

Abstract
Background
We investigated the protective effects of mitochondrial-targeted antioxidant and protective peptides, SS31 and SS20, on cardiac function, proteomic remodeling and signaling pathways.
Methods and Results
We applied an improved label-free shotgun proteomics approach to evaluate the global proteomics changes in transverse aortic constriction (TAC) induced heart failure, and the associated signaling pathway changes using Ingenuity Pathway Analysis (IPA). We found 538 proteins significantly changed after TAC, which mapped to 53 pathways. The top pathways were in the categories of actin cytoskeleton, mitochondrial function, intermediate metabolism, glycolysis / gluconeogenesis and citrate cycle. Concomitant treatment with SS31 ameliorated the congestive heart failure phenotypes and mitochondrial damage induced by TAC, in parallel with global attenuation of mitochondrial proteome changes, with an average of 84% protection of mitochondrial and 69% of non-mitochondrial protein changes. This included significant amelioration of All the IPA pathways noted above. SS20 had only modest effects on heart failure and this tracked with only partial attenuation of global proteomics changes; furthermore, while actin cytoskeleton pathways were significantly protected in SS20, mitochondrial and metabolic pathways essentially were not.
Conclusions
This study elucidates the signaling pathways significantly changed in pressure-overload induced heart failure. The global attenuation of TAC-induced proteomic alterations by the mitochondrial targeted peptide SS-31 suggests that perturbed mitochondrial function may be an upstream signal to many of pathway alterations in TAC and supports the potential clinical application of mitochondrial-targeted peptide drugs for the treatment heart failure.
Keywords: heart failure, mitochondria, proteomics, signal transduction
Pressure overload by chronic hypertension is a common cause of congestive heart failure1. Although multiple studies have reported several signaling mechanisms involved in cardiac hypertrophy and heart failure2, studies at the level of global proteomic changes offer the potential to provide much more comprehensive insights. Proteomics analysis has been applied in the investigation of human and experimental models of heart failure 3–5. A conventional two-dimensional gel electrophoresis coupled with isotope-coded affinity tag (iCAT) has identified 123 proteins differentially expressed during left ventricular hypertrophy4. Another recent study applying isobaric tags for relative and absolute quantification (iTRAQ) found 62 proteins significantly changed by 1.5 fold after transverse aortic constriction (TAC)5.
Mitochondrial damage and dysfunction is well documented in patients and experimental animal models of heart failure, which include mitochondrial oxidative damage6, impaired mitochondrial respiration7 impaired fatty acid utilization and increased glucose metabolism8. We have previously demonstrated that mitochondrial reactive oxygen species (ROS) play a central role in mitochondrial damage and the development of cardiac hypertrophy and failure in aging9, hypertensive cardiomyopathy6, 10 and pressure overload11. Scavenging mitochondrial ROS by catalase targeted to cardiomyocyte mitochondria (mCAT) significantly attenuated the hypertrophy and failure phenotypes6 as well as mitochondrial proteome remodeling associated with TAC11.
While the mCAT model provides a useful experimental model, translation to clinical practice requires a pharmacologic intervention. The Szeto-Schiller (SS)-tetrapeptides belong to a family of aromatic cationic peptides that are selectively concentrated 1000-fold in the mitochondrial inner membrane12. The SS31 peptide (D-Arg-2′,6′-dimethyltyrosine-Lys-Phe-NH2) has a direct antioxidant capacity and has been shown to scavenge various free radicals13, reduce mitochondrial ROS in neonatal cardiomyocytes treated with Angiotensin II10, promote mitochondrial respiration and ATP production, and inhibit cytochrome c mediated apoptosis in several cell types13, as well as mitigating several animal disease models associated with mitochondrial oxidative stress, including ischemia-reperfusion injury14, insulin resistance induced by high-fat diet15 and hypertensive cardiomyopathy10. A recent study in the renal ischemia-reperfusion model demonstrates that SS31 binds cardiolipin to prevent cardiolipin peroxidation by cytochrome c peroxidase, preserve mitochondrial cristae integrity, accelerate restoration of ATP and mitigate apoptosis and renal dysfunction16. The SS20 peptide (Phe-D-Arg-Phe-Lys-NH2) does not have direct ROS scavenging activity in vitro 12, 13 but does enhance mitochondrial respiration and acts as an indirect antioxidant by reducing ROS production12. It has also been shown to be effective in reducing cardiac ischemia-reperfusion injury17; however, SS-20 is less effective than SS-31 in some animal models.
We have described algorithms for chromatographic alignment of LC-MS data that greatly improve the sensitivity to detection of differences in abundance between complex protein mixtures without the use of heavy label18. In the present study we: 1) applied a label-free method to characterize the global proteomics changes in the pressure-overload induced heart failure, 2) applied Ingenuity pathway analysis (IPA) to investigate the signaling pathways in heart failure, 3) compared the efficacy of mitochondrial targeted peptides to attenuate TAC-induced heart failure in parallel with changes in the cardiac proteome.
