

Summary
Mitochondrial dysfunction is usually associated with aging. To systematically characterize the compensatory stress signaling cascades triggered in response to muscle mitochondrial perturbation, we analyzed a Drosophila model of muscle mitochondrial injury. We find that mild muscle mitochondrial distress preserves mitochondrial function, impedes the age-dependent deterioration of muscle function and architecture, and prolongs lifespan. Strikingly, this effect is mediated by at least two pro-longevity compensatory signaling modules: one involving a muscle-restricted redox-dependent induction of genes that regulate the mitochondrial unfolded protein response (UPRmt); and another involving the transcriptional induction of the Drosophila ortholog of insulin-like growth factor binding protein 7, which systemically antagonizes insulin signaling, and facilitates mitophagy. Given that several secreted IGF-binding proteins (IGFBPs) exist in mammals, our work raises the possibility that muscle mitochondrial injury in humans may similarly result in the secretion of IGFBPs, with important ramifications for diseases associated with aberrant insulin signaling.
Introduction
Mitochondria are key organelles that integrate multiple environmental signals. Disruption of mitochondrial function results in the activation of signaling cascades that reflect attempts by the cell to compensate for the effect of perturbed mitochondria; a phenomenon referred to as retrograde or mitochondrial stress signaling (Liu and Butow, 2006). An emerging area of investigation focuses on characterizing the molecular mechanisms of the adaptive cytoprotective responses to low levels of stress, in particular, oxidative stress in the mitochondrion (also referred to as mitohormesis) (Ristow and Zarse, 2010); and how interfering with these responses impact various metabolic disorders and ultimately aging.
Although severe mitochondrial dysfunction is detrimental, the salutary effects of mild mitochondrial distress have been reported in multiple organisms (Copeland et al., 2009; Dillin et al., 2002; Kirchman et al., 1999; Liu et al., 2005). For example, reducing the expression of some mitochondrial proteins in C. elegans and Drosophila significantly prolongs lifespan (Copeland et al., 2009; Dillin et al., 2002; Hamilton et al., 2005). In addition, mice mutant for the mitochondrially-localized electron transfer redox enzyme p66shc (Migliaccio et al., 1999; Orsini et al., 2004) or the cytochrome c oxidase assembly factor (SURF1) (Dell'agnello et al., 2007) have markedly extended lifespans; and importantly, the latter are also resistant to Ca2+-dependent neurodegeneration. While the exact mechanisms for the beneficial effects of mild mitochondrial distress are unclear, there is growing evidence in the literature to suggest that compensatory stress signaling networks may be a contributing factor. For instance, the increased replicative lifespan associated with mitochondrial perturbation in yeast is abrogated when genes involved in the retrograde response are disrupted (Kirchman et al., 1999); and the prolonged lifespan resulting from electron transport chain (ETC) perturbation in C. elegans is reversed by inactivation of some components of the UPRmt (Durieux et al., 2011). Because a complex signaling network is induced in response to mitochondrial perturbation in yeast (Liu and Butow, 2006), it is possible that a broad and diverse set of molecules may be involved in triggering the beneficial effects observed when mitochondrial function is compromised.
We reasoned that by partially disrupting mitochondrial function, it should be possible to examine the stress signaling networks associated with mitochondrial distress in Drosophila muscles, and the extent to which muscles co-ordinate with other organs to mount an integrated response. We find that muscle ETC perturbation can retard muscle and mitochondrial functional decay; and prolong lifespan, due to compensatory activation of components of the UPRmt, as well as ImpL2 (an ortholog to human IGFBP7) that can bind and inhibit Drosophila insulin-like peptides (Alic et al., 2011; Evdokimova et al., 2012; Honegger et al., 2008). Forced expression of UPRmt genes specifically in muscles, is sufficient to preserve mitochondrial function and delay age-related locomotory impairment. In addition, muscle mitochondrial distress triggers the upregulation of ImpL2, which represses whole-organism insulin signaling, and augments mitophagy by enhancing lysosome biogenesis. Thus, the autonomous induction of the UPRmt, coupled with non-autonomous repression of insulin signaling in response to mild muscle mitochondrial injury, promotes longevity and delays age-dependent muscle functional decline.
Results
Mild Mitochondrial Perturbation in the Larval Musculature Increases Lifespan and Preserves Muscle Function
To examine whether ETC disruption in Drosophila muscles can be beneficial, we knocked down the expression of NDUFS1/ND75, a component of complex I. Two different transgenic RNA interference (RNAi) lines targeting ND75 were crossed to the Dmef2-Gal4 driver (Dmef2 >) to allow muscle-specific knockdown (see Extended Experimental Procedures and Figure S1A). One of the lines (i.e ND75RNAi-strong) produced Dmef2>UAS-ND75RNAi animals which became developmentally arrested around 1.5 to 2 days after egg deposition (AED) with severe myonuclear disintegration (Figure 1A); while the other line (i.e ND75RNAi-weak) gave a weaker phenotype, with larvae arresting after approximately 10 days AED (Figure 1A and Figure S1). Although the latter exhibited smaller muscles, overall muscle integrity was maintained (Figures S1B, S1C and S2). Interestingly, the delayed muscle growth was associated with the induction of stress response factors such as 4E-BP and Superoxide Dismutase 2 (SOD2) (Figures S1B-S1D). We note that while compensatory stress response factors were induced in larval muscles, they experienced an extended developmental delay, and failed to develop into adults (Figure S1E). Thus, to examine whether temporary mitochondrial distress (rather than permanent stress), would allow the larvae to recover to adulthood, we used tub-Gal80ts; Dmef2-Gal4 (hereafter referred to as TD) to control ND75RNAi-weak expression (referred to as TDN), as the temperature sensitivity of the Gal80 inactivates Gal4 at 18C but not at 29C (McGuire et al., 2004). Larvae were collected at 27C, and shifted to 29C for different time intervals (Figure S1F). At the end of 120 hours at 29C (Gal4 fully active), muscle fibers were smaller and sarcomere assembly was severely impaired in muscles with perturbed mitochondria (Figures 1B and 1C). However, when returned to 18C (to reduce the extent of mitochondrial distress), they recovered to adults, albeit with reduced adult size and lifespan (Figures 1D, 1E and Table S1). When maintained at 29C for a shorter duration (24 hours) and then returned to 18C, the adults that emerged had a markedly increased lifespan relative to controls and maintained their climbing ability longer than age-matched wildtype flies (Figure 1F and Table S1). Importantly, the additional expression of Gal80 specifically in the muscles (to impair muscle-restricted expression of the RNAi) suppressed the growth inhibition evident in TDN flies after shifting to 29C, indicating that the phenotypes associated with the transgenic RNAi line are due to muscle-specific expression (Figure S1G). Taken together, these results suggest that while severe and/or persistent mitochondrial injury in muscles is detrimental, mild mitochondrial perturbation has hormetic effects as it delays age-related locomotory impairment and increases lifespan.
Figure 1. Mitochondrial Perturbation Increases Lifespan and Preserves Locomotory Ability.
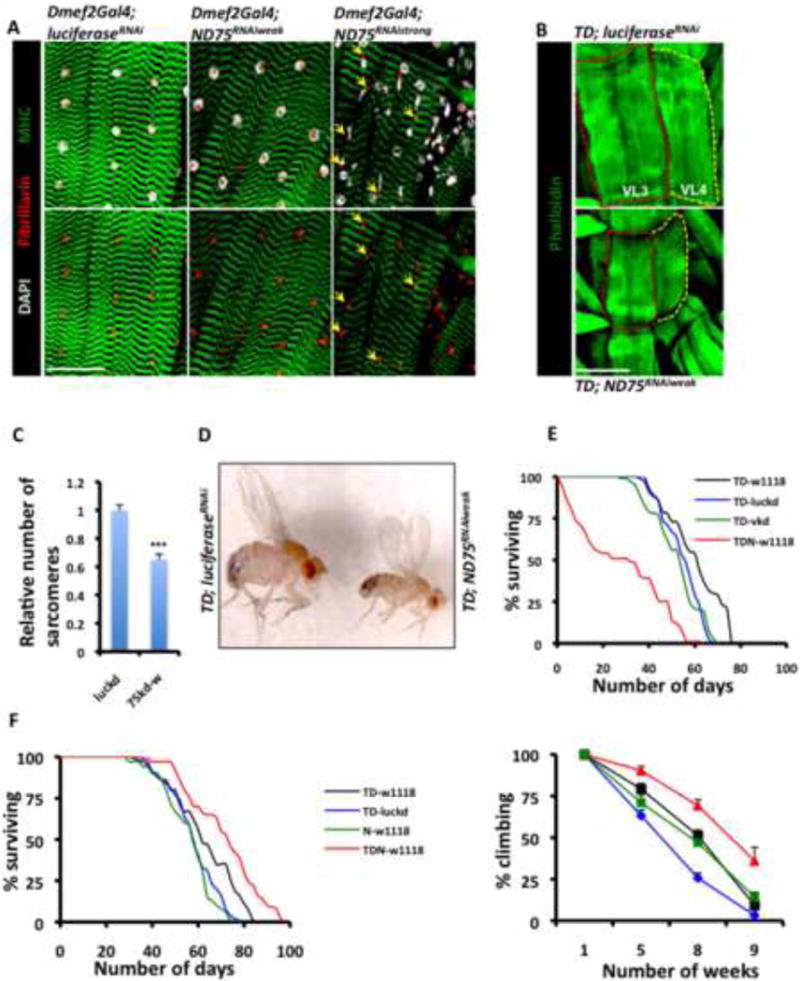
(A) Dmef2-Gal4 larval muscles expressing LuciferaseRNAi (control), ND75RNAi-weak and ND75RNAi-strong. The sarcomere marker Mhc-GFP, the nucleolar marker Fibrillarin, and the nuclear marker, DAPI are shown in green, red and grey, respectively. Note the nuclear disintegration (yellow arrows) of muscles expressing ND75RNAi-strong. Scale bar is 75μm.
