

Abstract
Fiji is a distribution of the popular Open Source software ImageJ focused on biological image analysis. Fiji uses modern software engineering practices to combine powerful software libraries with a broad range of scripting languages to enable rapid prototyping of image processing algorithms. Fiji facilitates the transformation of novel algorithms into ImageJ plugins that can be shared with end users through an integrated update system. We propose Fiji as a platform for productive collaboration between computer science and biology research communities.
Introduction
Much primary biological data is acquired as images. In recent years, with the adoption of automated microscopy technologies, the volume and complexity of image data has increased to the point that it is no longer feasible to extract information without employing computers. Thus biologists increasingly rely on computer scientists to come up with new solutions and on software to apply those solutions. Starting with Alan Turing’s seminal paper on morphogenesis1, through the development of efficient algorithms for sequence search2 and to computational whole genome assembly3, computer scientists have had a profound impact on biological research. In its simplest form, computerized image analysis overcomes the limitations and bias of the human observer. More importantly, computers have been essential for processing the images generated during high-throughput microscopy of large numbers and varieties of biological samples under a variety of conditions4–6. Additionally, imaging of single intact small organisms is now feasible with high resolution in 2d, 3d and across time7, resulting in massive image data sets that are well beyond the scale accessible by subjective observation8,9. The analysis of both types of image data requires computer vision techniques for reconstructing image volumes from numerous overlapping parts (registration)10,11; for searching biologically relevant features (segmentation)12; for following relevant objects across space and time (tracking)13,14; and for comparing specimens through mapping to anatomical or cellular digital atlases15,16.
Algorithms to achieve these tasks have been developed for natural images such as an urban street view, and must be adapted for biological image data. The mathematical formulations of the algorithms need to be transferred into an environment that is accessible to biologists. Such an environment must provide intuitive and comprehensive mechanisms to apply an algorithm to biological data and visualize the results in a user-friendly fashion. At the same time, the sheer size of many image data sets demands that algorithms run fast. These needs are tackled by the emerging interdisciplinary field of Bioimage Informatics that applies computer vision and other image processing approaches to biological research questions17,18.
There are several commercial (for example Imaris, Volocity, Amira) and Open Source (for instance ImageJ19, CellProfiler20, Vaa3D21, BioImageXD, Icy22, KNIME23 etc.) platforms for biological image analysis. Commercial platforms often focus on ease-of-use and broad coverage of image processing tasks, targeting relatively inexperienced users. Almost invariably the principle details of the available image processing algorithms are hidden, which is unsuitable for solving frontier research questions. Conversely, these details are completely transparent in Open Source platforms such as ImageJ whose long existence, wide adoption and extensible plugin architecture has made it a tool of choice for scientists from a broad range of disciplines. But ImageJ was developed primarily by biologists for biologists and its architecture does not follow modern software engineering principles. This makes the platform less attractive for computer scientists to use it to deliver new solutions to biologists.
To address this deficiency in ImageJ we started a new Open Source software project Fiji (Fiji Is Just ImageJ) that updates the underlying architecture of ImageJ and allows researchers to focus on the process of developing innovative, cutting edge solutions for biological image analysis. Fiji introduces powerful software libraries for rapid transfer from algorithmic discoveries to practical image analysis tools. Core algorithms available in Fiji can be exploited through a broad range of scripting languages that are familiar to bioinformaticians and further simplify the prototyping of new biomage solutions. Finally, Fiji provides a robust distribution system, which ensures that new algorithms reach its broad user base as soon as possible, initiating an iterative refinement based on communication between developers and users. In summary, Fiji is designed to serve as a software engineering ecosystem where computer science and biology research communities can collaborate to turn algorithms into usable programs for solving biological research questions.
How Fiji enhances ImageJ
ImageJ was created by Wayne Rasband at NIH19, and provides easy installation on arbitrary platforms and a simple user interface. Although it is primarily targeted at researchers with minimal computer skills, because ImageJ’s functionality can be easily extended with plugins (software components that can be separately installed to add functionality) ImageJ has also been attractive to researchers with training in software development. Over the years an impressive assortment of ImageJ plugins has been developed, touching on most areas of biological image analysis (http://imagej.nih.gov/ij/).
Fiji maintains compatibility with ImageJ and supplements it with additional core functionality. The Fiji project was created in the first place to support the installation and maintenance of one of the more complex ImageJ plugins, TrakEM2, which provides comprehensive solutions for management, registration, segmentation and annotation of large electron microscopy datasets (Cardona A, et al., unpublished results). TrakEM2 outgrew in its complexity and software infrastructure demands the facilities offered by classical ImageJ. Subsequently, several other advanced plugins (4D Viewer24, SPIM registration25, Trainable Segmentation and many more; see Supplementary Table 1) came to rely on Fiji’s modern software engineering practices such as a software versioning system, inclusion of third party libraries, automatic updates and straightforward compilation.
The combination of advanced image analysis solutions and the simplicity and familiarity of ImageJ’s user interface has attracted a substantial number of users to the platform. Fiji is effectively an Open Source distribution of ImageJ that includes a great variety of organized libraries, plugins relevant for biological research (Supplementary Table 1), scripting languages and extensive tutorials and documentation. The overproliferation and redundancy of plugins in ImageJ can make it difficult to identify solutions suitable for a particular biological problem. Fiji addresses this issue by offering a curated selection of plugins that are organized into categories within the plugin menu (Supplementary Fig. 1).
