

Abstract
Sucralose is a synthetic organochlorine sweetener (OC) that is a common ingredient in the world's food supply. Sucralose interacts with chemosensors in the alimentary tract that play a role in sweet taste sensation and hormone secretion. In rats, sucralose ingestion was shown to increase the expression of the efflux transporter P-glycoprotein (P-gp) and two cytochrome P-450 (CYP) isozymes in the intestine. P-gp and CYP are key components of the presystemic detoxification system involved in first-pass drug metabolism. The effect of sucralose on first-pass drug metabolism in humans, however, has not yet been determined. In rats, sucralose alters the microbial composition in the gastrointestinal tract (GIT), with relatively greater reduction in beneficial bacteria. Although early studies asserted that sucralose passes through the GIT unchanged, subsequent analysis suggested that some of the ingested sweetener is metabolized in the GIT, as indicated by multiple peaks found in thin-layer radiochromatographic profiles of methanolic fecal extracts after oral sucralose administration. The identity and safety profile of these putative sucralose metabolites are not known at this time. Sucralose and one of its hydrolysis products were found to be mutagenic at elevated concentrations in several testing methods. Cooking with sucralose at high temperatures was reported to generate chloropropanols, a potentially toxic class of compounds. Both human and rodent studies demonstrated that sucralose may alter glucose, insulin, and glucagon-like peptide 1 (GLP-1) levels. Taken together, these findings indicate that sucralose is not a biologically inert compound.
The organochlorine (OC) sweetener sucralose is a synthetic trichlorinated disaccharide with the chemical name 1,6-dichloro-1,6-dideoxy-β-D-fructofuranosyl-4-chloro-4-deoxy-α-D-galactopyranoside (Merck Index, 2006). Its sweetness potency is approximately 385- to 650-fold higher than sucrose (table sugar) by weight, depending upon the specific food or beverage application (DuBois et al., 1991; Schiffman and Gatlin, 1993; Schiffman et al., 2008). In 1991, sucralose was first approved for use in Canada, followed by Australia in 1993, and New Zealand in 1996 (Davies, 2010). In 1998, the U.S. Food and Drug Administration (FDA) approved sucralose for use in 15 food and beverage categories (U.S. FDA, 1998) that included water-based as well as fat-based products (e.g., frozen dairy desserts, baked goods, confections, puddings, chewing gum, fats and oils, beverages, and sugar substitutes such as Splenda). In 1999, the FDA expanded its approval beyond the original 15 categories for use as a general-purpose sweetener in all categories of foods and beverages (U.S. FDA, 1999). In 2004, sucralose (termed food additive E 955) was approved in the European Union (EU) in a variety of products including water- and fat-based desserts, certain alcoholic beverages, fat-based sandwich spreads, breakfast cereals, marinades, and chewing gum (EU, 2004). Sucralose accounts for 27.9% of the $1.146 billion global high-potency sweetener market (Leatherhead Food Research, 2011) and is utilized in thousands of food, beverage, and pharmaceutical products in North America, Latin America, Europe, the Middle East, and the Asia-Pacific region (Goldsmith, 2000a; Davies, 2010). The range of product utilization is more extensive for sucralose than for other artificial sweeteners due to its physicochemical properties. For example, sucralose is readily soluble in ethanol, methanol, and water (Bennett et al., 1992; Anderson et al., 2006; Merck Index, 2006; Li et al., 2010), and this solubility profile contributes to its versatility in both fat- and water-based food and beverage applications including alcoholic drinks. Other common artificial sweeteners such as aspartame, sodium saccharin, and acesulfame-potassium (ace-K) are only slightly or sparingly soluble in ethanol and/or methanol (Schiffman and Gatlin, 1993; Merck Index, 2006) and have more limited product applications. The Acceptable Daily Intake (ADI) level for sucralose was set at 5 mg/kg body weight per day (mg/kg/d) in the United States (U.S. FDA, 1998) and 15 mg/kg/d in the EU as recommended by Scientific Committee on Food of the European Commission (SCF, 2000), and there are no exclusions or restrictions for vulnerable population groups, including pregnant women, nursing mothers, infants, children, elderly, persons with medical conditions, and patients taking multiple medications.
Although sucralose is used globally in reduced-calorie and diet foods and beverages, issues regarding its biological effects and, therefore, its health profile have raised concerns. These issues include the following:
-
1.
Effects of sucralose on glucose transport and other parameters involved in body weight regulation. Sucralose modulates physiological parameters involved in normal body weight regulation, including faster intestinal glucose transport (Mace et al., 2007, 2009) via interaction with sweet taste receptors located in the gastrointestinal tract (GIT) (Margolskee et al., 2007; Jang et al., 2007), increased insulin secretion via activation of sweet taste receptors on pancreatic ß cells (Nakagawa et al., 2009), and altered sweet taste receptor expression in the hypothalamus (Ren et al., 2009) in rodents. Sucralose initiated the release of glucagon-like peptide-1 (GLP-1) in vitro (Jang et al., 2007; Margolskee et al., 2007), and ingestion of a sucralose-ace-K sweetened beverage increased GLP-1 secretion in healthy individuals as well as subjects with type 1 diabetes (Brown et al., 2012). GLP1 is an incretin hormone secreted in the gut that (1) induces glucose-dependent stimulation of insulin by the pancreas, (2) reduces glucagon secretion by the liver, (3) delays gastric emptying, and (4) increases satiety (Freeman, 2009). Further, sucralose was shown to elevate glucose and insulin levels in a small study of obese women (Pepino et al., 2013), who are at increased risk for further weight gain and development of diabetes.
-
2.
Effects of sucralose on presystemic detoxification mechanisms and impact on bioavailability of therapeutic drugs. Pharmacokinetic studies in animals and humans designed to characterize the metabolic fate of sucralose concluded that the majority of sucralose consumed (approximately 65–95% dependent on species) is not absorbed from the GIT but rather reportedly passes from the body unchanged in the feces (Grice and Goldsmith, 2000; John et al., 2000a, 2000b; Roberts et al., 2000; Sims et al., 2000; Wood et al., 2000). The low reported bioavailability of this OC sweetener is unexpected given that it is an amphiphilic molecule (Hough and Khan, 1978, 1989) with appreciable lipid solubility. The low absorption from the GIT suggested to Abou-Donia et al. (2008) that sucralose might be actively recirculated into the intestinal lumen by the efflux transporter P-glycoprotein (P-gp) and/or metabolized by intestinal cytochrome P-450 (CYP) enzymes presystemically during first-pass metabolism. Both P-gp and CYP play major roles in the pharmacokinetics of a broad range of OC compounds as described in this review. Abou-Donia et al. (2008) reported that sucralose (administered as the commercial sucralose product Splenda) at doses approved by the FDA and EU elevated the expression of P-gp and CYP enzymes in male rats to levels previously associated with significant reductions in the bioavailability of therapeutic drugs (Dürr et al., 2000).
-
3.
Metabolic fate and safety profile of sucralose metabolites. Thin-layer chromatograms (TLC) of methanolic fecal extracts following sucralose administration to rats (Sims et al., 2000) and humans (Roberts et al., 2000) revealed at least two closely eluting peaks that suggest sucralose is metabolized in the GIT. The identity and biological effects of the metabolites of sucralose associated with these peaks are not known at this time (Schiffman, 2012; Schiffman and Abou-Donia, 2012). The TLC data of Sims et al. (2000) and Roberts et al. (2000) appear inconsistent with the claim that sucralose is not metabolized in the GIT as asserted by Grice and Goldsmith (2000), Molinary and Quinlan (2006), and Grotz and Munro (2009).
-
4.
Effect of sucralose on the number and relative proportions of different intestinal bacterial types. Early studies of sucralose with bacteria in culture indicated that sucralose was not utilized as a carbon source by oral bacteria (Young and Bowen, 1990) or by bacteria from environmental samples (Labare and Alexander, 1994). Abou-Donia et al. (2008) extended these studies to bacteria cultured from the GIT of rats that had been administered sucralose daily. An overall reduction of the existing microflora was found (≥50%) at sucralose doses that were lower than the human ADI. Beneficial bacteria including lactobacilli and bifidobacteria were disproportionately affected compared to pathogenic bacteria including enterobacteria. Further, the reduction in fecal microflora was not fully reversible even 3 mo after cessation of sucralose. Sucralose was also reported to exhibit antimicrobial activity against two oral bacterial species involved in periodontal disease (Prashant et al., 2012).
-
5.
Potential toxicity from habitual sucralose ingestion. Historical in vitro genotoxicity tests found that sucralose was weakly mutagenic in a mouse lymphoma mutation assay, and that one of its hydrolysis products was weakly mutagenic in both the Ames test and mouse lymphoma mutation assay (WHO, 1989; U.S. FDA, 1998). A subsequent comet test by Sasaki et al. (2002) found that sucralose induced DNA damage in mouse GIT. Three independent labs showed that sucralose undergoes thermal decomposition at temperatures used in baking (Hutchinson, 1996; Hutchinson et al., 1999; Bannach et al., 2009; Rahn and Yaylayan, 2010), and heating sucralose with glycerol, the backbone of triglycerides, generated chloropropanols, a potentially toxic class of compounds (Rahn and Yaylayan, 2010).
Each of these five issues is addressed in more detail in this review.
EFFECTS OF SUCRALOSE ON GLUCOSE TRANSPORT AND OTHER PARAMETERS INVOLVED IN BODY WEIGHT REGULATION
Sucralose Modulates Physiological Processes Relevant to Body Weight
In the last several years, numerous studies in rodents reported that sucralose modulates physiological processes involved in nutrient absorption and body weight regulation via its interaction with sweet taste receptors (called T1R2/T1R3) located in enteroendocrine cells of the GIT (Jang et al., 2007; Margolskee et al., 2007), pancreatic ß cells (Nakagawa et al., 2009), and the hypothalamus (Ren et al., 2009). Sucralose in vitro enhanced glucose-stimulated insulin secretion in the mouse insulinoma (ß cell) MIN6 line (Nakagawa et al., 2009), isolated mouse islets (Nakagawa et al., 2009), and isolated rat islets (Corkey, 2012). Sucralose also initiated the release of glucagon-like peptide-1 (GLP-1) in several GIT model systems, including the human L cell line NCI-H716 (Jang et al., 2007) and mouse enteroendocrine cell line GLUTag (Margolskee et al., 2007). In vivo, oral ingestion of sucralose-sweetened water increased the expression of SGLT-1, the classical active Na+-glucose cotransporter, as well as glucose absorption in mice (Margolskee et al., 2007). Intestinal infusion of sucralose elevated glucose absorption via the diffusive apical GLUT2 pathway in rats (Mace et al., 2007, 2009). However, sucralose given by oral gavage to rats in combination with an intraperitoneal (ip) glucose tolerance test exerted no marked effect on GLP-1 (Fujita et al., 2009).
Although the majority of in vitro and in vivo studies in rodents detected significant effects of sucralose on physiological processes involved in nutrient absorption, findings of similar effects in humans have been inconsistent. Sucralose delivered in isolation (e.g., without co-administration of glucose) to human volunteers by oral (Ford et al., 2011) or by intragastric routes (Ma et al., 2009; Steinert et al., 2011) exerted no marked effect on GLP-1. Oral consumption of sucralose without co-administration of glucose (Brown et al., 2011) produced no significant effect on blood glucose levels. Sucralose delivered by intraduodenal infusion in combination with glucose also exerted no marked effect on blood glucose or plasma GLP-1 (Ma et al., 2010). However, GLP-1 was elevated in human subjects (both in healthy volunteers and in individuals with type 1 diabetes) who drank a caffeine-free diet soda sweetened with sucralose and ace-K when compared with a carbonated water control; in both conditions subjects consumed the test solution (diet soda or carbonated water) 10 min prior to a glucose load (Brown et al., 2009, 2012). Under similar test conditions, obese women displayed elevated glucose and insulin levels when exposed to sucralose alone (Pepino et al., 2013). Recent animal data suggest that habitual oral ingestion of high-potency sweeteners that do not provide calories may blunt GLP-1 release and produce hyperglycemia in response to subsequent oral glucose tolerance tests (Swithers et al., 2012).
Ren et al. (2009) found that sucralose modulated the expression of the gene for the sweet taste receptor T1R2 in cells from the hypothalamus, a nutrient-sensing region of the brain, in rodents. Data showed that T1R2 expression was elevated when the hypothalamic cells were exposed to low (compared to high) extracellular glucose concentrations, and this was reversed with addition of sucralose to the low-glucose medium. That is, addition of sucralose resulted in expression levels of T1R2 that would be expected if higher extracellular glucose levels were actually present. Thus, activation of the sweet taste receptor in the brain by nonnutritive sucralose might potentially affect appetite regulation by providing an inaccurate signal regarding the actual levels of extracellular glucose in the brain (Schiffman, 2012). However, it is not yet known if the portion of sucralose that is absorbed into the systemic circulation can traverse the blood–brain barrier to reach the hypothalamus.
Association Between Artificial Sweetener Use, Body Weight, and Obesity
Major contributors to the rising obesity epidemic are excess consumption of energy-dense foods and reduced physical activity (WHO, 2013). Many additional factors have also been reported to contribute to the obesity epidemic, including OC compound exposure, use of artificial sweeteners, consumption of soft drinks (both sugar-sweetened and diet), alterations of the microbiome, use of medications, epigenetic changes due to an obesogenic environment in utero, environmental exposure to endocrine disruptors, indoor air conditioning, and chronic sleep deprivation (Cizza and Rother, 2012; La Merrill and Birnbaum, 2011; Decherf and Demeneix, 2011; Birnbaum, 2013; Swithers et al., 2010). For many years, exposure to OC pesticides including lindane (Chadwick et al., 1988), DDT (Deichmann et al., 1972), aldrin (Deichmann et al., 1972), and hexachlorobenzene (Villeneuve et al., 1977) was associated with elevated body weight gain in rodent models. In humans, prenatal exposure to DDE (dichlorodiphenyl-dichloroethylene), a metabolite of DDT, was linked to increased body weight of female offspring (Karmaus et al., 2009). In addition, the prevalence of diabetes in humans has been positively correlated to OC exposure (Lee et al., 2006; Porta, 2006; Vasiliu et al., 2006). Epidemiological studies in humans (Fowler et al., 2008; Stellman and Garfinkel, 1986; Dhingra et al., 2007; Lutsey et al., 2008; Yang, 2010) and lab studies in animals (Swithers and Davidson, 2008; Swithers et al., 2009, 2010) both suggest an association between use of artificial sweeteners and body weight gain. Epidemiological studies also demonstrated that artificial sweetener use increased the risk for metabolic syndrome, type 2 diabetes, hypertension, and cardiovascular disease (Swithers, 2013). Most human epidemiological studies have not distinguished among the different types of artificial sweeteners (e.g., sucralose, saccharin, acesulfame-K, aspartame, neotame, stevioside, and rebaudioside) but rather treated them as a group. One exception is the Nurses’ Health Study cohort (Colditz et al., 1990), which did specifically associate saccharin use with weight gain. This finding is consistent with recent animal experiments (Swithers and Davidson, 2008; Swithers et al., 2009) in which saccharin intake was related to weight gain in rats. Because OC compounds and artificial sweeteners have both been associated with weight gain, and because sucralose is a member of both categories, it is important to determine its effect on mechanisms that regulate body weight.
Effect of Sucralose on Body Weight in Humans
To date, the effect of chronic oral consumption of sucralose on body weight at levels approved by the FDA and EU has not been studied prospectively in adults. In a lab setting, ingestion of sucralose tends to increase intake after a 60-min interval (Anderson et al., 2002); however, it is not yet known whether this finding transfers into chronic body weight gain in free-living adult populations. In an 18-mo trial with children, participants were randomly assigned to receive an 8-oz can per day of either a noncalorically sweetened or a sugar-sweetened beverage that provided 104 kcal (de Ruyter et al., 2012). Each can of sugar-free beverage contained 34 mg sucralose and 12 mg acesulfame-K, and the mean sucralose dosage for the sugar-free group was 1.1 mg/kg/d. The mean duration of the study was 541 d (77.3 wk), during which 477 of 641 children completed the intervention by consuming an average of 5.8 beverages per week. Measurement of urinary sucralose levels was one of several markers used to monitor compliance with the protocol. The calorie consumption from these beverages was 46,627 kcal greater for children in the sugar-sweetened group than in the sucralose-sweetened group (5.8 × 77.3 × 104). In spite of this highly significant difference in calories consumed from the beverages, the total weight gain over this 18-mo study was only 1 kg greater for children in the sugar-sweetened group compared to sucralose group. No explanation was provided to account for the small difference in weight gain given the large difference in caloric consumption from the beverages. However, one possible explanation is that the children who consumed the sugar-sweetened beverages compensated by reducing their food intake. A control group that ingested water as a comparison was not included. Another study in adolescents showed no consistent reduction of weight gain at a 2-year follow-up when their families were supplied with artificially sweetened beverages in order to reduce their consumption of sugar-sweetened sodas (Ebbeling et al., 2012).
Effect of Sucralose on Body Weight in Animals
In rats, a sucralose dosage of 1.1 mg/kg/d (equivalent to that provided to children by de Ruyter et al., 2012) produced a significant body weight gain (10.9% greater than controls) over a 12-wk period (Abou-Donia et al., 2008). However, weight gain relative to control was not observed in rats at higher dosages of 3.3, 5.5, and 11 mg/kg/d sucralose. After a 12-wk recovery period from sucralose treatment, there were significant increases in body weight for rats previously treated with sucralose relative to control animals for 2 of 4 sucralose dosage groups. After recovery from 1.1 mg/kg/d, there was a 17.1% rise in body weight relative to control animals, and after recovery from 5.5 mg/kg/d, a 21.3% elevation. In the human nutrition literature, a weight difference ≥5% is considered clinically significant (Wengreen and Moncur, 2009). Two possible explanations (described in more detail later in this review) for the significant body weight gain at the lowest (not highest) dosage of sucralose by the end of treatment period include: (1) Sucralose may no longer be bioavailable as an intact molecule at higher dosages due to increased intestinal expression of (and metabolism by) CYP enzymes, and (2) intestinal bacteria that normally reclaim calories from undigested dietary nutrients in the intestines are suppressed to a greater degree at the higher dosages. The finding of a weight gain after a 12-wk recovery from sucralose may be due to a partial rebound in the number of bacteria that ferment complex dietary carbohydrates to form short-chain fatty acids that are subsequently absorbed.
Animal data regarding body weight gain reported in historical subchronic and chronic toxicity studies utilized sucralose dosages that far exceed levels approved for use in foods by the FDA (U.S. FDA, 1998) and EU (2004), and the results on body weight gain in these toxicity studies are inconsistent and conflicting. Increases in mean body weight gain were found in sucralose-treated beagle dogs relative to controls at 0.3 to 3% of the diet (approximately 90 to 900 mg/kg/d respectively), and this weight gain was accompanied by a rise in food consumption (Goldsmith, 2000b). Body weight gain in rats, however, appeared to be dependent in part on the route of delivery of sucralose. Rats that consumed high doses of sucralose in toxicity tests by the oral route (dietary administration) showed significant decreases in body weight gain relative to controls in spite of relatively small reductions in food consumption. These disproportionately large declines in body weight gain despite only small decreases in intake were attributed to low palatability of sucralose at high concentrations. Body weight changes in rats that received high dosages of sucralose by gavage (which circumvents taste issues associated with dietary administration of an unpalatable test material) were variable with losses, gains and no marked changes reported (Goldsmith, 2000b; U.S. FDA, 1998). These animal feeding results are not directly transferable to humans, however, because the dosages delivered to animals in these toxicity tests were significantly higher than usage levels approved for humans.
Learned Associations and High-Potency Sweeteners as a Group
A learning paradigm has been proposed to explain how high-potency sweeteners as a group may influence body weight regulation and glucose homeostasis (Swithers et al., 2010, 2012; Swithers, 2013). This paradigm is based on the principle that humans and non-human animals learn the relationships between the sensory properties of foods (e.g., taste, smell, texture) and their postingestive nutritive consequences to maintain energy balance (Warwick and Schiffman, 1991; Swithers and Davidson, 2008; Swithers et al., 2009, 2010). Throughout evolutionary history (at least until the recent introduction of high-potency sweeteners), learned sweet taste cues have been reliable predictors of the energy density of food. However, the introduction of high-potency sweeteners that have negligible utilizable calories into the food supply reduced the validity of sweet taste as a signal for calories. Uncoupling the relationship between sensory properties of foods and their caloric content was shown to contribute to weight gain in rats (Warwick and Schiffman, 1991). Using learning paradigms with a rat model, Swithers and colleagues (Swithers and Davidson, 2008; Swithers et al., 2009, 2010) found that consumption of foods and fluids containing high-potency sweeteners (saccharin, acesulfame-K, and/or steviol glycosides were used in these experiments) interfered with the ability of sweet taste to predict caloric consequences and thus disrupted energy regulation. Further, the nonpredictive sweet–calorie relationship was persistent and difficult to reverse even after predictive sweet–calorie training with glucose (Swithers et al., 2009). Thus, exposing rats to high-potency sweeteners degraded the consistent and predictive relationship between taste, energy intake, and body weight regulation.
The degree to which these animal-learning studies of sweeteners can be extrapolated to humans and to sucralose in particular has not yet been determined. Humans can distinguish among many different sweetener types by taste (Schiffman et al., 1979, 1981, 1995; Schiffman and Gatlin, 1993), and functional magnetic resonance (fMRI) imaging studies also indicate that the human brain signals distinguish caloric sweeteners (e.g., sucrose) from noncaloric sweeteners (e.g., sucralose and saccharin) (Frank et al., 2008; Haase et al., 2008). However, neuroimaging studies by Rudenga and Small (2012) revealed that routine use of high-potency sweeteners as a group altered brain responses to sucrose in the amygdala and insula as determined by fMRI scanning. Data suggested that these alterations in brain activity were due to degradation or uncoupling of the predictive relationship between sweet taste and its postingestive consequences, as reported previously in rat models (Swithers et al. 2010). The findings of Rudenga and Small (2012) are consistent with those of Green and Murphy (2012), who reported that higher order reward regions of the brain are activated to a greater extent by sweeteners in young adult diet-soda drinkers compared to nondrinkers. These brain imaging studies in conjunction with epidemiological data that associate artificial sweetener use with weight gain suggest that high-potency sweeteners interfere with learned sweet–calorie relationships in humans as well as animals (Swithers, 2013).
Effect of Sucralose in Patients With Diabetes
A study of patients with diabetes (Grotz et al., 2003) reported no significant effect of sucralose on glycosylated hemoglobin (HbA1c) as a marker of the average plasma glucose concentrations over approximately 3 mo. Grotz et al. (2003) instructed patients with diabetes to self-administer capsules of sucralose twice daily over a 3-mo period; however, data on compliance with self-administration (e.g., urinary sucralose levels) and body weight changes were not reported. Daily supervision of capsule administration, rather than self-administration, is scientifically prudent for studies of patients with diabetes because noncompliance with treatment regimens is reportedly high in this population (WHO, 2003). Further, no apparent information of dissolution characteristics of the capsule was provided; thus, it is unknown where sucralose was released in the GIT. Future studies of sucralose on diabetes management will require randomized trials that are carefully supervised and control for the variables involved.
EFFECTS OF SUCRALOSE ON PRESYSTEMIC DETOXIFICATION MECHANISMS AND IMPACT ON BIOAVAILABILITY OF THERAPEUTIC DRUGS
Background: Role of Intestinal Transporters and CYP Metabolizing Enzymes in Bioavailability of Xenobiotics
Orally administered xenobiotics such as therapeutic drugs and artificial food additives reach the small intestine, where they cross the intestinal membranes to be transported into the hepatic portal system and ultimately to the systemic circulation. During their transit from the GIT to the systemic circulation, xenobiotics interact with metabolic enzymes and transporters in the GIT and liver that limit their systemic bioavailability. The decrease in the concentration of a xenobiotic compound as it passes through the GIT and liver is termed the “first-pass effect” (Riegelman and Rowland, 1973; Iwamoto and Klaassen, 1977; Shimomura et al., 2002; Watkins, 1997; Paine and Thummel, 2003; Paine, 2009). The intestinal component of the first-pass effect is especially significant in the disposition of sucralose because the majority of this OC sweetener is reportedly unabsorbed from the small intestine after ingestion (Grice and Goldmith, 2000) even though it is an amphiphilic compound with appreciable lipid solubility (Hough and Khan, 1978; 1989; Anderson et al., 2006; Li et al., 2010). Further, higher doses of orally administered sucralose in humans are associated with lower urinary excretion that suggests sucralose absorption is reduced at increased concentrations (Roberts et al., 2000). The findings by Abou-Donia et al. (2008) described in the next section suggest that the efflux transporter P-gp and metabolism by intestinal CYP limit oral absorption of sucralose (and/or its metabolites) from the GIT. P-gp is an energy-dependent “pump” that is highly expressed at the luminal surface of enterocytes (Lin and Yamazaki, 2003; Marchetti et al., 2007). It is a member of the ATP-binding cassette (ABC) transporter superfamily and a product of the ABCB1 gene (also known as MDR1). P-gp serves as a barrier to harmful chemicals (as well as many therapeutic drugs) by transporting them out of enterocytes, back into the intestinal lumen (Sparreboom et al., 1997; Masuda et al., 2000; Westphal et al., 2000; Suzuki and Sugiyama, 2000; Drescher et al., 2003; Abu-Qare et al., 2003; Fang et al., 2009). P-gp interacts with many structurally diverse hydrophobic and amphiphilic compounds (Garrigues et al., 2002; Marchetti et al., 2007; Yang et al., 2009) including OC drugs (Polli et al., 2001; Boulton et al., 2002; Wang et al., 2001; 2002; 2008) and OC pesticides (Bain and LeBlanc, 1996).
Intestinal metabolism by members of the CYP superfamily of heme-containing enzymes contributes significantly to the first-pass effect (Kolars et al., 1991; Watkins, 1992, 1997; Paine et al., 1996, 2006; Thummel et al., 1997; Lampen et al., 1998; Hall et al., 1999; Wacher et al., 2001; von Richter et al., 2001). The CYP superfamily is divided into families (including CYP1, CYP2, and CYP3, which are responsible for metabolism of drugs and xenobiotics) and into subfamilies labeled with a letter, such as CYP3A or CYP2D. CYP3A has a broad substrate specificity and is a predominant isoform of CYP expressed in the upper intestine (Watkins et al., 1987; Kolars et al., 1994; Guengerich, 1999; Zhang et al., 1999; Mitschke et al., 2008; Takara et al., 2003; Matsubara et al., 2004). Organochlorine drugs such as midazolam undergo significant intestinal metabolism by the CYP3A subfamily (Paine et al., 1996; Thummel et al., 1996; Higashikawa et al., 1999; Bruyère et al., 2009). The catalytic activities of intestinal and hepatic CYP3A are independently regulated so that measures of CYP-mediated metabolism of xenobiotics in the liver do not necessarily predict the effects of CYP-mediated metabolism in the intestine (Hakkak et al., 1993; Lown et al., 1994; Aiba et al., 2003; Paine and Thummel, 2003; van Herwaarden et al., 2009).
Although the roles of P-gp and CYP in limiting the bioavailability of xenobiotics are different, there is significant overlap in compounds that are effluxed by P-gp and those metabolized by CYP3A (Watkins, 1997; Wacher et al., 1995, 2001, Schuetz et al., 1996; Benet et al., 1999). While P-gp effluxes xenobiotics back into the intestinal lumen, CYP enzymes promote elimination of xenobiotics through oxidation and reduction reactions that render chemicals more polar and water-soluble. Studies of xenobiotic detoxification and clearance indicate that co-localization of P-gp and CYP3A within enterocytes facilitates an interactive process by which the bioavailability of compounds that are dual P-gp/CYP3A substrates is reduced. P-gp enhances presystemic metabolism in the intestine by increasing the duration of exposure to CYP3A via repeated efflux and reabsorption into enterocytes. This repetitive recycling process that engages both intestinal P-gp and CYP3A provides a barrier to absorption (Benet, 2009), and may account for the low systemic oral bioavailability of sucralose.
Splenda (Active Ingredient: Sucralose) Increases the Expression of Intestinal P-gp and Two CYP Isoforms
Abou-Donia et al. (2008) reported that Splenda, a commercially available form of sucralose, increased the expression of intestinal P-gp and CYP in male Sprague-Dawley rats. Splenda contains the high-potency OC sweetener sucralose along with the fillers, maltodextrin and glucose. Splenda was administered for 12 wk by oral gavage at sucralose dosages equivalent to 0 (vehicle control), 1.1, 3.3, 5.5, or 11 mg/kg/d. All sucralose dosages tested fell below the ADI approved for use in the food supply by the European Union (EU, 2004). The two lower dosages fell below the ADI for sucralose approved by the FDA (1998). After 12 wk, half of the animals from each group were sacrificed to measure the expression of intestinal P-gp, CYP3A, and CYP2D in the jejunum and ileum. The other half of the animals were allowed to recover from treatment (no Splenda) for another 12 wk, after which expression of intestinal P-gp, CYP3A, and CYP2D was again measured. P-gp and CYP expression were assessed using antibodies that detected P-gp, CYP3A protein, and CYP2D1, the rat isozyme analogous to human CYP2D6 (Laurenzana et al., 1995). These antibodies were selected because the CYP3A and CYP2D subfamilies are collectively involved in the metabolism of more than 70% of medications (Dantzig et al., 1999; Felmlee et al., 2008; Ingelman-Sundberg, 2005).
