

Abstract
Human 8-oxoguanine DNA glycosylase (OGG1) excises the mutagenic oxidative DNA lesion 8-oxo-7,8-dihydroguanine (8-oxoG) from DNA. Kinetic characterization of OGG1 is undertaken to measure the rates of 8-oxoG excision and product release. When the OGG1 concentration is lower than substrate DNA, time courses of product formation are biphasic; a rapid exponential phase (i.e. burst) of product formation is followed by a linear steady-state phase. The initial burst of product formation corresponds to the concentration of enzyme properly engaged on the substrate, and the burst amplitude depends on the concentration of enzyme. The first-order rate constant of the burst corresponds to the intrinsic rate of 8-oxoG excision and the slower steady-state rate measures the rate of product release (product DNA dissociation rate constant, koff). Here, we describe steady-state, pre-steady-state, and single-turnover approaches to isolate and measure specific steps during OGG1 catalytic cycling. A fluorescent labeled lesion-containing oligonucleotide and purified OGG1 are used to facilitate precise kinetic measurements. Since low enzyme concentrations are used to make steady-state measurements, manual mixing of reagents and quenching of the reaction can be performed to ascertain the steady-state rate (koff). Additionally, extrapolation of the steady-state rate to a point on the ordinate at zero time indicates that a burst of product formation occurred during the first turnover (i.e. y-intercept is positive). The first-order rate constant of the exponential burst phase can be measured using a rapid mixing and quenching technique that examines the amount of product formed at short time intervals (<1 sec) before the steady-state phase and corresponds to the rate of 8-oxoG excision (i.e. chemistry). The chemical step can also be measured using a single-turnover approach where catalytic cycling is prevented by saturating substrate DNA with enzyme (E>S). These approaches can measure elementary rate constants that influence the efficiency of removal of a DNA lesion.
Keywords: Chemistry, Issue 78, Biochemistry, Genetics, Molecular Biology, Microbiology, Structural Biology, Chemical Biology, Eukaryota, Amino Acids, Peptides, and Proteins, Nucleic Acids, Nucleotides, and Nucleosides, Enzymes and Coenzymes, Life Sciences (General), enzymology, rapid quench-flow, active site titration, steady-state, pre-steady-state, single-turnover, kinetics, base excision repair, DNA glycosylase, 8-oxo-7, 8-dihydroguanine, 8-oxoG, sequencing
Introduction
An aerobic environment hastens genomic instability. A major promutagenic DNA lesion resulting from oxidative stress is 7,8-dihydro-8-oxoguanine (8-oxoG). This is due to the ambiguous coding potential of 8-oxoG. Human 8-oxoguanine DNA glycosylase (OGG1) is responsible for initiating base excision repair of 8-oxoG. The glycosylase activity of OGG1 excises the 8-oxoG base resulting in product DNA with an apurinic site (AP-site). A weak lyase activity of OGG1 can incise the AP-site in some instances.
Kinetic characterization of DNA glycosylases generally finds that they exhibit biphasic time courses. After an initial fast phase of product formation (i.e. burst), a linear steady-state phase is observed1-3. This behavior is indicative of a step following chemistry (i.e. conformational change or product release) being rate-limiting during the linear portion of the time course, whereas the burst phase, often referred to as the transient phase, corresponds to product formation at the enzyme active site during the first cycle of the reaction. When product release is rate limiting during the steady-state phase, activity measurements provide a qualitative measure of product DNA binding affinity, but do not provide kinetic information concerning events at the enzyme active site (i.e. chemistry). Accordingly, methods to isolate and measure the exponential pre-steady-state burst phase are needed to probe events during the first enzymatic turnover at the enzyme's active site4.
There are three standard kinetic approaches to characterize the catalytic behavior of OGG1, (1) steady-state, (2) pre-steady-state, and (3) single-turnover. These approaches differ from one another by the concentration of enzyme in the reaction mixture and the enzyme to substrate ratio utilized in each approach. In a typical steady-state approach, sometimes referred to as multiple turnover kinetics, low concentrations of enzyme are used to follow product formation. The substrate concentration greatly exceeds the enzyme concentration so that multiple enzymatic turnovers do not significantly affect substrate concentration. In this situation, time courses should be linear and it is often difficult to discern whether a burst occurred during the first turnover due to the low enzyme concentration used in this approach; note that burst amplitude is equivalent to the enzyme concentration. This can be overcome by using a higher enzyme concentration and extrapolating the linear time course to zero time to detect whether the first enzymatic turnover occurred quickly. The intercept on the ordinate (y-axis) should be proportional to the enzyme concentration and provides a measure of the enzyme actively engaged with substrate. Although this approach can in principle provide evidence for the existence of a burst phase, a different approach is required to measure the kinetics of the burst phase. In many instances the burst phase is too fast to measure by manual mixing and quenching techniques. In this situation, pre-steady-state and single-turnover kinetic (i.e. transient kinetic) approaches often require a rapid-mixing and quenching instrument to follow early time points of a reaction5. In a pre-steady-state approach high concentrations of enzyme are used so that a significant amount of product is formed during the first turnover. Since multiple turnovers are followed to observe catalytic cycling (i.e. the linear phase that follows the burst), substrate concentration is greater than the enzyme concentration ([enzyme] < [substrate]). To isolate events at the active site of the enzyme without catalytic cycling, single-turnover conditions are utilized. In this case, substrate is saturated with enzyme (E>>S) so that all of the substrate will participate in the 'single turnover' and will typically exhibit a single-exponential time course.
