

Abstract
Synthetic hydrogels containing covalently-integrated soft and deformable drug depots capable of releasing therapeutic molecules in response to mechanical forces are attractive candidates for the treatment of degenerated tissues that are normally load bearing. Herein, radically crosslinkable block copolymer micelles (xBCM) assembled from an amphiphilic block copolymer consisting of hydrophilic poly(acrylic acid) (PAA) partially modified with 2-hydroxyethyl acrylate, and hydrophobic poly(n-butyl acryclate) (PnBA) were employed as the drug depots and the microscopic crosslinkers for the preparation of hyaluronic acid (HA)-based, hydrogels. HA hydrogels containing covalently integrated micelles (HAxBCM) were prepared by radical polymerization of glycidyl methacrylate (GMA)-modified HA (HAGMA) in the presence of xBCMs. When micelles prepared from the parent PAA-b-PnBA without any polymerizable double bonds were used, hydrogels containing physically entrapped micelles (HApBCM) were obtained. The addition of xBCMs to a HAGMA precursor solution accelerated the gelation kinetics and altered the hydrogel mechanical properties. The resultant HAxBCM gels exhibit an elastic modulus of 847 ± 43 Pa and a compressive modulus of 9.2 ± 0.7 kPa. Diffusion analysis of Nile Red (NR)-labeled xBCMs employing fluorescence correlation spectroscopy confirmed the covalent immobilization of xBCMs in HA networks. Covalent integration of dexamethasone (DEX)-loaded xBCMs in HA gels significantly reduced the initial burst release and provided sustained release over a prolonged period. Importantly, DEX release from HAxBCM gels was accelerated by intermittently-applied external compression in a strain-dependent manner. Culturing macrophages in the presence of DEX-releasing HAxBCM gels significantly reduced cellular production of inflammatory cytokines. Incorporating mechano-responsive modules in synthetic matrices offers a novel strategy to harvest mechanical stress present in the healing wounds to initiate tissue repair.
Keywords: Block copolymer micelles, Mechano-responsive, Hyaluronic acid, Hydrogels, Anti-inflammatory, Drug delivery
1. Introduction
Hydrogels are interconnected networks of macroscopic dimensions, consisting of hydrophilic (or amphiphilic) building blocks that are rendered insoluble due to the presence of crosslinks.1 Over the past few decades, a great deal of effort has been dedicated to the development of smart hydrogels that respond to a variety of stimuli such as pH,2 temperature, 3 ions,4 saccharides,5 antigens,6 enzymes,7 DNA,8 light,9 electric,10 and magnetic fields.11 Very few studies explore the synthesis and characterization of hydrogels that are responsive to mechanical forces.12 Most tissues in the body are subjected to mechanical stimuli; thus biomaterials and engineered tissues, when implanted, are inevitably exposed to mechanically stressed environments.13 Cartilage in the knee, for example, is routinely exposed to compression, tension, shear and torsion associated with the movement of the joint.14 Once damaged, as in the case of osteoarthritis (OA), patients’ locomotive function is severely compromised. OA-induced knee degeneration is manifested as pain and stiffness in the affected joints, as a result of synovial inflammation, cartilage erosion, soft tissue fibrosis and subchondral bone sclerosis.15 While normal physical activities do not evoke any discomfort in the healthy cartilage, they cause severe pain in OA patients and further exasperate the problem. Restricting OA patients to long-term bed rest is impractical, however, if the mechanical stress present in the moving joints can be harvested and converted to benign and conducive biochemical signals,16,17 more effective OA treatments can be developed. Biomaterials that dynamically release anti-inflammatory drugs in response to tissue stress may offer an attractive alternative for pain management and tissue repair for OA patients.
We are interested in developing mechano-responsive hydrogels with anti-inflammatory functions for cartilage repair purposes. Hyaluronic acid (HA), a non-sulfated glycosaminoglycan (GAG) present in the extracellular matrix (ECM) of all connective tissues, was chosen as a hydrogel building block owing to its biocompatibility, biodegradability and lack of immunogenicity.18–21 HA not only contributes significantly to cell proliferation and growth but also mediates early inflammatory response crucial for wound healing.22–24 Intraarticular injection of HA is a widely used therapy for symptomatic relief of pain and stiffness in OA patients.25 Separately, HA-based hydrogels have been utilized for the controlled release of anti-inflammatory drugs. For example, Hoffman and coworkers synthesized divinyl sulfone-crosslinked HA gels loaded with vitamin E succinate (VES).26 VES was released from the HA gels with a burst during the first few hours, and the drug release continued gradually for several days. The authors showed that the released VES reduced the production of anti-inflammatory cytokine, tumor necrosis factor-alpha (TNF-α). This type of drug release system relies on the passive diffusion of drug molecules from the drug reservoirs; an initial burst release is inevitable and the encapsulated drug molecules cannot be completely exhausted within the desired therapeutic window.27 HA hydrogels have also been used to release a hydrophobic, anti-inflammatory drug, dexamethasone (DEX).28 Because of the inherent incompatibility of the hydrophilic HA hydrogel network and the hydrophobic drug molecule, DEX had to be covalently conjugated to the HA network, and its release was mediated by hydrogel degradation. None of these HA hydrogels exhibited the ability to release anti-inflammatory drugs on demand by physiologically relevant mechanical forces. From a therapeutic perspective, it is highly desirable that the anti-inflammatory drug be released at an accelerated rate when the cartilage is compressed and the pain is most severe for OA patients.
Strategic incorporation of nanoscopic, mechano-responsive drug depots in synthetic hydrogels offers opportunity not only to fine-tune the gel mechanics but also to effectively convert mechanical forces exerted on the gel matrix to biochemical signals with a desired spatial distribution. Our group has created a new type of hydrogel material using self-assembled block copolymer micelles (BCMs) as the dynamic building blocks and microscopic crosslinkers.13,29,30 Crosslinkable BCMs (xBCMs) were assembled from amphiphilic block copolymer of poly(n-butyl acrylate) (PnBA) and 2-hydroxyethyl acrylate-modified poly(acrylic acid) (PAA). Radical polymerization of acrylamide in the presence of micellar crosslinkers gave rise to elastomeric hydrogels whose mechanical properties can be tuned by varying the xBCM composition. Transmission electron microscopy (TEM) imaging revealed that the covalently integrated BCMs underwent strain-dependent reversible deformation. A model hydrophobic drug, pyrene, loaded into the core of xBCMs prior to the hydrogel formation, was dynamically released in response to externally applied mechanical forces, through force-induced reversible micelle deformation and the penetration of water molecules into the micelle core, leading to the weakening of hydrophobic association between pyrene and the micelle core.
Herein, we extended the utility of BCM-crosslinked hydrogels by replacing polyacrylamide with biocompatible HA and pyrene with an anti-inflammatory drug, DEX. Specifically, glycidyl methacrylate (GMA)-modified HA (HAGMA, 1, Figure 1A) was photochemically crosslinked in the presence of DEX-loaded xBCM. The resultant HA gels (HAxBCM) contain covalently integrated micellar compartments with DEX being sequestered in the hydrophobic core. Compared to the traditional HA gels prepared by radical crosslinking of HAGMA, HAxBCM gels exhibited significantly improved drug loading and release capacity. Moreover, compressive forces exerted on the gels were transmitted to the crosslinked BCMs, resulting in a force-modulated DEX release on demand. The mechanical properties of HAxBCM gels, along with various control samples, were characterized collectively using a rheometer and a dynamic mechanical analyzer. Micelle mobility in the crosslinked networks was analyzed by fluorescence correlation spectroscopy (FCS) using Nile Red (NR)-loaded BCMs. The anti-inflammatory activities of DEX-releasing HAxBCM gels were evaluated via the in vitro culture of lipopolysaccharide (LPS)-activated macrophages.
Figure 1.
