

Abstract
We report here a method that utilizes photoactivatable drug-caged fluorophore conjugate to quantify intracellular drug trafficking processes at single cell resolution. Photoactivation is performed in labeled cellular compartments to visualize intracellular drug exchange at physiologic conditions, without the need for washing, facilitating its translation to in vivo cancer models.
Introduction
Multidrug resistance of cancer cells, existing at the time of diagnosis or acquired during chemotherapy exposure, is a major contributor of failed therapeutic response in the treatment of cancer.1 Resistance of tumor cells to chemotherapeutic agents such as taxanes, platinum (Pt) compounds and others has been associated with overexpression of membrane transporter proteins, drug redistribution within the cell and pathway alterations.2, 3 Particularly, drug sequestration in organelles has been shown to reduce drug efficacy by limiting access to the targeted cell compartment, often the nucleus. A method to directly visualize intracellular drug trafficking would thus be tremendously helpful in deciphering serial kinetics and thus elucidating mechanism of drug resistance.
To date, most transport assays employ fluorescent small molecules, such as rhodamine 123 and 3,3′-Diethyloxacarbocyanine Iodide (DiOC2(3)), as substrates to determine drug transport across cellular membranes.4 In these approaches, cells are first loaded with fluorescent substrate by incubation with substrate-containing media. Drug exchange is then quantified by measuring cellular substrate retention as a function of time after substrate washout (i.e. after replacing the substrate-containing media with a substrate-free media). However, because measurement contrast require substrate washout, a steep substrate concentration gradient between the intra- and extra-cellular spaces dominates the kinetic measurement. Consequently, washout assays do not accurately quantify drug exchange across cellular membranes at physiologic conditions and in vivo translation is limited by obvious practicalities. A further concern is that these fluorchromes are just surrogates and do not structurally resemble actual therapeutic drugs.
In this article, we developed a strategy to photoactivate caged drug-fluorophore conjugates inside single cells to thus study single cell drug transport at steady state conditions and without the need for washes. As summarized in the Scheme 1, cells are first incubated with non-fluorescent drug-caged fluorophore conjugate to allow substrate accumulation inside the cell. At steady state conditions, the drug-caged fluorophore conjugate is converted to its fluorescent conjugate within a fluorescently labeled cellular compartment using 405 nm laser light from a confocal microscope. Subsequent serial time-lapse imaging of the activated drug provides information about its intracellular transport.
Scheme 1.
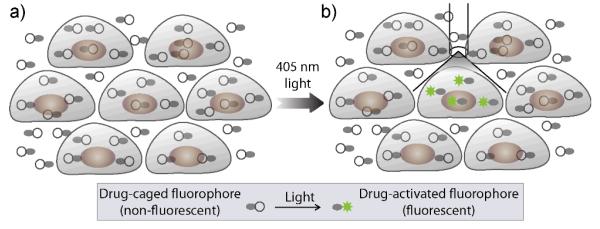
Strategy for direct quantification of single cell drug transport using photoactivatable drug-caged fluorophore conjugate. (a) Steady state accumulation of drug-fluorophore conjugate. (b) Photoactivate drug-caged fluorophore conjugate in single cell or labeled, intracellular compartment with 405 nm laser for monitor drug transport using fluorescence microscopy.