(B) Ventral longitudinal 3 and 4 muscle fibers (i.e VL3 and VL4) expressing ND75RNAi-weak (lower panel) are smaller than aged-matched fibers expressing LuciferaseRNAi (top panel), Scale bar is 75μm.
(C) Relative number of sarcomeres in VL3 and VL4 muscle fibers of the larval genotypes described in (B). luckd and 75kd-w refer to TD; LuciferaseRNAi and TD; ND75RNAi-weak respectively. Error bars denote mean ± standard error of the mean (s.e.m), n= 12 muscle fibers, and *** denotes p<0.001.
(D) 120 hours of ND75RNAi-weak expression in larval muscles results in smaller adult flies (right) relative to controls with larval expression of LuciferaseRNAi (left).
(E) Lifespan analysis of the adult flies described in (D). Lifespan curves are for flies that emerged from larvae expressing ND75RNAi-weak (red), vermillionRNAi (green) and LuciferaseRNAi (blue) in their muscles. The black curve represents flies from a cross between the TD driver (see text) and wildtype (i.e., w1118) flies.
(F) Lifespan analysis (left panel) and climbing ability (right panel) of adult flies that developed from larvae that were collected at 27C, shifted to 29C for a day and returned to 25C. Color codes are as in D, except that the green in these panels refers to offspring from a cross between w1118 and ND75RNAi-weak. Error bars in climbing graph indicate mean ± s.e.m; n=4 independent cohorts of flies, with the starting number of flies for each genotype in each cohort = 100 and p < 0.05 for TD versus TDN at 5, 8 or 9 weeks. Because LuciferaseRNAi flies had reduced climbing ability as they aged, comparisons are between TDN-w1118 (red) and TD-w1118 (black).
See Figure S1 and S2; and Table S1 for log-rank test, mean, median and maximum lifespan.
Antioxidant Expression Suppresses the Increased Lifespan and Preserved Locomotory Activity Associated with Mild Muscle Mitochondrial Distress
We further observed that TDN muscles had elevated ROS levels relative to controls (Figures 2A and 2B). Depending on the context, elevated ROS levels can be beneficial or cytocidal (Peternelj and Coombes, 2011; Powers et al., 2010). To examine the functional significance of the elevated ROS in this context, we expressed antioxidant enzymes (i.e., enzymes that scavenge ROS) in muscles with ND75 function impaired and examined the effect on longevity and climbing ability. Interestingly, forced expression of catalase (which reduces Hydrogen Peroxide to water) abolished the enhanced locomotory ability and lifespan of these flies (Figure 2C and Table S2). As catalase overexpression in the mitochondrial mutant suppresses the lifespan beyond that seen in the wild-type strain, it appears that scavenging ROS uncovers the deleterious effect of complex I perturbation. Similar results were obtained with Glutathione Peroxidase I (GTPx-1), which also reduces Hydrogen Peroxide to water (Lubos et al., 2011; Missirlis et al., 2003) (Figure 2D and Table S2). These results indicate that at least one of the signaling axes that confers tolerance to mitochondrial distress in this context involves a redox-mediated pathway.
Figure 2. Antioxidant Enzymes Suppress the Enhanced Longevity and Locomotory Behavior Associated with Complex I Perturbation.
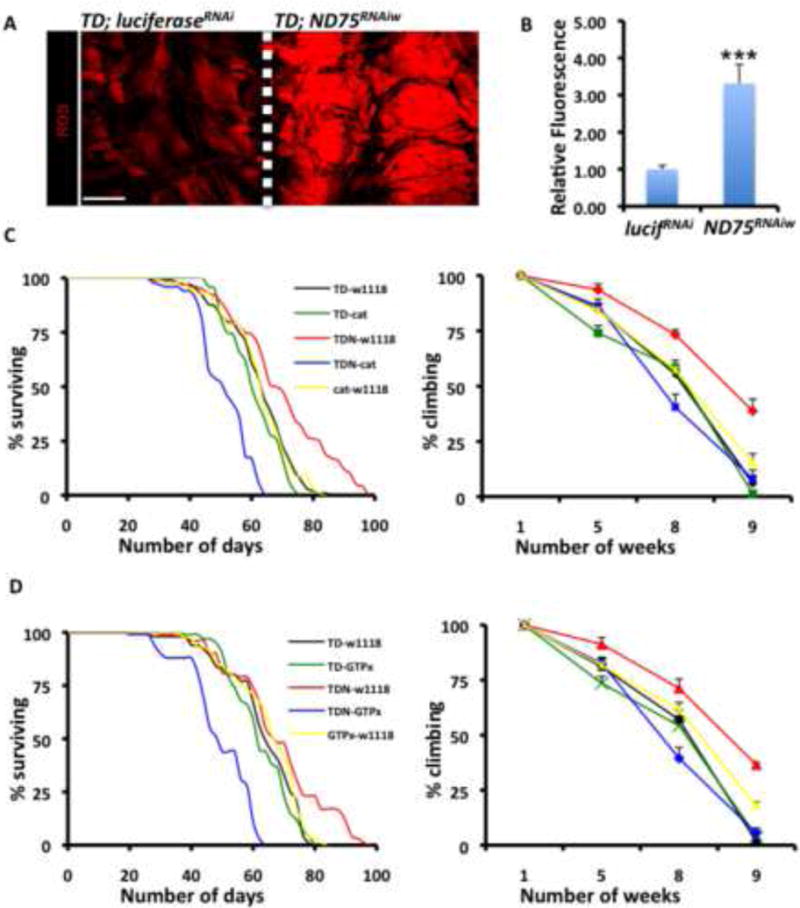
(A) ROS levels as assessed by Dihydroethidium (DHE) are elevated in third instar larval muscles with ND75 disrupted (right panel) relative to controls (left panel). Scale bar is 150μm.
(B) Mean signal intensities of DHE-stained muscle fibers described in A; Error bars denote mean ± s.e.m, n = 10 samples pooled from two independent experiments, and *** denotes p<0.001.
(C) Lifespan (left panel) and climbing ability (right panel) of flies that emerged from larvae collected at 27C, shifted to 29C for a day and returned to 25C. Genotypes are TDN crossed to w1118 (red), TDN crossed to catalase (blue), TD crossed to w1118 (black), TD crossed to catalase (green) and catalase crossed to w1118 (yellow). Error bars in right panel indicate mean ± s.e.m; n=4 independent cohorts with the starting number of flies for each genotype in each cohort = 100, and p < 0.01 for TDN-w1118 versus TDN-cat at 8 or 9 weeks. p value at 5 weeks is 0.129 (not significant).
(D) Similar to C, but with an alternate antioxidant enzyme GTPx-1, instead of catalase. p < 0.01 for TDN-w1118 versus TDN-GTPx-1 at 8 or 9 weeks. Differences observed at 5 weeks are not statistically significant.
See Table S2 for additional lifespan data.
Synthetic Lethal Screen to Identify Genes Required for Conferring Tolerance to Muscle Mitochondrial Injury
We designed a synthetic lethal screen to identify RNAi lines that (while viable when crossed to TD) produce a lethal phenotype in combination with TDN (see Extended Experimental Procedures and schematic in Figure S3). Significantly, RNAi lines targeting well-established cytoprotective factors such as AMP-activated protein kinase (AMPK), and PTEN-induced kinase 1 (PINK1), which is essential for mitochondrial quality control (Clark et al., 2006), satisfied these two criteria (Table S3). We also identified Hsp60C as a gene that is preferentially required in muscles with perturbed mitochondrial function (Table S3).
