

Abstract
The B subunit of cholera toxin (CTB) has been used as adjuvant to improve oral vaccine delivery in type 1 diabetes. The effect of CTB/peptide formulations on antigen-specific CD4 T cells has remained largely unexplored. We investigated by tetramer analysis how oral delivery of CTB fused to 2 CD4 T cell epitopes, the BDC-2.5 T cell 2.5mi mimotope and glutamic acid decarboxylase (GAD) 286–300, affected diabetogenic CD4 T cells in NOD mice. CTB-2.5mi activated 2.5mi+ T cells when administered intraperitoneally and generated Ag-specific Foxp3+ Treg and Th2 cells following intragastric delivery. While 2.5mi+ and GAD-specific T cells were tolerized in diabetes resistant NODxB6.Foxp3EGFP F1 and NOR mice, this did not occur in NOD mice. This indicated NOD mice had a recessive genetic resistance to induce oral tolerance to both CTB-fused epitopes. Contrarily to NODxB6.Foxp3EGFP F1 mice, oral treatment in NOD mice lead to strong 2.5mi+ T cell activation and the sequestration of these cells to the effector-memory pool. Oral treatment of NOD mice with CTB-2.5mi failed to prevent diabetes. These findings underline the importance of investigating the effect of oral vaccine formulations on diabetogenic T cells as in selected cases they may have counterproductive consequences in human patients.
Keywords: Type 1 diabetes, oral vaccine, MHC tetramer
INTRODUCTION
The induction of peripheral tolerance is the principal strategy of clinical and preclinical trials in order to battle against autoimmune diseases. Amongst many approaches, oral tolerance induction stands out as the method of choice as it is the least invasive method and therefore has a high potential to find wide acceptance in patients. In the past, a series of adjuvants have been used in order to increase oral vaccine delivery with the goal to minimize dosage of antigen. One of them, the subunit B of cholera toxin (CTB), has been shown to efficiently support the transmucosal transport of covalently linked protein by binding to its receptor, the monosialoganglioside GM1 [1].
Several approaches have been used to apply CTB for the design of oral vaccines to prevent type 1 diabetes (T1D). T1D is a result of the selective and progressive destruction of pancreatic, insulin-producing beta cells by autoreactive T cells [2]. The non-obese diabetic (NOD) mouse is a non-induced animal model that develops T1D with pathology similar to the human disease [3]. In this model, prevention of T1D by oral administration of CTB fusion proteins, conjugates, unconjugated adjuvant or included in recombinant vaccinia viruses using different autoantigens such as insulin and glutamic acid decarboxylase has been reported [4–8]. CTB as fusion protein or for conjugation has been generated in several systems including bacteria [9], silk worms [10] or plants [11]. In the vast majority of reports, no detailed T cell analyses were conducted, if at all. Disease prevention by transfer studies into lymphocyte deficient NOD.scid mice was traced to the generation of either regulatory T cells, a shift of a Th1 to a Th2 response, or both [4, 5]. However, in most documented attempts, prevention of T1D by orally administered CTB-fusion proteins has remained only partial. So far, no clinical trial in humans using any sort of oral vaccine has shown to provide solid protection against the disease [12]. Although CTB has been found to be a safe adjuvant in humans [13], in T1D prevention trials this adjuvant has not yet been used, most likely because detailed reports on the fate of Ag-specifc T cells following oral treatment with CTB-conjugate vaccines are still at large in relevant animal models.
In order to trace antigen-specific autoreactive T cells in the NOD mouse, we have previously generated Ag7-based MHC class II tetramers using a strong agonist peptide mimotope, 2.5mi, that reliably stain the diabetogenic T cell clone BDC-2.5 [14] as well as a natural T cell population in NOD mice, termed by us 2.5mi+ T cells [15, 16]. This T cell population is generated early in life, expands during T1D pathogenesis and infiltrates pancreatic islets. We previously explored to which degree manipulation of these cells by a DNA vaccination approach designed to target the 2.5 mimotope to the MCH class II loading compartment might induce general tolerance in NOD mice. Diabetes onset was delayed and partially reduced, demonstrating that appropriate delivery of the 2.5 mimotope can induce T1D protection in this model [17].
In light of these studies and in order to provide further mechanistic insights into oral tolerance induction in the NOD mouse model, we wondered whether a different formulation and application route of the 2.5 mimotope might improve tolerance. In the present report, we fused the mimotope to CTB and applied it to prediabetic mice intragastrically. Treatment led to the generation of Ag-specific Foxp3+ Treg and the secretion of the Th2 cytokine IL-4. Unexpectedly however, in vivo parenteral challenge with the mimotope after oral treatment revealed that while 2.5mi+ T cells were tolerized in diabetes resistant NODxB6 F1 and NOR mice, this did not occur in the disease susceptible NOD mouse strain. These findings were not limited to the mimotope but also extended to the glutamic acid decarboxylase (GAD) derived peptide 286–300. Indeed, contrarily to the tolerogenic effect observed in diabetes resistant control strains, oral treatment in NOD mice leads to strong T cell activation and the sequestration of 2.5mi+ T cells to the effector-memory T cell pool. As a consequence, diabetes was not prevented by this fusion protein. Therefore, our study uncovers a recessive impairment in the generation of oral tolerance in the NOD mouse when CTB is used as adjuvant, at least in combination with selected antigens such as the strong agonist 2.5 mimotope peptide or the GAD-derived peptide, but instead leads to T cell activation. These findings should have important consequences when planning equivalent vaccine formulations for clinical trials to combat the human disease.
RESEARCH DESIGN AND METHODS
Reagents and cells
Abs were purchased from Biolegend (La Jolla, CA, USA) or Beckton Dickinson (Franklin Lakes, NJ, USA), and cell culture media from Lonza (Barcelona, Spain). Streptavidin-PE was obtained from Columbia Bioscience (Columbia, MD, USA). cDNAs for positive interleukin PCR controls were purchased from Thermo Scientific (Epsom, UK). The BDC-2.5 T cell hybridoma was generously provided to us by Dr. K. Haskins (University of Colorado). Unless otherwise mentioned, all other reagents were obtained from Fluka or Sigma (Madrid, Spain).
Mouse strains
NOD/LtJ and B6.Foxp3EGFP mice [18] were originally purchased from The Jackson Laboratory (Bar Harbor, Maine, USA) and further bred in our special pathogen-free animal facility (Parc Cientific Barcelona, Spain). NOD/LtJxB6.Foxp3EGFP F1 mice were bred in-house. The T1D resistant NOR control strain has been previously described [19]. NOD versus NOR experimental comparisons were carried out at The Jackson Laboratory. NOD.Foxp3EGFP mice were generated in-house using a speed congenic approach by backcrossing NOD/LtJxB6.Foxp3EGFP F1 animals into the NOD background while monitoring 15 independent Idd loci. Mice homozygous after 4 generations for all NOD Idd loci were further backcrossed for 2 generations with NOD mice and then intercrossed to generate homozygous NOD.Foxp3EGFP (F6) animals.
Generation of recombinant proteins
The mature peptide of the subunit B from cholera toxin (amino acids T11-N124, Genbank access number X58785) was PCR amplified from Vibrio cholerae (Spanish Tissue Collection Type CECT 569) and subcloned into pET28a in between a vector-encoded 5′ met-val sequence and a 3′ sequence coding for a 9 aa linker ASGPGPGMV (linker 1), followed by a hexa-histidine tag and a C-terminal GS sequence. To generate CTB-2.5mi and CTB-GAD286–300 [20], the construct was further modified by introducing the 2.5mi peptide AHHPIWARMDA or GAD286–300 followed by a second, 6 aa linker GLDPGM in-between linker 1 and the hexahistidine tag (Fig. 1 A and supplemental Fig. S1 A). The proteins were expressed in E. coli BL21 and purified and refolded from inclusion bodies using a published protocol [21].
FIGURE 1. Generation of the CTB-2.5mi fusion protein and in vitro stimulation of BDC-2.5.
