

Abstract
Spindle poison-based therapy is of only limited benefit in acute myeloid leukemia while lymphoblastic leukemia/lymphoma responds well. In this study, we demonstrated that the spindle assembly checkpoint protein BubR1 was down-regulated in the vast majority of cases of acute myeloid leukemia whereas its expression was high in lymphoblastic cells. Correct function of the spindle assembly checkpoint is pivotal in mediating mitotic delay in response to spindle poisons. Mitotic delay by the spindle assembly checkpoint is achieved by inhibition of anaphase-promoting complex-dependent proteolysis of cyclin B and securin. We demonstrated a link between the repression of the spindle assembly checkpoint protein BubR1 in acute myeloid leukemia and the limited response to spindle poison. In accordance with its established role as an anaphase-promoting complex-inhibitor, we found that repression of BubR1 was associated with enhanced anaphase-promoting complex activity and cyclin B and securin degradation, which leads to premature sister-chromatid separation and failure to sustain a mitotic arrest. This suggests that repression of BubR1 in acute myeloid leukemia renders the spindle assembly checkpoint-mediated inhibition of the anaphase-promoting complex insufficient, which facilitates completion of mitosis in the presence of spindle poison. As both direct and BubR1-mediated restoration of cyclin B expression enhanced response to spindle poison, we propose that the downstream axis of the spindle assembly checkpoint is a promising target for tailored therapies for acute myeloid leukemia.
Introduction
Spindle poisons are an important part of therapy for acute lymphoblastic leukemia and Burkitt’s lymphoma. Acute myeloblastic leukemia (AML), however, is less sensitive to spindle poison-based therapy, as already described in early reports from the mid-1970s.1,2 Despite the relevance of this marked difference, the molecular basis of why lymphoblastic neoplasms respond well to spindle poison–based therapy while AML does not, is not understood.
Spindle poisons, such as vinca alkaloids, are classic chemotherapeutics for cancer therapy and exert their effect via interference with microtubule kinetics.3 Disturbed micro-tubule kinetics prevent satisfaction of the spindle assembly checkpoint (SAC) and hence arrest cells at metaphase. The SAC is a mitotic surveillance mechanism that senses improper attachment of chromosomes to the mitotic spindle. The mitotic checkpoint proteins BubR1, Bub3 and Mad2 are recruited to the kinetochores of unattached/misaligned chromosomes to form the mitotic checkpoint complex.4,5 This complex, along with the mitotic kinase Bub1, inhibits the E3 ubiquitin ligase anaphase-promoting complex/cyclosome (APC/C). Cdc20 activates the APC/C in mitosis and Cdh1 from the end of mitosis throughout the G1 phase of the cell cycle. The mitotic checkpoint complex inhibits the APC/C by binding to its activator Cdc20.5 Thereby APC/C-dependent ubiquitinylation of cyclin B and securin is prevented, which inhibits mitotic progression.5 Failure to satisfy the SAC by poisoning the mitotic spindle induces mitotic arrest and supports cell death.4,6–8 The extent to which cells are vulnerable to cell death depends on the balance between pro- and anti-mitotic factors. The degradation of the anti-apoptotic regulator Mcl-1 by APC/C- and Fbxw7-dependent ubiquitination was demonstrated to enhance the susceptibility to death in mitosis in the presence of antimitotic agents.9–11 The ability of cells to exit from mitosis in the presence of spindle poison and to survive limits the therapeutic success of such drugs and is regarded as a predictor of poor response.7 A mechanism that allows cells to escape from mitosis in the absence of a functional mitotic spindle is known as mitotic slippage, in which continued low-level degradation of cyclin B throughout the mitotic block triggers exit from mitosis and thus counteracts induction of cell death in mitosis.8,12,13 A weakened SAC would allow tumor cells to exit mitosis even in the presence of chromosome non-attachment/misalignment by mitotic slippage and acquire chromosomal instability.6,8 The mitotic checkpoint protein BubR1 is frequently deregulated and to a lesser extent mutated in neoplasias, pre-neoplastic lesions and the human cancer predisposition syndrome mosaic variegated aneuploidy, which causes impaired SAC.14–17
Here we demonstrate a link between the deregulated expression of the mitotic checkpoint protein BubR1 in AML and the response to spindle-poison-based therapy. We found low expression of BubR1 in the vast majority of primary AML blasts investigated. By performing functional studies both in non-responsive myeloblastic and responsive lymphoblastic cells, we investigated how reconstitution of the SAC and interference with SAC activity translate into response to spindle poison. Using live-cell imaging, retrovirus-delivered inducible knockdown and overexpression, we demonstrate that re-expression of BubR1 in myeloblastic cells confers an improved response to spindle poison.