Hsp60C is an ortholog of the bacterial GroEL. Orthologs of GroEL in C. elegans have been implicated in the UPRmt; which is a protein quality control mechanism where mitochondrial distress activates the transcription of nuclear-encoded chaperones and proteases to restore protein homeostasis in the mitochondrial matrix (Haynes and Ron, 2010). Other nuclear-encoded mitochondria-localized chaperones are the Hsp70 family member mtHsp70, and the chaperonin Hsp10, an ortholog of bacterial GroES (Haynes and Ron, 2010). There is at least one Drosophila ortholog of mtHsp70 (Hsc70-5), and two orthologs of Hsp10 (CG11267 and CG9920). In addition, the protease ClpP resides in the matrix of mitochondria and is involved in the UPRmt in C. elegans (Haynes and Ron, 2010). ClpP functions in concert with ClpX (CG4538 in Drosophila). Because Hsp60C disruption is synthetic lethal in mitochondrial mutant flies (Table S3), we hypothesized that the UPRmt might be an essential adaptive response process that conveys some of the beneficial effects of flies with perturbed mitochondrial function. To test this hypothesis, we performed a synthetic lethal test with the Drosophila ortholog of ClpX in the mitochondrial mutant muscles. Similar to Hsp60C, disruption of ClpX in muscles with perturbed mitochondrial function potently compromised viability, while sparing wildtype flies (Table S3).
Adult-onset Muscle Mitochondrial Perturbation Increases Lifespan and Preserves Mitochondrial and Muscle Function
Because of the developmental delay observed when the mitochondrial mutant larvae are shifted to 29C, we wondered whether the effects of disrupting the UPRmt genes were simply developmental, or perhaps degenerative. To resolve these concerns, we analyzed the effect of mitohormesis in adult flies. We compared TD flies (with or without UAS-ND75RNAi-weak expression, referred to as experimentals and controls respectively) raised at 18C during development but shifted to 27C as adults to allow disruption of ND75 in muscles during the adult stage (Figure S1F). No overt phenotypic differences or developmental delays were observed when both flies were raised at 18C (or even up to 25C) before eclosion. However, when shifted to 27C, lifespan was significantly increased in flies with ND75 (or other complex I proteins) disrupted in their muscles (Tables S4 and S5). In addition, the elevated temperature at the adult stage accelerated the appearance of mitochondrial degenerative phenotypes; such that by 6 weeks at 27C, mitochondrial DNA copy number of control flight muscles had fallen to about 60% of the value for the experimentals (Figure 3A). The preservation of mitochondrial mass was even more evident in transmission electron micrographs (TEM). Indeed, in control muscles, mitochondrial degeneration was extensive, resulting in large hollow interstitial spaces between remnants of mitochondria and myofibrils. However, mitochondrial morphology in the experimental muscles was largely preserved, leaving very few voids between the myofibrils and mitochondria (Figures 3B, 3C, S4A and S4B). In addition, the remaining mitochondria in control muscles had fragmented cristae, in stark contrast to the well-arranged and compact cristae of mitochondria in the experimentals (Figures 3B, 3C, S4A and S4B). ATP levels were significantly elevated in the experimentals relative to controls (Figure 3D). Finally, Citrate Synthase (CS) activity, which serves as a surrogate indicator of mitochondrial matrix integrity was elevated in the mitochondrial mutant muscles relative to control (Figure 3E). Similar results were obtained when Dihydrolipoamide Dehydrogenase (DLD, another mitochondrial matrix enzyme) activity was assessed (Figure 3F). Finally, by 7 weeks at 27C muscle architecture had become severely disordered in control flies, to the point where transected myofibers and severely disrupted sarcomeres were frequently seen (Figures 3G-3J); however, none of these features were evident in aged-matched muscles expressing ND75RNAi (Figures 3G-3J).
Figure 3. Adult-onset Muscle Mitochondrial Perturbation Increases Lifespan and Preserves Mitochondrial and Muscle Function.
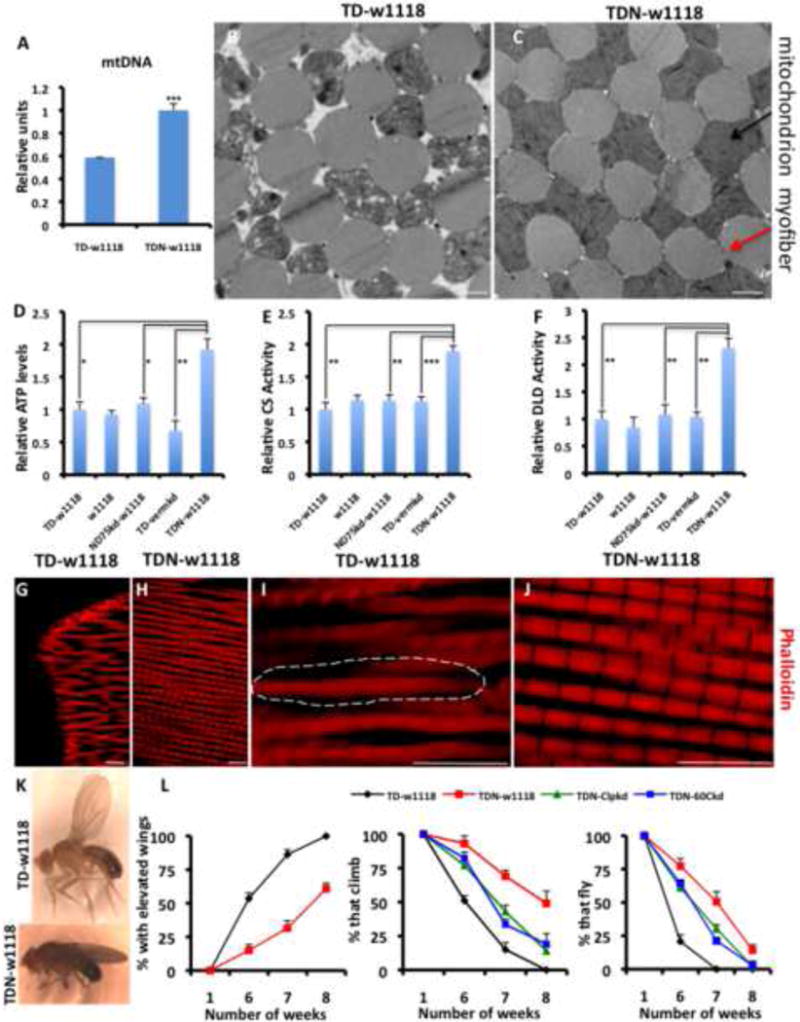
(A) Mitochondrial mass assessed as relative mitochondrial DNA levels, is higher in TDN muscles relative to TD. n=4.
(B and C) TEMs of TD (B) and TDN (C) muscles. Note the large interstitial spaces between remnants of degenerating mitochondria (black arrow) and myofibrils (red arrow) in the TD muscles. Scale bar is 1.0μm
(D-F) Relative ATP levels (D), Citrate Synthase (CS) activity levels (E), and Dihydrolipoamide Dehydrogenase (DLD) activity levels (F). Figure labeling is as follows: (i) tub-Gal80ts;Dmef2-Gal4 crossed to w1118 labeled as TD-w1118 (negative control), (ii) w1118 (i.e as wildtype flies), (iii) The ND75RNAi-weak line crossed to w1118 labeled as ND75kd-w1118 (control for insertion site), (iv) tub-Gal80ts;Dmef2-Gal4 crossed to a vermillionRNAi line labeled as TD-vermkd (RNAi negative control), and (v) tub-Gal80ts,ND75RNAi-weak;Dmef2-Gal4 crossed to w1118 labeled as TDN-w1118. n=4.
(G-J) Indirect Flight Muscles of seven-week old TD (G and I) and TDN (H and J) flies stained with phalloidin (red) to mark the sarcomeres. (I and J) are higher magnifications of (G and H), respectively (see scale bars). Note the severely disorganized sarcomere structure of TD flight muscles; the broken lines surround an area where the sarcomeres are indistinct, in contrast to the well-arranged sarcomeres of TDN flies. In addition, TD myofibers were frequently found to be transected, as shown in G. Scale bar is 10.0μm
(K) An example of a TD fly with elevated wings (top) compared to a TDN fly (lower panel).
(L) In all panels, TD flies are shown in black, TDN in red, TDN with ClpX knockdown, green, and TDN with Hsp60C knockdown is shown in blue. Left panel; most TD flies have elevated wings relative to TDN flies. Middle panel; climbing ability is preserved in TDN flies as they age, compared to TD flies. Knockdown of ClpX or Hsp60C suppresses the enhanced climbing ability. Far right panel shows the flight ability of the flies, which is preserved in TDN flies. n=4 independent cohorts with the starting number of flies for each genotype in each cohort = 100, p<0.001 for all comparisons between TD and TDN at 6, 7 and 8 weeks.
Error bars denote s.e.m; and * = p<0.05, ** = p<0.01 and *** = p<0.001. Also see Figures S3-S5 and Tables S3-S5.