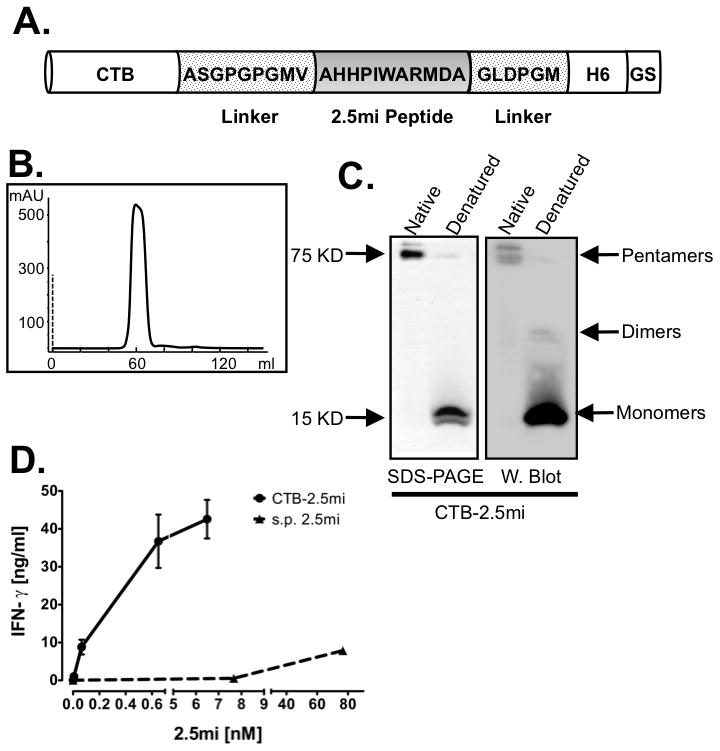
(A) Schematic representation of the CTB-2.5mi construct. The 2.5mi peptide is fused C-terminally to CTB and is embeded by 2 linker sequences, followed by a hexahistidine tag and a vector-encoded 2 aa tail (GS). The aa sequences of the linkers and the peptide are indicated.
(B) Elution profile of size exclusion chromatography of CTB-2.5mi.
(C) SDS-PAGE and Western blot analysis of purified CTB-2.5mi. The monomeric (15 KD) and pentameric (75KD) forms are shown by SDS-PAGE analysis under denaturing (reduced with β-mercaptoethanol and heated) and native (without β-mercaptoethanol and not heated) conditions, respectively, and the recombinant protein identity revealed by Western blot using an anti-hexahistidine tag antibody.
(D) Comparison of T cell activation by CTB-2.5mi versus the synthetic 2.5mi peptide (s.p. 2.5mi). The BDC-2.5mi T cell hybridoma was incubated with NOD-derived splenocytes as APCs at the indicated molar concentrations of the respective protein or peptide and T cell activation quantified by IFN-γ detection through ELISA. Error bars represent SEM of 2 independent experiments; each point was collected in triplicate per experiment.
Soluble Ag7 MHC molecules complexed to the 2.5mi, GAD286–300 or the GPI control peptide were expressed in D. melanogaster derived SC2 cells, purified and used for the generation of tetramers as published [15, 22, 23].
In vitro T cell stimulation and cytokine analysis by ELISA
BDC-2.5 T cell hybridomas (2x104 cell per well) were stimulated with the recombinant proteins for 48 hrs in complete RPMI media. IFN-γ secretion was detected by sandwich ELISA following the manufacture’s instructions (Ready-set-Go Mouse-IFN-γ Femto-HS, eBioscience).
Nested RT-PCR
Nested RT-PCR was carried out as published [24]. Briefly, splenocytes of mice treated orally with CTB-2.5mi were stained by tetramers and anti-CD4-APC, anti-CD8-PerCP, anti-CD19-PerCP as well as PI. Twenty CD4+2.5mi+Foxp3− or CD4+2.5mi+Foxp3+ T cells were sorted using a FACSaria II sorter (Becton Dickinson) directly into 50 μl of cDNA reaction mixture. Generated cDNA (2 μl per PCR reaction) was used for nested PCR as published [24].
ELISPOT Assays
ELISPOT assays were carried out using antibody pairs for mouse IFN-γ and IL-4 (BioLegend) as indicated by the manufacture. Total splenocytes from orally treated (CTB-2.5mi or PBS) female NOD mice were isolated 4 days after the last immunization as described above and 5x105 cells/well were plated in RPMI+10%FBS in the absence or presence of 10 or 100 ng/ml of CTB-2.5mi or CTB only to establish the response against the 2.5 mi peptide versus CTB on ELISPOT plates (Millipore). After 48 hrs of incubation at 37°C, ELISPOT assays were processed as indicated by the manufacturer using peroxidase-labeled streptavidin for secondary Ab detection and 3-amino-9-ethylcarbazole for color reaction (AEC, Sigma-Aldrich, Mo, USA). Spots were quantified using an automated ELISPOT reader system (C.T.L. Cellular Technology Ltd.).
Treatment of mice
Oral treatment was typically initiated at 5 to 8 weeks of age. Mice were administered 200 μg of CTB-2.5mi or CTB per dose intragastrically every 3 to 4 days, 5 or 10 doses total. In some cases, 4 days after the last dose, mice were immunized with 50 μg of CTB-2.5mi intraperitoneally. T cell responses were typically analyzed 4 days later.
All experiments were performed in accordance with the Animal Care and Veterinary Services and approved by the Ethics Committee of Animal Experimentation of the Barcelona Science Park.
Flow cytometry analysis
Antigen-specific T cell analysis was carried out as previously described [15, 17]. Briefly, single-cell suspensions were blocked with avidin (Sigma) in FACS buffer (PBS containing 2% FCS and 0.04% NaN3) and stained with PE-labeled MHC/peptide tetramers on ice. Depending on the combination of surface marker analysis, FITC-, APC- as well as Alexa Fluor-700–anti-CD4, anti-CD8-PE-Cy5, anti-CD19-PE-Cy5, anti-CD44-Pacific Blue, and anti-CD62L-APC-Cy7 were used (BioLegend, CA, USA). Dead cells were excluded by addition of propidium iodide. Flow cytometry was performed using a FACScan, a FACScalibur or a FACSAria II instrument (Becton Dickinson Immunocytometry Systems, Mountain View, California, USA), and data were analyzed using the FlowJo software (Tree Star Inc, Ashland OR, USA).
Statistical analysis
Bar diagrams were analyzed using the GraphPad Prism software and the Bonferroni test for selected pairs of columns or unpaired t test. Cumulative incidence of diabetes was determined by Kaplan-Meier estimates using the GraphPad Prism software version 5.0, and curves analyzed by log-rank test for statistical differences.
RESULTS
Generation of CTB-2.5mi fusion protein
Previous studies have shown that oral administration of CTB fused or conjugated with insulin led to T1D protection in the NOD mouse [9, 10]. We investigated how oral administration of the strong 2.5mi peptide agonist [15, 16], fused to CTB affected 2.5mi+ T cells. To this end, we generated a construct that encoded CTB, followed by the 2.5mi or the GAD286–300 peptide embedded in-between 2 linker peptides and a C-terminal histidine tag (Fig. 1A). Expression of CTB fusion proteins in E. coli and subsequent purification led to homogenous protein complexes as revealed by a single peak during size exclusion chromatography (Fig. 1B, only CTB-2.5mi is shown). SDS-PAGE analysis under denaturing and non-denaturing conditions indicated that the protein contained a functional CTB moiety as indicated by the formation of pentamers that were disrupted in presence of SDS and β-mercaptoethanol (Fig. 1C). Proper refolding of CTB was further indicated by binding to immobilized D-galactose ([25]; not shown).
CTB-2.5mi stimulates Ag-specific T cell in vitro and in vivo
We next investigated the capacity of CTB-2.5mi to be processed and presented to BDC-2.5 responder T cells in vitro. The BDC-2.5 hybridoma and total NOD-mouse derived splenocytes as source of antigen presenting cells were incubated in presence of increasing amounts of CTB-2.5mi or a synthetic version of the 2.5mi peptide and IFN-γ production was measured after 48 hrs of cultivation. The potency of CTB-2.5mi to stimulate IFN-γ production was more than 80-fold compared to the synthetic peptide (Fig. 1D). Therefore, CTB-2.5mi was properly processed and suitable to strongly stimulate Ag-specific T cells.