As noted above for enzymes that exhibit a burst phase, product release (koff) often limits the rate of the steady-state phase of the time course. The rate of product release (vss, conc./time) can be determined from the slope of the linear steady-state phase. The active enzyme concentration (E) is needed to convert the rate of product release to an intrinsic rate constant where koff = vss/[E]. Importantly, the active enzyme concentration is typically lower than the measured protein concentration due to impurities, inactive enzyme, enzyme non-productively bound to substrate, and the method used to determine protein concentration. The active enzyme concentration can be determined from the burst amplitude when product release is slow. Thus, extrapolating a steady-state time course to zero time provides an estimate of active enzyme needed to calculate koff (product release) from the observed steady-state rate.
To measure the kinetics of the burst, a pre-steady-state approach is necessary to follow product formation during the first turnover that occurs prior to the linear steady-state phase. The burst kinetics follows the formation of enzyme-product intermediate. Once the reaction is initiated by mixing enzyme with substrate, the amount of the enzyme-product rapidly increases until the reaction reaches a steady state phase. If catalysis is much more rapid than product release, the amplitude of the burst is equal to actively engaged enzyme and the observed exponential approach to equilibrium (kobs) corresponds to the rate of chemical conversion of substrate to product, assuming that the reverse rate of chemistry is negligible.
In some instances catalytic cycling interferes with a pre-steady-state analysis, such as when the magnitudes of rates for chemistry and product release are not significantly different. In this case, employing excess enzyme relative to substrate prevents catalytic cycling and limits substrate bound with enzyme to a single turnover. Accordingly, the first chemical step of the reaction can be isolated and accurately determined as the first-order rate constant (kobs). This rate constant should be similar to kobs determined from the pre-steady-state approach described above.
Here we describe how these kinetic approaches can be used to analyze the glycosylase activity of OGG1.
Protocol
1. Preparation of Enzyme and DNA Substrate
Over-express OGG1 as a GST-fusion protein in E. coli, utilize the GST-tag for purification, and then remove the GST-tag by cleavage with HRV-3C protease (Figure 1)6.
Purchase the chemically synthesized 5'-6-carboxyfluorescein (6-FAM) labeled oligonucleotide containing a single 8-oxoG residue and its complementary unlabeled oligonucleotide strand. The 34-mer oligonucleotides contain an 8-oxoG at position 17 from the 5'-end. Purify these oligonucleotides by polyacrylamide gel electrophoresis.
Prepare double-stranded DNA substrate containing 8-oxoG by mixing 5'-6-FAM labeled oligonucleotide with its complementary strand at a molar ratio of 1:1.2 in annealing buffer (10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 1 mM EDTA) in a 1.5 ml microfuge tube. Place the tube in a float in boiling water for 5 min. Leave the tube in place and then let the water cool slowly to room temperature (this takes approximately 2 hr).
Perform sample preparation and the experiment under low or minimal light conditions so as to minimize photobleaching of the fluorescent label.
2. Measurement of Steady-State Time Course and Active Site Titration of OGG1
2.1. Sample preparation and steady-state time course
Prepare DNA substrate and OGG1 solutions separately in reaction buffer (50 mM HEPES, pH 7.5, 20 mM KCl, 0.5 mM EDTA, and 0.1% bovine serum albumin) in 1.5 ml microfuge tubes on ice. The concentration of DNA is 400 nM and the apparent concentration of OGG1 is 30, 60, 90, or 120 nM as determined by a Bradford protein assay. Place the reaction tubes in a heat block set at 37 °C. Pre-incubate enzyme and DNA substrate solutions separately at 37 °C for 1 min.
Start the reaction by mixing equal volumes of the OGG1 and DNA substrate solutions by pipetting. After mixing these solutions 1:1 (v/v), the final concentrations are 200 nM DNA and 15, 30, 45, or 60 nM OGG1, respectively. Remove aliquots (10 μl) at time intervals and quench the reaction by mixing with 1 μl of 1 M NaOH. As the control for the time courses, 10 μl of the reaction mixture without enzyme is mixed with 1 μl of 1 M NaOH.
