

Abstract
The brain is astonishing in its complexity and capacity for change. This has fascinated scientists for more than a century, filling the pages of this journal for the past 25 years. But, a paradigm shift is underway. It seems likely that the plasticity that drives our ability to learn and remember can only be meaningful in the context of otherwise stable, reproducible, and predictable baseline neural function. Without the existence of potent mechanisms that stabilize neural function, our capacity to learn and remember would be lost in the chaos of daily experiential change. This underscores two great mysteries in neuroscience. How are the functional properties of individual neurons and neural circuits stably maintained throughout life? And, in the face of potent stabilizing mechanisms, how can neural circuitry be modified during neural development, learning and memory? Answers are emerging in the rapidly developing field of homeostatic plasticity.
INTRODUCTION
It has become clear that homeostatic signaling systems act throughout the central and peripheral nervous systems to stabilize the active properties of nerve and muscle (Davis, 2006; Marder, 2011; Turrigiano, 2011). Evidence for this has accumulated by measuring how nerve and muscle respond to the persistent disruption of synaptic transmission, ion channel function or neuronal firing. In systems ranging from Drosophila to human, cells have been shown to restore baseline function in the continued presence of these perturbations by rebalancing ion channel expression, modifying neurotransmitter receptor trafficking and modulating neurotransmitter release (Frank, 2013; Maffei and Fontanini, 2009; Watt and Desai, 2010). In each example, if baseline function is restored in the continued presence of a perturbation, then the underlying signaling systems are considered homeostatic (Figure 1).
Figure 1. Evidence for the homeostatic control of cellular excitation.

Top) The firing properties of central neurons are determined by a balance of synaptic excitation (red vesicles and red receptors), synaptic inhibition (blue vesicles and blue receptors), and the densities of ion channels that mediate either cellular depolarization (red ovals) or that oppose cellular depolarization (blue ovals). In response to chronic suppression of neural activity, central neurons can alter the relative abundance of ion channels and receptors at the cell surface to reestablish a set point level of activity. Bottom) At the neuromuscular junction (NMJ), chronic impairment of postsynaptic neurotransmitter receptor sensitivity or receptor abundance leads to a compensatory increase in presynaptic neurotransmitter release that precisely counteracts the change in receptor function to achieve normal synaptic depolarization of the muscle. Modified from Davis, 2006.
This is a rapidly growing field of investigation that can be subdivided into three areas that are defined by the way in which a cell responds to activity perturbation, including the homeostatic control of intrinsic excitability, neurotransmitter receptor expression and presynaptic neurotransmitter release. Each area is introduced below. An exciting prospect is that the logic of homeostatic signaling systems, if not specific molecular pathways, will be evolutionarily conserved. The nervous systems of all organisms confront perturbations ranging from genetic and developmental errors to changing environmental conditions. In this relatively short review, it is not possible to achieve a comprehensive description of the molecular advances in each system. Rather, an attempt is made to draw parallels across systems where conserved processes are emerging.
THE HOMEOSTATIC CONTROL OF INTRINSIC EXCITABILITY
The homeostatic control of intrinsic excitability was brought to the forefront by experiments that followed the fate of a neuron that was removed from its circuit and placed in isolated cell culture (Turrigiano et al., 1994). Over a period of days, the isolated neuron rebalanced ion channel surface expression and restored intrinsic firing properties that were characteristic of that cell in vivo. The effect was shown to be both activity and calcium dependent. This phenomenon has now been extended to the function of entire neural circuits in both invertebrates (Haedo and Golowasch, 2006; see Figure 2) and the developing vertebrate spinal cord (Gonzalez-Islas and Wenner, 2006).
Figure 2. Accurate restoration of cellular activity through homeostatic signaling.

A) Sample traces from cortical pyramidal neurons from wild type and Kv4.2 knockout mice (Nerbonne et al., 2008, data from figure 8 therein). The knockout mice lack the Kv4.2 protein and current. Although acute pharmacological inhibition severely potentiates neuronal excitability, homeostatic rebalancing of potassium channel expression accurately restores firing properties to wild type levels. B) Data are shown for recordings made at the Drosophila NMJ. Presynaptic release (quantal content) is plotted against spontaneous miniature amplitudes (mEPSP). Each data point is average data from a single NMJ from control NMJ (open black) or NMJ to which philanthotoxin 433 (PhTX) was applied for 10min prior to recording (open red). The line represents ideal homeostatic compensation where any change in mEPSP is offset by an identical percent change in quantal content. The modulation of presynaptic release accurately offsets a broad range of postsynaptic perturbation. C) Data are presented for the Drosophila NMJ plotting excitatory postsynaptic current (EPSC) amplitude versus extracellular calcium concentration. Larvae treated with PhTx (wt + PhTX) accurately retarget control (wt) EPSC amplitudes across an order of magnitude change in extracellular calcium. Animals harboring a loss of function mutation in rim show reduced EPSCs at all calcium concentrations. Application of PhTX to rim mutant larvae demonstrates a failure of homeostatic compensation at all calcium concentrations (Muller et al., 2012). D) Intracellular recordings from a stomatogastric neuron in the intact ganglion (control), following removal of the ganglion and placement in organ culture for 10 minutes (Decentralized) and after four days in culture (4 days). After 4 days, the firing properties of the identified neuron are remarkably similar to that observed in the intact animal. Scale bars 1 sec / 10mV. Data modified from Khorkova and Golowasch, 2007. E) Example traces from Xenopus central neurons including control and a neuron expressing transgenic Kir2.1 (Pratt et al., 2007). Expression of Kir2.1 induces a change in the underlying current densities including the sodium current (quantified at right) that help sustain firing properties at control levels.
A related phenomenon has been revealed in animals harboring mutations in genes that encode ion channels. Several studies provide evidence that deleting an ion channel gene invokes compensatory changes in the expression of other ion channels in both invertebrate and mammalian systems (MacLean et al., 2003; Swenson and Bean, 2005; Muraro et al., 2008; Andrásfalvy et al., 2008; Nerbonne et al., 2008; Van Wart and Matthews, 2006; Bergquist et al., 2010). In many cases, ion channel expression is rebalanced and cell-type specific firing properties are restored (Figure 2). One conclusion is that homeostatic signaling systems enable a neuron to compensate for the absence of an ionic current and re-establish cell type specific firing properties through altered expression of other ion channels. A second conclusion is that ion channel expression is not a fixed parameter associated with cell fate. Rather, a given cell type can maintain characteristic firing properties using different combinations of ion channel densities. The homeostatic rebalancing of ion channel expression is astonishing, in part, because of its staggering complexity (Marder and Prinz, 2002). There can be thousands of synaptic inputs and dozens of different channels controlling the firing properties of an individual cell. The molecular mechanisms that achieve the homeostatic rebalancing of ion channel expression remain virtually unknown (but see Muraro et al., 2008; Driscoll et al., 2013; Temporal et al., 2012; Korkhova and Golowasch,2007)
HOMEOSTATIC CONTROL OF SYNAPTIC EFFICACY: SYNAPTIC SCALING AT THE POSTSYNAPTIC DENSITY
Synaptic scaling was revealed by experiments examining the effects of chronic activity suppression in cultured mammalian neurons (Turrigiano et al., 1998; O’Brien et al., 1998). It is now clear, both in vitro and in vivo, that chronic manipulation of neural activity drives counteracting changes in neurotransmitter receptor abundance that help to restore neural activity to baseline levels (Thiagarajan et al., 2005; Zhao et al., 2011; Garcia-Bereguiain et al., 2013; Mrsic-Flogel et al., 2007; Echegoyen et al., 2007; Deeg and Aizenman, 2011; Gainey et al., 2009; Keck et al., 2013; Hengen et al., 2013; see also Tyagarajan et al., 2010). The bidirectional modulation of neurotransmitter receptor abundance was initially termed ‘synaptic scaling’ because the measured amplitudes of spontaneous miniature release events are modified in a multiplicative manner, presumably through proportional changes in receptor abundance at every individual synapse (Turrigiano et al., 1998; Turrigiano, 2011; see also Kim and Tsien, 2008). This effect has the property of preserving the relative differences in efficacy among the numerous synapses on a single postsynaptic target. Because of this, it has been proposed that synaptic scaling stabilizes neuronal excitability while preserving learning-related information contained in relative synaptic weights. Synaptic scaling can be induced over the course of hours to days (Turrigiano, 2011). It requires transcription and in some cases may be achieved through local protein translation (Sutton et al., 2006; Sutton et al., 2007). Synaptic scaling is generally studied in response to alterations in global neural activity. However, manipulating the activity of individual neurons (Goold and Nicoll, 2010; Ibata et al., 2008) can be sufficient to induce synaptic scaling (Figure 3). Even more remarkable is evidence that synaptic scaling can be input specific (Deeg and Aizenman, 2011) and even synapse specific (Béïque et al., 2011) when the manipulation of neural activity is restricted to subsets of inputs contacting a given postsynaptic neuron (Figure 3). It is not yet clear whether the magnitude of the scaling response is matched to the magnitude of the perturbation. This is impossible to determine in experiments using tetrodotoxin (TTX) to block neural activity. In experiments where neural activity is modulated, synaptic scaling participates in the restoration of baseline firing properties in vivo (Keck et al., 2013; Hengen et al., 2013). However, synaptic scaling is often is observed to act in concert with other compensatory changes including changes in presynaptic neurotransmitter release (Burrone et al., 2002; Kim and Tsien 2008; Lu et al,. 2013) or intrinsic excitability (Lambo and Turrigiano, 2013). It remains entirely unknown how multiple homeostatic effectors are coordinated to restore cell type specific firing properties.