Methods
Cell cultures
Cell lines were cultured as described in the DSMZ (Human and Animal Cell Lines Database, Braunschweig, Germany) datasheets. Synchronization procedures, interference with microtubule kinetics, proteasome inhibition and antibiotic selection were performed as outlined in the Online Supplementary Appendix.
Primary blasts from AML patients were isolated and cultured as described in the Online Supplementary Appendix. The analyses were carried out after approval by the local Ethics Committee according to the guidelines of the Declaration of Helsinki and good clinical practice. Informed consent was obtained from the patients.
Live-cell imaging kinetics
Cells were seeded in eight-well microscope chambers (Ibidi) that were coated with fibrinogen or collagen IV, at a concentration of 30,000 and 60,000 cells per well and imaged as previously described.13
Retroviral transduction
Retroviral particles were produced using Phoenix alpha packaging cells and inducible cell lines were established according to the manufacturer’s instructions. Detailed information concerning oligonucleotide sequences and the backbones used is provided in the Online Supplementary Appendix.
In vitro ubiquitination
The APC/C was immunopurified from DG-75 and Kasumi-1 cells using an anti-Cdc27 antibody (Sigma-Aldrich) and Protein G-agarose. Ubiquitination reactions using precipitated APC/C were performed in the presence of in vitro transcribed/translated 35S-marked cyclin B as described in detail in the Online Supplementary Appendix.
Western blot analysis
Western blotting was performed as described previously13 using the antibodies specified in the Online Supplementary Appendix.
Flow cytometry
Cell cycle distribution and BubR1 expression in single cells were determined using a FACSCalibur (Becton Dickinson).
Microarray data and data processing
Microarray datasets were obtained from the open-access NIH Gene-Expression Omnibus (GEO) database. Datasets used in our analysis were GSE3002918 and GSE13204.19
Data analysis and statistical analysis
TIFF image stacks were analyzed using LSM Image Browser (v.2.80.1123) (Carl Zeiss). Calculations and statistics were done in Microsoft Excel 2002 and/or GraphPad Prism V5.03 (GraphPad Software Inc.). P values <0.05 were considered statistically significant. The statistical test used was an unpaired t-test (two-tailed) with a confidence interval of 95%.
Results
The mitotic checkpoint protein BubR1 is repressed in acute myeloid leukemia
To address the expression levels of the regulatory proteins BubR1, Bub1, Bub3, Mad2, Cdc20, Cdh1, Fbxw7 and Mcl1 in lymphoblastic and myeloblastic neoplasias, we performed western blotting (Figure 1A). Protein expression levels were assessed in four asynchronously growing lymphoblastic Burkitt’s lymphoma cell lines (DG-75, Daudi, Raji and Ramos) and seven myeloblastic AML cell lines (HL-60, Kasumi-1, SKNO-1, Molm16, SKM1, Oci AML2 and NB4). Mean expression levels in lymphoblastic and myeloblastic cells were calculated following densitometric quantification. While mean expression levels of Bub1, Bub3, Mad2, Cdc20, Cdh1 and Fbxw7 were found at comparable levels in the lymphoblastic and myeloblastic cell lines, the levels of expression of BubR1 were significantly reduced in most AML cell lines, with the exception of HL-60 in which BubR1 levels were comparable to those in the lymphoblastic lines and SKNO-1 which had intermediate levels (Figure 1A,B). Moreover, we found a significant decrease in expression of the anti-apoptotic regulator Mcl1 in AML cell lines (Figure 1A,B). The finding of BubR1 down-regulation in AML cell lines was in line with microarray data showing down-regulation of BubR1 mRNA in AML cells derived from patients’ bone marrow. In contrast, normal bone marrow cells and t(8;14)-positive lymphoblastic cells derived from patients’ bone marrow (B-cell acute lymphoblastic leukemia) had high BubR1 mRNA levels (Figure 1C). Comparing BubR1 mRNA expression of normal CD34+ bone marrow cells with CD34+ AML cells, we also found much lower expression of BubR1 in CD34+ AML blasts.