Methods
Please see online supplement for details
TAC surgery, echocardiography and tissue analysis
All animal experiments were approved by the University of Washington Institutional Animal Care and Use Committee. Twenty nine 16–20 week-old C57Bl6 (Charles River) male mice were included in the experiment: 5 saline-sham, 9 saline-TAC, 6 SS20-TAC and 9 SS31-TAC. surgeries were performed as described11. Briefly, mice were anesthetized with pentobarbital (75mg/kg,ip). TAC was created using a 7–0 ligature around the vessel and a 27-gauge blunt needle. Osmotic minipumps (Alzet 1004) with each SS-peptides (3 mg/kg/d of SS20 or SS31, kindly provided by Peter W. Schiller, Universite de Montreal) or saline were inserted subcutaneously on the flank area right after the TAC surgery. M-mode echocardiography was performed at baseline and at the end of experiments (4 weeks after TAC) using Acuson CV70 (13 MHz probe), under isoflurane 0.5%9. Quantitative PCR was performed using Taqman Gene Expression Assays. Cardiac mitochondrial protein carbonyls were measured by OxiSelect ELISA (Cell Biolabs, San Diego, CA).
Electron microscopy was performed on a JEOL JEM 1200EXII transmission electron microscope. Quantitative analysis was performed blindly from 8–10 images per sample (10,000× magnification). Cristae per μm were measured perpendicular to the cristae axis19. Autophagosomes or autolysosomes were identified by the characteristic double or multi-lamellar membrane structure surrounding compressed mitochondria or membrane bound electron-dense material, and were counted per 100 μm2.
Proteomics Analysis
Proteomics analysis was performed for 5–7 mice from each group. Left ventricles were homogenized in cold buffer (250 mM sucrose, 1 mM EGTA, 10 mM HEPES, 10 mM Tris-HCl pH7.4). Homogenate was suspended with 0.1% Rapigest (Waters Corporation, Milford, MA), boiled for 5 min, treated with 5 mM DTT (60°C, 30 minutes), then 15 mM iodoacetamide (22°C, 30 minutes), followed by trypsin (1/100 protein) digestion at 37°C for 2 hours. The digested samples were treated with HCl (200 mM, 37°C, 30 minutes), centrifuged for 10 minutes at 20,000 × g and the supernatant cleansed using Waters Oasis MCX extraction columns. The samples were analyzed by liquid chromatography (LC, Waters nanoAcquity) and mass spectrometry (MS/MS, Thermo Scientific LTQ-FT Ultra).
High resolution MS data was processed by Bullseye20 to assign the accurate precursor ion mass to the low resolution tandem mass spectrometry data. MS/MS spectra were searched by SEQUEST (ver.27) against a mouse IPI database. Peptide spectrum match false discovery rates were determined by the Percolator algorithm21 with a q-value threshold of 0.01. Parsimonious protein inference was determined using the IDPicker algorithm 22. To reduce the effect of chromatographic drift during MS and allow comparisons of low abundance analytes that may be detected in one but not the other samples, we aligned the LC-MS/MS chromatograms. Briefly, for two given LC-MS/MS chromatograms, the MS/MS scan number for peptides identified in both samples were plotted against each other in a scatter plot. A LOESS regression was used to find the best fit line through the data points (Supplemental Figure 1). For peptides that were identified in one sample, we used the identified peptide’s MS/MS scan number and the LOESS regression to identify an approximate time range in the other samples. A matching chromatographic peak was then searched for within that time range. The data is available at https://sites.google.com/a/uw.edu/maccoss/home/supplementary_data/sstac
Statistical analysis
Data in Figures 1–3 are presented as means ± SEM. One-way ANOVA was used to compare differences among groups, followed by post-hoc tests for significance, with Bonferroni adjustment. P<0.05 was considered significant. For proteomics data, in order to determine statistically significant changes of proteins between experimental groups, we used a linear model of peptide abundance to calculate fold changes of proteins between experimental groups in the same manner as a two-sample t-test using the R/Bioconductor software. For the cases where a protein consisted of more than one peptide, the linear model was modified to account for the multiple peptides by using a blocking factor. This gave p-values that were adjusted for multiplicity with the Bioconductor package q-value, which allows for selecting statistically significant genes while controlling the estimated false discovery rate. MITOP2 protein list was applied to identify mitochondrial proteins (http://www.mitop.de:8080/mitop2/).
Figure 1. Echocardiography measurement after 4 weeks of TAC.

(A) Left ventricular mass index (LVMI). (B) Fractional shortening significantly declined in saline-TAC; this was significantly ameliorated by SS20 and more so by SS31, but not by SS48. (C) Interventricular septum (IVS), left ventricular posterior wall (LVPW) thickness and left ventricular end-diastolic dimension (LVEDD). *p<0.05 compared with sham.
Figure 3. Mitochondrial damage in TAC-induced heart failure.

