table 3). Taken together, these results highlight the differences in the inhibitor binding sites of these two apicomplexan enzymes.
Homology models of the TgDHOD amino acid sequence were built using SWISS-MODEL [62] in the automatic modeling mode, using as templates each of the available type 2 DHOD structures. The TgDHOD model with the most complete sequence coverage (based on PfDHOD 3I68) was generated after residue TgDHOD A174 (Fig. S2); however, no consensus was observed in the length of the first α-helix of the small domain (helix αA, Fig. 1) predicted by different secondary structure prediction algorithms. This α-helix is the binding site of many inhibitors and the predicted binding site of the ubiquinone cofactor, and the uncertainty in its predicted length suggests a possible structural dissimilarity between the TgDHOD and other type 2 DHODs which could be exploited to design T. gondii-specific inhibitors, and which may have implications for the binding of electron acceptors in vivo.
Enzymes catalyzing the first, second, third, fifth, and sixth steps of de novo pyrimidine biosynthesis are soluble with primarily cytosolic localization in mammals [63], and with chloroplast localization in plants [64]. In the fourth step, electrons from the dihydroorotate oxidation, catalyzed by family 2 DHODs localized in the inner mitochondrial membrane, are transferred to ubiquinone, thus contributing to oxidative phosphorylation. Questions remain about the importance of oxidative phosphorylation in different stages of T. gondii. The mitochondrial electron transport chain in T. gondii is composed of homologs of respiratory complexes II, III, and IV [65], while complex I is replaced by a rotenone-insensitive type-II NADH dehydrogenase [66]. Experiments with mitochondrial dyes demonstrate mitochondrial membrane potential in both extra- and intracellular parasites [67, 68]. In extracellular tachyzoites, under conditions when the electron transport chain is inhibited by KCN, or in absence of oxygen, parasites retain 60%–70% motility, suggesting that glycolysis is the major source of ATP in this non-replicating motile stage [69].
As is the case in other organisms, T. gondii respiratory chain-linked dehydrogenases, for example, glycerol-3-phosphate dehydrogenase, and dihydroorotate dehydrogenase are sources of electrons. Differences in the abilities of these two dehydrogenases to reduce the electron transport chain have been observed between extracellular and intracellular tachyzoites. In extracellular tachyzoites, oxygen consumption is stimulated by addition of ADP when malate or succinate is added as a substrate; however, no stimulation was observed in the presence of dihydroorotate or glycerol-3-phosphate [65]. Nevertheless, others have shown that in intracellular tachyzoites dihydroorotate or glycerol-3-phosphate (as well as malate or succinate) is able to generate a mitochondrial inner membrane potential [68].
While resting mammalian cells meet their needs for pyrimidine nucleotides primarily through salvage pathways, proliferating cells require de novo biosynthesis, and the enzymes of the de novo pathway are controlled at transcriptional and post-transcriptional levels [70]. Post-transcriptional control of the first step in the pathway, catalyzed by carbamoyl phosphate synthetase II (CPSII), leads to an eight-fold increase in flux through the pathway in exponentially growing mammalian cells compared to resting cells [63]. In T. gondii, less is known about the regulation of the pyrimidine biosynthetic pathway. Asai and co-workers [71] found that TgCPSII partially purified from parasite extracts was inhibited by UTP, but not activated by PRPP. Studies with TgCPSII minigenes show that mutations or deletions in the proposed regulatory regions of the CPSII decrease, and in some cases eliminate, the ability of the minigene to complement CPSII knockout parasites [72].