Place the reaction samples in a 90 °C heat block for 5 min to cleave the resulting product AP-site. After heating, add 1 μl of 1 M HCl to neutralize each sample.
Add an equal volume (12 μl) of gel loading buffer (95 % formamide, 20 mM EDTA, 0.02% bromophenol, and 0.02% xylene cyanol) to each reaction sample, and then place the mixture in a heat block set at 95 °C for 2 min, then immediately place the tube on ice.
Load the samples (5 μl) onto a 15% denaturing polyacrylamide gel containing 8 M urea in 89 mM Tris-HCl, pH 8.8, 89 mM boric acid, and 2 mM EDTA. The substrate and the cleaved products are well separated after running the gel.
2.2. Imaging of the gel
Scan the gel using an imager that can detect the fluorescently labeled DNA and visualize the substrate and product bands.
Quantify the bands after imaging the gel. Note that some background cleavage may be observed after treatment of the substrate itself with NaOH (Figure 2A). Subtract this background from the measured amount of each reaction product.
2.3. Data analysis
Plot the amount of the product formed at each reaction time (t) (Figure 2B). Analyze the raw data using Equation 1 to determine the amplitude of the burst (A0, y-intercept) and the slope of the linear steady-state phase (vss).
![]()
Plot the y-intercept relative to the total protein concentration (i.e. apparent enzyme concentration; Figure 2C), providing a measure of the active fraction of enzyme. Use a linear fit with a zero intercept to provide the correction factor for determining the fraction of active enzyme.
Plot the steady-state rate relative to the active enzyme concentration (Figure 2D), providing the product dissociation rate constant (i.e. slope of the fitted line).
3. Measurement of Pre-steady-state Time Course
As shown in Figure 2, product formation during the burst phase is too fast to measure by manual mixing and quenching. Hence, a rapid quench-flow instrument can provide a powerful method to measure rapid reactions that occur on the millisecond time scale (Figure 3)5. The instrument uses a computer-controlled drive motor to rapidly mix and quench reactions after specified reaction times. For example, use a Kintek RQF-3 Rapid Quench-Flow Instrument to measure the initial burst phase and subsequent steady-state phase of product formation catalyzed by OGG1. Rapid Quench-Flow Instruments are available from several manufacturers.
3.1. Preparation of sample
Separately prepare DNA substrate and OGG1 solutions in 1.5 ml tubes as described in 2-1. The concentrations of DNA and active OGG1 are 400 nM and 80 nM, respectively. This results in final concentrations of 200 nM DNA and 40 nM active OGG1 after mixing 1:1 (v/v).
3.2. Preparation of rapid quench-flow instrument
Connect a circulating water bath to the rapid quench-flow Instrument for temperature control (37 °C).
Adjust the instrument parameters and select the appropriate reaction loop for the desired time points according to the manufacturer's instructions. The reaction loops are of variable lengths to provide alternate reaction times.
Prepare reaction buffer and NaOH quench solutions in 10 ml Luer Lock disposable syringes and attach these syringes to the Drive Ports and load the Drive Reservoirs with reaction buffer (syringes B) and 142 mM NaOH (syringe Q) (Syringe Load Valves in LOAD position).To remove the air bubbles from the syringe, work the solution back and forth several times.
Lower the stepper motor until it contacts the top of the syringes (Syringe Load valves and Sample Load valves in FIRE position).
Flush the Sample Loops, the Reaction Loop, and the Exit Line with water and methanol. Dry the flushed areas completely (valves in FLUSH position).
3.3. Pre-steady-state time course
Make a hole in the top of a capped 1.5 ml tube using a 16-gauge needle. Attach a tube at the Exit Line to collect the quenched reaction.
Set the desired reaction time (in seconds) using the keypad. The Stepper Motor will back up to adjust for loop volume so as to maintain constant quench volume. Position plungers for syringe B against the Stepper Motor platform by adding buffer with the disposable syringes attached to the Drive Ports.
Fill 1 ml Luer Lock disposable syringes with DNA substrate and OGG1 solutions, respectively (sample valves in LOAD position). Attach syringes with DNA substrate and OGG1 solutions to each of the Sample Load Ports, and then fill the Sample Loops with DNA substrate and OGG1 solutions, respectively (Sample Load Valves in LOAD position).
Set all Syringe Load Valves and Sample Load Valves to the FIRE position. Start the reaction by a keypad stroke (e.g. press of "G" or "START" key). DNA substrate and OGG1 solutions (18 μl each) are immediately mixed in the Reaction Loop.
Wait until the reaction is automatically quenched at the desired reaction time by mixing 36 μl of the reaction mixture with 86 μl of 142 mM NaOH. After quenching the reaction, the sample is discharged from the Exit Line.