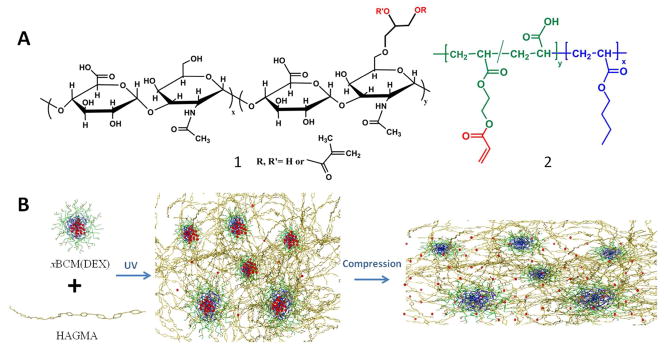
Construction of BCM-integrated HA hydrogels with force-modulated DEX release capacity. (A): Chemical structures of hydrogel building blocks: (1) glycidyl methacrylate-modified HA (HAGMA) and (2) the precursor of crosslinkable block copolymer micelles (xBCM), P(AA100-g-HEA20)-b-PnBA40. (B): Photocrosslinking of HAGMA in the presence of DEX-loaded xBCMs [xBCM(DEX)] resulted in the covalent immobilization of xBCMs in the crosslinked HA network. Compressive stress imposed on the hydrogel was transmitted to the integrated xBCMs, resulting in the release and re-distribution of DEX via the deformation of the rubbery PnBA core.
2. Materials and Methods
2.1. Materials
Hyaluronic acid (HA, sodium salt, ~600 kDa) was generously donated by Genzyme Corporation (Cambridge, MA). Tert-butyl acrylate (t-BA), n-butyl acrylate (n-BA), glycidyl methacrylate (GMA) and 2-hydroxyethyl acrylate (HEA) were purchased from Sigma-Aldrich (St Louis, MO) and were purified by passing through an inhibitor removal column (Aldrich). Ethyl 2-bromopropionate (EBP), copper (I) bromide, trifluoroacetic acid, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC), N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA), hyaluronidase (HAase, 30,000 U/mg), (N,N-dimethylamino) pyridine (DMAP), tetrabutylammonium bromide (TBAB), 2,2-dimethoxy-2-phenylacetophenone (DMPA), 1-vinyl-2-pyrrolidinone (NVP), and Nile Red (NR) were purchased from Aldrich and were used without further purification. Dexamethasone (DEX) was purchased from Tocris Biosciences (Minneapolis, MN). Deionized water was obtained through a NANOpure Diamond water purification system (Thermo Scientific, Barnstead, NH).
2.2. Synthesis and characterization of hydrogel precursors
HAGMA with an estimated 8 mol% modification (relative to the disaccharide repeats) was synthesized by reacting HA with a large excess of GMA in the presence of DMAP and TBAB. The product was obtained as a white fluffy solid after repeated precipitation, extensive dialysis and freeze drying.22,31 Separately, block copolymer of PAA and PnBA with a composition of PAA100-b-PnBA40 was synthesized via the PtBA100-b-PnBA40 intermediate, prepared by sequential atom transfer radical polymerization (ATRP) of tBA and nBA using EBP as the initiator, CuBr as the catalyst and PMDETA as the ligand, as described previously.30 Approximately 20 mol% AA repeats were subsequently acrylated via EDC-mediated reaction of PAA100-b-PnBA40 with HEA to afford P(AA100-g-HEA20)-b-PnBA40 (2, Figure 1A). All chemical transformations were monitored by 1H NMR using a Bruker AV400 spectrometer in CDCl3, d6-DMSO or D2O, and tetramethylsilane (TMS) was added as the internal reference.
2.3. Micelle preparation and DEX loading
Micelles were prepared by dialyzing a DMF solution of PAA100-b-PnBA40 or P(AA100-g-HEA20)-b-PnBA40 against DI water for 3 days,30 and the resultant micelles are referred to as BCM and xBCM. DEX-loaded micelles were prepared by slowly injecting a DEX/DMSO solution (10 mg/mL, 500 μL) into a stirred aqueous micelle solution (2.5 mg/mL, 10 mL). After overnight equilibration in the dark, the mixture was centrifuged (4,000 rpm, 30 min) using a Millipore centrifugal filter unit (15 mL, MWCO 3,000) and the collected particles were washed with copious DI water. The filtrate was pooled and the DEX content in the filtrate was analyzed using a UV-Vis spectrometer. DEX loading and encapsulation efficiency were calculated based on the initial drug added and the amount found in the filtrate. Micelle morphology was examined using bright field TEM on a FEI Tecnai 12 microscope operating at an accelerating voltage of 120 KV. TEM samples were prepared by applying a drop of micelle solution onto a carbon coated copper TEM grid (300 mesh) and allowing the solvent to evaporate under ambient conditions. Afterwards, a drop of freshly prepared saturated uranyl acetate aqueous solution was deposited onto the dried samples. After approximately 1 min, the excess solution was wicked away by a piece of filter paper, and the sample was allowed to dry for TEM observation. Particle size and size distribution were analyzed by dynamic light scattering (DLS) using a Malvern Zetasizer nanoZS instrument (Malvern Instruments, UK) at 25 °C with a scattering angle of 173°. Micelle solutions (concentrations ranging from 0.1 to 10 mg/mL in DI H2O or in 1 wt% aqueous solution of HAGMA) were passed through a 0.22 μm PVDF filter prior to analysis, and measurements were made in triplicate. When an HAGMA solution was used as the dispersion phase in place of DI H2O, the intensity size distribution was corrected by taking into account the higher viscosity of HAGMA solution, determined using a rheometer (AR-G2, TA Instrument, New Castle, DE) with a 25 mm aluminum parallel plate geometry at ambient temperature at a constant shear rate of 1 rad/s. Data were analyzed by Malvern’s DTS software using a cumulant analysis with a single exponential fit.
2.4. Hydrogel synthesis and characterization
HA hydrogels containing physically entrapped (HApBCM) or covalently integrated (HAxBCM) micelles were prepared using BCMs and xBCMs, respectively. Stock solutions of HAGMA (2 wt%) and micelles (2 wt%) in DI H2O prepared separately were mixed at a volume ratio of 1:1. After the addition of 6 μL initiator solution (30% DMPA in NVP), the mixture was subjected to UV irradiation (365 nm, 10 mW/cm2) for 10 min. Control gels free of micelles prepared similarly using 1 wt% and 2 wt% HAGMA are designated as HAGMA1 and HAGMA2. DEX-containing HAxBCM and HApBCM gels were prepared using DEX-loaded xBCMs and BCMs, respectively. DEX-loaded HA gels free of micelles were prepared by adding a DEX/DMSO solution (40 mg/mL, 50 μL) to an aqueous solution of HAGMA (2 wt%) prior to UV irradiation. The crosslinked hydrogel samples, prepared in D2O/H2O (v/v = 1/1) for HAxBCM or in D2O for HAGMA2, were analyzed by 1H NMR operated on a Bruker AV600 using a high resolution magic angle spinning (HR-MAS) probe. The respective precursor solutions were analyzed by regular NMR as described above. For equilibrium swelling ratio (SW) and sol fraction (SF) measurements, hydrogels were incubated at 37 °C until a constant weight was observed and the initial dry weight (Wi) was recorded. After equilibrating in DI water at 37 °C for 2 days, the wet weight of the swollen gels (Ws) was recorded. After the removal of excess liquid, the swollen gels were dried again at 37 °C for 2 days and the final dry weight (Wf) was recorded. SW was determined as and SF was calculated according to . Three repeats were tested for each hydrogel composition.
2.5. Fluorescence correlation spectroscopy (FCS)
Nile Red (NR) was employed as the fluorescent probe for FCS measurements. NR-loaded xBCMs [xBCM(NR)] were prepared by slowly adding a NR/methanol solution (0.8 mg/mL, 10 μL) to a stirred micelle solution (10 mg/mL, 1 mL). NR-containing HAxBCM gels were prepared as described above using xBCM(NR), and NR-containing, BCM-free HA gels were prepared by adding a NR/methanol solution (0.8 mg/mL) to HAGMA (1 wt% in PBS) prior to UV irradiation. FCS measurements were performed on a Zeiss 780 confocal scanning microscope at 25 °C. NR was excited by a 561-nm laser diode that was focused 20 μm into the bulk solution using a 40×1.2 N.A. water immersion objective. The confocal pinhole was set to one Airy unit for all measurements. The confocal volume was calibrated using a Rhodamine B standard, with a published diffusion coefficient of 427 μm2/s.32 Raw intensity data was recorded at 15 Mhz for 10 sec using a 32 channel GaAsP detector, with a fluorescence bandpass set to 565–700 nm using an acousto-optical tunable filter. FCS measurements were repeated 20 times for each sample and each data set was autocorrelated using the software provided by the manufacturer (ZEN Black 2011). Software written in LabView was used to average the autocorrelated data and to perform fits to the normalized autocorrelation function, G′(τ), based on the biophysical models described in the text. All FCS experiments were performed inside custom polydimethylsiloxane microchannels bonded between a #1.5 coverslip and microscope slide. Hydrogel samples were prepared by injecting thoroughly mixed hydrogel components into the microchannels, followed by UV crosslinking.
