

Abstract
Antisense oligonucleotides are powerful tools to regulate gene expression in cells and model organisms. However, a transfection or microinjection is needed for efficient delivery of the antisense agent. We report the conjugation of multiple HIV TAT peptides to a hairpin-protected antisense agent through a light-cleavable nucleobase caging group. This conjugation allows for the facile delivery of the antisense agent without a transfection reagent and photochemical activation offers precise control over gene expression. The developed approach is highly modular, as demonstrated by the conjugation of folic acid to the caged antisense agent. This enabled targeted cell delivery through cell-surface folate receptors followed by photochemical triggering of antisense activity. Importantly, the presented strategy delivers native oligonucleotides after light-activation, devoid of any delivery functionalities or modifications that could otherwise impair their antisense activity.
INTRODUCTION
The regulation of gene expression is an important biological process that occurs in many cellular pathways. The misregulation of a single gene can be detrimental to a physiological process, as it can cause a variety of diseases, ranging from muscular dystrophy1 and cancer2 to neurological3 and heart diseases.1, 4 Several biomolecular tools have been developed to perturb and study gene expression in a variety of different cellular pathways, including antisense agents (phosphorothioate DNA, peptide nucleic acids, locked nucleic acids, and others), small interfering RNAs (siRNAs), DNA decoys, and triplex-forming oligonucleotides.5-7 In addition to being excellent molecular probes, these reagents have the potential to be highly specific therapeutic agents. However, poor bioavailability and in vivo delivery of the oligonucleotides limit their applicability.
Cell penetrating peptides (CPPs), short (8-30 amino acid) synthetic peptides that facilitate cellular uptake of the peptide and its cargo,8-10 have been conjugated to a variety of biomacromolecules including plasmids,11 oligonucleotides,12 and proteins13 for efficient delivery into mammalian cells. Different CPPs have been discovered, including heparan derived CPPs (DPV3 and DPV1047), HIV TAT, penetratin, K-FGF, Bac7, and others.9 Even though the mechanism of cellular uptake for each CPP has not been fully elucidated, the two favored models for cell delivery are direct translocation and endocytosis.14 While CPPs can have very diverse sequences, within a specific class of CPPs sequence similarities and similar structural features have been identified, e.g., repeats of positively charged amino acids.9 CPPs can be covalently attached to their cargo or can be used to form noncovalent complexes.8 Covalently linked CPPs have been shown to potentially reduce the bioactivity of the cargo, depending on the CPP, the cargo molecule, and the covalent linker.15 For example, the conjugation of an HIV TAT peptide to a siRNA oligonucleotide led to a significant decrease in its gene silencing ability.16 We speculated that this limitation could be completely prevented through the use of a light-cleavable linker between the CPP and the cargo oligonucleotide. Importantly, this approach not only solves delivery and activity issues of oligonucleotides, but also enables the precise spatial and temporal regulation of oligonucleotide function using light as a minimally invasive external control element.
Photochemical control of cellular processes can be achieved through the introduction of caging groups into biologically active molecules.17-22 Caging technology stems from the use of organic protecting groups to mask biological activity until light as an external trigger is applied, thereby removing the protecting group and restoring biological function. Previously, we and others, have introduced caging groups to the photochemical control of a variety of biological macromolecules, including antisense agents,23-27 DNA decoys,28 triplex-forming DNA,29 siRNAs,30-32 and proteins.33, 34 However, all reported reagents need to be either transfected or injected into cells and organisms.
Building on the developed caging approach, we designed new caged phosphoramidites containing an alkyne handle, introduced them into synthetic antisense agents, and conjugated HIV TAT peptides to the nucleobase-caged oligonucleotides in order to allow for 1) efficient delivery of the antisense agent into mammalian cells without the need for injection or transfection, and 2) the photochemical release of the native antisense agent from the CPP and its concomitant activation. Thus, a propargyl-6-nitroveratryloxymethyl (PNVOM) caging group was developed and installed on the nucleobase of a thymidine phosphoramidite. This PNVOM-caged thymidine was incorporated into antisense agents and conjugated with an azide-containing CPPs in a copper-catalyzed 1,3-dipolar cycloaddition reaction. The CPP-caged antisense conjugate was then simply added to the cell culture media in order to be taken up by the cells (Scheme 1). Due to the presence of the caging groups, the antisense agent is unable to hybridize to the target mRNA and hence is inactive, allowing expression of the target gene. After a brief UV irradiation, the caging groups together with the linked CPPs, were cleaved, activating the antisense agent and resulting in the silencing of the target gene.
Scheme 1.
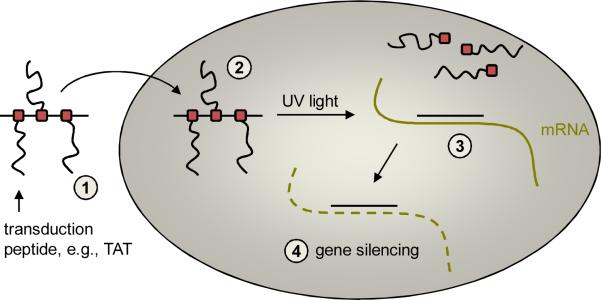
Delivery and light-activation of antisense agents containing caging groups conjugated with cell-penetrating peptides. PNVOM-caged thymidine nucleotides are site-specifically incorporated into antisense agents and are bioconjugated with azido-CPPs. The CPP-caged antisense agent conjugates are (1) simply added to the cell culture media and (2) are taken up by mammalian cells, but remain inactive due to the presence of the caging groups. (3) UV irradiation triggers decaging, cleaving the CPPs from the antisense agent and inducing sequence-specific mRNA binding. This leads to (4) the silencing of gene expression either by blocking the ribosome or by inducing RNase H-mediated mRNA degradation.
RESULTS and DISCUSSION
In order to conduct a site-specific DNA conjugation, an azido-modified HIV TAT peptide and a PNVOM-caged thymidine phosphoramidite were synthesized. The five-step synthesis of the phosphoramidite 6 started with the known aldehyde 1,35 which was prepared from vanillin (Scheme 2). Nitration of 1 with concentrated HNO3 gave the corresponding ortho, nitrobenzaldehyde 2 as a yellow solid in 71% yield. Methylation of 2 with AlMe3 in DCM at 0 °C provided the alcohol 3 in 70% yield, which was subsequently reacted with methylsulfide in the presence of benzoyl peroxide to furnish the thiomethylene derivative 4 in 78% yield. The sulfide 4 was activated in situ with sulfuryl chloride at –78 °C as the corresponding propargyl-6-nitroveratryloxymethyl chloride (PNVOM-Cl), and was directly reacted with DMT-protected thymidine in the presence of DBU in DMF to obtain PNVOM-caged thymidine 5 in 67% yield. Finally, reaction of the caged nucleoside 5 with 2-cyanoethyl-N-N-diisopropylchlorophosphoramidite delivered the caged thymidine phosphoramidite 6 as a white solid in 90% yield.
Scheme 2.

Synthesis of the propargyl-6-nitroveratryloxymethyl (PNVOM) caged thymidine phosphoramidite 6. Dichloromethane (DCM), di(p-methoxyphenyltrityll-methyl (DMTr), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), dimethylformamide (DMF), N-N-diisopropylethylamine (DIPEA).
Hairpin-protected antisense agents have previously been shown to be efficient in inhibiting gene expression without the need for additional backbone modifications.23, 28, 36 The hairpin loop structure protects the phosphodiester antisense agents from intracellular degradation without inhibiting their silencing activity. Based on previous literature reports,36 a GC-rich stem region of the hairpin structure was designed based on strong Watson-Crick base pair interactions to generate stable DNA duplex formation. Additionally, the hairpin-protected antisense agents contain no stabilizing chemical modifications (e.g., phosphorothioates) that could elicit an immune response37 or increase non-specific protein binding.38 PNVOM-caged thymidine nucleotides were site-specifically incorporated into hairpin-protected antisense agents using standard DNA synthesis conditions (Table 1). Two previously reported antisense agent targets were selected to demonstrate light-activation of TAT-conjugated antisense agents: Renilla luciferase39 as a classical reporter gene that can be easily assayed, and Eg540 as an endogenous gene that generates a distinct phenotype when silenced. PNVOM-caged thymidine nucleotides were strategically installed throughout the antisense sequence for the two target genes. Equal distribution of the PNVOM-caged thymidine within the antisense sequence will ensure that hybridization to its complementary mRNA is fully inhibited and that multiple alkyne moieties are available for conjugation. Additionally, we have previously shown that the use of three evenly distributed caging groups throughout 18-19mers is sufficient to prevent DNA:RNA hybridization.24, 41
Table 1.