(A) Mitochondrial protein carbonyl content (nmol/mg) measured by enzyme immunoassay. (B) Western blots of microtubule associated protein light chain 3 (LC3) demonstrated significant increase in LC3-II, a marker of activated autophagosomes, in saline-TAC hearts, which was significantly attenuated in SS31-TAC and partially attenuated in SS20-TAC hearts. Representative electron micrograph of cardiac apices in sham (C), saline-TAC (D) and SS31-TAC (E) demonstrated increased numbers of damaged mitochondria and autophagosomes (arrows) in saline-TAC hearts. (F) Higher magnification of mitochondria showed disrupted cristae in saline-TAC hearts (left) compared to normal cristae in SS31-TAC hearts (right panel). (G) Quantitative analysis of the number of autophagosomes (per 100μm2) and mitochondrial cristae density (cristae/μm). *p<0.05 for sham vs. saline-TAC, #p<0.05 SS31-TAC vs. saline-TAC.
The networks and canonical pathways were generated by Ingenuity Pathway Analysis (IPA, Ingenuity Systems, www.ingenuity.com) using inputs of gene identifiers, log2 fold-changes and p-values between two-group comparisons. A p<0.05 cutoff and absolute fold change >1.5 was set to identify molecules whose expression was significantly differentially regulated. Canonical pathway analysis identified the pathways from the IPA library of canonical pathways that were significant to the dataset by following methods: 1) A ratio of the number of molecules from the dataset that map to the pathway divided by the total number of molecules that map to the canonical pathway is displayed. 2) Fisher’s exact test was used to calculate a p-value reflecting the probability that the association between the genes in the dataset and the canonical pathway is explained by chance alone.
Results
Mitochondrial-targeted SS31 peptide ameliorates pressure-overload induced heart failure
Pressure overload by TAC for 4 weeks induced significant LV hypertrophy in mice, as shown by a greater than 2-fold increase in echocardiographic LV mass index (LVMI, p=0.03, Figure1A). Systolic function is markedly impaired, indicated by a significant decline of fractional shortening (FS) in saline-TAC, when compared with sham (28.3±1.1 vs. 53.2 ± 2.2, respectively, p<0.001, Figure1B). The decline in FS was significantly ameliorated by concomitant treatment with either SS20 or SS31 (SS20: 43.5 ± 5.7, p=0.013; SS31: 50.4 ± 2.2, p<0.001, both compared with saline-TAC), to levels comparable to sham. The LV chamber was significantly dilated after TAC (p=0.005 compared with sham), and the increase in LV end-diastolic diameter (LVEDD) in saline-TAC was significantly mitigated by either SS20 or SS31 (p=0.03 and p=0.003, respectively, Figure1C).
TAC induced marked cardiac enlargement, which was protected by SS31 treatment (Figure 2A). Heart weight normalized to tibia length significantly increased in saline-TAC compared with sham (p<0.001) and this was significantly attenuated by SS31 (p=0.03), but not by SS20 (Figure2B). Likewise, lung weight dramatically increased after TAC, suggestive of lung congestion due to heart failure (p=0.02) and this was significantly ameliorated only by SS31 (p=0.03), but not by SS20. Masson trichrome stains demonstrated marked interstitial fibrosis in saline-TAC myocardium (Figure 2C left). Quantitative image analysis revealed that TAC induced ~4-fold increase in myocardial fibrosis (% of blue stain), which was significantly prevented only in SS31-TAC (p=0.005; Figure 2C–D). Consistent with this, quantitative PCR revealed that collagen1a2, a major component of fibrosis in the heart, was increased in TAC hearts by ~4-fold, and this was significantly mitigated by SS31 (p=0.04), but not by SS20 (Figure 2E). Thus, SS31 is significantly more effective than SS20 in reducing cardiac fibrosis and lung congestion (Figure 2B–E).
Figure 2. Pathology of TAC-induced heart failure.

(A) Representative images of sham, saline-TAC and SS31-TAC treated hearts. (B) Heart weight and lung weight normalized to tibia length (mg/mm). (C) Representative Masson trichrome stain of the heart displayed marked interstitial fibrosis (blue) in saline-TAC hearts. (D) Quantitative analysis of fibrosis (% myocardial area) revealed that fibrosis in saline-TAC was significantly attenuated by SS31, but not by SS20 or SS48. (E) Quantitative PCR of collagen1a2 normalized to 18s. *p<0.05 for sham vs. saline-TAC, #p<0.05 TAC-SS vs. saline-TAC.
Mitochondrial damage in heart failure and protection by SS-peptides
TAC resulted in mitochondrial oxidative damage, as indicated by a large increase in mitochondrial protein carbonyl content after TAC, and this was significantly protected by both SS31 and SS20 (Figure 3A). Autophagy was concomitantly induced, with a ~2-fold increase in light chain 3-II (LC3-II), a major constituent of activated autophagosomes, in saline-TAC compared to sham (Figure 3B). This was significantly attenuated in SS31-TAC (p<0.05 for bothLC3 II/I and LC3 II), and slightly attenuated in SS20-TAC (p<0.05 for LC3 II/I only). Electron microscopy revealed a greater than 2-fold increase in autophagosome number with saline-TAC, and this was mitigated by SS31 (p<0.01, Figure 3C–E, G). As a structural indicator of mitochondrial quality19, mitochondrial cristae density significantly declined by ~30% after TAC (p<0.01); this was ameliorated by SS31 (p<0.01, Figure 3F,H). Mitochondrial DNA (mtDNA) copy number measured by qPCR as the mitochondrial to nuclear DNA ratio showed a 49% reduction in Saline-TAC compared to sham (p=0.02). Concomitant SS31 treatment protected the heart from this decline: there was a 27% non-significant increase in mtDNA copy number in SS31-TAC compared to sham (p=0.09).