The Fiji project aims to provide useful functionality for a broad range of researchers, starting from programming agnostic biologists, through bioinformaticians and software engineers to professional computer vision researchers (Fig. 1a). Fiji enhances ImageJ’s ease of installation by bundling all required components into a self-contained package that will run on any computer platform. In addition, programatically skilled biologists can use its many familiar scripting languages to build custom image processing pipelines. For professional software engineers Fiji offers source code management, high-performance implementation of algorithms and simple worldwide deployment. Finally, Fiji could become useful to computer vision researchers as it bundles standard libraries, builds bridges to other platforms (for instance MATLAB through the Miji plugin) and provides facilities for rapid prototyping of data-type agnostic, generic algorithms.
Figure 1. Fiji as a high-powered distribution of ImageJ.
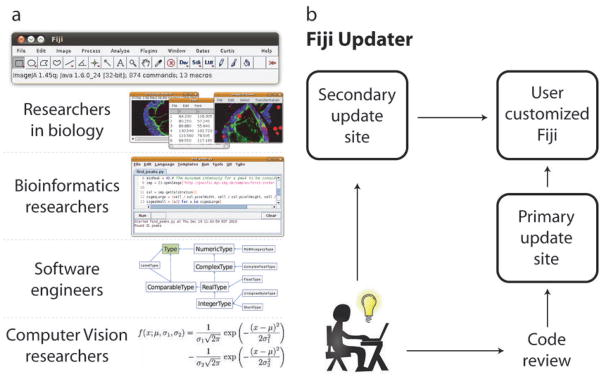
(a) The Fiji platform enables interaction with multidimentional image data in point-and-click interface identical to ImageJ. It simplifies transition from mathematical formulations coming from computer vision researchers to functional programs written by software engineers using version control and algorithmic libraries (schematic diagram of ImgLib design) and allows bioinformaticians to construct powerful image processing pipelines using scripting languages (Script Editor plugin screenshot). (b) The Fiji Updater provides a unique mechanism for releasing new algorithmic developments to the user community. Every time Fiji is launched it offers to download available updates from the main server in Dresden, Germany or from alternative update sites. New plugins approved by the Fiji developer community are instantaneously delivered to thousands of Fiji users worldwide or can be offered directly through user initiated update sites.
Fiji’s source code is hosted in a version controlled source code repository (Git) for people to download and tinker with and compiles in one step (http://fiji.sc/cgi-bin/gitweb.cgi). A special “contrib” branch is accessible for anybody to publish their plugin, library or script. Upon successful review, the code is included in the Fiji package system and immediately released to thousands of worldwide Fiji users via an integrated Fiji Updater (Fig. 1b). This mechanism provides a formalized way to release new image analysis solutions to biological researchers while also ensuring quality control of the contributed code and its long-term maintenance. Research groups can set up their own update sites and offer new plugins to the community by following simple instructions (http://fiji.sc/Adding_Update_Sites). Consequently, users can select which set of plugins they obtain from the various update sites, forming their own customized Fiji installation that suits their image analysis needs (Fig. 1b).
The Fiji project puts special emphasis on communication among users and developers. Fiji plugins are extensively documented via an active Wiki representing the definitive guide to all aspects of Fiji’s installation, maintenance, programming and usage (http://fiji.sc and Supplementary Fig. 2). Feedback and feature requests are posted to a dedicated mailing list. Fiji developers engage in active discussions online and the core Fiji developers meet regularly for coding sprees called ‘hackathons’ where they work collaboratively on the Fiji source code. These events typically result in dramatic improvement and extension of the Fiji code base, the so called ‘hackathon effect’ (Supplementary video 1).
In short, Fiji focuses on the process of making new solutions practical and bringing them immediately to the users. In the following section we discuss how scripting languages in Fiji empower biologists to do complex image analysis by themselves and how the Fiji library ImgLib simplifies transfer of abstract algorithms into usable programs.
Scripting and ImgLib for implementing algorithms
Analysis of biological image data typically requires applying multiple algorithms in sequence to many images. Scripting uses a series of simple programming commands (the “script”) to define a sequence of algorithmic operations and then apply them to an image collection.
The simple macro language that allows recording of commands and construction of basic programs made scripting popular among ImageJ users. Fiji builds on that functionality by supporting a broad range of scripting languages (Jython, Clojure, Javascript, JRuby, Beanshell), providing the most comprehensive selection of programming tools on both the Open Source and commercial bioimaging platforms. These languages are usable without knowledge of Java, and compared to the macro language offer more advanced programming syntax while retaining relative simplicity for the casual programmer.
In Fiji, individual scripting commands can be quickly tested by applying them interactively to opened images using the appropriate Interpreter plugin (for example Jython Interpreter). Beyond interactive execution of commands on current images Fiji also provides a Script Editor that enables writing, debugging, testing and running of arbitrarily complex scripts in all the above languages and additionally Java itself. With the Script Editor users can perform complex tasks with a few lines of code requiring minimal programming knowledge. It becomes relatively simple (16 lines of code) to load a multi-channel 3d image stack, identify all cells in one channel and visualize the results in the 4d Viewer plugin (Fig. 2a). Similarly users can implement a simple task such as swapping fluorescent channels in multidimensional images and apply it to a large collection of images in a directory using scripting commands for file manipulation (Supplementary Fig. 3). The details of the code are unimportant (extensive tutorials are available at http://fiji.sc/wiki/index.php/Category:Scripting) however it is crucial to note that all the scripting languages enable users to access Fiji’s extensive algorithmic libraries that implement advanced image analysis techniques in Java. Thus researchers do not need to become fluent in Java programming and can use their existing scripting skills to apply complex algorithms to their data. Scripts are seamlessly included in Fiji’s menu structure and can be publicly distributed through the Fiji Updater. These publishing mechanisms ensure validity and long-term viability, and are more convenient for biologists compared with closed scripting environments such as MATLAB.