Our group has recently described a number of BODIPY labeled targeted anticancer drugs,5 however without photoactivation capabilities. As a model systems to test the photoactivation strategy we chose a prototype poly(ADP-ribose)polymerase inhibitor (PARPi) based on the Olaparib (AZD-2281). BODIPY (boron-dipyrromethene) was selected as an ideal fluorophore conjugate because it is cell permeable and stable over a wide range of pH, and consequently, within various intracellular compartments. The PARPi-BODIPY is a useful model systems in itself and has previously been validated for in vivo imaging.5
We first synthesized a photocaged version of BODIPY fluorophore (see Figure 1a), where a 2,6-dinitrobenzyl (DNB) group is attached to the 8-phenyl-1,3,5,7-tetramethyl BODIPY skeleton to render the BODIPY non-fluorescent.6 Appending the DNB caging group through an ether bond also provides a biologically stable linkage between the fluorophore and the caging group.7,8 Figure 1a shows the synthesis of PARPi-BODIPYc conjugate through amide coupling between carboxyl-functionalized caged BODIPY and piperazine functionality of PARPi. Irradiation of long wavelength UV irradiation (~350-410 nm) to this PARPi-BODIPYc conjugate removes the DNP caging group through a photolytic cleavage, generating the fluorescent derivative of PARPi (namely PARPi-BODIPYa, see Figure 1a). The identity of the photocleaved products was characterized by high-performance liquid chromatography-mass spectrometry (HPLC-MS) analysis. As shown in Figure 1b, formation of PARPi-BODIPYa upon light irradiation was verified by the appearance of a second trace in the HPLC chromatogram corresponding to the molecular mass of PARPi-BODIPYa. Next, we determined the fluorescence property of PARPi-BODIPYc conjugate, both before and after light activation, using a fluorescence microplate reader. Prior photoactivation, PARPi-BODIPYc conjugate showed undetectable fluorescence, indicating efficient fluorescent quenching by DNB caging group. However, irradiation of ~365 nm light from a hand-held UV lamp (energy density 1.6 mW/cm2, photon energy 3.4 eV, number of photons per second per cm2 2×1015) resulted in an immediate recovery in fluorescence. Figure 2a shows the fluorescence enhancement after different duration of light illumination. Drastic change in fluorescence can also be observed by naked eye (inset Figure 2a). Fluorescence change during the time course of photochemical reaction plotted in Figure 2b showed the completion of the fluorescence activation within ~45 min of light irradiation. Overall these characterizations indicate that the PARPi-BODIPYc conjugate provides a non-fluorescent substrate that can be efficiently converted to a fluorescent substrate by light irradiation. Additionally, the fluorescent derivative of PARPi (PARPi-BODIPYa) showed minimal loss of fluorescence with repeated signal acquisition (Supporting figure 3). The photobleaching effect is thus minimal and simplifies measurement of efflux rates (vide infra).
Figure 1.

a) Synthesis of PARPi-BODIPYc conjugate and activation using 365 nm UV light. b) HPLC-MS chromatograms showing the quantitative formation of PARPi-BODIPYa conjugate from PARPi-BODIPYc upon UV irradiation.
Figure 2.

a) Fluorescence recovery during the uncaging of PARPi-BODIPYc. Uncaging was performed by irradiating 1 μM solution of PARPi-BODIPYc with ~365 nm UV light (from a hand held UV lamp). Fluorescence spectra were collected after exciting the fluorophore conjugate at 485 nm. b) Photocleavage kinetics showing the change in fluorescence at 519 nm over UV irradiation time.
We next determined the inhibitory potential of the PARPi derivatives using an in vitro PARP1 activity assay.9 The half maximal inhibitory concentration (IC50) values of different PARPi derivatives along with the parent drug are presented in Figure 3. We observed higher IC50 values for the fluorophore conjugates as compared to the parent drug, presumably due to steric reasons. However, the IC50 values are still in the low nanomolar range for the derivatives, indicating good potency of the derivatives towards PARP enzyme binding.10 To further verify the therapeutic efficacy of the derivatives, we performed cell viability assay. Toxicity profiles of the derivatives, assessed in MDA-MB-436 (a BRCA1-mutant breast cancer cell) cell culture studies, showed that the derivatives were slightly more toxic to the cells than the parent drug (Supporting figure 2). The observed higher cytotoxicity for the BODIPY-labeled drugs may result from amplified cellular retention of the labeled compounds due to increased hydrophobicity.11 Lysosomal or microsomal metabolism of the derivatives can also contribute to toxicity, as the cell toxicity assay was performed over several days.12 Nevertheless, these results indicate that fluorophore conjugation appears to have minimal adverse effect on the target binding capability of PARPi with the resultant derivatives still maintaining therapeutic efficacy in cells.