We next examined how CTB-2.5mi stimulated 2.5mi+ T cells in vivo in comparison to the synthetic peptide. In order to simultaneously assess the generation of Ag-specific Foxp3+ T cells, we used NODxB6.Foxp3EGFP F1 reporter mice as we previously showed that these mice generate 2.5mi+ T cells as well as Foxp3+ Treg with the same specificity (2.5mi+Foxp3+ CD4 T cells; [17]). Females were injected with equimolar amounts of either CTB-2.5mi or the synthetic 2.5mi peptide i.p. in the absence of adjuvant and proliferation of Ag-specific T cells was analyzed at day 4 p.i. by tetramer staining. A 10-fold stronger T cell proliferation was detected in the spleen when the CTB fusion protein was used compared to the synthetic peptide in both effector T cells (2.5mi+Foxp3−) and iTreg (2.5mi+Foxp3+) (Fig. 2 A and B; see supplemental Table S1 for absolute cell numbers). Taken together, our studies show that fusion of the 2.5mi peptide to CTB favors 2.5mi+ T cells expansion as well as their feeding into the iTreg pool when administered i.p.
FIGURE 2. In vivo expansion of 2.5mi+ T Cells by CTB-2.5mi.
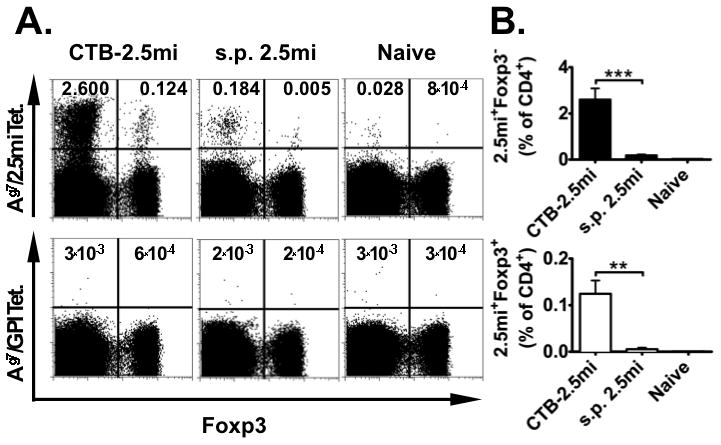
NODxB6.Foxp3EGFP F1 females (n=3) were immunized i.p. with 6.5 nmol of synthetic peptide 2.5m or CTB-2.5mi. Four days later, expansion of antigen-specific CD4 T cells was analyzed by staining with Ag7/2.5mi or Ag7/GPI282–292 control tetramers and analyzed by FACS.
(A) Representative FACS profiles showing the percentage of splenic 2.5mi+ effector T cells and 2.5mi+Foxp3+ Treg. T cells were gated on B220−, CD8−, PI− and CD4+.
(B) Bar diagram of the same experiment showing the mean ± SEM of the percentage 2.5mi+ effector T cells (top) and 2.5mi+Foxp3+ Treg (bottom; **p<0.01, ***p<0.0001). The diagram was generated from 2 independent experiments, using 3 mice per experiment and type of treatment.
CTB-2.5mi induces Ag-specific T cell tolerance in NODxB6.Foxp3EGFP F1, but not in NOD mice
It was previously shown that oral tolerance is generated via Th2 cells, iTreg and T cell anergy [26–29]. To investigate whether CTB-2.5mi induced Ag-specific tolerance, we first treated NOD mice 5 times by i.g. delivery of the fusion protein. Four days after the last dose, we either tested the presence of 2.5mi+ T cells, or injected CTB-2.5mi intraperitoneally (i.p.) and then analyzed for 2.5mi+ T cells 4 days later. As controls, we analyzed either age-matched naive littermates, or littermates that had received CTB-2.5mi i.p. without prior i.g. treatment. The frequencies of 2.5mi+ T cells detected in naive versus i.g. treated mice were essentially undistinguishable in the spleen and mesenteric LN (MLN) (Fig. 3A, B and C; see supplemental Table S2 for absolute cell numbers). I.p. administration of CTB-2.5mi led to a strong proliferation of 2.5mi+ T cells, regardless of i.g. treatment. We corroborated these findings in a second NOD colony (NOD/LtDvs) in a different laboratory (D. Serreze, Jackson) (Fig. 4, J and K). Therefore, CTB-2.5mi was unable to induce oral tolerance in NOD mice as defined by the inability to prevent Ag-specific T cell expansion after i.p. challenge with Ag.
FIGURE 3. 2.5mi+ T cells are not tolerized by oral treatment with CTB-2.5mi in NOD mice.
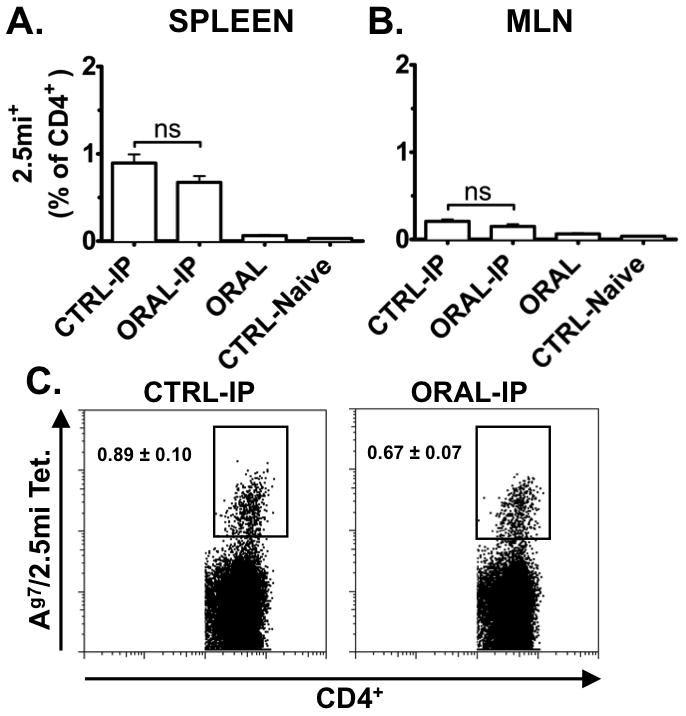
Five-to-6-week-old NOD females were treated with 5 doses of 200 μg CTB-2.5mi every 3 to 4 days i.g. (ORAL) and analyzed 4 days after the last dose for Ag-specific 2.5mi+ T cell expansion in the spleen (A) and in mesenteric lymph nodes (B; MLN) as explained in the legend for Fig. 2 by tetramer staining and FACS analysis. Unmanipulated littermates served as baseline control (CTRL-Naive). Alternatively, 4 days after the last oral dose, mice received a boost immunization i.p. with 50 μg of CTB-2.5mi, were rested for an additional 4 days, and next analyzed as above (ORAL-IP). As controls, littermates were immunized i.p. without receiving the protein i.g. (CTRL-IP). Mean values ± SEM of 2 independent experiments including 6 animals per group and treatment are shown. Values represent percentages of 2.5mi+CD4+ T cells within total CD4+ T cells, gated on B220−, CD8−, PI− and CD4+ cells.
(C) Representative FACS profiles of splenocytes of NOD mice treated and analyzed as explained above.
FIGURE 4. Tolerization of 2.5mi+ T cells by oral treatment with CTB-2.5mi in NODxB6.Foxp3EGFP F1 and NOR mice.