Global cardiac proteomes in TAC-Induced Heart Failure and the effect of mitochondrial targeted peptides
Using LC-MS/MS analysis with chromatographic alignment 961 proteins were compared between experimental groups. As shown in Supplemental Table 1, the number of significantly changed proteins (unadjusted p-values <0.05) in TAC hearts between two group comparison were: 538 proteins significantly changed after TAC (sham vs. saline-TAC, 340 with q<0.05); 628 proteins significantly changed for SS31-TAC vs. saline-TAC (432 with q<0.05); and 412 proteins significantly changed for SS20-TAC vs. saline-TAC (221 with q<0.05). Of the 538 proteins significantly changed after TAC, 161(29.8%) belong to mitop2 mitochondrial proteins.
To elucidate the magnitude of attenuation in the proteomic changes in heart failure by SS-peptides, we plotted the fold-change of the 538 significantly changed proteins in sham vs. saline-TAC (x-axis) vs. the fold-change of these same proteins in SS31-TAC or SS20-TAC (y-axis). Changes seen in sham vs. saline-TAC that are attenuated by SS-peptide treatment during TAC will be seen in upper right and lower left quadrants. Figure 4A demonstrates that 98% of proteins significantly changed by TAC fell in these two quadrants. The great majority (88%) of mitochondrial proteins declined in abundance after TAC, whereas 67% of non-mitochondrial proteins increased in abundance after TAC. The slope of the regressions in Figure 4 indicates the average strength of the attenuation of TAC-induced proteomic changes by SS peptides; as TAC with SS peptide becomes more similar to sham, the slope will approach 1.0). SS31 attenuated TAC proteome remodeling by an average magnitude of 84% for the 161 mitochondrial proteins (slope 0.84), and an average of 69% for the 379 non-mitochondrial protein (Figure 4A). The difference in the slope between mitochondrial and non-mitochondrial proteins was significant in SS31 (p<0.01 by ANCOVA). The magnitude of protection by SS20 was not significantly different between mitochondrial proteins and non mitochondrial proteins (an average of 63% for mitop vs. 56% for non mitop, Figure 4B). We should note that while the linear fits in Figure 4 use the assumption of independent “observations” and identically distributed errors, the reader is cautioned that these assumptions may not be strictly true due to possible relationships between proteins.
Figure 4. Scatter plots of protein changes by proteomics analysis.
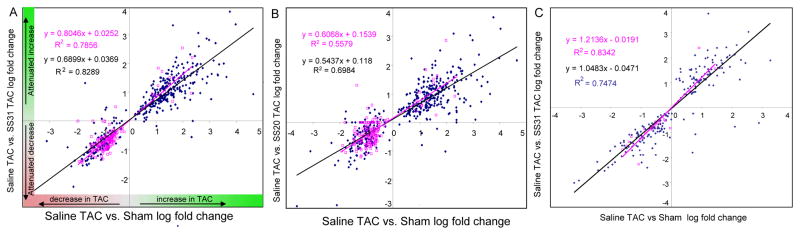
The fold change (log2) in 538 significantly altered proteins in sham vs. saline-TAC hearts is shown on the x-axis (A–C). The y axis shows the corresponding change of each protein in each TAC-SS-peptide vs. saline-TAC comparison: SS31 (A) or SS20 (B). Proteins are shown as mitochondrial (red) or non-mitochondrial (blue) in panels A–D. (C) shows the fold changes in 277 proteins significantly altered only in SS31-TAC vs. saline-TAC that are not significant in the sham vs. saline-TAC comparison.
Ingenuity Pathway Analysis of significantly changed proteins
To investigate the biological pathways affected by pressure overload induced heart failure, we applied Ingenuity Pathway Analysis23. As shown in Supplemental Table 2, there were 53 canonical pathways significantly affected by TAC (sham vs. saline-TAC), 52 of which were significantly attenuated by SS31.
The 20 most significantly changed IPA pathways and the average magnitude of change in proteins within the pathways are shown in the Table. TAC-induced heart failure significantly increased actin cytoskeleton pathways (2.8 to 3.2 fold). Intermediate metabolism pathways, including metabolism of β-alanine, fatty acid, gluconeogenesis and BCAA (branch-chain amino acid) degradation, significantly declined in heart failure (2.1–2.6 fold). Mitochondrial function and citrate cycle pathways decreased by 1.8–2.1 fold. SS31 prevents the TAC-induced changes in mitochondrial function, gluconeogenesis and citrate cycle pathways (fold changes of SS31-TAC vs.saline-TAC are almost the same as those of Sham vs. saline-TAC) and substantially attenuates the magnitude of changes in actin cytoskeleton and intermediate metabolism pathways. Compared with SS31, SS20 has weaker effect on TAC-induced increase in actin cytoskeleton pathways. Most notable, however, is that the protection of mitochondrial function, intermediate metabolism and gluconeogenesis by SS20 does not reach statistical significance (Table).