Figure 3. Cell autonomous and synapse specific homeostatic plasticity.

A) Experimental configuration is diagrammed for stimulation and simultaneous recording from adjacent CA1 pyramidal neurons in hippocampal organotypic culture that are either untransfected (Record:Ctrl, black) or transfected with channel rhodopsin 2 and photostimulated in slice culture for 24hours, 50ms light pulses at 3Hz (Record: Photostim, blue). Below, representative data are shown for AMPA-mediated currents. Below is a scatter plot of recording pairs with the mean shown in red. Photostimulation causes a decrease in AMPA current due to downscaling and a decrease in synapse number (Goold and Nicoll, 2010). B) Two neighboring spines with or without overlay of the Syn-YFP terminals from a Syn-YFP/Kir2.1- overexpressing cell. 2P uncaging of MNI-glutamate was elicited at the tip of these spines (yellow crossed lines) and the resulting AMPAR-mediated synaptic current (2P-EPSC) is shown (Vh = −60 mV) (Béïque et al., 2011). A postsynaptic potentiation is seen in response to synapse specific presynaptic expression of Kir2.1. Data and images take from Goold and Nicoll (2010, A) and Béïque et al., (2011, B).
HOMEOSTATIC CONTROL OF PRESYNAPTIC NEUROTRANSMITTER RELEASE
The homeostatic modulation of presynaptic neurotransmitter release was brought to the forefront through studies at the genetically tractable Drosophila neuromuscular junction (NMJ; Davis and Goodman, 1998a). Genetic manipulations that alter postsynaptic glutamate receptor function (Petersen et al., 1996; Davis et al., 1997; Frank et al., 2006), muscle innervation (Davis and Goodman, 1998b) or muscle excitability (Paradis et al., 2001) were shown to induce large compensatory changes in presynaptic neurotransmitter release that precisely restore set point muscle depolarization in response to nerve stimulation. This phenomenon has been referred to as ‘synaptic homeostasis’ but will be referred to here as ‘presynaptic homeostasis’. This form of homeostatic signaling is evolutionarily conserved from fly to human at the NMJ (Cull-Candy et al., 1980; Plomp et al., 1992). As with other forms of homeostatic plasticity, this is a quantitatively accurate form of neuromodulation (Figure 2B; Frank et al., 2006). It can be induced in seconds to minutes, during which its expression is independent of transcription or translation (Frank et al., 2006). It can also be stably maintained, a process that requires transcription (Marie et al., 2010). Presynaptic homeostasis at the NMJ is bi-directional and can be synapse specific (Davis and Goodman, 1998; Daniels et al., 2006).
Importantly, presynaptic homeostasis has also been observed at mammalian central synapses in vitro in response to differences in target innervation (Liu and Tsien, 1995), altered postsynaptic excitability (Burrone et al., 2002; Thiagarajan et al., 2005; but see Goold and Nicoll, 2010; Deeg and Aizenman, 2011), and following chronic inhibition of neural activity (Kim and Ryan 2010; Zhao et al., 2011). Regardless of the system being studied, the expression of presynaptic homeostasis is remarkable because it involves the rapid, persistent and accurate modulation of presynaptic vesicle fusion.
HOMEOSTATIC DESIGN
The homeostatic modulation of neural function is distinct from other forms of neural plasticity because it is a quantitatively accurate form of modulation. For example, the homeostatic rebalancing of ion channel expression precisely counteracts the loss of the Kv4.2 potassium channel in pyramidal neurons and achieves firing properties that are almost indistinguishable from controls (Figure 2A). It should be pointed out, however, that compensation is not perfect because it is constrained by the unique subcellular localization and functional properties of the compensating ion channels (see also Bergquist et al., 2010). In Kv4.2 knockout pyramidal neurons, somatic excitability is precisely restored but dendrites remain hyper-excited (Chen et al., 2006; Nerbonne et al., 2008; see also Van Wart and Matthews, 2006).
Another example of quantitative accuracy is found at the NMJ. The magnitude of postsynaptic glutamate receptor inhibition is accurately offset, over a wide range, by a graded increase in presynaptic neurotransmitter release (Figure 2B). The accurate modulation of presynaptic release is apparent when measured over a 10-fold range of extracellular calcium (0.3–3mM; Figure 2C). A similarly accurate modulation of presynaptic release is observed following muscle-specific expression of an inward rectifying potassium channel (Kir2.1), which induces a non-linear disruption of excitability because Kir2.1 inactivates during EPSP depolarization. Nonetheless, a precise increase in presynaptic release offset the disruption of muscle excitation caused by Kir2.1 expression and restored peak EPSP amplitude to control levels (Paradis et al., 2001). Again, compensation is accurate but imperfect since EPSP decay remains more rapid than controls, which will alter summation during a stimulus train (Paradis et al., 2001), an effect similar to that observed at the NMJ of lobster (Pulver et al., 2004). Other examples of accurate compensation are highlighted in Figure 2(D and E). One of the greatest challenges in the field of homeostatic signaling is to define how accurate modulation achieved.
There are several features that are commonly employed in both natural and engineered homeostatic signaling systems that achieve quantitative accuracy (Stelling et al., 2004). First, homeostatic systems require a set point that precisely defines the output of the system. This is also the state to which the system returns following a perturbation. Second, homeostatic systems generally require feedback control to precisely retarget the system set point. Homeostatic systems require sensors that detect a given perturbation. By analogy with engineered systems, it is hypothesized that homeostatic signaling systems will require an error signal, representing the difference between the system set point and steady state activity reported by the sensors. Finally, the error signal is used to promote a change in homeostatic effectors that drive compensatory changes in the process being studied. These signaling features are often invoked in discussions of neuronal homeostatic signaling. However, at a molecular level, our understanding remains rudimentary. The challenge is to begin assembling an emerging molecular ‘parts list’ into a complete homeostatic signaling system(s) that can explain how quantitatively control of neural activity is achieved.