Table 1 shows the changes in P-gp and CYP expression relative to control (vehicle, no Splenda) after daily treatment at each of 4 dosage levels for 12 wk. At the lowest dose (1.1 mg/kg/d sucralose), there was no significant change in the expression of P-gp or CYP. However, at 3.3 mg/kg/d sucralose, there was a marked increase in P-gp as well as significant elevation in CYP. [Note: For a 130-lb (58.9 kg) woman, the 3.3-mg/kg/d dosage of sucralose is equivalent to approximately two 12-oz (340 g) servings of a diet soda. For a 70-lb (31.8 kg) child, the 3.3-mg/kg/d dosage is equivalent to only one 12-oz (340 g) serving.] At 5.5 mg/kg/d sucralose delivered in Splenda, the expression of P-gp and CYP remained elevated with expression of CYP growing in magnitude with increasing dosage. [Note: For a 130-lb woman, the 5.5-mg/kg/d dosage is equivalent to approximately two 12-oz (340 g) servings of a sucralose-sweetened soft drink and two pieces of sucralose-sweetened cake.] At 11 mg/kg/d sucralose, expression of P-gp decreased significantly rather than increasing, while CYP expression continued to rise. Residual effects on P-gp and CYP expression at some dose levels remained at the end of the 12-wk recovery period in the absence of Splenda. Thus, Splenda enhanced the expression of P-gp and CYP at sucralose dosages commonly ingested by consumers, and some changes in expression persisted 3 mo after cessation of intake.
TABLE 1.
Significant Changes in the Expression of P-gp, CYP3A, and CYP2D Subsequent to Sucralose Ingestion (Delivered in Splenda) Relative to Control (No Sucralose) in Rat
| Sucralose dosage (mg/kg/d) | P-gp | CYP3A | CYP2D | |
|---|---|---|---|---|
| Treatment | 1.1 | — | — | — |
| 3.3 | ↑ | ↑ | ↑ | |
| 143.5% | 43.5% | 36.7% | ||
| 5.5 | ↑ | ↑ | ↑ | |
| 122.6% | 70.0% | 152.1% | ||
| 11 | ↓ | ↑ | ↑ | |
| 64% | 151.3% | 249.3% | ||
| Recovery | 1.1 | — | — | — |
| 3.3 | ↑ | — | — | |
| 16% | ||||
| 5.5 | ↑ | — | ↑ | |
| 56.8% | 32.9% | |||
| 11 | ↑ | ↑ | ↑ | |
| 82.2% | 22.4% | 22.1% |
Note. Data from Abou-Donia et al. (2008).
The results in Table 1 indicate that the magnitude of elevation for both CYP3A and CYP2D expression increased in a linear, dose-dependent manner as the dosage of sucralose increased from 3.3 to 5.5 to 11 mg/kg/d. This finding of significant and parallel increases in expression of two different CYP enzymes does not support the claim made by Brusick et al. (2009) that increases in CYP from sucralose ingestion were only normal biological variations. Significant changes in P-gp expression, like CYP expression, also occurred at sucralose dosages 3.3, 5.5, and 11 mg/kg/d; however, the magnitude of the elevation of P-gp expression was maximum at 3.3 mg/kg/d sucralose and decreased monotonically at increasing dosages; that is, as CYP expression rose, P-gp expression decreased. Two possible explanations for this relationship between CYP and P-gp are the following. First, Abou-Donia et al. (2008) and Schiffman and Abou-Donia (2012) suggested that the dose-dependent rise in CYP expression increased the metabolism of sucralose and reduced the concentration of intact sucralose molecules in the GIT. At an oral dosage of 11 mg/kg/d, the concentration of intact sucralose in the GIT was reduced to levels that were insufficient to elevate the expression of P-gp. Second, saturation of P-gp may have occurred as the dosage of sucralose increased. Nonlinear pharmacokinetics, including saturation, is characteristic of the intestinal component of the xenobiotic metabolism (Tachibana et al., 2012; Tamai et al., 1997; Harrison et al., 2004; Saitoh et al., 2007; Rowland and Tozer, 2011).
The rise in CYP expression reported by Abou-Donia et al. (2008) may result from “autoinduction,” by which sucralose enhances it own metabolism. Autoinduction is a well-known biological phenomenon by which xenobiotics induce proteins involved in their own detoxification (Schuetz et al., 1996). Autoinduction of CYP isozymes was reported after administration of both OC (Zhu et al., 2009; Chang et al., 1997) and non-organochlorine (Bertilsson et al., 1980, 1986; Strolin Benedetti et al., 1990; Strolin Benedetti and Dostert, 1994; Chen and Raymond, 2006) drugs. The fillers in Splenda did not contribute to the expression of P-gp and CYP enzymes because glucose (as well as the maltodextrin component, which is hydrolyzed to glucose) is rapidly absorbed in the duodenum (Booth, 1994). The expression of P-gp and CYP was quantified by Abou-Donia et al. (2008) in the jejunum and ileum rather than the duodenum because P-gp expression is greater in the more distal region of the small intestine (Iida et al., 2005). Abou-Donia et al. (2008) postulated that the elevated expression of intestinal P-gp and CYP subsequent to sucralose ingestion may increase the clearance of co-administered medications (and hence reduce their bioavailability) due to the critical role of P-gp and CYP in the pharmacokinetics of therapeutic drugs (Custodio et al. 2008; Shugarts and Benet 2009). An example is the upregulation of P-gp and CYP by St. John's wort, which led to renal transplant rejections due to sub-therapeutic levels of the immunosuppressant cyclosporine (Bauer et al., 2003).
The findings of increased intestinal expression of P-gp and CYP by Abou-Donia et al. (2008) do not support claims by Grotz and Munro (2009) and Brusick et al. (2009) that CYP is not induced by sucralose. These claims were based on an unpublished report in 1987 that trichlorogalactosucrose, the previous name for sucralose, did not induce hepatic CYP expression in rats (Hawkins et al., 1987). However, no methodological descriptions, data, or results from this report have been published in the peer-reviewed literature. Further, lack of hepatic induction does not preclude induction of intestinal CYP, because, as noted previously, the correlation between intestinal and hepatic metabolic rates in both rats and humans is low (Aiba et al., 2003; Lown et al., 1994).
Splenda Effects on P-gp and CYP Are Consistent With the Literature on Other Organochlorine Compounds
The finding by Abou-Donia et al. (2008) that the sucralose (delivered as Splenda) interacts with efflux and metabolizing proteins is consistent with an extensive scientific literature that indicates OC compounds characteristically interact with CYP (and in some cases P-gp). Table 2 provides a list of 89 representative OC therapeutic drugs; all of these drugs were shown to interact with CYP (and some with P-gp) as substrates, inhibitors, and/or inducers. A given drug may act as an inhibitor or inducer (as well as a substrate) depending on a variety of factors, including drug concentration and time course (Matheny et al., 2001). The most frequent interactions for OC drugs in Table 2 were with CYP3A (62%) and CYP2D (36%), that is, members of the same two subfamilies of CYP involved in the disposition of sucralose (Abou-Doniaet al., 2008). Although the majority of the drugs in Table 2 were substrates of CYP and/or P-gp, some of these OC drugs, like sucralose, were reported to increase CYP and/or P-gp expression. The OC drugs midazolam (Schuetz et al., 1996), nefazodone (Störmer et al., 2001), trazodone (Störmer et al., 2001), clotrimazole (Schuetz et al., 1996), and efavirenz (Störmer et al., 2002) were reported to be inducers of P-gp. The OC drugs clotrimazole (Schuetz et al., 1996), griseofulvin (Yasuda et al., 2008), and efavirenz (Flockhart, 2013) were found to be inducers of CYP3A. Both P-gp and CYP3A were induced by clotrimazole and efavirenz.
TABLE 2.
Representative Organochlorine Drugs Reported to Be Substrates, Inhibitors, or Inducers of CYP isozymes and/or P-gp Using One or More Assay Types
P-gp and CYP also play a role in the disposition of nonpharmaceutical OC compounds such as OC pesticides, herbicides, and other industrial chemicals. Pesticides such as chlorpyrifos, tetrachlorohydroquinone, chlordecone, heptachlor, chlorthiophos, dicapthon, fluvalinate, and permethrin interact with P-gp (Lanning et al., 1996; Bain and LeBlanc, 1996). Organochlorine insecticides and pesticides that are metabolized by CYP isozymes include chlorpyrifos (Tang et al., 2001), p,p‘-DDT [1,1,1-trichloro-2,2-bis(p-chlorophenyl)ethane] (Abou-Donia and Menzel, 1968a; 1968b; Kitamura et al., 2002), and methoxychlor (Hu and Kupfer, 2002). The herbicides acetochlor, butachlor, and metolachlor are metabolized by CYP (Coleman et al., 2000), as are many industrial OC compounds (Gonzalez and Gelboin, 1994) including chlorinated benzenes (Bogaards et al., 1995), chloroform (Gemma et al., 2003), chlorofluorocarbons (Dekant et al., 1995), and trichloroethylene (Lipscomb et al., 1997). Similar to OC drugs, OC pesticides and industrial compounds may be inducers and inhibitors of CYP in addition to being substrates (Pang et al., 1999; Coumoul et al., 2002). Overall, the preponderance of data indicates that P-gp and CYP are routinely involved in the disposition of a vast array of OC compounds including drugs, pesticides, and industrial chemicals, as well as the OC sweetener sucralose.
Additional Intestinal Transporters and CYP Isozymes May Be Involved in the Disposition of Sucralose
The finding that only a fraction of relatively high concentrations of ingested sucralose is absorbed from the intestine raises the possibility that efflux transporters or CYP isozymes, in addition to P-gp, CYP3A, and CYP2D, may play a role in limiting the bioavailability of sucralose (Beringer and Slaughter, 2005; Custodio et al., 2008; Endres et al., 2006; Shugarts and Benet, 2009; U.S. FDA, 2011, for reviews of efflux and uptake transporters). Organochlorine drugs such as clotrimazole and glibenclamide were shown to interact with numerous types of efflux transporters at physiological barriers including the intestine (and liver, kidney, and blood–brain barrier). The antifungal drug clotrimazole is an inducer of MRP2 (multidrug resistance-related protein 2) (Kauffmann et al., 2002), and the antidiabetic drug glibenclamide is a substrate of the efflux transporter BCRP (breast cancer resistance protein) (Gedeon et al., 2006). MRP2 and BCRP, like P-gp, are members of the ABC transporter superfamily and shunt substrates out of the cytoplasm of eukaryotic cells, including enterocytes. In order to determine and identify transporters in addition to P-gp that may play a role in the disposition of sucralose, testing strategies similar to those utilized in pharmaceutical research (International Transporter Consortium et al. 2010) can be applied. Evaluation of CYP isoforms in addition to the subfamilies CYP3A and CYP2D may also be warranted to determine which other CYP subfamilies also contribute to sucralose metabolism.
General Mechanisms Responsible for Increases in P-gp and CYP Expression
Increases in P-gp and CYP expression result from modulation of gene transcription as well as from posttranscriptional and posttranslational modification (Kliewer et al., 2002; Handschin and Meyer, 2004; Matheny et al., 2004; Chen et al., 2012). Increased expression of both P-gp and CYP3A occurs through a sequence of steps involving transcriptional activation of genes (initiated by nuclear receptors including the pregnane X receptor, PXR) followed by enhanced production of messenger RNA (mRNA) and protein synthesis (Kliewer et al., 2002; Matheny et al., 2004). PXR is a xenobiotic sensor that is directly activated by xenobiotic (including OC) compounds. PXR coordinately upregulates genes and induces the expression of proteins involved in detoxification (including P-gp and CYP3A); this results in levels of transporters and metabolic proteins that are in excess of those normally present. PXR plays a major role in the induction of CYP3A from exposure to OC pesticides, such as DDT, chlordane, dieldrin, and endosulfam, as well as OC drugs such as clotrimazole (Ihunnah et al., 2011; Coumol et al., 2002). Unlike induction of P-gp and CYP3A, the CYP2D subfamily has traditionally been considered refractory to induction via gene activation involving nuclear receptors (Ingelman-Sundberg, 2005). While enhanced expression and activity of CYP2D occur in humans, monkeys, and rodents after exposure to drugs and herbs (Mrozikiewicz et al., 2010; Hellum et al., 2007; Flockhart, 2013; Miksys et al., 2002; Warner and Gustafsson, 1994; Yue et al., 2008; Mann et al., 2008), this elevated expression was typically attributed to posttranscriptional events such as increased translation efficiency or stabilization of protein (Yue et al., 2008). In summary, our current state of knowledge suggests that the predominant mechanism responsible for increased expression of P-gp and CYP3A from sucralose ingestion is transcriptional activation involving PXR, while the enhanced expression of CYP2D occurs via a posttranscriptional mechanism.
Factors in addition to PXR, such as activation of taste receptors and microRNAs, may also contribute to increased expression of P-gp and CYP3A from sucralose ingestion. Induction of P-gp was reported in intestinal cells via signaling from gut “taste” receptors (Jeon et al., 2011). In addition, OC compounds can interact with microRNAs (Tilghman et al., 2012), and recent studies indicate that microRNAs are involved in the regulation of CYP3A enzymes (Takagi et al., 2008; Pan et al., 2009). Nuclear mechanisms in addition to PXR activation may also play a role in P-gp and CYP3A induction (Pan et al., 2009; Tachibana et al., 2009).
Potential Adverse Sucralose–Drug Interactions
The increased expression of P-gp and CYP in rats reported by Abou-Donia (2008) raises the possibility of potential adverse sucralose-drug interactions in humans. The scientific literature on drug–drug interactions indicates that the magnitude of the elevated expression of P-gp and CYP3A from Splenda in rats is comparable to or greater than the increases found to reduce the bioavailability of some drugs in humans (Dürr et al., 2000). Several studies suggested that rat data on P-gp kinetics might be extrapolated to humans (Stephens et al., 2001; Hsiao et al., 2006). Co-administration of drugs that increase the expression of intestinal P-gp and/or CYP has emerged as an important factor in drug–drug interactions (Hebert et al., 1992; 1999; Holtbecker et al., 1996; Backman et al., 1996; Greiner et al., 1999; Johne et al., 1999; Ruschitzka et al., 2000; Dürr et al., 2000; Westphal et al., 2000; Dresser et al., 2003; Lin and Yamazaki, M. 2003; Bauer et al., 2003; Park et al., 2004; Matheny et al., 2004; Pelkonen et al., 2008; Xie et al., 2005; Chen and Raymond, 2006; Tapaninen et al., 2010). The elevated expression of P-gp and CYP by consumption of sucralose is of particular concern for patients who take medications that are substrates of P-gp and CYP proteins. Overexpression of P-gp from anticancer agents such as anthracyclines (doxorubicin and daunorubicin) and Vinca alkaloids (vinblastine and vincristine) is associated with multidrug resistance; that is, elevated P-gp efflux may reduce intracellular concentrations of therapeutic drugs in neoplastic tissues (Gottesman and Pastan, 1988; Endicott and Ling, 1989; Mechetner et al., 1998; Gottesman et al., 2002). Overexpression of P-gp by two- to fivefold is typical in drug-resistant human tumors and hence clinically relevant in cancer treatment (Maitra et al., 2001). Data suggest that the 2.43-fold (or 143.5%) rise in P-gp expression (Table 1) after daily consumption of Splenda with a sucralose dosage of 3.3 mg/kg/d (equivalent to two 12-oz soft drinks) may be clinically relevant.
METABOLIC FATE AND SAFETY PROFILE OF SUCRALOSE METABOLITES
Metabolites of sucralose have been detected in the feces and urine of rats and humans by thin-layer chromatographic (TLC) methods, but the chemical identities of these metabolites have not yet been established (Sims et al. 2000; Roberts et al. 2000). Thin-layer chromatograms (TLC) of methanolic fecal extracts following administration of 14C-sucralose to rats (Sims et al. 2000) and humans (Roberts et al. 2000) suggested that sucralose is metabolized in the GIT. A comparison of TLC radiochromatographic profiles of methanolic fecal extracts following a single intravenous (iv) and a single oral administration of 14C sucralose in two rats is shown in Figures 1a and 1b, respectively (from Sims et al., 2000). The profile in Figure 1b (the peaks are enlarged to the right) from the rat that received oral 14C-sucralose yielded a broader trace that contained multiple, closely eluting peaks of approximately equal height when compared to the thinner profile with one peak from the rat that received the iv dose. The multiple peaks in the trace in Figure 1b indicate the presence of at least two radioactive chemicals in the fecal material; that is, sucralose underwent metabolism and was not excreted unchanged in the feces. In addition, the Rf values (i.e., relative distance) of the peaks in Figure 1b from the oral dose are shifted somewhat to the left of the Rf value of the putative sucralose peak from the IV dose in Figure 1a. This suggests that at least one of the peaks in Figure 1b represents a chemical other than sucralose itself. The rat that received the oral dose had been maintained on a sucralose-containing diet for 85 wk that, according to the findings of Abou-Donia et al. (2008), would most likely have enhanced the expression of CYP. TLC radiochromatographic profiles of fecal extracts from a single human subject using two different solvents also indicate the presence of several peaks (Roberts et al., 2000). Although TLC is a straightforward technique to separate component compounds in mixtures, it cannot be used to identify specific metabolites. Use of liquid chromatography–mass spectrometry (LC-MS) enables the chemical identification of sucralose metabolites. After the metabolites have been systematically identified, they can be evaluated for safety. Metabolites of drugs with exposures that are >10% of the administered dose or systemic exposure are recommended for safety assessment by regulatory agencies (Robison and Jacobs, 2009).
FIGURE 1.
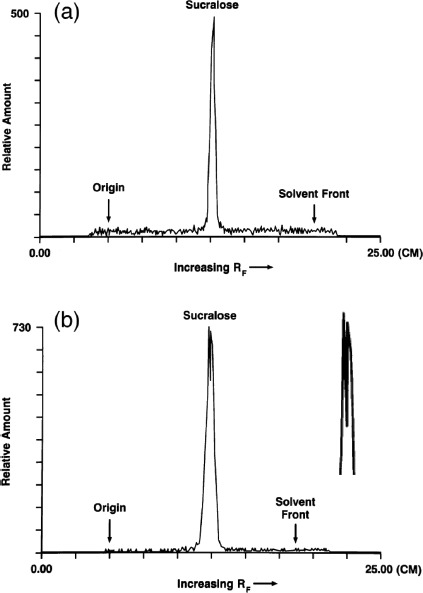
Thin-layer radiochromatographic profile of methanolic fecal extracts following both intravenous (iv) and oral administration of 14C-sucralose: (a) 0–24 h fecal sample from a male rat given an iv dose of 14C-sucralose (2 mg/kg); (b) 0–24 h fecal sample from a male rat maintained on a diet containing 30,000 ppm sucralose for 85 wk before receiving an oral dose of 14C-sucralose (100 mg/kg). An enlargement of the peak profile is given to the right. (TLC traces from Sims et al., 2000).
Sims et al. (2008) reported that TLC scans of urine samples from rats given either an iv dose or oral administration of 14C-sucralose showed one large peak and several smaller peaks. They maintained that the large peak corresponded to unchanged sucralose while the smaller peaks were chromatographically more polar than sucralose. Historical reviews claimed that the small peaks are glucuronide conjugates of sucralose and not products of CYP metabolism in the liver (Grice and Goldsmith, 2000; Molinary and Quinlan, 2006; Grotz and Munro, 2009). That is, the small peaks, which are chemically more polar than sucralose, have historically been considered glucuronide adducts of sucralose (with sucralose itself remaining intact) rather than actual metabolites. Several factors, however, limit these conclusions regarding the compounds excreted in the urine after 14C-sucralose administration. First, the statement that the large peak from urine in rats was a single compound cannot be confirmed because the TLC trace presented in a figure by Sims et al. (2000) was truncated before reaching the crest. Second, the putative sucralose peak from 0–24 h urine samples after both iv and oral administration of 14C-sucralose had an Rf value that did not coincide with the Rf value from pure 14C-sucralose spiked into control rat urine. Third, the minor metabolites were reportedly resistant to hydrolysis by classical enzymatic techniques (Wood et al., 2000; Roberts et al., 2000), and this raises questions about the identity of at least one of the minor components. If the metabolites found in the urine were indeed glucuronide conjugates, one would have anticipated hydrolysis by ß-glucuronidase.
The glucuronidation reaction, a detoxification pathway in which glucuronic acid is added to a substrate (such as sucralose), is catalyzed by the conjugating enzyme UDP-glucuronosyltransferase (UGT). The expression of UGT, like CYP3A isoforms and P-gp, is induced through the interaction of xenobiotics with the PXR (Chen et al., 2003b, 2012). Organochlorine compounds induce UGT with a variety of adverse effects, including alterations in thyroid function (Langer, 1998; Kato et al., 2003, 2010; Yanagiba et al., 2009). UGT activity also confers resistance to several chemotherapeutic drugs (Meijerman et al., 2008). Given the finding that P-gp and CYP3A are induced by sucralose (Abou-Donia et al., 2008) and that PXR is known to initiate P-gp and CYP3A induction, it is possible that activation of PXR may also induce UGT and affect the disposition of drugs that are substrates for glucuronidation.
The finding of multiple peaks in the TLC traces of fecal extracts from rats and humans is consistent with the finding by Abou-Donia et al. (2008) that oral consumption of sucralose increases the expression of CYP isozymes in the intestine known to metabolize xenobiotics including drugs and other foreign substances. The identity of the metabolites has not yet been established, but known sucralose reaction products include its two hydrolysis products (e.g., the organochlorine monosaccharides 4-chloro-4-deoxygalactose [4-CG] and 1,6-dichloro-1,6-dideoxyfructose [1,6-DCF]; Grice and Goldsmith, 2000), an unsaturated aldehyde of sucralose (Labare and Alexander, 1994), and 3’,6’-anhydro-4,1’-dichlorogalactosucrose, which is gradually produced under aqueous, alkaline conditions (Barndt and Jackson, 1990). Overall, the TLC and CYP findings in aggregate do not support the historical contention that sucralose is not metabolized in the GIT as claimed by Sims et al. (2000) and Roberts et al. (2000) and in review papers by Grice and Goldsmith (2000), Molinary and Quinlan (2006), and Grotz and Munro (2009).
EFFECT OF SUCRALOSE ON THE NUMBER AND RELATIVE PROPORTIONS OF DIFFERENT INTESTINAL BACTERIAL TYPES
Studies of bacteria in culture media suggest that sucralose is not utilized as a growth substrate by microorganisms from the oral cavity (Young and Bowen, 1990) or from soil (Lappin-Scott et al., 1987; Labare and Alexander, 1993, 1994). These findings raise the question of whether the presence of unabsorbed sucralose or its metabolites in the GIT affect the metabolic activity and composition of GIT microflora of humans and non-human animals. Gut microflora perform many useful functions, including fermentation of complex dietary carbohydrates with concomitant formation of short-chain fatty acids (SCFA), synthesis of vitamins (e.g., B and K), modulation of immune responses, regulation of postnatal gut development, inhibition of pathogens, absorption of calcium and magnesium, and metabolism of drugs (Albert et al., 1980; Cummings and Macfarlane, 1991, 1997; Shearer, 1995; Hill, 1997; Bauer, 1998; Holzapfel et al., 1998; Chonan et al., 2001; Topping and Clifton, 2001; Hart et al., 2002; Teitelbaum and Walker, 2002; Fooks and Gibson, 2002; Guarner and Malagelada, 2003). Intestinal microbiota may modulate the expression of CYP, conjugating enzymes including UGT, and P-gp (Nicholson et al., 2005; Ueyama et al., 2005; Jia et al., 2008; Claus et al., 2008; Björkholm et al., 2009; Meinl et al., 2009), which were reported to play a role in the disposition of sucralose. Intestinal bacterial composition was also shown to play a role in obesity (Duncan et al., 2007; Ley et al., 2006; Turnbaugh et al., 2006; Turnbaugh and Gordon, 2009).
Sucralose Administered as Splenda Reduces Bacterial Counts in the Gastrointestinal Tract and Alters Their Relative Proportions
Abou-Donia et al. (2008) found that sucralose delivered as Splenda reduced the numbers and altered the composition of microbiota in the GIT of male Sprague-Dawley rats. Fecal samples were collected each week during the 12 wk of treatment and for 12 wk of recovery from treatment for bacterial culture studies, quantification of fecal pH, and histopathological studies of the colon. Data showed that bacterial counts in the GIT from daily sucralose ingestion decreased progressively and monotonically in a methodical pattern during each successive week of sucralose treatment. Table 3 shows the percent difference in bacterial counts for rats treated with sucralose relative to counts from control rats at the end of the 12-wk treatment period and at the end of the 12-wk recovery. The numbers of total anaerobes, bifidobacteria, lactobacilli, Bacteroides, clostridia, and total aerobic bacteria were significantly decreased at the end of the 12-wk treatment period with losses up to 79.7% for lactobacilli; there was no significant treatment-related effect on enterobacteria. These changes in bacterial counts were accompanied by intermittent incidences of unformed or soft feces as well as histopathological changes in the colon, including lymphocytic infiltrates into epithelium, epithelial scarring, mild depletion of goblet cells, and glandular disorganization. At the end of the 12-wk recovery period, the total anaerobes and bifidobacteria were still significantly lowered. Fecal pH increased during the treatment period (up to 7.4%) and remained significantly elevated at the end of the 12-wk recovery period. Abou-Donia et al. (2008) concluded that sucralose (administered as Splenda) reduced the number of indigenous intestinal bacteria, with significantly greater suppression for the generally beneficial anaerobes (e.g., lactobacilli, and bifidobacteria), and with less inhibition for more detrimental bacteria (e.g., enterobacteria). Further, the numbers of total anaerobes remained partially suppressed after a 3-mo recovery period.
TABLE 3.
Percent Differences in Bacterial Counts for Sucralose-Treated Rats Relative to Untreated Control at the End of the 12-wk Treatment Period and at the End of the 12-wk Recovery
| Sucralose dosage delivered daily in Splendaa |
||||
|---|---|---|---|---|
| Percent change at 1.1 mg/kgbc | Percent change at 3.3 mg/kgbc | Percent change at 5.5 mg/kgbc | Percent change at 11 mg/kgbc | |
| End of treatmentd | ||||
| Total anaerobes | −49.8 | −72.2 | −73.7 | −78.9 |
| Bifidobacteria | −36.9 | −71.9 | −76.0 | −77.7 |
| Lactobacilli | −39.1 | −62.8 | −66.8 | −79.7 |
| Bacteroides | −67.5 | −75.6 | −74.1 | −77.5 |
| Clostridia | — | −47.4 | −55.3 | −50.5 |
| Total aerobes | — | −51.2 | −51.2 | −67.8 |
| Enterobacteria | — | — | — | — |
| End of recoverye | ||||
| Total anaerobes | −53.9 | −76.6 | −56.7 | −48.6 |
| Bifidobacteria | — | −74.6 | −61.1 | — |
| Lactobacilli | — | — | — | — |
| Bacteroides | — | — | — | — |
| Clostridia | — | — | — | — |
| Total aerobes | — | — | — | — |
| Enterobacteria | — | — | — | — |
Note. Data from Abou-Donia et al. (2008).
Rats treated by oral gavage with Splenda containing sucralose doses of 1.1, 3.3, 5.5, and 11 mg/kg/d for 12 wk. Fecal samples were collected weekly over a 24-wk experimental period (12 wk of treatment and 12 wk of recovery).
Percent difference in the mean values of bacterial counts of Splenda/sucralose-treated groups relative to untreated controls. Numerical values are significantly different from the control group according to Student's t-test.
—Indicates not significantly different from the control group.
Percent difference after the 12-wk treatment.
Percent difference after 12-wk discontinuation of Splenda/sucralose treatment.