Figure 1.

The spindle assembly checkpoint (SAC) component BubR1 is frequently down-regulated in AML. (A) Expression levels of BubR1, Bub1, Bub3, Mad2, Cdc20, Cdh1, Fbxw7 and Mcl1 were determined in four asynchronously growing lymphoblastic and seven myeloblastic cell lines by western blotting. (B) Protein levels were assessed by densitometry: mean expression levels of BubR1 and the anti-apoptotic protein Mcl1 were significantly reduced in AML cell lines. (C) GEO microarray datasets from the open-access NIH database were analyzed for BubR1 expression. AML bone marrow (BM) samples exhibited much lower BubR1 mRNA levels than normal BM or t(8;14)-positive acute lymphoblastic leukemia (ALL) BM samples as indicated by the mean signal intensity ± standard deviation. Left panel: data from GSE 13204; right panel data from GSE30029. *P<0.05, **P<0.001, ***P<0.0001. (D) The G2/M-specific expression levels of BubR1 were analyzed by dual-color flow cytometry in cytokine-stimulated primary AML blast populations and normalized to the level of DG-75 (lymphoblastic) expression. Fluorescence intensities indicate the expression level of BubR1 as measured per cell. All 38 tested primary AML blast populations exhibited lower expression levels than DG-75 cells. Thirty-two out of 38 (84.2%) primary AML blast populations showed even lower expression levels than the myeloblastic cell line Kasumi-1. HeLa cells with and without BubR1 knockdown were included to validate the accuracy of the approach.
To investigate whether BubR1 is also repressed in primary AML blast populations at the protein level, we analyzed BubR1 expression in the G2/M population by fluorescence activated cell sorting (FACS) in order to measure the BubR1 fluorescence intensity per cell (Figure 1D, Online Supplementary Figure S1). DG-75 and Kasumi-1 cells were included in each experiment as internal standards, to prove that the assay correlates well with the western blot results. The analyzed set of 38 primary AML samples included all French-American-British (FAB) groups, except for AML M6 and AML M7 (Online Supplementary Table S1). Calculated BubR1 fluorescence intensities were normalized to the BubR1 fluorescence intensity of the lymphoblastic DG-75 cells. We found that the vast majority of primary AML cells (32/38, 84.2%) expressed low levels of BubR1 and only 6/38 (15.8%) primary blast populations expressed BubR1 at levels higher than or equal to those in the myeloblastic Kasumi-1 cell line. We could not detect any differences in expression in subsets according to cytogenetics or percentage of blasts. Of note, all of the primary AML populations investigated showed lower levels of BubR1 expression than those in the lymphoblastic DG-75 cells.
Acute myeloid leukemia cells fail to induce a sustained mitotic arrest, undergo sister-chromatid separation in the presence of spindle disruption, and degrade the APC/C substrates cyclin B and securin in a proteasome-dependent manner
To test whether AML cells are capable of inducing a sustained mitotic arrest, lymphoblastic and myeloblastic cell lines were exposed to spindle-disruptive doses of nocodazole. While lymphoblastic cell lines exhibited a prominent G2/M peak, myeloblastic cell lines accumulated in G2/M to a lesser degree (Figure 2A). Kasumi-1 cells that were pre-synchronized by a thymidine block exhibited accumulation in G2/M within 12 h after release from the thymidine block. However, this initial response was followed by a remarkable decrease of G2/M cells 24 and 36 h after release (Figure 2B). In contrast, DG-75 cells still exhibited a notable G2/M peak even 36 h after release from the thymidine block (Figure 2B). Moreover, failure of Kasumi-1 cells to induce a stable mitotic block was associated with a higher rate of premature sister-chromatid separation as determined by metaphase spreads of lymphoblastic and myeloblastic cells. A higher frequency of myeloblastic cells carried single chromatids despite spindle disruption, suggesting sister-chromatid separation in the absence of a functional mitotic spindle (Figure 2C).
Figure 2.