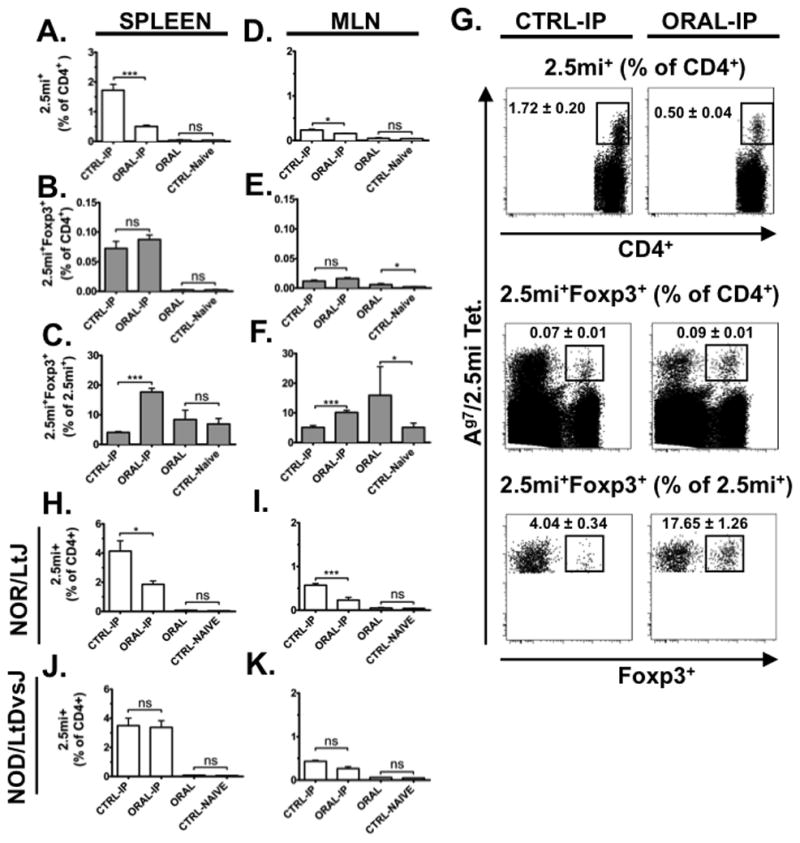
Five-to-6-week-old NOD, NODxB6.Foxp3EGFP F1, or NOR females were treated with 5 doses of 200 μg CTB-2.5mi every 3 to 4 days i.g. (ORAL) or PBS at the same times (CTRL-Naive ) and analyzed 4 days after the last dose for Ag-specific 2.5mi+ T cell expansion in the spleen as explained in the legend for Fig. 2 by tetramer staining and FACS analysis. Alternatively, 4 days after the last oral dose, mice received a boost immunization i.p. with 50 μg of CTB-2.5mi, were rested for an additional 4 days, and next analyzed as above (ORAL-IP versus CTRL-IP). Mean values ± SEM of 2 independent experiments including 6 animals per group and treatment are shown (***p <0.0001; *p <0.05).
(A, D) Bar diagrams represent percentages of 2.5mi+CD4+ T cells within total CD4+ T cells in the spleens or MLNs of variously treated NODxB6.Foxp3EGFP F1 mice.
(B, E) Bar diagrams represent percentages of 2.5mi+Foxp3+ T cells among total CD4+ T cells within spleens or MLNs of variously treated NODxB6.Foxp3EGFP F1 mice.
(C, F) Bar diagrams represent percentages of 2.5mi+Foxp3+ T cells among total 2.5mi+ T cells within spleens or MLNs of variously treated NODxB6.Foxp3EGFP F1 mice.
(G) Representative FACS data from the same experimental analyses of NODxB6.Foxp3EGFP F1 mice. Analysis of the spleen is shown. Top: Ag7/2.5mi tetramer+ T cells are analyzed within total CD4+ T cells. Middle: Ag7/2.5mi tetramer+ Foxp3+ T cells are analyzed within total CD4+ T cells. Bottom: Foxp3+ T cells are analyzed within Ag7/2.5mi tetramer+ T cells. Left: Mice received CTB-2.5mi i.p. only. Right: Mice were treated first with CTB-2.5mi i.g. and next received the protein i.p.
(H, I, J and K) Comparison of 2.5mi+CD4+ T cells within total CD4+ T cells in the spleens or MLNs of NOD and NOR mice (colonies D. Serreze, Jackson Laboratories) within each designated treatment group. H and J, spleen, I and K, MLN.
For the analysis, cells were gated on B220−, CD8−, PI− and CD4+ cells.
Despite possessing 2.5mi+ T cells [17], NODxB6 F1 mice do not develop T1D and are tolerant to self. In order to analyze whether the failure of CTB-2.5mi to induce oral tolerance was intrinsic to the protein or to the NOD genetic background, we repeated the same experiments using NODxB6.Foxp3EGFP F1 reporter mice. This allowed simultaneously monitoring 2.5mi+ T effector cells and Ag-specific iTreg. Compared to PBS treated controls, expansion of 2.5mi+ T cells was significantly suppressed in the spleen and to a less extent the MLN of CTB-2.5mi orally treated NODxB6.Foxp3EGFP F1 mice that became evident when both groups were boosted i.p. with CTB-2.5mi (Fig. 4A, D and G). Oral treatment led to a significant increase of 2.5mi+Foxp3+ T cells among total CD4 T cells in the MLN but not in the spleen (Fig. 4B and E). Following i.p. boost challenge with the protein after i.g. treatment, the proportion of Foxp3+ T cells within the 2.5mi+ T cell subpopulation increased significantly in both tissues (Fig. 4C and F). Therefore, contrarily to NOD mice, T1D resistant NODxB6.Foxp3EGFP F1 controls were able to mount oral tolerance to CTB-2.5mi. While Foxp3+ Ag-specific Treg were generated, due to the rather small amounts, however, their role is questionable. These results suggest that NOD mice have a recessive genetic resistance to induce oral tolerance to CTB-2.5mi that can be overcome by a single outcross into the B6 background. The differential response to CTB-2.5mi was restricted to the oral route since we did not detect any difference in NOD versus NODxB6.Foxp3EGFP F1 reporter mice in either the percentage or duration of 2.5mi+ Tconv and Treg cell expansion after i.p. challenge only (M.P. and T.S., manuscript in preparation). We carried out a further control study to test if the ability to establish tolerance to orally administered CTB-2.5mi in NODxB6.Foxp3EGFP F1, but not NOD mice was a phenomenon restricted to a comparison of these particular two strains. Thus, we compared the ability to establish tolerance to orally administered CTB-2.5mi in NOD mice to the closely related H2g7 matched, but T1D resistant NOR control strain [19]. Following previous oral treatment with CTB-2.5mi, subsequent i.p. primed CD4 T cell responses to this antigen were significantly less in spleens and MLNs of NOR than NOD mice (Fig. 4H and I). It should be pointed out that NOR background mice carrying the Foxp3EGFP reporter are currently not available. For this reason it was not possible to determine if the greater ability to establish tolerance to orally administered CTB-2.5mi in NOR than NOD mice was due to variable conversion of CD4 T cells recognizing this antigen into the Treg compartment. To analyze whether these findings were limited to the 2.5mi agonist peptide, we repeated the same set of experiments with GAD286–300 fused to CTB in NOD versus NOR mice and analyzed Ag-specific T cells by tetramers complexed to this peptide (Supplemental Fig. S1 A and B). Again, NOD but not NOR mice were incapable to mount oral tolerance to CTB-GAD286–300. These collective results indicate the susceptibility of NOD mice to development of autoimmune T1D is associated with the strain characteristic of an impaired ability to establish CD4 T cell tolerance to an orally administered antigen.
Restoring the NOD background in Foxp3EGFP reporter mice should eliminate the ability to establish oral tolerance. We thus further backcrossed the Foxp3EGFP reporter from the F1 hybrids for 6 generations to the NOD strain to generate NOD.Foxp3EGFP animals. While oral treatment led to a weak but significant reduction of 2.5mi+ T cells in MLN, the mice were unable to suppress T cell expansion in the spleen as predicted (Fig. 5A and D). Interestingly, compared to controls NOD.Foxp3EGFP females produced significantly more 2.5mi+Foxp3+ T cells in the spleen and MLN (Fig. 5B, C, E and F). Therefore, the inability of NOD mice to be rendered tolerant to orally administered CTB-2.5mi is not due to a failure of this antigen to induce an expansion of Foxp3+ Tregs. Instead, the current findings would be consistent with previous reports that diabetogenic effector T cells in NOD mice become refractive to the suppressive effects of Tregs [30].
FIGURE 5. Failure to achieve tolerance of 2.5mi+ T cells by oral treatment with CTB-2.5mi in NOD.Foxp3EGFP mice.