Figure 2. Scripting and ImgLib.

(a) An example of a simple Jython script that achieves complex segmentation and 3d visualization drawing on the Fiji libraries steered by simple scripting commands. The task is to open a 3d RGB image (line 5) of a Drosophila first instar brain where cortex and neuropile glia are labeled in green by Nirvana-Gal4 and UASmcd8GFP, surface glial cells are labeled red with anti-repo antibody, and all nuclei are labeled blue with Sytox. The goal is to automatically count red surface glial cells using the Difference of Gaussian (DoG) detector (line 9) applying the constraints for cell size and labeling intensity (lines 3 and 4). These constraints are expressed as DoG sigma parameters (lines 7 and 8) by extracting image dimensions from metadata (line 6). The cell count is printed in the dialog box (line 10) and cells are subsequently displayed in the 4d viewer as red spheres of fixed diameter, overlaid with orthogonal view of the raw 3d images (lines 13–16). (b–j) Algorithms implemented using generic ImgLib constructs operate on images regardless of dimensionality. The figure shows the output of two ImgLib algorithms: Maximally Stable Extremal Regions (MSER) and again DoG. The 3d input image is a confocal stack of a C. elegans worm expressing nuclear marker. Scale bar 10 μm. (h). A slice from the stack is used as the 2d input image. Scale bar 10 μm. (e). A line segment from the slice is used as the 1d input image (b). The results of the DoG algorithm for 1d, 2d, and 3d are visualized in (c), (f), and (i). The results of the MSER algorithm for 1d, 2d, and 3d are shown in (d), (g), and (j). The algorithms are run on 1d (c,d), 2d (f,g), and 3d (i,j) input without changing a single line of code (see Supplementary Fig. 4). The nested MSER regions representing competing segmentation hypotheses for the nuclei are color coded (green, red, blue and magenta).
A key feature of the Fiji project is ImgLib (Fig. 2b–j)26, a library for type-, dimension-, and storage-independent representation of image data that enables writing generic algorithms in a high-performance manner. In ImgLib, image processing algorithms are implemented just once and can be applied without change to most kinds of images. In other words, ImgLib enables users to apply a complex image processing algorithm to any image regardless of how many dimensions the image has (1d, 2d, 3d, 2d+t, 3d+t, etc.), what the underlying data type is (8-bit, 16-bit, etc.) or how the image is stored (in memory, paged to disc, distributed over the net).
We demonstrate the dimensionality independence of ImgLib when applying segmentation algorithms to confocal scans of C. elegans larvae expressing fluorescent markers of cell nuclei (Fig. 2b–j). Difference of Gaussian (DoG) and Maximally Stable Extremal Regions (MSER)27 are two examples of blob detection algorithms inspired by computer vision literature that are suitable for detecting cells in biological images. The DoG is computed by subtracting two consecutive Gaussian convolutions of the image. Intensity maxima and minima in the DoG represent bright and dark blob detections, respectively. The MSER tree provides a nested hierarchy of blob segmentation hypotheses, which is particularly relevant in densely packed cellular field. Both algorithms are applicable for processing of 1d curves (Fig. 2b–d), 2d image slices (Fig. 2e–g) and 3d image volumes (Fig. 2h–j). With ImgLib a single, simple piece of computer code (Supplementary Fig. 4) is capable of running the algorithms on images of arbitrary dimensions without any modification (DoG Fig. 2c,f,i and MSER Fig. 2d,g,j for 1d, 2d, 3d respectively).
ImgLib is and should be invisible to casual users. It is an “under the hood” advanced software engineering concept that makes programming easier. ImgLib empowers users and computer vision researchers with the abstraction necessary to seamlessly translate mathematical description of an advanced idea or algorithm into concise code that will run efficiently on large bioimages. Similar libraries that separate the algorithm essence from the actual implementation are available on other platforms such as ITK/VTK28 and VIGRA29.
Both examples of scripts mentioned above use ImgLib to do the processing steered by simple scripting commands. Equivalent programs in ImageJ’s macro language or Java would be much more complex or not possible. This is due to the combination of scripting language simplicity and the dimension-, type- and storage-strategy independence of ImgLib. In the next section we showcase how the synergy of Fiji, ImgLib and other libraries results in transformation of abstract algorithms into usable biological image analysis applications.
Synergy as a design principle
The growing collection of modern image analysis algorithms in Fiji is a product of interactions between Fiji projects (Supplementary Fig. 5) that use computer vision algorithms to support ongoing biological research. We briefly outline three advanced image processing pipelines for stitching multidimensional images from tiles (Fig. 3a–e), reconstruction of massive serial section transmission electron microscopy (ssTEM) mosaics (Fig. 3f–j) and reconstruction of long-term, time-lapse, multi-view recordings of development with Selective Plane Illumination Microscopy (SPIM) (Fig. 3k–o). Many more examples of projects and synergies can be found on the Fiji Wiki (http://fiji.sc) and we invite programmatically skilled biologists, professional developers with interest in biology and computer vision researchers with a novel algorithm looking for applications to join the Fiji movement and contribute new approaches, ideas and plugins.