Recent work shows that deletions of pyrimidine salvage enzymes in a type II strain do not affect cyst development [42]. In contrast, deletion of orotate phosphoribosyl transferase (OPRT), the enzyme catalyzing the fifth step of pyrimidine biosynthesis, causes type II parasites to become auxotrophic for uracil, thus this pathway appears to be essential in bradyzoites, as well as tachyzoites [42]. The activity of the pathway would presumably require a functioning electron transport chain to permit reduction of ubiquinone by TgDHOD. Although inhibitors of mitochondrial respiration induce in vitro differentiation from tachyzoites to bradyzoites [73, 74], the latter must have at least low levels of mitochondrial activity, and this would explain the apparent activity of atovoquone against bradyzoites in vitro [75]. A survey of ToxoDB supports the presence of expressed sequence tags (ESTs) in bradyzoites for complexes II, III, IV, and cytochorome c, ESTs for the pyrimidine biosynthetic enzymes catalyzing steps 1, 3, 4, and 6, and the salvage enzyme, UPRT.
Our genetic and biochemical studies suggest that TgDHOD is an excellent target for the development of anti-Toxoplasma drugs. The availability of an active, recombinant protein will make possible future crystallization and structure determinations for this enzyme.
Supplementary Material
Highlights.
A recombinant Toxoplasma gondii dihydroorotate dehydrogenase (TgDHOD) was kinetically characterized.
TgDHOD was not significantly affected by known inhibitors of human DHOD.
Three Plasmodium falciparum DHOD inhibitors were tested and exhibited one to two orders of magnitude higher IC50s for TgDHOD.
C-terminally tagged TgDHODs in transgenic T. gondii colocalize with the parasite’s mitochondrion.
Gene targeting suggested an essential role for TgDHOD that was not rescued by uracil supplementation.
Acknowledgments
The authors gratefully acknowledge support from the Facultad de Ciencias, Universidad de los Andes, from the Fundación para la promoción de la investigación y la tecnología (FPIT)-Banco de la República, Colciencias (MAHT), and from the National Institutes of Health R01AI041930 (DJB, subaward to BHZ). We would like to thank Dr. Dave R. Evans for his guidance and support of MAHT during her visit to Wayne State University (Detroit, MI, USA). We would also like to thank Melissa Serrano (University of Puerto Rico School of Medicine, San Juan, PR, USA) for her excellent technical assistance.
Abbreviations
- DAPI
4′, 6-diamino-2-phenylindole
- EDTA
ethylenediaminetetraacetic acid
- PBS
phosphate buffered saline
- PMSF
phenylmethanesulfonyl fluoride
- PVDF
polyvinylidene difluoride
abbreviations for quinones and inhibitors are listed in the Materials section
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weiss LM, Dubey JP. Toxoplasmosis: A history of clinical observations. Int J Parasitol. 2009;39:895–901. doi: 10.1016/j.ijpara.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Montoya JG, Remington JS. Management of Toxoplasma gondii infection during pregnancy. Clin Infect Dis. 2008;47:554–66. doi: 10.1086/590149. [DOI] [PubMed] [Google Scholar]
- 3.Haverkos HW. Assessment of therapy for toxoplasma encephalitis. The TE Study Group. Am J Med. 1987;82:907–14. doi: 10.1016/0002-9343(87)90151-3. [DOI] [PubMed] [Google Scholar]
- 4.Luft BJ, Remington JS. Toxoplasmic encephalitis in AIDS. Clin Infect Dis. 1992;15:211–22. doi: 10.1093/clinids/15.2.211. [DOI] [PubMed] [Google Scholar]