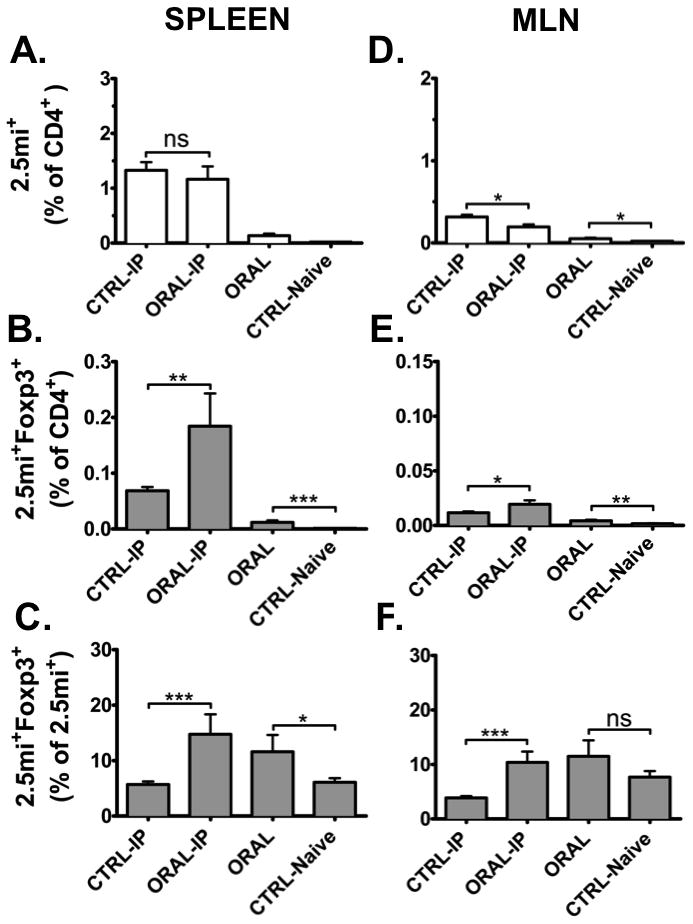
Five-to-6-week-old NOD.Foxp3EGFP females (Foxp3EGFP reporter backcrossed for 6 generations into the NOD strain) were treated with 5 doses of 200 μg CTB-2.5mi every 3 to 4 days i.g. and analyzed 4 days after the last dose for Ag-specific 2.5mi+ T cell expansion in the spleen as explained in the legend for Fig. 2 by tetramer staining and FACS analysis (ORAL and CTRL-Naive). Alternatively, 4 days after the last oral dose, mice received a boost immunization i.p. with 50 μg of CTB-2.5mi, were rested for an additional 4 days, and next analyzed as above (ORAL-IP and CTRL-IP). Mean values ± SEM of 2 independent experiments including 6 animals per group and treatment are shown (*p<0.05, **p<0.01, ***p<0.0001).
(A, D) Bar diagrams represent percentages of 2.5mi+CD4+ T cells among total CD4+ T cells within spleens or MLNs of variously treated NOD.Foxp3EGFP F1 mice.
(B, E) Bar diagrams represent percentages of 2.5mi+Foxp3+ T cells among total CD4+ T cells within spleens or MLNs of variously treated NOD.Foxp3EGFP F1 mice.
(C, F) Bar diagrams represent percentages of 2.5mi+Foxp3+ T cells among total 2.5mi+ T cells within spleens or MLNs of variously treated NOD.Foxp3EGFP F1 mice. For the FACS analysis, cells were gated on B220−, CD8−, PI− and CD4+ cells.
Oral administration of CTB-2.5mi results in a strong induction of IFN-γ and IL-4 in NOD mice
Since CTB-2.5mi did not induce Ag-specific oral tolerance in the NOD background, we next investigated whether this was associated with a particular Th1 to Th2 cytokine production balance. NOD.Foxp3EGFP and NODxB6 F1 mice received 5 doses of CTB-2.5mi or PBS i.g., and sorted 2.5mi+ Teff cells were analyzed for cytokine expression by RT-PCR. In F1 animals neither Th1 (IFN-γ) or Th2 (IL-4) cytokine expression by 2.5mi+ T cells was detected. However, these cells expressed both cytokines in NOD.Foxp3EGFP mice (Fig 6A). ELISPOT assays carried out in parallel revealed that in both strains, oral treatment with CTB-2.5mi significantly increased IFN-γ as well as IL-4 cytokine production. However, NOD mice generated significantly more cells producing either of these cytokines compared to NODxB6 F1 mice (Fig. 6, B and C and supplemental Fig. S2). The in vitro response was specific for the 2.5mi peptide as control cultures containing CTB only as a source of Ag generated negligible amounts of IFN-γ or IL-4 (Fig. 6, B and C). Therefore, a failure to induce a Th1 to Th2 cytokine production shift could not explain the impaired tolerogenic effect of oral CTB-2.5mi administration in NOD mice as Th2 cells were amply generated but could not prevent the generation of IFN-γ producing Th1 cells.
FIGURE 6. Th1 and Th2 cytokine analysis in NOD.Foxp3EGFP and NODxB6.Foxp3EGFP F1 mice.
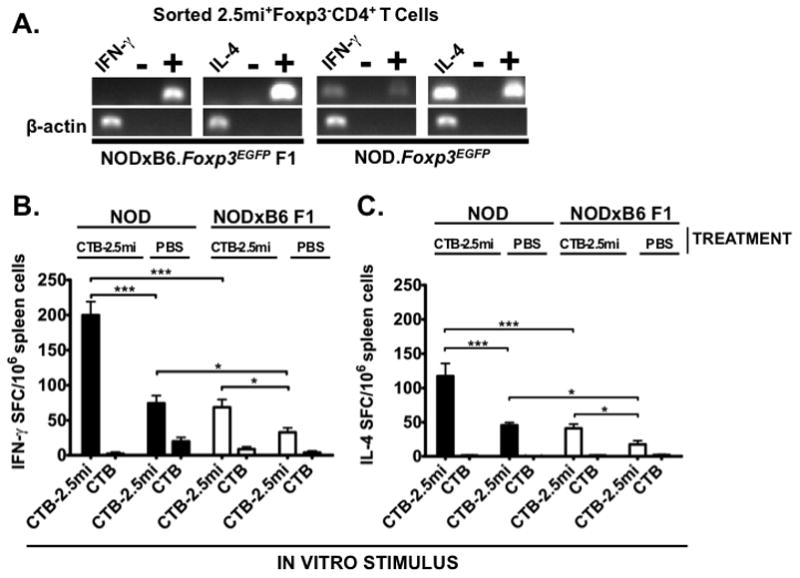
Five-to-6-week-old NOD.Foxp3EGFP and NODxB6.Foxp3EGFP F1 females were treated with 5 doses of 200 μg CTB-2.5mi (A, B and C) or PBS (B and C) every 3 to 4 days i.g. Four days after the last dose, T cells were analyzed for cytokine production.
(A) Ag-specific 2.5mi+Foxp3− T cells were sorted by FACS directly ex vivo without further manipulation into a RT-PCR mixture and analyzed for transcription of IFN-γ and IL-4. Left panels, NODxB6.Foxp3EGFP F1 mice; right panels, NOD.Foxp3EGFP mice; + positive PCR control using IFN-γ and IL-4 coding DNA plasmid templates, -no template PCR control.
(B) and (C) ELISPOT analysis. Total splenocytes from treated mice were incubated with 100 ng/ml of CTB-2.5mi or CTB and cytokine secretion quantified 2 days later (B, IFN-γ; C, IL-4). Values indicate spot forming cells (SFC) per 106 splenocytes; mean values ± SEM are indicated after subtraction of SFC in unstimulated wells. n=6 and 12, resulting from 2 and 4 independent experiments for NOD.Foxp3EGFP and NODxB6.Foxp3EGFP F1 mice, respectively (*p<0.05, **p<0.01, ***p<0.0001).
Oral antigen administration results in a greater proportion of naive 2.5mi+ T cells in NODxB6 F1 than NOD mice
Based on their greater level of cytokine production we tested if 2.5mi+ T cells are activated more readily in response to orally administered antigen in NOD than NODxB6 F1 mice. To determine the activation status of 2.5mi+ T cells in NOD.Foxp3EGFP and NODxB6.Foxp3EGFP F1 mice, animals received either 5 doses of CTB-2.5mi i.g. or mock-treatment with PBS, and splenic 2.5mi+ T cells were analyzed for simultaneous CD44 and CD62L expression. In mock-treated NODxB6.Foxp3EGFP F1 mice, the majority of 2.5mi+Foxp3− T cells had a naive phenotype (CD44−CD62L+) and oral treatment did not significantly affect the composition of the naive, central memory (CD44+CD62L+) or effector memory (CD44+CD62L−) populations (Fig. 7A, see supplemental Table S3 for absolute cell numbers). In contrast, oral treatment of NOD.Foxp3EGFP animals had a drastic effect on 2.5mi+Foxp3− T cells. CTB-2.5mi treatment induced strong activation and a significant shift towards the effector memory T cell pool (Fig. 7B, see supplemental Table S3 for absolute cell numbers). The same effect was observed in pancreatic lymph nodes (not shown). Therefore, rather than inducing tolerance, oral treatment with CTB-2.5mi induced T cell activation in the diabetes-prone NOD background but not in disease-resistant NODxB6.Foxp3EGFP F1 mice.