Brusick et al. (2009) claimed that the reduced numbers of microbiota reported by Abou-Donia et al. (2008) were simply the result of normal variation. Three lines of evidence do not support this claim. First, the progressive and methodical weekly decrements in bacterial counts are not consistent with normal variation. If the decrements were due to normal variation for a single bacterial type such as bifidobacteria, the probability of systematic weekly reductions over 12 wk for a single dosage would be p <.0002. Further, the likelihood that this systematic pattern in bacterial reduction occurred simultaneously for multiple bacterial types and sucralose dosages as a result of chance from normal biological variation is infinitesimal based on calculations using a Poisson distribution (Schiffman and Abou-Donia, 2012; Brownlee, 1960; Schiffman et al., 1979). Second, while changes in specific microbial species may occur over time in response to environmental factors and aging, many phylogenetic groups in the GIT such as Bifidobacterium are stable in both short-term intervals (<1 yr) and long-term periods (>10 yr) in humans (Rajiliác-Stojanoviác et al., 2012). The average values of similarity for Bifidobacterium over different time spans reported by Rajiliác-Stojanoviác et al. (2012) were 0.92 at 3 mo, 0.91 at 2 yr, and 0.93 at 10 yr. In the Abou-Donia et al. (2008) study, however, bifidobacteria were reduced by 71.9%, 76%, and 77.7% at dosages of 3.3, 5.5, and 11 mg/kg/d sucralose, respectively, and did not fully rebound during recovery. Abou-Donia et al. (2008) also found decrements in Bacteroides (67.5%, 75.6%, 74.1%, and 77.5% at dosages of 1.1, 3.3, 5.5, and 11 mg/kg sucralose, respectively) that were statistically significant for all dosages. This perturbation of Bacteroides raises the possibility that sucralose may impact the stability of the entire bacterial ecosystem in the GIT because Bacteroides play a critical role in the stability and resilience of gut colonization (Lee et al., 2013). Third, the finding that total anaerobes remained partially suppressed after a 3-mo recovery period would not be expected if the changes from sucralose were simply random variation. Incomplete recovery of the bacterial composition in the GIT was also reported after exposure to antibiotics (Rajiliác-Stojanoviác et al., 2012).
The reduction in the number of intestinal bacteria subsequent to sucralose ingestion in rats reported by Abou-Donia et al. (2008) is consistent with the finding that sucralose exhibited antimicrobial activity against two oral bacteria involved in periodontal disease, Aggregatibacter actinomycetemcomitans and Porphyromonas gingivalis, in an in vitro study (Prashant et al., 2012). Many chlorinated compounds such as triclosan, chlortetracycline, clotrimazole, clofazimine, chloramphenicol, and vancomycin in addition to sucralose are also known to possess antibacterial properties. Triclosan, like sucralose, is a trichlorinated compound and is used in many consumer products such as toothpaste; further, triclosan also enhances the expression of several CYP isozymes and interacts with PXR (Hanioka et al., 1997; Jinno et al., 1997; Calafat et al., 2008). Chronic exposure to chlorinated as well as unchlorinated compounds with antibacterial properties may lead to antibiotic resistance (Middleton and Salierno, 2013; Anderson, 1975; Russell, 1998, 2004), in which antimicrobial agents lose their initial efficacy over time.
Bacteria in Soil Metabolize Sucralose
Although sucralose is apparently not used as a sole carbon source by bacteria for growth, it might be transformed microbiologically in soil and sewage (Lappin-Scott et al., 1987; Labare and Alexander, 1993, 1994). Labare and Alexander (1994), as noted previously, found that metabolism of sucralose by microorganisms in environmental samples generated numerous metabolic by-products, including an unsaturated aldehyde of sucralose, 1,6-DCF, and possibly the uronic acid of sucralose. Currently there is no evidence in the open scientific literature that sucralose is metabolized to 1,6-DCF or other aldehydes in vertebrate intestines. If 1,6-DCF and/or the unsaturated aldehyde are ultimately found to be by-products of intestinal metabolism of sucralose, this suggests possible mechanisms responsible for the reduced levels of bacteria in the GIT as reported by Abou-Donia et al. (2008). The sucralose hydrolysis product 1,6-DCF is an alkylating agent (Schiffman, 2012) and may damage bacterial DNA. Aldehydes are highly reactive molecules that adversely affect a wide spectrum of biological processes in bacterial as well as mammalian cells (Voulgaridou et al., 2011; O'Brien et al., 2005). Aldehydes such as glutaraldehyde, formaldehyde, and ortho-phthalaldehyde (OPA) possess antibacterial properties and have been used as disinfectants (Russell, 1998, 2004; Fraud et al., 2001).
Consequences of Reduction in Beneficial Bacterial Counts
Reductions and imbalances in the composition of intestinal bacteria play a role in numerous medical conditions, including allergies, gastric cancer, Crohn's disease, obesity, and inflammatory bowel disease (IBD) (Blaser and Falkow, 2009; Clemente et al., 2012; Cho and Blaser, 2012). Qin (2011, 2012) recently proposed that inhibition of intestinal microflora by sucralose is a causative factor in IBD based on epidemiological trends. Treatment of IBD includes the oral administration of probiotic bacteria such as lactobacilli and bifidobacteria (Borchers et al., 2009); thus, the reduction in these probiotic bacterial types by sucralose may be detrimental to IBD patients. Overall, the scientific literature on ingestion of probiotic bacteria in foods such as yogurt suggests that the magnitude of the decrease in bacterial counts from sucralose is biologically significant. That is, the magnitude of the reduction in intestinal bacteria from sucralose administered in Splenda is similar to or greater than the magnitude of the increase (not decrease) in probiotic bacteria noted to produce significant health benefits including protection against infection, maintenance of the intestinal epithelial barrier, and treatment of IBD (Gill et al., 2001; Villena et al., 2005; Wang et al., 2004; Borchers et al., 2009; O'Flaherty and Klaenhammer, 2010; Ross et al., 2010; Sherman et al., 2009). Although concerns about potential adverse effects of sucralose on GIT bacteria were first raised several decades ago by the Joint FAO/WHO Expert Committee on Food Additives (WHO, 1991), the effects of sucralose on bacteria in the GIT in the general population and at-risk groups with vulnerable colonic ecosystems (e.g., IBD [Swidsinski et al., 2009], diarrhea [Jafari et al., 2009], immune deficiencies [Hooper et al., 2012], and the elderly [Woodmansey, 2007; O'Toole and Claesson, 2010]) have not yet been performed.
POTENTIAL TOXICITY FROM HABITUAL SUCRALOSE INGESTION
Although several adverse behavioral effects including abnormal locomotion were reported in nonmammalian species after short-term low-dose sucralose exposure (Wickland et al., 2012), no toxic effects were attributed to sucralose in historical single-dose or short-term sucralose studies in humans (Mezitis et al., 1996; Baird et al., 2000). However, the potential chronic effects of habitual sucralose consumption in humans have not been systematically investigated. Results from several recent studies raise a number of issues regarding potential safety of chronic sucralose intake in humans.
Genotoxicity
Sucralose is slowly hydrolyzed to its two constituent OC monosaccharides 4-CG and 1,6-DCF in acidic solutions (e.g., sodas) over time (Grice and Goldsmith, 2000). 1,6-DCF was found to be weakly mutagenic in both the Ames test and the L5178Y TK+/− assay, and sucralose itself was found to be weakly mutagenic in the mouse lymphoma mutation assay (WHO, 1989; U.S. FDA, 1998). Other historical tests of genotoxicity were inconclusive, such as those for clastogenic activity of sucralose in a mouse micronucleus (MN) test and a chromosomal aberration (CA) test in cultured human lymphocytes (U.S. FDA, 1998). Sucralose also damaged DNA in gastrointestinal organs of the mouse in a comet test (Sasaki et al., 2002). The comet test is a single-cell gel electrophoresis assay that is used to detect DNA damage in various organs of experimental animals resulting from exposure to food additives, industrial chemicals, and pharmaceuticals (Speit et al., 2009; Pfuhler et al., 2011). Two-year rodent bioassays found significant increases in the incidence of non-neoplastic findings; however, no evidence of elevated carcinogenic activity for either unprocessed sucralose (e.g., uncooked) or its hydrolysis products was reported in historical studies (Mann et al., 2000a, 2000b; WHO, 1989). The clinical relevance of the findings from the Ames test, the L5178Y TK+/− assay, and the comet test for humans who consume sucralose on a continuous basis is not yet known.
Although the genome is the fundamental substrate of genetic content, it is the expression of genes that determines phenotype and clinical outcome, including any potential pathology. Genetic expression is shaped and regulated by epigenetic mechanisms that turn genes on and off without changing the underlying sequence of DNA (Hatzimichael et al., 2008; Hou et al., 2012). These mechanisms include altered DNA methylation (addition of a methyl group to the 5’ position of the cytosine ring), histone modification (chemical alteration of histone proteins—the genetic packing material), and altered microRNA expression (posttranslational regulation of genes by targeting messenger RNA) (Hou et al., 2012). Studies using animal models and in vitro assays found that exposure to OC compounds as well as dietary factors induce epigenetic events. Organochlorine compounds including vinclozolin, methoxyclor, and dichloro- and trichloroacetic acid alter DNA methylation patterns (Tao et al., 2000a, 2000b; Anway et al., 2005; Zama and Uzumcu, 2009; Guerrero-Bosagna et al., 2010). Vinclozolin was reported to induce persistent epigenetic reprogramming that may be transmitted transgenerationally (Stouder and
Paoloni-Giacobino, 2010; Zama and Uzumcu, 2010). Further, dietary factors have also been implicated in epigenetic changes (Burdge et al., 2007; Vucetic et al., 2010; McKay and Mathers, 2011; Feil and Fraga, 2012). It is not yet known whether epigenetic events might be induced by habitual use of the OC sweetener sucralose or its hydrolysis product (and alkylating agent) 1,6-DCF in a dietary regimen.
Safety of Sucralose That Has Been Heated
Recently, there has been renewed scientific interest in the safety of by-products generated by sucralose at elevated temperatures. Historically, sucralose was reported to be heat stable at temperatures used in cooking (Barndt and Jackson, 1990; Miller, 1991). However, this conclusion is not supported by data presented by Barndt and Jackson (1990) nor is it consistent with thermal degradation data from three other independent labs (Hutchinson, 1996; Hutchinson et al., 1999; Bannach et al., 2009; Rahn and Yaylayan, 2010). Barndt and Jackson (1990) incorporated radioactive sucralose (14C-sucralose) into recipes for yellow cake, cookies, and graham crackers. After baking, the radio-labeled material was extracted from the baked goods and analyzed by TLC. Although the authors concluded that no peaks other than sucralose were detected in the TLC scans of the recovered material, examination of the TLC trace of an extract from cookies shows multiple, closely eluting peaks that suggest thermal degradation (Rahn and Yaylayan, 2010; Schiffman, 2012; Schiffman and Abou-Donia, 2012).
Subsequent to the publication by Barndt and Jackson (1990), Hutchinson (1996) and Hutchinson et al. (1999) studied the thermal decomposition of sucralose in aqueous solutions at pH 3, 7, and 11 heated to 100, 140, and 180°C for 1 h. Data showed that the stability of sucralose decreased as the temperature and pH increased; at 180°C sucralose completely degraded at all pH levels with the release of chloride ions. In addition, Hutchinson et al. (1999) analyzed the volatile compounds released and concluded that dehydrochlorination steps accompanied their production. Bannach et al. (2009) used thermo-analytic techniques to study the effect of temperature on the stability of sucralose. They concluded that thermal decomposition of sucralose commences at 119°C with liberation of HCl and water. More recently, Rahn and Yaylayan (2010) raised new concerns about the stability and safety of sucralose in heated applications. Like Hutchinson et al. (1999) and Bannach et al. (2009), Rahn and Yaylayan (2010) concluded that sucralose undergoes thermal degradation. Further, Rahn and Yaylayan (2010) found that chloropropanols were generated when sucralose was heated in the presence of glycerol. Chloropropanols comprise a group of contaminants that include known genotoxic, carcinogenic, and tumorigenic compounds (Biles and Piper, 1983; Cho et al., 2008, Tritscher, 2004; SCF, 2001; WHO, 2002). Rahn and Yaylayan (2010) concluded that “caution should be exercised in the use of sucralose as a sweetening agent during baking of food products containing glycerol and or lipids due to the potential formation of toxic chloropropanols.” Other chlorinated compounds including dibenzo-p-dioxins and dibenzofurans, dioxin-like polychlorinated biphenyls, and polychlorinated naphthalenes were also generated by heating sucralose in the presence of foods (Dong et al., 2011, 2013; Wu et al., 2011). Overall, these studies indicate that sucralose is not stable at elevated temperatures, and that the compounds generated were affected by the other ingredients in the mixture.
Potential for Bioaccumulation
Many OC compounds tend to bioaccumulate over time in a variety of animal species and tissue types (Stellman et al., 1998; Screnci et al., 2000; Weisbrod et al., 2001; Aronson et al., 2000). For this reason, information on the potential bioaccumulation of sucralose and its metabolites is vital for determining the chronic safety of this OC sweetener. Sucralose is an amphiphilic molecule that is comprised of hydrophobic domains (-C-CH2Cl) as well as hydrophilic domains (hydroxyl groups); that is, it possesses both lipophilic and hydrophilic properties. However, Grotz and Munro (2009) claimed that sucralose is “not lipophilic, and not expected to be bioaccumulative.” This publication failed to consider the breadth of the scientific literature or provide adequate data to support their claim. The contention by Grotz and Munro (2009) that sucralose is not lipophilic conflicts with the following lines of scientific evidence.
Basic Principles of Chemistry
The scientific rationale for the synthesis of sucralose was the basic principle that incorporation of halogen atoms into a lead compound results in analogs with increased lipophilicity and hence improved penetration of lipid membranes and tissues, elevated bioavailability, and potential for enhanced potency depending on the sites of the halogen substituents in the derived molecule (Thomas, 2007).
Publications by the Discoverers of Sucralose
Hough and Khan (1978) stated, “The introduction of chloro groups into the sucrose molecule clearly increases its lipophilicity, an important factor in sweetness intensification.” Further, they specifically underscored the amphiphilic nature of sucralose (termed 1’,4,6’-trichlorogalactosucrose in early publications) by stating that it had “the optimum hydrophilic-lipophilic structure” of the chlorinated sweeteners that they synthesized. In the case of disaccharides, Hough and Khan (1989) reported that selective replacement of certain hydrophilic hydroxyl groups with “hydrophobic halogeno substituents” enhanced sweetness and led to the discovery of a “galacto-sucrose” compound ultimately named sucralose.
Historical Publications Relevant to the Solubility of Sucralose
Grice and Goldsmith (2000) noted that a fraction of ingested sucralose is “absorbed from the upper part of the gastrointestinal tract by passive diffusion” in humans and animal models (up to 35–40%, depending on the species). That is, sucralose was sufficiently lipophilic to diffuse through the hydrophobic interior of the phospholipid membrane bilayers of enterocytes and sufficiently water-soluble to be distributed in the systemic circulation—that is, sucralose is an amphiphilic molecule. The passive diffusion of sucralose through membranes of enterocytes may be explained by the fact that phospholipids are themselves amphiphilic molecules that arrange themselves into bilayers with their lipophilic chains toward the inside of the bilayer and their polar groups towards the surrounding aqueous medium. Other amphiphilic molecules, such as cholesterol and glycolipids, also interact with this lipophilic bilayer. The polar nature of brush border membranes is one factor that explains in part why log P (log of the octanol/water partition coefficient) of a compound was found to be an unreliable predictor of intestinal absorption (van Breemen and Li, 2005; Kansy et al., 1998); that is, the polarities of the brush border membrane and octanol are different.
The amphiphilic nature of sucralose is reflected in the choice of solvents such as methanol, butanol, diethylether, ethyl acetate, and ethyl methyl ketone used historically to recover sucralose from excreta (Sims et al., 2000; Roberts et al., 2000). These particular solvents are often used to extract lipid components from mixtures (Ramesh et al., 1979; Goldsmith et al., 1988; Mao-Qiang et al., 1996; Cabrini et al., 1992; Crabbe et al., 2001; Lin et al., 2004). The fact that sucralose is readily soluble in alcohols was corroborated by others (Bennett et al., 1992; Anderson et al., 2006; Li et al., 2010).
Possible Role of Glucuronidation in Sucralose Metabolism
Glucuronide conjugates are reportedly formed during sucralose metabolism (Grice and Goldsmith, 2000; Roberts et al., 2000; Wood et al., 2000). Although certain aspects of the claim of glucuronidation are unresolved as noted earlier, this would provide further evidence for the lipophilicity of sucralose. The process of glucuronidation would render xenobiotics (e.g., sucralose and its metabolites) more water-soluble by linking them to glucuronic acid.
Lipophilicity Requirements for Interaction With P-gp and CYP
Verification of the amphiphilic nature of sucralose comes from the finding of Abou-Donia et al. (2008) that sucralose interacts with P-gp and CYP isozymes. This interaction with P-gp and CYP could not have occurred unless sucralose had diffused through domains of the phospholipid membrane bilayer of enterocytes. A requisite property of xenobiotic compounds to interact with P-gp and CYP is that they are amphiphilic or lipophilic (Garrigues et al., 2002; Marchetti et al., 2007; Yang et al., 2009; Wang et al., 2002).
Recognition of Amphiphilic Properties by Regulatory Agencies
The U.S. FDA (1998) and the EU (2004) approvals of sucralose for use in both fat- and water-based products reflect their understanding of the amphiphilic nature of sucralose.
The potential bioaccumulation of sucralose and its metabolites after long-term exposure in the presence and absence of P-gp and CYP inhibitors has not yet been investigated. Three lines of evidence suggest, however, that bioaccumulation of sucralose and/or its metabolites may occur. First, the expression of P-gp and CYP reported by Abou-Donia et al. (2008) was elevated 12 wk following withdrawal from sucralose (at 5.5 mg/kg/d and 11 mg/kg/d) (see Table 1). This finding suggests that bioaccumulated sucralose or its metabolites may have been released during the 12-wk recovery because P-gp and CYP typically return to baseline after induction within 1–2 wk (Imai et al., 2008; Shimada et al., 2002). Further, Abou-Donia et al. (2008) also observed that bacterial counts and fecal pH were still significantly affected in sucralose-treated animals compared with controls at the conclusion of the 12-wk recovery period. The second line of evidence is that excretion of sucralose and/or its metabolites was prolonged in two of 8 unmedicated human subjects who consumed a single oral dose of 1 mg/kg 14C-sucralose which is less sucralose than in one 12-oz drink (Roberts et al., 2000). Approximately 12% of the radioactivity had not yet been excreted 5 d following 14C-sucralose administration. Although this prolonged GIT transit time (beyond the norm of ∼1–3 d in healthy individuals) may be attributed to individual variation, it may also result from bioaccumulation. The third line of evidence is based on the temporal sensory profile of sucralose that has a delayed onset of sweetness and a lingering of sweetness perception when ingested into the oral cavity. This temporal profile is characteristic of sweetener compounds that undergo nonspecific binding to sites other than (or distinct from) the taste receptor that binds the sweetener (DuBois, 2011; DuBois and Prakash, 2012). Nonspecific binding is a factor known to play a role in bioaccumulation (Bae et al., 2001). Future studies need to be performed to determine whether the co-administration of sucralose (and/or its metabolites and breakdown products) with drugs that are inhibitors of P-gp or CYP enhances the potential for bioaccumulation because sucralose, in the absence of inhibitors, is normally effluxed by P-gp and metabolized by CYP in the gut.
Significant Biological Effects Occur From Sucralose Ingestion Below the ADI
As noted earlier, the FDA established an ADI for humans at 5 mg/kg/d in 1998 based on toxicity studies in the rat. The ADI is an estimate of the maximum amount of a substance that a person can ingest daily over a lifetime without harmful effects. Prior to the derivation of the rat-based ADI, the FDA reviewed studies of sucralose and/or its hydrolysis products in several animal species including rat, mouse, dog, rabbit, and monkey for potential toxicological effects (see Table 4 for published accounts of some adverse effects at high doses). After their review of these animal studies submitted by the petitioner, the FDA concluded that the appropriate animal model for establishing an ADI for sucralose was the rat. The FDA determined that the no-observed-effect level (NOEL) in rat was 500 mg/kg/d; this number was based on a combined chronic toxicity/carcinogenicity study, a diet restriction study, and a 26-wk gavage study. After applying a 100-fold safety factor, the ADI was set at 5 mg/kg/d. The ADI adopted by the Scientific Committee on Food in Europe (SCF, 2000) was determined to be 15 mg/kg/d.
TABLE 4.
Representative Examples of Adverse Effects Reported in Toxicity Studies of Sucralose and Its Hydrolysis Products at High Dosages
| Reference | Species (and number of animals affected when given) | Adverse effect |
|---|---|---|
| Finn and Lord (2000) | Mouse (1 of 10) | Death during 21 d study: dose (150 mg/kg/d of sucralose hydrolysis products administered by gavage) |
| Mouse (2 of 10) | Deaths during 21 d study: dose (1000 mg/kg/d of sucralose hydrolysis products administered by gavage) | |
| Mouse (increased frequency) | Unkempt coats during 21 d study: dose (500 and 1000 mg/kg/d of sucralose hydrolysis products administered by gavage) | |
| Mouse (1 of 10) | Slow righting reflex (neurological finding) during 21 d study: dose (500 mg/kg/d of sucralose hydrolysis products administered by gavage) | |
| Mann etal. (2000b) | Mouse | Lower erythrocyte counts in females after 104 wk: dose (3% dietary sucralose); chronic nephropathy in females and chronic inflammation of the liver in males: dose (3.0% dietary sucralose); degeneration of the testicular tubular germinal epithelium: dose (0.3–3.0% dietary sucralose) |
| Finn and Lord (2000) | Marmoset | Altered salivary responses during second half of 28 d study: dose (1000 mg/kg/d of sucralose and its hydrolysis products administered by gavage) |
| Marmoset (2 of 3) | Depressed postural reactions at 13 d into a 28 d study: dose (1000 mg/kg/d of sucralose administered by gavage); reactions reportedly remitted by d 27 | |
| Marmoset (2 of 3) | Depressed postural responses on d 27 of 28 d study: dose (1000 mg/kg/d of sucralose hydrolysis products administered by gavage) | |
| Kille et al. (2000) | Rabbit, pregnant | Gastrointestinal tract disturbances, such as peri-anal soiling, scouring and caecal enlargement: dose (750 to 1000 mg/kg/d of sucralose administered by gavage) |
| Rabbit, presumed pregnant (7 of 15) | Mild to severe gastrointestinal distress during d 6 to 19 of gestation: dose (700 mg/kg/d administered by gavage) | |
| Kille et al. (2000); U.S. FDA, 1998 | Rabbit, pregnant (12 of 13 deaths occurred in sucralose-treated animals) | Deaths in four groups of 16 to 18 pregnant rabbits: dose (0, 175, 350, and 700 mg/kg/d administered by gavage during d 6 to 19 of gestation); deaths of 11 rabbits (1 control, 4 receiving 175 mg/kg/d group, 2 receiving 350 mg/kg/d group, and 4 receiving 700 mg/kg/d) were not attributed to sucralose by the experimenters although 10 of these 11 deaths occurred during treatment with sucralose. Only two deaths at 700 mg/kg/d were attributed to sucralose by the experimenters. |
| Rabbit (4 of 9) | Abortions: dose (700 mg/kg/d administered by gavage) | |
| Goldsmith (2000b) | Rat | Decrease in total leukocyte and lymphocyte counts of females after 4 wk: dose (5.0% dietary sucralose) Decrease in alanine aminotransferase (ALT) after 4 wk: dose (5.0% dietary sucralose; also 2.5% in females) Decreased serum phosphorus in all male treatment groups after 4 wk: dose (1 %, 2.5%, and 5% dietary sucralose) Increased urinary excretion of calcium and magnesium in females: dose (5% dietary sucralose) Dose-related increase in cecal weight with and without contents: dose (2.5 and 5.0% dietary sucralose) Decreases in organ weights including spleen, thymus, and heart after 8 wk: dose (5.0% dietary sucralose) |
| Mann et al. (2000a) | Rat | Decrease in alanine aminotransferase (ALT) activity after 11, 38, 51, 77 and 103 wk of treatment; dose (3% dietarysucralose) Thyroxine levels were decreased in male rats at 11 wk in a dose-dependent manner: dose (0.3% to 3.0% dietary sucralose) Higher phosphorus excretion after 77 wk: dose (0.3% to 3.0% dietary sucralose) Non-neoplastic findings in females including renal pelvic epithelial hyperplasia, renal pelvic mineralization, and adrenal cortical haemorrhagic degeneration; cataracts in males: dose (3.0% dietary sucralose) |
| Goldsmith (2000b) | Rat | Increased body weight-relative kidney weights during 28-d study: dose (3000 mg/kg/d by gavage) Increased cecum weights during 28-d study: dose (1500 and 3000 mg/kg/d by gavage) Elongated or distended caecum during 28-d study: dose (1500 and 3000 mg/kg/d by gavage) |
| Goldsmith (2000b) | Dog | Lower lymphocyte counts at 3 mo: dose (1 % dietary sucralose); lower lymphocyte counts at 6 mo; dose (1 % and 3% dietary sucralose); lower lymphocyte counts at 12 mo; dose (0.3% dietary sucralose) |
Subsequent data from Abou-Donia et al. (2008) described earlier in this review indicate that the NOEL for sucralose in rats is actually much lower than the ADI set by the FDA and EU. Significant alterations of the rat microbiome occurred at 1.1 mg/kg/d sucralose (administered as Splenda), and significant increases in intestinal P-gp and CYP were observed at 3.3 mg/kg/d. If the disposition of sucralose in rat and human is the same, then significant biological effects would be expected in humans below the ADI. These effects may include intestinal disorders such as IBD as suggested by Qin (2011, 2012), in addition to altered bioavailability of drugs due to enhanced expression of P-gp and CYP. Additional research is required to determine whether the metabolites of sucralose generated in humans and rats are identical and in the same ratio. If not, then the historical rat safety assessment of sucralose (Goldsmith 2000; Mann et al. 2000b) may not be fully applicable to humans.
Positive Allosteric Modulators Ramp Up Physiological Responses to Sucralose
Positive allosteric modulators (PAMs) have recently been discovered that increase the function and biological activity of G protein-coupled receptors (GPCRs) including the T1R2/T1R3 sweet taste receptor (DuBois and Prakash, 2012). Co-administration of PAMs with sucralose (Servant et al., 2010, 2011; Zhang et al., 2010) was shown to amplify activity and functioning of T1R2/T1R3. PAMs are chemicals that interact with geographical regions of the receptor that are topographically distinct from active “orthosteric” binding sites. Allosteric molecules subtly change the conformation of a receptor and allow a ligand (e.g., sucralose) to bind more effectively to its active site (Wenner, 2009). An example of a PAM that is currently used as a sucralose enhancer in the food supply is the chemical 4-amino-5,6-dimethylthieno[2,3-d]pyrimidin-2(1H)-one (ADTP) (Servant et al., 2010; Leffingwell, 2011; Smith et al., 2011; U.S. FDA, 2012; DuBois and Prakash, 2012). ADTP significantly enhanced responses to sucralose in intact cells, in which activation of the human sweet taste receptor coupled to a G protein ultimately led to a net rise in intracellular calcium mobilization. It also enhanced the perceived sweetness of sucralose by up to eightfold in humans as determined by behavioral/sensory responses even though ADTP has no sweet taste of its own (Servant et al., 2010).
ADTP, which has a Chemical Abstract Service Registry number of 121746-18-7, was added to the GRAS (Generally Regarded as Safe) list (Leffingwell, 2011; Smith et al., 2011; U.S. FDA, 2012) after review by the Expert Panel of the Flavor and Extract Manufacterers Association (FEMA), an organization of flavor manufacturers and ingredient suppliers that is responsible for the GRAS evaluation program and oversees safety assessments of flavoring compounds added to the food supply (Smith et al., 2005). The European Food Safety Authority (EFSA, 2011) panel also gave a positive assessment regarding addition of ADTP to the European food supply but noted that “it is not possible to conclude” that ADTP “would be metabolised to innocuous products at the reported levels of intake as flavouring substances” The panel also suggested that additional exposure and toxicological data might be necessary (EFSA, 2011). A WHO report (WHO, 2012) noted that several clinical chemistry parameters were altered by ADTP, including increased creatinine, total chlolesterol, total triglycerides, glucose, and protein levels, but considered these changes to be an adaptive response to treatment rather than toxicologically important. Further studies are needed to determine whether sucralose in the presence of PAMs including ADTP induces clinically significant alterations in physiological processes because allosteric modulation of GPCRs may contribute to unexpected clinical outcomes (Wootten et al., 2012).
FINAL COMMENTS AND CONCLUSIONS
Although sucralose is utilized globally as a sweetener in thousands of food and beverage products, further scientific research is warranted in several areas due to the following potentially significant findings:
-
1.
Sucralose alters metabolic parameters and its chronic effects on body weight are unknown. Sucralose modulates glucose transport and insulin secretion in rodents (Margolskee et al., 2007; Mace et al., 2007; Nakagawa et al., 2009). It was reported to increase glucose and insulin levels in obese women (Pepino et al., 2013) and, when given together with ace-K, to enhance GLP-1 secretion in healthy individuals and persons with type 1 diabetes (Brown and Rother, 2012).
-
2.