Set the Syringe Load Valves to the LOAD position and the Sample Load Valves to the FLUSH position. Flush the Sample Loops, the Reaction Loop, and the Exit Line with water and methanol and dry as described in 3.2. Repeat the above procedure for each time point.
Perform a control experiment without enzyme to determine a background correction, which can be done manually or with the rapid-quench instrument. To perform a control with the rapid-quench instrument, fill the Sample Loop for DNA with DNA substrate but keep the Sample Loop for OGG1 empty. Set the reaction time, perform the mix and quench as described in 3.3.4-3.3.6.
Wash the Sample Loops and the Reaction Loop with 2 M NaOH, 2 M HCl, water, and methanol and then dry the washed lines. After setting the Stepper Motor to home position, switch the Syringe Load Valves to the LOAD position and wash the Drive Reservoirs with water.
Treat the quenched reaction samples with heat, and then separate substrate and product DNA by 15% denaturing polyacrylamide gel electrophoresis as described in 2.1.
Visualize and quantify the bands on the gel as described in 2.2.
3.4. Data analysis
Fit the time courses of product formation by non-linear regression analysis to an equation with a rising exponential and linear terms (Equation 2) providing the first-order rate constant (kobs), the amplitude of the burst (A0), and a linear rate (vss).
![]()
4. Single-turnover Time Course
Prepare DNA substrate and OGG1 in separate 1.5 ml tubes as described in 2-1. The concentrations of DNA and active OGG1 are 100 nM and 500 nM, respectively; this yields final concentrations of 50 nM DNA and 250 nM OGG1 after mixing 1:1 (v/v).
Prepare the rapid quench-flow instrument and prepare the reaction samples as described in 3.2 and 3.3.
Treat the quenched reaction samples with heat and subject them to 15% denaturing polyacrylamide gel electrophoresis to separate substrate and products as described in 2.1.
Visualize and quantify the bands on the gel as described in 2.2.
Fit the time courses of product formation to a single exponential to determine the first-order rate constant (kobs) as given in Equation 3.
![]()
Representative Results
Steady-state kinetic analysis was performed by using 200 nM DNA substrate and four different apparent concentrations of OGG1 (15, 30, 45, and 60 nM) as determined by a Bradford protein assay2. The time courses of product formation were fit to a linear equation to determine the y-intercept, which were 2.2, 11, 15, and 26 nM, respectively, relative to each protein concentration (Figure 2B). The y-intercepts were further plotted relative to each actual protein concentration (Figure 2C). The fraction of active enzyme was determined to be 38 % from the slope of the line in Figure 2C. To determine the steady-state rate, vss determined from the linear fits in Figure 2B were plotted relative to the y-intercepts (Figure 2D). The slope of the line in Figure 2D was 0.0028 sec-1, which is equivalent to the dissociation rate constant for the product AP-site and incised AP-site formed by DNA glycosylase/lyase activities.
A pre-steady-state time course was followed by using 200 nM DNA substrate and 40 nM active OGG1. The time courses of the product formation can be fit to an equation with rising exponential and linear terms. As shown in Figure 4, kobs and koff were determined to be 0.75 sec-1 and 0.0055 sec-1, respectively2. The amplitude for the burst phase (33 nM) was slightly lower than predicted from the concentration of the active OGG1 added (40 nM). This may be due to nonspecific binding of OGG1 to DNA substrate that reduces the available numbers of enzyme molecules.
Under single-turnover conditions, the 8-oxoG excision reaction was finished within 6 sec (Figure 5)2. The time course of product formation could be fit to a single exponential equation and yielded kobs of 0.74 sec-1. Notably, the amplitude for product formation was lower than the expected 50 nM added DNA substrate. This could have been due in part to background cleavage of the DNA by NaOH-treatment (Figure 2A) or the presence of unannealed substrate oligonucleotides in the reaction mixture.
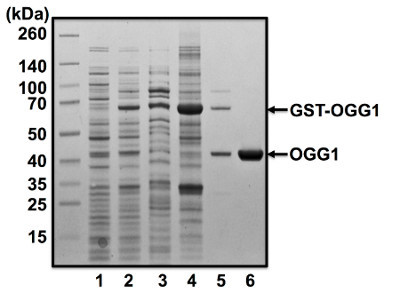 Figure 1. Purification of human OGG1 from E. coli. The human OGG1 gene was cloned into pGEX-6P-1 and overexpressed in BL21 (DE3) cells. Lane 1; uninduced cells, lane 2; IPTG-induced cells, lane 3; soluble protein fraction, lane 4; glutathione sepharose 4B after incubation with soluble protein fraction, lane 5; flow through fraction after incubation of glutathione sepharose 4B with HRV 3C protease, lane 6; flow through fraction after removal of contaminants by Mono Q column. A photograph of the Coomassie Blue stained gel is shown.