FIGURE 7. Activation and memory markers of 2.5mi+ effector T cells after oral treatment with CTB-2.5mi in NOD.Foxp3EGFP and NODxB6.Foxp3EGFP F1 mice.
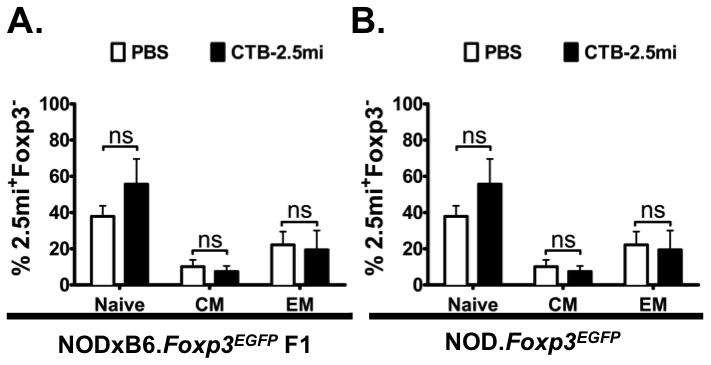
NODxB6.Foxp3EGFP F1 (A) and NOD.Foxp3EGFP mice (B) were treated with 5 doses of 200 μg CTB-2.5mi and 4 days after the last dose 2.5mi+Foxp3− T cells from the spleen were analyzed for CD62L and CD44 expression. Mean values ± SEM of 6 mice per group analyzed in 2 independents experiments are shown. Naive, CD62Lhi CD44lo; CM (central memory), CD62Lhi CD44hi; EM (effector memory), CD62Llo CD44hi.
Oral administration of CTB-2.5mi does not prevent T1D
It was possible that an incompatibility of the 2.5mi strong agonist peptide together with CTB as carrier molecule was responsible for the oral administration of this agent inducing T cell activation rather than tolerance induction in NOD mice. To test this hypothesis, 5-week-old female NOD mice were treated every 3–4 days by i.g. delivery with a total of 5 doses of CTB-2.5mi, CTB only, or PBS, and diabetes onset was monitored. Disease was neither delayed nor reduced by CTB-2.5mi. If anything, treatment slightly accelerated disease (Supplemental Fig. S3). Incrementing the dose and time of treatment to a total of 10 i.g. injections did not change this outcome. The failure of CTB-2.5mi to induce systemic tolerance was unlikely due to the 2.5mi epitope itself since this peptide could prevent disease when administered in form of a DNA vaccine [17]. Neither was it likely due to the route of administration since CTB-2.5mi, when administered i.p., likewise was unable to prevent disease (M.P. and T.S., unpublished). Collectively, the results point towards an incompatibility of CTB in combination with a strong peptide agonist such as 2.5mi to induce tolerogenic responses in the NOD background.
DISCUSSION
The success of immunological tolerance induction in the NOD mouse depends on many factors such as the route of Ag applications, the nature of the Ag itself as well as the adjuvant. To study oral tolerance in the NOD mouse, we reduced the antigenic complexity of an oral vaccine to 2 peptides, the 2.5 mimotope and a GAD derived peptide, since we could trace the outcome on the corresponding CD4 T cell populations using MHC tetramers. The 2.5mi epitope can induce T1D protective tolerance in the NOD mouse when given as targeted DNA vaccine [17] or as soluble Ag7/peptide dimer [31]. Therefore, the 2.5mi epitope is a good candidate to compare the fate of Ag-specific, autoreactive T cells depending on the routes and adjuvants used for application. In this report, we focused on CTB due to its reported adjuvant effect [32, 33] and its capacity to suppress diabetes with antigens such as insulin or GAD-derived peptides [6, 8]. However, none of these studies have analyzed in detail the fate of Ag-specific T cells by MHC tetramers. I.p. delivery of CTB strongly enhanced the expansion of 2.5mi+ T cells in comparison to administration of the synthetic peptide. However, oral treatment of NOD mice with CTB-2.5mi neither prevented nor delayed disease but rather slightly accelerated it. Several mechanisms have been reported to be responsible for oral tolerance induction, including the generation of Foxp3+ Treg [34, 35], Th3 cells [36], a shift from a Th1 to a Th2 cytokine production profile [4], and T cell anergy [37, 38]. In murine diabetes models, the generation of Treg as well as of Th2 protective T cells have been reported, but due to lacking tools neither T cell anergy nor T cell deletion have been experimentally approached. Treatment of NOD mice with CTB-2.5mi did not delete 2.5mi+ T cells but induced Ag-specific Foxp3+ Treg, Th2, and also Th1 cells. Neither Treg not Th2 cells were sufficient to protect from disease, possibly since 2.5mi+ T cell with a Th2 profile might be as diabetogenic as Th1 cells as previously demonstrated [39].
Since NODxB6.Foxp3EGFP F1 and NOR mice were fully capable of suppressing the expansion of 2.5mi+ T cells when treated orally, sufficient CTB-2.5mi protein survives in the intestine to be presented by APCs to these T cells. The problem therefore did not lie in the inappropriate degradation of CTB-2.5mi. When the NOD background was reestablished by further backcrossing the Foxp3EGFP reporter into NOD, the ability to generate oral tolerance was lost in the spleen and only a weak effect was detected in the MLN. The difference in the MLN between our NOD and NOD.Foxp3EGFP mice might be due to minor animal-to-animal variations or to the fact that NOD.Foxp3EGFP mice still carried some parts of the B6 specific background. Together, this clearly demonstrates a recessive constraint in oral tolerance induction of the NOD strain when using CTB in combination with a strong agonist peptide. The inability of the NOD strain to mount oral tolerance was not limited to the 2.5 mimotope which might follow different rules considering that it is a strong agonist peptide but held also true for a natural epitope, in this case GAD286–300.
Levels of 2.5mi responsive Th1 and Th2 cells increased significantly in both NOD and NODxB6.Foxp3EGFP F1 mice orally treated with this antigen. However, there was a remarkable difference: the numbers of IFN-γ as well as IL-4 producing cells after oral immunization were significantly higher in NOD versus NODxB6 F1 mice. These observations corroborate a previous report showing that NOD-derived CD4 T cells are more prone than those from B6.H2g7 mice to produce IFN-γ [40]. We show here that this is not limited to IFN-γ but also holds true for IL-4.
A striking difference between the NOD and the NODxB6 F1 background was found in naive versus orally treated mice in the conventional (Foxp3−) 2.5mi+ T cell population. Following oral CTB-2.5mi treatment, CD4 T cells recognizing this antigen remained mostly naive in NODxB6 F1 mice, but were diverted primarily into the effector memory pool in NOD mice. These data are in accordance with a recent report showing that when generated in B6.H2g7 mice, BDC-2.5 TCR transgenic T cells proliferate less in vitro and in vivo upon stimulation in comparison to the same clonotype originating in the NOD strain [30]. The difference was traced to strain dependent variations in BDC-2.5 effector rather than to Treg activity [30]. Our data expand this observation to polyclonal 2.5mi+ T cells and show that this is a recessive trait of the NOD background.