Figure 3.

IC50 determination by inhibition of PARP histone with unlabeled and labeled variants of PARPi (● PARPi. ■PARPi-Bodipy Fl, ◆PARPi-BODIPYc, ○ PARPi-BODIPYa). Chemical structures of the unlabeled and labeled variants of PARPi, used in this assay, are presented in the Supporting figure 1.
Finally, we demonstrate photo-activation and time-lapse monitoring of single cell drug loss in live cells at steady state conditions. In this experiment, a tumor cell line expressing fluorescent histone H2B-apple (HT1080) was incubated with a 1μM concentration of PARPi-BODIPYc conjugate for 1h to allow substrate accumulation inside the cells. Subsequently, bound and sequestered drug was photoactivated within single cells without any prior washout steps. Photoactivation and imaging were performed at 37° in the presence of phenol red-free culture medium using a 20× water immersion objective and a Confocal Laser Scanning Microscope (CLSM; Olympus FV1000). As shown in Figure 4a, cells imaged before photoactivation showed no appreciable substrate signal and fluorescence was observed only from the nuclear expression of the H2B-apple fluorescent protein (pseudo colored red). Drug conjugate within cells outlined by the visual window (dotted square in Figure 4a) were then activated with a 405 nm laser in less than one minute. Fluorescence images acquired after photoactivation showed bright BODIPY fluorescence (pseudo colored green) at single cell resolution, validating fluorescence labeling of the PARPi-BODIPYc and showing a high signal-to-background ratio (Figure 4a). Subsequently, fluorescent drug loss from the cells was quantified by time-lapse microscopy (see Figure 4a). Drug efflux kinetics, measured from single cells, is quantified in Figure 4b. Additionally, population efflux rates were compared for sampling rates varying from (0.02-0.8Hz) to further validate the negligible effect of photobleaching in vivo (Supporting figure 4). Overall, we demonstrate that PARPi-BODIPYc conjugate provides a cell permeable substrate that may be used to quantify cellular drug exchange at steady state condition based on its photoactivatable fluorescence property. We also investigated the target binding ability of the PARPi conjugate after fluorescent activation. As shown in Figure 5, observed nuclear localization of the BODIPY fluorescence after substrate washout indicated successful target binding.
Figure 4.

a) Pre- and post-activation time lapse images of HT1080 cell line expressing H2B-apple after 1h incubation to PARPi-BODIPYc. b) Quantification of single cell drug efflux.
Figure 5.
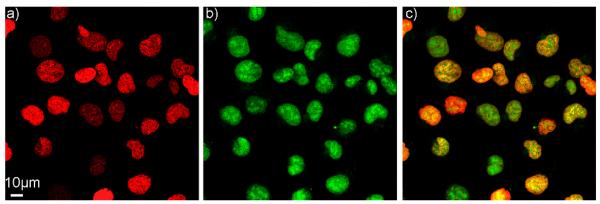
PARPi-BODIPYa localizes in nucleus 3h after drug washout, (a) Histone H2B-Apple (pseudo colored red), (b) PARPi-BODIPYa (pseudo colored green), (c) merge image.
Conclusions
In conclusion, we have developed a method that utilizes a photoactivatable drug-caged fluorophore conjugate to directly quantify localized drug trafficking. The primary advantage of this method is that drug exchange is measured at physiologic conditions (washing steps not needed for contrast) and consequently, may be translated in vivo. A PARPi conjugated caged BODIPY probe was developed as a proof of concept, but this synthesis strategy may readily extend to other therapeutics for direct visualization of drug transport from specific, cellular compartments. We envision this method may be used to improve quantification of drug sequestration and trafficking during perfusion with in vivo cancer models.