Sucralose alters P-gp and CYP expression. Sucralose (delivered as Splenda) increased the expression of the efflux transporter P-gp and CYP metabolizing enzymes that control the bioavailability of drugs (Abou-Donia et al., 2008). Significant elevation in the expression of P-gp and CYP occured at dosages approved by regulatory agencies, including the FDA (U.S. FDA, 1998) and the EU (2004). The magnitude of enhanced expression of P-gp and CYP3A4 produced by sucralose in rats is similar to that induced by the herbal antidepressant St. John's wort (Dürr et al., 2000) that interferes with bioavailability of therapeutic drugs for heart failure, HIV infection, and organ rejection (Johne et al., 1999; Piscitelli et al., 2000; Ruschitzka et al., 2000; Dürr et al., 2000; Tian et al., 2005).
-
3.
The metabolic fate and health profile of sucralose metabolites are currently unknown. Metabolites of sucralose were detected in the feces and urine of rats and humans using TLC (Sims et al. 2000; Roberts et al., 2000), but the identity and health profile of these metabolites have not yet been determined. The detection of metabolites of sucralose by TLC in the GIT and the increased expression of intestinal CYP reported by Abou-Donia et al. (2008) do not support claims by Grotz and Munro (2009) and Brusick et al. (2009) that sucralose is “stable in vivo” and eliminated “unchanged” in the feces. Thus, the pharmacokinetics and safety of sucralose and its metabolites need to be revisited to determine how sucralose is handled by the body.
-
4.
Sucralose alters indigenous bacterial balance in the GIT. Sucralose (delivered as Splenda) reduced the number of indigenous bacteria in the GIT with significantly greater suppression for the generally beneficial anaerobes (e.g., lactobacilli, bifidobacteria) and with less inhibition for more detrimental bacteria (e.g., enterobacteria). Further, the numbers of total anaerobes did not return to baseline after a 3-mo recovery period (Abou-Donia et al., 2008). Alterations in the number and composition of bacteria were accompanied by elevation of fecal pH and histopathological changes in intestinal epithelial barrier. Given that alterations in gut microflora contribute to numerous medical conditions (Guarner and Malagelada, 2003; Hart et al., 2002; Turnbaugh and Gordon, 2009), further investigation of the impact of sucralose on gut microflora is warranted.
-
5.
Numerous toxicological issues regarding long-term exposure to sucralose are unresolved. Several issues that warrant additional investigation to determine their clinical relevancy include potential damage to DNA (Sasaki et al., 2002), generation of chloropropanols during baking (Rahn and Yaylayan, 2010), and potential epigenetic alterations, particularly for the sucralose hydrolysis product 1,6-DCF which is an alkylating agent. The possibility of bioaccumulation of sucralose and/or its metabolites in the presence and absence of medications has not yet been studied. The health effects of co-administration of sucralose with the taste enhancer ADTP are not known.
Finally, artificial sweeteners, including sucralose, are regulated as food additives and are not required to undergo screening for interactions with drug-metabolizing enzymes prior to approval. While food additives do undergo toxicology, teratogenicity, and carcinogenicity testing, the screening overall is less stringent than that required for drugs (U.S. FDA, 2007; U.S. FDA 2011; Robison and Jacobs, 2009). Further, thousands of flavoring substances are excluded from mandatory premarket approval by the U.S. FDA and reach the food supply after review by the Expert Panel of the Flavor and Extract Manufacturers Association (Smith et al., 2011). Given the fact that sucralose (Abou-Donia et al., 2008) and certain foods and herbs interact with drug-metabolizing enzymes and transporters (Nowack et al., 2009; Zhou and Lai, 2008), guidelines need to be developed for the appropriate use and labeling of food ingredients based on their pharmacokinetic parameters and biological activity to ensure the safety of the food supply for all segments of the population.
REFERENCES
- Abou-Donia M. B., Menzel D. B. Chick microsomal oxidases. Isolation, properties, and stimulation by embryonic exposure to 1,1,1-trichloro-2,2-bis(p-chlorophenyl)ethane. Biochemistry. 1968a;7:3788–3794. doi: 10.1021/bi00851a002. [DOI] [PubMed] [Google Scholar]
- Abou-Donia M. B., Menzel D. B. The metabolism in vivo of 1,1,1-trichloro-2,2-bis(p-chlorophenyl)ethane (DDT), 1,1-dichloro-2,2-bis(p-chlorophenyl)ethane (DDD) and 1,1-dichloro-2,2-bis(p-chlorophenyl)ethylene (DDE) in the chick by embryonic injection and dietary ingestion. Biochem. Pharmacol. 1968b;17:2143–2161. doi: 10.1016/0006-2952(68)90189-5. [DOI] [PubMed] [Google Scholar]
- Abou-Donia M. B., El-Masry E. M., Abdel-Rahman A. A., McLendon R. E., Schiffman S. S. Splenda alters gut microflora and increases intestinal P-glycoprotein and cytochrome P-450 in male rats. J. Toxicol. Environ. Health A. 2008;71:1415–1429. doi: 10.1080/15287390802328630. [DOI] [PubMed] [Google Scholar]
- Abu-Qare A. W., Elmasry E., Abou-Donia M. B. A role for P-glycoprotein in environmental toxicology. J. Toxicol. Environ. Health B. 2003;6:279–288. doi: 10.1080/10937400306466. [DOI] [PubMed] [Google Scholar]
- Adachi Y., Suzuki H., Sugiyama Y. Comparative studies on in vitro methods for evaluating in vivo function of MDR1 P-glycoprotein. Pharm. Res. 2001;18:1660–1668. doi: 10.1023/a:1013358126640. [DOI] [PubMed] [Google Scholar]
- Adas F., Berthou F., Picart D., Lozac'h P., Beaugé F., Amet Y. Involvement of cytochrome P450 2E1 in the (ω-1)-hydroxylation of oleic acid in human and rat liver microsomes. J. Lipid Res. 1998;39:1210–1219. [PubMed] [Google Scholar]
- Aiba T., Takehara Y., Okuno M., Hashimoto Y. Poor correlation between intestinal and hepatic metabolic rates of CYP3A4 substrates in rats. Pharm. Res. 2003;20:745–748. doi: 10.1023/a:1023429401738. [DOI] [PubMed] [Google Scholar]
- Albert M. J., Mathan V. I., Baker S. J. Vitamin B12 synthesis by human small intestinal bacteria. Nature. 1980;283:781–782. doi: 10.1038/283781a0. [DOI] [PubMed] [Google Scholar]
- Anderson E. S. The problem and implications of chloramphenicol resistance in the typhoid bacillus. J. Hyg. (Lond). 1975;74:289–299. doi: 10.1017/s0022172400024360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson G. H., Catherine N. L., Woodend D. M., Wolever T. M. Inverse association between the effect of carbohydrates on blood glucose and subsequent short-term food intake in young men. Am. J. Clin. Nutr. 2002;76:1023–1030. doi: 10.1093/ajcn/76.5.1023. [DOI] [PubMed] [Google Scholar]
- Anderson M., Opawale F., Rao M., Delmarre D., Anyarambhatla G. Excipients for oral liquid formulations. In: Katdare A., Chaubal M. V., editors. Excipient development for pharmaceutical biotechnology, and drug delivery systems. New York, NY: Informa Healthcare USA; 2006. pp. 155–180. [Google Scholar]
- Anway M. D., Cupp A. S., Uzumcu M., Skinner M. K. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308:1466–1469. doi: 10.1126/science.1108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronson K. J., Miller A. B., Woolcott C. G., Sterns E. E., McCready D. R., Lickley L. A., Fish E. B., Hiraki G. Y., Holloway C., Ross T., Hanna W. M., SenGupta S. K., Weber J. P. Breast adipose tissue concentrations of polychlorinated biphenyls and other organochlorines and breast cancer risk. Cancer Epidemiol. Biomarkers Prev. 2000;9:55–63. [PubMed] [Google Scholar]
- Backman J. T., Olkkola K. T., Neuvonen P. J. Rifampin drastically reduces plasma concentrations and effects of oral midazolam. Clin. Pharmacol. Ther. 1996;59:7–13. doi: 10.1016/S0009-9236(96)90018-1. [DOI] [PubMed] [Google Scholar]
- Bae W., Mehra R. K., Mulchandani A., Chen W. Genetic engineering of Escherichia coli for enhanced uptake and bioaccumulation of mercury. Appl. Environ Microbiol. 2001;67:5335–5338. doi: 10.1128/AEM.67.11.5335-5338.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain L. J., LeBlanc G.A. Interaction of structurally diverse pesticides with the human MDR1 gene product P-glycoprotein. Toxicol. Appl. Pharmacol. 1996;141:288–298. doi: 10.1006/taap.1996.0286. [DOI] [PubMed] [Google Scholar]
- Baird I. M., Shephard N. W., Merritt R. J., Hildick-Smith G. Repeated dose study of sucralose tolerance in human subjects. Food Chem. Toxicol. 2000;38(suppl. 2):S123–S129. doi: 10.1016/s0278-6915(00)00035-1. [DOI] [PubMed] [Google Scholar]
- Bannach G., Almeida R. R., Lacerda L. G., Schnitzler E., Ionashiro M. Thermal stability and thermal decomposition of sucralose. Ecl. Quím. São Paulo. 2009;34:21–26. [Google Scholar]
- Bapiro T. E., Egnell A. C., Hasler J. A., Masimirembwa C. M. Application of higher throughput screening (HTS) inhibition assays to evaluate the interaction of antiparasitic drugs with cytochrome P450s. Drug Metab. Dispos. 2001;29:30–35. [PubMed] [Google Scholar]
- Barndt R. L., Jackson G. Stability of sucralose in baked goods. Food Technol. 1990;44(Jan):62–66. [Google Scholar]
- Bauer S., Störmer E., Johne A., Krüger H., Budde K., Neumayer H. H., Roots I., Mai I. Alterations in cyclosporin A pharmacokinetics and metabolism during treatment with St John's wort in renal transplant patients. Br. J. Clin. Pharmacol. 2003;55:203–211. doi: 10.1046/j.1365-2125.2003.01759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer T. M. The role of gut bacteria in drug metabolism. In: Blum H. E., Bode C., Bode J. C., Sartor R. B., editors. Gut and the liver. Hingham, MA: Kluwer Academic; 1998. pp. 177–184. [Google Scholar]
- Baumhäkel M., Kasel D., Rao-Schymanski R. A., Böcker R., Beckurts K. T., Zaigler M., Barthold D., Fuhr U. Screening for inhibitory effects of antineoplastic agents on CYP3A4 in human microsomes. Int. J. Clin. Pharmacol. Ther. 2001;39:517–528. doi: 10.5414/cpp39517. [DOI] [PubMed] [Google Scholar]
- Baune B., Flinois J. P., Furlan V., Gimenez F., Taburet A. M., Becquemont L., Farinotti R. Halofantrine metabolism in microsomes in man: Major role of CYP 3A4 and CYP 3A5. J. Pharm. Pharmacol. 1999;51:419–426. doi: 10.1211/0022357991772628. [DOI] [PubMed] [Google Scholar]
- Becquemont L., Mouajjah S., Escaffre O., Beaune P., Funck-Brentano C., Jaillon P. Cytochrome P-450 3A4 and 2C8 are involved in zopiclone metabolism. Drug Metab. Dispos. 1999;27:1068–1073. [PubMed] [Google Scholar]
- Benet L. Z., Izumi T., Zhang Y., Silverman J. A., Wacher V. J. Intestinal MDR transport proteins and P-450 enzymes as barriers to oral drug delivery. J. Control Release. 1999;62:25–31. doi: 10.1016/s0168-3659(99)00034-6. [DOI] [PubMed] [Google Scholar]
- Benet L. Z. The drug transportermetabolism alliance: Uncovering and defining the interplay. Mol. Pharm. 2009;6:1631–1643. doi: 10.1021/mp900253n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett C., Dordick J. S., Hacking A. J., Cheetham P. S. J. Biocatalytic synthesis of disaccharide high-intensity sweetener sucralose via a tetrachlororaffinose intermediate. Biotechnol. Bioeng. 1992;39:211–217. doi: 10.1002/bit.260390213. [DOI] [PubMed] [Google Scholar]
- Beringer P. M., Slaughter R. L. Transporters and their impact on drug disposition. Ann. Pharmacother. 2005;39:1097–1108. doi: 10.1345/aph.1E614. [DOI] [PubMed] [Google Scholar]
- Berson A., Descatoire V., Sutton A., Fau D., Maulny B., Vadrot N., Feldmann G., Berthon B., Tordjmann T., Pessayre D. Toxicity of alpidem, a peripheral benzodiazepine receptor ligand, but not zolpidem, in rat hepatocytes: Role of mitochondrial permeability transition and metabolic activation. J. Pharmacol. Exp. Ther. 2001;299:793–800. [PubMed] [Google Scholar]
- Berthou F., Dreano Y., Belloc C., Kangas L., Gautier J. C., Beaune P. Involvement of cytochrome P450 3A enzyme family in the major metabolic pathways of toremifene in human liver microsomes. Biochem. Pharmacol. 1994;47:1883–1895. doi: 10.1016/0006-2952(94)90319-0. [DOI] [PubMed] [Google Scholar]
- Bertilsson L., Höjer B., Tybring G., Osterloh J., Rane A. Autoinduction of carbamazepine metabolism in children examined by a stable isotope technique. Clin. Pharmacol. Ther. 1980;27:83–88. doi: 10.1038/clpt.1980.13. [DOI] [PubMed] [Google Scholar]
- Bertilsson L., Tomson T., Tybring G. Pharmacokinetics: Time-dependent changes—Autoinduction of carbamazepine epoxidation. J. Clin. Pharmacol. 1986;26:459–462. doi: 10.1002/j.1552-4604.1986.tb03558.x. [DOI] [PubMed] [Google Scholar]
- Biles R. W., Piper C. E. Mutagenicity of chloropropanol in a genetic screening battery. Fundam. Appl. Toxicol. 1983;3:27–33. doi: 10.1016/s0272-0590(83)80169-9. [DOI] [PubMed] [Google Scholar]
- Birnbaum L. S. When environmental chemicals act like uncontrolled medicine. Trends Endocrinol. Metab. 2013;24:321–323. doi: 10.1016/j.tem.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkholm B., Bok C. M., Lundin A., Rafter J., Hibberd M. L., Pettersson S. Intestinal microbiota regulate xenobiotic metabolism in the liver. PLoS One. 2009;4:e6958. doi: 10.1371/journal.pone.0006958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser M. J., Falkow S. What are the consequences of the disappearing human microbiota? Nat. Rev. Microbiol. 2009;7:887–894. doi: 10.1038/nrmicro2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogaards J. J., van Ommen B., Wolf C. R., van Bladeren P. J. Human cytochrome P450 enzyme selectivities in the oxidation of chlorinated benzenes. Toxicol. Appl. Pharmacol. 1995;132:44–52. doi: 10.1006/taap.1995.1085. [DOI] [PubMed] [Google Scholar]
- Bonnet U. Moclobemide: Therapeutic use and clinical studies. CNS Drug Rev. 2003;9:97–140. doi: 10.1111/j.1527-3458.2003.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth D. A. Psychology of nutrition. London, UK: Taylor & Francis; 1994. pp. 56–57. [Google Scholar]
- Borchers A. T., Selmi C., Meyers F. J., Keen C. L., Gershwin M. E. Probiotics and immunity. J. Gastroenterol. 2009;44:26–46. doi: 10.1007/s00535-008-2296-0. [DOI] [PubMed] [Google Scholar]
- Bort R., Macé K., Boobis A., Gómez-Lechón M. J., Pfeifer A., Castell J. Hepatic metabolism of diclofenac: Role of human CYP in the minor oxidative pathways. Biochem. Pharmacol. 1999;58:787–796. doi: 10.1016/s0006-2952(99)00167-7. [DOI] [PubMed] [Google Scholar]
- Bort R., Ponsoda X., Carrasco E., Gómez-Lechón M. J., Castell J. V. Metabolism of aceclofenac in humans. Drug Metab. Dispos. 1996;24:834–841. [PubMed] [Google Scholar]
- Boulton D. W., DeVane C. L., Liston H. L., Markowitz J. S. In vitro P-glycoprotein affinity for atypical and conventional antipsychotics. Life Sci. 2002;71:163–169. doi: 10.1016/s0024-3205(02)01680-6. [DOI] [PubMed] [Google Scholar]
- Brannan M. D., Affrime M. B., Radwanski E., Cayen M. N., Banfield C. Effects of various cytochrome P450 inhibitors on the metabolism of loratadine. Clin. Pharmacol. Ther. 1995;57:193. (OII-A-4). [Google Scholar]
- Brown A. W., Bohan Brown M. M., Onken K. L., Beitz D. C. Short-term consumption of sucralose, a nonnutritive sweetener, is similar to water with regard to select markers of hunger signaling and short-term glucose homeostasis in women. Nutr. Res. 2011;31:882–888. doi: 10.1016/j.nutres.2011.10.004. [DOI] [PubMed] [Google Scholar]
- Brown R. J., Rother K. I. Nonnutritive sweeteners and their role in the gastrointestinal tract. J. Clin. Endocrinol. Metab. 2012;97:2597–2605. doi: 10.1210/jc.2012-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown R. J., Walter M., Rother K. I. Ingestion of diet soda before a glucose load augments glucagon-like peptide-1 secretion. Diabetes Care. 2009;32:2184–2186. doi: 10.2337/dc09-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown R. J., Walter M., Rother K. I. Effects of diet soda on gut hormones in youths with diabetes. Diabetes Care. 2012;35:959–964. doi: 10.2337/dc11-2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee K. A. Statistical theory and methodology in science and engineering. New York, NY: John Wiley & Sons; 1960. [Google Scholar]
- Brusick D., Borzelleca J. F., Gallo M., Williams G., Kille J., Hayes A. W., Pi-Sunyer F. X., Williams C., Burks W. Expert panel report on a study of Splenda in male rats. Regul. Toxicol. Pharmacol. 2009;55:6–12. doi: 10.1016/j.yrtph.2009.06.013. [DOI] [PubMed] [Google Scholar]
- Bruyère A., Declevès X., Bouzom F., Proust L., Martinet M., Walther B., Parmentier Y. Development of an optimized procedure for the preparation of rat intestinal microsomes: Comparison of hepatic and intestinal microsomal cytochrome P450 enzyme activities in two rat strains. Xenobiotica. 2009;39:22–32. doi: 10.1080/00498250802517714. [DOI] [PubMed] [Google Scholar]
- Burdge G. C., Hanson M. A., Slater-Jefferies J. L., Lillycrop K. A. Epigenetic regulation of transcription: A mechanism for inducing variations in phenotype (fetal programming) by differences in nutrition during early life? Br. J. Nutr. 2007;97:1036–1046. doi: 10.1017/S0007114507682920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrini L., Landi L., Stefanelli C., Barzanti V., Sechi A.M. Extraction of lipids and lipophilic antioxidants from fish tissues: A comparison among different methods. Comp. Biochem. Physiol. Part B. 1992;101:383–386. doi: 10.1016/0305-0491(92)90016-k. [DOI] [PubMed] [Google Scholar]
- Calafat A. M., Ye X., Wong L. Y., Reidy J. A., Needham L. L. Urinary concentrations of triclosan in the U.S. population: 2003–2004. Environ. Health Perspect. 2008;116:303–307. doi: 10.1289/ehp.10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick R. W., Cooper R. L., Chang J., Rehnberg G. L., McElroy W. K. Possible antiestrogenic activity of lindane in female rats. J. Biochem. Toxicol. 1988;3:147–158. doi: 10.1002/jbt.2570030303. [DOI] [PubMed] [Google Scholar]
- Chang T. K., Yu L., Maurel P., Waxman D. J. Enhanced cyclophosphamide and ifosfamide activation in primary human hepatocyte cultures: Response to cytochrome P-450 inducers and autoinduction by oxazaphosphorines. Cancer Res. 1997;57:1946–1954. [PubMed] [Google Scholar]
- Chen C., Hanson E., Watson J. W., Lee J. S. P-glycoprotein limits the brain penetration of nonsedating but not sedating H1-antagonists. Drug Metab. Dispos. 2003a;31:312–318. doi: 10.1124/dmd.31.3.312. [DOI] [PubMed] [Google Scholar]
- Chen C., Staudinger J. L., Klaassen C. D. Nuclear receptor, pregname X receptor, is required for induction of UDP-glucuronosyltranferases in mouse liver by pregnenolone-16 alpha-carbonitrile. Drug Metab. Dispos. 2003b;31:908–915. doi: 10.1124/dmd.31.7.908. [DOI] [PubMed] [Google Scholar]
- Chen J., Raymond K. Roles of rifampicin in drug-drug interactions: Underlying molecular mechanisms involving the nuclear pregnane X receptor. Ann. Clin. Microbiol. Antimicrob. 2006;5:3. doi: 10.1186/1476-0711-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Tang Y., Guo C., Wang J., Boral D., Nie D. Nuclear receptors in the multidrug resistance through the regulation of drug-metabolizing enzymes and drug transporters. Biochem. Pharmacol. 2012;83:1112–1126. doi: 10.1016/j.bcp.2012.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho I., Blaser M. J. The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 2012;13:260–270. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W. S., Han B. S., Lee H., Kim C., Nam K. T., Park K., Choi M., Kim S. J., Kim S. H., Jeong J., Jang D. D. Subchronic toxicity study of 3-monochloropropane-1,2-diol administered by drinking water to B6C3F1 mice. Food Chem. Toxicol. 2008;46:1666–1673. doi: 10.1016/j.fct.2007.12.030. [DOI] [PubMed] [Google Scholar]
- Chonan O., Takahashi R., Watanuki M. Role of activity of gastrointestinal microflora in absorption of calcium and magnesium in rats fed ß1–4 linked galactooligosaccharides. Biosci. Biotechnol. Biochem. 2001;65:1872–1875. doi: 10.1271/bbb.65.1872. [DOI] [PubMed] [Google Scholar]
- Choo E. F., Leake B., Wandel C., Imamura H., Wood A. J., Wilkinson G. R., Kim R. B. Pharmacological inhibition of P-glycoprotein transport enhances the distribution of HIV-1 protease inhibitors into brain and testes. Drug Metab. Dispos. 2000;28:655–660. [PubMed] [Google Scholar]
- Cizza G., Rother K. I. Beyond fast food and slow motion: Weighty contributors to the obesity epidemic. J. Endocrinol. Invest. 2012;35:236–242. doi: 10.3275/8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus S. P., Tsang T. M., Wang Y., Cloarec O., Skordi E., Martin F. P., Rezzi S., Ross A., Kochhar S., Holmes E., Nicholson J. K. Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Mol. Syst. Biol. 2008;4:219. doi: 10.1038/msb.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement B., Demesmaeker M. Formation of guanoxabenz from guanabenz in human liver. A new metabolic marker for CYP1A2. Drug Metab. Dispos. 1997;25:1266–1271. [PubMed] [Google Scholar]
- Clemente J. C., Ursell L. K., Parfrey L. W., Knight R. The impact of the gut microbiota on human health: An integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colditz G. A., Willett W. C., Stampfer M. J., London S. J., Segal M. R., Speizer F. E. Patterns of weight change and their relation to diet in a cohort of healthy women. Am. J. Clin. Nutr. 1990;51:1100–1105. doi: 10.1093/ajcn/51.6.1100. [DOI] [PubMed] [Google Scholar]
- Coleman S., Linderman R., Hodgson E., Rose R. L. Comparative metabolism of chloroacetamide herbicides and selected metabolites in human and rat liver microsomes. Environ. Health Perspect. 2000;108:1151–1157. doi: 10.1289/ehp.001081151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corkey B. E. Banting lecture 2011: Hyperinsulinemia: Cause or consequence? Diabetes. 2012;61:4–13. doi: 10.2337/db11-1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coumoul X., Diry M., Barouki R. PXR-dependent induction of human CYP3A4 gene expression by organochlorine pesticides. Biochem. Pharmacol. 2002;64:1513–1519. doi: 10.1016/s0006-2952(02)01298-4. [DOI] [PubMed] [Google Scholar]
- Crabbe E., Nolasco-Hipolito C., Kobayashi G., Sonomoto K., Ishizaki A. Biodiesel production from crude palm oil and evaluation of butanol extraction and fuel properties. Process Biochem. 2001;37:65–71. [Google Scholar]
- Cummings J. H., Macfarlane G. T. The control and consequences of bacterial fermentation in the human colon. J. Appl. Bacteriol. 1991;70:443–459. doi: 10.1111/j.1365-2672.1991.tb02739.x. [DOI] [PubMed] [Google Scholar]
- Cummings J. H., Macfarlane G. T. Role of intestinal bacteria in nutrient metabolism. J. Parenter. Enteral. Nutr. 1997;21:357–365. doi: 10.1177/0148607197021006357. [DOI] [PubMed] [Google Scholar]
- Custodio J. M., Wu C. Y., Benet L. Z. Predicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorption. Adv. Drug Deliv. Rev. 2008;60:717–733. doi: 10.1016/j.addr.2007.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzig A. H., Shepard R. L., Law K. L., Tabas L., Pratt S., Gillespie J. S., Binkley S. N., Kuhfeld M. T., Starling J. J., Wrighton S. A. Selectivity of the multidrug resistance modulator, LY335979, for P-glycoprotein and effect on cytochrome P-450 activities. J. Pharmacol. Exp. Ther. 1999;290:854–862. [PubMed] [Google Scholar]
- Davies E. Sweets for my sweet. Chem. World. 2010;7:46–49. [Google Scholar]
- Decherf S., Demeneix B. A. The obesogen hypothesis: A shift of focus from the periphery to the hypothalamus. J. Toxicol. Environ. Health B. 2011;14:423–448. doi: 10.1080/10937404.2011.578561. [DOI] [PubMed] [Google Scholar]
- Deichmann W. B., MacDonald W. E., Cubit D. A., Beasley A. G. Effects of starvation in rats with elevated DDT and dieldrin tissue levels. Int. Arch. Arbeitsmed. 1972;29:233–252. doi: 10.1007/BF00539251. [DOI] [PubMed] [Google Scholar]