In light of these results, how may the reported success of other groups to induce T1D protection using CTB coupled autoantigens in the NOD mouse be explained? Differences in the treatment protocol are an unlikely explanation since in the case of CTB conjugated to insulin, even a single i.g. dose has been reported to protect from the disease [9]. The strong adjuvant effect of CTB together with a strong agonist peptide such as 2.5mi might be the underlying problem why diabetes was not prevented. We did not carry out long-term diabetes prevention trials with the GAD peptide. However, the inability of the NOD strain to mount oral tolerance against CTB-coupled peptides was not restricted to the BDC2.5 mimotope, but also extended to the GAD peptide. A previous report has shown that GAD531–545 when fused as trimer to CTB can induce oral tolerance [6]. An intensive oral treatment (4 times weekly, from 5 – 35 weeks of age) was necessary to achieve a 60% reduction of diabetes in NOD mice. It has been previously reported that GAD524–538 induces a regulatory T cell population upon immunization [41]. It may depend on the constellation of the preexisting T cell population (effector versus regulatory) which of them will preferentially expand and tip the balance from tolerance to effector function. When we treated NOD mice with three doses of a lysosome-targeted DNA vaccine, the same mimotope could induce protection and delay of the disease [17]. Therefore, the combination of the mimotope together with CTB is less efficient than when applied in a DNA vaccine formulation where the peptide is targeted intracellularly to lysosomes. Recent experiments in our laboratory have demonstrated that CTB-2.5mi is equally unable to prevent disease when administered i.p. (not shown). Over the last 15 years, however, the progress on diabetes suppression with CTB based oral fusion vaccines has been modest. Since early reports where 50% diabetes suppression was demonstrated [9, 11], to our knowledge the strongest suppression to date (60%) has been achieved using GAD peptide 531–545 fused as trimer to CTB as mentioned above [6]. These data indicate that solid diabetes suppression using CTB as fusion partner has remained difficult to obtain.
In conclusion, the genetic background is essential when trying to elaborate oral antigen treatment protocols since autoimmune-prone strains behave very differently compared to well-established standard models. The NOD strain has a clear impairment to mount oral tolerance, at least with the two peptides analyzed when fused to CTB, in comparison to disease-resistant strains. It will be important to consider these observations when engaging into the much more complex world of human trials.
Supplementary Material
Acknowledgments
We would like to thank Dr. Kathy Haskins for providing the BDC-2.5 hybridoma, Anuska Llano for assistance with the ELISPOT reader and Dr. Paloma Mas (CRAG) for critical reading of the manuscript. We thank the cytometry facility of the Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS) for the technical help.
This work was supported by grants from the Spanish Ministry of Science and Innovation and European Regional Development Fund (FEDER; SAF2004-02666, SAF2007-65291, SAF2010-18548, SAF2011-29319 to T.S.; SAF2007-31050E and SAF2008-02536 to C.M.), from the Spanish Ministry of Health and Consumption (PI040587 to T.S. and P041310 to C.M.), and from the European Community (MIRG-CT-2004-012692 to T.S.). T.S. was also supported by the Ramón y Cajal program from the Spanish Ministry of Science and Innovation. DS is supported by NIH grants DK46266, DK97610, DK72473, as well as by grants from the Juvenile Diabetes Research Foundation and the American Diabetes Association.
ABBREVIATIONS
- NOD
non-obese diabetic
- T1D
type 1 diabetes
- GPI
glucose-6-phosphate isomerase
- CTB
cholera toxin subunit B
- MLN
mesenteric lymph nodes
- 2.5mi
2.5 mimotope
- GAD
glutamic acid decarboxylase
Footnotes
AUTHORSHIP
M.P. generated CTB-fusion proteins and constructs, and carried out cellular and animal studies; N.G., A.Z.O. and C.I. generated MHC tetramers and carried out animal breeding, L.T. provided MHC tetramer constructs and cell lines, C.M. provided intellectual input and assisted with experimental design and interpretation, D.S. provided intellectual input, T.S. led the investigation, devised the project and wrote the manuscript.
The authors declare no potential conflict of interest relevant to this article.
References
- 1.Sanchez J, Holmgren J. Cholera toxin structure, gene regulation and pathophysiological and immunological aspects. Cell Mol Life Sci. 2008;65:1347–1360. doi: 10.1007/s00018-008-7496-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mathis D, Vence L, Benoist C. Beta-Cell death during progression to diabetes. Nature. 2001;414:792–798. doi: 10.1038/414792a. [DOI] [PubMed] [Google Scholar]
- 3.Shoda LKM, Young DL, Ramanujan S, Whiting CC, Atkinson MA, Bluestone JA, Eisenbarth GS, et al. A Comprehensive Review of Interventions in the NOD Mouse and Implications for Translation. Immunity. 2005;23 :115–126. doi: 10.1016/j.immuni.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 4.Ploix C, Bergerot I, Durand A, Czerkinsky C, Holmgren J, Thivolet C. Oral administration of cholera toxin B-insulin conjugates protects NOD mice from autoimmune diabetes by inducing CD4+ regulatory T-cells. Diabetes. 1999;48:2150–2156. doi: 10.2337/diabetes.48.11.2150. [DOI] [PubMed] [Google Scholar]
- 5.Aspord C, Czerkinsky C, Durand A, Stefanutti A, Thivolet C. α4 Integrins and L-selectin Differently Orchestrate T-cell Activity During Diabetes Prevention Following Oral Administration of CTB-insulin. J Autoimmunity. 2002;19:223–232. doi: 10.1006/jaut.2002.0610. [DOI] [PubMed] [Google Scholar]
- 6.Gong Z, Pan L, Le Y, Liu Q, Zhou M, Xing W, Zhuo R, et al. Glutamic acid decarboxylase epitope protects against autoimmune diabetes through activation of Th2 immune response and induction of possible regulatory mechanism. Vaccine. 2010;28:4052–4058. doi: 10.1016/j.vaccine.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 7.Denes B, Krausova V, Fodor N, Timiryasova T, Henderson D, Hough J, Yu J, et al. Protection of NOD Mice From Type 1 Diabetes After Oral Inoculation with Vaccinia Viruses Expressing Adjuvanted Islet Autoantigens. J Immunother. 2005;28:438–448. doi: 10.1097/01.cji.0000171315.82997.9a. [DOI] [PubMed] [Google Scholar]
- 8.Bregenholt S, Wang M, Wolfe T, Hughes A, Bærentzen L, Dyrberg T, Von Herrath MG, et al. The Cholera Toxin B Subunit is a Mucosal Adjuvant for Oral Tolerance Induction in Type 1 Diabetes. Scand J Immunol. 2003;57:432–438. doi: 10.1046/j.1365-3083.2003.01248.x. [DOI] [PubMed] [Google Scholar]
- 9.Bergerot I, Ploix C, Petersen J, Moulin V, Rask C, Fabien N, Lindblad M, et al. A cholera toxoid-insulin conjugate as an oral vaccine against spontaneous autoimmune diabetes. Proc Natl Acad Sci. 1997;94:4610–4614. doi: 10.1073/pnas.94.9.4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gong Z, Jin Y, Zhang Y. Suppression of diabetes in non-obese diabetic (NOD) mice by oral administration of a cholera toxin B subunit-insulin B chain fusion protein vaccine produced in silkworm. Vaccine. 2007;25:1444–1451. doi: 10.1016/j.vaccine.2006.10.039. [DOI] [PubMed] [Google Scholar]
- 11.Arakawa T, Yu J, Chong DKX, Hough J, Engen PC, Langridge WHR. A plant-based cholera toxin B subunit-insulin fusion protein protects against the development of autoimmune diabetes. Nat Biotech. 1998;16:934–938. doi: 10.1038/nbt1098-934. [DOI] [PubMed] [Google Scholar]
- 12.Achenbach P, Barker J, Bonifacio E. Modulating the natural history of type 1 diabetes in children at high genetic risk by mucosal insulin immunization. Curr Diabetes Reports. 2008;8:87–93. doi: 10.1007/s11892-008-0017-y. [DOI] [PubMed] [Google Scholar]