We next examined various behavioral phenotypes that correlate with disrupted muscle or mitochondrial function (Clark et al., 2006; Greene et al., 2003). Control flies developed the elevated wing phenotype (a property of flies with severe disruption of indirect flight muscle mitochondria (Clark et al., 2006)) after 6 weeks at 27C (Figures 3K and 3L). In contrast, less than 20% of flies expressing ND75RNAi developed this phenotype after 6 weeks (Figures 3K and 3L). In addition, flies expressing ND75RNAi displayed better climbing ability and flight performance when compared to controls (Figures 3L). We note that while RNAi targeting Hsp60C or ClpX suppressed the increased climbing/flight performance and lifespan of TDN flies, they also affected wildtype flies (Figures 3L and Table S5). These results show that basal levels of UPRmt genes are required to sustain normal mitochondrial function as flies age, similar to what has been observed for other critical mitochondrial quality control proteins such as PINK1 and Parkin (Clark et al., 2006; Greene et al., 2003).
Forced Expression of UPRmt Genes is Sufficient to Preserve Muscle and Mitochondrial Function
We examined whether forced expression of UPRmt genes is sufficient to positively modulate longevity and/or mitochondrial and muscle function. Because there are 4 paralogs of Hsp60 in Drosophila (Hsp60, Hsp60B, Hsp60C and Hsp60D), we performed synthetic lethal tests to examine if the other 3 are required in TDN muscles for survival. Although RNAi to Hsp60B or Hsp60D did not affect either TDN or TD, disruption of Hsp60 suppressed the survival of both TDN and TD flies. However, induction of Hsp60 in larval TDN muscles (perhaps as a compensatory response), suggests that it may have a cytoprotective function in addition to its apparent developmental role (Figure S4C). To test this hypothesis and to delineate a cytoprotective role for the UPRmt in muscles, we generated transgenic UAS lines of Hsp60 and Hsp60C and expressed them in muscles using the Mhc-Gal4 driver at 25C. No lifespan-promoting effects were observed when the UAS lines were crossed to w1118 flies (Figure 4A). However, although less than 40% of Mhc-Gal4/w1118 flies survived beyond 80 days, 60-80% of the flies expressing two independent UAS constructs of either Hsp60 or Hsp60C survived beyond 80 days, suggesting that they had markedly increased lifespans relative to wildtype controls (Figure 4B). In addition, ATP levels, CS, and DLD activities were elevated in muscles expressing Hsp60 after 5 weeks at 25C (Figures 4C-4E); and this was associated with a preservation of locomotory activity of such flies (Figure 4F). These results show that the induction of the UPRmt genes, such as Hsp60 and Hsp60C in muscles, preserves mitochondrial function, promotes longevity and delays age-dependent muscle functional decline.
Figure 4. Forced Expression of UPRmt Genes is Sufficient to Preserve Muscle and Mitochondrial Function.
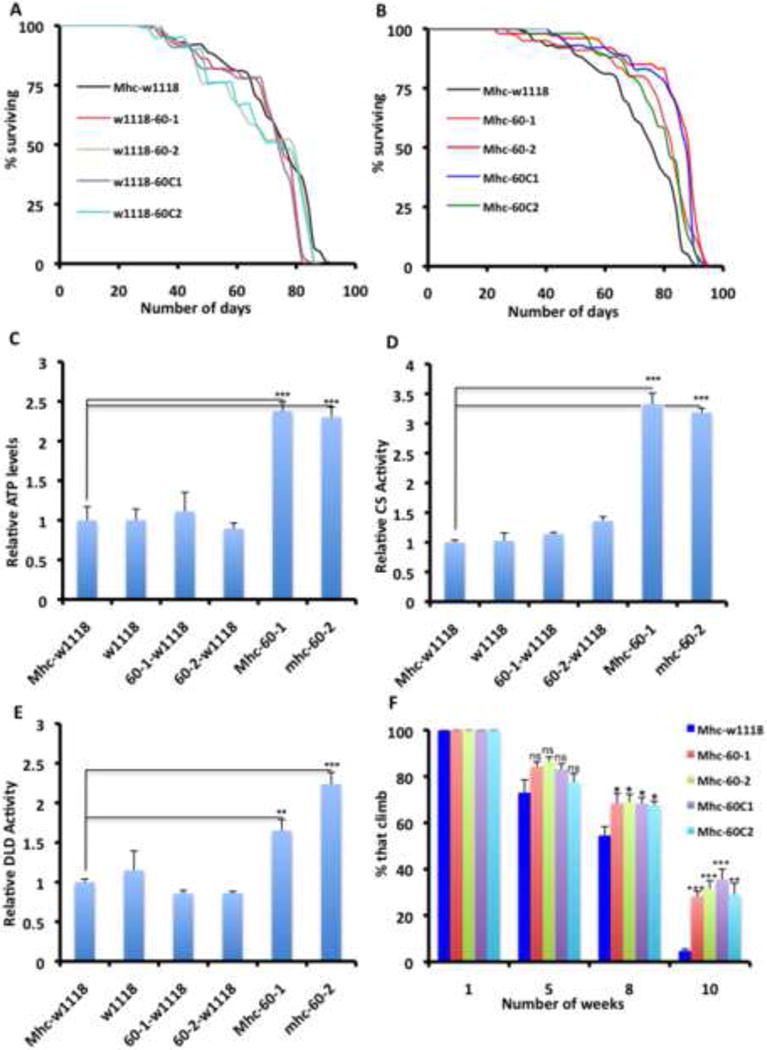
(A) Lifespan curves of flies expressing Hsp60 and Hsp60C outcrossed to w1118 flies, compared to Mhc-Gal4 flies outcrossed to w1118 flies.
(B) Lifespan curves of flies expressing Hsp60 or Hsp60C crossed to Mhc-Gal4, compared to Mhc-Gal4 flies crossed to w1118 flies. All transgenes result in an increase in lifespan relative to the background control. Note that the Mhc-w1118 curves are the same in A and B, as the lifespan assays in A and B were performed concurrently.
(C-E) Relative ATP levels, CS activity levels and DLD activity levels respectively, of thoraxes from flies with the genotypes listed, after 5 weeks at 25C. Figure labeling is as follows: (i) Mhc-Gal4 crossed to w1118 labeled as Mhc-w1118 (negative control), (ii) w1118 (i.e as wildtype flies), (iii) UAS-hsp60 insertion #1 crossed to w1118 labeled as 60-1-w1118 (control for insertion site #1), (iv) UAS-hsp60 insertion #2 crossed to w1118 labeled as 60-2-w1118 (v) Mhc-Gal4 crossed to UAS-hsp60 insertion #1 or #2, labeled as Mhc-60-1 and Mhc-60-2 respectively. n=4, error bars denote s.e.m; and * = p<0.05, ** = p<0.01 and *** = p<0.001.
(F) Climbing ability of flies expressing Hsp60 or Hsp60C under the control of the Mhc-Gal4 driver. Data shown is for three independent experiments, and is expressed as the mean ± s.e.m. The starting number of flies for each genotype in each experiment = 100, and * = p<0.05, ** = p<0.01 and *** = p<0.001. p values shown refer to comparisons between the particular genotype and Mhc-Gal4/w1118 flies (deep blue columns). Climbing ability was not preserved when the Hsp60/60C transgenic flies were crossed to w1118 flies (not shown).
See Figure S4C and Table S6R.
Redox Signaling Autonomously Regulates the Mitochondrial Unfolded Protein Response
We explored whether UPRmt genes are induced in TDN muscles as an adaptive compensatory response to mitochondrial injury. Interestingly, within a week of shifting to 27C most UPRmt markers were induced in TDN muscles (Figure 5A); an event which was recapitulated when another complex I protein (CG9762), which also results in increased lifespan, was disrupted (Figures 5B and S5, and Table S4). Mitochondrial stress signaling typically involves kinases activating specific transcription factors that culminate in the upregulation of genes that compensate for the effect of the dysfunctional mitochondria (Finley and Haigis, 2009). Because overexpression of antioxidant enzymes suppressed the hormetic effects associated with complex I perturbation (Figure 2 and Table S2), we hypothesized that at least part of the mechanism that results in increased lifespan and sustained locomotory activity in TDN flies likely involves a redox-sensitive signaling cascade. Accordingly, we searched for redox-sensitive signaling pathways that are concurrently active when UPRmt genes are induced. We observed that the JNK target puckered (puc) was induced concurrently with UPRmt markers (Figure 5C). Importantly, overexpression of antioxidant enzymes (either GTPx-1 or catalase) abrogated the induction of the JNK target puc (Figures 5D and 5E), and several of the UPRmt markers (Figure 5D), indicating that redox signaling regulates UPRmt induction. Additionally, forced expression of the JNK effector transcription factor D-Jun, resulted in induction of several of the UPRmt markers (Figure 5F); and disruption of Jun (D-Jun) abrogated the enhanced lifespan of TDN flies (Table S5). Overexpression of the redox-sensitive NF-кB transcription factor, Relish, also resulted in induction of several of the UPRmt markers (Figure 5G); and we note that although antioxidant expression suppressed the induction of several UPRmt genes, Hsp10 expression was refractory to antioxidant expression (Figure 5D). Thus, there may be additional signaling modules for regulating UPRmt induction independent of the JNK pathway. Nevertheless, these results show that in response to mitochondrial complex I perturbation, a redox-mediated mitochondrial stress signaling network impinging in part on the JNK pathway, results in activation of the UPRmt.