ESTABLISHING A HOMEOSTATIC SET POINT
A set point is operationally defined as the physiological state that is held constant by a homeostatic signaling system. It seems that the establishment of set point of neuronal activity must be related to the specification of cell identity. For example, the firing properties of a neuron can be as diagnostic of cell identity as any other anatomical attribute including cell size, dendrite shape or the biochemical choice of neurotransmitter. Yet, as emphasized above, ion channel expression, which shapes intrinsic excitability and neural activity, is not a fixed parameter associated with cell fate. How then is a set point for neuronal activity determined?
It is well established that combinatorial transcription factor codes specify cell fate in the nervous system (Jessell, 2000). Data from C. elegans suggest that cell fate is subsequently maintained through the action of ‘terminal selector’ transcription factors (Hobert, 2011). Terminal selectors are expressed throughout life and control the expression of effector genes that define cellular identity, including ion channels. If a terminal selector is deleted, cell fate is not maintained (Hobert, 2011). Perhaps, rather than rigidly controlling ion channel expression, a terminal selector defines which ion channels can be expressed, but allows channel expression levels to vary according to homeostatic feedback.
The idea of a terminal selector remains consistent with groundbreaking theoretical and computational work from the stomatogastric ganglion examining how a set point is retargeted through homeostatic feedback (Prinz et al,. 2004; Marder, 2011). This work proposes that individual neurons can express different combinations of ion channels and synaptic strengths in order to arrive at cell-type specific firing properties. Given the diversity of channels and synapses that participate in neuronal firing, the number of physiologically relevant combinations channel densities and synaptic weights that can generate a specific firing pattern is demonstrated to be vast in silico (Prinz et al,. 2004; Marder, 2011). Experimental evidence for this type of variation includes the demonstration that anatomically identical cells can have similar firing properties that are driven by diverse combinations of underlying current densities and synaptic weights (Swenson and Bean, 2005; Schulz et al., 2007; Shulz et al., 2006; Andrásfalvy et al., 2008; Goaillard et al., 2009; Temporal et al., 2012).
Mechanistically, it is clear that altered ion channel transcription is involved in the homeostatic rebalancing of ion channel expression (Shulz et al., 2007; Bergquist et al., 2010). Interesting data from the lobster stomatogastric system has shown that neuromodulators influence the transcription of ion channels in a coordinated fashion (Korkhova and Golowasch, 2007; Temporal et al., 2012). These data not only highlight the importance of neuromodulation, but provide insight into how the homeostatic rebalancing of ion channel expression might be constrained. Another idea is that ion channel translation could also be a key modulatory step, downstream of the terminal selector. For example, a homeostatic change in sodium channel expression following chronic manipulation of synaptic activity requires the translational regulator pumillio, a mechanism that is conserved in both flies and mice (Driscoll et al., 2013). Finally, it is also well established that extrinsic factors can influence cell phenotype, one example being neurotransmitter switching (Dulcis et al., 2013). It remains possible that ion channel rebalancing reflects a similar phenotypic switch, albeit more complex. Ultimately, even though we are gaining information about how a cell rebalances ion channel expression, a clear model for how the genome defines a cell-type specific set point for neural activity remains elusive.
SENSORS
How cells detect a change in neural activity to initiate homeostatic plasticity remains unknown. Homeostatic signaling can be induced cell autonomously (Goold and Nicoll, 2010; Burrone et al., 2002) and through focal application of TTX to the soma (Ibata et al., 2008). These data are consistent with a somatic sensor of cell-wide activity. As expected, calcium-dependent signaling is essential. For example, both synaptic upscaling and downscaling have been shown to require the activity of CamKK and CamKIV (Goold and Nicoll, 2010, Ibata et al., 2008). But, the link between altered activity and the induction of a homeostatic response still remains unclear. Many experiments utilize dramatic activity alterations, either blocking activity with TTX or inducing seizure-like network activity, which will invoke changes in calcium-dependent signaling and transcription. However, there are examples where moderate and graded changes in neural activity and muscle depolarization are detected. For example, the magnitude of glutamate receptor inhibition at the NMJ is precisely offset by an increase in presynaptic release (Figures 1 and 2) implying a sensor that is able to detect graded changes in muscle excitation. Similarly, moderate changes in neuronal firing measured in visual cortex following visual deprivation can invoke homeostatic plasticity, leading to the restoration of baseline firing rates (Keck et al., 2013; Hengen et al., 2013; see also Deeg and Aizenman, 2011). Early efforts to model homeostatic plasticity in the stomatogastric system have emphasized that multiple activity sensors are necessary to discriminate quantitative differences in neuronal firing (Liu et al., 1998). Yet, biologically, a system of coordinated sensors with the fidelity to follow neural activity remains unknown.
An interesting possibility is that metabolic sensors may be employed in addition to, or in parallel with, changes in intracellular calcium. In dissociated hippocampal culture, eukaryotic elongation factor 2 (eEF2) has been implicated as a sensor that can detect disruption of glutamatergic transmission (Sutton et al 2004; Sutton et al., 2007). Additional work implicates a function for TOR-dependent signaling downstream of AMPA receptor blockade (Henry et al., 2012). The potential importance of this signaling system for homeostasis in vivo is emphasized in experiments demonstrating that TOR signaling is essential for balanced network excitation and inhibition (Bateup et al., 2013). The importance of TOR is also emphasized by work at the Drosophila NMJ in vivo, showing that genetic disruption of TOR and S6 Kinase signaling blocks the sustained expression of presynaptic homeostasis (Penney et al., 2012). In many systems, TOR signaling is used to detect qualitative changes in the cellular environment and, thereby, regulates cellular homeostasis and growth (Laplante and Sabatini, 2012). As such, it is a candidate for detecting qualitative changes in neural function and stimulating downstream homeostatic plasticity.
HOMEOSTATIC EFFECTORS: SYNAPTIC SCALING
Synaptic scaling is expressed as a change in neurotransmitter receptor abundance. Although a great deal has been discovered about the transcription, assembly and trafficking of glutamate receptors, the mechanisms that control receptor trafficking in a homeostatic fashion remain largely unknown. Many key issues remain to be molecularly defined, including how synaptic scaling is achieved in a cell-wide fashion with proportional effects at every active zone. Similarly, there is very little information to explain how the synaptic scaling mechanism becomes limited as neuronal firing properties are restored toward baseline, set point levels and how the system is eventually shut off (but see Tatavarty et al., 2013). In attempting to define how the homeostatic control of glutamate receptor trafficking is achieved, it is useful to make comparisons to non-neuronal systems where homeostatic control of surface transporters and ion channels has been defined without the added complexity of cell diversity. These systems include trafficking of ENaC channels during the systemic control of salt balance and trafficking of the Glut4 glucose transporter during glucose homeostasis (Lifton et al., 2001; Martin et al., 2006; Leto and Saltiel 2012). Several advances are highlighted here that provide insight into emerging homeostatic control of glutamate receptor trafficking.
The induction of synaptic scaling has been an area of considerable progress. An emerging theme is the activity-dependent induction of immediate early gene signaling including Homer1a, Arc (Arg3.1), Narp and Polo-like kinase 2 (Plk2) (Seeburg et al., 2008; Hu et al., 2010; Chang et al., 2010; Béïque et al., 2011; Shepherd et al., 2006). In one study, enhanced network activity was shown to stimulate expression of Homer1a, which subsequently activates mGluR signaling in an agonist independent manner (Hu et al., 2010). This model is intriguing because the control of mGluR subcellular localization has the potential to define the spatial extent of the homeostatic response. In a separate set of studies, enhanced network activity induces Plk2, which phosphorylates the postsynaptic scaffolding protein SPAR in a CDK5-dependent manner. Subsequent SPAR degradation weakens the retention of AMPA receptors at the postsynaptic membrane, facilitating synaptic down-scaling (Seeburg et al., 2008; Seeburg and Sheng, 2008). Finally, although not an immediate early gene, retinoic acid has been shown to be required for synaptic upscaling, in this case following postsynaptic glutamate receptor inhibition (Wang et al., 2011; Sarti et al., 2012). In this model a decrease in dendritic calcium following AMPA receptor blockade induces retinoic acid synthesis and subsequent AMPA receptor production (Wang et al., 2011). Retinoic acid acts via the retinoic acid receptor (RAR-α) (Sarti et al., 2012) and could, potentially, signal cell autonomously (Wang et al., 2011).