Figure 1. Purification of human OGG1 from E. coli. The human OGG1 gene was cloned into pGEX-6P-1 and overexpressed in BL21 (DE3) cells. Lane 1; uninduced cells, lane 2; IPTG-induced cells, lane 3; soluble protein fraction, lane 4; glutathione sepharose 4B after incubation with soluble protein fraction, lane 5; flow through fraction after incubation of glutathione sepharose 4B with HRV 3C protease, lane 6; flow through fraction after removal of contaminants by Mono Q column. A photograph of the Coomassie Blue stained gel is shown.
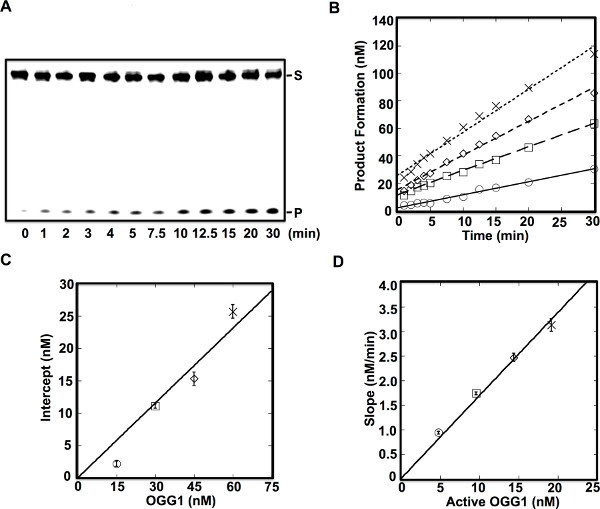 Figure 2. Steady-state kinetics and active site titration of purified OGG12. OGG1 (15 nM, ○; 30 nM, □; 45 nM, ◊; or 60 nM, ×) was incubated with 200 nM DNA substrate (5'-CTGCAGCTGATGCGCCXTACGGATCCCCGGGTAC-3', where X is 8-oxoG paired with C) at 37 °C for 1-30 min as indicated. These enzyme concentrations represent apparent protein concentrations based on a Bradford protein assay, quantitated from a BSA standard curve. A, Denaturing polyacrylamide gels showing separated substrates (S) and products (P). OGG1 (30 nM) and 200 nM DNA substrate was used for the reaction. B, Time courses of product formation. The data were fit to a linear equation to determine the amplitude of the burst phase (y-intercept) and an apparent rate of product release (slope). C, Plot of the intercepts determined from the linear fits in panel B relative to that determined by the Bradford assay. The slope of the line corresponds to the fraction of active enzyme. The error bar denotes the error in the amplitude inferred from linear fits to the product formation in panel B. D, Plot of the slopes determined from the linear fits in panel B relative to the active enzyme concentration. The error bar denotes the error in the slope inferred from linear fits to the product formation in panel B. The slope of the line corresponds to the steady-state rate (koff), 0.0028 sec-1.
Figure 2. Steady-state kinetics and active site titration of purified OGG12. OGG1 (15 nM, ○; 30 nM, □; 45 nM, ◊; or 60 nM, ×) was incubated with 200 nM DNA substrate (5'-CTGCAGCTGATGCGCCXTACGGATCCCCGGGTAC-3', where X is 8-oxoG paired with C) at 37 °C for 1-30 min as indicated. These enzyme concentrations represent apparent protein concentrations based on a Bradford protein assay, quantitated from a BSA standard curve. A, Denaturing polyacrylamide gels showing separated substrates (S) and products (P). OGG1 (30 nM) and 200 nM DNA substrate was used for the reaction. B, Time courses of product formation. The data were fit to a linear equation to determine the amplitude of the burst phase (y-intercept) and an apparent rate of product release (slope). C, Plot of the intercepts determined from the linear fits in panel B relative to that determined by the Bradford assay. The slope of the line corresponds to the fraction of active enzyme. The error bar denotes the error in the amplitude inferred from linear fits to the product formation in panel B. D, Plot of the slopes determined from the linear fits in panel B relative to the active enzyme concentration. The error bar denotes the error in the slope inferred from linear fits to the product formation in panel B. The slope of the line corresponds to the steady-state rate (koff), 0.0028 sec-1.
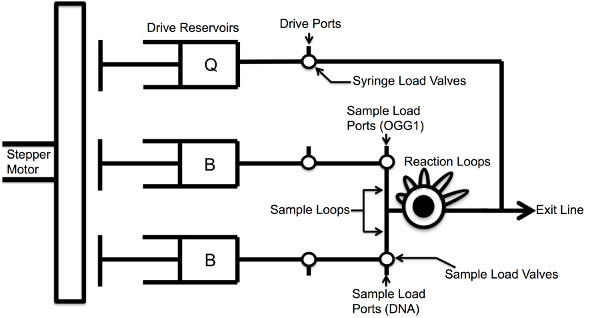 Figure 3. Overview of Rapid Quench-Flow Instrument.