- Dekant W., Assmann M., Urban G. The role of cytochrome P450 2E1 in the species-dependent biotransformation of 1,2-dichloro-1,1,2-trifluoroethane in rats and mice. Toxicol. Appl. Pharmacol. 1995;135:200–207. doi: 10.1006/taap.1995.1224. [DOI] [PubMed] [Google Scholar]
- de Ruyter J. C., Olthof M. R., Seidell J. C., Katan M. B. A trial of sugar-free or sugar-sweetened beverages and body weight in children. N. Engl. J Med. 2012;367:1397–1406. doi: 10.1056/NEJMoa1203034. [DOI] [PubMed] [Google Scholar]
- Desta Z., Soukhova N., Mahal S. K., Flockhart D. A. Interaction of cisapride with the human cytochrome P450 system: Metabolism and inhibition studies. Drug Metab. Dispos. 2000;28:789–800. [PubMed] [Google Scholar]
- Desta Z., Wu C. M., Morocho A. M., Flockhart D. A. The gastroprokinetic and antiemetic drug metoclopramide is a substrate and inhibitor of cytochrome P450 2D6. Drug Metab. Dispos. 2002;30:336–343. doi: 10.1124/dmd.30.3.336. [DOI] [PubMed] [Google Scholar]
- Dhingra R., Sullivan L., Jacques P. F., Wang T. J., Fox C. S., Meigs J. B., D'Agostino R. B., Gaziano J. M., Vasan R. S. Soft drink consumption and risk of developing cardiometabolic risk factors and the metabolic syndrome in middle-aged adults in the community. Circulation. 2007;116:480–488. doi: 10.1161/CIRCULATIONAHA.107.689935. [DOI] [PubMed] [Google Scholar]
- Dong S., Wu J., Liu G., Zhang B., Zheng M. Unintentionally produced dioxin-like polychlorinated biphenyls during cooking. Food Control. 2011;22:1797–1802. [Google Scholar]
- Dong S., Liu G., Zhang B., Gao L., Zheng M. Formation of polychlorinated naphthalenes during the heating of cooking oil in the presence of high amounts of sucralose. Food Control. 2013;32:1–5. [Google Scholar]
- Doran A., Obach R. S., Smith B. J., Hosea N. A., Becker S., Callegari E., Chen C., Chen X., Choo E., Cianfrogna J., Cox L. M., Gibbs J. P., Gibbs M. A., Hatch H., Hop C. E., Kasman I. N., Laperle J., Liu J., Liu X., Logman M., Maclin D., Nedza F. M., Nelson F., Olson E., Rahematpura S., Raunig D., Rogers S., Schmidt K., Spracklin D. K., Szewc M., Troutman M., Tseng E., Tu M., Van Deusen J. W., Venkatakrishnan K., Walens G., Wang E. Q., Wong D., Yasgar A. S., Zhang C. The impact of P-glycoprotein on the disposition of drugs targeted for indications of the central nervous system: Evaluation using the MDR1A /1B knockout mouse model. Drug Metab. Dispos. 2005;33:165–174. doi: 10.1124/dmd.104.001230. [DOI] [PubMed] [Google Scholar]
- Draper A. J., Madan A., Parkinson A. Inhibition of coumarin 7-hydroxylase activity in human liver microsomes. Arch. Biochem. Biophys. 1997;341:47–61. doi: 10.1006/abbi.1997.9964. [DOI] [PubMed] [Google Scholar]
- Drescher S., Glaeser H., Mürdter T., Hitzl M., Eichelbaum M., Fromm M. F. P-glycoprotein-mediated intestinal and biliary digoxin transport in humans. Clin. Pharmacol. Ther. 2003;73:223–231. doi: 10.1067/mcp.2003.27. [DOI] [PubMed] [Google Scholar]
- Dresser G. K., Schwarz U. I., Wilkinson G. R., Kim R. B. Coordinate induction of both cytochrome P4503A and MDR1 by St John's wort in healthy subjects. Clin. Pharmacol. Ther. 2003;73:41–50. doi: 10.1067/mcp.2003.10. [DOI] [PubMed] [Google Scholar]
- DuBois G. E. Validity of early indirect models of taste active sites and advances in new taste technologies enabled by improved models. Flav. Fragr. J. 2011;26:239–253. [Google Scholar]
- DuBois G. E., Prakash I. Noncaloric sweeteners, sweetness modulators, and sweetener enhancers. Annu. Rev. Food Sci. Technol. 2012;3:353–380. doi: 10.1146/annurev-food-022811-101236. [DOI] [PubMed] [Google Scholar]
- DuBois G. E., Walters D. E., Schiffman S. S., Warwick Z. S., Booth B. J., Pecore S. D., Gibes K., Carr B. T., Brands L. M. Concentration-response relationships of sweeteners: A systematic study. In: Walters D. E., Orthoefer F. T., DuBois G. E., editors. Sweeteners. Discovery, molecular design, and chemoreception, ACS Symposium Series 450. Washington, DC: American Chemical Society; 1991. pp. 261–276. [Google Scholar]
- Duncan S. H., Belenguer A., Holtrop G., Johnstone A. M., Flint H. J., Lobley G. E. Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl. Environ. Microbiol. 2007;73:1073–1078. doi: 10.1128/AEM.02340-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dürr D., Stieger B., Kullak-Ublick G. A., Rentsch K. M., Steinert H. C., Meier P. J., Fattinger K. St John's Wort induces intestinal P-glycoprotein /MDR1 and intestinal and hepatic CYP3A4. Clin. Pharmacol. Ther. 2000;68:598–604. doi: 10.1067/mcp.2000.112240. [DOI] [PubMed] [Google Scholar]
- Ebbeling C. B., Feldman H. A., Chomitz V. R., Antonelli T. A., Gortmaker S. L., Osganian S. K., Ludwig D. S. A randomized trial of sugar-sweetened beverages and adolescent body weight. N. Engl. J. Med. 2012;367:1407–1416. doi: 10.1056/NEJMoa1203388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids (CEF) of the European Food Safety Authority. Scientific Opinion on Flavouring Group Evaluation 301 (FGE.301): A sulphur substituted pyrimidin-derivative and its hydrochloride salt from Chemical Group 30. EFSA J. 2011;9:1994. [28 pp.]. [Google Scholar]
- Ehrhardt M., Lindenmaier H., Burhenne J., Haefeli W. E., Weiss J. Influence of lipid lowering fibrates on P-glycoprotein activity in vitro. Biochem. Pharmacol. 2004;67:285–292. doi: 10.1016/j.bcp.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Ekins S., Kim R. B., Leake B. F., Dantzig A. H., Schuetz E. G., Lan L. B., Yasuda K., Shepard R. L., Winter M. A., Schuetz J. D., Wikel J. H., Wrighton S. A. Three-dimensional quantitative structure-activity relationships of inhibitors of P-glycoprotein. Mol. Pharmacol. 2002;61:964–973. doi: 10.1124/mol.61.5.964. [DOI] [PubMed] [Google Scholar]
- Endicott J. A., Ling V. The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu. Rev. Biochem. 1989;58:137–171. doi: 10.1146/annurev.bi.58.070189.001033. [DOI] [PubMed] [Google Scholar]
- Endres C. J., Hsiao P., Chung F. S., Unadkat J. D. The role of transporters in drug interactions. Eur. J. Pharm. Sci. 2006;27:501–517. doi: 10.1016/j.ejps.2005.11.002. [DOI] [PubMed] [Google Scholar]
- European Union. Directive 2003/115/EC of the European Parliament and of the Council of 22 December 2003 amending Directive 94/35/EC on sweeteners for use in foodstuffs. Off. J. Eur. Union. 2004;47(L24):65–71. http://eur-lex.europa.eu/JOHtml.do?uri=OJ:L:2004:024:SOM:en:HTML (accessed February 6, 2013). [Google Scholar]
- Faassen F., Vogel G., Spanings H., Vromans H. Caco-2 permeability, P-glycoprotein transport ratios and brain penetration of heterocyclic drugs. Int. J. Pharm. 2003;263:113–122. doi: 10.1016/s0378-5173(03)00372-7. [DOI] [PubMed] [Google Scholar]
- Fang H. M., Xu J. M., Mei Q., Diao L., Chen M. L., Jin J., Xu X. H. Involvement of cytochrome P450 3A4 and P-glycoprotein in first-pass intestinal extraction of omeprazole in rabbits. Acta Pharmacol. Sin. 2009;30:1566–1572. doi: 10.1038/aps.2009.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faucette S. R., Hawke R. L., Lecluyse E. L., Shord S. S., Yan B., Laethem R. M., Lindley C. M. Validation of bupropion hydroxylation as a selective marker of human cytochrome P450 2B6 catalytic activity. Drug Metab. Dispos. 2000;28:1222–1230. [PubMed] [Google Scholar]
- Faucette S. R., Wang H., Hamilton G. A., Jolley S. L., Gilbert D., Lindley C., Yan B., Negishi M., LeCluyse E. L. Regulation of CYP2B6 in primary human hepatocytes by prototypical inducers. Drug Metab. Dispos. 2004;32:348–358. doi: 10.1124/dmd.32.3.348. [DOI] [PubMed] [Google Scholar]
- Feil R., Fraga M. F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012;13:97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- Felmlee M. A., Lon H. K., Gonzalez F. J., Yu A. M. Cytochrome P450 expression and regulation in CYP3A4/CYP2D6 double transgenic humanized mice. Drug Metab. Dispos. 2008;36:435–441. doi: 10.1124/dmd.107.018838. [DOI] [PubMed] [Google Scholar]
- Finn J. P., Lord G. H. Neurotoxicity studies on sucralose and its hydrolysis products with special reference to histopathologic and ultrastructural changes. Food Chem. Toxicol. 2000;38(suppl. 2):S7–S17. doi: 10.1016/s0278-6915(00)00024-7. [DOI] [PubMed] [Google Scholar]
- Flaherty K. T., Lathia C., Frye R. F., Schuchter L., Redlinger M., Rosen M., O'Dwyer P. J. Interaction of sorafenib and cytochrome P450 isoenzymes in patients with advanced melanoma: A phase I/II pharmacokinetic interaction study. Cancer Chemother. Pharmacol. 2011;68:1111–1118. doi: 10.1007/s00280-011-1585-0. [DOI] [PubMed] [Google Scholar]
- Flockhart D. A. Drug interactions: Cytochrome P450 drug interaction table. Indiana University School of Medicine; 2013. http://medicine.iupui.edu/clinpharm/ddis/table.asp (accessed February 6, 2013). [Google Scholar]
- Fooks L. J., Gibson G. R. Probiotics as modulators of the gut flora. Br. J. Nutr. 2002;88(suppl. 1):S39–S49. doi: 10.1079/BJN2002628. [DOI] [PubMed] [Google Scholar]
- Ford H. E., Peters V., Martin N. M., Sleeth M. L., Ghatei M. A., Frost G. S., Bloom S. R. Effects of oral ingestion of sucralose on gut hormone response and appetite in healthy normal-weight subjects. Eur. J. Clin. Nutr. 2011;65:508–513. doi: 10.1038/ejcn.2010.291. [DOI] [PubMed] [Google Scholar]
- Fowler S. P., Williams K., Resendez R. G., Hunt K. J., Hazuda H. P., Stern M.P. Fueling the obesity epidemic? Artificially sweetened beverage use and long-term weight gain. Obesity. 2008;16:1894–1900. doi: 10.1038/oby.2008.284. [DOI] [PubMed] [Google Scholar]
- Frank G. K., Oberndorfer T. A., Simmons A. N., Paulus M. P., Fudge J. L., Yang T. T., Kaye W. H. Sucrose activates human taste pathways differently from artificial sweetener. Neuroimage. 2008;39:1559–1569. doi: 10.1016/j.neuroimage.2007.10.061. [DOI] [PubMed] [Google Scholar]
- Fraud S., Maillard J. Y., Russell A. D. Comparison of the mycobactericidal activity of ortho-phthalaldehyde, glutaraldehyde and other dialdehydes by a quantitative suspension test. J. Hosp. Infect. 2001;48:214–221. doi: 10.1053/jhin.2001.1009. [DOI] [PubMed] [Google Scholar]
- Freeman J. S. Role of the incretin pathway in the pathogenesis of type 2 diabetes mellitus. Cleve. Clin. J. Med. 2009;76(suppl. 5):S12–S19. doi: 10.3949/ccjm.76.s5.03. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Wideman R. D., Speck M., Asadi A., King D. S., Webber T. D., Haneda M., Kieffer T. J. Incretin release from gut is acutely enhanced by sugar but not by sweeteners in vivo. Am. J. Physiol. Endocrinol. Metab. 2009;296:E473–E479. doi: 10.1152/ajpendo.90636.2008. [DOI] [PubMed] [Google Scholar]
- Gardiner S. J., Begg E. J. Pharmacogenetics, drug-metabolizing enzymes, and clinical practice. Pharmacol. Rev. 2006;58:521–590. doi: 10.1124/pr.58.3.6. [DOI] [PubMed] [Google Scholar]
- Garrigues A., Escargueil A. E., Orlowski S. The multidrug transporter, P-glycoprotein, actively mediates cholesterol redistribution in the cell membrane. Proc. Natl. Acad. Sci. USA. 2002;99:10347–10352. doi: 10.1073/pnas.162366399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedeon C., Behravan J., Koren G., Piquette-Miller M. Transport of glyburide by placental ABC transporters: Implications in fetal drug exposure. Placenta. 2006;27:1096–1102. doi: 10.1016/j.placenta.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Geick A., Eichelbaum M., Burk O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J. Biol. Chem. 2001;276:14581–14587. doi: 10.1074/jbc.M010173200. [DOI] [PubMed] [Google Scholar]
- Gemma S., Vittozzi L., Testai E. Metabolism of chloroform in the human liver and identification of the competent P450s. Drug Metab. Dispos. 2003;31:266–274. doi: 10.1124/dmd.31.3.266. [DOI] [PubMed] [Google Scholar]
- Gerebtzoff G. In silico prediction of blood-brain barrier permeation and P-glycoprotein activity. 2006. Dissertation zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel. Liège, Belgium. Basel. [DOI] [PubMed]
- Ghosal A., Gupta S., Ramanathan R., Yuan Y., Lu X., Su A. D., Alvarez N., Zbaida S., Chowdhury S. K., Alton K. B. Metabolism of loratadine and further characterization of its in vitro metabolites. Drug Metab. Lett. 2009;3:162–170. doi: 10.2174/187231209789352067. [DOI] [PubMed] [Google Scholar]
- Gill H. S., Rutherfurd K. J., Cross M. L., Gopal P. K. Enhancement of immunity in the elderly by dietary supplementation with the probiotic Bifidobacterium lactis HN019. Am. J. Clin. Nutr. 2001;74:833–839. doi: 10.1093/ajcn/74.6.833. [DOI] [PubMed] [Google Scholar]
- Giraud C., Tran A., Rey E., Vincent J., Tréluyer J. M., Pons G. In vitro characterization of clobazam metabolism by recombinant cytochrome P450 enzymes: Importance of CYP2C19. Drug Metab. Dispos. 2004;32:1279–1286. [PubMed] [Google Scholar]
- Goldsmith L. A. Forward. Food Chem. Toxicol. 2000a;38(suppl. 2):iii. doi: 10.1016/s0278-6915(00)00028-4. [DOI] [PubMed] [Google Scholar]
- Goldsmith L. A. Acute and subchronic toxicity of sucralose. Food Chem. Toxicol. 2000b;38(suppl. 2):S53–S69. doi: 10.1016/s0278-6915(00)00028-4. [DOI] [PubMed] [Google Scholar]
- Goldsmith L. B., Friberg S. E., Wahlberg J. E. The effect of solvent extraction on the lipids of the stratum corneum in relation to observed immediate whitening of the skin. Contact Dermatitis. 1988;19:348–350. doi: 10.1111/j.1600-0536.1988.tb02949.x. [DOI] [PubMed] [Google Scholar]
- Golstein P. E., Boom A., van Geffel J., Jacobs P., Masereel B., Beauwens R. P-glycoprotein inhibition by glibenclamide and related compounds. Pflugers Arch. 1999;437:652–660. doi: 10.1007/s004240050829. [DOI] [PubMed] [Google Scholar]
- Gonzalez F. J., Gelboin H. V. Role of human cytochromes P450 in the metabolic activation of chemical carcinogens and toxins. Drug Metab. Rev. 1994;26:165–183. doi: 10.3109/03602539409029789. [DOI] [PubMed] [Google Scholar]
- Gorski J. C., Hall S. D., Jones D. R., VandenBranden M., Wrighton S. A. Regioselective biotransformation of midazolam by members of the human cytochrome P450 3A (CYP3A) subfamily. Biochem. Pharmacol. 1994;47:1643–1653. doi: 10.1016/0006-2952(94)90543-6. [DOI] [PubMed] [Google Scholar]
- Gorski J. C., Jones D. R., Wrighton S. A., Hall S. D. Contribution of human CYP3A subfamily members to the 6-hydroxylation of chlorzoxazone. Xenobiotica. 1997;27:243–256. doi: 10.1080/004982597240578. [DOI] [PubMed] [Google Scholar]
- Gottesman M. M., Pastan I. The multidrug transporter, a double-edged sword. J. Biol. Chem. 1988;263:12163–12166. [PubMed] [Google Scholar]
- Gottesman M. M., Fojo T., Bates S. E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- Granfors M. T., Backman J. T., Laitila J., Neuvonen P. J. Tizanidine is mainly metabolized by cytochrome P450 1A2 in vitro. Br. J. Clin. Pharmacol. 2004;57:349–353. doi: 10.1046/j.1365-2125.2003.02028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green E., Murphy C. Altered processing of sweet taste in the brain of diet soda drinkers. Physiol. Behav. 2012;107:560–567. doi: 10.1016/j.physbeh.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner B., Eichelbaum M., Fritz P., Kreichgauer H. P., von Richter O., Zundler J., Kroemer H. K. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J. Clin. Invest. 1999;104:147–153. doi: 10.1172/JCI6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice H. C., Goldsmith L. A. Sucralose—An overview of the toxicity data. Food Chem. Toxicol. 2000;38(suppl. 2):S1–S6. doi: 10.1016/s0278-6915(00)00023-5. [DOI] [PubMed] [Google Scholar]
- Grotz V. L., Munro I. C. An overview of the safety of sucralose. Regul. Toxicol. Pharmacol. 2009;55:1–5. doi: 10.1016/j.yrtph.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Grotz V. L., Henry R. R., McGill J. B., Prince M. J., Shamoon H., Trout J. R., Pi-Sunyer F. X. Lack of effect of sucralose on glucose homeostasis in subjects with type 2 diabetes. J. Am. Diet. Assoc. 2003;103:1607–1612. doi: 10.1016/j.jada.2003.09.021. [DOI] [PubMed] [Google Scholar]
- Guarner F., Malagelada J. R. Gut flora in health and disease. Lancet. 2003;361:512–519. doi: 10.1016/S0140-6736(03)12489-0. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P. Cytochrome P-450 3A4: Regulation and role in drug metabolism. Annu. Rev. Pharmacol. Toxicol. 1999;39:1–17. doi: 10.1146/annurev.pharmtox.39.1.1. [DOI] [PubMed] [Google Scholar]
- Guerrero-Bosagna C., Settles M., Lucker B., Skinner M. K. Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome. PLoS One. 2010;5:e13100. doi: 10.1371/journal.pone.0013100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y., Zhang Y., Wang Y., Chen X., Si D., Zhong D., Fawcett J. P., Zhou H. Role of CYP2C9 and its variants (CYP2C9∗3 and CYP2C9∗13) in the metabolism of lornoxicam in humans. Drug Metab. Dispos. 2005;33:749–753. doi: 10.1124/dmd.105.003616. [DOI] [PubMed] [Google Scholar]
- Haase L., Cerf-Ducastel B., Murphy C. Cortical activation in response to pure taste stimuli during the physiological states of hunger and satiety. Neuroimage. 2009;44:1008–1021. doi: 10.1016/j.neuroimage.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakkak R., Ronis M. J., Badger T. M. Effects of enteral nutrition and ethanol on cytochrome P450 distribution in small intestine of male rats. Gastroenterology. 1993;104:1611–1618. doi: 10.1016/0016-5085(93)90636-q. [DOI] [PubMed] [Google Scholar]
- Hall S. D., Thummel K. E., Watkins P. B., Lown K. S., Benet L. Z., Paine M. F., Mayo R. R., Turgeon D. K., Bailey D. G., Fontana R. J., Wrighton S. A. Molecular and physical mechanisms of first-pass extraction. Drug Metab. Dispos. 1999;27:161–166. [PubMed] [Google Scholar]
- Hamelin B. A., Bouayad A., Drolet B., Gravel A., Turgeon J. In vitro characterization of cytochrome P450 2D6 inhibition by classic histamine H1 receptor antagonists. Drug Metab. Dispos. 1998;26:536–539. [PubMed] [Google Scholar]
- Handschin C., Meyer U. A. Induction of drug metabolism: The role of nuclear receptors. Pharmacol. Rev. 2003;55:649–673. doi: 10.1124/pr.55.4.2. [DOI] [PubMed] [Google Scholar]
- Hanioka N., Jinno H., Nishimura T., Ando M. Effect of 2,4,4’-trichloro-2’-hydroxydiphenyl ether on cytochrome P450 enzymes in the rat liver. Chemosphere. 1997;34:719–730. doi: 10.1016/s0045-6535(97)00464-5. [DOI] [PubMed] [Google Scholar]
- Harrison A., Betts A., Fenner K., Beaumont K., Edgington A., Roffey S., Davis J., Comby P., Morgan P. Nonlinear oral pharmacokinetics of the α-antagonist 4-amino-5-(4-fluorophenyl)-6,7-dimethoxy-2-[4-(morpholinocarbonyl)-perhydro-1,4-diazepin-1-yl]quinoline in humans: Use of preclinical data to rationalize clinical observations. Drug Metab. Dispos. 2004;32:197–204. doi: 10.1124/dmd.32.2.197. [DOI] [PubMed] [Google Scholar]
- Harrison T. S., Perry C. M. Aripiprazole: A review of its use in schizophrenia and schizoaffective disorder. Drugs. 2004;64:1715–1736. doi: 10.2165/00003495-200464150-00010. [DOI] [PubMed] [Google Scholar]
- Hart A. L., Stagg A. J., Frame M., Graffner H., Glise H., Falk P., Kamm M. A. The role of the gut flora in health and disease, and its modification as therapy. Aliment. Pharmacol. Ther. 2002;16:1383–1393. doi: 10.1046/j.1365-2036.2002.01310.x. [DOI] [PubMed] [Google Scholar]
- Hatzimichael E., Stebbing J., Crook T. Methylation changes in neoplasia: Diagnostic and therapeutic implications. In: Tollefsbol T., editor. Cancer epigenetics. Boca Raton, FL: CRC Press; 2008. pp. 399–414. [Google Scholar]
- Hawkins D. R., Wood S. W., Waller A. R., Jordan M. C. Enzyme induction studies of TGS and TGS-HP in the rat. 1987. Unpublished report from Huntingdon Research Centre, Huntingdon, U.K. Submitted to the World Health Organization by Tate & Lyle. Referenced in WHO (World Health Organization). 1989. Trichlorogalactosucrose. In Toxicological evaluation of certain food additives and contaminants WHO Food Additives Series, no. 24. Cambridge University Press, 1989, no. 655. http://www.inchem.org/documents/jecfa/jecmono/v024je05.htm (accessed February 6, 2013).
- Hebert M. F., Fisher R. M., Marsh C. L., Dressler D., Bekersky I. Effects of rifampin on tacrolimus pharmacokinetics in healthy volunteers. J. Clin. Pharmacol. 1999;39:91–96. doi: 10.1177/00912709922007499. [DOI] [PubMed] [Google Scholar]
- Hebert M. F., Roberts J. P., Prueksaritanont T., Benet L. Z. Bioavailability of cyclosporine with concomitant rifampin administration is markedly less than predicted by hepatic enzyme induction. Clin. Pharmacol. Ther. 1992;52:453–457. doi: 10.1038/clpt.1992.171. [DOI] [PubMed] [Google Scholar]
- Hellum B. H., Hu Z., Nilsen O. G. The induction of CYP1A2, CYP2D6 and CYP3A4 by six trade herbal products in cultured primary human hepatocytes. Basic Clin. Pharmacol. Toxicol. 2007;100:23–30. doi: 10.1111/j.1742-7843.2007.00011.x. [DOI] [PubMed] [Google Scholar]
- Higashikawa F., Murakami T., Kaneda T., Kato A., Takano M. Dose-dependent intestinal and hepatic first-pass metabolism of midazolam, a cytochrome P450 3A substrate with differently modulated enzyme activity in rats. J. Pharm. Pharmacol. 1999;51:67–72. doi: 10.1211/0022357991771971. [DOI] [PubMed] [Google Scholar]
- Hijazi Y., Boulieu R. Contribution of CYP3A4, CYP2B6, and CYP2C9 isoforms to N-demethylation of ketamine in human liver microsomes. Drug Metab. Dispos. 2002;30:853–858. doi: 10.1124/dmd.30.7.853. [DOI] [PubMed] [Google Scholar]
- Hill M. J. Intestinal flora and endogenous vitamin synthesis. Eur. J. Cancer Prev. 1997;6(suppl. 1):S43–S45. doi: 10.1097/00008469-199703001-00009. [DOI] [PubMed] [Google Scholar]
- Holtbecker N., Fromm M. F., Kroemer H. K., Ohnhaus E. E., Heidemann H. The nifedipine-rifampin interaction. Evidence for induction of gut wall metabolism. Drug Metab. Dispos. 1996;24:1121–1123. [PubMed] [Google Scholar]
- Holzapfel W. H., Haberer P., Snel J., Schillinger U., Huis in't Veld J. H. Overview of gut flora and probiotics. Int. J. Food Microbiol. 1998;41:85–101. doi: 10.1016/s0168-1605(98)00044-0. [DOI] [PubMed] [Google Scholar]
- Hooper L.V., Littman D. R., Macpherson A. J. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins J. M., Shenfield G. M., Gross A. S. Relationship between proguanil metabolic ratio and CYP2C19 genotype in a Caucasian population. Br. J. Clin. Pharmacol. 1998;46:499–504. doi: 10.1046/j.1365-2125.1998.00807.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L., Zhang X., Wang D., Baccarelli A. Environmental chemical exposures and human epigenetics. Int. J. Epidemiol. 2012;41:79–105. doi: 10.1093/ije/dyr154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough L., Khan R. Intensification of sweetness. Trends Biochem. Sci. 1978;3:61–63. [Google Scholar]
- Hough L., Khan R. Enhancement of the sweetness of sucrose by conversion into chloro-deoxy derivatives. In: Grenby T. H., editor. Progress in sweeteners. London, UK: Elsevier Applied Science; 1989. pp. 97–120. [Google Scholar]
- Hsiao P., Sasongko L., Link J. M., Mankoff D. A., Muzi M., Collier A. C., Unadkat J. D. Verapamil P-glycoprotein transport across the rat blood-brain barrier: Cyclosporine, a concentration inhibition analysis, and comparison with human data. J. Pharmacol. Exp. Ther. 2006;317:704–710. doi: 10.1124/jpet.105.097931. [DOI] [PubMed] [Google Scholar]
- Hu S., Chen Z., Franke R., Orwick S., Zhao M., Rudek M. A., Sparreboom A., Baker S. D. Interaction of the multikinase inhibitors sorafenib and sunitinib with solute carriers and ATP-binding cassette transporters. Clin. Cancer Res. 2009;15:6062–6069. doi: 10.1158/1078-0432.CCR-09-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Kupfer D. Metabolism of the endocrine disruptor pesticide-methoxychlor by human P450s: Pathways involving a novel catechol metabolite. Drug Metab. Dispos. 2002;30:1035–1042. doi: 10.1124/dmd.30.9.1035. [DOI] [PubMed] [Google Scholar]
- Hutchinson S. A. The effect of pH, temperature and reactants on the thermal and non-thermal degradation of the high-intensity sweeteners: Alitame and sucralose. 1996. PhD dissertation. New Brunswick, NJ: Rutgers University.