Other advances center on how surface delivery and synaptic retention of AMPA receptors is controlled so that a homeostatic response can be graded and potentially matched to the magnitude of a perturbation. For example, PICK1 (protein interacting with C-kinase) scaffolds an intracellular AMPA receptor pool. There is evidence that PICK1 levels are decreased in a graded fashion in response to chronic activity inhibition, releasing AMPA receptors for translocation to the plasma membrane (Anggono et al., 2011). Other work focuses on how AMPA receptors are retained at the postsynaptic density by PSD95, PSD93 and SAP102. It has been shown that PSD95 and SAP102 levels are modulated bi-directionally by neural activity (Sun and Turrigiano, 2011). In this study, PSD95 is shown to be necessary but not sufficient for synaptic scaling, acting through the regulated organization of the postsynaptic scaffold (Sun and Turrigiano, 2011). Clearly, there will be additional complexity as an increasing number of molecules are shown to be necessary for synaptic scaling including MHC1 (Goddard et al., 2001), BDNF (Rutherford et al., 1998; Jakawich et al., 2010; Correa et al., 2013), and Beta3-integrins (McGeachie et al., 2013).
HOMEOSTATIC EFFECTORS: PRESYNAPTIC HOMEOSTASIS
The power of model system forward genetics in Drosophila has opened the door to a mechanistic understanding of presynaptic homeostasis. An electrophysiology-based forward genetic screen is ongoing, based on intracellular recordings of neuromuscular transmission, to identify mutations that prevent the homeostatic enhancement of presynaptic neurotransmitter release following pharmacological inhibition of postsynaptic glutamate receptors (Dickman and Davis, 2009; Muller et al., 2011; Younger et al., 2013). To date, more than 1000 mutations and RNAi have been tested (Dickman and Davis, 2009; Muller et al., 2011; Younger et al., 2013). Based largely on the results of this forward genetic approach, a model has emerged to explain how synaptic vesicle release is precisely potentiated at the NMJ.
Two presynaptic processes converge to potentiate vesicle fusion during presynaptic homeostasis: 1) potentiation of presynaptic calcium influx and 2) potentiation of the readily releasable pool of synaptic vesicles (RRP) (Figure 4). First, a combination of calcium imaging and genetic data demonstrate that an increase in presynaptic calcium influx through the CaV2.1 calcium channel is necessary to achieve a homeostatic increase in vesicle release (Muller et al., 2011; Muller et al, 2012). A surprising mechanism is employed to modulate presynaptic calcium influx. The involvement of a presynaptic DEG/ENaC sodium leak channel was uncovered in the aforementioned genetic screen. In the emerging model, presynaptic DEG/ENaC channel insertion at or near the nerve terminal causes low voltage modulation of the presynaptic resting potential due to sodium leak and subsequent potentiation of presynaptic calcium influx (Figure 5). This model is attractive because it provides an analogue mechanism that could fine tune presynaptic calcium influx according to the demands of the homeostatic signaling system. Low-voltage modulation of neurotransmitter release has been observed in systems ranging from the crayfish NMJ to the rodent hippocampus (Wojtowicz and Atwood, 1983; Awatrimani et al., 2005; Christie et al., 2011) although links to homeostatic plasticity have not been made in these systems. Interestingly, ENaC channels can be considered as homeostatic effector proteins during the systemic control of salt balance (Lifton, 2001).
Figure 4. Presynaptic homeostasis is achieved by parallel changes in the size of the readily releasable vesicle pool and presynaptic calcium influx.

A–B) Variance-mean analysis supports a RIM-dependent modulation of the RRP during synaptic homeostasis. Left, example EPSC traces for a WT NMJ at the indicated extracellular calcium concentration (millimolar). B) Example EPSC amplitude variance-mean plots of two WT synapses in the absence (gray) and presence (black) of PhTX with parabola fits (solid lines) that were extrapolated to the x-intercept (dashed lines; see Muller et al., 2012). C) Representative traces for measurement of the spatially averaged calcium signal within synaptic boutons at the NMJ in a wild type (control) and GluRIIA mutant. The homeostatic enhancement of presynaptic release is correlated with a statistically significant increase in the peak amplitude of the spatially averaged calcium signal (see Muller and Davis, 2011).
Figure 5. Emerging model for the expression of presynaptic homeostasis.
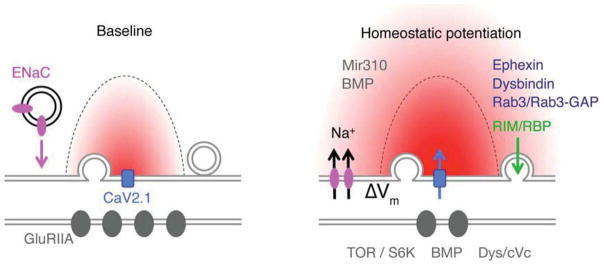
Schematic of an active zone at the Drosophila NMJ shown presynaptic CaV2.1 calcium channels (blue), the action potential triggered calcium microdomain (red), postsynaptic glutamate receptors (GluRIIA, dark gray) and presynaptic ENaC channel (pink). Two genes, pickpocket11 and pickpocket16 were discovered to be necessary, presynaptically, for the rapid induction and sustained expression of presynaptic homeostasis (Younger et al., 2013). These genes encode subunits of an Epithelial/Degenerin (ENaC) sodium leak channel. These genes are co-transcribed and transcriptionally upregulated following persistent disruption of postsynaptic glutamate receptor function. These and other data support a model in which ENaC channel insertion drives a modest depolarization of the presynaptic resting potential (ΔV), which enhances presynaptic calcium and neurotransmitter release. A parallel change in the readily releasable pool of vesicles is also necessary for synaptic homeostasis, which relies on the presynaptic adaptor proteins RIM (Muller et al., 2012) and RIM binding protein (RBP) (green). When both process are enabled, a homeostatic enhancement of release is observed. A number of additional synaptic proteins have been shown to be required for presynaptic homeostasis (dark blue text). Among them, there is evidence for the involvement of micro-RNA-based signaling (Mir10, gray; Tsurudome et al., 2011) and permissive BMP mediated signaling released from muscle (gray) and acting at the motoneuron soma (gray, Goold et al., 2007). Postsynaptically, there is evidence for the required function of Tor and S6K as well as Dystrobrevin-dependent scaffolding (Penney et al,. 2012; Pilgram et al., 2011). Figure modified from Younger et al., (2013). Additional molecular mechanisms involved in presynaptic homeostasis have been recently reviewed (Frank, 2013).
Remarkably, the potentiation of presynaptic calcium influx alone is not sufficient to drive a homeostatic change in synaptic vesicle fusion. A parallel increase in the readily releasable pool of synaptic vesicles (RRP) is required (Weyhersmüller et al., 2011; Muller et al., 2012). An analysis of mutations in RIM (Rab3 Interacting Molecule), which block presynaptic homeostasis (Figure 2C) was particularly informative. When the homeostatic modulation of presynaptic calcium influx and RRP size were tested in RIM mutants, only the modulation of the RRP was blocked and synaptic homeostasis was prevented (Muller et al., 2012). The means by which RIM mediates this activity has yet to be determined. The RIM interacting molecules Rab3 and Rab3-GAP also participate in presynaptic homeostasis (Muller et al., 2011). In mammalian systems, these molecules establish a biochemical bridge between the calcium channel and the synaptic vesicle (Han et al., 2011; Kaeser et al., 2011). This may represent a central, regulated scaffold that coordinates the homeostatic modulation of the RRP with calcium entry. Additional genes have been found to be essential for presynaptic homeostasis including postsynaptic scaffolding (Pilgram et al., 2011), postsynaptic TOR/S6K (Penney et al., 2012) and micro-RNA signaling (Tsurudome et al., 2010), all nicely summarized in a recent review of homeostatic plasticity at the Drosophila NMJ (Frank, 2013).