Figure 3. Overview of Rapid Quench-Flow Instrument.
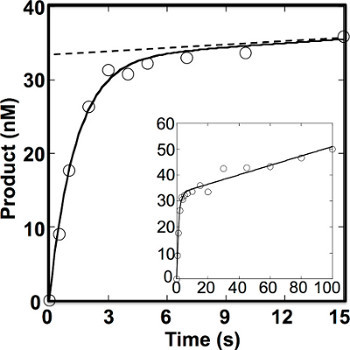 Figure 4. Pre-steady-state kinetics of 8-oxoG excision by OGG12. OGG1 (40 nM active enzyme) was incubated with 200 nM DNA substrate (5'-CATGGGCGGCATGAACCXGAGGCCCATCCTCACC-3', where X is 8-oxoG paired with C) at 37 °C for 0-100 sec. The dotted line indicates an extrapolation of the steady-state phase. The data were fit to the burst equation with an amplitude equal to 33 ± 0.89 nM, kobs equal to 0.75 ± 0.083 sec-1, vss equal to 0.18 ± 0.018 nM sec-1 and koff equal to 0.0055 ± 0.020 sec-1.
Figure 4. Pre-steady-state kinetics of 8-oxoG excision by OGG12. OGG1 (40 nM active enzyme) was incubated with 200 nM DNA substrate (5'-CATGGGCGGCATGAACCXGAGGCCCATCCTCACC-3', where X is 8-oxoG paired with C) at 37 °C for 0-100 sec. The dotted line indicates an extrapolation of the steady-state phase. The data were fit to the burst equation with an amplitude equal to 33 ± 0.89 nM, kobs equal to 0.75 ± 0.083 sec-1, vss equal to 0.18 ± 0.018 nM sec-1 and koff equal to 0.0055 ± 0.020 sec-1.
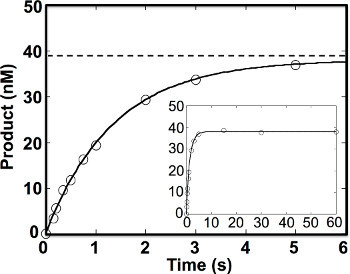 Figure 5. Single turnover kinetics of 8-oxoG excision by OGG12. OGG1 (250 nM active enzyme) was incubated with 50 nM DNA substrate (5'-CTGCAGCTGATGCGCCXTACGGATCCCCGGGTAC-3', where X is 8-oxoG paired with C) at 37 °C for 0-60 sec. The data were fit to the single exponential equation with kobs equal to 0.74 ± 0.015 sec-1. The dotted line indicates the extrapolated amplitude (i.e. product at infinite time).
Figure 5. Single turnover kinetics of 8-oxoG excision by OGG12. OGG1 (250 nM active enzyme) was incubated with 50 nM DNA substrate (5'-CTGCAGCTGATGCGCCXTACGGATCCCCGGGTAC-3', where X is 8-oxoG paired with C) at 37 °C for 0-60 sec. The data were fit to the single exponential equation with kobs equal to 0.74 ± 0.015 sec-1. The dotted line indicates the extrapolated amplitude (i.e. product at infinite time).
Discussion
The kinetic approaches described here outline methods to define elementary kinetic constants. If a time course of product formation is biphasic with the first enzymatic turnover occurring rapidly, then a step after chemistry is rate-limiting during subsequent catalytic turnovers. In the case of OGG1, the first turnover can be measured using high enzyme concentrations with either limiting (S<E) or high (S>E) DNA concentrations. In the first case, the reaction is limited to a 'single-turnover' and provides a measure of the catalytic rate at the active site of the enzyme. In the latter case, multiple enzymatic turnovers are assessed. The burst of product formation occurring during the first turnover provides a measure of the chemical event at the enzyme's active site, while subsequent turnovers provide a measure of the step that limits catalytic cycling. In addition, the amplitude of the burst of product formation provides a measure of actively engaged enzyme. This must be known in order to calculate the product dissociation rate constant from the steady-state velocity (koff = vss/E). For OGG1, the release of product DNA (i.e. product DNA dissociation rate constant) limits catalytic cycling and its steady-state rate7,8. As shown in Figure 4, the DNA product dissociation rate constant (koff = 0.0055 sec-1) for OGG1 is two orders of magnitude lower than the observed catalytic rate (kobs = 0.75 sec-1). It is a common kinetic feature of DNA glycosylases that excise damaged DNA bases to bind their products (DNA with an abasic site) tightly so that product release is rate limiting during steady-state measurements1,8-10. Importantly, steady-state activity measurements thereby provide a simple and straightforward way to determine product DNA binding affinity; lower activity suggests tighter product binding.