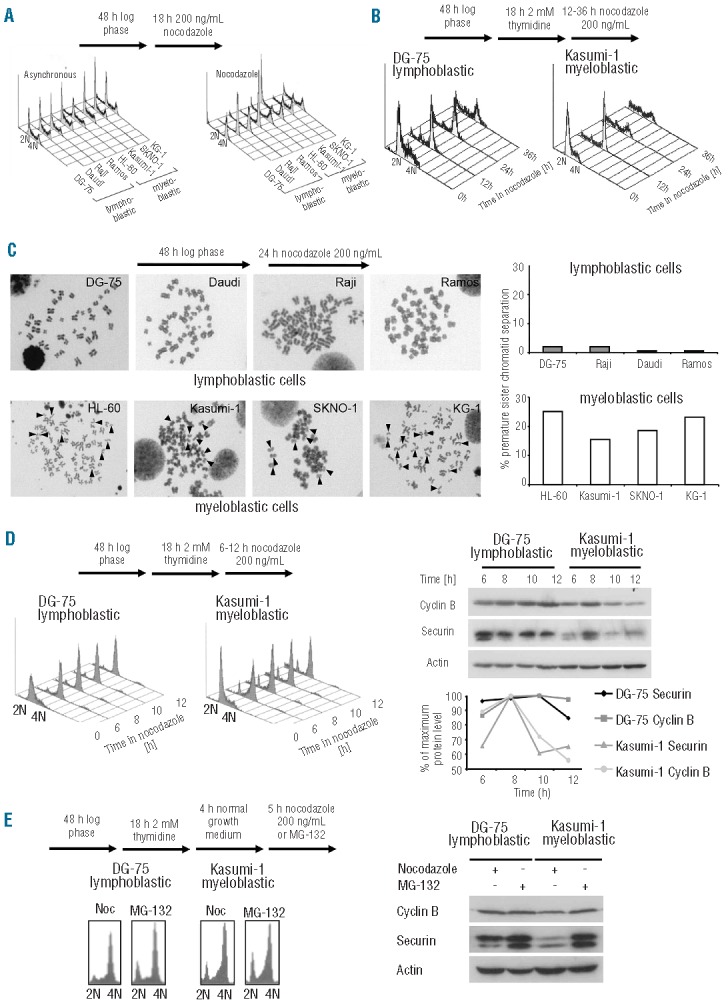
AML cells fail to induce sustained mitotic arrest, undergo sister-chromatid separation in the presence of spindle disruption, and degrade the APC/C substrates cyclin B and securin in a proteasome-dependent manner. (A) The ability to undergo a mitotic arrest in response to spindle poison was assessed in asynchronously growing cells that were exposed to nocodazole and compared to untreated cells. Myeloblastic cells only exhibited a limited G2/M peak upon spindle disruption compared to lymphoblastic cells. (B) For detailed insights into the differences in timing, lymphoblastic and myeloblastic cells were presynchronized in S-phase by a thymidine block to enter synchronously into mitotic block. DG-75 cells were able to maintain a mitotic block over 36 h, while the G2/M-peak in Kasumi-1 cells was barely detectable any more after 24 h. (C) Continued proteolysis during a mitotic block translated into sister-chromatid separation. Metaphase spreads of nocodazole-arrested myeloblastic cells exhibited a high number of metaphases containing separated or single chromatids (indicated by black arrowheads). Lymphoblastic cells appeared to have tightly connected sister-chromatids. (D) To assess the stability of APC/C target proteins during the first hours of a mitotic block, lymphoblastic and myeloblastic cells were allowed to enter a mitotic block following thymidine pre-synchronization. Cyclin B and securin expression levels were stabilized in DG-75 cells, while the levels of expression of both proteins rapidly declined following an initial rise with mitotic entry associated accumulation in Kasumi-1 cells. (E) To investigate proteasome-dependence of the decline of cyclin B and securin expression levels during a mitotic block, lymphoblastic and myeloblastic cells were exposed to the proteasome inhibitor MG-132. Proteasome inhibition in DG-75 led to similar expression levels as observed during mitotic block. Kasumi-1 cells showed stable cyclin B and securin expression levels upon proteasome-inhibition whereas expression levels of both proteins were remarkably lower during the mitotic block.
To study the expression of APC/C target proteins during a mitotic block, DG-75 and Kasumi-1 cells were synchronized in early S-phase by thymidine block. Cells were released from the S-phase block to enter mitosis synchronously in the presence of spindle-disruptive doses of nocodazole (200 ng/mL). We found that proteasome-dependent proteolysis of cyclin B and securin took place in the presence of spindle disruption in pre-synchronized Kasumi-1 cells as indicated by a decline in expression levels that could be rescued by the presence of the proteasome inhibitor MG-132 (Figure 2D, E).