Sequences of the hairpin protected antisense agents used in this study. pT represents the PNVOM-caged thymidine nucleotide, installed through coupling of the phosphoramidite 6 during solid-phase DNA synthesis.
| Sequence | |
|---|---|
| CNTRL |
|
| Luc-AA |
|
| CLuc-AA |
|
| Eg5-AA |
|
| CEg5-AA |
|
One of the first transduction proteins discovered was the HIV TAT trans-activating factor. The protein transduction domain responsible for cellular delivery was later elucidated to be the peptide sequence Tat48-60.42 The conjugation of this HIV TAT CPP to macromolecules has shown efficient cellular delivery and thus was selected for this study. In order to conduct a copper-catalyzed [3+2] cycloaddition for the bioconjugation of the HIV TAT peptide to the antisense agents, 6-azidohexanoic acid was prepared43 and added at the N-terminus of the CPP during solid-phase peptide synthesis (GenScript, Inc). The antisense agents containing three PNVOM groups were then reacted with the N3-HIV TAT peptide in an aqueous environment under mild conditions (Scheme 2).44 Addition of a tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) ligand showed enhanced reaction rates and improved water solubility over an unsuccessfully tested tris(benzyltriazolylmethyl)amine (TBTA) ligand.45, 46 The HIV TAT conjugated antisense agents were immediately purified using a NAP5 column in order to minimize potential DNA degradation caused by any residual copper catalyst.45, 47
Scheme 2.

Bioconjugation of the PNVOM caged antisense agent to the azido-HIV TAT peptide and decaging reaction of the HIV TAT conjugated antisense agent. HIV TAT = GRKKRRQRRRPPQ.
Activation of the antisense agent is achieved by photolysis of the CPP conjugates through a brief UV irradiation at 365 nm. In order to confirm that the HIV TAT peptide is indeed photochemically cleaved, thus releasing the native antisense agent, a 5′ radiolabeled caged oligonucleotide-TAT conjugate was analyzed via gel shift assay (Figure 1) before and after UV irradiation. First, the gel reveals a complete conversion of the caged antisense agent (lane 1) to the TAT conjugate (lane 2) through three [3+2] cycloaddition reactions. Upon UV irradiation (365 nm, 2 min), the HIV TAT peptide is readily cleaved resulting in the generation of the free oligonucleotide (lane 3). These in vitro experiments thus set the stage for the photochemical activation of antisense activity in mammalian cells.
Figure 1.

Gel shift assay of the radiolabeled, caged HIV TAT conjugated antisense agent before and after UV irradiation. A substantial gel shift was observed for the conjugated CLuc-AA-TAT in comparison to CLuc-AA. However, a brief irradiation (2 min, 365 nm) restored the free, non-caged antisense agent. Lane 1: CLuc-AA, Lane 2: CLuc-AA-TAT –UV, Lane 3: CLuc-AA,TAT +UV. Minimal CLuc-AA-TAT spillover from lane 2 is most likely causing the small high molecular weight band in lane 1.
The HIV TAT-conjugated antisense agents were subsequently evaluated in mammalian cell culture. Human embryonic kidney (HEK) 293T cells were treated with the CNTRL (negative control), Luc-AA (positive control), caged CLuc-AA, and caged HIV TAT-conjugated CLuc-AA-TAT by simple addition directly to the cell culture media, in the absence of any transfection reagent, and incubated overnight. The cells were either irradiated (2 min, 365 nm) or kept in the dark and were then co-transfected with the reporter plasmids pGL3 and pRL-TK, encoding firefly luciferase and Renilla luciferase, respectively. A double-transfection was performed in order to normalize reporter gene expression in a dual-luciferase assay and thus account for cell viability and transfection efficiency. The scrambled antisense agent (CNTRL) and a non-caged Renilla luciferase antisense agent (Luc-AA) were also transfected as negative and positive controls, respectively. All transfections were conducted using X-tremeGENE according to the manufacturer's instructions. As expected, the transfected CNTRL antisense agent did not inhibit luciferase expression, while transfected Luc-AA reduced Renilla luciferase expression by 85% relative to the CNTRL antisense agent (Figure 2). The control antisense agents, CNTRL and Luc-AA, led to little or no inhibition of luciferase expression when just added to the cell culture media, confirming that they are not sufficiently taken up by the cells. Additionally, CLuc-AA induced no inhibition of luciferase expression in the presence or absence of UV irradiation. Together, this indicates that the non-conjugated antisense agents did not enter the cells in the absence of a transfection agent, thus no silencing of luciferase expression was observed. When the CPP conjugate CLuc-AA-TAT was added to the cell culture media, no inhibition of gene expression was observed when the cells were kept in the dark. However, after a brief UV irradiation (2 min, 365 nm), the caging groups were removed resulting in a 92% inhibition of Renilla luciferase expression, even exceeding the results obtained when using a transfection reagent. A dose-response titration of CLuc-AA-TAT was performed to achieve optimal inhibition of luciferase expression after UV irradiation (see Supporting Figure 1). Therefore, the covalent linkage of the HIV TAT peptide allowed the antisense agent to enter the cells; however, only after UV irradiation were the HIV TAT peptide sequences removed through nucleobase decaging, activating the antisense agent and inducing gene silencing. These results confirm the ability to deliver light-activated antisense agents into live cells through the generation of transduction peptide conjugates at nucleobase-caging groups, obviating the need for a transfection reagent.
Figure 2.

Light-activation of antisense agents targeting Renilla luciferase. Antisense agents (CNTRL, Luc-AA, CLuc-AA, and CLuc-AA-TAT) were simply added to HEK 293T cell culture media or actively transfected. Cells were either irradiated (365 nm, 2 min, 25 W) or kept in the dark. The cells were expressing firefly and Renilla luciferase from pGL3 and pRL-TK plasmids, respectively, and a dual-luciferase assay was performed after 48 h. Renilla luciferase expression was normalized to firefly luciferase expression and the negative control antisense agent (CNTRL) was set to 100%. All experiments were performed in triplicate and error bars represent standard deviations. ** p value < 0.005.
Light-activation of antisense agents enables precise temporal control over their activity and thus over the activity of the specific gene that they are targeting. In order to explore the ability to regulate the function of the HIV TAT conjugated antisense agent CLuc-AA-TAT with temporal resolution, it was added to the cell culture media of HEK 293T cells. After an overnight incubation, the media was removed and the cells were transfected with the pGL3 and pRL-TK reporter constructs followed by UV irradiation after 0, 12, 24, 36, 40, or 46 h. The scrambled negative control antisense agent CNTRL and the non-caged positive control antisense agent Luc-AA were transfected into cells as well. Luciferase expression was measured after a total incubation period of 48 h. As expected, the CNTRL antisense agent showed no inhibition of luciferase expression, while Luc-AA inhibited 90% luciferase function regardless of the duration of irradiation (Figure 3). Prior to irradiation, CLuc-AA-TAT showed no inhibition of luciferase expression. Irradiation immediately after reporter plasmid transfection showed 93% inhibition of luciferase activity and displayed a steady increase in luciferase expression for later time points of irradiation. At time points 40 and 46 h, no inhibition of luciferase activity was observed, presumably due to the short incubation period of the activated antisense agent before luciferase readout at 48 h and the already present abundance of luciferase mRNA and protein.
Figure 3.

Temporal activation of luciferase antisense agents. CLuc-AA-TAT (100 nM) was added directly to cell culture media of HEK 293T cells. After an overnight incubation, the media was removed and the cells were transfected with the reporter plasmids pGL3 and pRL-TK and selected wells were also transfected with negative and positive control antisense agents, CNTRL and Luc-AA, respectively. The cells were irradiated at 0, 12, 24, 36, 40 or 46 h post-transfection (365 nm, 2 min, 25 W) and a dual-luciferase assay was performed after 48 h. Renilla luciferase expression was normalized to firefly luciferase expression and the CNTRL –UV Renilla luciferase expression was set to 100%. All experiments were performed in triplicate and error bars represent standard deviations.