2.6. Enzymatic degradation
The enzymatic stability of various HA-based hydrogels was evaluated in the presence of HAase following our previously published procedures.31 Individual hydrogel disks (~1 mg dry weight) were separately immersed in a HAase solution (1 mL, 5 U mL−1) in PBS at 37 °C. The supernatant was aspirated every other day and stored at −80 °C until further analysis, and the degradation medium was replenished with freshly prepared enzyme solution. The amount of HA degraded was quantified by the carbazole assay 33. The apparent degradation was calculated by dividing the amount of HA released up to a chosen time by the initial dry weight of the gel disks. The HA degradation was normalized by the initial HA content in the gel disks.
2.7. Mechanical properties
Rheological tests were performed on an AR-G2 rheometer with UV curing accessories (TA Instruments, New Castle, DE) using a standard stainless steel parallel-plate 8-mm geometry. Oscillatory strain, time and frequency sweeps were performed at ambient temperature, and the storage (G′) and loss (G″) moduli were recorded. A 2.5 μL aliquot of hydrogel precursor solution containing photoinitiator was pipetted onto the bottom plate, and the top plate was lowered to a set gap size of 50 μm; the edge of the geometry was covered with mineral oil to prevent water evaporation. The solution was further mixed at 1% strain for 2 min before the UV irradiation was initiated. Experiments were performed on at least three samples and the results were averaged. Compression tests were performed using a Rheometrics Mechanical Analyzer (RSA-G2, TA Instruments, New Castle, DE) at 25 °C. Various gel disks (height: 4.0 mm; diameter: 6.3 mm) were prepared in standard cell culture inserts (Millipore, Bedford, MA), and compression tests were performed immediately upon gelation. All samples were compressed at a rate of 20% per min until fracture. Compression tests were performed in triplicate for all samples and representative stress-strain curves are shown. The modulus was calculated using the initial 0–15% linear portion of the stress–strain curve.
2.8. In vitro DEX release
Hydrogel disks containing an estimated 1 mg DEX per disk, prepared as described above, were incubated in 30 mL PBS at 37 °C under gentle stirring at 90 rpm. At pre-determined times, three mL release buffer was withdrawn, and the media was replenished with an equal amount of fresh PBS. Dynamic release experiments were carried out using a RSA-G2 DMA. The hydrogel disks, sandwiched between the two DMA plates, were immersed in 30 mL DI H2O for 3 h. Immediately before the compression was initiated, H2O was replaced with fresh PBS. A 1-h-on-1-h-off cyclic compression regimen (12 cycle/h) was applied to the samples at a rate of 0.333 mm/s to a strain of 15 or 30% for a total of 8 h. Three mL of release buffer was withdrawn every hour, and the media was replenished with an equal amount of PBS. Control experiments were conducted under static conditions. The DEX content in the collected buffer was measured by UV/Vis at 242 nm.
2.9. Macrophage culture and cytokine production
RAW264.7 murine macrophages were purchased from American Type Culture Collection (ATCC, Manassas, VA) and were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum (Denville Scientific, Metuchen, NJ), 1% antibiotic/antimycotic (GIBCO, Grand Island, NY), 10 mM HEPES, and 55 μM β-mercaptoethanol at 37°C. Upon reaching 60–80% confluency, cells were lifted off the plates by gentle scraping and were subsequently seeded on a 24-well plate at a density of 105 cells per well in 800 μL cell culture media. After overnight incubation, 100 μL gel disks, prepared using sterile-filtered components, were added to the cell culture inserts suspended above the cell monolayer. After 1-h incubation, gel disks were removed and lipopolysaccharide (LPS, 50 ng/mL in cell culture media, 800 μL) (Invitrogen, Carlsbad, CA) was added to each well. After additional 8-h incubation, media were collected and stored at −20 °C until further analysis. Upon completion of the 8-h LPS treatment, the cell layer was rinsed with cold PBS, digested with 250 μL papain digestion buffer (200 μg/mL, Sigma-Aldrich) for 18 h at 63 °C.34 The digested DNA-containing solution was then centrifuged at 4 °C for 5 min at 10,000 rpm, and the supernatant was aliquoted (20 μL each) for the Picogreen DNA assay (Invitrogen, Carlsbad, CA), following the manufacture’s procedure. TNF-α concentration in collected media samples was measured via an enzyme-linked immunosorbent assay (ELISA) (R&D Systems, Minneapolis, MN). Phase contrast images of cells cultured under different conditions were acquired using a Nikon Eclipse Ti microscope.
2.10. Statistical analysis
All quantitative measurements were performed on 3 replicates. All values are expressed as means ± standard deviations (SDs). Statistical significance was determined using a two-tailed student t-test. A p value of less than 0.05 was considered to be statistically different.
3. Results and Discussion
Our design of mechano-responsive, anti-inflammatory hydrogels is motivated by the need to mediate inflammatory responses in pathologically comprised tissues (e.g. degenerated cartilage) that are mechanically active or mechanically stressed. We hypothesize that force-induced release and redistribution of anti-inflammatory drugs from a hydrogel matrix derived from a biologically relevant glycosaminoglycan (GAG), will cooperatively and synergistically facilitate tissue repair and regeneration. In our design (Figure 1B), HA, a non-sulfated GAG found in the connective tissues in all higher animals with well-known anti-inflammatory properties,35 was chemically modified with GMA to permit facile network formation via a photochemical process.22,31 Reactive micelles capable of sequestering hydrophobic drug molecules, such as DEX, were employed as crosslinkable modules and nanoscale compartments to be integrated in the HAGMA gels. In this study, we characterized micelle-integrated HA gels in terms of the micelle diffusivity, hydrogel mechanical properties, DEX release capability and anti-inflammatory functions.
3.1. Hydrogel synthesis
In an aqueous environment, amphiphilic block copolymers self-assemble into nanoscale structures composed of a hydrophobic core stabilized by a hydrophilic shell.36,37 In our study, poly(acrylic acid) (PAA) was chosen as the hydrophilic block owing to the susceptibility of COOH groups to chemical modification. Poly (n-butyl acrylate) (PnBA) was chosen as the hydrophobic block because of its low glass transition (−49 to −55 °C).38,39 Thus at 37 °C, the polymer chains are flexible and dynamic.38 Our previous work has confirmed the ability of this type of BCMs to encapsulate pyrene and to crosslink poly(acrylamide) chains to form a mechano-responsive hydrogel that releases pyrene in response to the dynamic tensile stretch.30
Block copolymers with estimated 40 nBA repeats and 100 AA repeats, 20% of which modified with HEA, were employed as the microscopic crosslinkers in this study due to the desired micelle stability and the need for longer PnBA blocks for drug encapsulation purposes. TEM imaging of densely packed micelles revealed that individual particles had a bright core and a dark shell (Figure 2A). The average diameter of xBCMs, based on 100 counts of particles from the TEM image, was estimated to be 35 ± 3 nm. On the other hand, DLS analysis (Figure 2D) revealed an estimated particle diameter of 42 ± 5 nm. Consistent with our previous observations, particle size estimated by TEM was smaller than the corresponding hydrodynamic size determined by DLS, because TEM reveals the actual dimensions of the micelles in a collapsed, dry state, while DLS reports the intensity-average dimensions of the micelles in aqueous solution.
Figure 2.

Characterization of self-assembled xBCMs by TEM (A, B, C) and DLS (D). (A–C): TEM images of xBCM (A), xBCM(DEX) (B) and xBCM/HAGMA mixture (C). Samples were negatively stained by uranyl acetate prior to imaging. (D): DLS size distribution profiles for xBCM in water (square), xBCM(DEX) in water (cross) and xBCM in an aquoues solution of HAGMA (circle, xBCM: 1wt%, HAGMA: 1wt%). DLS profiles from 3 separate measurements for each mixture are shown.
