

Abstract
Wnt proteins are critical to mammalian brain development and function. The canonical Wnt signaling pathway involves the stabilization and nuclear translocation of β-catenin; however, Wnt also signals through alternative, noncanonical pathways. To gain a systems-level, genome-wide view of Wnt signaling, we analyzed Wnt1-stimulated changes in gene expression by transcriptional microarray analysis in cultured human neural progenitor (hNP) cells at multiple time points over a 72-hour time course. We observed a widespread oscillatory-like pattern of changes in gene expression, involving components of both the canonical and the noncanonical Wnt signaling pathways. A higher-order, systems-level analysis that combined independent component analysis, waveform analysis, and mutual information–based network construction revealed effects on pathways related to cell death and neurodegenerative disease. Wnt effectors were tightly clustered with presenilin1 (PSEN1) and granulin (GRN), which cause dominantly inherited forms of Alzheimer’s disease and frontotemporal dementia (FTD), respectively. We further explored a potential link between Wnt1 and GRN and found that Wnt1 decreased GRN expression by hNPs. Conversely, GRN knockdown increased WNT1 expression, demonstrating that Wnt and GRN reciprocally regulate each other. Finally, we provided in vivo validation of the in vitro findings by analyzing gene expression data from individuals with FTD. These unbiased and genome-wide analyses provide evidence for a connection between Wnt signaling and the transcriptional regulation of neurodegenerative disease genes.
INTRODUCTION
Wnts constitute a large family of secreted, but spatially restricted, lipophilic signaling molecules. They exert wide-ranging biological activities by initiating equally varied signaling cascades (1, 2). The best-studied (“canonical”) Wnt signaling pathway involves the disheveled (DVL) and Axin-mediated inactivation of glycogen synthase kinase–3β (GSK-3β), producing an attendant increase in β-catenin, which activates lymphoid enhancer factor (LEF) and T cell factor (TCF) (TCF/LEF)–dependent transcription. Negative feedback, most prominently through increased expression of the intracellular canonical signaling antagonists AXIN2, naked-1 (NKD1), and naked-2 (NKD2), tightly controls the overall activity of this pathway (1–3). An emergent property of this complex regulatory machinery is that the activity of the Wnt signaling pathway oscillates in the face of sustained Wnt binding (4–7).
First identified as potent oncogenes, Wnts encode master regulators of fetal brain development and continue to govern neuronal growth and survival in the adult brain (8, 9). For example, Wnt1 is essential for neural crest induction and proper development of the midbrain dopaminergic system (9–11) and inhibits apoptosis in various cell types (12–15). Furthermore, Wnt1, through the canonical signaling pathway, maintains the multipotency of human neural stem cells (16) and enhances neural progenitor proliferation and differentiation (17).
Wnt directly binds to more than two dozen partners, activating at least six different second messenger cascades in addition to the canonical pathway (1). As the number of signaling pathways known to be initiated by Wnt has increased, so too has the number of identified steps in each pathway and the number of components involved in mediating each step [for instance, β-catenin alone has more than three dozen known binding partners (18)]. Furthermore, the potential for crosstalk between Wnt and other signaling cascades has grown so extensive that these cascades resemble signaling webs more than directed pathways. The richness of Wnt signaling has also frustrated attempts to discern the most biologically important roles for this pathway from a myriad of plausible scenarios. Thus, it is surprising that few studies have systematically studied this pathway from a genomic standpoint.
Wnt signaling has also been implicated in several forms of neurodegenerative disease, most prominently Alzheimer’s disease (AD) and frontotemporal dementia (FTD), through various direct and indirect mechanisms (19–23). For example, Wnt modifies the activity of presenilin and microtubule-associated protein tau (tau; MAPT), proteins encoded by two genes that when mutated cause either AD or FTD, respectively. Presenilin deficiency leads to β-catenin stabilization (24), and AD-causing presenilin mutations also disrupt β-catenin function (25), providing a direct link between canonical Wnt signaling and genetic factors that cause AD. Pathological tau phosphorylation is also a hallmark of both AD and a subset of FTD cases, and GSK-3β is a tau kinase (26, 27). GSK-3β–mediated tau phosphorylation accelerates tau-induced neurodegeneration in animal models of tauopathy (21), whereas GSK-3β inhibitors have been touted as potential therapeutic agents in tau-related dementias, such as FTD and AD (28, 29). Wnt signaling therefore may provide a bridge between neurodevelopment and neurodegeneration (30, 31).
To develop an unbiased understanding of the global effects of Wnt signaling in the nervous system from a systems biology perspective, we measured higher-order gene expression patterns in primary human neural progenitor (hNP) cells during the early phases of Wnt signaling. This analysis revealed a complex pattern of transcriptional changes occurring over a 72-hour period, which uncovered a strong connection between Wnt1-dependent signaling networks and expression of genes implicated in the etiology of both AD and FTD, as well as other neurodegenerative conditions. Haploinsufficiency of GRN (granulin), one of the genes down-regulated by the Wnt1 signal, causes a dominantly inherited FTD; however, virtually nothing is known about GRN regulation or how the progranulin (PGRN) protein it encodes affects neuronal survival. As an experimental validation of our network predictions at the level of a single gene, we investigated the potential mechanism of this interaction. We discovered a reinforcing feedback relationship between the control of Wnt and PGRN abundance, where decreased PGRN increased WNT1 expression, whereas Wnt1 repressed GRN expression and decreased PGRN abundance. This further implicates aberrant Wnt signaling in the etiology of FTD and supports investigation of Wnt pathway modulation in the treatment of neurodegenerative disease.
RESULTS
A genome-wide time course of Wnt-induced changes in transcript abundance
The initial aim of this study was to obtain an unbiased view of the time-dependent changes in gene expression caused by Wnt signaling. Because different Wnts can potentially activate multiple distinct signaling cascades, we focused on the effects of Wnt1, which is a consistent activator of the canonical pathway (32, 33). Our experimental design eliminates many of the limitations encountered in previous studies of Wnt-mediated transcription, including using few time points (34), relying on transformed or immortalized cell lines that may poorly reflect normal human tissue (35), or surveying only a small fraction of the transcriptome (36). Specifically, we measured genome-wide mRNA abundance with Illumina Human RefSeq-8 BeadArrays at multiple time points (2, 4, 6, 8, 24, and 72 hours) to optimally capture different early epochs after the initial Wnt signal (36).
After Wnt1 application, the individual time course of these changes in gene expression—which could reflect changes in transcription, mRNA abundance, or both—showed various patterns, including transients, monotonic changes, and oscillations (Fig. 1A). Although the expression of only a minor fraction (0.3 to 20%) of genes was changed at any given time point (Fig. 1B; t test, P ≤ 0.05), the expression of nearly two-thirds of sampled genes changed at some point during the 72-hour time course examined. Overall, the largest changes in gene expression, whether up or down, were apparent at 2, 6, and 24 hours; fewer changes were observed at 4 and 72 hours.
Fig. 1.

Wnt1-induced changes in the human neural progenitor transcriptome vary across time. (A) Representative genes showing common patterns of changes in gene expression over 24 hours, normalized to initial values. (B) Genes with significantly (P ≤ 0.05; t test) increased (magenta) or decreased (cyan) message abundance after Wnt1 application. (C and D) qPCR validation of microarray data. (C) Bars are range-normalized changes in expression (2 hours versus control) showing high correlation in directionality of expression, array (ILMN) versus qPCR (ρ = 0.48, n = 24 genes; one-tailed P ≤ 0.01). (D) Time series plots showing high correlation between qPCR- and microarray-quantified transcript. (E) Canonical Wnt reporter: Wnt1 applied to hNPs stably expressing a LEF/TCF-dsGFP reporter (n = 4 cultures) (139) causes a time-dependent increase in canonical activity that peaks between 4 and 6 hours. (F) Overlap in gene expression changes across time. (Upper left) Plot showing the fraction of genes whose message increased at 2 hours, but consistently decreased at subsequent time points. (Top, left to right) Venn diagrams showing the overlap of genes whose message increased (magenta) at both 2 and 8 hours or decreased (cyan) at 6 and 24 hours. (Bottom) Venn diagrams of genes whose message increased at 2 hours and then down-regulated at 6 hours (left), down at 6 hours then up at 8 hours (middle), or up at 8 hours and down again by 24 hours (right).
At 2 hours, increases in gene expression predominated, whereas at 6 and 24 hours, most differentially expressed genes showed decreased mRNA abundance compared to untreated cells. After the initial wave of increased transcript abundance at 2 hours, we found that quantities of most of these mRNAs had declined at 6 hours, then increased again at 8 hours, only to return to near-baseline activity by 24 to 72 hours. Next, using real-time quantitative polymerase chain reaction (qPCR), we examined whether we could confirm the initial burst of increased gene expression and the overall patterns of changes in gene expression recorded by the microarrays. First, we examined changes in the expression of 25 genes at t = 2 hours. The genes that compose this sample showed a wide range of changes in expression at t = 2 hours rather than changing in lock-step with the bulk of genes at this time (Fig. 1B). Eighteen of 24 genes (75%) identified as changing in gene expression by microarray showed a consistent pattern of changes when analyzed by qPCR (Fig. 1C). After range normalization, the microarray and reverse transcription–PCR (RT-PCR) data exhibited a significant Pearson correlation coefficient (ρ = 0.48, n = 24; one-tailed P ≤ 0.01). Next, we examined the changes in abundance for a subset of six mRNAs across all time points (Fig. 1D). From time point to time point, the direction of expression changes was the same 83% of the time, with a high collective correlation of 0.68 ± 0.14 (median ± SD) between qPCR and microarray results. Together, these data indicated a high degree of reproducibility of the microarray-based measurements of changes in mRNA abundance across time.
Because Wnt1 is the archetypical ligand for the canonical pathway, we tested the temporal relationship between activation of this pathway and the observed pattern of transcriptional changes. In hNPs stably expressing a TCF/LEF reporter (16, 37), signaling through the canonical pathway peaked 4 to 6 hours after Wnt1 application, returning to baseline by 24 hours (Fig. 1E). This monophasic rise and fall in activity of the canonical pathway coincides predictably with the initial changes in gene expression (Fig. 1B), but contrasts with those changes at later time points. These data suggest that canonical signaling may initiate a more complex cascade of changes in either transcription or mRNA stabilization, so that later events are mediated by both canonical and noncanonical signaling. Consistent with this hypothesis, we observed substantial changes in the expression of components of the noncanonical Wnt signaling pathways (fig. S1).
Successive waves of increases and decreases in transcription could arise from a set of coherently oscillating genes or represent a more complex summation of asynchronously changing sets of genes. The latter interpretation is supported by a closer inspection of the specific genes changing at each time point. For example, of the 2848 genes showing increased expression at 2 hours, less than half showed a decrease in mRNA abundance at later time points, with most of those showing decreases at 6 hours (Fig. 1F, upper left trace, and table S1). There is also a limited overlap of the genes participating in the successive bursts of increased expression. For example, only 153 genes showed increased expression at both 2 and 8 hours, representing 5 and 14% of the total number of genes changed at those time points, respectively. Together, these data suggest the presence of multiple superimposed transcriptional programs that sum to form apparent waves of altered gene expression.
Within this complex picture, several time-dependent functional patterns emerge, including the expected Wnt1-induced transcription of many genes implicated in the canonical Wnt signaling pathway, including WNT5B, FZD (frizzled) 2, 4, and 8, AXIN1, GSK3B, CTNNB1 (β-catenin), TCF7L2, NKD1, and AXIN2 (table S2). The products of many of these genes stimulate proliferation, repress differentiation, or are direct feedback regulators of the Wnt pathway itself (for example, Axin2). These apparent waves of Wnt1-mediated transcription are consistent with the oscillatory-like patterns of Wnt target gene expression that underlie operation of the embryonic vertebrate segmentation clock (38, 39).
Pathway analysis
We performed a Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis (40) as a first step in unbiased characterization of the function of differentially expressed Wnt1-induced genes. This analysis confirmed early enrichment (t = 2 hours) for genes involved in Wnt signaling (Fig. 2A, Table 1A, and table S2). Two-thirds of the genes in the canonical Wnt pathway represented in the KEGG differentially regulated after the Wnt1 signal, compared to the control condition, validating the activation of Wnt signaling in these experiments.