To demonstrate applicability of the developed delivery and light-activation approach beyond the regulation of reporter genes, a caged TAT-conjugated antisense agent targeting the endogenous Eg5 gene was synthesized and tested in cell culture. Eg5 is a member of the kinesin-5 family and is an essential gene involved in the regulation of the cell cycle, specifically for bipolar spindle formation.48, 49 Inhibition of Eg5 results in the arrest of a multitude of cellular functions including spindle formation, mitosis, and cell proliferation.40, 50 Due to its importance in cell cycle regulation and concomitant therapeutic potential in cancer,51, 52 a caged Eg5 antisense agent (CEg5-AA, Table 1) was designed based on a literature-reported sequence40 and covalently labeled with the N3-HIV TAT peptide, generating CEg5-AA-TAT. The CEg5-AA and CEg5-AA-TAT antisense agents were added to the cell culture media of HeLa cells, followed by an overnight incubation and a brief UV irradiation (365 nm, 2 min). The scrambled oligonucleotide CNTRL and the non-caged Eg5 antisense agent Eg5-AA were transfected into HeLa cells as negative and positive controls, respectively. After 48 h incubation, the cells were fixed and the actin filaments were stained with FITC-phalloidin and the nucleus was counterstained with DAPI. Confocal imaging revealed a binucleated phenotype in cells transfected with Eg5-AA (Figure 4), in agreement with previous reports that knockdown of Eg5 arrests HeLa cells in mitosis.49, 50 In contrast, cells transfected with the CNTRL antisense agent displayed no obvious phenotypic changes. In agreement with our results discussed above, CEg5-AA directly added to the media also did not result in a phenotype change either before or after UV irradiation, confirming the necessity of a covalently linked HIV TAT transduction peptide for delivery of the antisense agent. The presence of the CEg5-AA-TAT antisense agent in the cell culture media did not induce a change in cell morphology until the three nucleobase-caging groups were cleaved through UV exposure, activating the Eg5 antisense agent and generating the same binucleated cell phenotype. The frequency of the binucleated phenotype was assessed by scoring 100 cells per experiment. Cells transfected with CNTRL showed no binucleated cells, whereas 40% of cells transfected with the positive control agent Eg5-AA displayed an Eg5 knockdown phenotype (see Supporting Figure S2). Only a minimal number (<3%) of binucleated cells were observed when CEg5-AA was added directly to the cell culture media in the presence and absence of subsequent UV light exposure, again indicating a lack of cellular uptake of a non-bioconjugated caged antisense agent. When CEg5-AA-TAT was added to the cell culture media, a small number of binuclear cells were observed (12%; some background activity could be the result of conducting the decaging experiment in the same chamber slide; no leakiness was observed in the luciferase and qRT PCR experiments shown in Figures 2, 3, and 5), until UV irradiation restore antisense activity leading to an Eg5 inhibition phenotype in 37% of observed cells. These results demonstrate, for the first time, the ability to generate reagents that display facile cellular uptake followed by photochemical regulation of an endogenous gene.
Figure 4.

Light-activated Eg5 inhibition in HeLa cells. CEg5-AA and its corresponding HIV TAT peptide conjugate CEg5-AA-TAT were added to the cell culture media. After an overnight incubation the cells were briefly irradiated (365 nm, 2 min) or kept in the dark. Negative (CNTRL) and positive (Eg5-AA) control antisense agents were transfected into HeLa cells. All cells were subsequently fixed and stained with FITC–phallodin and DAPI, and imaged using a Zeiss 710 confocal microscope (40x oil objective).
Figure 5.

Quantitative RT PCR analysis of Eg5 gene expression. CEg5-AA-TAT was added directly to the cell culture media and incubated overnight at 37 °C and 5% CO2. The media was replaced and the cells were irradiated (365 nm, 2 min, 25 W). A negative CNTRL antisense agent and a non-caged Eg5 antisense agent Eg5-AA were transfected in HeLa cells. After 48 h incubation, RNA was isolated and subjected to qRT PCR analysis. Eg5 expression was normalized to GAPDH expression and the CNTRL was set to 100%. All experiments were performed in triplicate and error bars represent standard deviations.
Quantitative RT PCR was performed to confirm that the observed phenotypic change was indeed the result of Eg5 inhibition. The cell culture experiments were conducted in an identical fashion as before, but instead of fixing, staining, and imaging, total RNA was isolated and subjected to qRT PCR. Eg5 expression was normalized to GAPDH expression and the transfected negative control antisense agent CNTRL was set to 100%. Transfection of the non-caged, positive control Eg5 antisense agent Eg5-AA into HeLa cells resulted in 60% inhibition of Eg5 expression (Figure 5). Additionally, no change in Eg5 expression was observed in the presence of UV irradiation. Upon treatment of HeLa cells with CEg5-AA-TAT without UV irradiation, no inhibition of Eg5 expression was detected due to the complete inactivity of the antisense agents taken up by the cells due to the presence of the nucleobase caging groups. However, silencing of Eg5 expression to the same level as the positive control was observed after exposure to UV light, in agreement with the luciferase reporter results and the phenotypic assays shown above. Additionally, the reduction of Eg5 mRNA levels indicates that the synthesized hairpin-modified antisense agents induce gene silencing through an RNase H-mediated mRNA degradation mechanism, as expected for unmodified phosphodiester DNA.53, 54 Furthermore, Eg5 visualization by Western blot is in agreement with the qRT PCR results, as HeLa cells treated with CEg5-AA-TAT exhibited a 45% inhibition of Eg5 expression after UV irradiation (see Supplementary Figure S3). These results confirmed that the CEg5-AA-TAT conjugate was taken up by cells and that activation of the antisense agent occurred only after UV irradiation, allowing temporal control over gene expression. Moreover, the phenotypic change observed by the Eg5-AA antisense agent is supported by an expression analysis of Eg5, demonstrating the applicability of the developed oligonucleotide delivery and light-activation methodology to any reporter or endogenous gene.
The mechanism of CPP delivery is still under debate, while passive diffusion and endocytosis have been proposed as the two main models.55 Each mode of cellular uptake depends on several parameters, including CPP concentration, cargo size, CPP type, and cell type.56, 57 Several studies have demonstrated that the HIV TAT CPP undergoes an endocytosis mechanism.58, 59 A potential drawback of this mechanism is that the cargo can be trapped within endosomes, thereby limiting its activity. However, in addition to transduction peptides, macromolecules can be successfully delivered into cells via natural vitamin endocytosis pathways, for example through folate receptors.60 The abundance of folate receptors in certain cancer cells, while only low levels are present in healthy cells,61 allows for the selective delivery. For this reason, cancer drugs such as maytansine and desacetylvinblastine have recently been conjugated to folate for targeted delivery.60 Additionally, folic acid has previously been conjugated to an antisense agent through the 3′ terminus for selective delivery into human ovarian cancer.62 Thus, we hypothesized that conjugating folic acid molecules to the NPVOM-caged antisense agents will allow for selective uptake of the antisense agents into cancer cells and photoregulation of gene function.
In order to achieve targeted delivery of NPVOM-caged antisense agents, the folic acid azide 8 was synthesized (Scheme 3). First, the 6-azidohexan-1-amine (7) was prepared in 2 steps from commercially available 1-6-dibromohexane.63 Subsequent coupling of folic acid with 7 was performed with N-N′-dicyclohexylcarbodiimide (DCC) and pyridine in DMSO to deliver the desired azido-folic acid probe 8 in 72% yield.64 Bioconjugations of 8 and CLuc-AA were conducted under the same Cu-catalyzed [3+2] cycloaddition conditions as described for the HIV TAT peptide (see Scheme 2), thus setting the stage for targeted cell delivery and light activation.
Scheme 3.
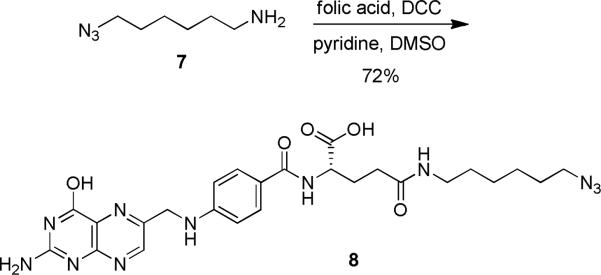
Synthesis of the azido folate 8 in three steps from commercially available starting materials. N-N’-Dicyclohexylcarbodiimide (DCC), dimethyl sulfoxide (DMSO).
The cellular delivery of the folic acid-conjugated antisense agent was evaluated in mammalian cell culture using the previously described luciferase assay (see Figure 2). The conjugated antisense agent, CLuc-AA,FA was added directly to HeLa cells due to the overexpression of folate receptors in this cell line.65, 66 As a negative control, CLuc-AA was also directly added to the cell culture media. As a positive control Luc-AA was transfected into HeLa cells using X-tremeGENE according to the manufacturer's protocol (Roche). As expected, in cells transfected with Luc-AA, 80% inhibition of Renilla luciferase was detected regardless of UV irradiation (Figure 6A). In agreement with the previous assay (see Figure 2), CLuc-AA did not inhibit luciferase expression in the presence or absence of UV irradiation due to the lack of a transfection reagent, a CPP or a small molecule carrier. Addition of the folic acid conjugate CLuc-AA-FA directly to the cell culture media did not lead to a decrease in luciferase expression when cells were kept in the dark, as expected. A brief UV irradiation (2 min, 365 nm) activated the antisense agent by cleaving the folic acid residues, resulting in 76% inhibition of Renilla luciferase expression. Thus, the covalent linkage of the folic acid to the antisense agent facilitated the direct uptake of the antisense agent into HeLa cells and only after UV irradiation were the antisense agents activated to inhibit luciferase expression. Finally, to show the selectivity of the folic acid conjugate to certain cancer cells, the above experiment was performed in MCF7 cells, which lack folate receptors.67 As a positive control, MCF7 cells transfected with Luc-AA displayed 50% inhibition of luciferase expression (Figure 6B). CLuc-AA added directly to the cell culture media resulted in no inhibition of luciferase expression regardless of UV irradiation, in agreement with the previous assays. Additionally, the folic acid conjugated antisense agent, CLuc-AA-FA, also showed no inhibition of luciferase expression when added directly to the cell culture media either before or after UV irradiation. The lack of gene silencing shown by the folic acid conjugate confirms the ability to selectively deliver light-activated antisense agents into cells.