Figure 3. Fiji projects.

(a–e) Stitching plugin for globally optimal registration of tiled 3d confocal images. The source data of Drosophila first instar larval nervous system (a) were registered using phase correlation with global optimization (b) and visualized in Fiji 4d Viewer (c). Four labeled neurons (colour coded in 4d Viewer (d)) were segmented using a manual segmentation plugin (Segmentation Editor) and their volumes were measured (e). (f–j) Globally optimal reconstruction of large ssTEM mosaics using TrakEM2 plugin. Schematic view of ssTEM mosaic, each square is an individual image tile and the independent sections are color coded (f). (g) Screenshot of a video visualizing the progress of global optimization for a single section. SIFT features are in cyan and the residual error signifying displacement of corresponding SIFT features at the current iteration is depicted by red lines. (h) Dual color overlay of two registered consecutive sections showing the entire hemisphere of the larval brain. Scale bar 10 μm. (i) Axonal profiles within a small part of a single section in the ssTEM dataset were manually segmented using TrakEM2. Each profile is labeled with a different color. Scale bar 0.5 μm. (j) Relationship between numbers of pre-synaptic partners and post-synaptic sites extracted manually with TrakEM2 from a micro-cube of the registered data. (k–o) Plugin suite for processing of multi-view SPIM data. (k) Schematic representation of multi-view SPIM imaging showing the 3d stacks of the same specimen acquired from different angles. (l) Progress of the global optimization of multi-view SPIM acquisition of Drosophila embryo. Corresponding geometric bead descriptors are colored according to their residual displacement at the current iteration of the optimizer. (m) The resulting reconstructed embryo at the 12th (top) and 13th (bottom) nuclear division cycle shown as 3d rendering in Fiji’s 3d viewer. Scale bar 50 μm. (n) Results of Difference of Gaussian segmentation of the nuclei marked with His-YFP; same stages as in (m). Each nucleus is marked with a different color. (o) Quantification of the nuclear counts at the 12th and 13th nuclear division in the embryo shown in (m) and (n).
Stitching of Confocal Stacks
Large biological samples are frequently imaged with high-resolution microscopy techniques as sets of overlapping images or image stacks (Fig. 3a). The reconstruction of the entire specimen involves pairwise registration of the overlapping images, with a strategy to minimize the displacement of the corresponding image content. The Stitching plugin uses phase correlation to compute the optimal translation among pairs of overlapping 2d or 3d image tiles (Fig. 3b). Subsequently, the plugin uses a global optimization procedure that evenly spreads the error of the registration steps, avoiding the introduction of artifactual deformations and delivering an undistorted representation of the specimen30. The reconstructed volume is visualized in the 4d Viewer24 (Fig. 3c), structures are segmented using manual segmentation tools such as Segmentation Editor (Fig. 3d) allowing measurements of lengths, areas, volumes of segmented elements (Fig. 3e) and quantification of their overlap by Colocalization Analysis.
ssTEM Reconstruction
ssTEM is experiencing a renaissance as a tool of choice for mapping brain micro-circuitry31,32. Large sections are typically imaged as sets of overlapping image tiles (Fig. 3f) and datasets consisting of hundreds or even thousands of such sections are becoming available. Fiji users can first apply lens deformation model to pre-process the ssTEM images before registration using the Distortion Correction plugin33. Relatively simple ssTEM datasets can be reconstructed using standalone plugins such as Register Virtual Stack Slices, Elastic Alignment and Montage (Saalfeld, S., et al. unpublished data) or bUnwarpJ34. Arbitrarily large ssTEM datasets consisting of tens of thousands images are best assembled using the integrative TrakEM2 plugin for registration, segmentation and analysis of electron microscopy data. One of the many avenues for reconstructing ssTEM data in TrakEM2 applies scale invariant feature transform (SIFT)35 to detect corresponding image content and then solves the globally optimal configuration of massive tile systems11 (Fig. 3g,h and Supplementary video 2 and 3).
Reconstructed volumes can be segmented into a series of profiles following individual neurons either using the powerful manual segmentation tools (Fig. 3i) or automatically with Active Contours36 and Level Sets37 algorithms integrated into TrakEM2. Alternatively, in order to extract the neuronal tracks from electron microscopy data, users can employ the Trainable Segmentation plugin that leverages the Waikato Environment for Knowledge Analysis (WEKA) library for machine learning38 to extract statistical properties from user-provided examples and apply them to classify the rest of the image or other similar images39. The analysis of the reconstructed ssTEM scans of parts of the nervous system requires quantitative comparisons across different samples40 such as counting individual synaptic connections between segmented neurons (Fig. 3j). Ultimately high-resolution TEM scans need to be registered to low-resolution light microscopy data for correlative analysis and tools such as the Simple Neurite Tracer plugin41, the VIB Protocol and Interactive Image Transformations using various Landmark Correpondences are ready to be adapted for this purpose. The flexibility and integrative nature of TrakEM2 with its unique ability to reconstruct very large electron microscopy datasets on commodity hardware cannot be found on any other bioimaging platform.
Processing of Multi-view SPIM Data
Modern developmental biologists want to image developing embryos in toto with resolution sufficient to distinguish and track individual labeled cells throughout development. SPIM is an emerging technique to achieve these goals and Fiji provides a comprehensive pipeline for acquisition, reconstruction, segmentation, tracking and analysis of terabyte-size long-term, time-lapse, multi-view SPIM datasets.