As described in Figure 2 and Representative Results, the active fraction of enzyme is determined to be 38%. The active fraction is lower than that determined from the protein concentration, even though the prepared enzyme is >95% homogeneous (Figure 1). This can be due the non-specific binding of the enzyme to the substrate and/or the method of protein determination that may not accurately assess the true protein concentration (e.g. Bradford protein assay typically uses bovine serum albumin as a standard that may not be a good mimic for the enzyme of interest). Alternatively, protein concentration can be determined by other methods or estimated from a calculated molar extinction coefficients at 280 nm such as that developed by Gill and von Hippel11.
As shown in Figures 4 and 5, the magnitude of kobs determined from the pre-steady-state time course should be consistent with that determined from a single-turnover experiment. The value of koff should also be experimentally consistent between alternate kinetic methods. If, however, these rate constants are different (e.g. koff is different between steady-state and pre-steady-state conditions), different steps may be measured by each method and a new model should be considered. Alternatively, product inhibition or substrate depletion can influence the apparent steady-state rate in a pre-steady-state time course that contaminates the linear portion.
In the situation where the burst and the steady-state phases are not well separated (i.e. kobs ~ koff), the amplitude of the burst and the observed rate constants are composites of elementary rate constants for multiple steps12. Furthermore, if product release is rapid (i.e. koff > kobs), the time course of product formation is not biphasic (first turnover and subsequent turnovers are limited by the same step; chemistry). In this case, a single-turnover experiment can provide a measure of catalysis.
If a single-turnover approach is undertaken to determine an intrinsic catalytic rate, sufficient enzyme must be used to assure that binding is not partially rate limiting. Likewise, substrate binding must also be fast so that the burst rate is not limited by substrate binding. To verify that enzyme binding is not rate limiting during a single-turnover experiment, the time course is repeated with a different enzyme concentration to confirm that the exponential time course is not altered. In addition, since the amplitude of the burst or single-turnover time course is directly proportional to enzyme or substrate concentrations, respectively, these concentrations should be chosen so they can be reliably measured. Finally, it should be noted that when the burst and steady-state rates are not well-separated, the burst amplitude underestimates the true active enzyme concentration12.
The kinetics approaches described above provide a reliable method to isolate key steps during the catalytic cycling of DNA glycosylases. With these reference kinetic constants, the influence of altered DNA sequence/structure1,13-18 , active site residues19,20, metal ion21, or cellular accessory protein on catalysis or enzyme cycling can now be evaluated22-24.
Disclosures
Authors have nothing to disclose.
Acknowledgments
We thank Dr. Julie K. Horton for critical reading of the manuscript and Dr. Rajendra Prasad for helpful suggestions and discussions. Portions of this research were originally published in The Journal of Biological Chemistry, Sassa A et. al., "DNA Sequence Context Effects on the Glycosylase Activity of Human 8-Oxoguanine DNA Glycosylase." J Biol Chem. 287, 36702-36710 (2012) 2. This work was supported, in whole or in part, by National Institutes of Health Research Project Grant Z01-ES050158 in the Intramural Research Program, NIEHS.
References
- Porello SL, Leyes AE, David SS. Single-turnover and pre-steady-state kinetics of the reaction of the adenine glycosylase MutY with mismatch-containing DNA substrates. Biochemistry. 1998;37:14756–14764. doi: 10.1021/bi981594+. [DOI] [PubMed] [Google Scholar]
- Sassa A, Beard WA, Prasad R, Wilson SH. DNA sequence context effects on the glycosylase activity of human 8-oxoguanine DNA glycosylase. The Journal of Biological Chemistry. 2012;287:36702–36710. doi: 10.1074/jbc.M112.397786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong I, Lundquist AJ, Bernards AS, Mosbaugh DW. Presteady-state analysis of a single catalytic turnover by Escherichia coli uracil-DNA glycosylase reveals a "pinch-pull-push" mechanism. The Journal of Biological Chemistry. 1074;277:19424–19432. doi: 10.1074/jbc.M201198200. [DOI] [PubMed] [Google Scholar]
- Johnson KA. Advances in transient-state kinetics. Current Opinion in Biotechnology. 1998;9:87–89. doi: 10.1016/s0958-1669(98)80089-x. [DOI] [PubMed] [Google Scholar]