Figure 6.

Targeted delivery and light-activation of antisense agents. Antisense agents (CLuc-AA, and CLuc-AA-FA), were added directly to cell culture media of (A) HeLa cells or (B) MCF7 cells, or alternatively transfected. Cells were either irradiated (2 min, 365 nm, 25 W) or kept in the dark. Firefly and Renilla luciferase were expressed from pGL3 and pRL-TK plasmids, respectively, and a dual-luciferase assay was performed after 48 h. Renilla luciferase expression was normalized to firefly luciferase expression and the negative control antisense agent (CLuc-AA – UV) was set to 100%. All experiments were performed in triplicate and error bars represent standard deviations. ** = p value <0.005
Conclusion
In summary, a new methodology was developed that simultaneously enables cellular uptake and precise optochemical control of antisense oligonucleotide function in mammalian tissue culture. This was achieved by developing a new nucleotide caging group that not only inhibits oligonucleotide hybridization but also provides a functional handle for the bioconjugation with cell penetrating peptides (CPPs) and small molecule carriers. Specifically, [3+2] cycloaddition reactions were used to conjugate N3-HIV TAT peptides to antisense agents containing three propargyl-6-nitroveratryloxymethyl (PNVOM) groups at specific thymidine nucleobases. The HIV TAT peptide facilitates the delivery of the antisense agent directly into mammalian cells without the need of any transfection reagents or injection protocols. The antisense agent is completely inactive until a brief UV irradiation cleaves the caging group and thereby the peptide domains from the oligonucleotide. This photolysis then provides a regular, non-modified antisense agent that is fully active and efficiently and sequence-specifically silences the gene of interest. Since a non-modified oligonucleotide is generated through light activation, this methodology can be directly applied to any antisense agent type and modification without the need for newly establishing the gene silencing behavior of the CPP conjugates. We tested the developed approach in HEK 293T cells and in HeLa cells targeting two different genes, a transiently transfected luciferase reporter gene and an endogenous gene under control of its native promoter. Through luciferase and phenotypic assays, as well as quantitative real-time PCR and Western blot analysis, it was shown that HIV TAT antisense agent conjugates can be used to photoregulate gene expression with excellent off → on switching behavior. The developed methodology displays full activation of the conjugated antisense agent after UV irradiation – with comparable or better gene silencing activity as a commercially available, transfected antisense agent. Moreover, the direct covalent attachment of a CPP to the antisense agent alleviates the need for any non-covalent liposomal CPP formation. We expect that this methodology can is generally applicable to any antisense agent, with any CPP, and in any cell type. In addition to the direct conjugation of an HIV TAT peptide, we showed that this approach allows for the selective targeting of cervical cancer cells via conjugation of folic acid residues to the caged antisense agent. This approach will greatly facilitate the spatio-temporal investigation of gene function, as it does not require any transfection reagents or other design of oligonucleotide delivery vehicles. The developed technology can be easily adapted to any antisense agent or biomolecule through direct conjugation to a carrier peptide or small molecule ligand for targeted delivery through a cell surface receptor (e.g., the sigma receptor68). Additionally, conjugating other tags to the antisense agent (e.g., fluorophores, peptides, aptamers, etc.) in conjunction with or without a CPP can be envisioned in order to visualize cellular trafficking of the oligonucleotide or for selective delivery to certain cell types. Two-photon caging groups, such as 3-nitro-2-ethyldibenzofuran, modified with alkyne handles could be used for activation with IR light, allowing for optochemical activation in deeper tissue and with high spatial resolution.69-71
METHODS
Synthesis of PNVOM-caged antisense agents
DNA synthesis was performed using an Applied Biosystems Model 394 automated DNA/RNA Synthesizer using standard β-cyanoethyl phosphoramidite chemistry. The caged antisense agents were synthesized using 40 nmole scale, low volume solid phase supports obtained from Glen Research. Reagents for automated DNA synthesis were also obtained from Glen Research. Standard synthesis cycles provided by Applied Biosystems were used for all normal bases using 2 min coupling times. The coupling time was increased to 10 min for the positions at which the caged-T modified phosphoramidites were incorporated. Each synthesis cycle was monitored by following the release of dimethoxy trityl (DMT) cations after each deprotection step. No significant loss of DMT was noted following the addition of the caged-T to the DNA, so 10 min was sufficient to allow maximal coupling of the caged nucleotides. Yields of caged DNAs were close to theoretical yields routinely obtained.
Conjugation of the TAT-azide peptides and the folic acid azide to the antisense agents
A solution of the PNVOM-caged antisense agent (2 μM) in 0.1 M phosphate buffer was incubated at room temperature for 2 h with the azido-HIV TAT peptide (12 μM) or azido-folic acid (12 μM) in the presence of the tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) ligand (0.5 mM), CuSO4 (0.1 mM), and sodium ascorbate (5 mM). The reaction mixture was purified using a NAP 5 column (GE Healthcare) and concentrated with a speed vac.
Luciferase assay of the cellular activity of HIV TAT-conjugated antisense agents
HEK 293T cells were grown at 37 °C, 5% CO2 in Dulbecco's modified Eagle's medium (Hyclone), supplemented with 10% Fetal Bovine serum (Hyclone), and 10% streptomycin/penicillin (MP Biomedicals). Cells were passaged into a 96 well plate (200 μL per well, ~ 1 × 104 cells per well) and grown to ~70% confluence within 24 h. Conjugated antisense agents (100 nM) were added directly to the cell culture media and incubated overnight at 37 °C, 5% CO2. The media was removed and the cells were transfected with the reporter plasmids, pGL3 (Promega) and pRL-TK (Promega), and selected wells were also transfected with control antisense agents using X-tremeGENE siRNA transfection reagent (Roche). Cells were incubated at 37 °C, 5% CO2 for 4 h, the transfection medium was removed, and the cells were irradiated for 2 min on a UV transilluminator (365 nm, 25 W). DMEM media was added and the cells were incubated at 37 °C, 5% CO2 for 48 h. The media was removed and a dual-luciferase assay (Promega) was performed. Renilla luciferase was normalized to firefly luciferase. All experiments were performed in triplicate and the error bars represent the standard deviation in three independent experiments.
Temporal control of HIV TAT-conjugated antisense agents
HEK 293T cells were passaged into a 96 well plate (200 μL per well, ~1 × 104 cells per well) and grown to ~70% confluence within 24 h. Conjugated antisense agents (100 nM) were added directly to the cell culture media and incubated overnight at 37 °C, 5% CO2. The cells were transfected with the reporter plasmids, pGL3 and pRL-TK, and selected wells were also transfected with control antisense agents using X-tremeGENE siRNA transfection reagent (Roche). Cells were incubated at 37 °C for 4 h, and the transfection medium was replaced with DMEM media. The cells were then irradiated 0, 12, 24, 36, 40 or 46 h later (365 nm, 2 min, 25 W). After a total incubation period of 48 h after reporter plasmid transfection at 37 °C, 5% CO2, media was removed and a dual-luciferase assay (Promega) was performed. Renilla luciferase was normalized to firefly luciferase. All experiments were performed in triplicate and the error bars represent the standard deviation in three independent experiments.
Phenotypic assay of photochemical Eg5 inhibition
HeLa cells were grown at 37 °C, 5% CO2 in Dulbecco's modified Eagle's medium (Hyclone), supplemented with 10% Fetal Bovine serum (Hyclone), and 10% streptomycin/penicillin (MP Biomedicals). Cells were passaged into 4-well chamber slide (1 mL per well, ~4 × 104 cells per well) and grown to ~70% confluence within 24 h. Conjugated antisense agents (100 nM) were added directly to the cell culture media and incubated overnight at 37 °C, 5% CO2. The media was removed and the cells were irradiated for 2 min on a UV transilluminator (365 nm, 25 W). Control antisense agents were transfected into the cells with X-tremeGENE siRNA reagent (Roche). DMEM media was added and the cells were incubated at 37 °C, 5% CO2 for 48 h. The cells were fixed with formaldehyde (3.75%) and permeabilized with Triton-X100, strained with Alexa Fluor 488 Phalloidin (ex/em 488 nm/495-630 nm, Invitrogen) and DAPI (ex/em 405 nm/410-495 nm, Invitrogen). Cells were imaged on a Zeiss LSM 710 confocal microscope (40× oil objective).