For image acquisition with SPIM Fiji synergizes with another large ImageJ offshoot project Micro-Manager42 to provide Open Source steering software for custom OpenSPIM microscopes (Tomancak, P. unpublished results) to deliver 3d stacks of the same specimen imaged from different angles directly into the Fiji environment (Fig. 3k). Following acquisition, the multi-view data can be reconstructed into a continuous 3d volume using fluorescent beads in the rigid agarose medium as fiduciary markers (Bead-Based SPIM Registration plugin25). The constellations of fluorescent beads form local geometric descriptors that are matched across views and used to create a 3d image by globally optimizing local affine transformation models (Fig. 3l and Supplementary video 4). The reconstructed images can be optionally fused into a single output volume (Fig. 3m) using the Multi-view Fusion plugin. Interactive Difference of Gaussian segmentation that is part of the Bead-Based SPIM Registration plugin can be used to segment and count the nuclei in the reconstructed specimen (Fig. 3n). The registration process, the segmentation results (Fig. 3o) and the tracking of nuclei across time (Supplementary video 5) can be visualized by programatically steering the Fiji 4d Viewer24. All this is happening on top of truly massive long-term, time-lapse, multi-view SPIM datasets in the terabyte range and makes Fiji currently the only widely available solution for dealing with this type of data.
Fiji plugin integration
The various Fiji plugins interact on two principal levels. Trivially, the output of one plugin can serve as input for another since they all run in a common platform. More elaborate integration is possible because most Fiji methods are increasingly implemented as software libraries using ImgLib for data representation. For instance the confocal stack, ssTEM mosaic and multi-view SPIM registration plugins are using a common global optimizer capable of accepting different representations of image content (phase correlation, SIFT and local geometric descriptors) and minimizing the displacement of correspondences using the transformation model appropriate for the given imaging modality (translation only, rigid and affine). The integration of standalone plugins into tightly cooperating suites such as TrakEM2 is an ongoing trend in all Fiji projects. Casual users benefit from this level of integration by being able to seamlessly combine solutions from diverse areas of bioimage analysis and create efficient analysis pipelines to interpret their images.
Conclusion
ImageJ has for a long time been the tool of choice for biologists who need basic as well as advanced image analysis. Over the lifetime of ImageJ, a revolution in microscopy brought about an order of magnitude increase in typical image sizes and two or more orders of magnitude increase in throughput. The Fiji project provides biologists with powerful tools for the generation of advanced image processing pipelines, via scripting languages and feature-rich libraries for processing large quantities of large images, while building upon the simplicity of ImageJ.
Over the past two years the Fiji project has achieved remarkable international recognition (Fig. 4a). Analysis of Fiji updater records reveals that Fiji is used in every major academic research center throughout the world (Fig. 4b). The number of Fiji downloads and the access to the Fiji Wiki (Fig. 4a and Supplementary Fig. 6) has grown steadily over the past years with currently twenty thousand estimated users. Several prominent scientific institutions use Fiji as an Open Source solution for image processing needs of their researchers. As the community grows, new projects and new talented researchers join, expanding the ability of biologists to cope with image data.
Figure 4. Fiji usage.
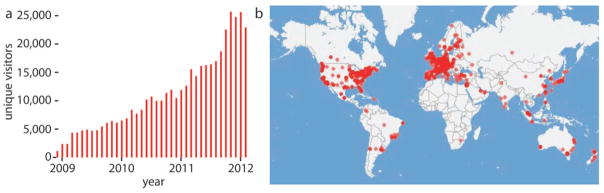
(a) A chart showing the number of unique visitors to Fiji wiki per month over the last three years. (b) World map overlaid with geolocations of computers that updated Fiji over a period of one week.
Many image analysis solutions available for Fiji were developed uniquely under Fiji and do not have a comparable alternative in other platforms. Some of the standard image processing or visualization tasks implemented in Fiji plugins are addressed, sometimes more comprehensively, by other platforms (for instance 3d visualization (Vaa3D21) or Trainable Segmentation (CellProfiler20)). However in Fiji these tools are embedded in a large ecosystem of plugins that can interact through a common infrastructure, for example by assembling diverse image processing steps into complex pipeline with the use of scripting languages.
The reliance on Java invites the question whether Fiji is able to deal efficiently with computing intensive tasks on top of large biological image data. It is true that careful algorithm implementations in the C/C++ programming language will sometimes execute faster than the equivalent Java program. In other cases, Java outperforms C/C++ (see for example http://fiji.sc/Why_Java). It is equally true that specialized implementations will typically outperform generic ones. Many of the flagship Fiji projects described above deal with massive terabyte-sized datasets and yet deliver interactive performance on standard hardware. Important advantages of Java are the ease of installation on a variety of platforms, portability, stability and backwards compatibility that are harder to achieve with packages built with C. Thus, Fiji strikes a balance between usability, performance and flexibility and as such it occupies a unique position in the landscape of biological image analysis where it synergistically complements other tools as well as provides unique and novel capabilities.