Supplementary Material
Acknowledgement
We thank Dr. Katherine Yang for providing HT1080 cell lines and Jenna Klubnick for providing PARPi derivative. We thank Dr. Eunha Kim, Dr. Jonathan Carlson and other colleagues in our laboratories for helpful advice and discussion. This research was supported in part by NIH grants P50CA086355, PO1-CA139980, R01CA164448 and RO1EB010011.
Notes and references
- 1.Gottesman MM, Fojo T, Bates SE. Nat. Rev. Cancer. 2002;2:48. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 2.Gottesman MM, Pastan I, Ambudkar SV. Curr. Opin. Genet. Dev. 1996;6:610. doi: 10.1016/s0959-437x(96)80091-8. [DOI] [PubMed] [Google Scholar]
- 3.(a) Chapuy B, Koch R, Radunski U, Corsham S, Cheong N, Inagaki N, Ban N, Wenzel D, Reinhardt D, Zapf A, Schweyer S, Kosari F, Klapper W, Truemper L, Wulf GG. Leukemia. 2008;22:1576. doi: 10.1038/leu.2008.103. [DOI] [PubMed] [Google Scholar]; (b) Goler-Baron V, Assaraf YG. PLoS One. 2011;6:e16007. doi: 10.1371/journal.pone.0016007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Forster S, Thumser AE, Hood SR, Plant N. PLoS One. 2012;7:e33253. doi: 10.1371/journal.pone.0033253. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Minderman H, Vanhoefer U, Toth K, Yin MB, Minderman MD, Wrzosek C, Slovak ML, Rustum YM. Cytometry. 1996;25:14. doi: 10.1002/(SICI)1097-0320(19960901)25:1<14::AID-CYTO2>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 5.(a) Keliher EJ, Reiner T, Earley S, Klubnick J, Tassa C, Lee AJ, Ramaswamy S, Bardeesy N, Hanahan D, Depinho RA, Castro CM, Weissleder R. Neoplasia. 2013;15:684. doi: 10.1593/neo.13276. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Reiner T, Lacy J, Keliher EJ, Yang KS, Ullal A, Kohler RH, Vinegoni C, Weissleder R. Neoplasia. 2012;14:169. doi: 10.1593/neo.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kobayashi T, Komatsu T, Kamiya M, Campos C, Gonzalez-Gaitan M, Terai T, Hanaoka K, Nagano T, Urano Y. J. Am. Chem. Soc. 2012;134:11153. doi: 10.1021/ja212125w. [DOI] [PubMed] [Google Scholar]
- 7.Lu X, Agasti SS, Vinegoni C, Waterman P, DePinho RA, Weissleder R. Bioconjugate Chem. 2012;23:1945. doi: 10.1021/bc300319c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Agasti SS, Kohler RH, Liong M, Peterson VM, Lee H, Weissleder R. Small. 2013;9:222. doi: 10.1002/smll.201201007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhao Y, Zheng Q, Dakin K, Xu K, Martinez ML, Li WH. J. Am. Chem. Soc. 2004;126:4653. doi: 10.1021/ja036958m. [DOI] [PubMed] [Google Scholar]
- 9.Reiner T, Earley S, Turetsky A, Weissleder R. ChemBioChem. 2010;11:2374. doi: 10.1002/cbic.201000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thurber GM, Yang KS, Reiner T, Kohler RH, Sorger P, Mitchison T, Weissleder R. Nat. Commun. 2013;4:1504. doi: 10.1038/ncomms2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Han F, Xu Y, Jiang D, Qin Y, Chen H. Anal. Biochem. 2013;435:106. doi: 10.1016/j.ab.2013.01.003. [DOI] [PubMed] [Google Scholar]; (b) Alford R, Simpson HM, Duberman J, Hill GC, Ogawa M, Regino C, Kobayashi H, Choyke PL. Molecular Imaging. 2009;8:341. [PubMed] [Google Scholar]
- 12.Lelong IH, Guzikowski AP, Haugland RP, Pastan I, Gottesman MM, Willingham MC. Mol. Pharmacol. 1991;40:490. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.