Fig. 2.

Wnt1 modulates genes implicated in Wnt signaling and AD. (A) KEGG pathway: graphical summary of the diversity of Wnt signaling–related genes (canonical and noncanonical), significantly enriched (FDR ≤ 5%) at 2 hours after Wnt1 application (n = 59 of 151 KEGG Wnt genes). Significantly increased (magenta) or decreased (cyan) mRNA abundance (t test; *P ≤ 0.05). (B) KEGG AD pathways highlighting genes (magenta) whose message was significantly increased by Wnt1 at t = 2 hours (t test; *P ≤ 0.05). (See fig. S9 for scalable version of this figure.)
Table 1.
GO analysis of changes in Wnt1-mediated gene expression reveals time-specific enrichment. (A) Selected KEGG pathways (129) or (B) selected highest-level (level 5) GO biological processes showing significant categorical enrichment at each time point (FDR ≤5%). GO analysis was performed in DAVID (40) on all significantly changed genes (t test, P ≤ 0.05), irrespective of the direction of the change. ATP, adenosine triphosphate; ncRNA, noncoding RNA; MAPK, mitogen-activated protein kinase; GTPase, guanosine triphosphatase; VEGF, vascular endothelial growth factor
| A |
B |
||||
|---|---|---|---|---|---|
| KEGG pathway | P | GO biological process level 5 | P | ||
| 2 Hours | Alzheimer’s disease | 1.47 × 10−6 | 2 Hours | RNA splicing | 7.95 × 10−9 |
| Chronic myeloid leukemia | 1.15 × 10−5 | mRNA processing | 9.72 × 10−9 | ||
| Colorectal cancer | 4.00 × 10−5 | Regulation of transcription, DNA-dependent | 7.79 × 10−6 | ||
| Huntington’s disease | 5.36 × 10−5 | ATP synthesis coupled proton transport | 1.10 × 10−4 | ||
| Oxidative phosphorylation | 9.16 × 10−5 | Energy coupled proton transport | 1.10 × 10−4 | ||
| Pyrimidine metabolism | 1.11 × 10−4 | Regulation of neuron projection development | 1.64 × 10−4 | ||
| Parkinson’s disease | 1.18 × 10−4 | Regulation of ubiquitin-protein ligase activity | 2.33 × 10−4 | ||
| Pancreatic cancer | 2.11 × 10−4 | Regulation of axonogenesis | 3.72 × 10−4 | ||
| Pathways in cancer | 5.35 × 10−4 | Regulation of cell morphogenesis involved | 4.67 × 10−4 | ||
| Apoptosis | 7.32 × 10−4 | in differentiation | |||
| Ubiquitin-mediated proteolysis | 8.74 × 10−4 | Chromatin modification | 9.76 × 10−4 | ||
| Insulin signaling pathway | 1.07 × 10−3 | Regulation of apoptosis | 1.32 × 10−3 | ||
| Renal cell carcinoma | 1.19 × 10−3 | ncRNA metabolic process | 1.80 × 10−3 | ||
| ErbB signaling pathway | 1.22 × 10−3 | 4 Hours | G protein-coupled receptor protein signaling | 4.65 × 10−4 | |
| RNA polymerase | 1.72 × 10−3 | Cell surface receptor linked signal transduction | 2.3 × 10−3 | ||
| MAPK signaling pathway | 2.31 × 10−3 | 6 Hours | ncRNA processing | 1.04 × 10−7 | |
| Prostate cancer | 2.48 × 10−3 | Mitochondrial transport | 3.10 × 10−5 | ||
| Lysine degradation | 3.23 × 10−3 | Regulation of ubiquitin-protein ligase activity | 2.69 × 10−4 | ||
| Wnt signaling pathway | 3.33 × 10−3 | ATP synthesis coupled proton transport | 4.11 × 10−4 | ||
| Pyruvate metabolism | 3.93 × 10−3 | Negative regulation of ubiquitin-protein ligase | 9.36 × 10−4 | ||
| Axon guidance | 5.50 × 10−3 | Sterol biosynthetic process | 1.25 × 10−3 | ||
| Cell cycle | 5.65 × 10−3 | 8 Hours | Negative regulation of apoptosis | 1.57 × 10−3 | |
| 6 Hours | Huntington’s disease | 5.09 × 10−8 | Negative regulation of programmed cell death | 1.80 × 10−3 | |
| Oxidative phosphorylation | 2.49 × 10−7 | ncRNA metabolic process | 2.42 × 10−3 | ||
| Alzheimer’s disease | 8.21 × 10−7 | Muscle tissue development | 2.6 × 10−3 | ||
| Pyrimidine metabolism | 1.23 × 10−5 | 24 Hours | Chromatin modification | 1.01 × 10−6 | |
| Parkinson’s disease | 4.96 × 10−5 | Mitosis | 4.04 × 10−5 | ||
| RNA polymerase | 5.42 × 10−4 | Small GTPase-mediated signal transduction | 5.60 × 10−5 | ||
| Pentose phosphate pathway | 1.37 × 10−3 | M phase of mitotic cell cycle | 7.28 × 10−5 | ||
| Purine metabolism | 2.10 × 10−3 | Protein kinase cascade | 7.79 × 10−5 | ||
| Glycosphingolipid biosynthesis | 3.04 × 10−3 | Regulation of apoptosis | 1.36 × 10−4 | ||
| Alanine and aspartate metabolism | 3.18 × 10−3 | Lipid oxidation | 1.33 × 10−3 | ||
| 24 Hours | VEGF signaling pathway | 8.92 × 10−5 | Retrograde vesicle-mediated transport | 1.43 × 10−3 | |
| Pathways in cancer | 3.51 × 10−4 | Negative regulation of cell growth | 1.67 × 10−3 | ||
| Base excision repair | 2.60 × 10−3 | Phospholipid biosynthetic process | 2.44 × 10−3 | ||
| Axon guidance | 3.45 × 10−3 | Central nervous system development | 2.50 × 10−3 | ||
| Non–small cell lung cancer | 4.47 × 10−3 | Regulation of axonogenesis | 2.79 × 10−3 | ||
Our analysis also revealed enrichment for genes and pathways involved in any of the following three broad categories (Table 1, A and B, and table S3): (i) cell proliferation or death, (ii) energy metabolism, and (iii) regulation of the transcriptional or translational machinery (representative KEGG pathways at early and late time points; figs. S2 and S3). Because more than 90% of genes that show changes in gene expression at each time point do so in the same direction, GO analysis was performed on all differentially expressed genes irrespective of the direction of the change. Few differences were noted when these changes were analyzed with respect to the direction of the change in expression (table S3B).
KEGG pathway analysis also revealed a significant enrichment of neurodegenerative pathways (Table 1A) including the AD (Fig. 2B), Huntington’s disease (HD) (fig. S3), and Parkinson’s disease (PD) pathways, consistent with the hypothesized role of Wnt in neurodegenerative disease (21, 23, 41–46). Moreover, we saw enrichment for genes associated with AD in the National Institutes of Health Genetic Association Database (table S4). Indeed, nearly one of three of the genes in the KEGG AD pathway were among the earliest group of genes showing increased Wnt1-mediated expression (t = 2 hours), a biologically meaningful and statistically significant result (see Materials and Methods). Because this analysis was based on an unbiased and genome-wide derived overlap, these data provide a first tier of systematic evidence for a causal connection between Wnt signaling and regulating expression of genes implicated in neurodegenerative disease.
Time series analysis
Our initial differential expression analysis revealed complex patterns of changes in gene expression. Analyzing this level of biological complexity across time is complicated by the nature of the time series data itself. For example, time series data are often mathematically nonstationary and show autocorrelation, creating nonlinearities or discontinuities in the data, which limit the use of many statistical techniques that assume fixed or Gaussian probability distributions (47). A further complication of analyzing time series data is that expression patterns in pairs of genes are often not monotonically correlated, rendering inappropriate commonly used nonparametric tests of association, such as the Spearman rank correlation (48, 49).
To reach a more complete biological understanding of Wnt1’s effects, we analyzed these data in three separate ways, using distinct statistical techniques that are relatively insensitive to the underlying data’s distribution: (i) dynamic time warping (DTW) (50–52), (2) independent components analysis (ICA) (53, 54), and (3) mutual information–based network analysis (55).
A systems-level analysis of Wnt1 expression trajectories using DTW
The signal processing method of DTW provides a flexible and robust metric for measuring the difference between two waveforms (50–52). DTW creates this distance measure by locally compressing or stretching (warping) one trace to best match the other and then summing the distances of individual aligned elements. In this way, DTW provides high power to detect differences in time series by accounting for the trajectory of changes in expression over time, unlike a purely statistical technique, such as analysis of covariance (ANCOVA). To better establish the applicability of DTW to microarray time series data, we analyzed how noise or phase shifting affected DTW-based gene ranking or GO enrichment, first with simulated data and then as applied to our Wnt1 data set. First, we found that DTW distance increased linearly with random noise and in proportion to the length of random traces being compared (fig. S4, A and B). Next, we assessed the effects of phase shifting of identical waveforms on the resulting DTW distance (DTWdist) and compared this to the Spearman correlation, a more common metric in microarray analysis. Because zero is the lower boundary of DTWdist, increased phase shifting resulted in monotonically increasing distances, unlike the correlation coefficient, which crossed the zero point (fig. S4, C and D). This relationship reduces the ambiguity in interpreting DTW-measured distances that are zero (or very small), an advantage of DTW over correlations measures (that is, a DTW distance of zero necessarily indicates that two traces are highly similar, not merely in phase). When comparing less idealized transients of gene expression, DTW distance again conforms better to the common sense notion of association than does either the Spearman or the Pearson correlation (fig. S4E).
We used DTW to identify those genes most strongly influenced by Wnt1 signaling by measuring the warping distance between the time course of changes in gene expression in cells treated with Wnt1 and that in untreated cells (longer DTW distances represent traces that are more Wnt1-specific). When these distances were ranked from most to least Wnt1-specific (high to low), they initially fell off exponentially (with an exponential decay constant, τ = 0.94, and correlation coefficient, R2 = 0.99; fig. S5A), where one decay constant is equivalent to 300 genes. Using this natural breakpoint, we limited further analysis to the top-ranked 300 genes (table S5A). Sensitivity analysis revealed that this ranking predictably degraded with the addition of increasing noise (fig. S5, B and C).
KEGG pathway analysis of the native data revealed an enrichment for cancer-related genes (fig. S6), consistent with Wnt1 being a protooncogene. Likewise, a GO analysis of these genes demonstrated enrichment for genes implicated in neurogenesis, angiogenesis, or axon and dendrite growth (table S5B), and genes associated with neuropsychiatric disease (disorders with both psychiatric and neurodegenerative features) including the dementia-related genes APP (amyloid precursor protein), MAPT, and GRN (table S5C).
A functional ICA analysis of the Wnt1 expression network
Our studies of differential gene expression and ontological enrichment rest on the following assumptions: (i) Perturbations (such as drug application) alter existing biological processes by changing patterns of gene expression across time. (ii) Each gene product may be involved in many different biological processes (for instance, β-catenin has different functions at cell surface and nucleus). (iii) Multiple genes may subserve a single biological process. (iv) Only a few genes are essential for a given biological process, although many other genes may contribute to it. On the basis of this biological model (fig. S7A), we can formulate an analytical model (fig. S7B) for the purposes of computation. By viewing the overall pattern of gene expression obtained from microarray analysis as resulting from the sum of many different biological processes, our analysis becomes a special case of blind source separation, that is to say, recovery of a set of mixed signals, with little or no information about source signals or the mixing process. ICA, which extracts non-Gaussian statistical structures from high-dimensional data (54, 56–59), is among the most widely used techniques for blind source separation and has been applied to the analysis of microarray data (58).
ICA can decompose the observed expression data into the most likely original independent sources of variation [a visual demonstration of ability of parallel decomposition ICA (pICA) to recover separate signals from highly convoluted data is illustrated in fig. S7C]. When applied to gene expression data, these independent components (the original sources of variation defined by ICA) will ideally correspond to a set of biologically coherent processes (that is, convergent physiological effects). For example, a “neuronal maturation” component might be dominated by groups of co-regulated genes involved in coordinating processes like neurotransmitter packaging, neurite outgrowth, and synapse formation.