Figure 5. Redox Signaling Autonomously Regulates the Mitochondrial Unfolded Protein Response.
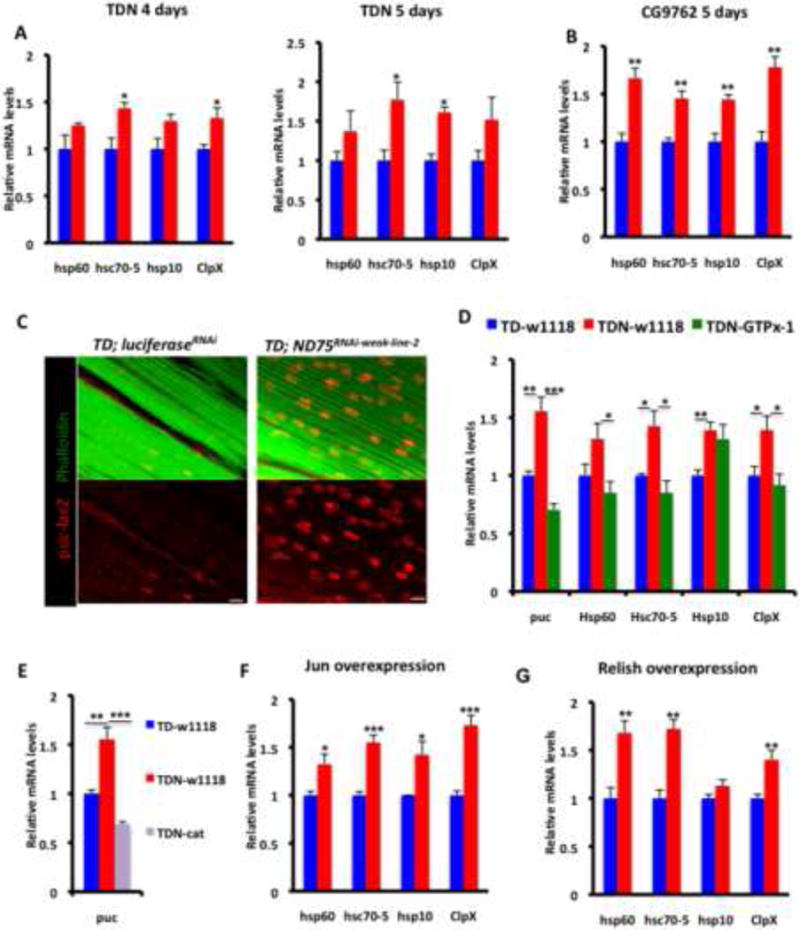
(A) Expression of UPRmt markers in TDN muscles (red) relative to TD muscles (blue) after 4 (left) or 5 (right) days at 27C.
(B) Expression of UPRmt markers in CG9762 muscles (red) relative to TD muscles (blue) after 5 days at 27C.
(C) puc-lacZ expression in TD-LuciferaseRNAi (left) and TDN (right) muscles. Scale bar is 10μm.
(D) GTPx-1 expression in TDN muscles suppresses the induction of the JNK target, puckered, and several of the UPRmt markers in TDN muscles. TDN (red), TDN-GTPx-1 (green) and TD control (blue).
(E) The expression of another antioxidant enzyme, catalase, in TDN muscles also suppresses the extent of induction of puckered,
(F) Expression of UPRmt markers in muscles expressing the AP-1 transcription factor Jun.
(G) Expression of UPRmt markers in muscles expressing Relish.
In all qPCR panels, fold induction shown refers to the mean ± s.e.m, n=4 independent experiments, and * = p<0.05, ** = p<0.01 and *** = p<0.001.
ImpL2 Secretion from Muscles with Mitochondrial Distress Triggers Non-Autonomous Repression of Insulin Signaling
TDN flies maintained at 29C for 120 hours, and then returned to 18C had muscles that were considerably smaller than wildtype controls (Figures 1B and 1C). However, wing length was shorter and the overall size of the flies was also smaller (Figures 1D and 6A), raising the possibility that the growth-inhibiting effect of mitochondrial injury in Drosophila muscles may be propagated to other organs/tissues. Interestingly, one of the genes that we identified in our synthetic lethal screen was ImpL2. This observation suggested the possibility that ImpL2 may be secreted from flight muscles in the thorax (the muscles with perturbed mitochondrial function) to cause systemic repression of insulin signaling, and thus contribute to the extended longevity of TDN flies. Strikingly, ImpL2 is induced in flight muscles of the thorax following mitochondrial injury (Figure 6B); an event that correlates with the induction of markers of insulin suppression, such as 4E-BP and InR (Figure 6B) (Karpac et al., 2011). Significantly, 4E-BP and InR were also induced non-autonomously in the abdomen and head (Figure 6B). To further confirm the non-autonomous repression of insulin signaling, we used the tGPH reporter – a fusion protein of GFP and the pleckstrin homology domain of general receptor for phosphoinositides-1 (GRP1), that is recruited to the plasma membrane when cellular levels of phosphatidylinositol-3,4,5-P3 (PIP3) are elevated in response to increased PI3-kinase activity. In the fat body of TD flies, we observed robust membrane localization of the tGPH reporter; however, in TDN flies, tGPH expression was largely restricted to the nucleus and cytoplasm (Figure 6C). Taken together, we conclude that disruption of complex I in muscles of TDN flies reduces insulin signaling non-autonomously.
Figure 6. ImpL2 Secretion from Muscles with Mitochondrial Distress Triggers Non-Autonomous Repression of Insulin Signaling.
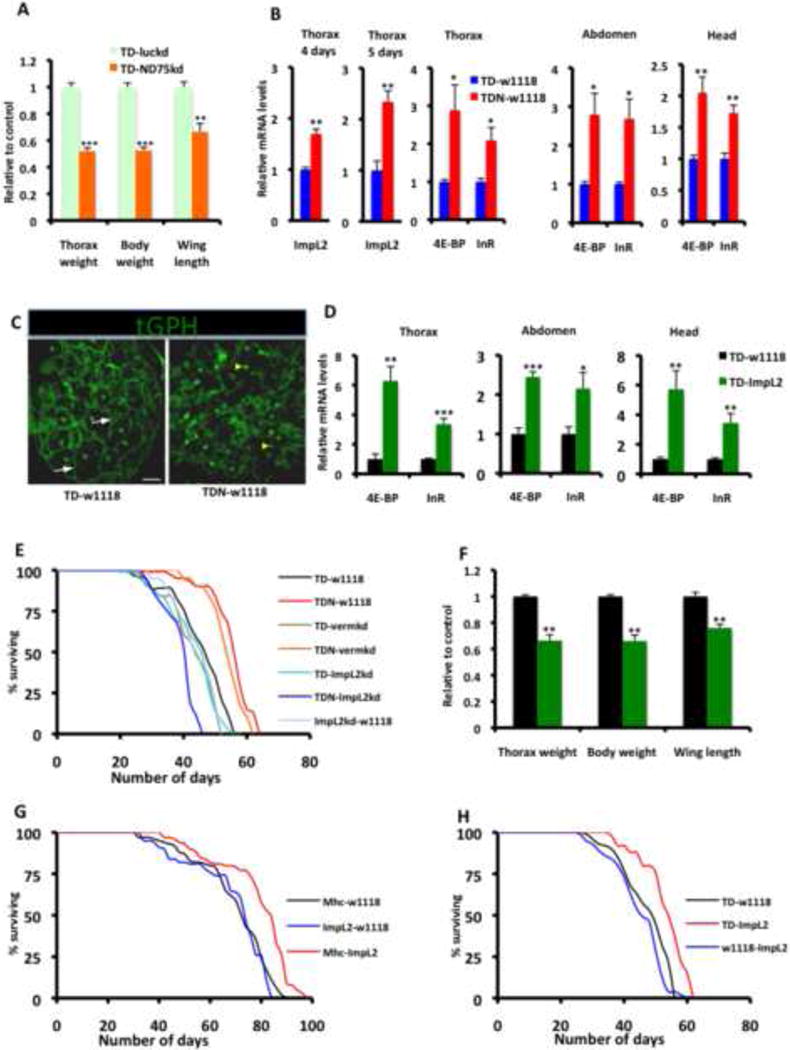
(A) Relative thorax weight, overall body weight and wing length of flies expressing RNAi to ND75 (TD-ND75kd) relative to control flies expressing RNAi to luciferase (TD-luckd). Larvae were collected at 29C for 120 hours and then returned to 18C until adults eclosed. In each instance, n = 4 cohorts, with each cohort consisting of 20 flies.