The block copolymer micelles were designed as the drug depot within the HA matrix for the controlled release of DEX. DEX is a hydrophobic drug (maximum solubility in PBS is ca. 0.1 mg/mL40) widely used as a potent anti-inflammatory and bone growth steroid.28,41 If administered without a control release mechanism, DEX can cause severe side effects that significantly compromise the quality of life.42,43 In our investigations, DEX was loaded into pre-assembled BCMs by injecting a concentrated DEX/DMSO solution into a stirred aqueous micelle solution. Overall, DEX loading in BCMs and xBCMs was calculated as 12.2 ± 0.7 % and 12.7 ± 1.2 %, respectively. Thus, partial esterification of PAA in the block copolymer did not compromise the ability of these polymers to form micelles29 and to encapsulate DEX. DEX loading to xBCMs did not alter the spherical micelle morphology (Figure 2B). Based on the TEM image, the average diameter of DEX-loaded xBCMs was estimated to be 54 ± 7 nm, larger than the drug-free xBCMs. DLS profile based on scattering intensity (Figure 2D) showed a similar trend, with DEX-loaded micelles having an average diameter of 58 ± 10 nm for the majority of the particles. The small peak at 6 ± 2 nm was probably from the small amount of DEX aggregates that were not encapsulated in the micelles but remained in the micelle solution.
Our goal is to incorporate xBCMs in the HA matrix covalently. Therefore, it is important to confirm HA does not negatively impact the stability of the assembled micelles. Figure 2C shows a representative TEM image of HAGMA physically mixed with xBCM. Abundant spherical objects with an estimated diameter of 41 ± 5 nm are present. Compared to the TEM image acquired from xBCM alone (Figure 2A), these particles were slightly larger. Fragmented interstitial, possibly from the dehydrated HA film was also observed. Under the experimental conditions, both HA and xBCMs are negatively charged, therefore both were stained by uranyl acetate. DLS analysis of a physical mixture of HAGMA (1 wt%) and xBCMs (1 wt%) (Figure 2D), after adjusting the solution viscosity, revealed the presence of particles with an average diameter of 43 ± 4 nm. The same experiments conducted on HAGMA solution without xBCMs did not reveal any particles within the instrument detection range (0.6 nm to 6,000 nm). Collectively, our results confirm that the addition of HAGMA to the micelle solution does not compromise the micelle integrity.
HA hydrogels containing covalently integrated xBCMs (HAxBCM) were prepared by UV irradiation of a precursor solution containing dissolved HAGMA (1 wt%) and dispersed xBCM (1 wt%) with added photoinitiator. Bulk gels free of any particulate matter prepared from 1 or 2 wt% HA-GMA are referred to as HAGMA1 and HAGMA2. 1H NMR experiments were carried out on hydrogel samples employing high resolution, magic angle spinning (HR-MAS); the corresponding precursor solutions were analyzed under normal solution phase conditions. Characteristic peaks associated with the vinyl protons (5.8 ppm to 6.4 ppm) in HAxBCM hydrogel precursors (Figure S1A) were found to completely disappear after 10-min UV irradiation (Figure S1B). On the other hand, the same HR-MAS experiment conducted on HAGMA2 gels free of xBCMs revealed the presence of vinyl proton peaks in the gel phase after UV crosslinking (Figure S1D). The moderate down-field shift for the vinyl protons in gel phase (Figure S1D) compared to those observed in solution phase (Figure S1C) could be a consequence of the different modes of data acquisition (HR-MAS vs normal solution phase NMR); the altered chemical environment introduced by the kinetics chains after crosslinking could also be a contributing factor.44 Our NMR results qualitatively show that the unsaturated double bonds are consumed more efficiently in HAxBCM gels than in HAGMA2 gels.
In general, radical chain polymerization may consume double bonds, but the growing chains may not be connected to the network, thereby generating the soluble fraction that is physically trapped in the network. As expected, the sol fraction for traditional, radically crosslinked hydrogels is high (Table S1). HAGMA1 gels had a sol fraction as high as 57.7 ± 14.2% and physical entrapment of passive BCMs in HA gels only slightly reduced the sol fraction to 48.9 ± 14.7%. For HAGMA2 gels, the measured sol fraction was 27.5 ± 7.8%. Covalent integration of an equal amount of xBCMs in HA gels reduced the sol fraction significantly (17.2 ± 3.7%). When immersed in water, HAxBCM gels swelled ca.30 times of its dry weight, whereas the measured equilibrium swelling ratio for HAGMA2 and HAGMA1 gels were 62.0 ± 3.6 and 158.6 ± 32.6, respectively. Despite the large difference in their sol fraction, hydrogels prepared using inert BCMs without any crosslinkable acrylates swelled to a similar degree as those prepared using xBCMs. While physically entrapped micelles and/or physically entangled polymer chains can imbibe water, thus contributing to gel swelling, they are partially removed by repetitive washing as the soluble fraction. Compared to the HAGMA2, HAxBCM gels had the same polymer content (both are 2 wt%), yet its swelling ratio is half of that for HA-GMA2 gels. Arguably, the crosslinked HAGMA chains might be more water-imbibing than the entrapped BCMs containing a hydrophobic core.
Collectively, compared to the linear HAGMA chains, the microscopic multifunctional micelles are more efficient crosslinkers. While changing the crosslinking chemistry could reduce the sol fraction,45 our results here show that incorporation of microscopic crosslinkers offers an alternative strategy to increase the crosslinking efficiency. Certainly, acrylates are more reactive than methacrylates.46 However, we emphasize that the nanoscale micelle objects containing reactive acrylates installed on the PAA shell might be more efficient crosslinkers than the soluble HAGMA macromer.29 If HA is modified with acrylate groups, instead of methacrylate groups, the sol fraction could be further reduced.
3.2. FCS analysis
Fluorescence correlation spectroscopy (FCS) is a robust experimental technique widely used for the determination of translational diffusion coefficients of mixed populations of solutes.47–49 In our system, NR was used to fluorescently label BCMs; the free fluorophore has a low quantum yield of fluorescence in hydrophilic environments, but exhibits a 10 to 100-fold increase in fluorescence when sequestered in the hydrophobic compartments of micelles.49 FCS was performed using confocal microscopy that limits the observation volume to a fraction of a femtoliter. NR fluorophores were excited by focusing a 561 nm laser into the bulk solution using a 63×1.2 N.A. water immersion objective. As the fluorophores transit through the confocal volume, their emitted fluorescence photons are collected and autocorrelated over lag times, τ. For a single diffusing specie such as NR in PBS buffer, the autocorrelation function, G′(τ) is described by the following biophysical model:47
| Eq. 1 |
The first term of the autocorrelation function denotes the triplet state photo-kinetics, with amplitude, T and characteristic decay constant, τT. The second term describes N molecules with diffusion coefficient, D translating through a 3D Gaussian confocal volume defined by a half-axis width, ωo and height, zo. Typically, G′(τ) is normalized by N when comparing molecular diffusivity across different species or solvents.
Figure 3A shows the averaged and normalized autocorrection function, <G′(τ)>, for NR molecules in PBS, HAGMA (1 wt%) solution and HAGMA1 gel. For NR in PBS, a single species diffusion model was used to fit <G′(τ)>, yielding a diffusion coefficient of 304 μm2/s, which is in good agreement with reported values.50,51 Similar measurements performed with NR in HAGMA solutions and HAGMA1 gels yielded slower diffusion coefficients of 277 μm2/s and 254 μm2/s, respectively. The reduced NR mobility is due to an increase in the solution viscosity introduced by the entangled HA chains, as well as possible van der Waals interactions between NR and HAGMA.
Figure 3.