Myeloblastic cells are less responsive to nocodazole and retain activity of the APC/C to ubiquitinylate cyclin B
To assess the sensitivity of lymphoblastic and myeloblastic cells to spindle poison, DG-75 and Kasumi-1 cells were grown asynchronously in the presence of an ascendant series of non-spindle-disruptive concentrations of nocodazole (later referred to as low/lower dose nocodazole, comparable to the continuous serum levels of spindle poison that are observed after the peak concentration following drug administration20). Lymphoblastic DG-75 cells already exhibited a strong mitotic arrest at a concentration of 20 ng/mL (Figure 3A) and exhibited only residual APC/C activity in low-dose nocodazole when compared to Kasumi-1 cells (Figure 3B). In contrast, Kasumi-1 cells displayed notable activity of the APC/C to ubiquitinylate cyclin B (Figure 3B) and failed to induce a detectable accumulation of cells in G2/M (Figure 3A). To confirm that myeloblastic cells in general are less sensitive to spindle poison, we tested the sensitivity of all seven AML cell lines analyzed in Figure 1 to low-dose nocodazole and included DG-75 cells as the SAC competent control. We found that all tested AML cell lines are less sensitive than DG-75 cells to nocodazole (Figure 3C).
Figure 3.
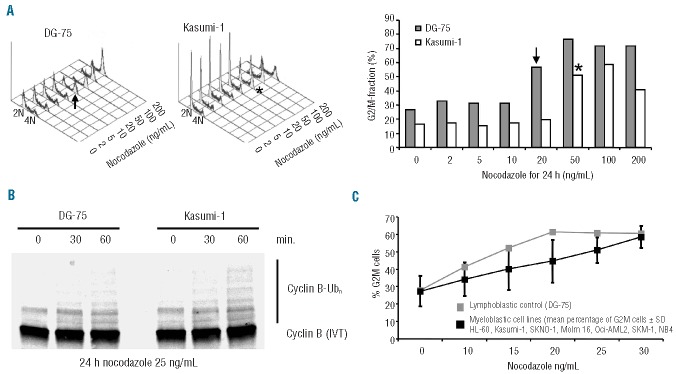
Myeloblastic cells are less responsive to nocodazole and retain activity of the APC/C to ubiquitinylate cyclin B during a mitotic block. (A) Log-phase DG-75 and Kasumi-1 cells were exposed to ascending concentrations of nocodazole. Accumulation in G2/M began at lower concentrations in lymphoblastic cells than in the more resistant myeloblastic cells. →Indicates the lowest dose causing distinct changes in cell cycle distribution in DG-75 cells. *Indicates the lowest dose causing distinct changes in cell cycle distribution in Kasumi-1 cells. (B) In-vitro ubiquitination of the APC/C substrate cyclin B was performed using immunopurified APC/C from DG-75 and Kasumi-1 cells that were grown in the presence of 25 ng/mL nocodazole for 24 h. The autoradiography indicates enhanced ubiquitination of 35 S-labeled cyclin B in Kasumi-1 cells as compared to DG-75 cells. (C) Seven AML cell lines and the lymphoblastic cell line DG-75 as a SAC-competent control were challenged with an ascending series of lower nocodazole doses for 24 h and accumulation in G2M was assessed by flow cytometry following propidium iodide staining.
Kasumi-1 cells pass through mitosis quickly and exhibit different fates during mitosis upon challenge with spindle poison
To address the processes during mitosis in lymphoblastic and myeloblastic cells in detail, we performed live-cell imaging of cells that were immobilized on fibrinogen-coated microscope slides. First, we analyzed asynchronously growing unperturbed cells and found that myeloblastic Kasumi-1 cells proceeded much faster through mitosis than did their lymphoblastic counterparts (Figure 4A). Of note, DG-75 cells also showed a larger standard deviation, indicating a greater variability in mitotic timing. We hypothesize that this greater variability was caused by a more sensitive response to disturbing factors during mitosis (Figure 4A). To visualize how Kasumi-1 cells respond to interference with microtubule kinetics in detail, we performed live-cell imaging in the presence of low-dose nocodazole (25 ng/mL). Under these conditions, Kasumi-1 cells displayed different fates during passage through mitosis, while DG-75 cells already exhibited a strong mitotic block at a slightly lower dose of nocodazole (20 ng/mL). Importantly, Kasumi-1 cells exhibited a delay in time from nuclear envelope breakdown until onset of anaphase in the presence of low-dose nocodazole indicating the existence of partial SAC function (Online Supplementary Figure S3A,B). Following a mitotic delay, we found that Kasumi-1 cells underwent chromosome segregation, without signs of genetic instability, anaphase bridging or cell death after anaphase (Figure 4B, first three picture series). We also observed cell death following a prolonged metaphase-like state, confirming that the SAC in Kasumi-1 cells was partially functioning (Figure 4B, fourth picture series; for quantification see Figure 5D). In contrast, DG-75 cells showed a strong accumulation in metaphase (data not shown). Due to this strong response and the permanent arrest, statistics concerning the fate of the cells could not be generated from DG-75 cells.