(B) ImpL2 is induced in TDN flight muscles relative to TD flight muscles at 4 (first panel, from left) and 5 (second panel) days after shifting adult flies to 27C. This correlates with the induction of 4E-BP and InR in flight muscles (thorax) of TDN flies (third panel). In addition, these markers are induced non-autonomously in the abdomen (fourth panel) and head (fifth panel) of TDN muscles
(C) Localization of tGPH (a PI3K activity reporter) in fat body from TD (left) and TDN (right) flies. Note the extensive membrane recruitment of tGPH (white arrows) in fat body from TD flies. However, in fat bodies from TDN flies, expression of the reporter was largely restricted to the nucleus (yellow arrowhead) and surrounding cytoplasm. Scale bar is 20μm
(D) Overexpression of ImpL2 in the thorax results in the induction of targets of insulin repression in the thorax (left panel) as well as in the abdomen (middle panel) and head (far right panel).
(E) Lifespan curves of flies expressing TDN or TD with various transgenes. The increased lifespan of TDN flies outcrossed to w1118 flies (red, TDN-w1118) or vermillionRNAi (orange, TDN-vermkd, which serves as a negative control for RNAi expression) is suppressed when TDN is crossed to ImpL2RNAi (blue, TDN-ImpL2kd).
(F) Thorax weight, total body weight and wing length of flies expressing ImpL2 (TD-ImpL2) relative to control flies (i.e TD-w1118). In each instance, n = 4 cohorts, with each cohort consisting of 20 flies. Note the considerably smaller thoraxes, total body weight, and shorter wing length of TD-ImpL2 flies relative to TD-w1118 controls.
(G) Lifespan curves of flies expressing UAS-ImpL2 outcrossed to w1118 flies compared to Mhc-Gal4 flies outcrossed to w1118 or UAS-ImpL2 flies. Lifespan was performed at 25C.
(H) Lifespan curves (performed at 27C) of flies expressing UAS-ImpL2 outcrossed to w1118 flies, compared to TD flies crossed to w1118 or UAS-ImpL2 flies.
In all panels, fold change shown refers to the mean ± s.e.m and * = p<0.05, ** = p<0.01 and *** = p<0.001. n=4 independent experiments. Also see Figure S6 and Table S7
Interestingly, forced expression of ImpL2 in flight muscles recapitulated both the autonomous and non-autonomous induction of 4E-BP and InR (Figure 6D); and disruption of ImpL2 function in TDN muscles, via either RNAi or a loss-of-function allele, abrogated the extended longevity of TDN flies, underscoring the importance of systemic repression of insulin activity in response to mitochondrial injury in muscles (Figures 6E, S6A and S6B; and Table S7A). Moreover, larval overexpression of ImpL2 in muscles using the Dmef2-Gal4 or Mhc-Gal4 drivers recapitulated the reduced organ size and overall body weight of flies with severely compromised mitochondrial function in their muscles (Figures 6F, also see Figures S6C and S6D). Finally, overexpression of ImpL2 in muscles using either Mhc-Gal4 or TD was sufficient to increase lifespan (Figure 6G and 6H; and Table S7B and S7C).
ImpL2 Expression Increases Lysosome Biogenesis, Possibly to Augment Autophagic Flux in TDN muscles
Because overepression of ImpL2 in muscles did not result in UPRmt induction (not shown), we hypothesized that ImpL2 likely induces an alternate cell maintenance/repair mechanism to confer tolerance to mitochondrial perturbation. Autophagy is an essential catabolic process that sequesters damaged organelles, misfolded proteins or pathogens in the lysosome for degradation; it generally has a pro-survival function when induced in response to stress (Rubinsztein et al., 2012). Mitophagy is a specific form of autophagy that culls damaged mitochondria into autophagosomes for eventual degradation after fusion with lysosomes. In view of the induction of the UPRmt in TDN muscles, we asked whether mitophagy – an alternate mitochondrial quality control process – is induced in TDN muscles. A transgenic line expressing GFP fused with a mitochondrial-targeting signal (mito-GFP) correctly labels mitochondria in flight muscles (Figure 7A). Because the mitochondrial matrix is enriched with endogenously biotinylated proteins, streptavidin (which binds to biotin) can be used to detect mitochondria, especially in tissues where mitochondria are abundant (Hollinshead et al., 1997). Indeed, in flight muscles, mito-GFP co-localizes with streptavidin (Figure 7A); thus, streptavidin is an appropriate marker for mitochondria in this tissue. Accordingly, to test whether mitophagy is induced in TDN muscles, we examined flight muscles of TDN and TD flies 5 days after shifting to 27C, for co-localization between autophagosomes, (which can be identified as Atg8-GFP punctae) (Rusten et al., 2004) and streptavidin. At these early stages of adult life, Atg8-GFP punctae were largely undetectable in TD flight muscles (Figure 7B). However, we detected co-localization between Atg8-GFP and streptavidin in TDN muscles (Figure 7C). To confirm these observations, we examined TEMs of TD and TDN flight muscles at the same time point that the Atg8-GFP punctae were detected. Under normal conditions, mitochondria in flight muscles can be observed in TEMs as long parallel stripes (in longitudinal sections) or as a reticular network (in transverse sections) that alternate with myofibrils. In TDN muscles, we reproducibly detected mitochondria that had separated from the reticular network and had become encased in autophagosomes at different stages of formation (i.e both fully formed and partially formed autophagosomes were observed); however, comparable sequestered structures were not observed in TD muscles (Figures 7D-7G). Because autophagosomes subsequently fuse with lysosomes, we examined whether there were changes in the number of lysosomes. TDN muscles clearly had more lysosomes than TD muscles (Figures 7H-7I); and ImpL2 overexpression potently increased the number of lysosomes, when compared to TD muscles (Figure 7J). Importantly, a loss-of-function allele of ImpL2, that suppresses the extended longevity of TDN flies (Figure S6B), partially suppressed the number of lysosomes formed in TDN muscles (Figure 7K). We note however, that forced expression of ImpL2 did not significantly alter the number of autophagosomes formed, as assessed by either Atg8-GFP or Hu-LC3-eGFP (not shown). On the other hand, disruption of Atg6, Atg8A or Atg12, which are core autophagy genes, abrogated the increased lifespan of TDN flies (Figures 7L and 7M; and Table S8). Collectively, these results support a model where mitophagy is upregulated in TDN muscles and that ImpL2 acts primarily to expand the number of lysosomes; possibly to augment autophagic capacity.
Figure 7. ImpL2 Expression Increase Lysosome Biogenesis, Possibly to Augment Autophagic Flux in TDN muscles.