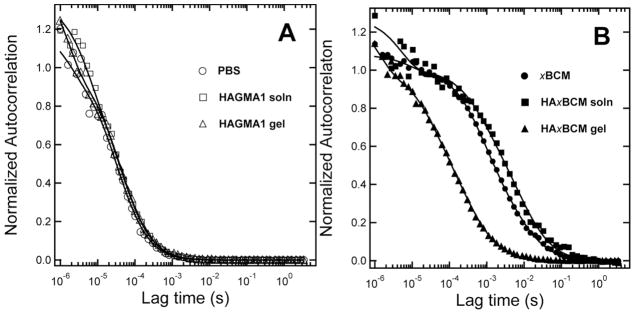
Molecular mobility of NR in different environments as revealed by FCS autocorrelation curves. (A): Free NR in PBS (open circle), HAGMA solution (open squares) or crosslinked HAGMA1 gel (open triangle). One-species model was used to calculate NR diffusivity. (B): xBCM-sequestered NR in PBS (filled circle), HAGMA solution (filled squares) or HAxBCM gel (filled triangle). Two-species model was used to calculate diffusivity of various NR-associated entities.
The autocorrelation of NR in xBCM and HAxBCM, both in the solution state and the gel state, is shown in Figure 3B. For samples containing xBCM, a one-species model did not accurately fit the data, thus, a two-species diffusion model was used:48
| Eq. 2 |
where m1 and m2 are the fractions of species 1 and species 2, and b1 and b2 represent the molecular brightness of species 1 and species 2. For xBCM in PBS (Figure 3B), the fit to <G′(τ)> yielded >99.9% population having D1 = 13 μm2/s and less than 0.1% with D2 = 1.0 μm2/s. The value of 13 μm2/s can be used to estimate the hydrodynamic radius of the diffusing species using the Stokes-Einstein equation: , where kB is the Boltzmann constant, T is the absolute temperature, η is the solution viscosity (of water) and D is diffusion coefficient. The calculated hydrodynamic radius of 19 nm is consistent with xBCM measurements from TEM micrographs. The slower diffusing species yields a mean hydrodynamic radius of 240 nm, which we interpret as xBCM aggregates. Micelles placed in a 1 wt% HAGMA solution diffused more slowly (D1 = 6.0 μm2/s, D2 = 0.6 μm2/s) compared to those in PBS (Figure 3B). The approximately two-fold reduction in diffusivity was expected because the viscosity of HAGMA solution is greater than PBS, and the secondary forces (e.g. H-bonds and hydrophobic interactions) between HA and micelles may further hinder the free diffusion of micelles.
In this analysis, it is important to take into account the relative brightness of the two species in order to correctly estimate their percentages, because the autocorrelation function varies as the square of the fluorescence intensity, or brightness. If we consider a mean radius of 19 nm for the micelles, the mean aggregate size is about (240 nm/19 nm)3-fold larger in volume, and therefore 12.63-fold brighter as compared to an individual xBCM. Thus, the autocorrelation function is strongly weighted by the relatively rare (<0.1%) xBCM aggregates. One particular strength of FCS analysis is the ability to detect such rare species, because millions of molecules can be sampled diffusing through the confocal volume in the 10 s observation time. Obtaining statistically meaningful data of rare species using TEM would require analyzing thousands of images.
The main motivation for using FCS analysis is to characterize the diffusive behavior of xBCMs in the crosslinked HA gel. Initially, all NR molecules were sequestered in the xBCMs. Once exposed to the aqueous media, some NR molecules were driven out of the micelle cores, as a result of the concentration gradient in and outside the micelles, thereby establishing an equilibrium state between the sequestered and the free NR molecules. Interestingly, the autocorrelation function obtained from HAxBCM gels (Figure 3B) was characterized by two diffusing species: a large population of fast moving species and a minor population of slow moving species. Fits to <G′(τ)> using the measured NR in crosslinked HAGMA1 gel as a fixed variable (254 μm2/s) yielded >99.9% free NR and <0.1% of a population that exhibited a diffusion coefficient of 25.4 μm2/s. The slower moving species corresponds to a particle with a 10 nm radius diffusing in PBS, or a ~5 nm particle diffusing in HAGMA gel, which is estimated by the apparent two-fold increase in viscosity observed with the BCMs in HAGMA solution. We interpret these results as evidence of xBCMs being immobilized in crosslinked HA networks; the crosslinked HA acts as a molecular sieve, allowing NR and smaller molecular species, possibly NR-associated unimers,52 to diffuse through the observation volume. Again, to accurately determine the abundance of each species, their relative brightness needs to be considered because free NR is weakly fluorescent, and in this analysis we used a conservative estimate of a 10-fold increase in brightness upon micelle association.49 Thus the FCS curve for BCMs entrapped in corsslinked HA networks suggests that the larger BCM particles are immobilized or severely restricted in diffusion, and the fluorescence fluctuations we observe arise from the diffusion of free NR through the gel matrix. If the BCM molecules were not immobilized and instead were diffusing though the gel, we would expect to see the autocorrelation function shifted towards 6.0 μm2/s (the diffusion coefficient for xBCM in HA-GMA solution) with as little as 1% mobile BCM particles. Collectively, our FCS results confirm the ability of xBCMs to sequester hydrophobic molecules and to react covalently with the polymerizing HAGMA chains.
3.3. Mechanical properties and enzymatic degradation
The viscoelastic properties of various hydrogels were first characterized by oscillatory rheometry using parallel-plate geometry with UV accessories. Prior to photocrosslinking, the viscosity of various hydrogel precursor solutions was analyzed (Figure S2). At the same polymer content of 2 wt%, the HAGMA2 solution is 3 times more viscous than the HAxBCM solution. Obviously, high molecular weight linear HAGMA chains are more prone to physical entanglement than the compact, similarly charged micelles of an equivalent weight. After the precursor solution was thoroughly mixed on the geometry for 2 min, the UV light was turned on and the elastic modulus (G′) instantly exceeded the loss modulus (G″) in less than 1 min for all types of hydrogels investigated (Figure 4A), with G′ reaching a plateau value within 5 min. Interestingly, the plateau modulus was reached much faster for HAxBCM gels (1.5 min) than the corresponding control gels (HAGMA2: 3.3 min) with an equal polymer mass but free of micelles. Owing to the low mobility of the HAGMA macromer, as well as the low reactivity of methacrylates as compared to acrylates, radicals formed by the dissociation of DMPA and subsequent polymerization of NVP react preferentially with the acrylate units on the more diffusible xBCMs.53 Consequently, xBCMs serve as accelerators for the crosslinking reaction. At frequencies of 0.01–10 Hz under a constant strain (1 %), the elastic and loss moduli for all four types of hydrogels are frequency-independent (Figure 4B), confirming the covalent nature of the networks. These solid-like, elastomeric materials exhibited G′ values of three orders of magnitude higher than the corresponding G″ values, with the damping ratio for all hydrogels tested being less than 0.02.
Figure 4.

Mechanical characterization of micelle-free (HAGMA1: square; HAGAM2: circle) and micelle-containing (HAxBCM: upward triangle; HApBCM: downward triangle) HA gels. (A, B): Rheological testing conducted in time sweep (A) and frequency sweep (B). G′: filled symbols; G″: open symbols. (C): Stress-strain curves obtained using a DMA under compression. Results shown are an average of three individal measurements.
Overall, the addition of xBCM to 1 wt% HAGMA doubled the gel modulus. In HAxBCM gels, xBCMs serve as covalent bridges to connect adjacent HA chains to establish elastically active chains in the network. In the absence of xBCMs, radicals formed by UV radiation react with the unsaturated methacrylates on HA to form the primary growing radicals. Because these radicals are attached to HA chains, they have lower mobility/reactivity and a significant fraction terminates by chain transfer to solvent (water) before they can react with GMA on other HA chains to form crosslinks. The bridging mechanism provided by xBCMs incorporates additional GMA units in the HA network and forms additional elastically active crosslinking points, resulting in higher modulus, lower swelling ratio and sol fraction. Of note, physical entrapment of BCMs in HAGMA gels compromised the gel mechanics. However, compared to HAGMA2 (G′ = 1104 ± 101 Pa), HAxBCM were softer (G′ = 847 ± 43 Pa). This is understandable because a covalently crosslinked HA network responds to mechanical perturbation differently than the BCM held together by hydrophobic interactions between PnBA chains; such physical association is easier to disrupt.