Figure 4.

Kasumi-1 cells pass through mitosis quickly and exhibit different fates during mitosis upon challenge with spindle poison. (A) Mitotic timing was assessed using live-cell imaging in hematopoietic cell lines stably expressing histone H2 fused to the fluorescent marker protein GFP (H2-GFP). Kasumi-1 cells passed through mitosis quickly as compared to DG-75 cells (n=2 independent experiments, at least 40 mitoses were analyzed). ***P<0.0001. (B) Myeloblastic cells divided in the presence of 25 ng/mL of nocodazole (low-dose nocodazole), a concentration sufficient to cause a mitotic block in DG-75 cells. For visualization of cell divisions in the presence of 25 ng/mL nocodazole, we performed live-cell imaging of partially immobilized cells. Exposure to nocodazole resulted in cell death either in mitosis or in the following G1 phase in some cases (third and fourth series of image). However, Kasumi-1 cells also divided, although sometimes with remarkable segregation errors, but remained viable throughout the time of observation (first and second series of images).
Figure 5.
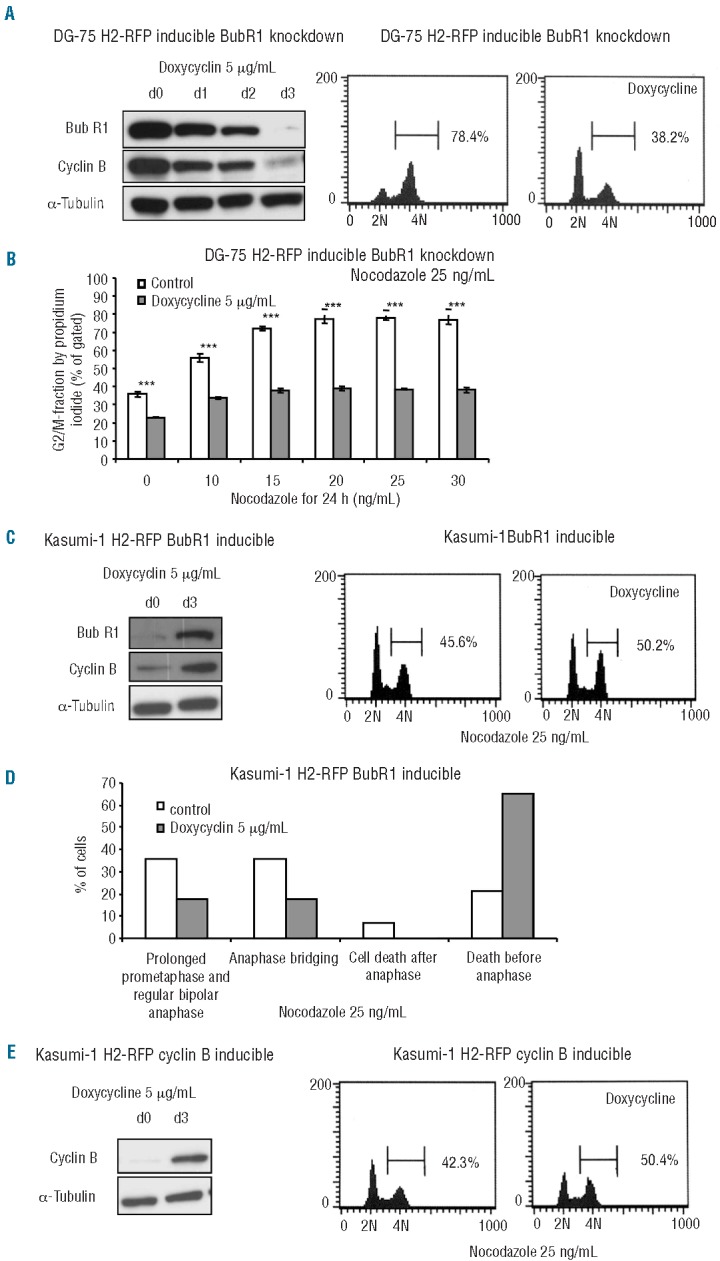
BubR1 fosters a mitotic arrest and cell death in mitosis via stabilization of cyclin B. (A) Reduction in BubR1 and associated cyclin B expression in DG-75 cells following induction of BubR1 knockdown was assessed by western blotting. Loss of accumulation in G2/M was measured by propidium iodide (PI) staining and represented in cell cycle histograms. The extent to which induced cells accumulated in G2/M was remarkably restricted (38.2% versus 78.4%) when compared to non-induced control cells. (B) The extent to which DG-75 cells accumulate in response to ascending concentrations of nocodazole was assessed by PI staining. Quantifications for an ascending series of nocodazole concentrations are shown for BubR1-knockdown-induced and non-induced DG-75 cells. (C) Increase in BubR1 expression levels in Kasumi-1 cells following induction was assessed by western blotting. Increase in cyclin B expression levels was associated with induction of BubR1 expression. Accumulation in G2/M was assessed by PI staining and is represented by cell cycle histograms. The extent to which induced cells accumulated was higher (50.2% versus 45.6%) when compared to non-induced control cells. Since only vital cells add to the G2/M peak, cells that underwent cell death in mitosis were not detected leading to potential underestimation of the effect of BubR1 reconstitution. (D) For determination of the mitotic fate in Kasumi-1 cells in low-dose nocodazole we performed live-cell imaging. The proportion of cells