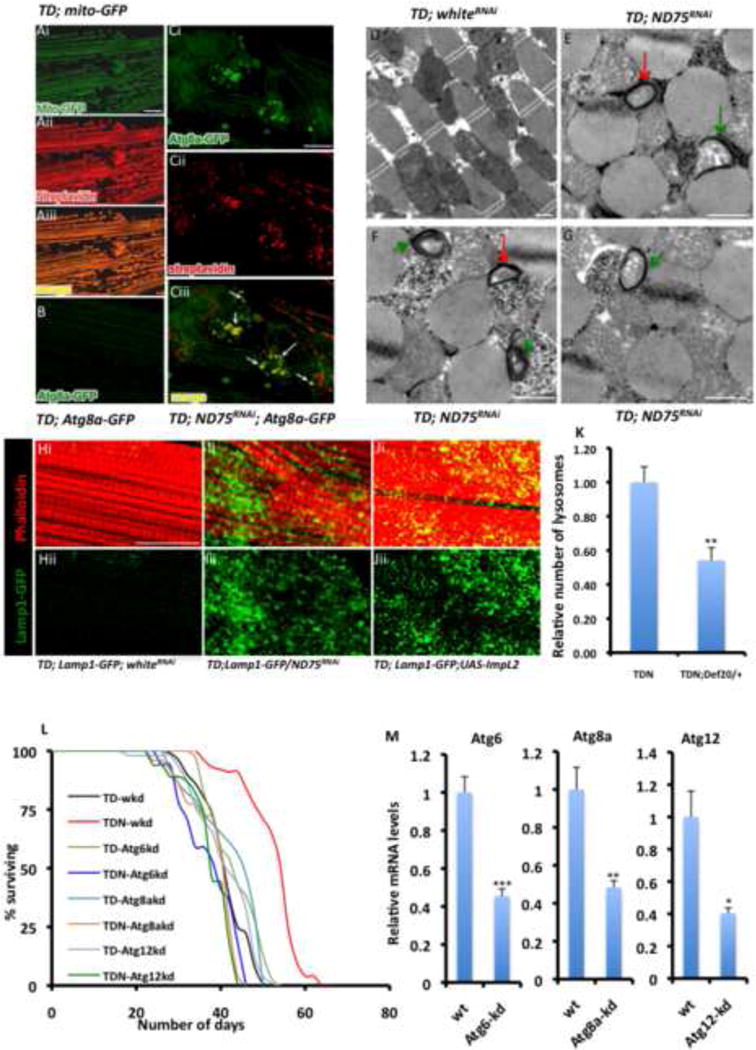
(A) A transgenic UAS-GFP line with a mitochondrial targeting sequence (mito-GFP) specifically labels mitochondria in flight muscles (i) and co-localizes with streptavidin AlexaFluor 594 staining (ii) as shown in (iii). Scale bar is 20μm
(B and C) Representative confocal images from control flies, TD (B) and TDN (Ci to Ciii) flight muscles showing UAS-Atg8-GFP expression, a marker of autophagosomes. (iii) is a merged image of i and ii. Note the extensive overlap between Atg8-GFP and streptavidin, denoted by the white arrows. Scale bar is 20μm
(D-G) Representative TEMs of TD (D) and TDN flight muscles (E-G) showing autophagosomes at different stages of formation after 5 days at 27C. Fully or partially formed autophagosomes, are denoted with red or green arrows respectively. Scale bar is 1μm
(H-J) Phalloidin staining is shown in red. The lysosomal marker, Lamp1-GFP (green) is very weakly expressed in TD - whiteRNAi muscles (Hi and ii), but is robustly expressed in TDN muscles (Ii and ii) or muscles overexpressing ImpL2 (Ji and ii). whiteRNAi controls for titration of Gal4; see Extended Experimental Procedures. Scale bar is 20μm
(K) Relative number of lysosomes in TDN and TDN;Def20/+ thoraxes after 5 days at 27C. The number of lysosomes in TDN;Def20/+, assessed as the relative number of punctae that stain for Lamp1-GFP in confocal z-stack images was normalized to the corresponding value for TDN. Error bars denote mean ± s.e.m, and ** denotes p<0.01; n= 6
(L) Lifespan curves at 27C of flies expressing TDN or TD with transgenic RNAi constructs to Atg6, Atg8a and Atg12 labeled as TDN-Atg6kd, TDN-Atg8akd or TDN-Atg12kd. Note that the increased lifespan of TDN flies outcrossed to whiteRNAi flies (red, TDN-wkd) is abrogated in TDN-Atg6kd, TDN-Atg8akd or TDN-Atg12kd flies. No lifespan-promoting effects were evident when any of the Atg RNAi lines were crossed to whiteRNAi flies.
(M) Transcript levels in adult TDN-wkd (labeled wt) thoraxes relative to TDN-Atg6kd, TDN-Atg8akd or TDN-Atg12kd following the induction of RNAi against Atg6, Atg8a and Atg12 respectively for 5 days at 27C. Fold change shown refers to the mean ± s.e.m, n=4 independent experiments, and * = p<0.05, ** = p<0.01 and *** = p<0.001.
See Table S8.
Discussion
We have shown that forced expression of genes that regulate the mitochondrial unfolded protein response is sufficient to retard age-dependent mitochondrial and muscle functional impairment; and overexpression of antioxidant enzymes abolish the protective effects of muscle mitohormesis due to complex I perturbation. It is noteworthy that exercise physiologists have long acknowledged that interventions to reduce the supposed redox damage in muscles following a bout of physical exercise, may actually result in unfavorable alterations to the expression of cytoprotective genes (Peternelj and Coombes, 2011). Our results indicate that a plausible explanation for this effect is that antioxidant treatment dampens the extent of activation of ROS-mediated signaling cascades, culminating in lower levels of mitochondrial repair/maintenance genes required for re-establishing muscle homeostasis following exercise. Our work, coupled with observations in other systems strengthen the emerging concept that ROS serve as signaling molecules that engage specific signal transduction cascades (Hamanaka and Chandel, 2010; Raimundo et al., 2012; Schulz et al., 2007).
A striking finding of our study is that muscle mitochondrial distress upregulates the insulin-antagonizing peptide (ImpL2), which non-autonomously represses insulin signaling. Under adverse environmental conditions, different organs must have the capacity to mount a coordinated adaptive response to the stressor. For instance, the recently discovered exercise-induced Irisin can increase energy expenditure to improve glucose homeostasis (Bostrom et al., 2012). It is interesting that ImpL2 secretion in response to muscle mitochondrial distress also acts in an adaptive manner by stimulating lysosome biogenesis, possibly to enhance pro-survival autophagy; an event that is critical for survival under stress. Notably, in an analogous situation in the heart, sub-lethal ischaemia is known to trigger the release of cardiomyokines that act in an adaptive manner to preserve myocardial tissue health (Glembotski, 2011).
The idea that mitochondrial respiratory chain deficiency in one tissue can alter events in another tissue has also been observed in C elegans, where RNAi-mediated knockdown of cytochrome c oxidase-1 subunit Vb in neurons activates the mitochondrial unfolded protein response autonomously (in neurons), and non-autonomously in the gut (Durieux et al., 2011). As a plausible explanation for the non-autonomous effect of mitochondrial respiratory chain deficiency, the authors hypothesized that specific signal(s) may be secreted in response to mitochondrial dysfunction in one tissue, to subsequently propagate the mitochondrial stress signal to other tissues. Our studies in Drosophila show that in addition to the mitochondrial unfolded protein response, other mitochondrial stress responses (in this case, insulin repression), can be transmitted between different organs as well. Accordingly, there may be multiple mechanisms and longevity-promoting signals required for propagating mitochondrial stress responses between organs/tissues.
It has been hypothesized that the various compensatory signaling modules activated in response to mitochondrial dysfunction are not necessarily independent processes, but rather part of a complex mitochondrial regulatory network with multiple axes operating in unison to enhance survival under stress (Andreux et al., 2013). Interestingly, our data supports this notion; we find that at least two mitochondrial quality control processes – the UPRmt and mitophagy – are concurrently active in complex I disrupted muscles. Importantly, while forced expression of UPRmt genes recapitulates many of the phenotypes associated with the preservation of mitochondrial function (Figure 4), lifespan increase was less robust, when compared to the effect of ImpL2 overexpression. Thus, it appears that the UPRmt and ImpL2-dependent pathways regulate different facets of an intricate mitochondrial stress response network: induction of the UPRmt serves primarily to preserve or restore mitochondrial function, while ImpL2 induction increases lysosome biogenesis which will enhance the clearance of damaged mitochondria through mitophagy. Interestingly, by selectively culling dysfunctional mitochondria from an otherwise normal pool, mitophagy may augment lifespan by ensuring the propagation of mitochondria with optimum function. The mechanism(s) triggering the longevity-enhancing effect of ImpL2 is likely to extend beyond mitophagy, as ImpL2 may help remove misfolded protein aggregates, as has been shown for FOXO/4E-BP signaling (Demontis and Perrimon, 2010). In addition, 4E-BP overexpression is sufficient to increase lifespan, and is required for regulating metabolism under stress (Demontis and Perrimon, 2010; Teleman et al., 2005); accordingly, it may play a role in this context as well. Undoubtedly, future studies should help resolve the full breadth of cytoprotective processes that contribute to the lifespan-promoting effect of ImpL2.
In summary, we uncover a mechanism by which mitochondrial perturbation in muscles can cause systemic effects. Given that the human ortholog of ImpL2 (IGFBP7) has recently been shown to bind to the IGF-1 receptor and blocks activation of insulin-like growth factors (Evdokimova et al., 2012), it is enticing to speculate that muscle mitochondrial injury in humans could also lead to upregulation of IGFBP7 (or other IGFBPs) to cause systemic repression of insulin signaling. Such an event will have implications for the known association between mitochondrial dysfunction and diseases associated with aberrant insulin signaling, such as type II diabetes. Interestingly, circulating IGFBP7 levels are elevated in patients with type II diabetes (Kutsukake et al., 2008); whether this elevation is due partly to muscle mitochondrial dysfunction remains to be tested. Unquestionably, future studies in Drosophila and other systems to identify additional signaling molecules elevated in response to mitochondrial perturbation, is likely to open up therapeutic opportunities for many metabolic diseases.
Experimental Procedures
Drosophila Strains and Genetics
For a list of stocks used, statistical analyses, and detailed experimental conditions, see Extended Experimental Procedures.
Immunostaining
Larval somatic muscles or adult thoraxes were dissected in PBS, fixed in 4% formaldehyde in PBS for about 30 minutes (1 hour for thoraxes) at room temperature, and stained following standard procedures.
Electron Microscopy
Electron microscopy was performed essentially as described by Bai et al. (Bai et al., 2007).
ROS Staining with DHE
ROS levels were monitored using the superoxide indicator Dihydroethidium, as described by Owusu-Ansah et al. (Owusu-Ansah and Banerjee, 2009; Owusu-Ansah et al., 2008).