- Hutchinson S. A., Ho G. S., Ho C. T. Stability and degradation of the high-intensity sweeteners: Aspartame, alitame, and sucralose. Food Rev. Int. 1999;15:249–261. [Google Scholar]
- Huwyler J., Wright M. B., Gutmann H., Drewe J. Induction of cytochrome P450 3A4 and P-glycoprotein by the isoxazolyl-penicillin antibiotic flucloxacillin. Curr. Drug Metab. 2006;7:119–126. doi: 10.2174/138920006775541534. [DOI] [PubMed] [Google Scholar]
- Ibrahim S., Peggins J., Knapton A., Licht T., Aszalos A. Influence of antipsychotic, antiemetic, and Ca2+ channel blocker drugs on the cellular accumulation of the anticancer drug daunorubicin: P-glycoprotein modulation. J. Pharmacol. Exp. Ther. 2000;295:1276–1283. [PubMed] [Google Scholar]
- Ieiri I., Kimura M., Irie S., Urae A., Otsubo K., Ishizaki T. Interaction magnitude, pharmacokinetics and pharmacodynamics of ticlopidine in relation to CYP2C19 genotypic status. Pharmacogenet. Genomics. 2005;15:851–859. doi: 10.1097/01213011-200512000-00003. [DOI] [PubMed] [Google Scholar]
- Ihunnah C. A., Jiang M., Xie W. Nuclear receptor PXR, transcriptional circuits and metabolic relevance. Biochim. Biophys. Acta. 2011;1812:956–963. doi: 10.1016/j.bbadis.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida A., Tomita M., Hayashi M. Regional difference in P-glycoprotein function in rat intestine. Drug Metab. Pharmacokinet. 2005;20:100–106. doi: 10.2133/dmpk.20.100. [DOI] [PubMed] [Google Scholar]
- Imai H., Kotegawa T., Tsutsumi K., Morimoto T., Eshima N., Nakano S., Ohashi K. The recovery time-course of CYP3A after induction by St John's wort administration. Br. J. Clin. Pharmacol. 2008;65:701–707. doi: 10.1111/j.1365-2125.2008.03120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): Clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005;5:6–13. doi: 10.1038/sj.tpj.6500285. [DOI] [PubMed] [Google Scholar]
- International Transporter Consortium. Giacomini K. M., Huang S. M., Tweedie D. J., Benet L. Z., Brouwer K. L., Chu X., Dahlin A., Evers R., Fischer V., Hillgren K. M., Hoffmaster K. A., Ishikawa T., Keppler D., Kim R. B., Lee C. A., Niemi M., Polli J. W., Sugiyama Y., Swaan P. W., Ware J. A., Wright S. H., Yee S. W., Zamek-Gliszczynski M. J., Zhang L. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010;9:215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoherranen N., Hachad H., Yeung C. K., Levy R. H. Qualitative analysis of the role of metabolites in inhibitory drug-drug interactions: Literature evaluation based on the metabolism and transport drug interaction database. Chem. Res. Toxicol. 2009;22:294–298. doi: 10.1021/tx800491e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaki K., Sakaeda T., Kakumoto M., Nakamura T., Komoto C., Okamura N., Nishiguchi K., Shiraki T., Horinouchi M., Okumura K. Haloperidol is an inhibitor but not substrate for MDR1/P-glycoprotein. J. Pharm. Pharmacol. 2006;58:1617, 1622. doi: 10.1211/jpp.58.12.0008. [DOI] [PubMed] [Google Scholar]
- Iwamoto K., Klaassen C. D. Firstpass effect of morphine in rats. J. Pharmacol. Exp. Ther. 1977;200:236–244. [PubMed] [Google Scholar]
- Jafari F., Garcia-Gil L. J., Salmanzadeh-Ahrabi S., Shokrzadeh L., Aslani M. M., Pourhoseingholi M. A., Derakhshan F., Zali M. R. Diagnosis and prevalence of enteropathogenic bacteria in children less than 5 years of age with acute diarrhea in Tehran children's hospitals. J. Infect. 2009;58:21–27. doi: 10.1016/j.jinf.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Jang H. J., Kokrashvili Z., Theodorakis M. J., Carlson O. D., Kim B. J., Zhou J., Kim H. H., Xu X., Chan S. L., Juhaszova M., Bernier M., Mosinger B., Margolskee R. F., Egan J. M. Gut-expressed gustducin and taste receptors regulate secretion of glucagon-like peptide-1. Proc. Natl. Acad. Sci. USA. 2007;104:15069–15074. doi: 10.1073/pnas.0706890104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean P., Lopez-Garcia P., Dansette P., Mansuy D., Goldstein J. L. Oxidation of tienilic acid by human yeast-expressed cytochromes P-450 2C8, 2C9, 2C18 and 2C19. Evidence that this drug is a mechanism-based inhibitor specific for cytochrome P450 2C9. Eur. J. Biochem. 1996;241:797–804. doi: 10.1111/j.1432-1033.1996.00797.x. [DOI] [PubMed] [Google Scholar]
- Jeon T. I., Seo Y. K., Osborne T. F. Gut bitter taste receptor signalling induces ABCB1 through a mechanism involving CCK. Biochem. J. 2011;438:33–37. doi: 10.1042/BJ20110009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia W., Li H., Zhao L., Nicholson J. K. Gut microbiota: A potential new territory for drug targeting. Nat. Rev. Drug Discov. 2008;7:123–129. doi: 10.1038/nrd2505. [DOI] [PubMed] [Google Scholar]
- Jinno H., Hanioka N., Onodera S., Nishimura T., Ando M. Irgasan® DP 300 (5-chloro-2-(2,4-dichlorophenoxy)-phenol) induces cytochrome P450s and inhibits haem biosynthesis in rat hepatocytes cultured on Matrigel. Xenobiotica. 1997;27:681–692. doi: 10.1080/004982597240271. [DOI] [PubMed] [Google Scholar]
- John B. A., Wood S. G., Hawkins D. R. The pharmacokinetics and metabolism of sucralose in the mouse. Food Chem. Toxicol. 2000a;38(suppl. 2):S107–S110. doi: 10.1016/s0278-6915(00)00032-6. [DOI] [PubMed] [Google Scholar]
- John B. A., Wood S. G., Hawkins D. R. The pharmacokinetics and metabolism of sucralose in the rabbit. Food Chem. Toxicol. 2000b;38(suppl. 2):S111–S113. doi: 10.1016/s0278-6915(00)00033-8. [DOI] [PubMed] [Google Scholar]
- Johne A., Brockmöller J., Bauer S., Maurer A., Langheinrich M., Roots I. Pharmacokinetic interaction of digoxin with an herbal extract from St John's wort (Hypericum perforatum) Clin. Pharmacol. Ther. 1999;66:338–345. doi: 10.1053/cp.1999.v66.a101944. [DOI] [PubMed] [Google Scholar]
- Kan W. M., Liu Y. T., Hsiao C. L., Shieh C. Y., Kuo J. H., Huang J. D., Su S. F. Effect of hydroxyzine on the transport of etoposide in rat small intestine. Anticancer Drugs. 2001;12:267–273. doi: 10.1097/00001813-200103000-00012. [DOI] [PubMed] [Google Scholar]
- Kansy M., Senner F., Gubernator K. Physicochemical high throughput screening: Parallel artificial membrane permeation assay in the description of passive absorption processes. J. Med. Chem. 1998;41:1007–1010. doi: 10.1021/jm970530e. [DOI] [PubMed] [Google Scholar]
- Karmaus W., Osuch J. R., Eneli I., Mudd L. M., Zhang J., Mikucki D., Haan P., Davis S. Maternal levels of dichlorodiphenyl-dichloroethylene (DDE) may increase weight and body mass index in adult female offspring. Occup. Environ. Med. 2009;66:143–149. doi: 10.1136/oem.2008.041921. [DOI] [PubMed] [Google Scholar]
- Kato Y., Haraguchi K., Ito Y., Fujii A., Yamazaki T., Endo T., Koga N., Yamada S., Degawa M. Polychlorinated biphenyl-mediated decrease in serum thyroxine level in rodents. Drug Metab. Dispos. 2010;38:697–704. doi: 10.1124/dmd.109.031153. [DOI] [PubMed] [Google Scholar]
- Kato Y., Haraguchi K., Yamazaki T., Ito Y., Miyajima S., Nemoto K., Koga N., Kimura R, Degawa M. Effects of polychlorinated biphenyls, kanechlor-500, on serum thyroid hormone levels in rats and mice. Toxicol. Sci. 2003;72:235–241. doi: 10.1093/toxsci/kfg025. [DOI] [PubMed] [Google Scholar]
- Katoh M., Nakajima M., Shimada N., Yamazaki H., Yokoi T. Inhibition of human cytochrome P450 enzymes by 1,4-dihydropyridine calcium antagonists: Prediction of in vivo drug-drug interactions. Eur. J. Clin. Pharmacol. 2000a;55:843–852. doi: 10.1007/s002280050706. [DOI] [PubMed] [Google Scholar]
- Katoh M., Nakajima M., Yamazaki H., Yokoi T. Inhibitory potencies of 1,4-dihydropyridine calcium antagonists to P-glycoprotein-mediated transport: Comparison with the effects on CYP3A4. Pharm. Res. 2000b;17:1189–1197. doi: 10.1023/a:1007568811691. [DOI] [PubMed] [Google Scholar]
- Katoh M., Nakajima M., Yamazaki H., Yokoi T. Inhibitory effects of CYP3A4 substrates and their metabolites on P-glycoprotein-mediated transport. Eur. J. Pharm. Sci. 2001;12:505–513. doi: 10.1016/s0928-0987(00)00215-3. [DOI] [PubMed] [Google Scholar]
- Kauffmann H. M., Pfannschmidt S., Zöller H., Benz A., Vorderstemann B., Webster J. I., Schrenk D. Influence of redox-active compounds and PXR-activators on human MRP1 and MRP2 gene expression. Toxicology. 2002;171:137–146. doi: 10.1016/s0300-483x(01)00570-4. [DOI] [PubMed] [Google Scholar]
- Kharasch E. D., Thummel K. E. Identification of cytochrome P450 2E1 as the predominant enzyme catalyzing human liver microsomal defluorination of sevoflurane, isoflurane, and methoxyflurane. Anesthesiology. 1993;79:795–807. doi: 10.1097/00000542-199310000-00023. [DOI] [PubMed] [Google Scholar]
- Kille J. W., Tesh J. M., McAnulty P. A., Ross F. W., Willoughby C. R., Bailey G. P., Wilby O. K., Tesh S. A. Sucralose: Assessment of teratogenic potential in the rat and the rabbit. Food Chem. Toxicol. 2000;38(suppl. 2):S43–S52. doi: 10.1016/s0278-6915(00)00027-2. [DOI] [PubMed] [Google Scholar]
- Kim K. A., Chung J., Jung D. H., Park J. Y. Identification of cytochrome P450 isoforms involved in the metabolism of loperamide in human liver microsomes. Eur. J. Clin. Pharmacol. 2004;60:575–581. doi: 10.1007/s00228-004-0815-3. [DOI] [PubMed] [Google Scholar]
- Kim K. Y., Mancano M. A. Fenofibrate potentiates warfarin effects. Ann. Pharmacother. 2003;37:212–215. doi: 10.1177/106002800303700210. [DOI] [PubMed] [Google Scholar]
- Kitamura S., Shimizu Y., Shiraga Y., Yoshida M., Sugihara K., Ohta S. Reductive metabolism of p,p'-DDT and o,p'-DDT by rat liver cytochrome P450. Drug Metab. Dispos. 2002;30:113–118. doi: 10.1124/dmd.30.2.113. [DOI] [PubMed] [Google Scholar]
- Kliewer S. A., Goodwin B., Willson T. M. The nuclear pregnane X receptor: A key regulator of xenobiotic metabolism. Endocr. Rev. 2002;23:687–702. doi: 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- Kolars J. C., Awni W. M., Merion R. M., Watkins P. B. First-pass metabolism of cyclosporin by the gut. Lancet. 1991;338:1488–1490. doi: 10.1016/0140-6736(91)92302-i. [DOI] [PubMed] [Google Scholar]
- Kolars J. C., Lown K. S., Schmiedlin-Ren P., Ghosh M., Fang C., Wrighton S. A., Merion R. M., Watkins P. B. CYP3A gene expression in human gut epithelium. Pharmacogenetics. 1994;4:247–259. doi: 10.1097/00008571-199410000-00003. [DOI] [PubMed] [Google Scholar]
- Kotlyar M., Brauer L. H., Tracy T. S., Hatsukami D. K., Harris J., Bronars C. A., Adson D. E. Inhibition of CYP2D6 activity by bupropion. J. Clin. Psychopharmacol. 2005;25:226–229. doi: 10.1097/01.jcp.0000162805.46453.e3. [DOI] [PubMed] [Google Scholar]
- Kudo S., Odomi M. Involvement of human cytochrome P450 3A4 in reduced haloperidol oxidation. Eur. J. Clin. Pharmacol. 1998;54:253–259. doi: 10.1007/s002280050455. [DOI] [PubMed] [Google Scholar]
- Kwara A., Lartey M., Sagoe K. W., Xexemeku F., Kenu E., Oliver-Commey J., Boima V., Sagoe A., Boamah I., Greenblatt D. J., Court M. H. Pharmacokinetics of efavirenz when co-administered with rifampin in TB/HIV co-infected patients: Pharmacogenetic effect of CYP2B6 variation. J. Clin. Pharmacol. 2008;48:1032–1040. doi: 10.1177/0091270008321790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Merrill M., Birnbaum L. S. Childhood obesity and environmental chemicals. Mt. Sinai J. Med. 2011;78:22–48. doi: 10.1002/msj.20229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labare M. P., Alexander M. Biodegradation of sucralose, a chlorinated carbohydrate, in samples of natural environments. Environ. Toxicol. Chem. 1993;12:797–804. [Google Scholar]
- Labare M. P., Alexander M. Microbial cometabolism of sucralose, a chlorinated disaccharide, in environmental samples. Appl. Microbiol. Biotechnol. 1994;42:173–178. doi: 10.1007/BF00170242. [DOI] [PubMed] [Google Scholar]
- Lampen A., Zhang Y, Hackbarth I., Benet L. Z., Sewing K. F., Christians U. Metabolism and transport of the macrolide immunosuppressant sirolimus in the small intestine. J. Pharmacol. Exp. Ther. 1998;285:1104–1112. [PubMed] [Google Scholar]
- Langer P. Polychlorinated biphenyls and the thyroid gland—Minireview. Endocr. Regul. 1998;32:193–203. [PubMed] [Google Scholar]
- Lanning C. L., Fine R. L., Sachs C. W, Rao U. S., Corcoran J. J., Abou-Donia M. B. Chlorpyrifos oxon interacts with the mammalian multidrug resistance protein, P-glycoprotein. J. Toxicol. Environ. Health. 1996;47:395–407. doi: 10.1080/009841096161726. [DOI] [PubMed] [Google Scholar]
- Lappin-Scott H. M., Holt G., Bull A. T. Microbial transformation of 1,6-dichloro-1,6-dideoxy-βD-fructofuranosyl-4-chloro-4-deoxy-αD-galactopyranoside (TGS) by soil populations. WorldJ. Microbiol. Biotechnol. 1987;3:95–102. [Google Scholar]
- Laurenzana E. M., Sorrels S. L., Owens S. M. Antipeptide antibodies targeted against specific regions of rat CYP2D1 and human CYP2D6. Drug Metab. Dispos. 1995;23:271–278. [PubMed] [Google Scholar]
- Le Guellec C, Lacarelle B., Catalin J., Durand A. Inhibitory effects of anticancer drugs on dextromethorphan-O-demethylase activity in human liver microsomes. Cancer Chemother. Pharmacol. 1993;32:491–495. doi: 10.1007/BF00685896. [DOI] [PubMed] [Google Scholar]
- Leatherhead Food Research. The global food additives market. 5th ed. Leatherhead, Surrey, UK: Leatherhead; September 2011. [Google Scholar]
- Lee D. H., Lee I. K., Song K., Steffes M., Toscano W., Baker B. A., Jacobs D. R., Jr. A strong dose-response relation between serum concentrations of persistent organic pollutants and diabetes: Results from the National Health and Examination Survey 1999–2002. Diabetes Care. 2006;29:1638–1644. doi: 10.2337/dc06-0543. [DOI] [PubMed] [Google Scholar]
- Lee S. M., Donaldson G. P., Mikulski Z., Boyajian S., Ley K., Mazmanian S. K. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature. 2013. doi:10.1038/nature12447. [DOI] [PMC free article] [PubMed]
- Leffingwell J. Flavor properties of FEMA GRAS LIST 25 flavor chemicals. Perfum. Flavorist. 2011;36(June):24–31. [Google Scholar]
- Lemke T. L., Williams D. A., Roche V. F., Zito S. W. Foye's principles of medicinal chemistry. Baltimore, MD: Lippincott Williams and Wilkins; 2008. pp. 267–268. [Google Scholar]
- Ley R. E., Turnbaugh P. J., Klein S., Gordon J. I. Microbial ecology: Human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Li X.-Q., Björkman A., Andersson T. B., Ridderström M., Masimirembwa C. M. Amodiaquine clearance and its metabolism to N-desethylamodiaquine is mediated by CYP2C8: A new high affinity and turnover enzyme-specific probe substrate. J. Pharmacol. Exp. Ther. 2002;300:399–407. doi: 10.1124/jpet.300.2.399. [DOI] [PubMed] [Google Scholar]
- Li X., Du Z., Huang X., Yuan W., Ying H. Solubility of sucralose in different solvents from (283.15 to 333.15) K. J. Chem. Eng. Data. 2010;55:2600–2602. [Google Scholar]
- Lilja J. J., Niemi M., Fredrikson H., Neuvonen P. J. Effects of clarithromycin and grapefruit juice on the pharmacokinetics of glibenclamide. Br. J. Clin. Pharmacol. 2007;63:732–740. doi: 10.1111/j.1365-2125.2006.02836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima S. A. C., Tavares J., Gameiro P., de Castro B., Cordeiro-da-Silva A. Flurazepam inhibits the P-glycoprotein transport function: An insight to revert multidrug-resistance phenotype. Eur. J. Pharmacol. 2008;581:30–36. doi: 10.1016/j.ejphar.2007.11.045. [DOI] [PubMed] [Google Scholar]
- Lin J. H., Yamazaki M. Role of P-glycoprotein in pharmacokinetics: Clinical implications. Clin. Pharmacokinet. 2003;42:59–98. doi: 10.2165/00003088-200342010-00003. [DOI] [PubMed] [Google Scholar]
- Lin J. H., Liu L. Y., Yang M. H., Lee M. H. Ethyl acetate/ethyl alcohol mixtures as an alternative to Folch reagent for extracting animal lipids. J. Agric. Food Chem. 2004;52:4984–4986. doi: 10.1021/jf049360m. [DOI] [PubMed] [Google Scholar]
- Lipscomb J. C., Garrett C. M., Snawder J. E. Cytochrome P450-dependent metabolism of trichloroethylene: Interindividual differences in humans. Toxicol. Appl. Pharmacol. 1997;142:311–318. doi: 10.1006/taap.1996.8040. [DOI] [PubMed] [Google Scholar]
- Lown K. S., Kolars J. C., Thummel K. E., Barnett J. L., Kunze K. L., Wrighton S. A., Watkins P. B. Interpatient heterogeneity in expression of CYP3A4 and CYP3A5 in small bowel. Lack of prediction by the erythromycin breath test. Drug Metab. Dispos. 1994;22:947–955. [PubMed] [Google Scholar]
- Lutsey P. L., Steffen L. M., Stevens J. Dietary intake and the development of the metabolic syndrome: The Atherosclerosis Risk in Communities study. Circulation. 2008;117:754–761. doi: 10.1161/CIRCULATIONAHA.107.716159. [DOI] [PubMed] [Google Scholar]
- Ma J., Bellon M., Wishart J. M., Young R., Blackshaw L. A., Jones K. L., Horowitz M., Rayner C. K. Effect of the artificial sweetener, sucralose, on gastric emptying and incretin hormone release in healthy subjects. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;296:G735–G739. doi: 10.1152/ajpgi.90708.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J., Chang J., Checklin H. L., Young R. L., Jones K. L., Horowitz M., Rayner C. K. Effect of the artificial sweetener, sucralose, on small intestinal glucose absorption in healthy human subjects. Br. J. Nutr. 2010;104:803–806. doi: 10.1017/S0007114510001327. [DOI] [PubMed] [Google Scholar]
- Mace O. J., Affleck J., Patel N., Kellett G. L. Sweet taste receptors in rat small intestine stimulate glucose absorption through apical GLUT2. J. Physiol. 2007;582:379–392. doi: 10.1113/jphysiol.2007.130906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace O. J., Lister N., Morgan E., Shepherd E., Affleck J., Helliwell P., Bronk J. R., Kellett G. L., Meredith D., Boyd R., Pieri M., Bailey P. D., Pettcrew R., Foley D. An energy supply network of nutrient absorption coordinated by calcium and T1R taste receptors in rat small intestine. J. Physiol. 2009;587:195–210. doi: 10.1113/jphysiol.2008.159616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahar Doan K. M., Humphreys J. E., Webster L. O., Wring S. A., Shampine L. J., Serabjit-Singh C. J., Adkison K. K., Polli J. W. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J. Pharmacol. Exp. Ther. 2002;303:1029–1037. doi: 10.1124/jpet.102.039255. [DOI] [PubMed] [Google Scholar]
- Maitra R., Halpin P. A., Karlson K. H., Page R. L., Paik D. Y., Leavitt M. O., Moyer B. D., Stanton B. A., Hamilton J. W. Differential effects of mitomycin C and doxorubicin on P-glycoprotein expression. Biochem J. 2001;355:617–624. doi: 10.1042/bj3550617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann A., Miksys S., Lee A., Mash D. C., Tyndale R. F. Induction of the drug metabolizing enzyme CYP2D in monkey brain by chronic nicotine treatment. Neuropharmacology. 2008;55:1147–1155. doi: 10.1016/j.neuropharm.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann S. W., Yuschak M. M., Amyes S. J. G., Aughton P., Finn J. P. A carcinogenicity study of sucralose in the CD-1 mouse. Food Chem. Toxicol. 2000a;38(suppl. 2):S91–S98. doi: 10.1016/s0278-6915(00)00030-2. [DOI] [PubMed] [Google Scholar]
- Mann S. W., Yuschak M. M., Amyes S. J. G., Aughton P., Finn J. P. A combined chronic toxicity/carcinogenicity study of sucralose in Sprague-Dawley rats. Food Chem. Toxicol. 2000b;38(suppl. 2):S71–S89. doi: 10.1016/s0278-6915(00)00029-6. [DOI] [PubMed] [Google Scholar]
- Mao-Qiang M., Feingold K. R., Wang F., Thornfeldt C. R., Elias P. M. A natural lipid mixture improves barrier function and hydration in human and murine skin. J. Soc. Cosmet. Chem. 1996;47:157–166. [Google Scholar]
- Marchetti S., Mazzanti R., Beijnen J. H., Schellens J. H. Concise review: Clinical relevance of drug drug and herb drug interactions mediated by the ABC transporter ABCB1 (MDR1, P-glycoprotein) Oncologist. 2007;12:927–941. doi: 10.1634/theoncologist.12-8-927. [DOI] [PubMed] [Google Scholar]
- Margolskee R. F., Dyer J., Kokrashvili Z., Salmon K. S. H., Ilegems E., Daly K., Maillet E. L., Ninomiya Y., Mosinger B., Shirazi-Beechey S. P. T1R3 and gustducin in gut sense sugars to regulate expression of Na+-glucose cotransporter 1. Proc. Natl. Acad. Sci. USA. 2007;104:15075–15080. doi: 10.1073/pnas.0706678104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda S., Uemoto S., Hashida T., Inomata Y., Tanaka K., Inui K. Effect of intestinal P-glycoprotein on daily tacrolimus trough level in a living-donor small bowel recipient. Clin. Pharmacol. Ther. 2000;68:98–103. doi: 10.1067/mcp.2000.107912. [DOI] [PubMed] [Google Scholar]
- Matheny C. J., Ali R. Y., Yang X., Pollack G. M. Effect of prototypical inducing agents on P-glycoprotein and CYP3A expression in mouse tissues. Drug Metab. Dispos. 2004;32:1008–1014. [PubMed] [Google Scholar]
- Matheny C. J., Lamb M. W., Brouwer K. R., Pollack G. M. Pharmacokinetic and pharmacodynamic implications of P-glycoprotein modulation. Pharmacotherapy. 2001;21:778–779. doi: 10.1592/phco.21.9.778.34558. [DOI] [PubMed] [Google Scholar]
- Matsubara T., Kim H. J., Miyata M., Shimada M., Nagata K., Yamazoe Y. Isolation and characterization of a new major intestinal CYP3A form, CYP3A62, in the rat. J. Pharmacol. Exp. Ther. 2004;309:1282–1290. doi: 10.1124/jpet.103.061671. [DOI] [PubMed] [Google Scholar]
- May-Manke A., Kroemer H., Hempel G., Bohnenstengel F., Hohenlöchter B., Blaschke G., Boos J. Investigation of the major human hepatic cytochrome P450 involved in 4-hydroxylation and N-dechloroethylation of trofosfamide. Cancer Chemother. Pharmacol. 1999;44:327–334. doi: 10.1007/s002800050985. [DOI] [PubMed] [Google Scholar]
- McKay J. A., Mathers J. C. Diet induced epigenetic changes and their implications for health. Acta Physiol. (Oxford) 2011;202:103–118. doi: 10.1111/j.1748-1716.2011.02278.x. [DOI] [PubMed] [Google Scholar]
- Mechetner E., Kyshtoobayeva A., Zonis S., Kim H., Stroup R., Garcia R., Parker R. J., Fruehauf J. P. Levels of multidrug resistance (MDR1) P-glycoprotein expression by human breast cancer correlate with in vitro resistance to taxol and doxorubicin. Clin. Cancer Res. 1998;4:389–398. [PubMed] [Google Scholar]
- Meijerman I., Beijnen J. H., Schellens J. H. Combined action and regulation of phase II enzymes and multidrug resistance proteins in multidrug resistance in cancer. Cancer Treat. Rev. 2008;34:505–520. doi: 10.1016/j.ctrv.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Meinl W., Sczesny S., Brigelius-Flohé R., Blaut M., Glatt H. Impact of gut microbiota on intestinal and hepatic levels of phase 2 xenobiotic-metabolizing enzymes in the rat. Drug Metab. Dispos. 2009;37:1179–1186. doi: 10.1124/dmd.108.025916. [DOI] [PubMed] [Google Scholar]
- Mezitis N. H., Maggio C. A., Koch P., Quddoos A., Allison D. B., Pi-Sunyer F. X. Glycemic effect of a single high oral dose of the novel sweetener sucralose in patients with diabetes. Diabetes Care. 1996;19:1004–1005. doi: 10.2337/diacare.19.9.1004. [DOI] [PubMed] [Google Scholar]
- Middleton J. H., Salierno J. D. Antibiotic resistance in triclosan tolerant fecal coliforms isolated from surface waters near wastewater treatment plant outflows (Morris County, NJ, USA) Ecotoxicol. Environ. Safety. 2013;88:79–88. doi: 10.1016/j.ecoenv.2012.10.025. [DOI] [PubMed] [Google Scholar]
- Miksys S., Rao Y., Hoffmann E., Mash D. C., Tyndale R. F. Regional and cellular expression of CYP2D6 in human brain: Higher levels in alcoholics. J. Neurochem. 2002;82:1376–1387. doi: 10.1046/j.1471-4159.2002.01069.x. [DOI] [PubMed] [Google Scholar]
- Miller D. B., Spence J. D. Clinical pharmacokinetics of fibric acid derivatives (fibrates) Clin. Pharmacokinet. 1998;34:155–162. doi: 10.2165/00003088-199834020-00003. [DOI] [PubMed] [Google Scholar]
- Miller G. A. Sucralose. In: O'Brien Nabors L., Gelardi R. C., editors. Alternative sweeteners. 2nd ed. New York, NY: Marcel Dekker; 1991. pp. 173–195. [Google Scholar]
- Mitschke D., Reichel A., Fricker G., Moenning U. Characterization of cytochrome P450 protein expression along the entire length of the intestine of male and female rats. Drug Metab Dispos. 2008;36:1039–1045. doi: 10.1124/dmd.107.019687. [DOI] [PubMed] [Google Scholar]
- Miura M., Otani K., Ohkubo T. Identification of human cytochrome P450 enzymes involved in the formation of 4-hydroxyestazolam from estazolam. Xenobiotica. 2005;35:455–465. doi: 10.1080/00498250500111612. [DOI] [PubMed] [Google Scholar]
- Miyama T., Takanaga H., Matsuo H., Yamano K., Yamamoto K., Iga T., Naito M., Tsuruo T., Ishizuka H., Kawahara Y., Sawada Y. P-glycoprotein-mediated transport of itraconazole across the blood-brain barrier. Antimicrob. Agents Chemother. 1998;42:1738–1744. doi: 10.1128/aac.42.7.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinary S. V., Quinlan M. E. Sucralose. In: Mitchell H., editor. Sweeteners and sugar alternatives in food technology. Oxford, UK: Blackwell; 2006. pp. 130–148. [Google Scholar]
- Mrozikiewicz P. M., Bogacz A., Karasiewicz M., Mikolajczak P. L., Ozarowski M., Seremak-Mrozikiewicz A., Czerny B., Bobkiewicz-Kozlowska T., Grzeskowiak E. The effect of standardized Echinacea purpurea extract on rat cytochrome P450 expression level. Phytomedicine. 2010;17:830–833. doi: 10.1016/j.phymed.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Najib J. Eszopiclone, a nonbenzo-diazepine sedative-hypnotic agent for the treatment of transient and chronic insomnia. Clin. Ther. 2006;28:491–516. doi: 10.1016/j.clinthera.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Nakagawa Y., Nagasawa M., Yamada S., Hara A., Mogami H., Nikolaev V. O., Lohse M. J., Shigemura N., Ninomiya Y., Kojima I. Sweet taste receptor expressed in pancreatic ß-cells activates the calcium and cyclic AMP signaling systems and stimulates insulin secretion. PLoS One. 2009;4:e5106. doi: 10.1371/journal.pone.0005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima M., Nakamura S., Tokudome S., Shimada N., Yamazaki H., Yokoi T. Azelastine N-demethylation by cytochrome P-450 (CYP)3A4, CYP2D6, and CYP1A2 in human liver microsomes: Evaluation of approach to predict the contribution of multiple CYPs. Drug Metab. Dispos. 1999b;27:1381–1391. [PubMed] [Google Scholar]
- Nakajima M., Ohyama K., Nakamura S., Shimada N., Yamazaki H., Yokoi T. Inhibitory effects of azelastine and its metabolites on drug oxidation catalyzed by human cytochrome P-450 enzymes. Drug Metab. Dispos. 1999a;27:792–797. [PubMed] [Google Scholar]
- Need A. C., Motulsky A. G., Goldstein D. B. Priorities and standards in pharmacogenetic research. Nat. Genet. 2005;37:671–681. doi: 10.1038/ng1593. [DOI] [PubMed] [Google Scholar]
- Nicholson J. K., Holmes E., Wilson I. D. Gut microorganisms, mammalian metabolism and personalized health care. Nat. Rev. Microbiol. 2005;3:431–438. doi: 10.1038/nrmicro1152. [DOI] [PubMed] [Google Scholar]
- Nishiya Y., Hagihara K., Ito T., Tajima M., Miura S., Kurihara A., Farid N. A., Ikeda T. Mechanism-based inhibition of human cytochrome P450 2B6 by ticlopidine, clopidogrel, and the thiolactone metabolite of prasugrel. Drug Metab. Dispos. 2009;37:589–593. doi: 10.1124/dmd.108.022988. [DOI] [PubMed] [Google Scholar]
- Niwa T., Shiraga T., Ishii I., Kagayama A., Takagi A. Contribution of human hepatic cytochrome p450 isoforms to the metabolism of psychotropic drugs. Biol. Pharm. Bull. 2005;28:1711–1716. doi: 10.1248/bpb.28.1711. [DOI] [PubMed] [Google Scholar]
- Nowack R., Andrassy J., Fischereder M., Unger M. Effects of dietary factors on drug transport and metabolism: The impact on dosage guidelines in transplant patients. Clin. Pharmacol. Ther. 2009;85:439–443. doi: 10.1038/clpt.2008.303. [DOI] [PubMed] [Google Scholar]
- Nyakutira C., Röshammar D., Chigutsa E., Chonzi P., Ashton M., Nhachi C., Masimirembwa C. High prevalence of the CYP2B6 516G->T(∗6) variant and effect on the population pharmacokinetics of efavirenz in HIV/AIDS outpatients in Zimbabwe. Eur. J. Clin. Pharmacol. 2008;64:357–365. doi: 10.1007/s00228-007-0412-3. [DOI] [PubMed] [Google Scholar]
- Obach R. S., Cox L. M., Tremaine L. M. Sertraline is metabolized by multiple cytochrome P450 enzymes, monoamine oxidases, and glucuronyl transferases in human: an in vitro study. Drug Metab. Dispos. 2005;33:262–270. doi: 10.1124/dmd.104.002428. [DOI] [PubMed] [Google Scholar]
- O'Brien P. J., Siraki A. G., Shangari N. Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health. Crit. Rev. Toxicol. 2005;35:609–662. doi: 10.1080/10408440591002183. [DOI] [PubMed] [Google Scholar]
- O'Flaherty S., Klaenhammer T. R. The role and potential of probiotic bacteria in the gut, and the communication between gut microflora and gut/host. Int. Dairy J. 2010;20:262–268. [Google Scholar]
- Olesen O. V., Linnet K. Identification of the human cytochrome P450 isoforms mediating in vitro N-dealkylation of perphenazine. Br. J. Clin. Pharmacol. 2000;50:563–571. doi: 10.1046/j.1365-2125.2000.00298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen O. V., Linnet K. Contributions of five human cytochrome P450 isoforms to the N-demethylation of clozapine in vitro at low and high concentrations. J. Clin. Pharmacol. 2001;41:823–832. doi: 10.1177/00912700122010717. [DOI] [PubMed] [Google Scholar]
- Ong C. E., Coulter S., Birkett D. J., Bhasker C. R., Miners J. O. The xenobiotic inhibitor profile of cytochrome P4502C8. Br. J. Clin. Pharmacol. 2000;50:573–580. doi: 10.1046/j.1365-2125.2000.00316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono S., Hatanaka T., Miyazawa S., Tsutsui M., Aoyama T., Gonzalez F. J., Satoh T. Human liver microsomal diazepam metabolism using cDNA-expressed cytochrome P450s: Role of CYP2B6, 2C19 and the 3A subfamily. Xenobiotica. 1996;26:1155–1166. doi: 10.3109/00498259609050260. [DOI] [PubMed] [Google Scholar]
- O'Toole P. W., Claesson M. J. Gut microbiota: Changes throughout the lifespan from infancy to elderly. Int. Dairy J. 2010;20:281–291. [Google Scholar]
- Paine M. F. Sites of extra hepatic metabolism, Part II: Gut. In: Pearson P. G., Wienkers L. C., editors. Handbook of drug metabolism. 2nd ed. New York, NY: Informa Healthcare USA; 2009. pp. 273–298. [Google Scholar]
- Paine M. F., Thummel K. E. Role of intestinal cytochromes P450 in drug disposition. In: Lee J. S., Obach R. S., Fisher M. B., editors. Drug metabolizing enzymes: cytochrome P450 and other enzymes in drug discovery and development. New York, NY: Marcel Dekker; 2003. pp. 421–452. [Google Scholar]
- Paine M. F., Hart H. L., Ludington S. S., Haining R. L., Rettie A. E., Zeldin D. C. The human intestinal cytochrome P450 “pie”. Drug Metab. Dispos. 2006;34:880–886. doi: 10.1124/dmd.105.008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine M. F., Shen D. D., Kunze K. L., Perkins J. D., Marsh C. L., McVicar J. P., Barr D. M., Gillies B. S., Thummel K. E. Firstpass metabolism of midazolam by the human intestine. Clin. Pharmacol. Ther. 1996;60:14–24. doi: 10.1016/S0009-9236(96)90162-9. [DOI] [PubMed] [Google Scholar]
- Pan Y. Z., Gao W., Yu A. M. MicroRNAs regulate CYP3A4 expression via direct and indirect targeting. Drug Metab. Dispos. 2009;37:2112–2117. doi: 10.1124/dmd.109.027680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang S., Cao J. Q., Katz B. H., Hayes C. L., Sutter T. R., Spink D. C. Inductive and inhibitory effects of non- ortho-substituted polychlorinated biphenyls on estrogen metabolism and human cytochromes P450 1A1 and 1B1. Biochem. Pharmacol. 1999;58:29–38. doi: 10.1016/s0006-2952(99)00070-2. [DOI] [PubMed] [Google Scholar]
- Park J. Y., Kim K. A., Kim S. L. Chloramphenicol is a potent inhibitor of cytochrome P450 isoforms CYP2C19 and CYP3A4 in human liver microsomes. Antimicrob. Agents Chemother. 2003;47:3464–3469. doi: 10.1128/AAC.47.11.3464-3469.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. Y., Kim K. A., Kang M. H., Kim S. L., Shin J. G. Effect of rifampin on the pharmacokinetics of rosiglitazone in healthy subjects. Clin. Pharmacol. Ther. 2004;75:157–162. doi: 10.1016/j.clpt.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Pelkonen O., Turpeinen M., Hakkola J., Honkakoski P., Hukkanen J., Raunio H. Inhibition and induction of human cytochrome P450 enzymes: Current status. Arch. Toxicol. 2008;82:667–715. doi: 10.1007/s00204-008-0332-8. [DOI] [PubMed] [Google Scholar]
- Pepino M. Y., Tiemann C. D., Patterson B. W., Wice B. M., Klein S. Sucralose affects glycemic and hormonal responses to an oral glucose load. Diabetes Care. 2013;36:2530–2535. doi: 10.2337/dc12-2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfuhler S., Fellows M., van Benthem J., Corvi R., Curren R., Dearfield K., Fowler P., Frötschl R., Elhajouji A., Le Hégarat L., Kasamatsu T., Kojima H., Ouédraogo G., Scott A., Speit G. In vitro genotoxicity test approaches with better predictivity: Summary of an IWGT workshop. Mutat. Res. 2011;723:101–107. doi: 10.1016/j.mrgentox.2011.03.013. [DOI] [PubMed] [Google Scholar]
- Pharmacogenomics Knowledge Base. 2013. http://www.pharmgkb.org (accessed February 6, 2013).