We first performed pICA on the expression data from Wnt1-treated cells, pooled across all seven time points and replicates, producing seven statistically independent components (ICA1-7). In each component, each gene is valued by its statistical contribution to that component, termed gene loading. In each component, genes with gene-loading values below a threshold of 3.0 were discarded from the component, creating modules (ICM1-7) that captured only the most relevant genes in that component. Examples of normalized time series data for the top 10 genes, by gene-loading value, in each of seven independent components are displayed in Fig. 3A. In creating these modules (that is, loading-thresholded components), we used a cutoff far more stringent than those previously reported so as to be conservative in our analysis (58).
Fig. 3.

Unsupervised parallel independent component analysis (pICA) blindly separates gene expression patterns by biological function. (A) Normalized, mean expression time course for the top 10 genes in each ICA component, ranked by gene loading. (B) GO enrichment via DAVID was performed on each independent component module after thresholding at a gene-loading level of 3.0. Colors delineate individual modules. Listed are the top nonredundant level 5 biological processes (129), disease ontologies (underlined), or KEGG pathways (boldface), with associated P values (n.s., not significant; n = number of genes per module). (C) Overlapping MiMI interactome networks built using the top 20 odd-ranked genes versus the top 20 even-ranked genes from ICM2 [Top Odd versus Top Even (left) or Bottom Odd versus Top Even (right)] networks built from the bottom 20 genes versus Top Even genes. The Top Odd network recovered significantly more Top Even genes (turquoise circles; n = 24) than did the Bottom network (magenta circles; n = 8). (D) Genes in each ICM were probed against the Broad Institute’s Molecular Signatures Database. The most highly enriched data set is presented for each module, as well as a representative sampling of other significantly enriched data sets. (See fig. S10 for scalable version of this figure. See table S8 for a full listing of GSEA-identified data sets.)
The individual genes that contributed most strongly to each component were subjected to GO and KEGG analysis. Using ICA, we observed modular ontologic enrichment, consistent with biologically meaningful gene clustering (Fig. 3B and table S6). For example, KEGG annotations delineated a single Wnt signaling–enriched component and a single AD-enriched component (ICM2). However, we also observed some redundancy, with multiple components enriched for genes related to altered translation or ribosomal function. Sensitivity analysis to evaluate the robustness of this technique revealed that the ability of ICA to recover relevant biological processes degraded gradually in the face of increasingly noisy expression data (table S7). For example, ICA still recovers a Wnt module even when the noise present in the underlying data is increased by a full SD.
As a further biological validation of ICA-based clustering, we sought to integrate our newly delineated expression modules into established gene-protein interaction networks, using the public interaction database MiMI (http://mimi.ncibi.org/MimiWeb/main-page.jsp) as a generator (60, 61). Starting with the gene ranking in the Wnt-specific ICA module ICM2, we created three interaction networks seeded with the (i) top 20 odd-ranked genes (genes ranked 1st, 3rd, 5th…19th; top odd), (ii) top 20 even-ranked genes, or (iii) the bottom 20 odd-ranked genes. We compared the degree of overlap between the odd versus even (top) networks and the top versus bottom networks, with the expectation that top genes (even or odd) should extract networks more similar to each other than to the network formed from lower-ranked genes (bottom). Compared to the bottom network, the top odd gene network recovered three times as many genes from the even network, including the seed gene granulin (GRN; Fig. 3C). Using a different experimental modality, these data confirm functional associations between highly ranked genes in our ICMs.
As a complement to the interaction network building, we sought to place these ICA-derived gene modules into the context of known changes in expression. We probed each gene set against the 3429 gene expression profiles available through the Broad Institute Gene Set Enrichment Analysis (GSEA) Molecular Signatures Database (Fig. 3D and table S8). The resulting gene set enrichment analysis was highly complementary to the GO analysis reported above. Consistent with Wnt1’s role in oncogenesis, the genes in each module were frequently overrepresented in cancer-related data sets. Similarly, there is good agreement between GSEA signatures and the more specific GO categories identified above (Fig. 3B). For example, module 1 was enriched for GO-annotated angiogenesis-related genes, and the genes from this module were overrepresented among genes expressed by regenerating blood vessels. In contrast, genes present in the Wnt-specific module (ICM2) showed the greatest overlap with genes showing increased expression in the brains of Alzheimer’s patients (Fig. 3D; Fisher’s exact test, P ≤ 5 × 10−11), strengthening the possible connection between Wnt1 signaling and dementia (Fig. 2).
The two methods, DTW and ICA, address two fundamentally different biological questions and were used to provide convergent lines of evidence for identifying genes mediating the Wnt1 response. DTW identified genes specifically influenced by Wnt1 treatment by comparing the trajectories of gene expression for Wnt1-treated hNPs versus untreated hNPs. As presented, DTW did not explicitly provide information about which genes might act together to mediate similar biological processes. Our ICA decomposed multivariate expression data from Wnt1-treated hNPs into parallel statistical components, a linear representation of maximally non-Gaussian source signals that are as statistically independent as possible. In effect, this parsed genes into groups that potentially serve similar biological processes. One would expect that the genes common to both lists would be enriched for those with the strongest biological relevance, even if the magnitude of the overlap is modest. That these lists do overlap indicates that the cohort of common genes plays a fundamental role in mediating Wnt1 signaling. Specifically, we observed a significant overlap (17%; hypergeometric test, P ≤ 10−14) in the gene lists obtained by DTW and ICA (table S9). Of the 14 genes identified by DTW that have a genetic association with neuropsychiatric illness (table S5C), 4 genes were shared by the Wnt-specific ICA module (ICM2): APP, GRN, AQP4, and CTSD, the first two being associated with early-onset dementia.
Evidence from our GO, MiMI interactome, and GSEA-based analyses pointed to the biological importance of the ICM2 module, because it was enriched for Wnt- or dementia-related genes, prompting us to more fully investigate the interplay among its constituent genes. First, we performed a weighted gene coexpression analysis (WGCNA) of the genes that compose ICM2 (62–65). WGCNA performs unsupervised clustering of co-expressed genes into modules on the basis of topological overlap (TOM), a measure of the degree to which a given pair of genes share common neighbors (64). When applied to ICM2, WGCNA clustered the constituent’s expression profiles into four distinct submodules, each being described by a characteristic module eigengene (Fig. 4, A and B). These eigengenes are composite gene expression vectors derived from the first principal component of the measured mRNA abundances that are used to concisely describe the overall variability of the data (66, 67). Moreover, GO analysis of each submodule revealed unique patterns of significant ontologic enrichment, despite the small number of genes present in each submodule (Fig. 4C).
Fig. 4.

Combined topological overlap–based clustering and dynamic Bayesian network construction links Wnt1 signaling with changes in dementia-related genes. (A and B) WGCNA clustering of ICM2 genes: Wnt1-stimulated expression time courses for the genes that compose the ICM2 module were averaged and then subjected to TOM-WGCNA–based clustering (A). This produced four submodules (Mustard, Brown, Blue, and Turquoise). (B) Submodule eigengenes, where singular value decomposition was used to extract a characteristic first principal component eigengene for each submodule. The y axis is eigengene expression. (C) GO analysis reveals functional uniqueness of individual submodules. (D and E) Dynamic Bayesian network (DBN) depicting causal relationships, within each module. (D) Overview of the DBN-based causality network. Edge color codes the original submodule. Node color indicates those genes identified by DTW analysis (magenta; n = 23). Outlined diamonds denote those genes whose expression was increased in the brains of Alzheimer’s patients (n = 20) (70). DLK1 forms the primary hub in this network. [Note: SORT1, like DLK1, is a binding partner for PGRN (73).] (E) A more detailed view of the DLK1 hub and its associated genes, revealing a significant overlap (hypergeometric P ≤ 0.001) with DTW-identified genes and a strong enrichment for genes with increased message in AD brains. Transcriptional drivers (blue dots), targets (magenta arrowheads). (See fig. S11 for scalable version of this figure.)
Advanced Bayesian methods allow one to assess causality among genes with covarying expression patterns. To further explore these relationships in ICM2, the Wnt-dementia module, we constructed a dynamic Bayesian network (DBN) for each WGCNA-clustered submodule, using the G1DBN package (68, 69). We then coded the resulting network to reflect overlap with either Wnt1-modulated genes previously identified by DTW or those ICM2 genes with increased expression in the hippocampus of Alzheimer’s brains (70). As an internal validation of this approach, we observed that the genes identified as “hubs” in the network were primarily drivers of, rather than targets of, “spoke” genes (Fig. 4, D and E). The two most prominent hubs were the delta-like 1 homolog (DLK1), which encodes a Notch antagonist, and the stem cell marker promonin-1 (PROM1). As illustrated, DLK1 forms a nexus connecting the “Blue” and “Turquoise” submodules. Isolating the genes forming connections to DLK1 (Fig. 4D) produced a subset of 103 genes, with statistically significant enrichment for both DTW-identified genes and genes with increased message in the hippocampus of Alzheimer’s patients (70). Closer inspection revealed that this submodule contains several canonical Wnt effectors (including DVL3, APC2, and TSC2), as well as two genes implicated in causing dementia [APP and GRN, which binds both DLK1 and SORT1 (sortilin-1; Turquoise submodule)], strengthening the link between Wnt1 signaling and dementia (71–73).
Coexpression network analysis
Our previous network analysis revealed a highly probable link between Wnt1 signaling and dementia but was limited to a small number of genes. To put these observed changes into a more complete systems biology context, we performed a mutual information–based network analysis (55, 74) as a means of systematically exploring the higher-order structure of Wnt1-induced changes in gene expression across a much larger fraction of the genome. Mutual information–based network analysis provides a robust measure of the statistical association of gene expression patterns between gene pairs, but makes no assumptions about the underlying probability distributions (55, 74–78). The first step in our analysis was to dimensionally reduce the data (and thereby decrease the data set) by pooling the genes identified by DTW, ICA, or strong deviation from statistical normality (that is, a Gaussian distribution), a common feature of genes that change expression over time, a procedure that yielded 4398 genes. This reduced data set comprises the union of the most specifically Wnt1-regulated genes and those showing the greatest changes in expression over time, thereby providing the context for Wnt1-specific changes. Next, we constructed the Wnt-induced mutual information–based network (see Materials and Methods and fig. S8), and then pruned it on the basis of MRnet, the information-theoretic method of maximum relevance–minimum redundancy (55). This revealed several prominent Wnt-related hubs, two of which contained known dementia-related genes (Fig. 5, A to C). For example, five Wnt genes, Wnt4, Wnt5B, Wnt6, Wnt8A, and Wnt8B, out of seven Wnts represented in this set, colocalized to a nine-gene cluster centered on the hub gene COL25A1. COL25A1 is a brain-specific membrane-bound collagen that inhibits fibrillization of β-amyloid and associates with senile plaques in AD brains (79–82). Another hub that revealed a relationship between Wnt signaling and regulation of dementia-related genes (Fig. 5C) consisted of a nine-gene cluster centered on APOA4, which is associated with AD (83–85) and is a paralog of the dementia gene APOA1 (85–88), and included two genes involved in Wnt/β-catenin signal transduction, DVL2 and CTNNBL1. This cluster also contains the AD-related genes, presenilin1 (PSEN1) and death receptor 6 [DR6; tumor necrosis factor receptor (TNFR) superfamily 21: TNFRSF21], the FTD gene GRN, and TROY (TNFRSF19), which has been implicated in failure of the adult nervous system to regenerate (89). This grouping is all the more pronounced in that PGRN reduces inflammation by binding to TNFRs and that PGRN degradation is blocked by APOA1 (90).
Fig. 5.