Table.
The 20 most significant Pathways in IPA comparisons
| Pathway | Saline-TAC vs. Sham | Saline-TAC vs. SS31-TAC | Saline-TAC vs. SS20-TAC | |||
|---|---|---|---|---|---|---|
|
| ||||||
| IPA Rank | Fold change | IPA Rank | Fold change | IPA Rank | Fold change | |
| Cytoskeleton | ||||||
| Rho: Actin-based Motility | 11 | +3.2 | 14 | +2.5 | 5 | +2.1 |
| Actin Cytoskeleton | ||||||
| Signaling | 7 | +3.2 | 7 | +3.3 | 1 | +2.3 |
| ILK Signaling | 21 | +3.0 | 16 | +3.8 | 3 | −2.4 |
| Integrin Signaling | 16 | +3.0 | 19 | +2.5 | 11 | +2.1 |
| RhoA Signaling | 15 | +2.9 | 15 | +2.4 | 4 | +2.1 |
| Signaling by Rho | ||||||
| GTPases | 20 | +2.9 | 9 | +2.2 | ||
| RhoGDI Signaling | 17 | +2.8 | 18 | +2.3 | 7 | +2.12 |
| Cdc42 signaling | 10 | +2.4 | ||||
| TIght Junction Signaling | 12 | +2.2 | ||||
| CXCR4 signaling | 20 | +2.2 | ||||
| Cellular effects of Silendafil | ||||||
| PAK signaling | 18 | +2.0 | ||||
| Intermediate metabolism | ||||||
| β-alanine Metabolism | 10 | −2.6 | 8 | −1.9 | ||
| Fatty Acid Metabolism | 12 | −2.3 | 17 | −1.8 | ||
| Lysine Degradation | 18 | −2.3 | ||||
| BCAA Degradation | 4 | −2.2 | 6 | −1.8 | ||
| Propanoate Metabolism | 5 | −2.2 | 5 | −1.8 | ||
| Butanoate Metabolism | 9 | −2.1 | 11 | −1.9 | ||
| Pyruvate Metabolism | 8 | −1.8 | 13 | −2.2 | ||
| Glycolysis | 14 | −1.7 | 10 | −1.5 | ||
| Gluconeogenesis | 13 | −1.5 | 9 | −1.4 | ||
| Citrate Cycle | 3 | −2.1 | 4 | −2.1 | ||
| Mitochondrial Function | ||||||
| Ubiquinone Biosynthesis Oxidative | 6 | −1.9 | 3 | −2.0 | ||
| Phosphorylation Mitochondrial | 2 | −1.8 | 1 | −2.2 | ||
| Dysfunction | 1 | −1.8 | 2 | −2.0 | ||
| Others | ||||||
| Sertoli Cell Junction Signal | 19 | +3.2 | 16 | +2.7 | ||
| Purine Metabolism | 20 | −3.0 | ||||
| EIF2 Signaling | 12 | −2.6 | 17 | +2.2 | ||
| Calcium Signaling | 21 | +2.8 | 2 | −2.2 | ||
| Hepatic Fibrosis | 14 | −2.6 | ||||
| Axonal Guidance signaling | 13 | +2.4 | ||||
| Protein Kinase A signaling | 6 | +2.1 | ||||
| Protein Ubiquitination | 15 | +2.0 | ||||
| Sertoli-Sertoli cell signal | 19 | +3.0 | ||||
Note: +: upregulation, −: downregulation
To better illustrate the effects of SS-peptides on IPA canonical pathway proteins, we plotted a heat map of the top 15 pathways and their component proteins (Figure 5, Supplemental Table 3). It is obvious that most proteins within each pathway changed concordantly in all three comparisons, that is, that SS31 and SS20 treated TAC hearts shared similarities with sham hearts, and that this concordance was greatest for the protective effect of SS31 (the third columns in Figure 5 looks very similar to second column). SS20 can be seen to have a weaker and variable attenuation of the proteome remodeling; for example, rows that are pale in the first columns (SS20) and stronger pink or blue in second and third columns (Sham and SS31) were poorly protected by SS20 treatment.
Figure 5. Heat map of abundance ratio of the top canonical pathways from Ingenuity Pathway Analysis.

The abundance ratio of each component proteins of a pathway is colored red when higher in the SS-20, sham or SS-31treatment (numerator) than in the saline-TAC (denominator) in the ratio comparison. The color is blue for the converse. The top 15 canonical pathways are ordered from most significant (top of left set) to least (bottom of right set) Any protein that was significant in any one or more of the three ratio comparisons is included in the heat map. The intensity of color indicates the magnitude of change, with the scale shown at the bottom.
The networks of mitochondrial dysfunction/oxidative phosphorylation and citrate cycle are illustrated in Figure 6. Most of these are significantly decreased in saline-TAC compared with sham (Figure 6A, C), while treatment with SS31 significantly attenuated most these changes, to the levels comparable to sham (Figure 6B, D).
Figure 6. Network of mitochondrial oxidative phosphorylation (A, B) and citrate cycle (C, D), derived from Ingenuity Pathway Analysis.