Citrate Synthase and Dihydrolipoamide Dehydrogenase Activity Assays
Citrate Synthase and Dihydrolipoamide Dehydrogenase activity assays were assessed essentially as described by Rera et al. (Rera et al., 2011) and Berger et al (Berger et al., 1996), respectively.
ATP Assay
ATP levels were assessed with the ATP assay kit from molecular probes (A22066), following the manufacturer's instructions.
qRT-PCR Analyses of Transcript Levels
RNA was extracted with Trizol reagent, digested with DNAse-1 and reverse transcribed using the iScript cDNA Synthesis kit (Bio-Rad); after which PCR was performed using iQ SYBR Green Supermix (Bio-Rad). Relative transcript levels were assessed using the Comparative CT method. RpL32 was used as an internal control.
Supplementary Material
Highlights.
Complex I (Cop I)-mediated redox signaling activates the UPRmt in Drosophila
Forced expression of UPRmt genes preserves muscle/mitochondrial function
Cop I disruption in muscles causes systemic repression of insulin signaling
Cop I disruption in muscles inhibits insulin signaling via an IGF-binding protein
Acknowledgments
EO-A conceived, designed and performed all experiments. WS helped with statistical analyses, and made the graphical abstract. EO-A and NP discussed results, and wrote the manuscript. We are grateful to Rexford Ahima for critical discussions; and Eileen Furlong, Ernst Hafen, Liqun Luo, Fannis Missirlis, Linda Partridge and David Walker for stocks and reagents. We thank the DSHB for antibodies; the BDSC, the NIG (Japan) and the TRiP for various fly strains; Christians Villalta for embryo injections, and Elizabeth Bennechi for assistance with electron microscopy. This work was supported by an F32 NRSA Postdoctoral Fellowship (5F32AR57291) and a career Development Grant from the Muscular Dystrophy Association (Award #202260) to EO-A; and RO1 grant # AR057352 to NP. NP is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alic N, Hoddinott MP, Vinti G, Partridge L. Lifespan extension by increased expression of the Drosophila homologue of the IGFBP7 tumour suppressor. Aging Cell. 2011;10:137–147. doi: 10.1111/j.1474-9726.2010.00653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreux PA, Houtkooper RH, Auwerx J. Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov. 2013;12:465–483. doi: 10.1038/nrd4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Hartwig JH, Perrimon N. SALS, a WH2-domain-containing protein, promotes sarcomeric actin filament elongation from pointed ends during Drosophila muscle growth. Dev Cell. 2007;13:828–842. doi: 10.1016/j.devcel.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Berger I, Elpeleg ON, Saada A. Lipoamide dehydrogenase activity in lymphocytes. Clin Chim Acta. 1996;256:197–201. doi: 10.1016/s0009-8981(96)06420-0. [DOI] [PubMed] [Google Scholar]
- Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Bostrom EA, Choi JH, Long JZ, et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481:463–468. doi: 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Copeland JM, Cho J, Lo T, Jr, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol. 2009;19:1591–1598. doi: 10.1016/j.cub.2009.08.016. [DOI] [PubMed] [Google Scholar]
- Dell'agnello C, Leo S, Agostino A, Szabadkai G, Tiveron C, Zulian A, Prelle A, Roubertoux P, Rizzuto R, Zeviani M. Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Hum Mol Genet. 2007;16:431–444. doi: 10.1093/hmg/ddl477. [DOI] [PubMed] [Google Scholar]
- Demontis F, Perrimon N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell. 2010;143:813–825. doi: 10.1016/j.cell.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evdokimova V, Tognon CE, Benatar T, Yang W, Krutikov K, Pollak M, Sorensen PH, Seth A. IGFBP7 binds to the IGF-1 receptor and blocks its activation by insulin-like growth factors. Sci Signal. 2012;5:ra92. doi: 10.1126/scisignal.2003184. [DOI] [PubMed] [Google Scholar]
- Finley LW, Haigis MC. The coordination of nuclear and mitochondrial communication during aging and calorie restriction. Ageing Res Rev. 2009;8:173–188. doi: 10.1016/j.arr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glembotski CC. Functions for the cardiomyokine, MANF, in cardioprotection, hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:512–517. doi: 10.1016/j.yjmcc.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton B, Dong Y, Shindo M, Liu W, Odell I, Ruvkun G, Lee SS. A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 2005;19:1544–1555. doi: 10.1101/gad.1308205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci. 2010;123:3849–3855. doi: 10.1242/jcs.075119. [DOI] [PubMed] [Google Scholar]
- Hollinshead M, Sanderson J, Vaux DJ. Anti-biotin antibodies offer superior organelle-specific labeling of mitochondria over avidin or streptavidin. J Histochem Cytochem. 1997;45:1053–1057. doi: 10.1177/002215549704500803. [DOI] [PubMed] [Google Scholar]
- Honegger B, Galic M, Kohler K, Wittwer F, Brogiolo W, Hafen E, Stocker H. Imp-L2, a putative homolog of vertebrate IGF-binding protein 7, counteracts insulin signaling in Drosophila and is essential for starvation resistance. J Biol. 2008;7:10. doi: 10.1186/jbiol72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpac J, Younger A, Jasper H. Dynamic coordination of innate immune signaling and insulin signaling regulates systemic responses to localized DNA damage. Dev Cell. 2011;20:841–854. doi: 10.1016/j.devcel.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchman PA, Kim S, Lai CY, Jazwinski SM. Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae. Genetics. 1999;152:179–190. doi: 10.1093/genetics/152.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsukake M, Ishihara R, Momose K, Isaka K, Itokazu O, Higuma C, Matsutani T, Matsuda A, Sasajima K, Hara T, et al. Circulating IGF-binding protein 7 (IGFBP7) levels are elevated in patients with endometriosis or undergoing diabetic hemodialysis. Reprod Biol Endocrinol. 2008;6:54. doi: 10.1186/1477-7827-6-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, Hekimi S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005;19:2424–2434. doi: 10.1101/gad.1352905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Butow RA. Mitochondrial retrograde signaling. Annu Rev Genet. 2006;40:159–185. doi: 10.1146/annurev.genet.40.110405.090613. [DOI] [PubMed] [Google Scholar]
- Lubos E, Loscalzo J, Handy DE. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2011;15:1957–1997. doi: 10.1089/ars.2010.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire SE, Mao Z, Davis RL. Spatiotemporal gene expression targeting with the TARGET and gene-switch systems in Drosophila. Sci STKE. 2004;2004:l6. doi: 10.1126/stke.2202004pl6. [DOI] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- Missirlis F, Rahlfs S, Dimopoulos N, Bauer H, Becker K, Hilliker A, Phillips JP, Jackle H. A putative glutathione peroxidase of Drosophila encodes a thioredoxin peroxidase that provides resistance against oxidative stress but fails to complement a lack of catalase activity. Biol Chem. 2003;384:463–472. doi: 10.1515/BC.2003.052. [DOI] [PubMed] [Google Scholar]
- Orsini F, Migliaccio E, Moroni M, Contursi C, Raker VA, Piccini D, Martin-Padura I, Pelliccia G, Trinei M, Bono M, et al. The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J Biol Chem. 2004;279:25689–25695. doi: 10.1074/jbc.M401844200. [DOI] [PubMed] [Google Scholar]
- Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009;461:537–541. doi: 10.1038/nature08313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu-Ansah E, Yavari A, Mandal S, Banerjee U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat Genet. 2008;40:356–361. doi: 10.1038/ng.2007.50. [DOI] [PubMed] [Google Scholar]
- Peternelj TT, Coombes JS. Antioxidant supplementation during exercise training: beneficial or detrimental? Sports Med. 2011;41:1043–1069. doi: 10.2165/11594400-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Powers SK, Duarte J, Kavazis AN, Talbert EE. Reactive oxygen species are signalling molecules for skeletal muscle adaptation. Exp Physiol. 2010;95:1–9. doi: 10.1113/expphysiol.2009.050526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimundo N, Song L, Shutt TE, McKay SE, Cotney J, Guan MX, Gilliland TC, Hohuan D, Santos-Sacchi J, Shadel GS. Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell. 2012;148:716–726. doi: 10.1016/j.cell.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rera M, Bahadorani S, Cho J, Koehler CL, Ulgherait M, Hur JH, Ansari WS, Lo T, Jr, Jones DL, Walker DW. Modulation of longevity and tissue homeostasis by the Drosophila PGC-1 homolog. Cell Metab. 2011;14:623–634. doi: 10.1016/j.cmet.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M, Zarse K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis) Exp Gerontol. 2010;45:410–418. doi: 10.1016/j.exger.2010.03.014. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusten TE, Lindmo K, Juhasz G, Sass M, Seglen PO, Brech A, Stenmark H. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev Cell. 2004;7:179–192. doi: 10.1016/j.devcel.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Teleman AA, Chen YW, Cohen SM. 4E-BP functions as a metabolic brake used under stress conditions but not during normal growth. Genes Dev. 2005;19:1844–1848. doi: 10.1101/gad.341505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.