- 5.Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363:1965–76. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- 6.Fox BA, Bzik DJ. De novo pyrimidine biosynthesis is required for virulence of Toxoplasma gondii. Nature. 2002;415:926–9. doi: 10.1038/415926a. [DOI] [PubMed] [Google Scholar]
- 7.Fox BA, Bzik DJ. Avirulent uracil auxotrophs based on disruption of orotidine-5′-monophosphate decarboxylase elicit protective immunity to Toxoplasma gondii. Infect Immun. 2010;78:3744–52. doi: 10.1128/IAI.00287-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hyde JE. Targeting purine and pyrimidine metabolism in human apicomplexan parasites. Curr Drug Targets. 2007;8:31–47. doi: 10.2174/138945007779315524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pfefferkorn ER. Toxoplasma gondii: the enzymic defect of a mutant resistant to 5-fluorodeoxyuridine. Exp Parasitol. 1978;44:26–35. doi: 10.1016/0014-4894(78)90077-2. [DOI] [PubMed] [Google Scholar]
- 10.Donald RG, Roos DS. Insertional mutagenesis and marker rescue in a protozoan parasite: cloning of the uracil phosphoribosyltransferase locus from Toxoplasma gondii. Proc Natl Acad Sci U S A. 1995;92:5749–53. doi: 10.1073/pnas.92.12.5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kovarik JM, Burtin P. Immunosuppressants in advanced clinical development for organ transplantation and selected autoimmune diseases. Expert Opin Emerg Drugs. 2003;8:47–62. doi: 10.1517/14728214.8.1.47. [DOI] [PubMed] [Google Scholar]
- 13.Baldwin J, Michnoff CH, Malmquist NA, White J, Roth MG, Rathod PK, Phillips MA. High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J Biol Chem. 2005;280:21847–53. doi: 10.1074/jbc.M501100200. [DOI] [PubMed] [Google Scholar]
- 14.Heikkilä T, Ramsey C, Davies M, Galtier C, Stead AM, Johnson AP, Fishwick CW, Boa AN, McConkey GA. Design and synthesis of potent inhibitors of the malaria parasite dihydroorotate dehydrogenase. J Med Chem. 2007;50:186–91. doi: 10.1021/jm060687j. [DOI] [PubMed] [Google Scholar]
- 17.Phillips MA, Gujjar R, Malmquist NA, White J, El Mazouni F, Baldwin J, Rathod PK. Triazolopyrimidine-Based Dihydroorotate Dehydrogenase Inhibitors with Potent and Selective Activity against the Malaria Parasite Plasmodium falciparum. J Med Chem. 2008;51:3649–53. doi: 10.1021/jm8001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gujjar R, El Mazouni F, White KL, White J, Creason S, Shackleford DM, Deng X, Charman WN, Bathurst I, Burrows J, Floyd DM, Matthews D, Buckner FS, Charman SA, Phillips MA, Rathod PK. Lead optimization of aryl and aralkyl amine-based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with antimalarial activity in mice. J Med Chem. 2011;54:3935–49. doi: 10.1021/jm200265b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies M, Heikkilä T, McConkey GA, Fishwick CW, Parsons MR, Johnson AP. Structure-based design, synthesis, and characterization of inhibitors of human and Plasmodium falciparum dihydroorotate dehydrogenases. J Med Chem. 2009;52:2683–93. doi: 10.1021/jm800963t. [DOI] [PubMed] [Google Scholar]
- 18.Phillips MA, Rathod PK. Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect Disord Drug Targets. 2010;10:226–39. doi: 10.2174/187152610791163336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel V, Booker M, Kramer M, Ross L, Celatka CA, Kennedy LM, Dvorin JD, Duraisingh MT, Sliz P, Wirth DF, Clardy J. Identification and characterization of small molecule inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J Biol Chem. 2008;283:35078–85. doi: 10.1074/jbc.M804990200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coteron JM, Marco M, Esquivias J, Deng X, White KL, White J, Koltun M, El Mazouni F, Kokkonda S, Katneni K, Bhamidipati R, Shackleford DM, Angulo-Barturen I, Ferrer SB, Jiménez-Díaz MB, Gamo FJ, Goldsmith EJ, Charman WN, Bathurst I, Floyd D, Matthews D, Burrows JN, Rathod PK, Charman SA, Phillips MA. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J Med Chem. 2011;54:5540–61. doi: 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rowland P, Nielsen FS, Jensen KF, Larsen S. The crystal structure of the flavin containing enzyme dihydroorotate dehydrogenase A from Lactococcus lactis. Structure. 1997;5:239–52. doi: 10.1016/s0969-2126(97)00182-2. [DOI] [PubMed] [Google Scholar]
- 22.Rowland P, Nørager S, Jensen KF, Larsen S. Structure of dihydroorotate dehydrogenase B: electron transfer between two flavin groups bridged by an iron-sulphur cluster. Structure. 2000;8:1227–38. doi: 10.1016/s0969-2126(00)00530-x. [DOI] [PubMed] [Google Scholar]
- 23.Nielsen FS, Rowland P, Larsen S, Jensen KF. Purification and characterization of dihydroorotate dehydrogenase A from Lactococcus lactis, crystallization and preliminary X-ray diffraction studies of the enzyme. Protein Sci. 1996;5:852–6. doi: 10.1002/pro.5560050506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nørager S, Jensen KF, Björnberg O, Larsen S. E. coli dihydroorotate dehydrogenase reveals structural and functional distinctions between different classes of dihydroorotate dehydrogenases. Structure. 2002;10:1211–23. doi: 10.1016/s0969-2126(02)00831-6. [DOI] [PubMed] [Google Scholar]
- 25.Sierra Pagan ML, Zimmermann BH. Cloning and expression of the dihydroorotate dehydrogenase from Toxoplasma gondii. Biochim Biophys Acta. 2003;1637:178–181. doi: 10.1016/s0925-4439(02)00226-0. [DOI] [PubMed] [Google Scholar]
- 26.Rawls J, Knecht W, Diekert K, Lill R, Löffler M. Requirements for the mitochondrial import and localization of dihydroorotate dehydrogenase. Eur J Biochem. 2000;267:2079–87. doi: 10.1046/j.1432-1327.2000.01213.x. [DOI] [PubMed] [Google Scholar]
- 27.Hansen M, Le Nours J, Johansson E, Antal T, Ullrich A, Löffler M, Larsen S. Inhibitor binding in a class 2 dihydroorotate dehydrogenase causes variations in the membrane-associated N-terminal domain. Protein Sci. 2004;13:1031–42. doi: 10.1110/ps.03533004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kow RL, Whicher JR, McDonald CA, Palfey BA, Fagan RL. Disruption of the proton relay network in the class 2 dihydroorotate dehydrogenase from Escherichia coli. Biochemistry. 2009;48:9801–9. doi: 10.1021/bi901024m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu S, Neidhardt EA, Grossman TH, Ocain T, Clardy J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure. 2000;8:25–33. doi: 10.1016/s0969-2126(00)00077-0. [DOI] [PubMed] [Google Scholar]
- 30.Baumgartner R, Walloschek M, Kralik M, Gotschlich A, Tasler S, Mies J, Leban J. Dual binding mode of a novel series of DHODH inhibitors. J Med Chem. 2006;49:1239–47. doi: 10.1021/jm0506975. [DOI] [PubMed] [Google Scholar]
- 31.Hurt DE, Widom J, Clardy J. Structure of Plasmodium falciparum dihydroorotate dehydrogenase with a bound inhibitor. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 3):312–23. doi: 10.1107/S0907444905042642. [DOI] [PubMed] [Google Scholar]
- 32.Hurt DE, Sutton AE, Clardy J. Brequinar derivatives and species-specific drug design for dihydroorotate dehydrogenase. Bioorg Med Chem Lett. 2006;16(6):1610–5. doi: 10.1016/j.bmcl.2005.12.029. Epub 2006 Jan 10. [DOI] [PubMed] [Google Scholar]