- 13.Sánchez J, Holmgren J. Cholera toxin - a foe & a friend. Ind J Med Res. 2011;133:153–163. [PMC free article] [PubMed] [Google Scholar]
- 14.Haskins K, McDuffie M. Acceleration of diabetes in young NOD mice with a CD4+ islet-specific T cell clone. Science. 1990;249:1433–1436. doi: 10.1126/science.2205920. [DOI] [PubMed] [Google Scholar]
- 15.Stratmann T, Martin-Orozco N, Mallet-Designe V, Poirot L, McGavern D, Losyev G, Dobbs CM, et al. Susceptible MHC alleles, not background genes, select an autoimmune T cell reactivity. J Clin Invest. 2003;112:902–914. doi: 10.1172/JCI18337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshida K, Martin T, Yamamoto K, Dobbs C, Munz C, Kamikawaji N, Nakano N, et al. Evidence for shared recognition of a peptide ligand by a diverse panel of non-obese diabetic mice-derived, islet-specific, diabetogenic T cell clones. Int Immunol. 2002;14:1439–1447. doi: 10.1093/intimm/dxf106. [DOI] [PubMed] [Google Scholar]
- 17.Rivas EI, Driver JP, Garabatos N, Presa M, Mora C, Rodriguez F, Serreze DV, et al. Targeting of a T Cell Agonist Peptide to Lysosomes by DNA Vaccination Induces Tolerance in the Nonobese Diabetic Mouse. J Immunol. 2011;186:4078–4087. doi: 10.4049/jimmunol.0902395. [DOI] [PubMed] [Google Scholar]
- 18.Haribhai D, Lin W, Relland LM, Truong N, Williams CB, Chatila TA. Regulatory T Cells Dynamically Control the Primary Immune Response to Foreign Antigen. J Immunol. 2007;178:2961–2972. doi: 10.4049/jimmunol.178.5.2961. [DOI] [PubMed] [Google Scholar]
- 19.Prochazka M, Serreze DV, Frankel WN, Leiter EH. NOR/Lt Mice: MHC-Matched Diabetes-Resistant Control Strain for NOD Mice. Diabetes. 1992;41:98–106. doi: 10.2337/diab.41.1.98. [DOI] [PubMed] [Google Scholar]
- 20.Chao C-C, McDevitt HO. Identification of immunogenic epitopes of GAD 65 presented by Ag7 in non-obese diabetic mice. Immunogenetics. 1997;46:29–34. doi: 10.1007/s002510050238. [DOI] [PubMed] [Google Scholar]
- 21.Arêas AP, Oliveira ML, Ramos CR, Sbrogio-Almeida ME, Raw I, Ho PL. Synthesis of cholera toxin B subunit gene: cloning and expression of a functional 6XHis-tagged protein in Escherichia coli. Prot Expr Purif. 2002;25:481–487. doi: 10.1016/s1046-5928(02)00026-8. [DOI] [PubMed] [Google Scholar]
- 22.Stratmann T, Apostolopoulos V, Mallet-Designe V, Corper AL, Scott CA, Wilson IA, Kang AS, et al. The I-Ag7 MHC Class II Molecule Linked to Murine Diabetes Is a Promiscuous Peptide Binder. J Immunol. 2000;165:3214–3225. doi: 10.4049/jimmunol.165.6.3214. [DOI] [PubMed] [Google Scholar]
- 23.Corper AL, Stratmann T, Apostolopoulos V, Scott CA, Garcia KC, Kang AS, Wilson IA, et al. A Structural Framework for Deciphering the Link Between I-Ag7 and Autoimmune Diabetes. Science. 2000;288:505–511. doi: 10.1126/science.288.5465.505. [DOI] [PubMed] [Google Scholar]
- 24.Panus JF, McHeyzer-Williams LJ, McHeyzer-Williams MG. Antigen-specific T Helper Cell Function: Differential Cytokine Expression in Primary and Memory Responses. J Exp Med. 2000;192:1301–1316. doi: 10.1084/jem.192.9.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tinker JK, Erbe JL, Holmes RK. Characterization of fluorescent chimeras of cholera toxin and Escherichia coli heat-labile enterotoxins produced by use of the twin arginine translocation system. Infect Immun. 2005;73:3627–3635. doi: 10.1128/IAI.73.6.3627-3635.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller A, Lider O, Roberts AB, Sporn MB, Weiner HL. Suppressor T cells generated by oral tolerization to myelin basic protein suppress both in vitro and in vivo immune responses by the release of transforming growth factor beta after antigen-specific triggering. Proc Natl Acad Sci. 1992;89:421–425. doi: 10.1073/pnas.89.1.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melamed D, Friedman A. Direct evidence for anergy in T lymphocytes tolerized by oral administration of ovalbumin. Eur J Immunol. 1993;23:935–942. doi: 10.1002/eji.1830230426. [DOI] [PubMed] [Google Scholar]
- 28.Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, Muller W, et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity. 2011;34:237–246. doi: 10.1016/j.immuni.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 29.Mucida D, Kutchukhidze N, Erazo A, Russo M, Lafaille JJ, Curotto de Lafaille MA. Oral tolerance in the absence of naturally occurring Tregs. J Clin Invest. 2005;115:1923–1933. doi: 10.1172/JCI24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D’Alise AM, Auyeung V, Feuerer M, Nishio J, Fontenot J, Benoist C, Mathis D. The defect in T-cell regulation in NOD mice is an effect on the T-cell effectors. Proc Natl Acad Sci. 2008;105:19857–19862. doi: 10.1073/pnas.0810713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masteller EL, Warner MR, Ferlin W, Judkowski V, Wilson D, Glaichenhaus N, Bluestone JA. Peptide-MHC Class II Dimers as Therapeutics to Modulate Antigen-Specific T Cell Responses in Autoimmune Diabetes. J Immunol. 2003;171:5587–5595. doi: 10.4049/jimmunol.171.10.5587. [DOI] [PubMed] [Google Scholar]
- 32.Grdic D, Ekman L, Schön K, Lindgren K, Mattsson J, Magnusson K-E, Ricciardi-Castagnoli P, et al. Splenic Marginal Zone Dendritic Cells Mediate the Cholera Toxin Adjuvant Effect: Dependence on the ADP-Ribosyltransferase Activity of the Holotoxin. J Immunol. 2005;175:5192–5202. doi: 10.4049/jimmunol.175.8.5192. [DOI] [PubMed] [Google Scholar]
- 33.Tarkowski A, Sun J-B, Holmdahl R, Holmgren J, Czerkinsky C. Treatment of experimental autoimmune arthritis by nasal administration of a type II collagen–cholera toxoid conjugate vaccine. Arthritis Rheum. 1999;42:1628–1634. doi: 10.1002/1529-0131(199908)42:8<1628::AID-ANR10>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 34.Broere F, Wieten L, Klein Koerkamp EI, van Roon JAG, Guichelaar T, Lafeber FPJG, van Eden W. Oral or Nasal Antigen Induces Regulatory T Cells That Suppress Arthritis and Proliferation of Arthritogenic T Cells in Joint Draining Lymph Nodes. J Immunol. 2008;181:899–906. doi: 10.4049/jimmunol.181.2.899. [DOI] [PubMed] [Google Scholar]
- 35.Fukaya T, Takagi H, Sato Y, Sato K, Eizumi K, Taya H, Shin T, et al. Crucial roles of B7-H1 and B7-DC expressed on mesenteric lymph node dendritic cells in the generation of antigen-specific CD4+Foxp3+ regulatory T cells in the establishment of oral tolerance. Blood. 116:2266–2276. doi: 10.1182/blood-2009-10-250472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science. 1994;265:1237–1240. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- 37.Ise W, Nakamura K, Shimizu N, Goto H, Fujimoto K, Kaminogawa S, Hachimura S. Orally Tolerized T Cells Can Form Conjugates with APCs but Are Defective in Immunological Synapse Formation. J Immunol. 2005;175:829–838. doi: 10.4049/jimmunol.175.2.829. [DOI] [PubMed] [Google Scholar]
- 38.Mirenda V, Millington O, Lechler RI, Scott D, Hernandez-Fuentes MP, Read J, Tan PH, et al. Tolerant T cells display impaired trafficking ability. Eur J Immunol. 2005;35:2146–2156. doi: 10.1002/eji.200425823. [DOI] [PubMed] [Google Scholar]
- 39.Poulin M, Haskins K. Induction of Diabetes in Nonobese Diabetic Mice by Th2 T Cell Clones from a TCR Transgenic Mouse. J Immunol. 2000;164:3072–3078. doi: 10.4049/jimmunol.164.6.3072. [DOI] [PubMed] [Google Scholar]
- 40.Koarada S, Wu Y, Ridgway WM. Increased Entry into the IFN-γ Effector Pathway by CD4+ T Cells Selected by I-Ag7 on a Nonobese Diabetic Versus C57BL/6 Genetic Background. J Immunol. 2001;167:1693–1702. doi: 10.4049/jimmunol.167.3.1693. [DOI] [PubMed] [Google Scholar]
- 41.Quinn A, McInerney B, Reich EP, Kim O, Jensen KP, Sercarz EE. Regulatory and Effector CD4 T Cells in Nonobese Diabetic Mice Recognize Overlapping Determinants on Glutamic Acid Decarboxylase and Use Distinct Vβ Genes. J Immunol. 2001;166:2982–2991. doi: 10.4049/jimmunol.166.5.2982. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.