Hydrogels were subsequently analyzed under compression using a DMA. Representative stress-strain curves are presented in Figure 4C. Linear viscoelastic regions were observed at strains <20%. The up-turn at high strains (>30%), commonly observed in hydrogel materials, can be attributed to the material densification and/or strain hardening.54,55 The compressive moduli, calculated from the slope of the stress–strain curve in the linear region, are summarized in Table S2. Gel stiffness was found to be in good agreement with the rheological testing, with HAGMA2 being the stiffest and HApBCM being the softest. Covalent integration of xBCMs to HAGMA1 increased the ultimate compression stress at break from 93.1 ± 19.7 kPa to 134.9 ± 13.4 kPa (p<0.05) (Table S2), at the same time slightly reducing the corresponding strain (from 68.9 ± 2.1 to 65.1 ± 2.2 %, p<0.05). The compression toughness of the HAxBCM gels is similar to HAGMA2 gels but is significantly higher than HApBCM and HAGMA1 gels (p<0.05). Physical entrapment of BCMs in HAGMA1 did not significantly alter the compressive properties of the hydrogels.
It is clear that hydrogels containing physically trapped BCMs are statistically softer (p <0.05) than those containing covalently integrated xBCMs. When HApBCM gels are compressed, the mechanical force applied to the gels cannot be directly transmitted to the entrapped particles due to the absence of particle–matrix coupling and the presence of a depletion zone around the particles.31 On the other hand, the presence of the covalent linkages between the individual particles and their surrounding matrix in HAxBCM gels permits the external forces to be directly transmitted to the particles, resulting in particle deformation without compromising the overall matrix strength. We have observed the force-induced BCM deformation in a polyacrylamide-based hydrogel.30
For tissue repair and regeneration purposes, it is desirable that the synthetic matrices be enzymatically degradable. When exposed to 5 U mL−1 HAase at pH 7.4, all three types of hydrogels, namely HAGMA2, HApBCM and HAxBCM, became smaller and more fragile. In terms of HA loss with respect to the total gel mass, the apparent degradation, as a function of time shown in Figure 5A suggests that HAxBCM and HApBCM are enzymatically more stable than the corresponding bulk gels free of BCMs. Uronic acid produced at day 10 accounted for approximately 40.8 ± 2.5 %, 46.2 ± 2.4 % and 88.2 ± 6.1 % of the gel mass, respectively. As discussed above, HAxBCM might be more efficiently crosslinked than HAGMA2. Consequently, the network is more restrictive to HAase. However, 50% of the gel mass (xBCM) is not degradable by HAase. When normalized to the percent HA disappearance (Figure 5B), it is obvious that the interconnected HA chains in HAxBCM and HApBCM gels were as susceptible to degradation as those in BCM-free gels. The ability of HAase to degrade the HA-based hydrogels suggests that the chemical modification and covalent crosslinking did not significantly alter the biological identity of HA.
Figure 5.

Enzymatic degradation of HAGMA2 (black square), HApBCM (red circle) and HAxBCM (blue triangle) gels quantified by carbazole assay. (A): Apparent degradation calculated from the absolute mass loss; (B): Normalized degradation calculated by normalizing to the HA content in each gel.
3.4. DEX release
DEX-releasing hydrogels were prepared by radical crosslinking of HAGMA in the presence of free DEX or BCM-sequestered DEX. The release study was first conducted statically under the sink condition. Our results showed that DEX release kinetics strongly depended on the hydrogel composition and microstructure (Figure 6). DEX release from HAGMA2 gels is characterized by an initial burst within 2 h, followed by a release at a slower rate over seven days. Overall, DEX molecules were released rapidly from HAGMA2, cumulating 84.2 ± 6.9% in 24 hour. By day 3, close to 91% DEX initially loaded was released. Direct loading of the hydrophobic DEX molecules in HA matrix does not offer a practical means for controlling DEX release owing to drug precipitation and aggregation in the network. Physical encapsulation of DEX-loaded BCMs in a secondary HA matrix retarded the drug release because an additional diffusion pathway (hydrophobic core) is present. After the initial burst release, a steady release of 20.3 ± 1.5 μg/day was observed, reaching a cumulative release of 53.3 ± 3.2% at day 7. DEX release from HAxBCM gels was further retarded, with the initial burst within 2 h reduced to 12.3 ± 1.5%. A steady release of 19.3 ± 0.6 μg/day over the course of seven days was achieved, reaching a cumulative level of 29.9 ± 1.5% by day 7. DEX release occurs by diffusion from the BCMs into the gel matrix, and subsequent diffusion through the gel into the external medium. For DEX to diffuse out of the hydrophobic core, one has to consider the degree of association of DEX with PnBA, in comparison to diffusion. Of note, inter-micellar crosslinking cannot be ruled out during the synthesis of HAxBCM gels, especially considering the close proximity of acrylates on the surface of individual micelles. Micelle shell crosslinking further inhibits DEX’s ability to diffuse out of the micelle. The kinetic chains established between the BCMs and the secondary HA matrix provided a reinforcement mechanism that is conducive to reaching a stable and slow release pattern of DEX. The release experiments were terminated at day 7 before 100% DEX release was achieved. The remaining DEX left in the gels was recovered by DMSO extraction (data not shown).
Figure 6.

In vitro release profiles of DEX from HAGMA2 (square), HAxBCM (circle) and HApBCM (triangle) gels immersed in PBS at ambient temperature under static conditions. Data shown are average values of three independent experiments.
In our previous proof-of-concept investigations, pyrene, a hydrophobic dye, entrapped in xBCM prior to the polymerization and crosslinking of polyacrylamide, was found to be released in a step-wise fashion by the external stretch.30 In moving cartilage, the predominant mode of mechanical force is compression. A 1 h-on-1-h-off cyclic compression regimen was imposed on hydrogel samples using a DMA for a total of 8 h at a strain of 0, 15 or 30% (Figure 7A). When the as-synthesized gels were used, the modulatory effects of the mechanical stimulation were not manifested (data not shown). We speculate that the initial burst release, resulting from the rapid swelling of the gel networks during the first 3 hours and drug molecules physically adsorbed on the BCM surface, masked out the mechanical effects. Consequently, hydrogel samples were immersed in H2O for 3 h until the DEX release reached the second steady phase of slow release (see above) before the dynamic release study was initiated. As shown in Figure 7B for HAxBCM gels, in the absence of compression, DEX was released from the gels at a rate of 5.9 ± 1.2 μg/h. DEX release was significantly increased when the HAxBCM gels were compressed. Within the first hour of dynamic compression up to 30% strain, DEX was released at a rate of 345.2 ± 19.7 μg/h, accounting for 37% of drug initially loaded, approximately 1.6 times faster than the rate observed from the same gel compressed to 15%. While the release rate remained unchanged for the static controls during the subsequent cycles, an accelerated release during compression and diminished release during the rest period was observed for the 30% and 15% dynamic compression. While the undulating pattern of release rate was maintained throughout the course of the release study, the relative difference in magnitude among the three samples tested gradually narrowed. Figure 7C shows the cumulative release profiles over the course of 8 h. When subjected to 30% compression for 8 h, DEX release from HAxBCM reached a cumulative level of 60%, approximately 20% more than those exposed to 15% strain.
Figure 7.

Effects of dynamic compression (A) on DEX release from HAxBCM (B, C) and HApBCM (D, E) gels. (A): Mechanical stimulations applied to the hydrogel samples. (B, D): DEX release rate as function of time under different release conditions. Release rate was measured every hour for up to 8 h, and dotted lines connecting each data point are used to reveal the trend. (C, E): Cumulative DEX release as function of time under different release conditions.
Control experiments were performed to analyze the effect of compression on DEX release from HApBCM and HAGMA. As shown in Figure 7 (D, E), DEX release from HApBCM was minimally affected by compression, although 30% compression within the first hour resulted in a 1.5 fold increase in DEX release rate relative to the static and controls subjected to 15% compression. Thereafter, HApBCM gels showed a minimal mechano-responsiveness. Similar results were observed for DEX release from HAGMA gels free of micelles (Figure S3). The moderate mechanical effects seen within the first hour for both HApBCM and HAGMA gels are attributed to a greater concentration gradient between the gels and the surrounding media. The lack of covalent attachment between BCMs and the HAGMA network in the case of HApBCM gels prohibits the macroscopic deformation from being transmitted to the physically entrapped micelles. HAGMA gels, on the other hand, lack mechano-responsive drug depots, thus unable to respond to the mechanical forces.