Quantitative RT-PCR
HeLa cells were passaged into 6-well plates (2 mL per well, ~ 2 × 105 cells per well) and grown to ~70% confluence in 24 h. Conjugated antisense agents (100 nM) were added directly to the cell culture media and incubated overnight at 37 °C and 5% CO2. The media was removed and the cells were irradiated for 2 min on a UV transilluminator (365 nm, 25 W). Control antisense agents were transfected into the cells with X-tremeGENE siRNA reagent (Roche). DMEM media was added and the cells were incubated at 37 °C and 5% CO2 for 48 h. RNA was isolated with TRIZOL reagent (Invitrogen). cDNAs were synthesized with the iScript cDNA Synthesis kit (Bio-Rad) and quantitative RT PCR was performed with SsoFast EvaGreen Supermix (Bio-Rad) using an Eg5 forward primer 5′ CAGCTGAAAAGGAAACAGCC, Eg5 reverse primer 5′ ATGAACAATCCACACCAGCA,50 GAPDH forward primer 5′ TGCACCACCAACTGCTTAGC and GAPDH reverse primer 5′ GGCATGGACTGTGGTCATGAG.72 The threshold cycles (Ct) of each sample were normalized to the GAPDH housekeeping gene and the inhibition of gene silencing is represented as percent of Eg5 expression. For each of the triplicates, the data were averaged and standard deviations were calculated.
Luciferase assay of the cellular activity of folic acid conjugated antisense agents
HeLa cells and MCF7 cells were grown at 37 °C, 5% CO2 in Dulbecco's modified Eagle's medium without folate (Sigma), supplemented with 10% Fetal Bovine serum (Hyclone), and 10% streptomycin/penicillin (MP Biomedicals). Cells were passaged into a 96 well plate (200 μL per well, ~1 × 104 cells per well) and grown to ~ 70% confluence within 24 h. Folate-conjugated antisense agents (100 nM) were added directly to the cell culture media and incubated overnight at 37 °C, 5% CO2. The cells were then transfected with the reporter plasmids, pGL3 and pRL-TK, and selected wells were also transfected with control antisense agents using X-tremeGENE siRNA transfection reagent (Roche). Cells were incubated at 37 °C for 4 h, the media was removed, and the cells were irradiated for 2 min on a UV transilluminator (365 nm, 25W). DMEM media was added and the cells were incubated at 37 °C, 5% CO2 for 48 h. The media was removed and a dual-luciferase assay (Promega) was performed. Renilla luciferase was normalized to firefly luciferase. All experiments were performed in triplicate and the error bars represent the standard deviation in three independent experiments.
5-Methoxy-2-nitro-4-(prop-2-yn-1-yloxy)benzaldehyde (2)
The benzaldehyde 1 (3.0 g, 15.7 mmol) was dissolved in ice-cold concentrated HNO3 (30 mL) for 4 hours and slowly warmed up to RT overnight. The reaction mixture was then poured into ice-cold water (200 mL) and the yellow precipitate was filtered off. The solid was re-dissolved in DCM (50 mL) and was washed with saturated aqueous NaHCO3 (20 mL) and with brine (20 mL), and dried with anhydrous sodium sulfate. After filtration, the solvent was evaporated under reduced pressure and the residue was purified by silica gel chromatography using DCM/hexane (7:3) to deliver the corresponding nitrobenzaldehyde 2 (2.56 g, yield 71%) as a yellow solid. 1H NMR (300 MHz, CDCl3): δ = 2.57 (s, 1H), 3.96 (s, 3H), 4.85 (s, 2H), 7.17 (s, 1H), 7.73 (s, 1H), 10.39 (s, 1H). 13C NMR (75 MHz, DMSO-d6): δ = 57.1, 78.5, 80.3, 109.8, 111.0, 126.3, 143.7, 150.0, 153.7, 189.2. HRMS: m/z calcd for C11H10NO5 [M+H]+: 236.0553; found 236.0063.
1-(5-Methoxy-2-nitro-4-(prop-2-yn-1-yloxy)phenyl)ethanol (3)
Trimethylaluminium (12.6 mmol, 2.0 M in hexane) was slowly added to a solution of the nitrobenzaldehyde 2 (1.9 g, 8.0 mmol) in DCM (30 mL) and stirred at 0 °C under an argon atmosphere. After 2 h, ice cold water (5 mL) was very slowly added to the reaction mixture in order to quench any unreacted reagent. Then, aqueous NaOH (40 mL, 1 M) was added and the reaction mixture was stirred for 1 h. The organic layer was separated and washed with 1 M NaOH (2 × 20 mL) and brine (20 mL) and, dried with anhydrous sodium sulfate and filtered. The solvent was evaporated under reduced pressure and the residue was purified by silica gel chromatography using CHCl3/CH3COCH3 (9:1) to furnish alcohol 3 (2.4 g, yield 70%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6): δ = 1.38 (t, J = 4.5 Hz, 3H), 3.65-3.63 (s, 1H), 3.91 (s, 3H), 4.91 (d, J = 1.2 Hz, 2H), 5.28-5.25 (m, 1H), 5.509 (d, J = 2.1 Hz, 2H), 7.38 (s, 1H), 7.66 (s, 1H). 13C NMR (75 MHz, acetone-d6): δ = 24.8, 55.9, 65.0, 77.2, 78.4, 109.6, 110.4, 124.2, 139.3, 145.4, 154.6. HRMS: m/z calcd for C12H13NO5 [M+H]+: 274.0686; found 274.0731.
((1-(5-Methoxy-2-nitro-4-(prop-2-yn-1-yloxy)phenyl)ethoxy)methyl)(methyl)sulfane (4)
Methyl sulfide (4.7 mL, 63.3 mmol) was added to a solution of the alcohol 3 (1.6 g, 6.36 mmol) in acetonitrile (20 mL) and stirred under a nitrogen atmosphere at 0 °C. Benzoyl peroxide (6.1 g, 25.4 mmol) was added in 4 portions to the reaction mixture with an interval of 30 min each. After the addition of the last portion, the mixture was stirred for an additional 3 h. Aqueous NaOH (100 mL, 1 M) was added and the mixture was vigorously stirred overnight. The aqueous layer was extracted with ether (3 × 30 mL) and the combined ether layers was washed with aqueous NaOH (2 × 20 mL, 1 M) and with brine (20 mL), and dried with anhydrous sodium sulfate. After filtration, the solvent was evaporated under reduced pressure and the residue was purified by silica gel chromatography using EtOAc/hexane (1:9) to obtain 4 (1.5 g, yield 78%) as a brown solid. 1H NMR (300 MHz, CDCl3): δ = 1.51 (d, J = 6.3 Hz, 3H), 2.14 (s, 3H), 2.57 (t, J = 2.4 Hz, 1H), 3.97 (s, 3H), 4.34 (d, J = 6.3 Hz, 1H), 4.46 (d, J = 6.3 Hz, 1H), 4.81 (d, J = 1.6 Hz, 2H), 5.54 (q, J = 6.3 Hz, 1H), 7.21 (s, 1H), 7.74 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 14.3, 23.5, 56.6, 57.2, 70.9, 73.5, 77.2, 77.4, 109.2, 110.5, 136.0, 140.4, 145.6, 154.5. HRMS: m/z calcd for C14H17NO5S [M+Na]+ : 334.0720; found 334.0789.