Wnt1 induces a gene expression architecture that correlates many well-known dementia genes with Wnt-related signal transduction. (A to C) Microarray-based gene expression data were generated from Wnt1-treated or untreated hNPs, followed at seven time points over 72 hours and repeated in triplicate. Displayed is the subset of the MINA (mutual information–based network analysis)–based network that contains connections that exceeded a threshold of 1.1 bits. (A) Multiple Wnts cluster around COL25A1, an AD-related gene. (B) CTNNB1 (β-catenin) and CXCR4 hubs. (C) Neighborhood containing a dense cluster of dementia [magenta; presenilin (PSEN1), GRN, APOA4, DR6 (death receptor 6)] and Wnt transduction–related genes. Nodes are color-coded to reflect genes implicated in neural development (orange), dementia (magenta), Wnt signaling (cyan), or diseases distinct from dementia (yellow). [Note: CCNT1 (cyclin-T1) is a binding partner of PGRN.] (D) Loss of APOA4 dysregulates dementia hub genes: qPCR of hippocampal gene expression among wild-type and APOA4-null mice reveals significant changes in the expression of connected genes (blue), relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH), but not the other Wnt-dementia hub gene COL25A1 (orange). Values are fold changes in gene expression calculated with the ΔΔCt method (125) [*P ≤ 0.05; n = 4 brains, PCR-ANCOVA (125)]. (See fig. S12 for scalable version of this figure.)
Experimental validation of network predictions
To test whether the in vitro network architecture predicted in vivo relationships—specifically the clustering of dementia- and Wnt-related genes—we examined the effects of loss of the hub gene APOA4. Using qPCR, we compared the expression of genes predicted to be highly correlated with APOA4 in the hippocampi of APOA4 knockout to wild-type mice. Loss of APOA4 substantially altered the expression of many neighborhood genes (that is, genes one to two degrees separated from APOA4), including PSEN1, TNFRSF21 (death receptor 6), TNFRSF19 (TROY), DVL2, and DVL3, validating network predictions in vivo (Fig. 5D).
Thus, these unbiased expression analyses validated, at the whole-genome level, previous hypothesis-driven work, suggesting a connection between Wnt signaling and neurodegenerative disease (43, 45, 46, 91–94). Furthermore, both analysis of DTW-differential expression and mutual information–based network analysis revealed a higher-order relationship between GRN and Wnt pathway genes in hNPs. Little is known about the biological function or regulation of GRN, except that haploinsufficiency causes a dominantly inherited form of FTD (95–98). Therefore, we further investigated the connection between Wnt and GRN.
Experimental characterization and validation of the Wnt-GRN (PGRN) relationship
We knocked down PGRN in hNPs using lentivirally transduced RNA interference (RNAi) hairpins (Fig. 6, A to C), observing a 50% decrease in PGRN abundance, using gene-specific RNAi, and no off-target effects on PGRN, using control RNAi (Fig. 6B). Decreasing PGRN abundance significantly altered the abundance of the mRNAs encoding several different Wnts, including WNT1 (Fig. 6C), which was increased nearly fourfold by PGRN knockdown.
Fig. 6.
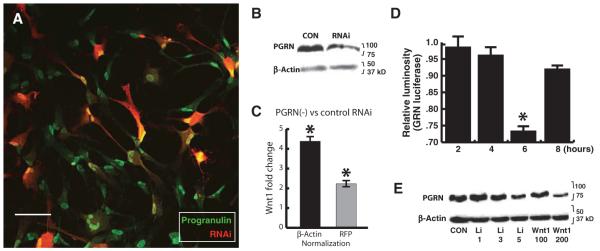
PGRN knockdown increases Wnt1 expression. (A) Lentiviral vector using CAG promoter is not silenced in hNPs and knocks down PGRN: hNPs were infected with lentiviral vector expressing DsRed2 and RNAi against GRN. The color plate illustrates hNPs immunolabeled for PGRN (green) and DsRed2 (red), under control conditions. Scale bar, 25 μm. (B) Western blot analysis of PGRN: At 2 weeks, cell lysates were collected, and PGRN abundance was analyzed by Western blot, demonstrating PGRN knockdown (n = 4). (C) qPCR for Wnt1 mRNA showing that GRN knockdown significantly alters Wnt expression: Bars indicate fold change in Wnt1 expression when normalized to either the general internal control β-actin (reflecting total cells) or the vector-derived DsRed (reflecting RNAiGRN expression; means ± SEM). n = 3; t test, *P ≤ 0.05. (D and E) Canonical Wnt signaling dose-dependently inhibits PGRN transcription and expression: (D) Wnt1 significantly represses baseline activity of GRN promoter in HEK293 cells transfected with a GRN luciferase reporter [n = 4; analysis of variance (ANOVA), *P ≤ 0.05]. (E) Western blot analysis reveals linear dose-dependent reduction of PGRN abundance in hNPs 4 hours after bath application of Wnt1 or lithium (Li) [n = 4; doses indicated; Pearson R2 = 0.99 (Wnt) and 0.94 (lithium)].
To determine whether Wnt’s effects on PGRN depended on changes in expression, we assessed Wnt1 regulation of GRN promoter activity. We found that Wnt1 significantly repressed the activity of a human GRN luciferase reporter overexpressed in a human cell line [human embryonic kidney (HEK) 293]. The time course of changes in reporter activity was parallel to that of the Wnt1-dependent changes in GRN expression observed in primary hNPs. Wnt1 repression of GRN-luciferase activity was maximal at 6 hours (Fig. 6D), consistent with studies demonstrating that β-catenin abundance peaks 4 to 6 hours after Wnt application in HEK293 cells (36).
Next, we examined the relationship between PGRN and Wnt1 signaling in primary hNPs. We found that both Wnt1 and lithium, a GSK-3β antagonist that mimics canonical Wnt signaling, decreased PGRN abundance in hNPs in a dose-dependent manner (Fig. 6E). Along with the microarray and qPCR results above, these data indicate that PGRN and Wnt reciprocally regulate each other’s expression in hNPs.
Finally, we sought to validate the connection between Wnt signaling and GRN expression by means of expression data collected from GRN-haploinsufficient demented patients. This data set (99) is composed of whole-genome expression data from the frontal cortex, hippocampus, and cerebellum of patients with either sporadic FTD or GRN-haploinsufficient FTD, or controls of similar sex and age distribution. We observed that various Wnt-related ligands and receptors, and all four LEF/TCF transcription factors implicated in the canonical pathway, were differentially expressed in tissue from either the frontal cortex (FTX), the hippocampus (HIP), or both, of GRN-haploinsufficient FTD patients, compared to their expression in tissue from the FTX and HIP of sporadic FTD patients (Fig. 7A). Moreover, WNT1 expression was increased in tissue from the FTX and HIP of GRN-haploinsufficient patients compared to tissue from the cerebellum, a region not affected by FTD (FTX: log-fold ratio, 1.6; P = 0.006; HIP: log-fold ratio, 2.42; P = 0.053). In contrast, WNT1 expression was not significantly increased in the FTX or HIP of sporadic FTD patients.
Fig. 7.

Canonical Wnt transcription factors are among genes most specifically correlated with frontocortical expression in GRN-haploinsufficient FTD. (A) Wnt signaling receptors and ligands differentially expressed in the frontal cortex of GRN-haploinsufficient FTD patients versus patients suffering from sporadic FTD (Bayesian t test, P ≤ 0.05). (B) Disease eigengene: This is a graphical representation of the frontal cortex GRN-specific vector used to correlate disease state and anatomical location with expression of individual genes in the full FTD data set. The data set comprises whole transcriptome gene expression in the frontal cortex (magenta), hippocampus (blue), or cerebellum (orange) of patients with either GRN-haploinsufficient FTD (dark) or sporadic FTD (medium) or controls (light) (99). (C) KEGG Wnt/β-catenin signaling–related genes among the total set of 392 genes significantly correlated either positively (orange; increased message) or negatively (blue; decreased message) with the synthetic FTX-GRN eigengene depicted in (B). Genes binding β-catenin (bold). Significance was based on Spearman rank correlations between each gene and the disease-specific synthetic eigengene (ρ ≥ 0.40, *P ≤ 0.001, n = 56 arrays).
Finally, we sought to identify the changes in gene expression that correlate most specifically with GRN-mediated FTD and determine whether Wnt-related genes were overrepresented in this set. To accomplish this, we first created a synthetic eigengene to represent those genes exclusively exhibiting differential expression in the frontal cortex of GRN-haploinsufficient FTD patients, but not in sporadic FTD patients, nor in the hippocampus or cerebellum or any patients (Fig. 7B). By comparing idealized pattern of gene (eigengene) expression against measured mRNA abundance of the other genes on the array, we found that 392 genes were highly correlated with GRN haploinsufficiency and frontocortical localization. DAVID-based GO analysis (40) of this gene set revealed significant enrichment for genes involved in Wnt receptor signaling (EASE P = 0.001). Ingenuity-based analysis yielded a similar enrichment for Wnt/β-catenin signaling genes (P = 0.004, Fisher’s exact test). Furthermore, among this gene set, we found several β-catenin binding transcription factors, including LEF1 and TCF7L2, two of the four best-described targets of canonical Wnt signaling (Fig. 7C). These data validate and extend the expression data, confirming a connection between Wnt signaling and neurodegenerative pathways, including those associated with PGRN deficiency in humans.
DISCUSSION
This study was designed to expand understanding of the role of Wnt signaling in human neural development using hNPs as a model system. Because Wnts can activate various intracellular signaling pathways, we used an unbiased systems biology approach to identify the Wnt1 targets most relevant to neural stem cells. We found that Wnt1 altered transcript abundance of a large number of genes in an oscillatory fashion. Although Wnt1 affected the expression of only a minority of genes at any given time point, it altered the expression of more than half of the hNP transcriptome during the 24-hour period after treatment. We showed that Wnt1 broadly affected most noncanonical Wnt signaling cascades, despite being the archetypal ligand for initialing canonical (β-catenin) signaling. Moreover, Wnt1 activation of these canonical and noncanonical Wnt pathways appeared to widely influence expression of genes mediating cellular metabolism (for example, steroid, purine, and ribosome synthesis) in addition to their more well-known functions, proliferation or cell fate. Using fetal progenitors as a model system, we found that Wnt1-responsive genes showed a strong collective association with sets of genes implicated in neurodegenerative diseases associated with aging, most prominently AD and FTD.
We used three complementary statistical workflows—DTW, ICA-WGCNA-DBN, and MI-MRnet—to generate a consensus network view of the Wnt1 transcriptional program. Individually and collectively, DTW, ICA-WGCNA-DBN, and MI-MRnet suggested a connection between Wnt1 and PGRN expression and, by extension, a potential connection between Wnt signaling and GRN-mediated FTD. Using a humanized inducible model of GRN deficiency, we found that PGRN and Wnt1 reciprocally regulated each other’s expression. Moreover, we found that the expression of numerous Wnt signaling genes was increased in the frontal cortices of GRN-haploinsufficient patients compared to other brain regions, or the frontal cortices of individuals without GRN mutations, indicating that this connection is clinically relevant. The variety of altered Wnt signaling genes suggests that they mediate a complex network of biological processes in GRN-haploinsufficient neurons. Further in silico and experimental work will be required to delineate these processes and which Wnt signaling genes are most relevant to them.
The Wnt1 gene expression network
Transcriptional oscillations have become increasingly recognized as a key feature of synchronized developmental programs (6, 7). We observed that a single application of Wnt1 induced multiple waves of changed expression in cells derived from early fetal neural progenitors over a 72-hour period. This is akin to what has been observed with the transcription factor hairy and enhancer of split1 (Hes1), which drives variable period oscillations in transcription within neural progenitors of the mouse telencephalon (100). Similarly, Wnt3a produces oscillations within developing limb buds (5–7) that control the rhythmic production of somites (precursors of the vertebrae) (6, 101), thereby linking the segmentation clock and signaling gradients within presomitic mesoderm. We identified a set of oscillating genes implicated in a wide range of biological pathways, including pathways relating to angiogenesis, immune modulation, cell proliferation, energy metabolism, and neuronal maturation (36, 38, 39). Thus, we propose that Wnt might serve a dual role in the developing nervous system: Wnt may act within neuroblasts to initiate the set of parallel developmental programs that are required for the formation of a mature neuron. In addition, Wnt secreted by neuroblasts could synchronize neural maturation with the development of necessary supporting tissue, such as vasculature and glia (102).