Symbols are colored red when significantly down regulated and green when significantly up-regulated; the intensity of color indicates the magnitude of change, with the scale shown in Figure 4A. (A) and (C) Sham vs. saline-TAC, (B) and (D) SS31-TAC vs. saline-TAC. Red in A and C indicates downregulation in TAC; red in B and D indicates SS31 attenuation of downregulation in TAC.
The interrelationships of significantly altered components of the actin cytoskeleton/RhoA signaling pathway are shown in Supplemental Figure 2. Integrin signaling appears to be the key input to this pathway. RhoA and Rac are the most central significantly modulated signaling molecules. Seven cellular functions are on the network “output”, most particularly actin polymerization, stabilization and assembly. Many of the components of these pathways are significantly increased after TAC and protected in SS31-TAC.
The effect of SS-peptides on cardiac proteome in normal hearts
In control experiments without TAC, comparing SS31 vs. saline controls there were 165 proteins with unadjusted p-values less than 0.05 (mean q =0.87), and comparing SS20 vs. saline controls there were 281 proteins with unadjusted p-values less than 0.05 (mean q=0.62); however, in both cases none of these changes were significant after multiple comparison adjustment (none has q-value < 0.05, Supplemental Table 4; note that the false discovery rate q-values were calculated for each contrast separately).
Discussion
Using an unbiased label-free shotgun proteomics and chromatogram alignment approach for measuring relative protein abundances, we detect 538 significant changes in the global cardiac proteome (of 961 analyzed proteins) after pressure overload induced heart failure. A previous study using 2D-electrophoresis followed by ICAT demonstrated 123 proteins differentially expressed during TAC induced LVH4, while another study applying iTRAQ found 62 proteins significantly changed after TAC5. To the best of our knowledge, the current report is the most comprehensive study of unbiased proteomics in a pressure-overload heart failure model. However, we should note that future studies that included cellular fractionation would further increase the sensitivity to detect small changes in different compartments, and that the current study is limited by the lack of analysis on post-translational modification, which plays an essential role in several signaling pathways..
We characterized the global proteomics changes by IPA and found 53 pathways significantly changed by TAC (Supplemental Table 2). While this study confirmed many key pathways involved in pressure-overload heart failure previously reported using non-proteomic approaches, including mitochondria, various metabolic pathways and actin cytoskeleton remodeling, several more novel pathways were also identified in our study, such as amino acid degradation. Although mitochondrial dysfunction has been implicated in heart failure, this is the first study using an unbiased proteomics approach to demonstrate that mitochondrial dysfunction and oxidative phosphorylation are the most significantly affected pathways in pressure-overload heart failure (IPA ranks 1 and 2 in Table). Most of the proteins in mitochondria and metabolism are concordantly downregulated in heart failure, in parallel with the decline in mtDNA copy number; this is in accordance with the concept of energy starvation in heart failure. We did not see any compensatory up-regulation of glycolysis or gluconeogenesis proteins after 4 weeks of TAC; this is consistent with previous data suggesting that changes in these metabolic pathways are not due to regulation of protein expression24.
The actin cytoskeleton/integrin/RhoA/Rac network has the largest magnitude of change among significantly affected pathways (Table, Supplemental Figure 2). Integrins link the extracellular matrix to the intracellular cytoskeleton25. Integrin signaling and cytoskeletal muscle LIM protein are internal stretch sensors26, 27, which transduce the biomechanical stretch signals (by TAC) and activate integrin-linked kinase (ILK)28, small GTPases like Rho, Ras and Rac2, 25, thereby regulating muscle contraction machinery. Integrins are recently shown to inhibit mitochondrial metabolism and increase ROS through Rho dependent mechanism29. Several defects in integrin signaling are linked to dilated cardiomyopathy because of defective contractility and the impairment of stretch response26, 27. Biomechanical stretch can also activate membrane receptors, particularly G-protein coupled receptors30, which led to activation of mitogen associated protein kinase (MAPK) and downstream hypertrophy signaling2, 30.
The BCAA (branch-chain amino acid) degradation pathway is among the top pathways in heart failure (Table). BCAAs accumulation are known to inhibit fatty acid utilization, activate the mTOR pathway, which promotes protein synthesis and cell growth31. Patients with propionic aciduria or methylmalonic aciduria (branched chain keto-acids) are known to have dilated cardiomyopathy in association with defects of mitochondrial respiratory chain32. Taken together, our study suggests the potential novel roles of BCAA in heart failure signaling.