- Johnson KA. Rapid kinetic analysis of mechanochemical adenosinetriphosphatases. Methods in Enzymology. 1986;134:677–705. doi: 10.1016/0076-6879(86)34129-6. [DOI] [PubMed] [Google Scholar]
- Kovtun IV, et al. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–452. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JW, Hazra TK, Izumi T, Mitra S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Research. 2001;29:430–438. doi: 10.1093/nar/29.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zharkov DO, Rosenquist TA, Gerchman SE, Grollman AP. Substrate specificity and reaction mechanism of murine 8-oxoguanine-DNA glycosylase. The Journal of Biological Chemistry. 2000;275:28607–28617. doi: 10.1074/jbc.M002441200. [DOI] [PubMed] [Google Scholar]
- Waters TR, Swann PF. Kinetics of the action of thymine DNA glycosylase. The Journal of Biological Chemistry. 1998;273:20007–20014. doi: 10.1074/jbc.273.32.20007. [DOI] [PubMed] [Google Scholar]
- Nilsen H, et al. Excision of deaminated cytosine from the vertebrate genome: role of the SMUG1 uracil-DNA glycosylase. The EMBO Journal. 2001;20:4278–4286. doi: 10.1093/emboj/20.15.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Analytical Biochemistry. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- Van de Berg , Beard BJ, A W, Wilson SH. DNA structure and aspartate 276 influence nucleotide binding to human DNA polymerase beta. Implication for the identity of the rate-limiting conformational change. The Journal of Biological Chemistry. 2001;276:3408–3416. doi: 10.1074/jbc.M002884200. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy N, Haraguchi K, Greenberg MM, David SS. Efficient removal of formamidopyrimidines by 8-oxoguanine glycosylases. Biochemistry. 2008;47:1043–1050. doi: 10.1021/bi701919u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipold MD, Muller JG, Burrows CJ, David SS. Removal of hydantoin products of 8-oxoguanine oxidation by the Escherichia coli DNA repair enzyme, FPG. Biochemistry. 2000;39:14984–14992. doi: 10.1021/bi0017982. [DOI] [PubMed] [Google Scholar]
- Zhao X, Krishnamurthy N, Burrows CJ, David SS. Mutation versus repair: NEIL1 removal of hydantoin lesions in single-stranded, bulge, bubble, and duplex DNA contexts. Biochemistry. 2010;49:1658–1666. doi: 10.1021/bi901852q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipold MD, Workman H, Muller JG, Burrows CJ, David SS. Recognition and removal of oxidized guanines in duplex DNA by the base excision repair enzymes hOGG1, yOGG1, and yOGG2. Biochemistry. 2003;42:11373–11381. doi: 10.1021/bi034951b. [DOI] [PubMed] [Google Scholar]
- Robey-Bond SM, Barrantes-Reynolds R, Bond JP, Wallace SS, Bandaru V. Clostridium acetobutylicum 8-oxoguanine DNA glycosylase (Ogg) differs from eukaryotic Oggs with respect to opposite base discrimination. Biochemistry. 2008;47:7626–7636. doi: 10.1021/bi800162e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarem DA, Wilson NR, Delaney S. Structure-dependent DNA damage and repair in a trinucleotide repeat sequence. Biochemistry. 2009;48:6655–6663. doi: 10.1021/bi9007403. [DOI] [PubMed] [Google Scholar]
- Livingston AL, Kundu S, Henderson Pozzi M, Anderson WD, David SS. Insight into the roles of tyrosine 82 and glycine 253 in the Escherichia coli adenine glycosylase MutY. Biochemistry. 2005;44:14179–14190. doi: 10.1021/bi050976u. [DOI] [PubMed] [Google Scholar]
- Kundu S, Brinkmeyer MK, Livingston AL, David SS. Adenine removal activity and bacterial complementation with the human MutY homologue (MUTYH) and Y165C, G382D, P391L and Q324R variants associated with colorectal cancer. DNA Repair. 2009;8:1400–1410. doi: 10.1016/j.dnarep.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zharkov DO, Rosenquist TA. Inactivation of mammalian 8-oxoguanine-DNA glycosylase by cadmium(II): implications for cadmium genotoxicity. DNA Repair. 2002;1:661–670. doi: 10.1016/s1568-7864(02)00074-5. [DOI] [PubMed] [Google Scholar]
- Jarem DA, Wilson NR, Schermerhorn KM, Delaney S. Incidence and persistence of 8-oxo-7,8-dihydroguanine within a hairpin intermediate exacerbates a toxic oxidation cycle associated with trinucleotide repeat expansion. DNA Repair. 2011;10:887–896. doi: 10.1016/j.dnarep.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokkapati SK, Wiederhold L, Hazra TK, Mitra S. Stimulation of DNA glycosylase activity of OGG1 by NEIL1: functional collaboration between two human DNA glycosylases. Biochemistry. 2004;43:11596–11604. doi: 10.1021/bi049097i. [DOI] [PubMed] [Google Scholar]
- Sidorenko VS, Nevinsky GA, Zharkov DO. Mechanism of interaction between human 8-oxoguanine-DNA glycosylase and AP endonuclease. DNA Repair. 2007;6:317–328. doi: 10.1016/j.dnarep.2006.10.022. [DOI] [PubMed] [Google Scholar]