Fiji is open not only with respect to its source code but also in its relationship with other platforms. It aims to communicate and integrate with other bioimage analysis software that outperform Fiji on a particular task. An example of such integration is the interface to MATLAB and ITK, the availability of ImgLib for adoption in any Java based platform (for instance KNIME and ImageJ2) and the participation of developers from diverse platforms in Fiji-centered hackathons (the last two hackathons were attended by representatives of ImageJ2, CellProfiler, Micro-Manager, ITK, KNIME, Icy, BioImageXD and OMERO). Most importantly, in the future, Fiji will reconnect to its roots by becoming the outer, application-oriented layer of the ImageJ2 (http://developer.imagej.net/) project which aims at redesigning the very core of ImageJ in order to meet the demands of next generation biology research. Fiji will focus on developing state-of-the-art plugins inspired by the needs of biological research projects while ImageJ2 will put emphasis on creating the infrastructure necessary to prototype, realize, distribute and deploy the novel algorithmic solutions in a sustainable manner. The two projects share a common starting point, the classical ImageJ enviroment and are committed to work together exchanging code and coordinating design decisions. The process of creating a powerful synergy between Fiji and ImageJ2 has already begun by incorporating the flagship Fiji plugins and libraries (such as the Fiji Updater and ImgLib) into ImageJ2 (Supplementary Fig. 7) forming a basis of a long-term partnership that will combine professional software engineering of the ImageJ2 core and biological application driven plugin development in Fiji.
Supplementary Material
Supplementary Figure 1. Organization of Fiji plugin folder
Supplementary Figure 2. Fiji Wiki
Supplementary Figure 3. Scripting in Fiji
Supplementary Figure 4. ImgLib code examples
Supplementary Figure 5. Fiji Projects
Supplementary Figure 6. An estimate of Fiji users based on wiki traffic
Supplementary Figure 7. The future of Fiji as application layer of ImageJ2
Supplementary Table 1. Overview of Fiji plugins
Supplementary Video 1. Visualization of Fiji development. The video produced using gource tool in Git visualizes the changes to Fiji source code repository from March 15th 2009 until May 16th 2009. The class hierarchy is visualized as a dynamic tree, the developers are flying pawns that extend rays to classes which they newly created or into which they introduced changes. Between 23rd of March to the 3rd of April there was a Fiji hackathon in Dresden marked by increased developer activity that carries over the period after the hackathon ended – the so called “hackathon effect”.
Supplementary Video 2. Visualization of SIFT mediated stitching of large ssTEM mosaics. The ventral nerve cord of Drosophila first instar larva was sectioned and imaged in electron microscope as a series of overlapping image tiles. The video visualizes the process of reconstruction of such large section series on seven exemplary sections. The corresponding SIFT features that connect images within section and across section are shown as green dots, the residual error of their displacement at a given iteration of the global optimizer is shown as cyan line (iteration number and minimal, average and maximal error are shown in lower left corner). The global optimization proceeds section by section and at each step distributes the registration error equally across the increasing set of tiles. In order to emphasize the visualization effect all tiles within section are initially placed at the same location discarding their known configuration within section.
Supplementary Video 3. Walk through reconstructed large ssTEM mosaic of third instar larval ventral nerve cord. The movie shows in each frame a single section from the rigidly aligned TEM section series, imaged as mosaic of overlapping tiles, of the D. melanogaster first instar larval ventral nerve cord at four different zoom levels (A–D where B shows the close up of the blue rectangled area in A etc.). The lateral resolution of the raw data is 4 nm/pixel. The number in the upper right corner indicates the section index.
Supplementary Video 4. Visualization of bead-based registration of multi-view microscopy scan of Drosophila embryo. Drosophila embryo expressing His-YFP marker has been imaged in a spinning disc confocal microscope from 18 different angles improvising rotation using custom made sample chamber. The video visualizes the global optimization that is using local geometric bead descriptor matches to recover the shape of the embryo specimen. The bead descriptors (representing constellations of sub-resolution fluorescent beads added to the rigid agarose medium in which the embryo was mounted) are colored according to their displacement at each iteration of the optimizer (red - maximum displacement, green – minimum displacement). The nuclei of the embryo specimen are shown in grey. The displacement at each iteration averaged across all descriptors is shown in lover left corner.
Supplementary Video 5. Segmentation and tracking of nuclei in Drosophila embryo. Cellular blastoderm stage Drosophila embryo expressing His-YFP marker in all cells has been imaged from five angles using SPIM throughout gastrulation. The video shows a result of segmentation and tracking algorithm that follows the movements of cells through the gastrulation process. The nuclei are colored according to the angle in which they were detected.
Acknowledgments
We thank W. Rasband for developing ImageJ and helping thousands of scientists around the world on a daily basis. We would like to thank the scientists who contributed to the Fiji movement by financing and organizing the hackathons. Namely G.M. Rubin for hackathons at Janelia Farm, I. Baines for hackathons at MPI-CBG, R. Douglas for a hackathon at INI, F. Peri and K. Miura for the EMBL hackathon, and INCF for Fiji Image Processing School. We thank W. Pereanu (UCLA) for the confocal image of the larval fly brain and M. Sarov (MPI-CBG) for the confocal scan of C. elegans larva. We thank all the numerous scientists who released their code under Open Source licenses and made the Fiji project possible. J. S. and P. T. were funded by Human Frontier Science Program Young Investigator Grant RGY0083. P.T. was additionally supported by The European Research Council Community’s Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement no. 260746.