Regulation of PGRN expression
PGRN, like Wnt, was first described for its role in oncogenesis, and later shown to have neurotrophin or growth factor–like activity (103–106). This led to the hypothesis that GRN haploinsufficiency leads to a relative deficit of PGRN and thereby the death of neurons within specific brain regions, such as the frontal cortex. However, more recent evidence suggests that active suppression of neuronal GRN expression, not overall reduced amounts of PGRN, precipitates frontal lobe degeneration (99, 107). Our findings that Wnt1 and PGRN reciprocally regulate each other’s expression provide a biological model that could potentially explain this active, neuron-specific decrease in PGRN.
We found that suppression of GRN expression, essentially mimicking FTD-associated human GRN haploinsufficiency, increased WNT1 expression, whereas Wnt1 suppressed GRN expression in otherwise normal neuronal cells. Hypothetically, a haploinsufficiency-induced reduction in PGRN could set up a negative feedback loop that actively suppressed GRN expression (beyond the decrease in GRN resulting from its haploinsufficiency). Wnts can act as autocrine or paracrine signals because they exhibit limited diffusion away from the source (108, 109). Therefore, increased neuronal Wnt production would selectively suppress expression of GRN in neighboring neurons, rendering them incapable of compensating for haploinsufficiency (that is, unable to increase expression from their unaffected allele). This model could explain several clinical pathophysiological observations including (i) the concentration of PGRN in the blood of individuals haploinsufficient of GRN levels can be far below the 50% that would be expected based on the decrease in gene dosage (110–112), and (ii) cells in different brain regions of affected patients vary in their ability to effectively maintain sufficient levels of PGRN synthesis (99, 107).
Whether increased Wnt signaling improves survival of GRN-haploinsufficient neurons remains an open question. However, it is possible that these cells engage Wnt as an alternative pro-survival signal. Compensating for the loss of PGRN-mediated tropism by means of Wnt could partially normalize function early in the disease process. However, it could ultimately contribute to cortical atrophy by further decreasing GRN expression. A similar model of “compensatory engagement” has been proposed to explain the indolent course and ultimate cognitive system failure in chronic neurodegenerative diseases like Alzheimer’s (113, 114). Because Wnt1 asynchronously activates functionally distinct transcriptional programs, it is possible that the pro-survival effects of Wnt1 could be pharmacologically separated from its repression of GRN expression. Therefore, for individuals with GRN haploinsufficiency, targeting the right subset of Wnt1 target genes very early in life could conceivably prevent the onset of disease, without affecting the protective effects of Wnt signaling.
In summary, our data support a role for Wnt signaling in clinical dementia in general, and FTD more specifically. Together, the combination of functional genomic, bioinformatic, and cell biological data supports a possible mechanism of GRN dysregulation in haploinsufficient patients. These data lay the preclinical foundation for developing Wnt modulators as potential treatments for PGRN-mediated FTD and other neurodegenerative conditions.
MATERIALS AND METHODS
Primary cell culture
We generated human fetal neural progenitors as previously described (16, 115, 116) and propagated them in human neural stem cell proliferation medium consisting of Neurobasal A, 10% BIT 9500 (Stem Cell Technology), fibroblast growth factor 2 (FGF2) (20 ng/ml; PeproTech), epidermal growth factor (EGF) (20 g/ml; PeproTech), leukemia inhibitory factor (LIF) (2 ng/ml), and heparin (2 μg/ml). To eliminate the potential confounding effects of FGF2 and EGF (117), we replaced proliferation medium with minimal growth factor medium [Neurobasal A, 5% BIT 9500, and heparin (2 μg/ml)] 48 hours before Wnt1 application. After this growth factor washout, the medium was again replaced 100% to eliminate autocrine factors that might have accumulated (16). hNPs were divided into treatment and control groups, and recombinant Wnt1 (400 ng/ml; PeproTech) was applied to the treatment groups. hNPs were harvested at time = 0, and then both Wnt1-treated and untreated control NPs were harvested at 2, 4, 6, 8, 24, and 72 hours. Three biological replicates were collected at each time point after Wnt1 treatment. For PGRN knockdown experiments, hNPs expressing pLCR-RNAiPGRN were differen-tiated in medium supplemented with 1% fetal calf serum (FCS; Hyclone), 500 μM all trans-retinoic acid (Sigma), 10 μM forskolin, NT3 (20 ng/ml), and brain-derived neurotrophic factor (BDNF). Functional confirmation of PGRN-related effects was performed on hNP lines derived from separate individuals than those whose cells were used for the time series microarray experiments. HEK293 cells were propagated in Dulbecco’s modified Eagle’s medium (DMEM), 10% FCS, and 1× Antibiotic-Antimycotic mix (Invitrogen). Recombinant human Wnt1 (PeproTech) was used at 200 ng/ml.
Viral preparation, infection, and knockdown
Lentivirus for overexpression or inducible knockdown of gene expression was produced as previously described (16, 118, 119) (see Supplementary Methods for details). Hairpins directed against GRN were designed, cloned into either pLCR or pTRIPZ (Open Biosystems) using the PCR-shagging protocol (120), and then tested for their ability to achieve greater than 50% knockdown. PGRN knockdown in TRIPZ-infected cells was induced by addition of doxycycline (2 μg/ml). APOA4 knockout mice were generated by homologous recombination and maintained as previously described (121, 122).
Immunodetection
Immunoblotting and immunocytochemistry of whole-cell lysates or cultures were performed by standard methods, essentially as previously described (123) (see Supplementary Methods).
Reporter assays
GRN promoter analysis was performed in HEK293T cells transected (1:50) with Renilla luciferase plasmid (pRL-EF) and either constitutively active firefly luciferase or pCMV-Tag4a PGRN expression plasmid (Switchgear Genomics) and then processed according to the manufacturer’s instructions (Dual-Luciferase Assay; Promega). Wnt activity was assessed essentially as previously described (16) (see Supplementary Methods for details).
Microarray analysis
Total RNA was harvested from hNPs with the RNeasy kit (Qiagen) according to the manufacturer’s protocol. Spectrophotometry [NanoDrop; A260/280 (absorbance at 260/280 nm), −1.8] and the Agilent Bioanalyzer were used to determine RNA concentrations and RNA quality. Total RNA (200 ng) was amplified, labeled, and hybridized to HumanRef-8v2 and v3 Expression BeadChip (Illumina). Data were preprocessed through BeadStudio (Illumina) to produce raw output files. All further processing was conducted in the R statistical computing environment (http://www.r-project.org/). When indicated, background correction, variance-stabilizing transformation (VST), and robust spline normalization (RSN) were applied using the Bioconductor package LUMI (http://www.bioconductor.org). Probes that varied in sequence between platforms were eliminated, leaving 18,396 probes that were examined in all experiments described herein.
Time point differential expression analysis
Microarray-based measurements of mRNA expression were log2-transformed, and then at each time point, the values from untreated control hNPs were subtracted from the measured values for Wnt-treated hNPs. By applying this procedure on a gene-by-gene basis, we created a data set in which genes whose expression was unaffected by Wnt1 treatment would be expected to have a normalized mean expression of zero. Significance of differential expression at individual time points was calculated with a zero-centered, one-way t test. Differential expression ratios and P values on the in vivo Human FTD data set [Gene Expression Omnibus (GEO) ID: GDS3459] were calculated in R with the Limma (Linear Models for Microarray) differential expression analysis package (124). The uncorrected P values were computed from a Bayesian moderated t test (124). For qRT-PCR, complementary DNA (cDNA) was generated from total RNA using random hexamers and SuperScript III (Invitrogen). Real-time PCR was conducted with SYBR-Rox SuperMix (Bio-Rad) and an HT-7900 (Applied Biosystems) and statistically analyzed in R with the t test or ANCOVA methods adapted from available SAS code (125).
GO analysis
GO analysis was performed with DAVID Functional Annotation Bioinformatics Microarray Analysis software (40), except for the sensitivity analysis where topGO was used (126, 127). In DAVID-based analyses, the reported P values are derived from the Expression Analysis Systematic Explorer (EASE) score probability, a modified Fisher’s exact test that is more conservative than the standard Fisher’s exact test in examining P values of gene lists (128). Where indicated, significance of overrepresentation was adjusted for multiple comparisons to control the false discovery rate (FDR) by means of the Benjamini-Hochberg step-down procedure for calculating the FDR in the case of independent tests or the approximated FDR tools provided in DAVID (40). All level 5 GO Biological Processes (BPs), the highest-level terms in the BP GOgraph structure (129), or KEGG pathways that remained significantly overrepresented (FDR = 5%) were reported. Ingenuity-based analysis reports enrichment probabilities based on the Fisher’s exact test.
Dynamic time warping
Whole time course analysis used a DTW-based approach (50, 51, 130) to calculate the distance between Wnt1-treated and control time series gene expression data on a gene-by-gene basis, using all 18,396 probes. In brief, DTW locally compresses or stretches (warping) one waveform to best match a reference waveform. The more warping that is required, the greater the difference between the two waveforms. Using this scheme, greater distances between trajectories for a given gene indicate a larger effect of Wnt1 treatment. Calculation of warping paths used the full data window, unconstrained endpoints, and the Euclidean distance to measure path differences (130). DTW sensitivity analysis: (Random traces) DTW distance was computed between randomly generated traces containing between 10 and 40 time points, and the same time course with increasing amounts of added Gaussian noise (μ = 0, 0 ≤ σ ≤ 1). DTWsimilarity (sDTW) was computed as follows: sDTW = 1/(1 + DTWDist).
Mutual information–based network analysis
Wnt1-induced networks were inferred in four steps on a refined gene list. (i) Dimensional reduction: The refined gene list was created by pooling the following: ICA-identified genes, the top 300 DTW-identified genes, and those genes with significantly nonnormal distributions, as assessed by either the Jarque-Bera skewness test or the Anscombe-Glynn kurtosis test (P ≤ 0.05). (ii) Network building: We calculated the weighted gene coexpression network on the basis of the entropy normalized mutual information (see below) distance (131) between each pair of genes in our refined list. First, data were discretized; that is, continuous variables were converted into discrete ones by equal frequency binning. Second, corrected marginal entropy (HX, HY) and joint entropy (HXY) were calculated from raw discrete marginal and joint probabilities with a James-Stein–type shrinkage estimator, which is robust for small sample sizes (132, 133). Raw mutual information (MI) measurements between gene pairs were calculated from the marginal and joint entropies according to the equation MI(X,Y) = H(X) + H(Y) − H(X,Y), expressed in natural bits (nats), where 1 nat = log2(e) binary bits. These were transformed into a maximum entropy-normalized dissimilarity metric (entropy-normalized MI) (131). This metric was calculated according to . Next, D’ was scaled according to MI(X,Y) = log2(e)*D’(Max – Min). (iii) Network pruning: We eliminated low probability connections from this network with the “maximum relevance, minimum redundancy” (MRnet) criteria (133). (iv) Final refinement: networks were further pruned by eliminating edges that fell below 1 bit of information (fig. S8). The resulting sparse network approximated a power-law distribution of connections, typical of the scale-free topology of gene-expression and protein networks (134, 135).
Parallel independent component analysis
Eigenarrays corresponding to parallel statistically independent components were calculated using an R implementation of the fastICA algorithm (136). pICA was used to extract seven independent components from the expression data for the Wnt1-treated hNPs. Because the ICA algorithm requires searching the maxima of a target function, the final output may reflect local rather than absolute maxima.
Modeling suggests that the ordering of genes in individual components is relatively, but not absolutely, stable for those genes whose gene loading exceeds a threshold of 3. To overcome this limitation (56), we created consensus components by reseeding the fastICA 250 times and then median-averaging the gene loading for each gene in each component. Because the fastICA output is not ordered, but the ranking of gene loadings for a given component was >90% identical from run to run, we used the Damerau-Levenshtein distance to match component lists to create seven consensus independent components.
Other network analyses
Topological overlap–based WGCNA networks and dynamic Bayesian network were performed as described (62–65) in R (http://www.r-project.org/).