1-((2R,4S,5R)-5-((bis(4-Methoxyphenyl)(phenyl)methoxy)methyl)-4-hydroxytetrahydrofuran-2-yl)-3-((1-(5-methoxy-2-nitro-4-(prop-2-yn-1-yloxy)phenyl)ethoxy)methyl)-5-methylpyrimidine-2,4(1H,3H)-dione (5)
Sulfuryl chloride (22 μL, 0.26 mmol) was slowly added to a solution of the thioether 4 (75 mg, 0.24 mmol) in DCM (0.4 mL) and the mixture was stirred at –78 °C under an argon atmosphere. After 20 min, the solvent was removed under high vacuum and the residue was dried for 5 h. The residue was dissolved in dry DMF (0.5 mL) and added to a solution of the DMT-protected thymidine73 (130 mg, 0.24 mmol) and DBU (107 μL, 0.72 mmol) in DMF (0.5 mL). The reaction mixture was stirred at under an argon atmosphere at RT for 12 h and then poured into saturated aqueous NaHCO3 (10 mL). The aqueous layer was extracted with DCM (2 × 10 mL) and the combined organic layer were washed with saturated aqueous NaHCO3 (2 × 10 mL) and with brine (10 mL), and dried with anhydrous sodium sulfate. After filtration, the solvent was evaporated under reduced pressure and the residue was purified by silica gel chromatography using DCM/acetone (9:1) with 1% TEA to provide 4 (1.5 g, yield 78%) as a brown solid. 1H NMR (300 MHz, CDCl3): δ = 1.36 (s, 1.5 H), 1.39 (s, 1.5H), 1.47-1.53 (m, 3H), 2.10-2.21 (m, 1.5H), 2.27-2.28 (m, 0.5H), 2.32-2.41 (m, 2H), 2.52 (t, J = 2.0 Hz, 0.5H), 2.57 (t, J = 2.0 Hz, 0.5H), 3.30-3.48 (m, 2H), 3.78 (s, 6H), 3.96 (s, 3H), 3.99-4.04 (m, 1H), 4.51 (br, 1 H), 4.74 (d, J = 4.0 Hz, 2 H), 5.26-5.29 (m, 1H), 5.38-5.49 (m, 2H), 6.20 (t, J = 6.6 Hz, 0.5H), 6.29 (t, J = 6.3 Hz, 0.5H), 6.82-6.85 (m, 4H), 7.23-7.43 (m, 13 H), 7.67 (s, 0.5H), 7.70 (s, 0.5H). 13CNMR (75 MHz, CDCl3): δ = 12.5, 24.1, 41.2, 55.5, 56.7, 56.9, 57.2, 63.6, 70.2, 72.2, 72.5, 73.6, 85.2, 85.5, 86.1, 87.1, 109.4, 109.7, 110.1, 110.3, 110.5, 113.5, 127.4, 128.3, 130.2, 134.5, 135.5, 135.6, 136.8, 137.1, 139.7, 140.0, 144.4, 145.4, 150.8, 151.0, 154.0, 154.3, 158.9, 163.3. LRMS: m/z calcd for C44H45N3O12 [M+Na]+ : 830.2803; found 830.2953.
(2R,3S,5R)-2-((bis(4-Methoxyphenyl)(phenyl)methoxy)methyl)-5-(3-((1-(5-methoxy-2-nitro-4-(prop-2-yn-1-yloxy)phenyl)ethoxy)methyl)-5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl (2-cyanoethyl) diisopropylphosphoramidite (6)
DIPEA (43 μL, 0.24 mmol) and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (28 μL, 0.12 mmol) were added to a solution of the alcohol 5 (35 mg, 0.04 mmol) in DCM (1.0 mL) and the mixture was stirred under an argon atmosphere for 2 h. The TLC indicated complete consumption of starting material and the solvent was evaporated under reduced pressure. The residue was purified by silica gel chromatography using CHCl3/acetone (20:1) with 1% TEA to furnish the caged phosphoramidite 6 (39 mg, yield 90%) as a white solid. 1H NMR (300 MHz, CDCl3): δ = 1.04 (d, J = 6.6 Hz, 3H), 1.14-1.18 (m, 9 H), 1.30-1.36 (m, 3H), 1.50-1.54 (m, 3H), 2.08-2.26 (m, 1H), 2.37-2.71 (m, 4H), 3.25-3.66 (m, 5H), 3.71-3.89 (m, 7H), 3.93 (s, 3H), 4.07-4.19 (m, 1H), 4.54-4.65 (m, 1H), 4.72-4.77 (m, 2H), 5.23-5.31 (m, 1H), 5.37-5.47 (m, 2H), 6.14-6.29 (m, 1 H), 6.81-6.85 (m, 4H), 7.20-7.50 (m, 11H), 7.65-7.68 (m, 1H). 13C NMR (75 MHz, CDCl3): δ = 12.3, 20.4, 20.5, 23.9, 24.7, 40.1, 43.2, 43.4, 55.3, 56.5, 56.8, 57.0, 57.1, 57.9, 58.2, 58.5, 58.8, 63.3, 70.2, 73.6, 73.85, 74.0, 85.2, 86.9, 109.2, 109.5, 109.9, 110.1, 110.2, 110.4, 113.3, 117.5, 117.8, 127.2, 128.0, 128.2, 130.1, 134.4, 135.3, 136.6, 136.8, 137.0, 139.6, 139.6, 139.8, 139.9, 140.1, 144.2, 145.2, 150.7, 150.8, 153.8, 154.0, 154.1, 158.7, 163.1. 31P NMR (121 MHz, CDCl3): δ = 148.5, 148.6, 149.0, 149.1.
(S)-2-(4-(((2-amino-4-hydroxypteridin-6-yl)methyl)amino)benzamido)-5-((6-azidohexyl)amino)-5-oxopentanoic acid (8)
To a solution of folic acid (118 mg, 0.26 mmol) in DMSO (2 mL) was added the known 6-azidohexan-1-amine 763 (38 mg, 0.26 mmol), DCC (138 mg, 0.67 mmol), and pyridine (0.53 mL). The reaction mixture was stirred at rt for 2 days. The mixture was then filtered and the filtrate was slowly poured into ice-cold Et2O (45 mL). The precipitates were collected by centrifugation and washed 3 times by centrifugation in MeOH:Et2O (3:30 mL). The solvent was removed and the precipitate was dried to dryness under vacuum to afford a yellow solid (106 mg, 72%). The compound was further purified by reverse phase HPLC (Hewlett Packard 1100, column Agilent Zorbax C18, ACN/H2O 0.1% TFA). 1H NMR (400 MHz, DMSO-d6) δ 8.63 (s, 1H), 7.94 (d, J = 7.9 Hz, 1H), 7.82 (d, J = 5.5 Hz, 1H), 7.64 (d, J = 8.5 Hz, 2H), 6.61 (d, J = 8.5 Hz, 4H), 4.47 (s, 2H), 4.34-4.24 (m, 1H), 3.28-3.24 (m, 3H), 3.03 , 2.98 (m, 2H), 2.26-2.18 (m, 2H), 1.49-1.43 (m, 2H), 1.37-1.31 (m, 2H), 1.29 , 1.17 (m, 5H). The analytical data matched previous report of azido folate with C4 azido linker.74 LRMS: m/z calcd for C23H31N11O5 [M+H]+: 566.3 found 566.6.
Supplementary Material
ACKNOWLEDGEMENTS
This research was supported in part by the National Institutes of Health (P30CA012197, R01GM079114), the Beckman Foundation (Beckman Young Investigator Award for AD), and the Research Corporation (Cottrell Scholar Award for AD).