Collectively, these results suggest that the release behavior of DEX encapsulated in HAxBCM gels can be tuned by compression forces. The hydrophobic drug molecules, encapsulated in the hydrophobic micelle cores through the hydrophobic association, could diffuse through the hydrogel matrix with a diffusion coefficient dependent on the concentration gradient. In the presence of compression, block copolymer micelles, which are covalently linked to the hydrogel matrix, may be deformed perpendicular to the compression direction. Consequently, the hydrophobic association between DEX and the micelle core became weaker and DEX release rate increased accordingly.
3.5. Anti-inflammatory functions
In order to evaluate the anti-inflammatory functions of DEX-loaded HA hydrogels, macrophages cultured on tissue culture plates were exposed to different hydrogel compositions for 1 h, followed by 8 h LPS activation in the absence of any gel disks. Such treatment regimen did not compromise cell viability, nor did it alter the overall cell density across different samples during the tests (Figure 8A). A comparison between cells without any treatments (negative control) and those treated with LPS only (positive control) confirms that the LPS activation procedure was effective and other culture protocols did not inadvertently activate the macrophages. The anti-inflammatory effect of the hydrogel carriers free of DEX (Group 1, Figure 8B) is evident. Compared to cells cultured in the absence of any vehicle controls, cells cultured with all three types of HA gels reduced the TNF-α level by approximately two-fold (p<0.05). Such inhibitory effects can be attributed to the anti-inflammatory properties of the soluble HA components in all three types of gels eluted into the media during culture.23 No statistical difference across the three types of HA gel samples was observed.
Figure 8.

Effects of hydrogel exposure and LPS treatment on macrophage viability and TNF-α production analyzed by Picogreen DNA assay (A) and ELISA (B), respectively. Macrophages were subjected to 1-h exposure to DEX-free hydrogel carriers (Group 1) and DEX-releasing hydrogels (Group 2), followed by 8-h LPS activation. Cells without any gel or DEX treatment but were subjected to the 8-h LPS activation were included as the positive controls, and cells without any treatment were included as the negative controls. Values reported are the average of three separate measurements; *: statistically different (p<0.05). TNF-α expressions in Group 1 and 2 are all significantly lower (higher) than the positive (negative) controls.
On the other hand, TNF-α secretion by cells treated with DEX-containing hydrogels (Group 2) was significantly lower (p<0.05) than those treated with DEX-free gel carriers (Group 1). Relative to the respective DEX-free gel carriers, DEX-releasing HAxBCM, HApBCM and HAGMA2 gels significantly reduced cellular production of TNF-α by 4.6- (p<0.05), 3.0- (p<0.05) and 4.1-fold (p<0.05), respectively. Within Group 2, various gel compositions caused cells to secret different amounts of TNF-α. Specifically, cells treated with DEX-containing HAxBCM gels produced the least amount of pro-inflammatory cytokine (242.4 ± 31.1 pg/mL), while cells treated with DEX-releasing HAGMA2 gels produced the highest amount of TNF-α (352.0 ± 62.4 pg/mL).
Importantly, identical amounts of DEX were loaded into various gels that were indirectly exposed to the cultured macrophages. Although no statistical significance was observed among cells in Group 2, in terms of their TNF-α production, our in vitro release study conducted in PBS revealed that after 1-h incubation, 35.6 ± 3.6%, 9.5 ± 1.7% and 14.6 ± 0.7 % of DEX was released from the HAGMA2, HAxBCM and HApBCM gels, respectively. Considering the fact that the BCMs are entrapped in a protein-repellant, hydrated HA network and are further stabilized via the covalent coupling with the surrounding network, in the case of HAxBCM, the destabilizing effects of serum proteins on the entrapped BCMs are minimal.56 Therefore, it is reasonable to suggest that DEX release in serum-containing cell culture media follows a similar trend as that observed in serum-free PBS. Thus, cells in Group 2 received different amounts of DEX during the first hour of incubation. Collectively, our results suggest that the pre-treatment of DEX-loaded hydrogels attenuated the macrophage inflammatory response markedly and offset the subsequent LPS activation effectively. Our results underscore the importance of controlling the temporal drug presentation to maximize the drug efficacy. Overall, we conclude that compared to HAGMA and HApBCM gels, the HAxBCM gel preserves the activity of DEX most effectively and is a superior mechano-responsive vehicle for DEX.
A limitation of the HAxBCM hydrogel platform is the need to remove the majority of burst-released drug before the force-modulated mechanism is manifested. Future studies will be directed at modifying the hydrogel composition to enhance drug loading and to reduce burst release. Due to technical challenges associated with performing macrophage culture in the presence of mechanically stimulated, DEX-releasing hydrogels, our studies only provide the anti-inflammatory effecacy of the released DEX. In our on-going in vivo studies, mice with OA-like symptoms are treated with intraarticular hydrogel injections, followed by regular treadmill running. Using this biologically relevant model, we are assessing the utility of our mechanically responsive hydrogels in symptomatic relief and disease modification.
4. Conclusion
A new type of mechano-responsive hydrogels containing covalently integrated drug depots was synthesized by radical co-polymerization of HAGMA with crosslinkable micelles (xBCMs) assembled from 2-hydroxylethyl acrylate-modified PAA-b-PnBA. Compared to the HAGMA macromer, xBCMs were more efficient crosslinkers and the resultant micelle-integrated hydrogels are highly elastic, exhibiting an elastic modulus of 847 ± 43 Pa and a damping ratio of <0.02. The covalent immobilization the BCMs in crosslinked HA networks severely restricted the mobility of the micelles, but permitted compressive force-modulated DEX release. The released DEX, in conjunction with the soluble HA components, synergistically primed macrophages against LPS activation. This type of novel hydrogels will find applications in the management and repair of pathologically compromised tissues that are normally mechanically active.
Supplementary Material
Acknowledgments
This work was supported in part by the National Science Foundation (DMR-0643226 to XJ; DMR-0906815 to DJP) and National Institutes of Health (NIDCD: 3R01DC008965 to XJ). CRS acknowledges support from the National Science Foundation (EPSCoR EPS-0814251) and the State of Delaware. We thank Dr. Chaoying Ni and Mr. Frank Kriss for TEM assistance. We also appreciate Dr. Steve Bai and Dr. Guangjin Hou for their help with magic angle NMR analyses.