Parallels have emerged at mammalian central synapses, consistent with the homeostatic modulation of both vesicle pools and presynaptic calcium influx. Chronic activity blockade has been shown to cause a correlated increase in both presynaptic release and calcium influx, imaged simultaneously through co-expression of transgenic reporters for vesicle fusion and calcium (Zhao et al., 2011). Mechanistically, presynaptic CDK5 has been implicated. Loss or inhibition of CDK5 potentiates presynaptic release by promoting calcium influx and enhanced access to a recycling pool of synaptic vesicles. Chronic activity suppression phenocopies these effects and causes a decrease in synaptic CDK5 implying a causal link (Kim and Ryan, 2010). The activity of CDK5 has been shown to be balanced by calcineurin A and, together, these molecules act via the CaV2.2 calcium channel (Kim and Ryan 2013). Remarkably, the CDK5/Calcineurin dependent modulation of presynaptic release has sufficient signaling capacity to cause the silencing and unsilencing of individual active zones in hippocampal cultures (Kim and Ryan, 2013).
ENABLING A HOMEOSTATIC RESPONSE THROUGH PERMISSIVE SIGNLING
Studies at the Drosophila NMJ and mammalian central synapses demonstrate that secreted factors create an environment that is necessary for the expression and/or maintenance of homeostatic plasticity including both presynaptic homeostasis and postsynaptic scaling. Since these factors do not dictate the timing or magnitude of the homeostatic response they are considered essential, permissive cues. At the Drosophila NMJ, bone morphogenetic protein (BMP) signaling from muscle to motoneuron drives NMJ growth during larval development (McCabe et al., 2003). Subsequently, it was demonstrated that genetic deletion of the BMP ligand, a presynaptic BMP receptor or downstream transcription all block synaptic homeostasis (Goold et al., 2007). Importantly, BMP signaling does not function at the NMJ to instruct a change in neurotransmitter release. Instead, BMP-dependent transcription permits the induction of synaptic homeostasis, which is expressed locally at the NMJ. The nature of the permissive signal downstream of BMP-dependent transcription remains unknown (Goold et al., 2007). A related function has been proposed for TNF-α at mammalian central synapses. TNF-α is required for synaptic scaling, is released from glia in response to prolonged activity blockade and is sufficient to drive synaptic scaling (Beattie et al., 2002; Stellwagen et al., 2005; Stellwagen and Malenka, 2006). But, it was recently demonstrated that the rapid induction of synaptic scaling following 4–6 hours of activity blockade is normal in the absence of TNF-α. Synaptic scaling is only blocked if TNF-α is removed >12 hours prior to activity blockade. It is argued, based on these data, that TNF-α is a permissive signal that maintains synapses in a state amenable to homeostatic modulation (Steinmetz and Turrigiano, 2010). While the relevance of permissive homeostatic signaling remains to be determined, permissive signaling could be used to control whether or not homeostatic plasticity is expressed at different times during development (Maffei and Fortanini, 2009; Echegoyen et al., 2007) and its induction in the context of stress or disease (Goold et al., 2007, Steinmetz and Turrigiano, 2010).
INTERFACE OF DEVELOPMENT AND HOMEOSTATIC PLASTICITY
The nervous system is remarkably plastic during development. Individual neurons and muscle change dramatically in size and complexity. Synaptic connectivity is refined through mechanisms of synaptic competition. New cells are integrated into fully functioning neural circuitry. Do these changes represent perturbations that are stabilized by homeostatic signaling? One approach has been to define the cellular parameters that are held constant during periods of developmental growth, but this has not defined whether constancy is achieved through homeostatic control (Boucher et al., 2005). Answering this question is likely to be important for understanding how homeostatic plasticity might participate in diseases including Autism Spectrum Disorders (ASD) and schizophrenia that can be traced back to alterations in early brain development (Ramrocki and Zoghbi, 2008; Bougeron, 2009). The construction of an embryo from a single cell is a tightly choreographed process that includes inductive signaling with both negative and positive feedback control to ensure a robust, reproducible outcome (Baumgardt et al., 2007). From this perspective, homeostatic plasticity might serve to correct developmental inaccuracies, but would not be invoked as part of normal developmental signaling. Data from the Drosophila NMJ support this idea.
The NMJ of Drosophila larvae grow ~100 fold in volume during a five-day period of postembryonic development. In Drosophila, as in most systems, when a muscle fiber grows the input resistance drops precipitously, requiring enhanced presynaptic release to achieve constant muscle depolarization (Davis and Goodman, 1998). However, presynaptic homeostasis does not appear to be involved. Forward genetic screening has identified several mutations that block presynaptic homeostasis without altering anatomical or functional neuromuscular development (Dickman and Davis, 2009; Muller et al., 2011; Younger et al., 2013). This observation was extended by recent work using the ENaC channel blocker benzamil to acutely disrupt presynaptic homeostasis. Benzamil was applied to the NMJ of animals lacking the muscle-specific GluRIIA glutamate receptor subunit, a perturbation that is persistent throughout development and induces presynaptic homeostasis. Benzamil erased the expression of presynaptic homeostasis leaving behind a synapse with unpotentiated wild type release and wild type anatomy (Younger et al., 2013). Together these data demonstrate that presynaptic homeostasis is uncoupled from the mechanisms that achieve anatomical and physiological NMJ growth. One possibility is that presynaptic homeostasis is only induced developmentally when a cellular set point differs from ongoing activity. If the set point is developmentally programmed to change along with the maturation of cell fate, then a developmental change in cellular function could occur without the induction of homeostatic plasticity.
In the mammalian CNS, homeostatic and developmental plasticity co-exist. This is nicely documented in a binocular region of visual cortex following monocular deprivation (Mrsic-Flogel et al., 2007). When cells receive input predominantly from an open eye, deprived eye input is diminished, consistent with classical synaptic competition. However, when cells receive input predominantly from the deprived eye, these inputs are strengthened, consistent with homeostatic plasticity. Binocular deprivation also induces homeostatic synaptic strengthening. Although these processes co-exist, it remains unclear whether homeostatic plasticity normally participates in ocular dominance independent of an experimental perturbation such as eye suturing. In other examples, cell-autonomous suppression of neural activity has been shown to induce changes in synaptic connectivity as well as homeostatic plasticity, but the effects are separated in time (Burrone et al., 2002).
DISEASE
There are emerging molecular links between homeostatic plasticity and neurological disease. The schizophrenia associated gene dysbindin was isolated in a forward genetic screen for mutations that block presynaptic homeostasis (Dickman and Davis, 2009). Homer and mGluR signaling are implicated in mouse models of Fragile X syndrome (Ronesi et al., 2012) as is retinoic acid (Soden and Chen, 2010). Others have speculated the involvement of disrupted homeostatic signaling in posttraumatic epilepsy (Houweling et al., 2005), Rett Syndrome (Ramrocki and Zoghbi 2008; Qiu et al., 2012) and autism spectrum disorders (Bougeron, 2009). A wealth of information is emerging regarding rare de novo mutations with strong effects in autism spectrum disorders and it is possible that further associations with homeostatic plasticity will emerge (Murdoch and State, 2013). Clearly, it will be important to advance our understanding of how homeostatic plasticity interfaces with the function and plasticity of neural circuitry in vivo, a topic that has been recently reviewed (Watt and Desai, 2010; Maffei and Fontanini 2009; Turrigiano, 2011). It will be just as important to gain a complete molecular understanding of the molecular design and implementation of neuronal homeostatic signaling within individual neurons. Once understood, the manipulation of homeostatic signaling could enable therapeutic manipulation of neuronal activity with far reaching implications. By extension, it might become possible for medicine to treat the compensated nervous system, rather than the underlying disruptions that initiate disease.