Disease eigengene analysis
The FTD data (99) were downloaded from the gene expression omnibus Web site (GEO ID: GDS3459) and then log2-transformed before further analysis. The disease-specific eigengene was calculated to reflect perfect up-regulation in the specimens from the frontal cortex of GRN-haploinsufficient patients, but unchanged expression in other regions and other disease states. The putative disease eigengene is itself a vector of length equal to the number of samples in the data set. Each element is limited to the values of {−1,0,1}, where thesevalues correspond to either up-regulation, no change, or down-regulation in a given sample. Then, this vector is pairwise-correlated with the expression of every gene using the Spearman rank correlation. [This measure is more robust than the previously used Pearson correlation (62, 63, 135), being less sensitive to outliers and other nonlinearities in the data.] A Spearman correlation (ρ ≥ 0.40) was chosen, which is equivalent to a raw *P ≤ 0.001, as calculated with R statistical software. The underlying algorithm used by the software calculates the P value as follows: The distribution of Spearman’s rank correlation coefficients (ρ) in the null case (zero correlation) can be approximated by several methods; however, for larger sample sizes (n ≥ 30), it is nearly the t distribution (137, 138). Using the relationship (137, 138), we calculated that a ρ = 0.4, n = 56 produces a Student’s t ≈ 3.21. In turn, this t value and n = 56 are roughly equivalent to a P = 0.001, using tables readily available in most introductory statistics textbooks.
Supplementary Material
Acknowledgments
We thank G. Konopka, K. Winden, R. Versano, and J. Miller for their technical expertise and invaluable discussions and J. Davis-Turak, F. Gao, and G. Coppola for assistance with BeadStudio data preprocessing.
Funding: This work was supported by the John Douglas French Alzheimer’s Foundation (E.M.W.), National Alliance for Research on Schizophrenia and Depression (E.M.W.), National Institute of Mental Health (NIMH) K08MH74362 (E.M.W.), NIMH R01MH060233 (D.H.G.), National Institute on Aging 5R01AG026938 (D.H.G.), and NIH/National Institute of Neurological Disorders and Stroke Neurobehavioral Genetics Training Grant 5T32NS048004-05 (E.R.).
Footnotes
Author contributions: D.H.G. and E.M.W. conceived the study. E.M.W. wrote the analytic software, collected the data, and wrote the initial draft of the manuscript. E.M.W. and D.H.G. provided financing, edited, and had final approval over the manuscript. D.L., E.R., and G.E.O. were responsible for qPCR, tissue culture, in vitro knockdown, and GRN expression experiments. E.M. and H.R. were responsible for the APOA4 knockout mouse colony and harvesting of brain tissue.
Competing interests: The authors declare that they have no competing interests.
Data availability: Microarray expression data files can be obtained from the NIH Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/).
REFERENCES AND NOTES
- 1.Chien AJ, Conrad WH, Moon RT. A Wnt survival guide: From flies to human disease. J. Invest. Dermatol. 2009;129:1614–1627. doi: 10.1038/jid.2008.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 2009;10:468–477. doi: 10.1038/nrm2717. [DOI] [PubMed] [Google Scholar]
- 3.Fleming HE, Janzen V, LoCelso C, Guo J, Leahy KM, Kronenberg HM, Scadden DT. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell. 2008;2:274–283. doi: 10.1016/j.stem.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wawra C, Kühl M, Kestler HA. Extended analyses of the Wnt/β-catenin pathway: Robustness and oscillatory behaviour. FEBS Lett. 2007;581:4043–4048. doi: 10.1016/j.febslet.2007.07.043. [DOI] [PubMed] [Google Scholar]
- 5.Aulehla A, Wehrle C, Brand-Saberi B, Kemler R, Gossler A, Kanzler B, Herrmann BG. Wnt3a plays a major role in the segmentation clock controlling somitogenesis. Dev. Cell. 2003;4:395–406. doi: 10.1016/s1534-5807(03)00055-8. [DOI] [PubMed] [Google Scholar]
- 6.Aulehla A, Pourquié O. Oscillating signaling pathways during embryonic development. Curr. Opin. Cell Biol. 2008;20:632–637. doi: 10.1016/j.ceb.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Aulehla A, Wiegraebe W, Baubet V, Wahl MB, Deng C, Taketo M, Lewandoski M, Pourquié O. A β-catenin gradient links the clock and wavefront systems in mouse embryo segmentation. Nat. Cell Biol. 2008;10:186–193. doi: 10.1038/ncb1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lie DC, Colamarino SA, Song HJ, Désiré L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437:1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- 9.Inestrosa NC, Arenas E. Emerging roles of Wnts in the adult nervous system. Nat. Rev. Neurosci. 2010;11:77–86. doi: 10.1038/nrn2755. [DOI] [PubMed] [Google Scholar]
- 10.Castelo-Branco G, Arenas E. Function of Wnts in dopaminergic neuron development. Neurodegener. Dis. 2006;3:5–11. doi: 10.1159/000092086. [DOI] [PubMed] [Google Scholar]
- 11.Prakash N, Wurst W. Genetic networks controlling the development of midbrain dopaminergic neurons. J. Physiol. 2006;575:403–410. doi: 10.1113/jphysiol.2006.113464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li F, Chong ZZ, Maiese K. Winding through the WNT pathway during cellular development and demise. Histol. Histopathol. 2006;21:103–124. doi: 10.14670/hh-21.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He B, You L, Uematsu K, Xu Z, Lee AY, Matsangou M, McCormick F, Jablons DM. A monoclonal antibody against Wnt-1 induces apoptosis in human cancer cells. Neoplasia. 2004;6:7–14. doi: 10.1016/s1476-5586(04)80048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.You Z, Saims D, Chen S, Zhang Z, Guttridge DC, Guan KL, MacDougald OA, Brown AM, Evan G, Kitajewski J, Wang CY. Wnt signaling promotes oncogenic transformation by inhibiting c-Myc–induced apoptosis. J. Cell Biol. 2002;157:429–440. doi: 10.1083/jcb.200201110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen S, Guttridge DC, You Z, Zhang Z, Fribley A, Mayo MW, Kitajewski J, Wang CY. Wnt-1 signaling inhibits apoptosis by activating β-catenin/T cell factor–mediated transcription. J. Cell Biol. 2001;152:87–96. doi: 10.1083/jcb.152.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wexler EM, Paucer A, Kornblum HI, Palmer TD, Geschwind DH. Endogenous Wnt signaling maintains neural progenitor cell potency. Stem Cells. 2009;27:1130–1141. doi: 10.1002/stem.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuwahara A, Hirabayashi Y, Knoepfler PS, Taketo MM, Sakai J, Kodama T, Gotoh Y. Wnt signaling and its downstream target N-myc regulate basal progenitors in the developing neocortex. Development. 2010;137:1035–1044. doi: 10.1242/dev.046417. [DOI] [PubMed] [Google Scholar]
- 18.Nusse R. Protein Interactions. The Wnt Homepage. 2010 http://www.stanford.edu/~rnusse/pathways/binding.html.
- 19.Lovestone S, Anderton B. Cytoskeletal abnormalities in Alzheimer’s disease. Curr. Opin. Neurol. Neurosurg. 1992;5:883–888. [PubMed] [Google Scholar]
- 20.Karsten SL, Sang TK, Gehman LT, Chatterjee S, Liu J, Lawless GM, Sengupta S, Berry RW, Pomakian J, Oh HS, Schulz C, Hui KS, Wiedau-Pazos M, Vinters HV, Binder LI, Geschwind DH, Jackson GR. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron. 2006;51:549–560. doi: 10.1016/j.neuron.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 21.Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, Massachi S, Geschwind DH. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 2002;34:509–519. doi: 10.1016/s0896-6273(02)00706-7. [DOI] [PubMed] [Google Scholar]
- 22.Behrens MI, Lendon C, Roe CM. A common biological mechanism in cancer and Alzheimer’s disease? Curr. Alzheimer Res. 2009;6:196–204. doi: 10.2174/156720509788486608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boonen RA, van Tijn P, Zivkovic D. Wnt signaling in Alzheimer’s disease: Up or down, that is the question. Ageing Res. Rev. 2009;8:71–82. doi: 10.1016/j.arr.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 24.Kang DE, Soriano S, Xia X, Eberhart CG, DeStrooper B, Zheng H, Koo EH. Presenilin couples the paired phosphorylation of β-catenin independent of axin: Implications for β-catenin activation in tumorigenesis. Cell. 2002;110:751–762. doi: 10.1016/s0092-8674(02)00970-4. [DOI] [PubMed] [Google Scholar]
- 25.Nishimura M, Yu G, Levesque G, Zhang DM, Ruel L, Chen F, Milman P, Holmes E, Liang Y, Kawarai T, Jo E, Supala A, Rogaeva E, Xu DM, Janus C, Levesque L, Bi Q, Duthie M, Rozmahel R, Mattila K, Lannfelt L, Westaway D, Mount HT, Woodgett J, Frazer PE, St George-Hyslop P. Presenilin mutations associated with Alzheimer disease cause defective intracellular trafficking of β-catenin, a component of the presenilin protein complex. Nat. Med. 1999;5:164–169. doi: 10.1038/5526. [DOI] [PubMed] [Google Scholar]
- 26.Mudher A, Lovestone S. Alzheimer’s disease—Do tauists and baptists finally shake hands? Trends Neurosci. 2002;25:22–26. doi: 10.1016/s0166-2236(00)02031-2. [DOI] [PubMed] [Google Scholar]
- 27.Geschwind DH. Tau phosphorylation, tangles, neurodegeneration: The chicken or the egg? Neuron. 2003;40:457–460. doi: 10.1016/s0896-6273(03)00681-0. [DOI] [PubMed] [Google Scholar]
- 28.Cohen P, Goedert M. GSK3 inhibitors: Development and therapeutic potential. Nat. Rev. Drug Discov. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- 29.Schaffer BA, Bertram L, Miller BL, Mullin K, Weintraub S, Johnson N, Bigio EH, Mesulam M, Wiedau-Pazos M, Jackson GR, Cummings JL, Cantor RM, Levey AI, Tanzi RE, Geschwind DH. Association of GSK3B with Alzheimer disease and frontotemporal dementia. Arch. Neurol. 2008;65:1368–1374. doi: 10.1001/archneur.65.10.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geschwind DH, Miller BL. Molecular approaches to cerebral laterality: Development and neurodegeneration. Am. J. Med. Genet. 2001;101:370–381. [PubMed] [Google Scholar]
- 31.Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 33.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 34.Klapholz-Brown Z, Walmsley GG, Nusse YM, Nusse R, Brown PO. Transcriptional program induced by Wnt protein in human fibroblasts suggests mechanisms for cell cooperativity in defining tissue microenvironments. PLoS One. 2007;2:e945. doi: 10.1371/journal.pone.0000945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen S, McLean S, Carter DE, Leask A. The gene expression profile induced by Wnt 3a in NIH 3T3 fibroblasts. J. Cell Commun. Signal. 2007;1:175–183. doi: 10.1007/s12079-007-0015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gujral TS, MacBeath G. A system-wide investigation of the dynamics of Wnt signaling reveals novel phases of transcriptional regulation. PLoS One. 2010;5:e10024. doi: 10.1371/journal.pone.0010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 38.Dequéant ML, Pourquié O. Segmental patterning of the vertebrate embryonic axis. Nat. Rev. Genet. 2008;9:370–382. doi: 10.1038/nrg2320. [DOI] [PubMed] [Google Scholar]