References
- 1.Turing AM. The chemical basis of morphogenesis. 1953. Bull Math Biol. 1990;52:153–197. doi: 10.1007/BF02459572. discussion 119–152. [DOI] [PubMed] [Google Scholar]
- 2.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 3.Myers EW, et al. A whole-genome assembly of Drosophila. Science. 2000;287:2196–2204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- 4.Neumann B, et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature. 2010;464:721–727. doi: 10.1038/nature08869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collinet C, et al. Systems survey of endocytosis by multiparametric image analysis. Nature. 2010;464:243–249. doi: 10.1038/nature08779. [DOI] [PubMed] [Google Scholar]
- 6.Shariff A, Kangas J, Coelho LP, Quinn S, Murphy RF. Automated image analysis for high-content screening and analysis. J Biomol Screen. 2010;15:726–734. doi: 10.1177/1087057110370894. [DOI] [PubMed] [Google Scholar]
- 7.Megason SG, Fraser SE. Imaging in systems biology. Cell. 2007;130:784–795. doi: 10.1016/j.cell.2007.08.031. [DOI] [PubMed] [Google Scholar]
- 8.Keller PJ, Schmidt AD, Wittbrodt J, Stelzer EH. Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science. 2008;322:1065–1069. doi: 10.1126/science.1162493. [DOI] [PubMed] [Google Scholar]
- 9.Fowlkes CC, et al. A quantitative spatiotemporal atlas of gene expression in the Drosophila blastoderm. Cell. 2008;133:364–374. doi: 10.1016/j.cell.2008.01.053. [DOI] [PubMed] [Google Scholar]
- 10.Anderson JR, et al. A computational framework for ultrastructural mapping of neural circuitry. PLoS Biol. 2009;7:e1000074. doi: 10.1371/journal.pbio.1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saalfeld S, Cardona A, Hartenstein V, Tomancak P. As-rigid-as-possible mosaicking and serial section registration of large ssTEM datasets. Bioinformatics. 2010;26:i57–63. doi: 10.1093/bioinformatics/btq219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murray JI, et al. Automated analysis of embryonic gene expression with cellular resolution in C. elegans. Nat Methods. 2008;5:703–709. doi: 10.1038/nmeth.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandez R, et al. Imaging plant growth in 4D: robust tissue reconstruction and lineaging at cell resolution. Nat Methods. 2010;7:547–553. doi: 10.1038/nmeth.1472. [DOI] [PubMed] [Google Scholar]
- 14.Bao Z, et al. Automated cell lineage tracing in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2006;103:2707–2712. doi: 10.1073/pnas.0511111103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Long F, Peng H, Liu X, Kim SK, Myers E. A 3D digital atlas of C. elegans and its application to single-cell analyses. Nat Methods. 2009;6:667–672. doi: 10.1038/nmeth.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng H, et al. BrainAligner: 3D registration atlases of Drosophila brains. Nat Methods. 2011;8:493–500. doi: 10.1038/nmeth.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swedlow JR, Eliceiri KW. Open source bioimage informatics for cell biology. Trends Cell Biol. 2009;19:656–660. doi: 10.1016/j.tcb.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng H. Bioimage informatics: a new area of engineering biology. Bioinformatics. 2008;24:1827–1836. doi: 10.1093/bioinformatics/btn346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rasband WS. ImageJ. U. S. National Institutes of Health; Bethesda, Maryland, USA: 1997–2008. http://rsb.info.nih.gov/ij/ [Google Scholar]
- 20.Carpenter AE, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng H, Ruan Z, Long F, Simpson JH, Myers EW. V3D enables real-time 3D visualization and quantitative analysis of large-scale biological image data sets. Nat Biotechnol. 2010;28:348–353. doi: 10.1038/nbt.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Chaumont F, Dallongeville S, Olivo-Marin J-C. ICY: A new open-source community image processing software. IEEE Int Symp on Biomedical Imaging. 2011:234–237. [Google Scholar]
- 23.Berthold MR, et al. Studies in Classification, Data Analysis, and Knowledge Organization. Springer; 2007. KNIME: The Konstanz Information Miner. (GfKL 2007) [Google Scholar]
- 24.Schmid B, Schindelin J, Cardona A, Longair M, Heisenberg M. A high-level 3D visualization API for Java and ImageJ. BMC Bioinformatics. 2010;11:274. doi: 10.1186/1471-2105-11-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Preibisch S, Saalfeld S, Schindelin J, Tomancak P. Software for bead-based registration of selective plane illumination microscopy data. Nat Methods. 2010;7:418–419. doi: 10.1038/nmeth0610-418. [DOI] [PubMed] [Google Scholar]
- 26.Preibisch S, Tomancak P, Saalfeld S. Proceedings of ImageJ User and Developer Conference. 2010;1:72–76. [Google Scholar]
- 27.Matas J, Chum O, Urban M, Pajdla T. Robust Wide Baseline Stereo from Maximally Stable Extremal Regions. Image and Vision Computing. 2004;22(10):761–767. [Google Scholar]
- 28.Ibanez L, Schroeder W, Ng L, Cates J. The ITK Software Guide. Kitware Inc; 2005. [Google Scholar]
- 29.Köthe U. Reusable Software in Computer Vision. In: Jähne B, Haussecker H, Geissler P, editors. Handbook of Computer Vision and Applications. Vol. 3. San Diego: Academic Press; 1999. pp. 103–132. [Google Scholar]
- 30.Preibisch S, Saalfeld S, Tomancak P. Globally Optimal Stitching of Tiled 3D Microscopic Image Acquisitions. Bioinformatics. 2009;25:1463–1465. doi: 10.1093/bioinformatics/btp184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cardona A, et al. An integrated micro- and macroarchitectural analysis of the Drosophila brain by computer-assisted serial section electron microscopy. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bock DD, et al. Network anatomy and in vivo physiology of visual cortical neurons. Nature. 471:177–182. doi: 10.1038/nature09802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaynig V, Fischer B, Müller E, Buhmann JM. Fully automatic stitching and distortion correction of transmission electron microscope images. Journal of Structural Biology. 2010;171:163–173. doi: 10.1016/j.jsb.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 34.Arganda-Carreras I, Sorzano COS, Marabini R, Carazo JM, Ortizde Solorzano C, Kybic J. Lecture Notes in Computer Science. Vol. 4241. Springer; Berlin/Heidelberg: 2006. Consistent and Elastic Registration of Histological Sections using Vector-Spline Regularization in Lecture Notes in Computer Science ser; pp. 85–95. [Google Scholar]