- Pirmohamed M., Williams D., Madden S., Templeton E., Park B. K. Metabolism and bioactivation of clozapine by human liver in vitro. J. Pharmacol. Exp. Ther. 1995;272:984–990. [PubMed] [Google Scholar]
- Piscitelli S. C., Burstein A. H., Chaitt D., Alfaro R. M., Falloon J. Indinavir concentrations and St John's wort. Lancet. 2000;355:547–548. doi: 10.1016/S0140-6736(99)05712-8. [DOI] [PubMed] [Google Scholar]
- Polli J. W., Wring S. A., Humphreys J. E., Huang L., Morgan J. B., Webster L. O., Serabjit-Singh C. S. Rational use of in vitro P-glycoprotein assays in drug discovery. J. Pharmacol. Exp. Ther. 2001;299:620–628. [PubMed] [Google Scholar]
- Porta M. Persistent organic pollutants and the burden of diabetes. Lancet. 2006;368:558–559. doi: 10.1016/S0140-6736(06)69174-5. [DOI] [PubMed] [Google Scholar]
- Prakash C., Kamel A., Cui D., Whalen R. D., Miceli J. J., Tweedie D. Identification of the major human liver cytochrome P450 isoform(s) responsible for the formation of the primary metabolites of ziprasidone and prediction of possible drug interactions. Br. J. Clin. Pharmacol. 2000;49(suppl. 1):35S–42S. doi: 10.1046/j.1365-2125.2000.00151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prashant G. M., Patil R. B., Nagaraj T., Patel V. B. The antimicrobial activity of the three commercially available intense sweeteners against common periodontal pathogens: An in vitro study. J. Contemp. Dent. Pract. 2012;13:749–752. doi: 10.5005/jp-journals-10024-1222. [DOI] [PubMed] [Google Scholar]
- Preissner S., Kroll K., Dunkel M., Senger C., Goldsobel G., Kuzman D., Guenther S., Winnenburg R., Schroeder M., Preissner R. SuperCYP: A comprehensive database on cytochrome P450 enzymes including a tool for analysis of CYP-drug interactions. Nucleic Acids Res. 2010;38(database issue):D237–D243. doi: 10.1093/nar/gkp970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior T. I., Chue P. S., Tibbo P., Baker G. B. Drug metabolism and atypical antipsychotics. Eur. Neuropsychopharmacol. 1999;9:301–309. doi: 10.1016/s0924-977x(98)00040-6. [DOI] [PubMed] [Google Scholar]
- Projean D., Baune B., Farinotti R., Flinois J. P., Beaune P., Taburet A. M., Ducharme J. In vitro metabolism of chloroquine: Identification of CYP2C8, CYP3A4, and CYP2D6 as the main isoforms catalyzing N-desethylchloroquine formation. Drug Metab. Dispos. 2003;31:748–754. doi: 10.1124/dmd.31.6.748. [DOI] [PubMed] [Google Scholar]
- Qin X. What caused the recent worldwide increase of inflammatory bowel disease: Should sucralose be added as a suspect? Inflamm. Bowel Dis. 2011;17:E139. doi: 10.1002/ibd.21823. [DOI] [PubMed] [Google Scholar]
- Qin X. Etiology of inflammatory bowel disease: A unified hypothesis. World J. Gastroenterol. 2012;18:1708–1722. doi: 10.3748/wjg.v18.i15.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn A., Yaylayan V. A. Thermal degradation of sucralose and its potential in generating chloropropanols in the presence of glycerol. Food Chem. 2010;118:56–61. [Google Scholar]
- Rajiliác-Stojanoviác M., Heilig H. G., Tims S., Zoetendal E. G., de Vos W. M. Long-term monitoring of the human intestinal microbiota composition. Environ. Microbiol. 2012. doi:10.1111/1462-2920.12023. [DOI] [PubMed]
- Ramesh B., Adkar S. S., Prabhudesai A. V., Viswanathan C.V. Selective extraction of phospholipids from egg yolk. J. Am. Oil Chem. Soc. 1979;56:585–587. doi: 10.1007/BF02660240. [DOI] [PubMed] [Google Scholar]
- Rao U. S., Fine R. L., Scarborough G. A. Antiestrogens and steroid hormones: substrates of the human P-glycoprotein. Biochem. Pharmacol. 1994;48:287–292. doi: 10.1016/0006-2952(94)90099-x. [DOI] [PubMed] [Google Scholar]
- Ren X., Zhou L., Terwilliger R., Newton S. S., de Araujo I. E. Sweet taste signaling functions as a hypothalamic glucose sensor. Front. Integr. Neurosci. 2009;3:12. doi: 10.3389/neuro.07.012.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegelman S., Rowland M. Effect of route of administration on drug disposition. J. Pharmacokinet. Biopharm. 1973;1:419–434. doi: 10.1007/BF01059666. [DOI] [PubMed] [Google Scholar]
- Roberts A., Renwick A. G., Sims J., Snodin D. J. Sucralose metabolism and pharmacokinetics in man. Food Chem. Toxicol. 2000;38(suppl. 2):S31–S41. doi: 10.1016/s0278-6915(00)00026-0. [DOI] [PubMed] [Google Scholar]
- Robison T. W., Jacobs A. Metabolites in safety testing. Bioanalysis. 2009;1:1193–1200. doi: 10.4155/bio.09.98. [DOI] [PubMed] [Google Scholar]
- Ross R. P., Mills S., Hill C., Fitzgerald G. F., Stanton C. Specific metabolite production by gut microbiota as a basis for probiotic function. Int. Dairy J. 2010;20:269–276. [Google Scholar]
- Rotzinger S., Fang J., Baker G. B. Trazodone is metabolized to m-chlorophenylpiperazine by CYP3A4 from human sources. Drug Metab. Dispos. 1998;26:572–575. [PubMed] [Google Scholar]
- Rowland M., Tozer T. N. Clinical pharmacokinetics and pharmacodynamics. Concepts and applications. 4th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2011. Nonlinearities; pp. 445–482. [Google Scholar]
- Rudenga K. J., Small D. M. Amygdala response to sucrose consumption is inversely related to artificial sweetener use. Appetite. 2012;58:504–507. doi: 10.1016/j.appet.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruschitzka F., Meier P. J., Turina M., Lüscher T. F., Noll G. Acute heart transplant rejection due to Saint John's wort. Lancet. 2000;355:548–549. doi: 10.1016/S0140-6736(99)05467-7. [DOI] [PubMed] [Google Scholar]
- Russell A. D. Bacterial resistance to disinfectants: Present knowledge and future problems. J. Hosp. Infect. 1998;43(suppl.):S57–S68. doi: 10.1016/s0195-6701(99)90066-x. [DOI] [PubMed] [Google Scholar]
- Russell A. D. Bacterial adaptation and resistance to antiseptics, disinfectants and preservatives is not a new phenomenon. J. Hosp. Infect. 2004;57:97–104. doi: 10.1016/j.jhin.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Saitoh R., Miyayama T., Mitsui T., Akiba Y., Higashida A., Takata S., Kawanishi T., Aso Y., Itoh Z., Omura S. Nonlinear intestinal pharmacokinetics of mitemcinal, the first acid-resistant non-peptide motilin receptor agonist, in rats. Xenobiotica. 2007;37:1421–1432. doi: 10.1080/00498250701668592. [DOI] [PubMed] [Google Scholar]
- Sasaki Y. F., Kawaguchi S., Kamaya A., Ohshita M., Kabasawa K., Iwama K., Taniguchi K., Tsuda S. The comet assay with 8 mouse organs: Results with 39 currently used food additives. Mutat. Res. 2002;519:103–119. doi: 10.1016/s1383-5718(02)00128-6. [DOI] [PubMed] [Google Scholar]
- Schiffman S. S. Rationale for further medical and health research on high-potency sweeteners. Chem. Senses. 2012;37:671–679. doi: 10.1093/chemse/bjs053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffman S. S., Abou-Donia M. B. Sucralose revisited: Rebuttal of two papers about Splenda safety. Regul. Toxicol. Pharmacol. 2012;63:505–508. doi: 10.1016/j.yrtph.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Schiffman S. S., Gatlin C.A. Sweeteners: State of knowledge review. Neurosci. Biobehav Rev. 1993;17:313–345. doi: 10.1016/s0149-7634(05)80015-6. [DOI] [PubMed] [Google Scholar]
- Schiffman S. S., Booth B. J., Losee M. L., Pecore S. D., Warwick Z. S. Bitterness of sweeteners as a function of concentration. Brain Res. Bull. 1995;36:505–513. doi: 10.1016/0361-9230(94)00225-p. [DOI] [PubMed] [Google Scholar]
- Schiffman S. S., Cahn H., Lindley M. G. Multiple receptor sites mediate sweetness: Evidence from cross adaptation. Pharmacol. Biochem. Behav. 1981;15:377–388. doi: 10.1016/0091-3057(81)90266-5. [DOI] [PubMed] [Google Scholar]
- Schiffman S. S., Hornack K., Reilly D. Increased taste thresholds of amino acids with age. Am. J. Clin. Nutr. 1979;32:1622–1627. doi: 10.1093/ajcn/32.8.1622. [DOI] [PubMed] [Google Scholar]
- Schiffman S. S., Reilly D. A., Clark T. B., 3rd Qualitative differences among sweeteners. Physiol. Behav. 1979;23:1–9. doi: 10.1016/0031-9384(79)90113-6. [DOI] [PubMed] [Google Scholar]
- Schiffman S. S., Sattely-Miller E. A., Bishay I. E. Sensory properties of neotame: Comparison with other sweeteners. In: Weerasinghe D. K., DuBois G. E., editors. Sweetness and sweeteners: Biology, chemistry and psychophysics. New York, NY: Oxford University Press; 2008. pp. 511–529. ACS Symposium Series. [Google Scholar]
- Schinkel A. H., Wagenaar E., Mol C. A., van Deemter L. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J. Clin. Invest. 1996;97:2517–2524. doi: 10.1172/JCI118699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz E. G., Beck W. T., Schuetz J. D. Modulators and substrates of P-glycoprotein and cytochrome P4503A coordinately up-regulate these proteins in human colon carcinoma cells. Mol. Pharmacol. 1996;49:311–318. [PubMed] [Google Scholar]
- Schwab D., Fischer H., Tabatabaei A., Poli S., Huwyler J. Comparison of in vitro P-glycoprotein screening assays: Recommendations for their use in drug discovery. J. Med. Chem. 2003;46:1716–1725. doi: 10.1021/jm021012t. [DOI] [PubMed] [Google Scholar]
- Scientific Committee on Food. Opinion of the Scientific Committee on Food on Sucralose (adopted by the SCF on 7 September 2000). SCF/CS/ADDS/EDUL/190 Final 12/9/2000. European Commission. Health & Consumer Protection Directorate-General; 2000. ec.europa.eu/food/fs/sc/scf/out68_en.pdf (accessed February 6, 2013). [Google Scholar]
- Scientific Committee on Food. Opinion of the Scientific Committee on Food on 3-monochloropropane-1,2-diol (3-MCPD) European Commission, Health and Consumer Protection Directorate-General; 2001. http://ec.europa.eu/food/fs/sc/scf/out91_en.pdf (accessed February 6, 2013). [Google Scholar]
- Screnci D., McKeage M. J., Galettis P., Hambley T. W., Palmer B. D., Baguley B. C. Relationships between hydrophobicity, reactivity, accumulation and peripheral nerve toxicity of a series of platinum drugs. Br. J. Cancer. 2000;82:966–972. doi: 10.1054/bjoc.1999.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sérée E., Villard P. H., Pascussi J. M., Pineau T., Maurel P., Nguyen Q. B., Fallone F., Martin P. M., Champion S., Lacarelle B., Savouret J. F., Barra Y. Evidence for a new human CYP1A1 regulation pathway involving PPAR-alpha and 2 PPRE sites. Gastroenterology. 2004;127:1436–1445. doi: 10.1053/j.gastro.2004.08.023. [DOI] [PubMed] [Google Scholar]
- Servant G., Tachdjian C., Li X., Karanewsky DS. The sweet taste of true synergy: positive allosteric modulation of the human sweet taste receptor. Trends Pharmacol. Sci. 2011;32:631–636. doi: 10.1016/j.tips.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Servant G., Tachdjian C., Tang X. Q., Werner S., Zhang F., Li X., Kamdar P., Petrovic G., Ditschun T., Java A., Brust P., Brune N., DuBois G. E., Zoller M., Karanewsky D. S. Positive allosteric modulators of the human sweet taste receptor enhance sweet taste. Proc. Natl. Acad. Sci. USA. 2010;107:4746–4751. doi: 10.1073/pnas.0911670107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer M. J. Vitamin K. Lancet. 1995;345:229–234. doi: 10.1016/s0140-6736(95)90227-9. [DOI] [PubMed] [Google Scholar]
- Sherman P. M., Ossa J. C., Johnson-Henry K. Unraveling mechanisms of action of probiotics. Nutr. Clin. Pract. 2009;24:10–14. doi: 10.1177/0884533608329231. [DOI] [PubMed] [Google Scholar]
- Shimada T., Terada A., Yokogawa K., Kaneko H., Nomura M., Kaji K., Kaneko S., Kobayashi K., Miyamoto K. Lowered blood concentration of tacrolimus and its recovery with changes in expression of CYP3A and P-glycoprotein after highdose steroid therapy. Transplantation. 2002;74:1419–1424. doi: 10.1097/00007890-200211270-00014. [DOI] [PubMed] [Google Scholar]
- Shimomura M., Masuda S., Saito H., Sakamoto S., Uemoto S., Tanaka K., Inui K. Roles of the jejunum and ileum in the first-pass effect as absorptive barriers for orally administered tacrolimus. J. Surg. Res. 2002;103:215–222. doi: 10.1006/jsre.2002.6359. [DOI] [PubMed] [Google Scholar]
- Shiraga T., Kaneko H., Iwasaki K., Tozuka Z., Suzuki A., Hata T. Identification of cytochrome P450 enzymes involved in the metabolism of zotepine, an antipsychotic drug, in human liver microsomes. Xenobiotica. 1999;29:217–229. doi: 10.1080/004982599238623. [DOI] [PubMed] [Google Scholar]
- Shon J. H., Yoon Y. R., Kim M. J., Kim K. A., Lim Y. C., Liu K. H., Shin D. H., Lee C.H., Cha I. J., Shin J. G. Chlorpropamide 2-hydroxylation is catalysed by CYP2C9 and CYP2C19 in vitro: Chlorpropamide disposition is influenced by CYP2C9, but not by CYP2C19 genetic polymorphism. Br. J. Clin. Pharmacol. 2005;59:552–563. doi: 10.1111/j.1365-2125.2005.02364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugarts S., Benet L. Z. The role of transporters in the pharmacokinetics of orally administered drugs. Pharm. Res. 2009;26:2039–2054. doi: 10.1007/s11095-009-9924-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simooya O. O., Sijumbil G., Lennard M. S., Tucker G. T. Halofantrine and chloroquine inhibit CYP2D6 activity in healthy Zambians. Br. J. Clin. Pharmacol. 1998;45:315–317. doi: 10.1046/j.1365-2125.1998.00671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims J., Roberts A., Daniel J. W., Renwick A. G. The metabolic fate of sucralose in rats. Food Chem. Toxicol. 2000;38(suppl. 2):S115–S121. doi: 10.1016/s0278-6915(00)00034-x. [DOI] [PubMed] [Google Scholar]
- Smith R. L., Cohen S. M., Doull J., Feron V. J., Goodman J. I., Marnett L. J., Munro I. C., Portoghese P. S., Waddell W. J., Wagner B. M., Adams T. B. Expert Panel of the Flavor and Extract Manufacturers Association. Criteria for the safety evaluation of flavoring substances. The Expert Panel of the Flavor and Extract Manufacturers Association. Food Chem. Toxicol. 2005;43:1141–1177. doi: 10.1016/j.fct.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Smith R. L., Waddell W. J., Cohen S. M., Fukushima S., Gooderham N. J., Hecht S. S., Marnett L. J., Portoghese P. S., Rietjens I. M. C. M., Adams T. B., Lucas Gavin C., McGowen M. M., Taylor S. V. GRAS Flavoring Substances 25. Food Technol. 2011;65:44–75. [Google Scholar]
- Soldner A., Benet L. Z., Mutschler E., Christians U. Active transport of the angiotensin-II antagonist losartan and its main metabolite EXP 3174 across MDCK-MDR1 and Caco-2 cell monolayers. Br. J. Pharmacol. 2000;129:1235–1243. doi: 10.1038/sj.bjp.0703150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparreboom A., van Asperen J., Mayer U., Schinkel A. H., Smit J. W., Meijer D. K., Borst P., Nooijen W. J., Beijnen J. H., van Tellingen O. Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc. Natl. Acad. Sci. USA. 1997;94:2031–2035. doi: 10.1073/pnas.94.5.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speit G., Vasquez M., Hartmann A. The comet assay as an indicator test for germ cell genotoxicity. Mutat. Res. 2009;681:3–12. doi: 10.1016/j.mrrev.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Spracklin D. K., Hankins D. C., Fisher J. M., Thummel K. E., Kharasch E. D. Cytochrome P450 2E1 is the principal catalyst of human oxidative halothane metabolism in vitro. J. Pharmacol. Exp. Ther. 1997;281:400–411. [PubMed] [Google Scholar]
- Steinert R. E., Frey F., Töpfer A., Drewe J., Beglinger C. Effects of carbohydrate sugars and artificial sweeteners on appetite and the secretion of gastrointestinal satiety peptides. Br. J. Nutr. 2011;105:1320–1328. doi: 10.1017/S000711451000512X. [DOI] [PubMed] [Google Scholar]
- Stellman S. D., Garfinkel L. Artificial sweetener use and one-year weight change among women. Prev. Med. 1986;15:195–202. doi: 10.1016/0091-7435(86)90089-7. [DOI] [PubMed] [Google Scholar]
- Stellman S. D., Djordjevic M. V., Muscat J. E., Gong L., Bernstein D., Citron M. L., White A., Kemeny M., Busch E., Nafziger A. N. Relative abundance of organochlorine pesticides and polychlorinated biphenyls in adipose tissue and serum of women in Long Island, New York. Cancer Epidemiol. Biomarkers Prev. 1998;7:489–496. [PubMed] [Google Scholar]
- Stephens R. H., O'Neill C. A., Warhurst A., Carlson G. L., Rowland M., Warhurst G. Kinetic profiling of P-glycoprotein-mediated drug efflux in rat and human intestinal epithelia. J. Pharmacol. Exp. Ther. 2001;296:584–591. [PubMed] [Google Scholar]
- Störmer E., von Moltke L. L., Perloff M. D., Greenblatt D. J. P-glycoprotein interactions of nefazodone and trazodone in cell culture. J. Clin. Pharmacol. 2001;41:708–714. doi: 10.1177/00912700122010609. [DOI] [PubMed] [Google Scholar]
- Störmer E., von Moltke L. L., Perloff M. D., Greenblatt D. J. Differential modulation of P-glycoprotein expression and activity by non-nucleoside HIV-1 reverse transcriptase inhibitors in cell culture. Pharm. Res. 2002;19:1038–1045. doi: 10.1023/a:1016430825740. [DOI] [PubMed] [Google Scholar]
- Stouder C., Paoloni-Giacobino A. Transgenerational effects of the endocrine disruptor vinclozolin on the methylation pattern of imprinted genes in the mouse sperm. Reproduction. 2010;139:373–379. doi: 10.1530/REP-09-0340. [DOI] [PubMed] [Google Scholar]
- Strolin Benedetti M., Dostert P. Induction and autoinduction properties of rifamycin derivatives: A review of animal and human studies. Environ. Health Perspect. 1994;102(suppl. 9):101–105. doi: 10.1289/ehp.94102s9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strolin Benedetti M., Efthymiopoulos C., Sassella D., Moro E., Repetto M. Autoinduction of rifabutin metabolism in man. Xenobiotica. 1990;20:1113–1119. doi: 10.3109/00498259009046832. [DOI] [PubMed] [Google Scholar]
- Suzuki H., Sugiyama Y. Role of metabolic enzymes and efflux transporters in the absorption of drugs from the small intestine. Eur. J. Pharm. Sci. 2000;12:3–12. doi: 10.1016/s0928-0987(00)00178-0. [DOI] [PubMed] [Google Scholar]
- Svecova L., Vrzal R., Burysek L., Anzenbacherova E., Cerveny L., Grim J., Trejtnar F., Kunes J., Pour M., Staud F., Anzenbacher P., Dvorak Z., Pavek P. Azole antimycotics differentially affect rifampicin-induced pregnane X receptor-mediated CYP3A4 gene expression. Drug Metab. Dispos. 2008;36:339–348. doi: 10.1124/dmd.107.018341. [DOI] [PubMed] [Google Scholar]
- Swidsinski A., Loening-Baucke V., Herber A. Mucosal flora in Crohn's disease and ulcerative colitis: An overview. J. Physiol. Pharmacol. 2009;60(suppl. 6):61–71. [PubMed] [Google Scholar]
- Swithers S. E. Artificial sweeteners produce the counterintuitive effect of inducing metabolic derangements. Trends Endocrinol. Metab. 2013;24:431–441. doi: 10.1016/j.tem.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swithers S. E., Davidson T. L. A role for sweet taste: Calorie predictive relations in energy regulation by rats. Behav. Neurosci. 2008;122:161–173. doi: 10.1037/0735-7044.122.1.161. [DOI] [PubMed] [Google Scholar]
- Swithers S. E., Baker C. R., Davidson T. L. General and persistent effects of high-intensity sweeteners on body weight gain and caloric compensation in rats. Behav. Neurosci. 2009;123:772–780. doi: 10.1037/a0016139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swithers S. E., Laboy A. F., Clark K., Cooper S., Davidson T. L. Experience with the high-intensity sweetener saccharin impairs glucose homeostasis and GLP-1 release in rats. Behav. Brain Res. 2012;233:1–14. doi: 10.1016/j.bbr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swithers S. E., Martin A. A., Davidson T. L. High-intensity sweeteners and energy balance. Physiol. Behav. 2010;100:55–62. doi: 10.1016/j.physbeh.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana S., Yoshinari K., Chikada T., Toriyabe T., Nagata K., Yamazoe Y. Involvement of vitamin D receptor in the intestinal induction of human ABCB1. Drug Metab. Dispos. 2009;37:1604–1610. doi: 10.1124/dmd.109.027219. [DOI] [PubMed] [Google Scholar]
- Tachibana T., Kato M., Sugiyama Y. Prediction of nonlinear intestinal absorption of CYP3A4 and P-glycoprotein substrates from their in vitro Km values. Pharm. Res. 2012;29:651–668. doi: 10.1007/s11095-011-0579-2. [DOI] [PubMed] [Google Scholar]
- Takagi S., Nakajima M., Mohri T., Yokoi T. Post-transcriptional regulation of human pregnane X receptor by micro-RNA affects the expression of cytochrome P450 3A4. J. Biol. Chem. 2008;283:9674–9680. doi: 10.1074/jbc.M709382200. [DOI] [PubMed] [Google Scholar]
- Takano M., Hasegawa R., Fukuda T., Yumoto R., Nagai J., Murakami T. Interaction with P-glycoprotein and transport of erythromycin, midazolam and ketoconazole in Caco-2 cells. Eur. J. Pharmacol. 1998;358:289–294. doi: 10.1016/s0014-2999(98)00607-4. [DOI] [PubMed] [Google Scholar]
- Takara K., Ohnishi N., Horibe S., Yokoyama T. Expression profiles of drug-metabolizing enzyme CYP3A and drug efflux transporter multidrug resistance 1 subfamily mRNAS in small intestine. Drug Metab. Dispos. 2003;31:1235–1239. doi: 10.1124/dmd.31.10.1235. [DOI] [PubMed] [Google Scholar]
- Takara K., Tanigawara Y., Komada F., Nishiguchi K., Sakaeda T., Okumura K. Cellular pharmacokinetic aspects of reversal effect of itraconazole on P-glycoprotein-mediated resistance of anticancer drugs. Biol. Pharm. Bull. 1999;22:1355–1359. doi: 10.1248/bpb.22.1355. [DOI] [PubMed] [Google Scholar]
- Tamai I., Saheki A., Saitoh R., Sai Y., Yamada I., Tsuji A. Nonlinear intestinal absorption of 5-hydroxytryptamine receptor antagonist caused by absorptive and secretory transporters. J. Pharmacol. Exp. Ther. 1997;283:108–115. [PubMed] [Google Scholar]
- Tang J., Cao Y., Rose R.L., Brimfield A. A., Dai D., Goldstein J. A., Hodgson E. Metabolism of chlorpyrifos by human cytochrome P450 isoforms and human, mouse, and rat liver microsomes. Drug Metab. Dispos. 2001;29:1201–1204. [PubMed] [Google Scholar]
- Tang W., Stearns R. A., Wang R. W., Chiu S. H., Baillie T. A. Roles of human hepatic cytochrome P450s 2C9 and 3A4 in the metabolic activation of diclofenac. Chem. Res. Toxicol. 1999;12:192–199. doi: 10.1021/tx9802217. [DOI] [PubMed] [Google Scholar]
- Tao L., Yang S., Xie M., Kramer P. M., Pereira M.A. Hypomethylation and overexpression of c-jun and c-myc protooncogenes and increased DNA methyl-transferase activity in dichloroacetic and trichloroacetic acid-promoted mouse liver tumors. Cancer Lett. 2000a;158:185–193. doi: 10.1016/s0304-3835(00)00518-8. [DOI] [PubMed] [Google Scholar]
- Tao L., Yang S., Xie M., Kramer P. M., Pereira M.A. Effect of trichloroethylene and its metabolites, dichloroacetic acid and trichloroacetic acid, on the methylation and expression of c-Jun and c-Myc protooncogenes in mouse liver: Prevention by methionine. Toxicol. Sci. 2000b;54:399–407. doi: 10.1093/toxsci/54.2.399. [DOI] [PubMed] [Google Scholar]
- Tapaninen T., Neuvonen P. J., Niemi M. Rifampicin reduces the plasma concentrations and the renin-inhibiting effect of aliskiren. Eur. J. Clin. Pharmacol. 2010;66:497–502. doi: 10.1007/s00228-010-0796-3. [DOI] [PubMed] [Google Scholar]
- Tassaneeyakul W., Birkett D. J., Miners J. O. Inhibition of human hepatic cytochrome P4502E1 by azole antifungals, CNS-active drugs and non-steroidal anti-inflammatory agents. Xenobiotica. 1998;28:293–301. doi: 10.1080/004982598239579. [DOI] [PubMed] [Google Scholar]
- Teitelbaum J. E., Walker W. A. Nutritional impact of pre- and probiotics as protective gastrointestinal organisms. Annu. Rev. Nutr. 2002;22:107–138. doi: 10.1146/annurev.nutr.22.110901.145412. [DOI] [PubMed] [Google Scholar]
- Thomas G. Medicinal chemistry: An introduction. 2nd ed. Chichester, UK: John Wiley & Sons; 2007. p. 83. [Google Scholar]
- Thummel K. E., Kunze K. L., Shen D. D. Enzyme-catalyzed processes of first-pass hepatic and intestinal drug extraction. Adv. Drug. Deliv. Rev. 1997;27:99–127. doi: 10.1016/s0169-409x(97)00039-2. [DOI] [PubMed] [Google Scholar]
- Thummel K. E., O'Shea D., Paine M. F., Shen D. D., Kunze K. L., Perkins J. D., Wilkinson G. R. Oral first-pass elimination of midazolam involves both gastrointestinal and hepatic CYP3A-mediated metabolism. Clin. Pharmacol. Ther. 1996;59:491–502. doi: 10.1016/S0009-9236(96)90177-0. [DOI] [PubMed] [Google Scholar]
- Tian R., Koyabu N., Morimoto S., Shoyama Y., Ohtani H., Sawada Y. Functional induction and de-induction of P-glycoprotein by St. John's wort and its ingredients in a human colon adenocarcinoma cell line. Drug Metab. Dispos. 2005;33:547–554. doi: 10.1124/dmd.104.002485. [DOI] [PubMed] [Google Scholar]
- Tilghman S. L., Bratton M. R., Segar H. C., Martin E. C., Rhodes L. V., Li M., McLachlan J. A., Wiese T. E., Nephew K. P., Burow M. E. Endocrine disruptor regulation of microRNA expression in breast carcinoma cells. PLoS One. 2012;7:e32754. doi: 10.1371/journal.pone.0032754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokairin T., Fukasawa T., Yasui-Furukori N., Aoshima T., Suzuki A., Inoue Y., Tateishi T., Otani K. Inhibition of the metabolism of brotizolam by erythromycin in humans: In vivo evidence for the involvement of CYP3A4 in brotizolam metabolism. Br. J. Clin. Pharmacol. 2005;60:172–175. doi: 10.1111/j.1365-2125.2005.02380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolle-Sander S., Rautio J., Wring S., Polli J. W., Polli J. E. Midazolam exhibits characteristics of a highly permeable P-glycoprotein substrate. Pharm. Res. 2003;20:757–764. doi: 10.1023/a:1023433502647. [DOI] [PubMed] [Google Scholar]
- Tomaszewski P., Kubiak-Tomaszewska G., Pachecka J. Cytochrome P450 polymorphism-molecular, metabolic, and pharmacogenetic aspects. II. Participation of CYP isoenzymes in the metabolism of endogenous substances and drugs. Acta Pol. Pharm. 2008;65:307–318. [PubMed] [Google Scholar]
- Topping D. L., Clifton P. M. Shortchain fatty acids and human colonic function: Roles of resistant starch and nonstarch polysaccharides. Physiol. Rev. 2001;81:1031–1064. doi: 10.1152/physrev.2001.81.3.1031. [DOI] [PubMed] [Google Scholar]
- Tritscher A. M. Human health risk assessment of processing-related compounds in food. Toxicol Lett. 2004;149:177–186. doi: 10.1016/j.toxlet.2003.12.059. [DOI] [PubMed] [Google Scholar]
- Turnbaugh P. J., Gordon J. I. The core gut microbiome, energy balance and obesity. J. Physiol. 2009;587:4153–4158. doi: 10.1113/jphysiol.2009.174136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh P. J., Ley R. E., Mahowald M. A., Magrini V., Mardis E. R., Gordon J. I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Ueyama J., Nadai M., Kanazawa H., Iwase M., Nakayama H., Hashimoto K., Yokoi T., Baba K., Takagi K., Takagi K., Hasegawa T. Endotoxin from various gram-negative bacteria has differential effects on function of hepatic cytochrome P450 and drug transporters. Eur. J. Pharmacol. 2005;510:127–134. doi: 10.1016/j.ejphar.2005.01.025. [DOI] [PubMed] [Google Scholar]
- U.S. Food and Drug Administration. Food additives permitted for direct addition to food for human consumption; sucralose. 21CFR Part 172 [Docket No. 87F-0086] Fed. Reg. 1998;63(64):16417–16433. http://www.gpo.gov/fdsys/pkg/FR-1998-04-03/pdf/98-8750.pdf (accessed February 6, 2013). [Google Scholar]
- U.S. Food and Drug Administration. Food additives permitted for direct addition to food for human consumption: Sucralose. 21CFR Part 172 [Docket No. 99F-0001] Fed. Reg. 1999;64(155):43908–43909. http://www.fda.gov/ohrms/dockets/98fr/081299b.pdf (accessed February 6, 2013). [Google Scholar]
- U.S. Food and Drug Administration. Guidance for industry and other stakeholders. Toxicological principles for the safety assessment of food ingredients. Redbook 2000. 2007. Revised July 2007 http://www.fda.gov/Food/GuidanceComplianceRegulatoryInformation/GuidanceDocuments/FoodIngredientsandPackaging/Redbook/default.htm (accessed February 6, 2013).