- 39.Goldbeter A, Pourquié O. Modeling the segmentation clock as a network of coupled oscillations in the Notch, Wnt and FGF signaling pathways. J. Theor. Biol. 2008;252:574–585. doi: 10.1016/j.jtbi.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 40.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 41.Wiedau-Pazos M, Wong E, Solomon E, Alarcon M, Geschwind DH. Wnt-pathway activation during the early stage of neurodegeneration in FTDP-17 mice. Neurobiol. Aging. 2009;30:14–21. doi: 10.1016/j.neurobiolaging.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeFerrari GV, Papassotiropoulos A, Biechele T, WavrantDe-Vrieze F, Avila ME, Major MB, Myers A, Saez K, Henriquez JP, Zhao A, Wollmer MA, Nitsch RM, Hock C, Morris CM, Hardy J, Moon RT. Common genetic variation within the low-density lipoprotein receptor-related protein 6 and late-onset Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 2007;104:9434–9439. doi: 10.1073/pnas.0603523104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inestrosa NC, Toledo EM. The role of Wnt signaling in neuronal dysfunction in Alzheimer’s disease. Mol. Neurodegener. 2008;3:9. doi: 10.1186/1750-1326-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosi MC, Luccarini I, Grossi C, Fiorentini A, Spillantini MG, Prisco A, Scali C, Gianfriddo M, Caricasole A, Terstappen GC, Casamenti F. Increased Dickkopf-1 expression in transgenic mouse models of neurodegenerative disease. J. Neurochem. 2010;112:1539–1551. doi: 10.1111/j.1471-4159.2009.06566.x. [DOI] [PubMed] [Google Scholar]
- 45.DeFerrari GV, Inestrosa NC. Wnt signaling function in Alzheimer’s disease. Brain Res. Brain Res. Rev. 2000;33:1–12. doi: 10.1016/s0165-0173(00)00021-7. [DOI] [PubMed] [Google Scholar]
- 46.Inestrosa N, DeFerrari GV, Garrido JL, Alvarez A, Olivares GH, Barria MI, Bronfman M, Chacon MA. Wnt signaling involvement in β-amyloid-dependent neurodegeneration. Neurochem. Int. 2002;41:341–344. doi: 10.1016/s0197-0186(02)00056-6. [DOI] [PubMed] [Google Scholar]
- 47.Olsen C, Meyer PE, Bontempi G. On the impact of entropy estimation on transcriptional regulatory network inference based on mutual information. EURASIP J. Bioinform. Syst. Biol. 2009;2009:308959. doi: 10.1155/2009/308959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.MacKay DJC. Information Theory, Inference, and Learning Algorithms. Cambridge Univ. Press; Cambridge, UK/New York: 2003. [Google Scholar]
- 49.Numata J, Ebenhoh O, Knapp EW. Measuring correlations in metabolomic networks with mutual information. Genome Inform. 2008;20:112–122. [PubMed] [Google Scholar]
- 50.Aach J, Church GM. Aligning gene expression time series with time warping algorithms. Bioinformatics. 2001;17:495–508. doi: 10.1093/bioinformatics/17.6.495. [DOI] [PubMed] [Google Scholar]
- 51.Criel J, Tsiporkova E. Gene Time Expression Warper: A tool for alignment, template matching and visualization of gene expression time series. Bioinformatics. 2006;22:251–252. doi: 10.1093/bioinformatics/bti787. [DOI] [PubMed] [Google Scholar]
- 52.Hermans F, Tsiporkova E. Merging microarray cell synchronization experiments through curve alignment. Bioinformatics. 2007;23:e64–e70. doi: 10.1093/bioinformatics/btl320. [DOI] [PubMed] [Google Scholar]
- 53.Kong W, Vanderburg CR, Gunshin H, Rogers JT, Huang X. A review of independent component analysis application to microarray gene expression data. Biotechniques. 2008;45:501–520. doi: 10.2144/000112950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lutter D, Langmann T, Ugocsai P, Moehle C, Seibold E, Splettstoesser WD, Gruber P, Lang EW, Schmitz G. Analyzing time-dependent microarray data using independent component analysis derived expression modes from human macrophages infected with F. tularensis holartica. J. Biomed. Inform. 2009;42:605–611. doi: 10.1016/j.jbi.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 55.Meyer PE, Kontos K, Lafitte F, Bontempi G. Information-theoretic inference of large transcriptional regulatory networks. EURASIP J. Bioinform. Syst. Biol. 2007;2007:79879. doi: 10.1155/2007/79879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chiappetta P, Roubaud MC, Torrésani B. Blind source separation and the analysis of microarray data. J. Comput. Biol. 2004;11:1090–1109. doi: 10.1089/cmb.2004.11.1090. [DOI] [PubMed] [Google Scholar]
- 57.Frigyesi A, Veerla S, Lindgren D, Höglund M. Independent component analysis reveals new and biologically significant structures in micro array data. BMC Bioinformatics. 2006;7:290. doi: 10.1186/1471-2105-7-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kong W, Mou X, Liu Q, Chen Z, Vanderburg CR, Rogers JT, Huang X. Independent component analysis of Alzheimer’s DNA microarray gene expression data. Mol. Neurodegener. 2009;4:5. doi: 10.1186/1750-1326-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng CH, Huang DS, Kong XZ, Zhao XM. Gene expression data classification using consensus independent component analysis. Genomics Proteomics Bioinformatics. 2008;6:74–82. doi: 10.1016/S1672-0229(08)60022-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao J, Ade AS, Tarcea VG, Weymouth TE, Mirel BR, Jagadish HV, States DJ. Integrating and annotating the interactome using the MiMI plugin for cytoscape. Bioinformatics. 2009;25:137–138. doi: 10.1093/bioinformatics/btn501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jayapandian M, Chapman A, Tarcea VG, Yu C, Elkiss A, Ianni A, Liu B, Nandi A, Santos C, Andrews P, Athey B, States D, Jagadish HV. Michigan Molecular Interactions (MiMI): Putting the jigsaw puzzle together. Nucleic Acids Res. 2007;35:D566–D571. doi: 10.1093/nar/gkl859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. Functional organization of the transcriptome in human brain. Nat. Neurosci. 2008;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
- 65.Konopka G, Bomar JM, Winden K, Coppola G, Jonsson ZO, Gao F, Peng S, Preuss TM, Wohlschlegel JA, Geschwind DH. Human-specific transcriptional regulation of CNS development genes by FOXP2. Nature. 2009;462:213–217. doi: 10.1038/nature08549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol. 2007;1:54. doi: 10.1186/1752-0509-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alter O, Brown PO, Botstein D. Singular value decomposition for genome-wide expression data processing and modeling. Proc. Natl. Acad. Sci. U.S.A. 2000;97:10101–10106. doi: 10.1073/pnas.97.18.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lèbre S. Inferring dynamic genetic networks with low order independencies. Stat. Appl. Genet. Mol. Biol. 2009;8 doi: 10.2202/1544-6115.1294. Article 9. [DOI] [PubMed] [Google Scholar]
- 69.Lèbre S. PhD thesis. Université d’Evry-Val-d’Essonne; Evry, France: 2007. Stochastic Process Analysis for Genomics and Dynamic Bayesian Networks Inference. [Google Scholar]
- 70.Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer’s disease: Microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. U.S.A. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baladrón V, Ruiz-Hidalgo MJ, Bonvini E, Gubina E, Notario V, Laborda J. The EGF-like homeotic protein dlk affects cell growth and interacts with growth-modulating molecules in the yeast two-hybrid system. Biochem. Biophys. Res. Commun. 2002;291:193–204. doi: 10.1006/bbrc.2002.6431. [DOI] [PubMed] [Google Scholar]
- 72.Baladron V, Ruiz-Hidalgo MJ, Nueda ML, Diaz-Guerra MJ, Garcia-Ramirez JJ, Bonvini E, Gubina E, Laborda J. dlk acts as a negative regulator of Notch1 activation through interactions with specific EGF-like repeats. Exp. Cell Res. 2005;303:343–359. doi: 10.1016/j.yexcr.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 73.Hu F, Padukkavidana T, Vægter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68:654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chumakov S, Ballesteros E, Sanchez J. E. Rodriguez, Chavez A, Zhang M, Pettit BM, Fofanov Y. Using mutual information to discover temporal patterns in gene expression data. Med. Phys. Mex. Symp. Med. Phys. 2006;854:25–30. doi: 10.1063/1.2356392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Daub CO, Steuer R, Selbig J, Kloska S. Estimating mutual information using B-spline functions—An improved similarity measure for analysing gene expression data. BMC Bioinformatics. 2004;5:118. doi: 10.1186/1471-2105-5-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Steuer R, Kurths J, Daub CO, Weise J, Selbig J. The mutual information: Detecting and evaluating dependencies between variables. Bioinformatics. 2002;18(Suppl. 2):S231–S240. doi: 10.1093/bioinformatics/18.suppl_2.s231. [DOI] [PubMed] [Google Scholar]
- 77.Zhou X, Wang X, Dougherty ER, Russ D, Suh E. Gene clustering based on clusterwide mutual information. J. Comput. Biol. 2004;11:147–161. doi: 10.1089/106652704773416939. [DOI] [PubMed] [Google Scholar]
- 78.Kim DC, Wang X, Yang CR, Gao J. Learning biological network using mutual information and conditional independence. BMC Bioinformatics. 2010;11(Suppl. 3):S9. doi: 10.1186/1471-2105-11-S3-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Forsell C, Björk BF, Lilius L, Axelman K, Fabre SF, Fratiglioni L, Winblad B, Graff C. Genetic association to the amyloid plaque associated protein gene COL25A1 in Alzheimer’s disease. Neurobiol. Aging. 2010;31:409–415. doi: 10.1016/j.neurobiolaging.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 80.Söderberg L, Dahlqvist C, Kakuyama H, Thyberg J, Ito A, Winblad B, Näslund J, Tjernberg LO. Collagenous Alzheimer amyloid plaque component assembles amyloid fibrils into protease resistant aggregates. FEBS J. 2005;272:2231–2236. doi: 10.1111/j.1742-4658.2005.04647.x. [DOI] [PubMed] [Google Scholar]
- 81.Hashimoto T, Wakabayashi T, Watanabe A, Kowa H, Hosoda R, Nakamura A, Kanazawa I, Arai T, Takio K, Mann DM, Iwatsubo T. CLAC: A novel Alzheimer amyloid plaque component derived from a transmembrane precursor, CLAC-P/collagen type XXV. EMBO J. 2002;21:1524–1534. doi: 10.1093/emboj/21.7.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tong Y, Xu Y, Scearce-Levie K, Ptáček LJ, Fu YH. COL25A1 triggers and promotes Alzheimer’s disease-like pathology in vivo. Neurogenetics. 2010;11:41–52. doi: 10.1007/s10048-009-0201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cui Y, Huang M, He Y, Zhang S, Luo Y. Genetic ablation of apolipoprotein A-IV accelerates Alzheimer’s disease pathogenesis in a mouse model. Am. J. Pathol. 2011;178:1298–1308. doi: 10.1016/j.ajpath.2010.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Papassotiropoulos A, Wollmer MA, Tsolaki M, Brunner F, Molyva D, Lütjohann D, Nitsch RM, Hock C. A cluster of cholesterol-related genes confers susceptibility for Alzheimer’s disease. J. Clin. Psychiatry. 2005;66:940–947. [PubMed] [Google Scholar]
- 85.Carter CJ. Convergence of genes implicated in Alzheimer’s disease on the cerebral cholesterol shuttle: APP, cholesterol, lipoproteins, and atherosclerosis. Neurochem. Int. 2007;50:12–38. doi: 10.1016/j.neuint.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 86.Vollbach H, Heun R, Morris CM, Edwardson JA, McKeith IG, Jessen F, Schulz A, Maier W, Kölsch H. APOA1 polymorphism influences risk for early-onset nonfamiliar AD. Ann. Neurol. 2005;58:436–441. doi: 10.1002/ana.20593. [DOI] [PubMed] [Google Scholar]
- 87.Raygani AV, Rahimi Z, Kharazi H, Tavilani H, Pourmotabbed T. Association between apolipoprotein E polymorphism and serum lipid and apolipoprotein levels with Alzheimer’s disease. Neurosci. Lett. 2006;408:68–72. doi: 10.1016/j.neulet.2006.08.048. [DOI] [PubMed] [Google Scholar]