- 33.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manuel. 2. Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 34.Knecht W, Löffler M. Species-related inhibition of human and rat dihydroorotate dehydrogenase by immunosuppressive isoxazol and cinchoninic acid derivatives. Biochem Pharmacol. 1998;56:1259–64. doi: 10.1016/s0006-2952(98)00145-2. [DOI] [PubMed] [Google Scholar]
- 35.Zameitat E, Gojković Z, Knecht W, Piskur J, Löffler M. Biochemical characterization of recombinant dihydroorotate dehydrogenase from the opportunistic pathogenic yeast Candida Albicans. FEBS J. 2006;273:3183–91. doi: 10.1111/j.1742-4658.2006.05327.x. [DOI] [PubMed] [Google Scholar]
- 36.Ullrich A, Knecht W, Piskur J, Löffler M. Plant dihydroortate dehydrogenase differs significantly in substrate specificity and inhibition from the animal enzymes. FEBS Lett. 2002;529:346–50. doi: 10.1016/s0014-5793(02)03425-7. [DOI] [PubMed] [Google Scholar]
- 37.Yoshino M. A graphical method for determining inhibition parameters for partial and complete inhibitors. Biochem J. 1987;248:815–20. doi: 10.1042/bj2480815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 39.Huynh MH, Carruthers VB. Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot Cell. 2009;8:530–9. doi: 10.1128/EC.00358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oldenburg KR, Vo KT, Michaelis S, Paddon C. Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res. 1997;25:451–2. doi: 10.1093/nar/25.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donald RG, Carter D, Ullman B, Roos DS. Insertional tagging, cloning, and expression of the Toxoplasma gondii hypoxanthine-xanthine-guanine phosphoribosyltransferase gene. Use as a selectable marker for stable transformation. J Biol Chem. 1996;271:14010–9. doi: 10.1074/jbc.271.24.14010. [DOI] [PubMed] [Google Scholar]
- 42.Fox BA, Falla A, Rommereim LM, Tomita T, Gigley JP, Mercier C, Cesbron-Delauw MF, Weiss LM, Bzik DJ. Type II Toxoplasma gondii KU80 knockout strains enable functional analysis of genes required for cyst development and latent infection. Eukaryot Cell. 2011;10:1193–206. doi: 10.1128/EC.00297-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fox BA, Ristuccia JG, Gigley JP, Bzik DJ. Efficient gene replacements in Toxoplasma gondii strains deficient for nonhomologous end joining. Eukaryot Cell. 2009;8:520–9. doi: 10.1128/EC.00357-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gubbels MJ, Li C, Striepen B. High-throughput growth assay for Toxoplasma gondii using yellow fluorescent protein. Antimicrob Agents Chemother. 2003;47:309–16. doi: 10.1128/AAC.47.1.309-316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim K, Soldati D, Boothroyd JC. Gene replacement in Toxoplasma gondii with chloramphenicol acetyltransferase as selectable marker. Science. 1993;262:911–4. doi: 10.1126/science.8235614. [DOI] [PubMed] [Google Scholar]
- 46.Huynh MH, Rabenau KE, Harper JM, Beatty WL, Sibley LD, Carruthers VB. Rapid invasion of host cells by Toxoplasma requires secretion of the MIC2-M2AP adhesive protein complex. EMBO J. 2003;22:2082–90. doi: 10.1093/emboj/cdg217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tawk L, Dubremetz JF, Montcourrier P, Chicanne G, Merezegue F, Richard V, Payrastre B, Meissner M, Vial HJ, Roy C, Wengelnik K, Lebrun M. Phosphatidylinositol 3-monophosphate is involved in toxoplasma apicoplast biogenesis. PLoS Pathog. 2011;7:e1001286. doi: 10.1371/journal.ppat.1001286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeRocher AE, Coppens I, Karnataki A, Gilbert LA, Rome ME, Feagin JE, Bradley PJ, Parsons M. A thioredoxin family protein of the apicoplast periphery identifies abundant candidate transport vesicles in Toxoplasma gondii. Eukaryot Cell. 2008;7:1518–29. doi: 10.1128/EC.00081-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gero AM, O’Sullivan WJ. Blood Cells. Purines and pyrimidines in malarial parasites. 1990;16:467–84. [PubMed] [Google Scholar]