We have previously shown that mitochondrial ROS plays a central role in cardiac aging9, angiotensin-induced hypertrophy, Gαq-overexpression induced cardiac failure and fibrosis6. Scavenging mitochondrial ROS by catalase targeted to mitochondria (mCAT) significantly attenuated heart failure and the altered mitochondrial proteome remodeling11. ROS induced mitochondrial damage is one of the mechanisms involved in cardiac hypertrophy and failure33. Indeed, ROS has been shown to interact with MAP kinases, calcium signaling and several ion channels that involve in cardiac hypertrophy signaling and its progression to heart failure2, 33, 34. Thus, mitochondrial protection is a potential target for novel therapeutics in cardiac hypertrophy and failure. In the current study we demonstrate that mitochondrial targeted SS31 peptide with a direct scavenging capacity and mitochondrial protective effects is effective in amelioration of TAC-induced heart failure and proteomic remodeling, and the protective effect is much superior to SS20. It was recently reported that SS31 prevents cyt c induced cardiolipin peroxidation16. Cardiolipin peroxidation and depletion are associated with heart failure35. Cardiolipin is required for optimal activity of all the respiratory complexes and especially for formation of supercomplexes, and cardiolipin depletion results in inhibition of oxidative phosphorylation36. SS20 has no scavenging capacity but can inhibit cyt c peroxidase activity (unpublished results). The reduced efficacy of SS20 in ameliorating heart failure (Figure 1) was entirely concordant with the more modest and variable effects on attenuation of TAC induced proteome remodeling (Figures 4 and 5, Table).
Since the SS-peptide is not cardiomyocyte selective, the effects of SS-peptides on non-cardiomyocytes can not be excluded. However, we have previously reported that SS-31 does not affect the increase of blood pressure in Angiotensin II induced hypertension, but it attenuates the phenotypes of Angiotensin-II induced cardiac hypertrophy and diastolic dysfunction10. The mitochondrial protective effect by SS31 was corroborated by the fact that it attenuates 84% of TAC-induced changes in mitochondrial proteins and 69% in non-mitochondrial protein changes (Figure 4A). As SS31 has little to no effect on the cardiac proteome in control hearts without TAC (Supplemental Table 4), this is consistent with our finding that there were very few proteins that were significantly altered by SS31 in a fashion different from that of simply attenuating the changes of TAC (Figure 4C). Thus, we conclude that SS31 appears to attenuate TAC while producing few independent effects on the proteome. The relative absence of off-target effects of SS31 would be an advantage in its potential use as a clinical therapeutic agent.
The importance of mitochondrial pathways in heart failure and the concordance of attenuation of non mitochondrial proteomic alterations by the mitochondrial protective peptide suggest that perturbed mitochondrial function may be an upstream signal to many of the non-mitochondrial protein and pathway alterations in heart failure. Since multiple time points were not assessed to show that correction of mitochondrial abnormalities occurred before the amelioration of physiology and other proteome remodeling, it remains possible that SS31 interrupts a critical amplification loop rather than inhibiting the initial inciting events. Nevertheless, our global proteomic analysis suggests that preserving mitochondrial function is an intervention that produces an extremely broad protective response. If this is indeed fundamental to the mechanisms of pressure overload induced heart failure, then protection of mitochondrial function might be the most effective strategy to prevent heart failure.
Conclusion
Improved shotgun-proteomics and IPA identified the top pathways significantly altered in TAC-induced heart failure, including mitochondrial function, actin cytoskeleton, intermediate metabolism and citrate cycle. Treatment with mitochondrial-targeted SS31 significantly attenuated the phenotype and the global proteomic remodeling. This study provides novel insights of how a mitochondrial protective strategy might affect non-mitochondrial signaling pathways in heart failure and provides impetus to study the potential clinical application of SS31 or similar mito-protective interventions.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by the National Institutes of Health grants number: HL101186, AG000751, AG013280.
Footnotes
Disclosures
H.H. Szeto and the Cornell Research Foundation (CRF) hold several patents covering the SS peptides. The University of Washington is co-holder of one of these, naming Peter S Rabinovitch and Dao-Fu Dai as co-inventors. CRF has commercially licensed the SS peptide technology, in which CRF and H.H. Szeto have financial interests.
References
- 1.Levy D, Larson MG, Vasan RS, Kannel WB, Ho KK. The progression from hypertension to congestive heart failure. JAMA. 1996;275:1557–1562. [PubMed] [Google Scholar]
- 2.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 3.Agnetti G, Husberg C, Van Eyk JE. Divide and conquer: the application of organelle proteomics to heart failure. Circ Res. 108:512–526. doi: 10.1161/CIRCRESAHA.110.226910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindsey ML, Goshorn DK, Comte-Walters S, Hendrick JW, Hapke E, Zile MR, Schey K. A multidimensional proteomic approach to identify hypertrophy-associated proteins. Proteomics. 2006;6:2225–2235. doi: 10.1002/pmic.200500013. [DOI] [PubMed] [Google Scholar]
- 5.Kocher T, Pichler P, Schutzbier M, Stingl C, Kaul A, Teucher N, Hasenfuss G, Penninger JM, Mechtler K. High precision quantitative proteomics using iTRAQ on an LTQ Orbitrap: a new mass spectrometric method combining the benefits of all. J Proteome Res. 2009;8:4743–4752. doi: 10.1021/pr900451u. [DOI] [PubMed] [Google Scholar]