CONCLUSIONS
This field is wide open for exploration. The excitement surrounding this emerging field is that it spans so many disciplines, from control theory and modeling to biophysics, developmental biology and human disease. How is cell type specific homeostatic plasticity achieved? Can the phenotypic modulation of neural activity be visualized in vivo while homeostatic plasticity is induced and executed? If so, might it be possible to identify enzymatic activities and protein interactions that precede, track or lag the phenotypic expression of a homeostatic response? These are just a few major questions, in addition to those raised throughout this review, that remain to be experimentally addressed.
Acknowledgments
This work is supported by NIH grant #NS39313 to GWD. I thank Meg Younger, Kevin Ford, Michael Gavino, Martin Muller and Santiago Archila for help with the preparation of this review.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrásfalvy BK, Makara JK, Johnston D, Magee JC. Altered synaptic and non-synaptic properties of CA1 pyramidal neurons in Kv4.2 knockout mice. J Neurophysiol. 2008;586:3881–3892. doi: 10.1113/jphysiol.2008.154336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggono V, Clem RL, Huganir RL. PICK1 loss of function occludes homeostatic synaptic scaling. J Neurosci. 2011;31:2188–2196. doi: 10.1523/JNEUROSCI.5633-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awatramani GB, Price GD, Trussell LO. Modulation of Transmitter Release by Presynaptic Resting Potential and Background Calcium Levels. Neuron. 2005;48:109–121. doi: 10.1016/j.neuron.2005.08.038. [DOI] [PubMed] [Google Scholar]
- Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, Sabatini BL. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron. 2013;78:510–522. doi: 10.1016/j.neuron.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgardt M, Miguel-Aliaga I, Karlsson D, Ekman H, Thor S. Specification of neuronal identities by feedforward combinatorial coding. PLoS biology. 2007;5:e37. doi: 10.1371/journal.pbio.0050037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Zastrow Von M, et al. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Béïque JC, Na Y, Kuhl D, Worley PF, Huganir RL. Arc-dependent synapse-specific homeostatic plasticity. PNAS. 2011;108:816–821. doi: 10.1073/pnas.1017914108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergquist S, Dickman DK, Davis GW. A hierarchy of cell intrinsic and target-derived homeostatic signaling. Neuron. 2010;66:220–234. doi: 10.1016/j.neuron.2010.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T. A synaptic trek to autism. Curr Op Neurosci. 2009;19:231–234. doi: 10.1016/j.conb.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Bucher D, Prinz AA, Marder E. Animal-to-animal variability in motor pattern production in adults and during growth. J Neurosci. 2005;25:1611–1619. doi: 10.1523/JNEUROSCI.3679-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, O’Byrne M, Murthy VN. Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature. 2002;420:414–418. doi: 10.1038/nature01242. [DOI] [PubMed] [Google Scholar]
- Chang MC, Park JM, Pelkey KA, Grabenstatter HL, Xu D, Linden DJ, et al. Narp regulates homeostatic scaling of excitatory synapses on parvalbumin-expressing interneurons. Nature Neurosci. 2010;13:1090–1097. doi: 10.1038/nn.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, et al. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Chiu DN, Jahr CE. Ca2+-dependent enhancement of release by subthreshold somatic depolarization. Nat Neurosci. 2010;14:62–68. doi: 10.1038/nn.2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrêa SAL, Hunter CJ, Palygin O, Wauters SC, Martin KJ, McKenzie C, et al. MSK1 regulates homeostatic and experience-dependent synaptic plasticity. J Neurosci. 2012;32:13039–13051. doi: 10.1523/JNEUROSCI.0930-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy SG, Miledi R, Trautmann A, Uchitel OD. On the release of transmitter at normal, myasthenia gravis and myasthenic syndrome affected human end-plates. The Journal of physiology. 1980;299:621–638. doi: 10.1113/jphysiol.1980.sp013145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Chen K, Gelfand MV, Featherstone DE, Diantonio A. A single vesicular glutamate transporter is sufficient to fill a synaptic vesicle. Neuron. 2006;49(1):11–16. doi: 10.1016/j.neuron.2005.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annual review of neuroscience. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Davis GW, Goodman CS. Genetic analysis of synaptic development and plasticity: homeostatic regulation of synaptic efficacy. Current opinion in neurobiology. 1998a;8(1):149–156. doi: 10.1016/s0959-4388(98)80018-4. [DOI] [PubMed] [Google Scholar]
- Davis GW, Goodman CS. Synapse-specific control of synaptic efficacy at the terminals of a single neuron. Nature. 1998b;392(6671):82–86. doi: 10.1038/32176. [DOI] [PubMed] [Google Scholar]
- Davis GW, Schuster CM, Goodman CS. Genetic analysis of the mechanisms controlling target selection: target-derived Fasciclin II regulates the pattern of synapse formation. Neuron. 1997;19:561–573. doi: 10.1016/s0896-6273(00)80372-4. [DOI] [PubMed] [Google Scholar]
- Deeg KE, Aizenman CD. Sensory modality-specific homeostatic plasticity in the developing optic tectum. Nature Neurosci. 2011;14:548–550. doi: 10.1038/nn.2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman DK, Davis GW. The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science (New York, NY) 2009;326(5956):1127–1130. doi: 10.1126/science.1179685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll HE, Muraro NI, He M, Baines RA. Pumilio-2 regulates translation of Nav1.6 to mediate homeostasis of membrane excitability. J Neurosci. 2013;33:9644–9654. doi: 10.1523/JNEUROSCI.0921-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulcis D, Jamshidi P, Leutgeb S, Spitzer NC. Neurotransmitter switching in the adult brain regulates behavior. Science. 2013;340:449–453. doi: 10.1126/science.1234152. [DOI] [PubMed] [Google Scholar]
- Echegoyen J, Neu A, Graber KD, Soltesz I. Homeostatic plasticity studied using in vivo hippocampal activity-blockade: synaptic scaling, intrinsic plasticity and age-dependence. PloS one. 2007;2:e700. doi: 10.1371/journal.pone.0000700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA. Homeostatic plasticity at the Drosophila neuromuscular junction. Neuropharmacology. 2013 doi: 10.1016/j.neuropharm.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Pielage J, Davis GW. A presynaptic homeostatic signaling system composed of the Eph receptor, ephexin, Cdc42, and CaV2.1 calcium channels. Neuron. 2009;61:556–569. doi: 10.1016/j.neuron.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainey MA, Hurvitz-Wolff JR, Lambo ME, Turrigiano GG. Synaptic scaling requires the GluR2 subunit of the AMPA receptor. J Neurosci. 2009;29:6479–6489. doi: 10.1523/JNEUROSCI.3753-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bereguiain MA, Gonzalez-Islas C, Lindsly C, Butler E, Hill AW, Wenner P. In vivo synaptic scaling is mediated by GluA2-lacking AMPA receptors in the embryonic spinal cord. J Neurosci. 2013;33:6791–6799. doi: 10.1523/JNEUROSCI.4025-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goaillard JM, Taylor AL, Schulz DJ, Marder E. Functional consequences of animal-to-animal variation in circuit parameters. Nature Neurosci. 2009;12:1424–1430. doi: 10.1038/nn.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard CA, Butts DA, Shatz CJ. Regulation of CNS synapses by neuronal MHC class I. PNAS. 2007;104:6828–6833. doi: 10.1073/pnas.0702023104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Chub N, Garcia-Bereguiain MA, Wenner P. GABAergic synaptic scaling in embryonic motoneurons is mediated by a shift in the chloride reversal potential. J Neurosci. 2010;30:13016–13020. doi: 10.1523/JNEUROSCI.1659-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold CP, Davis GW. The BMP ligand Gbb gates the expression of synaptic homeostasis independent of synaptic growth control. Neuron. 