- 50.McFadden DC, Camps M, Boothroyd JC. Resistance as a tool in the study of old and new drug targets in Toxoplasma. Drug Resist Updat. 2001;4:79–84. doi: 10.1054/drup.2001.0184. [DOI] [PubMed] [Google Scholar]
- 51.Iltzsch MH. Pyrimidine salvage pathways in Toxoplasma gondii. J Eukaryot Microbiol. 1993;40:24–8. doi: 10.1111/j.1550-7408.1993.tb04877.x. [DOI] [PubMed] [Google Scholar]
- 52.Ganesan SM, Morrisey JM, Ke H, Painter HJ, Laroiya K, Phillips MA, Rathod PK, Mather MW, Vaidya AB. Yeast dihydroorotate dehydrogenase as a new selectable marker for Plasmodium falciparum transfection. Mol Biochem Parasitol. 2011;177:29–34. doi: 10.1016/j.molbiopara.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Painter HJ, Morrisey JM, Mather MW, Vaidya AB. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature. 2007;446:88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- 54.de Macedo CS, Uhrig ML, Kimura EA, Katzin AM. Characterization of the isoprenoid chain of coenzyme Q in Plasmodium falciparum. FEMS Microbiol Lett. 2002;207:13–20. doi: 10.1111/j.1574-6968.2002.tb11021.x. [DOI] [PubMed] [Google Scholar]
- 55.Tonhosolo R, D’Alexandri FL, Genta FA, Wunderlich G, Gozzo FC, Eberlin MN, Peres VJ, Kimura EA, Katzin AM. Identification, molecular cloning and functional characterization of an octaprenyl pyrophosphate synthase in intra-erythrocytic stages of Plasmodium falciparum. Biochem J. 2005;392:117–26. doi: 10.1042/BJ20050441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ellis JE, Setchell KD, Kaneshiro ES. Detection of ubiquinone in parasitic and free-living protozoa, including species devoid of mitochondria. Mol Biochem Parasitol. 1994;65:213–24. doi: 10.1016/0166-6851(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 57.Knecht W, Bergjohann U, Gonski S, Kirschbaum B, Löffler M. Functional expression of a fragment of human dihydroorotate dehydrogenase by means of the baculovirus expression vector system, and kinetic investigation of the purified recombinant enzyme. Eur J Biochem. 1996;240:292–301. doi: 10.1111/j.1432-1033.1996.0292h.x. [DOI] [PubMed] [Google Scholar]
- 58.Ullrich A, Knecht W, Fries M, Löffler M. Recombinant expression of N-terminal truncated mutants of the membrane bound mouse, rat and human flavoenzyme dihydroorotate dehydrogenase. A versatile tool to rate inhibitor effects? Eur J Biochem. 2001;268:1861–8. [PubMed] [Google Scholar]
- 59.Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, Bartlam M, Rao Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell. 2005;121:1043–57. doi: 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 60.Kessl JJ, Lange BB, Merbitz-Zahradnik T, Zwicker K, Hill P, Meunier B, Pálsdóttir H, Hunte C, Meshnick S, Trumpower BL. Molecular basis for atovaquone binding to the cytochrome bc1 complex. J Biol Chem. 2003;278:31312–8. doi: 10.1074/jbc.M304042200. [DOI] [PubMed] [Google Scholar]
- 61.Jöckel J, Wendt B, Löffler M. Structural and functional comparison of agents interfering with dihydroorotate, succinate and NADH oxidation of rat liver mitochondria. Biochem Pharmacol. 1998;56:1053–60. doi: 10.1016/s0006-2952(98)00131-2. [DOI] [PubMed] [Google Scholar]
- 62.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–5. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Evans DR, Guy HI. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J Biol Chem. 2004;279:33035–8. doi: 10.1074/jbc.R400007200. [DOI] [PubMed] [Google Scholar]