- 6.Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, Marcinek DJ, Dorn GW, 2nd, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. Mitochondrial Oxidative Stress Mediates Angiotensin II-Induced Cardiac Hypertrophy and G{alpha}q Overexpression-Induced Heart Failure. Circ Res. 2011;108:837–846. doi: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN. Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol. 2000;32:2361–2367. doi: 10.1006/jmcc.2000.1266. [DOI] [PubMed] [Google Scholar]
- 8.Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996;94:2837–2842. doi: 10.1161/01.cir.94.11.2837. [DOI] [PubMed] [Google Scholar]
- 9.Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, Gollahon K, Martin GM, Loeb LA, Ladiges WC, Rabinovitch PS. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dai DF, Chen T, Szeto H, Nieves-Cintron M, Kutyavin V, Santana LF, Rabinovitch PS. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011;58:73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dai DF, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, MacCoss MJ, Rabinovitch PS. Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res. 2012;93:79–88. doi: 10.1093/cvr/cvr274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10:601–619. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- 13.Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 14.Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, Zhao Z, Ganger M, Tow CY, Seshan SV. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol. 2011;22:1041–1052. doi: 10.1681/ASN.2010080808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW, 3rd, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, Seshan SV, Pardee JD, Szeto H. Cardiolipin as a novel target to re-energize ischemic mitochondria. J Am Soc Nephrol. 2013;24:1250–1261. doi: 10.1681/ASN.2012121216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho J, Won K, Wu D, Soong Y, Liu S, Szeto HH, Hong MK. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis. 2007;18:215–220. doi: 10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- 18.Finney GL, Blackler AR, Hoopmann MR, Canterbury JD, Wu CC, MacCoss MJ. Label-free comparative analysis of proteomics mixtures using chromatographic alignment of high-resolution muLC-MS data. Anal Chem. 2008;80:961–971. doi: 10.1021/ac701649e. [DOI] [PubMed] [Google Scholar]
- 19.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsieh EJ, Hoopmann MR, MacLean B, MacCoss MJ. Comparison of database search strategies for high precursor mass accuracy MS/MS data. J Proteome Res. 2010;9:1138–1143. doi: 10.1021/pr900816a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods. 2007;4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 22.Zhang B, Chambers MC, Tabb DL. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J Proteome Res. 2007;6:3549–3557. doi: 10.1021/pr070230d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jimenez-Marin A, Collado-Romero M, Ramirez-Boo M, Arce C, Garrido JJ. Biological pathway analysis by ArrayUnlock and Ingenuity Pathway Analysis. BMC Proc. 2009;3 (Suppl 4):S6. doi: 10.1186/1753-6561-3-S4-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 25.Ross RS, Borg TK. Integrins and the myocardium. Circ Res. 2001;88:1112–1119. doi: 10.1161/hh1101.091862. [DOI] [PubMed] [Google Scholar]
- 26.Brancaccio M, Fratta L, Notte A, Hirsch E, Poulet R, Guazzone S, De Acetis M, Vecchione C, Marino G, Altruda F, Silengo L, Tarone G, Lembo G. Melusin, a muscle-specific integrin beta1-interacting protein, is required to prevent cardiac failure in response to chronic pressure overload. Nat Med. 2003;9:68–75. doi: 10.1038/nm805. [DOI] [PubMed] [Google Scholar]
- 27.Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, McKenna W, Yokoyama M, Schork NJ, Omens JH, McCulloch AD, Kimura A, Gregorio CC, Poller W, Schaper J, Schultheiss HP, Chien KR. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111:943–955. doi: 10.1016/s0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- 28.Bendig G, Grimmler M, Huttner IG, Wessels G, Dahme T, Just S, Trano N, Katus HA, Fishman MC, Rottbauer W. Integrin-linked kinase, a novel component of the cardiac mechanical stretch sensor, controls contractility in the zebrafish heart. Genes Dev. 2006;20:2361–2372. doi: 10.1101/gad.1448306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Werner E, Werb Z. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. J Cell Biol. 2002;158:357–368. doi: 10.1083/jcb.200111028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004;6:499–506. doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]
- 31.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–534. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Keyzer Y, Valayannopoulos V, Benoist JF, Batteux F, Lacaille F, Hubert L, Chretien D, Chadefeaux-Vekemans B, Niaudet P, Touati G, Munnich A, de Lonlay P. Multiple OXPHOS deficiency in the liver, kidney, heart, and skeletal muscle of patients with methylmalonic aciduria and propionic aciduria. Pediatr Res. 2009;66:91–95. doi: 10.1203/PDR.0b013e3181a7c270. [DOI] [PubMed] [Google Scholar]
- 33.Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012;110:1109–1124. doi: 10.1161/CIRCRESAHA.111.246140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burgoyne JR, Mongue-Din H, Eaton P, Shah AM. Redox signaling in cardiac physiology and pathology. Circ Res. 2012;111:1091–1106. doi: 10.1161/CIRCRESAHA.111.255216. [DOI] [PubMed] [Google Scholar]
- 35.Saini-Chohan HK, Holmes MG, Chicco AJ, Taylor WA, Moore RL, McCune SA, Hickson-Bick DL, Hatch GM, Sparagna GC. Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. J Lipid Res. 2009;50:1600–1608. doi: 10.1194/jlr.M800561-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lenaz G, Genova ML. Supramolecular organisation of the mitochondrial respiratory chain: a new challenge for the mechanism and control of oxidative phosphorylation. Adv Exp Med Biol. 2012;748:107–144. doi: 10.1007/978-1-4614-3573-0_5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.