- U.S. Food and Drug Administration. Drug development and drug interactions: Table of substrates, inhibitors and inducers. 2011. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm (accessed February 6, 2013).
- U.S. Food and Drug Administration. Everything Added to Food in the United States (EAFUS) 2012. http://www.accessdata.fda.gov/scripts/fcn/fcnNavigation.cfm?filter=121746-18-7&sortColumn=36%28%25%3AH!52ED0!AP%2C%2C0M%2F5Q9Q%2CF%40++%0D%0A&rpt=eafusListing (accessed February 6, 2013).
- van Breemen R. B., Li Y. Caco-2 cell permeability assays to measure drug absorption. Expert Opin. Drug Metab. Toxicol. 2005;1:175–185. doi: 10.1517/17425255.1.2.175. [DOI] [PubMed] [Google Scholar]
- van de Poll M. E., Relling M. V., Schuetz E. G., Harrison P. L., Hughes W., Flynn P. M. The effect of atovaquone on etoposide pharmacokinetics in children with acute lymphoblastic leukemia. Cancer Chemother. Pharmacol. 2001;47:467–472. doi: 10.1007/s002800000250. [DOI] [PubMed] [Google Scholar]
- van Herwaarden A. E., van Waterschoot R. A., Schinkel A. H. How important is intestinal cytochrome P450 3A metabolism? Trends Pharmacol. Sci. 2009;30:223–227. doi: 10.1016/j.tips.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Vasiliu O., Cameron L., Gardiner J., Deguire P., Karmaus W. Polybrominated biphenyls, polychlorinated biphenyls, body weight, and incidence of adult-onset diabetes mellitus. Epidemiology. 2006;17:352–359. doi: 10.1097/01.ede.0000220553.84350.c5. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan K., von Moltke L. L., Duan S. X., Fleishaker J. C., Shader R. I., Greenblatt D. J. Kinetic characterization and identification of the enzymes responsible for the hepatic biotransformation of adinazolam and N-desmethyladinazolam in man. J. Pharm. Pharmacol. 1998;50:265–274. doi: 10.1111/j.2042-7158.1998.tb06859.x. [DOI] [PubMed] [Google Scholar]
- Villena J., Racedo S., Agüero G., Bru E., Medina M., Alvarez S. Lactobacillus casei improves resistance to pneumococcal respiratory infection in malnourished mice. J. Nutr. 2005;135:1462–1469. doi: 10.1093/jn/135.6.1462. [DOI] [PubMed] [Google Scholar]
- Villeneuve D. C., Van Logten M. J., Den Tonkelaar E. M., Greve P. A., Vos J. G., Speijers G. J. A., Van Esch G. J. Effect of food deprivation on low level hexachlorobenzene exposure in rats. Sci. Total Environ. 1977;8:179–186. doi: 10.1016/0048-9697(77)90076-6. [DOI] [PubMed] [Google Scholar]
- von Moltke L. L., Greenblatt D. J., Harmatz J. S., Duan S. X., Harrel L. M., Cotreau-Bibbo M. M., Pritchard G. A., Wright C. E., Shader R. I. Triazolam biotransformation by human liver microsomes in vitro: Effects of metabolic inhibitors and clinical confirmation of a predicted interaction with ketoconazole. J. Pharmacol. Exp. Ther. 1996;276:370–379. [PubMed] [Google Scholar]
- von Richter O., Greiner B., Fromm M. F., Fraser R., Omari T., Barclay M. L., Dent J, Somogyi A. A., Eichelbaum M. Determination of in vivo absorption, metabolism and transport of drugs by the human intestinal wall and liver with a novel perfusion technique. Clin. Pharmacol. Ther. 2001;70:217–227. doi: 10.1067/mcp.2001.117937. [DOI] [PubMed] [Google Scholar]
- Voulgaridou G. P., Anestopoulos I., Franco R., Panayiotidis M. I., Pappa A. DNA damage induced by endogenous aldehydes: Current state of knowledge. Mutat. Res. 2011;711:13–27. doi: 10.1016/j.mrfmmm.2011.03.006. [DOI] [PubMed] [Google Scholar]
- Vucetic Z., Kimmel J., Totoki K., Hollenbeck E., Reyes T. M. Maternal high-fat diet alters methylation and gene expression of dopamine and opioid-related genes. Endocrinology. 2010;151:4756–4764. doi: 10.1210/en.2010-0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waade R. B., Christensen H., Rudberg I., Refsum H., Hermann M. Influence of comedication on serum concentrations of aripiprazole and dehydroaripiprazole. Ther. Drug Monit. 2009;31:233–238. doi: 10.1097/FTD.0b013e3181956726. [DOI] [PubMed] [Google Scholar]
- Wacher V. J., Salphati L., Benet L. Z. Active secretion and enterocytic drug metabolism barriers to drug absorption. Adv. Drug Deliv. Rev. 2001;46:89–102. doi: 10.1016/s0169-409x(00)00126-5. [DOI] [PubMed] [Google Scholar]
- Wacher V. J., Wu C. Y., Benet L. Z. Overlapping substrate specificities and tissue distribution of cytochrome P450 3A and P-glycoprotein: Implications for drug delivery and activity in cancer chemotherapy. Mol. Carcinogen. 1995;13:129–134. doi: 10.1002/mc.2940130302. [DOI] [PubMed] [Google Scholar]
- Wagstaff A. J., Keating G. M. Anagrelide: A review of its use in the management of essential thrombocythaemia. Drugs. 2006;66:111–131. doi: 10.2165/00003495-200666010-00006. [DOI] [PubMed] [Google Scholar]
- Walsky R. L., Gaman E. A., Obach R. S. Examination of 209 drugs for inhibition of cytochrome P450 2C8. J. Clin. Pharmacol. 2005a;45:68–78. doi: 10.1177/0091270004270642. [DOI] [PubMed] [Google Scholar]
- Walsky R. L., Obach R. S., Gaman E. A., Gleeson J. P., Proctor W. R. Selective inhibition of human cytochrome P4502C8 by montelukast. Drug Metab. Dispos. 2005b;33:413–418. doi: 10.1124/dmd.104.002766. [DOI] [PubMed] [Google Scholar]
- Wandel C., Kim R., Wood M., Wood A. Interaction of morphine, fentanyl, sufentanil, alfentanil, and loperamide with the efflux drug transporter P-glycoprotein. Anesthesiology. 2002;96:913–920. doi: 10.1097/00000542-200204000-00019. [DOI] [PubMed] [Google Scholar]
- Wang E. J., Casciano C. N., Clement R. P., Johnson W. W. Active transport of fluorescent P-glycoprotein substrates: Evaluation as markers and interaction with inhibitors. Biochem. Biophys. Res. Commun. 2001;289:580–585. doi: 10.1006/bbrc.2001.6000. [DOI] [PubMed] [Google Scholar]
- Wang E. J., Lew K., Casciano C. N., Clement R. P., Johnson W. W. Interaction of common azole antifungals with P glycoprotein. Antimicrob. Agents Chemother. 2002;46:160–165. doi: 10.1128/AAC.46.1.160-165.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. S., Zhu H. J., Gibson B. B., Markowitz J. S., Donovan J. L., DeVane C. L. Sertraline and its metabolite desmethylsertraline, but not bupropion or its three major metabolites, have high affinity for P-glycoprotein. Biol. Pharm. Bull. 2008;31:231–234. doi: 10.1248/bpb.31.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. Y., Li S. N., Liu C. S., Perng D. S., Su Y. C., Wu D. C., Jan C. M., Lai C. H., Wang T. N., Wang W. M. Effects of ingesting Lactobacillus- and Bifidobacterium-containing yogurt in subjects with colonized Helicobacter pylori. Am. J. Clin. Nutr. 2004;80:737–741. doi: 10.1093/ajcn/80.3.737. [DOI] [PubMed] [Google Scholar]
- Ward B. A., Morocho A., Kandil A., Galinsky R. E., Flockhart D. A., Desta Z. Characterization of human cytochrome P450 enzymes catalyzing domperidone N-dealkylation and hydroxylation in vitro. Br. J. Clin. Pharmacol. 2004;58:277–287. doi: 10.1111/j.1365-2125.2004.02156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner M., Gustafsson J. A. Effect of ethanol on cytochrome P450 in the rat brain. Proc. Natl. Acad. Sci. USA. 1994;91:1019–1023. doi: 10.1073/pnas.91.3.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warwick Z. S., Schiffman S. S. Flavor-calorie relationships: Effect on weight gain in rats. Physiol. Behav. 1991;50:465–470. doi: 10.1016/0031-9384(91)90531-r. [DOI] [PubMed] [Google Scholar]
- Watkins P. B. Drug metabolism by cytochromes P450 in the liver and small bowel. Gastroenterol. Clin. North Am. 1992;21:511–526. [PubMed] [Google Scholar]
- Watkins P. B. The barrier function of CYP3A4 and P-glycoprotein in the small bowel. Adv. Drug Deliv. Rev. 1997;27:161–170. doi: 10.1016/s0169-409x(97)00041-0. [DOI] [PubMed] [Google Scholar]
- Watkins P. B., Wrighton S. A., Schuetz E. G., Molowa D. T., Guzelian P. S. Identification of glucocorticoid-inducible cytochromes P-450 in the intestinal mucosa of rats and man. J. Clin. Invest. 1987;80:1029–1036. doi: 10.1172/JCI113156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver R. J., Webster D., Melvin W. T., Dickins M., Burke M. D. Human cytochrome P4502C metabolism. Br. J. Clin. Pharmacol. 1993;36:167P. [Google Scholar]
- Weisbrod A. V., Shea D., Moore M. J., Stegeman J. J. Species, tissue and gender-related organochlorine bioaccumulation in white-sided dolphins, pilot whales and their common prey in the northwest Atlantic. Mar. Environ. Res. 2001;51:29–50. doi: 10.1016/s0141-1136(00)00032-5. [DOI] [PubMed] [Google Scholar]
- Weiss J., Dormann S. M., Martin-Facklam M., Kerpen C. J., Ketabi-Kiyanvash N., Haefeli W. E. Inhibition of P-glycoprotein by newer antidepressants. J. Pharmacol. Exp. Ther. 2003;305:197–204. doi: 10.1124/jpet.102.046532. [DOI] [PubMed] [Google Scholar]
- Wen B., Ma L., Rodrigues A. D., Zhu M. Detection of novel reactive metabolites of trazodone: Evidence for CYP2D6-mediated bioactivation of m-chlorophenylpiperazine. Drug Metab. Dispos. 2008;36:841–850. doi: 10.1124/dmd.107.019471. [DOI] [PubMed] [Google Scholar]
- Wengreen H. J., Moncur C. Change in diet, physical activity, and body weight among young-adults during the transition from high school to college. Nutr. J. 2009;8:32. doi: 10.1186/1475-2891-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenner M. A new kind of drug target. Sci. Am. 2009;301:70–74. 76. doi: 10.1038/scientificamerican0809-70. [DOI] [PubMed] [Google Scholar]
- Westphal K., Weinbrenner A., Zschiesche M., Franke G., Knoke M., Oertel R., Fritz P., von Richter O., Warzok R., Hachenberg T., Kauffmann H. M., Schrenk D., Terhaag B., Kroemer H. K., Siegmund W. Induction of P-glycoprotein by rifampin increases intestinal secretion of talinolol in human beings: A new type of drug/drug interaction. Clin. Pharmacol. Ther. 2000;68:345–355. doi: 10.1067/mcp.2000.109797. [DOI] [PubMed] [Google Scholar]
- Wiklund A. K., Breitholtz M., Bengtsson B. E., Adolfsson-Erici M. Sucralose—An ecotoxicological challenger? Chemosphere. 2012;86:50–55. doi: 10.1016/j.chemosphere.2011.08.049. [DOI] [PubMed] [Google Scholar]
- Wood S. G., John B. A., Hawkins D. R. The pharmacokinetics and metabolism of sucralose in the dog. Food Chem. Toxicol. 2000;38(suppl. 2):S99–S106. doi: 10.1016/s0278-6915(00)00031-4. [DOI] [PubMed] [Google Scholar]
- Woodmansey E. J. Intestinal bacteria and ageing. J. Appl. Microbiol. 2007;102:1178–1186. doi: 10.1111/j.1365-2672.2007.03400.x. [DOI] [PubMed] [Google Scholar]
- Wootten D., Savage E. E., Valant C., May L. T., Sloop K. W., Ficorilli J., Showalter A. D., Willard F. S., Christopoulos A., Sexton P. M. Allosteric modulation of endogenous metabolites as an avenue for drug discovery. Mol. Pharmacol. 2012;82:281–290. doi: 10.1124/mol.112.079319. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Toxicological evaluation of certain food additives and contaminants. WHO Food Additives Series, no. 24. Cambridge University Press; 1989. Trichlorogalactosucrose. no. 655. http://www.inchem.org/documents/jecfa/jecmono/v024je05.htm (accessed February 6, 2013). [Google Scholar]
- World Health Organization. Thirty-seventh report of the Joint FAO/WHO Expert Committee on Food Additives. WHO Technical Report Series 806. Geneva, Switzerland: World Health Organization; 1991. Evaluation of certain food additives and contaminants, 21–23. whqlibdoc.who.int/trs/WHO_TRS_806.pdf (accessed February 6, 2013). [PubMed] [Google Scholar]
- World Health Organization. 3-Chloro-1,2-propanediol. Safety evaluation of certain food additives and contaminants. 2002. WHO Food Additives Series 48. http://www.inchem.org/documents/jecfa/jecmono/v48je18.htm (accessed February 6, 2013).
- World Health Organization. Adherence to long-term therapies: Evidence for action. Geneva (Switzerland): World Health Organisation; 2003. http://www.who.int/chp/knowledge/publications/adherence_full_report.pdf (accessed February 6, 2013). [Google Scholar]
- World Health Organization. Safety evaluation of certain food additives and contaminants. 2012. pp. 291–298. WHO Food Additives Series 67. http://www.who.int,9789241660679_eng.pdf (accessed February 6, 2013).
- World Health Organization. Obesity and overweight. 2013. Fact sheet number 311, Dupdated March 2013. http://www.who.int/mediacentre/factsheets/fs311/en.
- Wu J., Dong S., Liu G., Zhang B., Zheng M. Cooking process: A new source of unintentionally produced dioxins? J. Agric. Food Chem. 2011;59:5444–5449. doi: 10.1021/jf200216r. [DOI] [PubMed] [Google Scholar]
- Xie R., Tan L. H., Polasek E. C., Hong C, Teillol-Foo M., Gordi T., Sharma A., Nickens D. J., Arakawa T., Knuth D. W, Antal E. J. CYP3A and P-glycoprotein activity induction with St. John's Wort in healthy volunteers from 6 ethnic populations. J. Clin. Pharmacol. 2005;45:352–356. doi: 10.1177/0091270004273320. [DOI] [PubMed] [Google Scholar]
- Yanagiba Y, Ito Y, Kamijima M., Gonzalez F J., Nakajima T. Octachlorostyrene induces cytochrome P450, UDP-glucuronosyltransferase, and sulfotransferase via the aryl hydrocarbon receptor and constitutive androstane receptor. Toxicol. Sci. 2009;111:19–26. doi: 10.1093/toxsci/kfp130. [DOI] [PubMed] [Google Scholar]
- Yang Q. Gain weight by “going diet?” Artificial sweeteners and the neurobiology of sugar cravings: Neuroscience 2010. Yale J. Biol. Med. 2010;83:101–108. [PMC free article] [PubMed] [Google Scholar]
- Yang T. H., Tian L. Y, Shang H. F, Cheng X. W, Geng J., Chen L, Zhou D. Suppression of the multidrug transporter P-glycoprotein using RNA interference in cultured rat astrocytes induced by coriaria lactone. Neurol. Res. 2009;31:1084–1091. doi: 10.1179/174313208X319134. [DOI] [PubMed] [Google Scholar]
- Yasuda K., Ranade A., Venkataramanan R., Strom S., Chupka J., Ekins S., Schuetz E., Bachmann K. A comprehensive in vitro and in silico analysis of antibiotics that activate pregnane X receptor and induce CYP3A4 in liver and intestine. Drug Metab. Dispos. 2008;36:1689–1697. doi: 10.1124/dmd.108.020701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda S. U., Zannikos P., Young A. E., Fried K. M., Wainer I. W, Woosley R. L. The roles of CYP2D6 and stereose-lectivity in the clinical pharmacokinetics of chlorpheniramine. Br. J. Clin. Pharmacol. 2002;53:519–525. doi: 10.1046/j.1365-2125.2002.01578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui N., Otani K., Kaneko S., Ohkubo T., Osanai T., Sugawara K., Chiba K., Ishizaki T. A kinetic and dynamic study of oral alprazolam with and without erythromycin in humans: In vivo evidence for the involvement of CYP3A4 in alprazolam metabolism. Clin. Pharmacol. Ther. 1996;59:514–519. doi: 10.1016/S0009-9236(96)90179-4. [DOI] [PubMed] [Google Scholar]
- Yoshii K., Kobayashi K., Tsumuji M., Tani M., Shimada N., Chiba K. Identification of human cytochrome P450 isoforms involved in the 7-hydroxylation of chlorpromazine by human liver microsomes. Life Sci. 2000;67:175–184. doi: 10.1016/s0024-3205(00)00613-5. [DOI] [PubMed] [Google Scholar]
- Young D. A., Bowen W. H. The influence of sucralose on bacterial metabolism. J. Dent. Res. 1990;69:1480–1484. doi: 10.1177/00220345900690080601. [DOI] [PubMed] [Google Scholar]
- Yue J., Miksys S., Hoffmann E., Tyndale R. F. Chronic nicotine treatment induces rat CYP2D in the brain but not in the liver: An investigation of induction and time course. J. Psychiat. Neurosci. 2008;33:54–63. [PMC free article] [PubMed] [Google Scholar]
- Zama A. M., Uzumcu M. Fetal and neonatal exposure to the endocrine disruptor methoxychlor causes epigenetic alterations in adult ovarian genes. Endocrinology. 2009;150:4681–4691. doi: 10.1210/en.2009-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zama A. M., Uzumcu M. Epigenetic effects of endocrine-disrupting chemicals on female reproduction: An ovarian perspective. Front. Neuroendocrinol. 2010;31:420–439. doi: 10.1016/j.yfrne.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Klebansky B., Fine R. M., Liu H., Xu H., Servant G., Zoller M., Tachdjian C., Li X. Molecular mechanism of the sweet taste enhancers. Proc. Natl. Acad. Sci. USA. 2010;107:4752–4757. doi: 10.1073/pnas.0911660107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q. Y., Dunbar D., Ostrowska A., Zeisloft S., Yang J., Kaminsky L. S. Characterization of human small intestinal cytochromes P-450. Drug Metab. Dispos. 1999;27:804–809. [PubMed] [Google Scholar]
- Zhang W., Ramamoorthy Y., Kilicarslan T., Nolte H., Tyndale R. F., Sellers E. M. Inhibition of cytochromes P450 by antifungal imidazole derivatives. Drug Metab. Dispos. 2002;30:314–318. doi: 10.1124/dmd.30.3.314. [DOI] [PubMed] [Google Scholar]
- Zhang Y. F., Chen X. Y., Guo Y. J., Si D. Y., Zhou H., Zhong D. F. Impact of cytochrome P450 CYP2C9 variant allele CYP2C9 ∗ 3 on the pharmacokinetics of glibenclamide and lornoxicam in Chinese subjects. Yao Xue Xue Bao. 2005;40:796–799. [PubMed] [Google Scholar]
- Zhou S. F., Lai X. An update on clinical drug interactions with the herbal antidepressant St. John's wort. Curr. Drug Metab. 2008;9:394–409. doi: 10.2174/138920008784746391. [DOI] [PubMed] [Google Scholar]
- Zhou S. F., Zhou Z. W., Yang L. P., Cai J. P. Substrates, inducers, inhibitors and structure-activity relationships of human Cytochrome P450 2C9 and implications in drug development. Curr. Med. Chem. 2009;16:3480–3675. doi: 10.2174/092986709789057635. [DOI] [PubMed] [Google Scholar]
- Zhu M., Kaul S., Nandy P., Grasela D. M., Pfister M. Model-based approach to characterize efavirenz autoinduction and concurrent enzyme induction with carbamazepine. Antimicrob. Agents Chemother. 2009;53:2346–2353. doi: 10.1128/AAC.01120-08. [DOI] [PMC free article] [PubMed] [Google Scholar]