- 64.Zrenner R, Stitt M, Sonnewald U, Boldt R. Pyrimidine and purine biosynthesis and degradation in plants. Annu Rev Plant Biol. 2006;57:805–36. doi: 10.1146/annurev.arplant.57.032905.105421. [DOI] [PubMed] [Google Scholar]
- 65.Vercesi AE, Rodrigues CO, Uyemura SA, Zhong L, Moreno SN. Respiration and oxidative phosphorylation in the apicomplexan parasite Toxoplasma gondii. J Biol Chem. 1998;273:31040–7. doi: 10.1074/jbc.273.47.31040. [DOI] [PubMed] [Google Scholar]
- 66.Lin SS, Kerscher S, Saleh A, Brandt U, Gross U, Bohne W. The Toxoplasma gondii type-II NADH dehydrogenase TgNDH2-I is inhibited by 1-hydroxy-2-alkyl-4(1H)quinolones. Biochim Biophys Acta. 2008;1777:1455–62. doi: 10.1016/j.bbabio.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 67.Melo EJ, Attias M, De Souza W. The single mitochondrion of tachyzoites of Toxoplasma gondii. J Struct Biol. 2000;130:27–33. doi: 10.1006/jsbi.2000.4228. [DOI] [PubMed] [Google Scholar]
- 68.Lin SS, Gross U, Bohne W. Type II NADH dehydrogenase inhibitor 1-hydroxy-2-dodecyl-4(1H)quinolone leads to collapse of mitochondrial inner-membrane potential and ATP depletion in Toxoplasma gondii. Eukaryot Cell. 2009;8:877–87. doi: 10.1128/EC.00381-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pomel S, Luk FC, Beckers CJ. Host cell egress and invasion induce marked relocations of glycolytic enzymes in Toxoplasma gondii tachyzoites. PLoS Pathog. 2008;4:e1000188. doi: 10.1371/journal.ppat.1000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sigoillot FD, Evans DR, Guy HI. Growth-dependent regulation of mammalian pyrimidine biosynthesis by the protein kinase A and MAPK signaling cascades. J Biol Chem. 2002;277:15745–51. doi: 10.1074/jbc.M201112200. [DOI] [PubMed] [Google Scholar]
- 71.Asai T, O’Sullivan WJ, Kobayashi M, Gero AM, Yokogawa M, Tatibana M. Enzymes of the de novo pyrimidine biosynthetic pathway in Toxoplasma gondii. Mol Biochem Parasitol. 1983;7:89–100. doi: 10.1016/0166-6851(83)90037-3. [DOI] [PubMed] [Google Scholar]
- 72.Fox BA, Ristuccia JG, Bzik DJ. Genetic identification of essential indels and domains in carbamoyl phosphate synthetase II of Toxoplasma gondii. Int J Parasitol. 2009;39:533–9. doi: 10.1016/j.ijpara.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bohne W, Heesemann J, Gross U. Reduced replication of Toxoplasma gondii is necessary for induction of bradyzoite-specific antigens: a possible role for nitric oxide in triggering stage conversion. Infect Immun. 1994;62:1761–7. doi: 10.1128/iai.62.5.1761-1767.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tomavo S, Boothroyd JC. Interconnection between organellar functions, development and drug resistance in the protozoan parasite, Toxoplasma gondii. Int J Parasitol. 1995;25:1293–9. doi: 10.1016/0020-7519(95)00066-b. [DOI] [PubMed] [Google Scholar]
- 75.Ferguson DJ, Huskinson-Mark J, Araujo FG, Remington JS. An ultrastructural study of the effect of treatment with atovaquone in brains of mice chronically infected with the ME49 strain of Toxoplasma gondii. Int J Exp Pathol. 1994;75:111–6. [PMC free article] [PubMed] [Google Scholar]
- 76.Annoura T, Nara T, Makiuchi T, Hashimoto T, Aoki T. The origin of dihydroorotate dehydrogenase genes of kinetoplastids, with special reference to their biological significance and adaptation to anaerobic, parasitic conditions. J Mol Evol. 2005;60:113–27. doi: 10.1007/s00239-004-0078-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.