- 35.Lowe DG. Distinctive Image Features from Scale-Invariant Keypoints. International Journal of Computer Vision. 2004;60:91–110. [Google Scholar]
- 36.Sethian JA. Level Set Methods and Fast Marching Methods: Evolving Interfaces in Computational Geometry, Fluid Mechanics, Computer Vision, and Materials Science. Cambridge University Press; 1999. [Google Scholar]
- 37.Kass M, Witkin A, Terzopoulos D. Snakes: Active contour models. International Journal of Computer Vision. 1988;1:321–331. [Google Scholar]
- 38.Hall M, et al. The WEKA Data Mining Software: An Update. SIGKDD Explorations. 2009;11 [Google Scholar]
- 39.Kaynig V, Fuchs TJ, Buhmann JM. Geometrical consistent 3D tracing of neuronal processes in ssTEM data. Med Image Comput Comput Assist Interv. 2010;13:209–216. doi: 10.1007/978-3-642-15745-5_26. [DOI] [PubMed] [Google Scholar]
- 40.Cardona A, et al. Identifying neuronal lineages of Drosophila by sequence analysis of axon tracts. J Neurosci. 2010;30:7538–7553. doi: 10.1523/JNEUROSCI.0186-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Longair MH, Baker DA, Armstrong JD. Simple Neurite Tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics. 27:2453–2454. doi: 10.1093/bioinformatics/btr390. [DOI] [PubMed] [Google Scholar]
- 42.Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N. Current Protocols in Molecular Biology. John Wiley & Sons, Inc; 2010. Computer Control of Microscopes Using μManager. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Organization of Fiji plugin folder
Supplementary Figure 2. Fiji Wiki
Supplementary Figure 3. Scripting in Fiji
Supplementary Figure 4. ImgLib code examples
Supplementary Figure 5. Fiji Projects
Supplementary Figure 6. An estimate of Fiji users based on wiki traffic
Supplementary Figure 7. The future of Fiji as application layer of ImageJ2
Supplementary Table 1. Overview of Fiji plugins
Supplementary Video 1. Visualization of Fiji development. The video produced using gource tool in Git visualizes the changes to Fiji source code repository from March 15th 2009 until May 16th 2009. The class hierarchy is visualized as a dynamic tree, the developers are flying pawns that extend rays to classes which they newly created or into which they introduced changes. Between 23rd of March to the 3rd of April there was a Fiji hackathon in Dresden marked by increased developer activity that carries over the period after the hackathon ended – the so called “hackathon effect”.
Supplementary Video 2. Visualization of SIFT mediated stitching of large ssTEM mosaics. The ventral nerve cord of Drosophila first instar larva was sectioned and imaged in electron microscope as a series of overlapping image tiles. The video visualizes the process of reconstruction of such large section series on seven exemplary sections. The corresponding SIFT features that connect images within section and across section are shown as green dots, the residual error of their displacement at a given iteration of the global optimizer is shown as cyan line (iteration number and minimal, average and maximal error are shown in lower left corner). The global optimization proceeds section by section and at each step distributes the registration error equally across the increasing set of tiles. In order to emphasize the visualization effect all tiles within section are initially placed at the same location discarding their known configuration within section.
Supplementary Video 3. Walk through reconstructed large ssTEM mosaic of third instar larval ventral nerve cord. The movie shows in each frame a single section from the rigidly aligned TEM section series, imaged as mosaic of overlapping tiles, of the D. melanogaster first instar larval ventral nerve cord at four different zoom levels (A–D where B shows the close up of the blue rectangled area in A etc.). The lateral resolution of the raw data is 4 nm/pixel. The number in the upper right corner indicates the section index.
Supplementary Video 4. Visualization of bead-based registration of multi-view microscopy scan of Drosophila embryo. Drosophila embryo expressing His-YFP marker has been imaged in a spinning disc confocal microscope from 18 different angles improvising rotation using custom made sample chamber. The video visualizes the global optimization that is using local geometric bead descriptor matches to recover the shape of the embryo specimen. The bead descriptors (representing constellations of sub-resolution fluorescent beads added to the rigid agarose medium in which the embryo was mounted) are colored according to their displacement at each iteration of the optimizer (red - maximum displacement, green – minimum displacement). The nuclei of the embryo specimen are shown in grey. The displacement at each iteration averaged across all descriptors is shown in lover left corner.
Supplementary Video 5. Segmentation and tracking of nuclei in Drosophila embryo. Cellular blastoderm stage Drosophila embryo expressing His-YFP marker in all cells has been imaged from five angles using SPIM throughout gastrulation. The video shows a result of segmentation and tracking algorithm that follows the movements of cells through the gastrulation process. The nuclei are colored according to the angle in which they were detected.