2007;56:109–123. doi: 10.1016/j.neuron.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold CP, Nicoll RA. Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron. 2010;68:512–528. doi: 10.1016/j.neuron.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haedo RJ, Golowasch J. Ionic mechanism underlying recovery of rhythmic activity in adult isolated neurons. J Neurophysiol. 2006;96:1860–1876. doi: 10.1152/jn.00385.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Kaeser PS, Südhof TC, Schneggenburger R. RIM determines Ca2+ channel density and vesicle docking at the presynaptic active zone. Neuron. 2011;69:304–316. doi: 10.1016/j.neuron.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengen KB, Lambo ME, Van Hooser SD, Katz DB, Turrigiano GG. Firing Rate Homeostasis in Visual Cortex of Freely Behaving Rodents. Neuron. 2013 doi: 10.1016/j.neuron.2013.08.038. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry FE, McCartney AJ, Neely R, Perez AS, Carruthers CJL, Stuenkel EL, et al. Retrograde changes in presynaptic function driven by dendritic mTORC1. J Neurosci. 2012;32:17128–17142. doi: 10.1523/JNEUROSCI.2149-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. Regulation of terminal differentiation programs in the nervous system. Ann Rev Cell Biol. 2011;27:681–696. doi: 10.1146/annurev-cellbio-092910-154226. [DOI] [PubMed] [Google Scholar]
- Houweling AR, Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Homeostatic synaptic plasticity can explain post-traumatic epileptogenesis in chronically isolated neocortex. Cerebral. 2005;15:834–845. doi: 10.1093/cercor/bhh184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JH, Park JM, Park S, Xiao B, Dehoff MH, Kim S, et al. Homeostatic scaling requires group I mGluR activation mediated by Homer1a. Neuron. 2010;68:1128–1142. doi: 10.1016/j.neuron.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, et al. Local Presynaptic Activity Gates Homeostatic Changes in Presynaptic Function Driven by Dendritic BDNF Synthesis. Neuron. 2010;68:1143–1158. doi: 10.1016/j.neuron.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessell TM. Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nature Rev Genetics. 2000;1:20–29. doi: 10.1038/35049541. [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, Südhof TC. RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell. 2011;144:282–295. doi: 10.1016/j.cell.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck T, Keller GB, Jacobsen RIR, Eysel UT, Bonhoeffer T, Hübener M. Synaptic scaling and homeostatic plasticity in the mouse visual cortex invivo. Neuron. 2013 doi: 10.1016/j.neuron.2013.08.018. in press. [DOI] [PubMed] [Google Scholar]
- Khorkova O, Golowasch J. Neuromodulators, not activity, control coordinated expression of ionic currents. J Neurosci. 2007;27:8709–8718. doi: 10.1523/JNEUROSCI.1274-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Ryan TA. CDK5 serves as a major control point in neurotransmitter release. Neuron. 2010;67:797–809. doi: 10.1016/j.neuron.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Ryan TA. Balance of calcineurin Aα and CDK5 activities sets release probability at nerve terminals. J Neurosci. 2013;33:8937–8950. doi: 10.1523/JNEUROSCI.4288-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Tsien RW. Synapse-Specific Adaptations to Inactivity in Hippocampal Circuits Achieve Homeostatic Gain Control while Dampening Network Reverberation. Neuron. 2008;58:925–937. doi: 10.1016/j.neuron.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambo ME, Turrigiano GG. Synaptic and intrinsic homeostatic mechanisms cooperate to increase L2/3 pyramidal neuron excitability during a late phase of critical period plasticity. J Neurosci. 2013;33:8810–8819. doi: 10.1523/JNEUROSCI.4502-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nature Rev Mol Cell Biology. 2012;13:383–396. doi: 10.1038/nrm3351. [DOI] [PubMed] [Google Scholar]
- Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–556. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- Liu Z, Golowasch J, Marder E, Abbott LF. A model neuron with activity-dependent conductances regulated by multiple calcium sensors. J Neurosci. 1998;18:2309–2320. doi: 10.1523/JNEUROSCI.18-07-02309.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Tsien RW. Properties of synaptic transmission at single hippocampal synaptic boutons. Nature. 1995;375:404–408. doi: 10.1038/375404a0. [DOI] [PubMed] [Google Scholar]
- Lu W, Bushong EA, Shih TP, Ellisman MH, Nicoll RA. The cell-autonomous role of excitatory synaptic transmission in the regulation of neuronal structure and function. Neuron. 2013;78(3):433–439. doi: 10.1016/j.neuron.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean JN, Zhang Y, Johnson BR, Harris-Warrick RM. Activity-independent homeostasis in rhythmically active neurons. Neuron. 2003;37:109–120. doi: 10.1016/s0896-6273(02)01104-2. [DOI] [PubMed] [Google Scholar]
- Maffei A, Fontanini A. Network homeostasis: a matter of coordination. Curr Op Neurobiol. 2009;19:168–173. doi: 10.1016/j.conb.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E. Variability, compensation, and modulation in neurons and circuits. PNAS. 2011;108(Supplement_3):15542–15548. doi: 10.1073/pnas.1010674108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Prinz AA. Modeling stability in neuron and network function: the role of activity in homeostasis. BioEssays. 2002;24:1145–1154. doi: 10.1002/bies.10185. [DOI] [PubMed] [Google Scholar]
- Marie B, Pym E, Bergquist S, Davis GW. Synaptic homeostasis is consolidated by the cell fate gene gooseberry, a Drosophila pax3/7 homolog. J Neurosci. 2010;30:8071–8082. doi: 10.1523/JNEUROSCI.5467-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin OJ, Lee A, McGraw TE. GLUT4 distribution between the plasma membrane and the intracellular compartments is maintained by an insulin-modulated bipartite dynamic mechanism. JBC. 2006;281:484–490. doi: 10.1074/jbc.M505944200. [DOI] [PubMed] [Google Scholar]
- McGeachie AB, Cingolani LA, Goda Y. Stabilising influence: integrins in regulation of synaptic plasticity. Neuroscience Res. 2011;70:24–29. doi: 10.1016/j.neures.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrsic-Flogel TD, Hofer SB, Ohki K, Reid RC, Bonhoeffer T, Hübener M. Homeostatic regulation of eye-specific responses in visual cortex during ocular dominance plasticity. Neuron. 2007;54:961–972. doi: 10.1016/j.neuron.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Müller M, Davis GW. Transsynaptic control of presynaptic Ca2 influx achieves homeostatic potentiation of neurotransmitter release. Curr Biol. 2012;22:1102–1108. doi: 10.1016/j.cub.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Liu KSY, Sigrist SJ, Davis GW. RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. J Neurosci. 2012;32:16574–16585. doi: 10.1523/JNEUROSCI.0981-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Pym ECG, Tong A, Davis GW. Rab3-GAP controls the progression of synaptic homeostasis at a late stage of vesicle release. Neuron. 2011;69:749–762. doi: 10.1016/j.neuron.2011.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraro NI, Weston AJ, Gerber AP, Luschnig S, Moffat KG, Baines RA. Pumilio binds para mRNA and requires Nanos and Brat to regulate sodium current in Drosophila motoneurons. J Neurosci. 2008;28:2099–2109. doi: 10.1523/JNEUROSCI.5092-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch JD, State MW. Recent developments in the genetics of autism spectrum disorders. Curr Op Genetics and Development. 2013;23:310–315. doi: 10.1016/j.gde.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM, Gerber BR, Norris A, Burkhalter A. Electrical remodelling maintains firing properties in cortical pyramidal neurons lacking KCND2-encoded A-type K+ currents. J Physiol. 2008;586:1565–1579. doi: 10.1113/jphysiol.2007.146597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- Paradis S, Sweeney ST, Davis GW. Homeostatic control of presynaptic release is triggered by postsynaptic membrane depolarization. Neuron. 2001;30:737–749. doi: 10.1016/s0896-6273(01)00326-9. [DOI] [PubMed] [Google Scholar]