Footnotes
SUPPORTING INFORMATION. Antisense-conjugate titration, binucleated cell phenotype frequency, Western blot analysis, and NMR spectra of synthesized compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Tupler R, Perini G, Pellegrino MA, Green MR. Profound misregulation of muscle-genespecific gene expression in facioscapulohumeral muscular dystrophy. Proc. Natl. Acad. Sci. U. S. A. 1999;96:12650–12654. doi: 10.1073/pnas.96.22.12650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waterfall JJ, Meltzer PS. Targeting epigenetic misregulation in synovial sarcoma. Cancer Cell. 2012;21:323–324. doi: 10.1016/j.ccr.2012.02.023. [DOI] [PubMed] [Google Scholar]
- 3.Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 4.Tian Wang Q. Epigenetic regulation of cardiac development and function by polycomb group and trithorax group proteins. Dev. Dyn. 2012;241:1021–1033. doi: 10.1002/dvdy.23796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deleavey GF, Damha MJ. Designing chemically modified oligonucleotides for targeted gene silencing. Chem. Biol. 2012;19:937–954. doi: 10.1016/j.chembiol.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 6.Brennan P, Donev R, Hewamana S. Targeting transcription factors for therapeutic benefit. Mol. Biosyst. 2008;4:909–919. doi: 10.1039/b801920g. [DOI] [PubMed] [Google Scholar]
- 7.Jain A, Wang G, Vasquez KM. DNA triple helices: biological consequences and therapeutic potential. Biochimie. 2008;90:1117–1130. doi: 10.1016/j.biochi.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heitz F, Morris MC, Divita G. Twenty years of cell,penetrating peptides: from molecular mechanisms to therapeutics. Br. J. Pharmacol. 2009;157:195–206. doi: 10.1111/j.1476-5381.2009.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milletti F. Cell-penetrating peptides: classes, origin, and current landscape. Drug Discov. Today. 2012;15:850–860. doi: 10.1016/j.drudis.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt N, Mishra A, Lai GH, Wong GC. Arginine-rich cell, penetrating peptides. FEBS Lett. 2010;584:1806–1813. doi: 10.1016/j.febslet.2009.11.046. [DOI] [PubMed] [Google Scholar]
- 11.Mann A, Thakur G, Shukla V, Singh AK, Khanduri R, Naik R, Jiang Y, Kalra N, Dwarakanath BS, Langel U, Ganguli M. Differences in DNA condensation and release by lysine and arginine homopeptides govern their DNA delivery efficiencies. Mol. Pharm. 2011;8:1729–1741. doi: 10.1021/mp2000814. [DOI] [PubMed] [Google Scholar]
- 12.Turner JJ, Jones S, Fabani MM, Ivanova G, Arzumanov AA, Gait MJ. RNA targeting with peptide conjugates of oligonucleotides, siRNA and PNA. Blood Cells Mol. Dis. 2007;38:1–7. doi: 10.1016/j.bcmd.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 13.El-Andaloussi S, Johansson HJ, Holm T, Langel U. A novel cell-penetrating peptide, M918, for efficient delivery of proteins and peptide nucleic acids. Mol. Ther. 2007;15:1820–1826. doi: 10.1038/sj.mt.6300255. [DOI] [PubMed] [Google Scholar]
- 14.Madani F, Lindberg S, Langel U, Futaki S, Gräslund A. Mechanisms of cellular uptake of cell-penetrating peptides. J. Biophys. 2011;2011:414729. doi: 10.1155/2011/414729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juliano R, Alam MR, Dixit V, Kang H. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res. 2008;36:4158–4171. doi: 10.1093/nar/gkn342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiu YL, Ali A, Chu CY, Cao H, Rana TM. Visualizing a correlation between siRNA localization, cellular uptake, and RNAi in living cells. Chem. Biol. 2004;11:1165–1175. doi: 10.1016/j.chembiol.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 17.Deiters A. Light activation as a method of regulating and studying gene expression. Curr. Opin. Chem. Biol. 2009;13:678–686. doi: 10.1016/j.cbpa.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Young DD, Deiters A. Photochemical control of biological processes. Org. Biomol. Chem. 2007;5:999–1005. doi: 10.1039/b616410m. [DOI] [PubMed] [Google Scholar]
- 19.Deiters A. Principles and Applications of the Photochemical Control of Cellular Processes. Chembiochem. 2010;11:47–53. doi: 10.1002/cbic.200900529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riggsbee CW, Deiters A. Recent advances in the photochemical control of protein function. Trends Biotechnol. 2010;28:468–475. doi: 10.1016/j.tibtech.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dmochowski IJ, Tang XJ. Taking control of gene expression with light-activated oligonucleotides. Biotechniques. 2007;43:161–163. doi: 10.2144/000112519. [DOI] [PubMed] [Google Scholar]
- 22.Mayer G, Heckel A. Biologically active molecules with a “light switch”. Angew. Chem. Int. Ed. 2006;45:4900–4921. doi: 10.1002/anie.200600387. [DOI] [PubMed] [Google Scholar]
- 23.Young D, Lively M, Deiters A. Activation and deactivation of DNAzyme and antisense function with light for the photochemical regulation of gene expression in mammalian cells. J. Am. Chem. Soc. 2010;132:6183–6193. doi: 10.1021/ja100710j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Young DD, Lusic H, Lively MO, Yoder JA, Deiters A. Gene Silencing in Mammalian Cells with Light-Activated Antisense Agents. Chembiochem. 2008;9:2937–2940. doi: 10.1002/cbic.200800627. [DOI] [PubMed] [Google Scholar]
- 25.Tang X, Swaminathan J, Gewirtz AM, Dmochowski IJ. Regulating gene expression in human leukemia cells using light-activated oligodeoxynucleotides. Nucleic Acids Res. 2008;36:559–569. doi: 10.1093/nar/gkm1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deiters A, Garner RA, Lusic H, Govan JM, Dush M, Nascone-Yoder NM, Yoder JA. Photocaged morpholino oligomers for the light-regulation of gene function in zebrafish and Xenopus embryos. J. Am. Chem. Soc. 2010;132:15644–15650. doi: 10.1021/ja1053863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ouyang X, Shestopalov IA, Sinha S, Zheng G, Pitt CL, Li WH, Olson AJ, Chen JK. Versatile synthesis and rational design of caged morpholinos. J. Am. Chem. Soc. 2009;131:13255–13269. doi: 10.1021/ja809933h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Govan JM, Lively MO, Deiters A. Photochemical control of DNA decoy function enables precise regulation of nuclear factor κB activity. J. Am. Chem. Soc. 2011;133:13176–13182. doi: 10.1021/ja204980v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Govan JM, Uprety R, Hemphill J, Lively MO, Deiters A. Regulation of Transcription through Light-Activation and Light-Deactivation of Triplex-Forming Oligonucleotides in Mammalian Cells. ACS Chem. Biol. 2012;7:1247–1256. doi: 10.1021/cb300161r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mikat V, Heckel A. Light-dependent RNA interference with nucleobase, caged siRNAs. RNA. 2007;13:2341–2347. doi: 10.1261/rna.753407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jain PK, Shah S, Friedman SH. Patterning of Gene Expression Using New Photolabile Groups Applied to Light Activated RNAi. J. Am. Chem. Soc. 2010;133:440–446. doi: 10.1021/ja107226e. [DOI] [PubMed] [Google Scholar]
- 32.Casey JP, Blidner RA, Monroe WT. Caged siRNAs for Spatiotemporal Control of Gene Silencing. Mol. Pharm. 2009;6:669–685. doi: 10.1021/mp900082q. [DOI] [PubMed] [Google Scholar]
- 33.Edwards WF, Young DD, Deiters A. Light-Activated Cre Recombinase as a Tool for the Spatial and Temporal Control of Gene Function in Mammalian Cells. ACS Chem. Biol. 2009;4:441–445. doi: 10.1021/cb900041s. [DOI] [PubMed] [Google Scholar]
- 34.Chou CJ, Young DD, Deiters A. Photocaged T7 RNA Polymerase for the Light Activation of Transcription and Gene Function in Pro- and Eukaryotic Cells. Chembiochem. 2010;11:972–977. doi: 10.1002/cbic.201000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamal A, Prabhakar S, Janaki Ramaiah M, Venkat Reddy P, Ratna Reddy C, Mallareddy A, Shankaraiah N, Lakshmi Narayan Reddy T, Pushpavalli SN, Pal-Bhadra M. Synthesis and anticancer activity of chalcone, pyrrolobenzodiazepine conjugates linked via 1,2,3-triazole ring side,armed with alkane spacers. Eur. J. Med. Chem. 2011;46:3820–3831. doi: 10.1016/j.ejmech.2011.05.050. [DOI] [PubMed] [Google Scholar]
- 36.Abdelgany A, Wood M, Beeson D. Hairpin DNAzymes: a new tool for efficient cellular gene silencing. J. Gene Med. 2007;9:727–738. doi: 10.1002/jgm.1061. [DOI] [PubMed] [Google Scholar]
- 37.Agrawal S. Importance of nucleotide sequence and chemical modifications of antisense oligonucleotides. Biochim. Biophys. Acta. 1999;1489:53–68. doi: 10.1016/s0167-4781(99)00141-4. [DOI] [PubMed] [Google Scholar]
- 38.Brown DA, Kang SH, Gryaznov SM, DeDionisio L, Heidenreich O, Sullivan S, Xu X, Nerenberg MI. Effect of phosphorothioate modification of oligodeoxynucleotides on specific protein binding. J. Biol. Chem. 1994;269:26801–26805. [PubMed] [Google Scholar]