Footnotes
Supporting Information Available. 1H NMR characterization of the crosslinking reaction, viscosity measurements of various hydrogel precursor mixtures, the effect of dynamic compression on DEX release from HAGMA gels, a summary of hydrogel swelling and mechanical properties. This information is available free of charge via the Internet at http://pubs.acs.org/
References
- 1.Grieshaber S, Jha A, Farran AE, Jia X. In: Biomaterials for Tissue Engineering Applications. Burdick J, Mauck R, editors. Springer; Vienna: 2011. p. 9. [Google Scholar]
- 2.Akala EO, Kopečková P, Kopeček J. Biomaterials. 1998;19:1037. doi: 10.1016/s0142-9612(98)00023-4. [DOI] [PubMed] [Google Scholar]
- 3.Qiu Y, Park K. Adv Drug Delivery Rev. 2001;53:321. doi: 10.1016/s0169-409x(01)00203-4. [DOI] [PubMed] [Google Scholar]
- 4.Hu J, Zhang G, Liu S. Chem Soc Rev. 2012;41:5933. doi: 10.1039/c2cs35103j. [DOI] [PubMed] [Google Scholar]
- 5.Miyata T, Uragami T, Nakamae K. Adv Drug Delivery Rev. 2002;54:79. doi: 10.1016/s0169-409x(01)00241-1. [DOI] [PubMed] [Google Scholar]
- 6.Miyata T, Asami N, Uragami T. Nature. 1999;399:766. doi: 10.1038/21619. [DOI] [PubMed] [Google Scholar]
- 7.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Proc Natl Acad Sci U S A. 2003;100:5413. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murakami Y, Maeda M. Biomacromolecules. 2005;6:2927. doi: 10.1021/bm0504330. [DOI] [PubMed] [Google Scholar]
- 9.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Science. 2009;324:59. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glazer PJ, van Erp M, Embrechts A, Lemay SG, Mendes E. Soft Matter. 2012;8:4421. [Google Scholar]
- 11.Xulu PM, Filipcsei G, Zrinyi M. Macromolecules. 2000;33:1716. [Google Scholar]
- 12.Lee KY, Peters MC, Anderson KW, Mooney DJ. Nature. 2000;408:998. doi: 10.1038/35050141. [DOI] [PubMed] [Google Scholar]
- 13.Tong Z, Jia X. MRS Communications. 2012;2:31. doi: 10.1557/mrc.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang JH, Thampatty BP. Biomech Model Mechanobiol. 2006;5:1. doi: 10.1007/s10237-005-0012-z. [DOI] [PubMed] [Google Scholar]
- 15.Goldring MB, Goldring SR. Journal of Cellular Physiology. 2007;213:626. doi: 10.1002/jcp.21258. [DOI] [PubMed] [Google Scholar]
- 16.Shyy JY, Chien S. Circulation Research. 2002;91:769. doi: 10.1161/01.res.0000038487.19924.18. [DOI] [PubMed] [Google Scholar]
- 17.Mammoto A, Connor KM, Mammoto T, Yung CW, Huh D, Aderman CM, Mostoslavsky G, Smith LE, Ingber DE. Nature. 2009;457:1103. doi: 10.1038/nature07765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu X, Jha AK, Harrington DA, Farach-Carson MC, Jia XQ. Soft Matter. 2012;8:3280. doi: 10.1039/C2SM06463D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prestwich GD, Marecak DM, Marecek JF, Vercruysse KP, Ziebell MR. J Controlled Release. 1998;53:93. doi: 10.1016/s0168-3659(97)00242-3. [DOI] [PubMed] [Google Scholar]
- 20.Burdick JA, Prestwich GD. Adv Mater (Weinheim, Ger) 2011;23:H41. doi: 10.1002/adma.201003963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kogan G, Soltes L, Stern R, Gemeiner P. Biotechnol Lett. 2007;29:17. doi: 10.1007/s10529-006-9219-z. [DOI] [PubMed] [Google Scholar]
- 22.Jia X, Burdick JA, Kobler J, Clifton RJ, Rosowski JJ, Zeitels SM, Langer R. Macromolecules. 2004;37:3239. [Google Scholar]
- 23.Chen WYJ, Abatangelo G. Wound Repair Regen. 1999;7:79. doi: 10.1046/j.1524-475x.1999.00079.x. [DOI] [PubMed] [Google Scholar]
- 24.Etscheid M, Beer N, Dodt J. Cell Signal. 2005;17:1486. doi: 10.1016/j.cellsig.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 25.Divine JG, Zazulak BT, Hewett TE. Clin Orthop Relat Res. 2007;455:113. doi: 10.1097/BLO.0b013e31802f5421. [DOI] [PubMed] [Google Scholar]
- 26.Hahn SK, Jelacic S, Maier RV, Stayton PS, Hoffman AS. J Biomater Sci Polym Ed. 2004;15:1111. doi: 10.1163/1568562041753115. [DOI] [PubMed] [Google Scholar]
- 27.Patil NS, Dordick JS, Rethwisch DG. Biomaterials. 1996;17:2343. doi: 10.1016/s0142-9612(96)00089-0. [DOI] [PubMed] [Google Scholar]
- 28.Ito T, Fraser IP, Yeo Y, Highley CB, Bellas E, Kohane DS. Biomaterials. 2007;28:1778. doi: 10.1016/j.biomaterials.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 29.Xiao L, Liu C, Zhu J, Pochan DJ, Jia X. Soft Matter. 2010;6:5293. doi: 10.1039/C0SM00511H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao L, Zhu J, Londono JD, Pochan DJ, Jia X. Soft Matter. 2012;8:10233. doi: 10.1039/C2SM26566D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jha AK, Malik MS, Farach-Carson MC, Duncan RL, Jia X. Soft Matter. 2010;6:5045. doi: 10.1039/C0SM00101E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Culbertson CT, Jacobson SC, Michael Ramsey J. Talanta. 2002;56:365. doi: 10.1016/s0039-9140(01)00602-6. [DOI] [PubMed] [Google Scholar]
- 33.Bitter T, Muir HM. Anal Biochem. 1962;4:330. doi: 10.1016/0003-2697(62)90095-7. [DOI] [PubMed] [Google Scholar]
- 34.Tong Z, Sant S, Khademhosseini A, Jia X. Tissue Eng Part A. 2011;17:2773. doi: 10.1089/ten.tea.2011.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gerdin B, Hällgren R. J Intern Med. 1997;242:49. doi: 10.1046/j.1365-2796.1997.00173.x. [DOI] [PubMed] [Google Scholar]
- 36.Oerlemans C, Bult W, Bos M, Storm G, Nijsen JF, Hennink W. Pharm Res. 2010;27:2569. doi: 10.1007/s11095-010-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu J, Zhang S, Zhang F, Wooley KL, Pochan DJ. Adv Funct Mater. 2013;23:1767. [Google Scholar]
- 38.Colombani O, Ruppel M, Schubert F, Zettl H, Pergushov DV, Müller AHE. Macromolecules. 2007;40:4338. [Google Scholar]
- 39.Sperling LH. Introduction to Physical Polymer Science. Wiley; 2005. [Google Scholar]
- 40.Dilova V, Zlatarova V, Spirova N, Filcheva K, Pavlova A, Grigorova P. Boll Chim Farm. 2004;143:20. [PubMed] [Google Scholar]
- 41.Kim DH, Martin DC. Biomaterials. 2006;27:3031. doi: 10.1016/j.biomaterials.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 42.Webber MJ, Matson JB, Tamboli VK, Stupp SI. Biomaterials. 2012;33:6823. doi: 10.1016/j.biomaterials.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ward WK, Hansen JC, Massoud RG, Engle JM, Takeno MM, Hauch KD. J Biomed Mater Res A. 2010;94A:280. doi: 10.1002/jbm.a.32651. [DOI] [PubMed] [Google Scholar]
- 44.Burdick JA, Lovestead TM, Anseth KS. Biomacromolecules. 2002;4:149. doi: 10.1021/bm025677o. [DOI] [PubMed] [Google Scholar]
- 45.Pritchard CD, O’Shea TM, Siegwart DJ, Calo E, Anderson DG, Reynolds FM, Thomas JA, Slotkin JR, Woodard EJ, Langer R. Biomaterials. 2011;32:587. doi: 10.1016/j.biomaterials.2010.08.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Odian G. Principles of Polymerization. John Wiley & Sons, Inc; Hoboken, NJ: 2004. [Google Scholar]
- 47.Maiti S, Haupts U, Webb WW. Proc Natl Acad Sci U S A. 1997;94:11753. doi: 10.1073/pnas.94.22.11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thompson N. In: Topics in Fluorescence Spectroscopy. Lakowicz J, editor. Vol. 1. Springer US; 2002. p. 337. [Google Scholar]
- 49.Erhardt R, Zhang M, Böker A, Zettl H, Abetz C, Frederik P, Krausch G, Abetz V, Müller AHE. J Am Chem Soc. 2003;125:3260. doi: 10.1021/ja028982q. [DOI] [PubMed] [Google Scholar]
- 50.Fu Y, Ye F, Sanders WG, Collinson MM, Higgins DA. J Phys Chem B. 2006;110:9164. doi: 10.1021/jp054178p. [DOI] [PubMed] [Google Scholar]
- 51.Sutter M, Oliveira S, Sanders N, Lucas B, Hoek A, Hink M, Visser AWG, Smedt S, Hennink W, Jiskoot W. J Fluoresc. 2007;17:181. doi: 10.1007/s10895-007-0156-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hadjichristidis N, Pispas S, Floudas G. Block Copolymers: Synthetic Strategies, Physical Properties, and Applications. John Wiley & Sons, Inc; Hoboken, NJ: 2003. [Google Scholar]
- 53.Sarvestani AS, Xu W, He X, Jabbari E. Polymer. 2007;48:7113. doi: 10.1163/156856207782246821. [DOI] [PubMed] [Google Scholar]
- 54.Gent AN. J Rheol. 2005;49:271. [Google Scholar]
- 55.Miquelard-Garnier G, Creton C, Hourdet D. Soft Matter. 2008;4:1011. doi: 10.1039/b717460h. [DOI] [PubMed] [Google Scholar]
- 56.Lu J, Owen SC, Shoichet MS. Macromolecules. 2011;44:6002. doi: 10.1021/ma200675w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.