- Penney J, Tsurudome K, Liao EH, Elazzouzi F, Livingstone M, Gonzalez M, et al. TOR is required for the retrograde regulation of synaptic homeostasis at the Drosophila neuromuscular junction. Neuron. 2012;74:166–178. doi: 10.1016/j.neuron.2012.01.030. [DOI] [PubMed] [Google Scholar]
- Pilgram GSK, Potikanond S, van der Plas MC, Fradkin LG, Noordermeer JN. The RhoGAP crossveinless-c interacts with Dystrophin and is required for synaptic homeostasis at the Drosophila neuromuscular junction. J Neurosci. 2011;31:492–500. doi: 10.1523/JNEUROSCI.4732-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp JJ, van Kempen GT, Molenaar PC. Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. J Physiol. 1992;458:487–499. doi: 10.1113/jphysiol.1992.sp019429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt KG, Aizenman CD, Aizenman CD. Homeostatic Regulation of Intrinsic Excitability and Synaptic Transmission in a Developing Visual Circuit. J Neurosci. 2007;27:8268–8277. doi: 10.1523/JNEUROSCI.1738-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz AA, Bucher D, Marder E. Similar network activity from disparate circuit parameters. Nat Neurosci. 2004;7:1345–1352. doi: 10.1038/nn1352. [DOI] [PubMed] [Google Scholar]
- Pulver SR, Bucher D, Simon DJ, Marder E. Constant amplitude of postsynaptic responses for single presynaptic action potentials but not bursting input during growth of an identified neuromuscular junction in the lobster, Homarus americanus. J Neurobiol. 2005;62:47–61. doi: 10.1002/neu.20066. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Sylwestrak EL, Lieberman DN, Zhang Y, Liu XY, Ghosh A. The Rett syndrome protein MeCP2 regulates synaptic scaling. J Neurosci. 2012;32:989–994. doi: 10.1523/JNEUROSCI.0175-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455:912–918. doi: 10.1038/nature07457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG, et al. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nature neuroscience. 2012;15(3):431–40. S1. doi: 10.1038/nn.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarti F, Schroeder J, Aoto J, Chen L. Conditional RARα knockout mice reveal acute requirement for retinoic acid and RARα in homeostatic plasticity. Frontiers in Molecular Neurosci. 2012;5:16. doi: 10.3389/fnmol.2012.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz DJ, Goaillard JM, Marder E. Variable channel expression in identified single and electrically coupled neurons in different animals. Nat Neurosci. 2006;9:356–362. doi: 10.1038/nn1639. [DOI] [PubMed] [Google Scholar]
- Schulz DJ, Goaillard JM, Marder EE. Quantitative expression profiling of identified neurons reveals cell-specific constraints on highly variable levels of gene expression. PNAS. 2007;104:13187–13191. doi: 10.1073/pnas.0705827104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg DP, Feliu-Mojer M, Gaiottino J, Pak DTS, Sheng M. Critical role of CDK5 and Polo-like kinase 2 in homeostatic synaptic plasticity during elevated activity. Neuron. 2008;58:571–583. doi: 10.1016/j.neuron.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg DP, Sheng M. Activity-Induced Polo-Like Kinase 2 Is Required for Homeostatic Plasticity of Hippocampal Neurons during Epileptiform Activity. J Neurosci. 2008;28:6583–6591. doi: 10.1523/JNEUROSCI.1853-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, et al. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soden ME, Chen L. Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. J Neurosci. 2010;30:16910–16921. doi: 10.1523/JNEUROSCI.3660-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz CC, Turrigiano GG. Tumor necrosis factor-α signaling maintains the ability of cortical synapses to express synaptic scaling. J Neurosci. 2010;30:14685–14690. doi: 10.1523/JNEUROSCI.2210-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelling J, Sauer U, Szallasi Z, Doyle FJ, Doyle J. Robustness of cellular functions. Cell. 2004;118:675–685. doi: 10.1016/j.cell.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Sun Q, Turrigiano GG. PSD-95 and PSD-93 Play Critical But Distinct Roles in Synaptic Scaling Up and Down. J Neurosci. 2011;31:6800–6808. doi: 10.1523/JNEUROSCI.5616-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Taylor AM, Ito HT, Pham A, Schuman EM. Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron. 2007;55:648–661. doi: 10.1016/j.neuron.2007.07.030. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Wall NR, Aakalu GN, Schuman EM. Regulation of dendritic protein synthesis by miniature synaptic events. Science. 2004;304:1979–1983. doi: 10.1126/science.1096202. [DOI] [PubMed] [Google Scholar]
- Swensen AM, Bean BP. Robustness of burst firing in dissociated purkinje neurons with acute or long-term reductions in sodium conductance. J Neurosci. 2005;25:3509–3520. doi: 10.1523/JNEUROSCI.3929-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatavarty V, Sun Q, Turrigiano GG. How to scale down postsynaptic strength. J Neurosci. 2013;33:13179–13189. doi: 10.1523/JNEUROSCI.1676-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temporal S, Desai M, Khorkova O, Varghese G, Dai A, Schulz DJ, Golowasch J. Neuromodulation independently determines correlated channel expression and conductance levels in motor neurons of the stomatogastric ganglion. J Neurophysiol. 2012;107:718–727. doi: 10.1152/jn.00622.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to Synaptic Inactivity in Hippocampal Neurons. Neuron. 2005;47:725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Turrigiano G. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Ann Rev Neurosci. 2011;34:89–103. doi: 10.1146/annurev-neuro-060909-153238. [DOI] [PubMed] [Google Scholar]
- Turrigiano G, Abbott LF, Marder E. Activity-dependent changes in the intrinsic properties of cultured neurons. Science. 1994;264:974–977. doi: 10.1126/science.8178157. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Tsurudome K, Tsang K, Liao EH, Ball R, Penney J, Yang JS, et al. The Drosophila miR-310 cluster negatively regulates synaptic strength at the neuromuscular junction. Neuron. 2010;68:879–893. doi: 10.1016/j.neuron.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagarajan SK, Tyagarajan SK, Fritschy JM, Fritschy JM. GABAA receptors, gephyrin and homeostatic synaptic plasticity. J Physiol. 2010;588:101–106. doi: 10.1113/jphysiol.2009.178517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wart A, Matthews G. Impaired firing and cell-specific compensation in neurons lacking nav1.6 sodium channels. J Neurosci. 2006;26:7172–7180. doi: 10.1523/JNEUROSCI.1101-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL, Zhang Z, Hintze M, Chen L. Decrease in calcium concentration triggers neuronal retinoic acid synthesis during homeostatic synaptic plasticity. J Neurosci. 2011;31:17764–17771. doi: 10.1523/JNEUROSCI.3964-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt AJ, Desai NS. Homeostatic plasticity and STDP: keeping a neuron’s cool in a fluctuating world. Frontiers in Synaptic Neuroscience. 2010:2. doi: 10.3389/fnsyn.2010.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyhersmüller A, Hallermann S, Wagner N, Eilers J. Rapid active zone remodeling during synaptic plasticity. J Neurosci. 2011;31:6041–6052. doi: 10.1523/JNEUROSCI.6698-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtowicz JM, Atwood HL. Maintained depolarization of synaptic terminals facilitates nerve-evoked transmitter release at a crayfish neuromuscular junction. J Neurobiol. 1983;14:385–390. doi: 10.1002/neu.480140506. [DOI] [PubMed] [Google Scholar]
- Younger MA, Müller M, Tong A, Pym EC, Davis GW. A Presynaptic ENaC Channel Drives Homeostatic Plasticity. Neuron. 2013;79:1183–1196. doi: 10.1016/j.neuron.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Dreosti E, Lagnado L. Homeostatic Synaptic Plasticity through Changes in Presynaptic Calcium Influx. J Neurosci. 2011;31:7492–7496. doi: 10.1523/JNEUROSCI.6636-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]