- 88.Roher AE, Maarouf CL, Sue LI, Hu Y, Wilson J, Beach TG. Proteomics-derived cerebrospinal fluid markers of autopsy-confirmed Alzheimer’s disease. Biomarkers. 2009;14:493–501. doi: 10.3109/13547500903108423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Park JB, Yiu G, Kaneko S, Wang J, Chang J, He XL, Garcia KC, He Z. A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron. 2005;45:345–351. doi: 10.1016/j.neuron.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 90.Okura H, Yamashita S, Ohama T, Saga A, Yamamoto-Kakuta A, Hamada Y, Sougawa N, Ohyama R, Sawa Y, Matsuyama A. HDL/apolipoprotein A-I binds to macrophage-derived progranulin and suppresses its conversion into proinflammatory granulins. J. Atheroscler. Thromb. 2010;17:568–577. doi: 10.5551/jat.3921. [DOI] [PubMed] [Google Scholar]
- 91.Grilli M, Toninelli G. Ferrari, Uberti D, Spano P, Memo M. Alzheimer’s disease linking neurodegeneration with neurodevelopment. Funct. Neurol. 2003;18:145–148. [PubMed] [Google Scholar]
- 92.Caricasole A, Bakker A, Copani A, Nicoletti F, Gaviraghi G, Terstappen GC. Two sides of the same coin: Wnt signaling in neurodegeneration and neuro-oncology. Biosci. Rep. 2005;25:309–327. doi: 10.1007/s10540-005-2893-6. [DOI] [PubMed] [Google Scholar]
- 93.Caraci F, Busceti C, Biagioni F, Aronica E, Mastroiacovo F, Cappuccio I, Battaglia G, Bruno V, Caricasole A, Copani A, Nicoletti F. The Wnt antagonist, Dickkopf-1, as a target for the treatment of neurodegenerative disorders. Neurochem. Res. 2008;33:2401–2406. doi: 10.1007/s11064-008-9710-0. [DOI] [PubMed] [Google Scholar]
- 94.Maiese K, Li F, Chong ZZ, Shang YC. The Wnt signaling pathway: Aging gracefully as a protectionist? Pharmacol. Ther. 2008;118:58–81. doi: 10.1016/j.pharmthera.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 96.Cruts M, Gijselinck I, vander Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, DePooter T, Mattheijssens M, VandenBroeck M, Cuijt I, Vennekens K, DeDeyn PP, Kumar-Singh S, VanBroeckhoven C. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 97.Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, Crook R, Melquist S, Kuntz K, Petersen R, Josephs K, Pickering-Brown SM, Graff-Radford N, Uitti R, Dickson D, Wszolek Z, Gonzalez J, Beach TG, Bigio E, Johnson N, Weintraub S, Mesulam M, White CL, III, Woodruff B, Caselli R, Hsiung GY, Feldman H, Knopman D, Hutton M, Rademakers R. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- 98.Pickering-Brown SM, Baker M, Gass J, Boeve BF, Loy CT, Brooks WS, Mackenzie IR, Martins RN, Kwok JB, Halliday GM, Kril J, Schofield PR, Mann DM, Hutton M. Mutations in progranulin explain atypical phenotypes with variants in MAPT. Brain. 2006;129:3124–3126. doi: 10.1093/brain/awl289. [DOI] [PubMed] [Google Scholar]
- 99.Chen-Plotkin AS, Geser F, Plotkin JB, Clark CM, Kwong LK, Yuan W, Grossman M, VanDeerlin VM, Trojanowski JQ, Lee VM. Variations in the progranulin gene affect global gene expression in frontotemporal lobar degeneration. Hum. Mol. Genet. 2008;17:1349–1362. doi: 10.1093/hmg/ddn023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Goedert JJ, Eyster ME, Lederman MM, Mandalaki T, DeMoerloose P, White GC, II, Angiolillo AL, Luban NL, Sherman KE, Manco-Johnson M, Preiss L, Leissinger C, Kessler CM, Cohen AR, DiMichele D, Hilgartner MW, Aledort LM, Kroner BL, Rosenberg PS, Hatzakis A. End-stage liver disease in persons with hemophilia and transfusion-associated infections. Blood. 2002;100:1584–1589. [PubMed] [Google Scholar]
- 101.Dequéant ML, Glynn E, Gaudenz K, Wahl M, Chen J, Mushegian A, Pourquié O. A complex oscillating network of signaling genes underlies the mouse segmentation clock. Science. 2006;314:1595–1598. doi: 10.1126/science.1133141. [DOI] [PubMed] [Google Scholar]
- 102.Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008;322:1247–1250. doi: 10.1126/science.1164594. [DOI] [PubMed] [Google Scholar]
- 103.Liau LM, Lallone RL, Seitz RS, Buznikov A, Gregg JP, Kornblum HI, Nelson SF, Bronstein JM. Identification of a human glioma-associated growth factor gene, granulin, using differential immuno-absorption. Cancer Res. 2000;60:1353–1360. [PubMed] [Google Scholar]
- 104.Ryan CL, Baranowski DC, Chitramuthu BP, Malik S, Li Z, Cao M, Minotti S, Durham HD, Kay DG, Shaw CA, Bennett HP, Bateman A. Progranulin is expressed within motor neurons and promotes neuronal cell survival. BMC Neurosci. 2009;10:130. doi: 10.1186/1471-2202-10-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bateman A, Bennett HP. The granulin gene family: From cancer to dementia. Bioessays. 2009;31:1245–1254. doi: 10.1002/bies.200900086. [DOI] [PubMed] [Google Scholar]
- 106.DeMuynck L, VanDamme P. Cellular effects of progranulin in health and disease. J. Mol. Neurosci. 2011 doi: 10.1007/s12031-011-9553-z. 10.1007/s12031-011-9553-z. [DOI] [PubMed] [Google Scholar]
- 107.Chen-Plotkin AS, Xiao J, Geser F, Martinez-Lage M, Grossman M, Unger T, Wood EM, VanDeerlin VM, Trojanowski JQ, Lee VM. Brain progranulin expression in GRN-associated frontotemporal lobar degeneration. Acta Neuropathol. 2010;119:111–122. doi: 10.1007/s00401-009-0576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR, III, Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 109.Coudreuse D, Korswagen HC. The making of Wnt: New insights into Wnt maturation, sorting and secretion. Development. 2007;134:3–12. doi: 10.1242/dev.02699. [DOI] [PubMed] [Google Scholar]
- 110.Ahmed Z, Mackenzie IR, Hutton ML, Dickson DW. Progranulin in frontotemporal lobar degeneration and neuroinflammation. J. Neuroinflammation. 2007;4:7. doi: 10.1186/1742-2094-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Coppola G, Karydas A, Rademakers R, Wang Q, Baker M, Hutton M, Miller BL, Geschwind DH. Gene expression study on peripheral blood identifies progranulin mutations. Ann. Neurol. 2008;64:92–96. doi: 10.1002/ana.21397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Finch N, Baker M, Crook R, Swanson K, Kuntz K, Surtees R, Bisceglio G, Rovelet-Lecrux A, Boeve B, Petersen RC, Dickson DW, Younkin SG, Deramecourt V, Crook J, Graff-Radford NR, Rademakers R. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain. 2009;132:583–591. doi: 10.1093/brain/awn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006;443:768–773. doi: 10.1038/nature05289. [DOI] [PubMed] [Google Scholar]
- 114.Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc. Natl. Acad. Sci. U.S.A. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Svendsen CN, Borg M. G. ter, Armstrong RJ, Rosser AE, Chandran S, Ostenfeld T, Caldwell MA. A new method for the rapid and long term growth of human neural precursor cells. J. Neurosci. Methods. 1998;85:141–152. doi: 10.1016/s0165-0270(98)00126-5. [DOI] [PubMed] [Google Scholar]
- 116.Palmer TD, Schwartz PH, Taupin P, Kaspar B, Stein SA, Gage FH. Cell culture. Progenitor cells from human brain after death. Nature. 2001;411:42–43. doi: 10.1038/35075141. [DOI] [PubMed] [Google Scholar]
- 117.Ding VM, Ling L, Natarajan S, Yap MG, Cool SM, Choo AB. FGF-2 modulates Wnt signaling in undifferentiated hESC and iPS cells through activated PI3-K/GSK3β signaling. J. Cell. Physiol. 2010;225:417–428. doi: 10.1002/jcp.22214. [DOI] [PubMed] [Google Scholar]
- 118.Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2005;102:13212–13217. doi: 10.1073/pnas.0506306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Szulc J, Wiznerowicz M, Sauvain MO, Trono D, Aebischer P. A versatile tool for conditional gene expression and knockdown. Nat. Methods. 2006;3:109–116. doi: 10.1038/nmeth846. [DOI] [PubMed] [Google Scholar]
- 120.Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002;16:948–958. doi: 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Weinstock PH, Bisgaier CL, Hayek T, Aalto-Setala K, Sehayek E, Wu L, Sheiffele P, Merkel M, Essenburg AD, Breslow JL. Decreased HDL cholesterol levels but normal lipid absorption, growth, and feeding behavior in apolipoprotein A-IV knockout mice. J. Lipid Res. 1997;38:1782–1794. [PubMed] [Google Scholar]
- 122.Whited KL, Lu D, Tso P, Lloyd K. C. Kent, Raybould HE. Apolipoprotein A-IV is involved in detection of lipid in the rat intestine. J. Physiol. 2005;569:949–958. doi: 10.1113/jphysiol.2005.097634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nakano I, Joshi K, Visnyei K, Hu B, Watanabe M, Lam D, Wexler E, Saigusa K, Nakamura Y, Laks DR, Mischel PS, Viapiano M, Kornblum HI. Siomycin A targets brain tumor stem cells partially through a MELK-mediated pathway. Neuro. Oncol. 2011;13:622–634. doi: 10.1093/neuonc/nor023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article. [DOI] [PubMed] [Google Scholar]
- 125.Yuan JS, Reed A, Chen F, Stewart CN., Jr. Statistical analysis of real-time PCR data. BMC Bioinformatics. 2006;7:85. doi: 10.1186/1471-2105-7-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Alexa A, Rahnenführer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- 127.Hedegaard J, Arce C, Bicciato S, Bonnet A, Buitenhuis B, Collado-Romero M, Conley LN, Sancristobal M, Ferrari F, Garrido JJ, Groenen MA, Hornshoj H, Hulsegge I, Jiang L, Jiménez-Marin A, Kommadath A, Lagarrigue S, Leunissen JA, Liaubet L, Neerincx PB, Nie H, vander Poel J, Prickett D, Ramirez-Boo M, Rebel JM, Robert-Granié C, Skarman A, Smits MA, Sorensen P, Tøsser-Klopp G, Watson M. Methods for interpreting lists of affected genes obtained in a DNA microarray experiment. BMC Proc. 2009;3(suppl. 4):S5. doi: 10.1186/1753-6561-3-s4-s5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hosack DA, Dennis G, Jr., Sherman BT, Lane HC, Lempicki RA. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4:R70. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Giorgino T. Computing and visualizing dynamic time warping alignments in R: The dtw package. J. Stat. Softw. 2009;31:1–24. [Google Scholar]
- 131.Kraskov A, Grassberger P. In: MIC: Mutual information based hierarchical clustering, in Information Theory and Statistical Learning. Emmert-Streib F, Dehmer M, editors. Springer; New York: 2009. pp. 101–123. [Google Scholar]
- 132.Hausser J, Strimmer K. Entropy inference and the James-Stein estimator, with application to nonlinear gene association networks. J. Machine Learning Res. 2009;10:1469–1484. [Google Scholar]
- 133.Meyer PE, Lafitte F, Bontempi G. minet: A R/Bioconductor package for inferring large transcriptional networks using mutual information. BMC Bioinformatics. 2008;9:461. doi: 10.1186/1471-2105-9-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Barabási AL, Oltvai ZN. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 135.Horvath S, Dong J. Geometric interpretation of gene coexpression network analysis. PLoS Comput. Biol. 2008;4:e1000117. doi: 10.1371/journal.pcbi.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hyvärinen A, Oja E. Independent component analysis: Algorithms and applications. Neural Netw. 2000;13:411–430. doi: 10.1016/s0893-6080(00)00026-5. [DOI] [PubMed] [Google Scholar]
- 137.van de Wiel MA, Bucchianico AD. Fast computation of the exact null distribution of Spearman’s ρ and Page’s L statistic for samples with and without ties. J. Stat. Planning Infer. 2001;92:133–145. [Google Scholar]
- 138.Best DJ, Roberts DE. Algorithm AS 89: The upper tail probabilities of Spearman’s rho. J. R. Stat. Soc. Ser. C (Appl. Stat.) 1975;24:377–379. [Google Scholar]
- 139.Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–414. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.