- 39.Xu Y, Zhang HY, Thormeyer D, Larsson O, Du Q, Elmén J, Wahlestedt C, Liang Z. Effective small interfering RNAs and phosphorothioate antisense DNAs have different preferences for target sites in the luciferase mRNAs. Biochem. Biophys. Res. Commun. 2003;306:712–717. doi: 10.1016/s0006-291x(03)01024-6. [DOI] [PubMed] [Google Scholar]
- 40.Koller E, Propp S, Zhang H, Zhao C, Xiao X, Chang M, Hirsch SA, Shepard PJ, Koo S, Murphy C, Glazer RI, Dean NM. Use of a chemically modified antisense oligonucleotide library to identify and validate Eg5 (kinesin-like 1) as a target for antineoplastic drug development. Cancer Res. 2006;66:2059–2066. doi: 10.1158/0008-5472.CAN-05-1531. [DOI] [PubMed] [Google Scholar]
- 41.Young DD, Edwards WF, Lusic H, Lively MO, Deiters A. Light-triggered polymerase chain reaction. Chem. Commun. 2008:462–464. doi: 10.1039/b715152g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vivès E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 43.Grandjean C, Boutonnier A, Guerreiro C, Fournier JM, Mulard LA. On the preparation of carbohydrate-protein conjugates using the traceless Staudinger ligation. J. Org. Chem. 2005;70:7123–7132. doi: 10.1021/jo0505472. [DOI] [PubMed] [Google Scholar]
- 44.Lallana E, Riguera R, Fernandez-Megia E. Reliable and efficient procedures for the conjugation of biomolecules through Huisgen azide-alkyne cycloadditions. Angew. Chem. Int. Ed. 2011;50:8794–8804. doi: 10.1002/anie.201101019. [DOI] [PubMed] [Google Scholar]
- 45.Hong V, Presolski SI, Ma C, Finn MG. Analysis and optimization of copper,catalyzed azide-alkyne cycloaddition for bioconjugation. Angew. Chem. Int. Ed. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Presolski SI, Hong V, Cho SH, Finn MG. Tailored ligand acceleration of the Cu-catalyzed azide-alkyne cycloaddition reaction: practical and mechanistic implications. J. Am. Chem. Soc. 2010;132:14570–14576. doi: 10.1021/ja105743g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shelbourne M, Brown T, El-Sagheer AH. Fast and efficient DNA crosslinking and multiple orthogonal labelling by copper-free click chemistry. Chem. Commun. 2012;48:11184–11186. doi: 10.1039/c2cc35084j. [DOI] [PubMed] [Google Scholar]
- 48.Cole DG, Saxton WM, Sheehan KB, Scholey JM. A “slow” homotetrameric kinesin-related motor protein purified from Drosophila embryos. J. Biol. Chem. 1994;269:22913–22916. [PMC free article] [PubMed] [Google Scholar]
- 49.Ma N, Tulu US, Ferenz NP, Fagerstrom C, Wilde A, Wadsworth P. Poleward transport of TPX2 in the mammalian mitotic spindle requires dynein, Eg5, and microtubule flux. Mol. Biol. Cell. 2010;21:979–988. doi: 10.1091/mbc.E09-07-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu C, Zhao J, Bibikova M, Leverson JD, Bossy-Wetzel E, Fan JB, Abraham RT, Jiang W. Functional analysis of human microtubule-based motor proteins, the kinesins and dyneins, in mitosis/cytokinesis using RNA interference. Mol. Biol. Cell. 2005;16:3187–3199. doi: 10.1091/mbc.E05-02-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Xu W. Progress on kinesin spindle protein inhibitors as anti-cancer agents. Anticancer Agents Med. Chem. 2008;8:698–704. [PubMed] [Google Scholar]
- 52.Knight SD, Parrish CA. Recent progress in the identification and clinical evaluation of inhibitors of the mitotic kinesin KSP. Curr. Top. Med. Chem. 2008;8:888–904. doi: 10.2174/156802608784911626. [DOI] [PubMed] [Google Scholar]
- 53.Dias N, Stein C. Antisense oligonucleotides: basic concepts and mechanisms. Mol. Cancer Ther. 2002;1:347–355. [PubMed] [Google Scholar]
- 54.Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 55.Stewart KM, Horton KL, Kelley SO. Cell-penetrating peptides as delivery vehicles for biology and medicine. Org. Biomol. Chem. 2008;6:2242–2255. doi: 10.1039/b719950c. [DOI] [PubMed] [Google Scholar]
- 56.van den Berg A, Dowdy SF. Protein transduction domain delivery of therapeutic macromolecules. Curr. Opin. Biotechnol. 2011;22:888–893. doi: 10.1016/j.copbio.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 57.Juliano RL, Ming X, Nakagawa O. The chemistry and biology of oligonucleotide conjugates. Acc. Chem. Res. 2012;45:1067–1076. doi: 10.1021/ar2002123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guterstam P, Madani F, Hirose H, Takeuchi T, Futaki S, El Andaloussi S, Gräslund A, Langel U. Elucidating cell-penetrating peptide mechanisms of action for membrane interaction, cellular uptake, and translocation utilizing the hydrophobic counter-anion pyrenebutyrate. Biochim. Biophys. Acta. 2009;1788:2509–2517. doi: 10.1016/j.bbamem.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 59.Wolfrum C, Shi S, Jayaprakash KN, Jayaraman M, Wang G, Pandey RK, Rajeev KG, Nakayama T, Charrise K, Ndungo EM, Zimmermann T, Koteliansky V, Manoharan M, Stoffel M. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nature Biotechnol. 2007;25:1149–1157. doi: 10.1038/nbt1339. [DOI] [PubMed] [Google Scholar]
- 60.Vlahov IR, Leamon CP. Engineering folate,drug conjugates to target cancer: from chemistry to clinic. Bioconjug. Chem. 2012;23:1357–1369. doi: 10.1021/bc2005522. [DOI] [PubMed] [Google Scholar]
- 61.Leamon CP. Folate-targeted drug strategies for the treatment of cancer. Curr. Opin. Investig. Drugs. 2008;9:1277–1286. [PubMed] [Google Scholar]
- 62.Li S, Deshmukh HM, Huang L. Folate-mediated targeting of antisense oligodeoxynucleotides to ovarian cancer cells. Pharm. Res. 1998;15:1540–1545. doi: 10.1023/a:1011946915209. [DOI] [PubMed] [Google Scholar]
- 63.Romuald C, Busseron E, Coutrot F. Very contracted to extended co, conformations with or without oscillations in two- and three-station [c2]daisy chains. J. Org. Chem. 2010;75:6516–6531. doi: 10.1021/jo101234u. [DOI] [PubMed] [Google Scholar]
- 64.Schneider R, Schmitt F, Frochot C, Fort Y, Lourette N, Guillemin F, Müller JF, Barberi-Heyob M. Design, synthesis, and biological evaluation of folic acid targeted tetraphenylporphyrin as novel photosensitizers for selective photodynamic therapy. Bioorg. Med. Chem. 2005;13:2799–2808. doi: 10.1016/j.bmc.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 65.Willibald J, Harder J, Sparrer K, Conzelmann KK, Carell T. Click-Modified Anandamide siRNA Enables Delivery and Gene Silencing in Neuronal and Immune Cells. J. Am. Chem. Soc. 2012;134:12330–12333. doi: 10.1021/ja303251f. [DOI] [PubMed] [Google Scholar]
- 66.Shakeri-Zadeh A, Ghasemifard M, Mansoor G. Structural and optical characterization of folate-conjugated gold-nanoparticles. Physica E-Low-Dimensional Systems & Nanostructures. 2010;42:1272–1280. [Google Scholar]
- 67.Lee SM, Chen H, O'Halloran TV, Nguyen ST. Clickable polymer-caged nanobins as a modular drug delivery platform. J. Am. Chem. Soc. 2009;131:9311–9320. doi: 10.1021/ja9017336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nakagawa O, Ming X, Huang L, Juliano RL. Targeted intracellular delivery of antisense oligonucleotides via conjugation with small-molecule ligands. J. Am. Chem. Soc. 2010;132:8848–8849. doi: 10.1021/ja102635c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lusic H, Uprety R, Deiters A. Improved synthesis of the two-photon caging group 3-nitro-2-ethyldibenzofuran and its application to a caged thymidine phosphoramidite. Org. Lett. 2010;12:916–919. doi: 10.1021/ol902807q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Momotake A, Lindegger N, Niggli E, Barsotti RJ, Ellis-Davies GC. The nitrodibenzofuran chromophore: a new caging group for ultra-efficient photolysis in living cells. Nat. Methods. 2006;3:35–40. doi: 10.1038/nmeth821. [DOI] [PubMed] [Google Scholar]
- 71.Ando H, Furuta T, Tsien RY, Okamoto H. Photo-mediated gene activation using caged RNA/DNA in zebrafish embryos. Nat. Genet. 2001;28:317–325. doi: 10.1038/ng583. [DOI] [PubMed] [Google Scholar]
- 72.Nielsen R, Courtoy PJ, Jacobsen C, Dom G, Lima WR, Jadot M, Willnow TE, Devuyst O, Christensen EI. Endocytosis provides a major alternative pathway for lysosomal biogenesis in kidney proximal tubular cells. Proc. Natl. Acad. Sci. U. S. A. 2007;104:5407–5412. doi: 10.1073/pnas.0700330104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Denny WA, Leupin W, Kearns DR. Simplified Liquid-Phase Preparation of Four Decadeoxyribonucleotides and their Preliminary Spectroscopic Characterization. Helvetica Chemica. Acta. 1982;65:2372–2393. [Google Scholar]
- 74.Mindt TL, Müller C, Melis M, de Jong M, Schibli R. “Click-to, chelate”: in vitro and in vivo comparison of a 99mTc(CO)3-labeled N(tau)-histidine folate derivative with its isostructural, clicked 1,2,3-triazole analogue. Bioconjug. Chem. 2008;19:1689–1695. doi: 10.1021/bc800183r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.