undergoing cell death in metaphase was more than 3-fold higher in BubR1-induced cells and bipolar divisions were less frequently observed (a representative experiment of two independent experiments is shown). (E) The increase in cyclin B expression levels in Kasumi-1 cells following induction was assessed by western blotting. Accumulation in G2/M was quantified by PI staining and is represented by cell cycle histograms. The extent to which induced cells accumulated was higher (50.4% versus 42.3%) when compared to non-induced control cells.
Inducible knockdown of BubR1 in lymphoblastic DG-75 cells reduces response to spindle poison
To determine whether repression of BubR1 is able to render a hematopoietic cell line that is per se sensitive to spindle poison less responsive to antimitotic therapy, we performed BubR1 knockdown in lymphoblastic DG-75 cells. To achieve a strong knockdown effect, we established inducible retrovirus-delivered BubR1 knockdown. Maximal knockdown of BubR1 was observed by day 3 after induction with doxycycline and was accompanied by a considerable destabilization of cyclin B (Figure 5A). In the presence of low concentrations of nocodazole, we observed a strong accumulation of non-induced DG-75 cells in G2M (Figure 5B). In contrast, induction of BubR1 knockdown with doxycycline led to a severely reduced accumulation in G2/M, confirming that BubR1 is an important factor in mediating G2/M arrest of lymphoblastic cells in response to spindle-poison-based therapy (Figure 5A,B).
Inducible re-expression of BubR1 in myeloblastic Kasumi-1 cells stabilizes cyclin B and sensitizes cells to spindle-disrupting drugs
Based on the foregoing, we asked whether re-expression of BubR1 can re-sensitize myeloblastic cells to spindle poison. To answer this question, we established inducible retrovirus-delivered BubR1 expression in Kasumi-1 cells (showing low BubR1 expression) and HL-60 cells (an AML cell line showing almost normal levels of BubR1 expression). Upon induction by doxycycline, we found a strong up-regulation of BubR1 expression in Kasumi-1 cells which reached a maximum by day 3 (Figure 5C). Induction of BubR1 was associated with significant stabilization of cyclin B in myeloblastic cells (Figure 5C), a finding which is in line with the reported role of BubR1 as an inhibitor of the APC/C. Unlike in Kasumi-1 cells, in HL-60 cells which already show high BubR1 expression levels, we did not note a further increase in BubR1 expression following induction by doxycycline (Online Supplementary Figure S4A). Using live-cell imaging, we showed that BubR1-induced Kasumi-1 cells progressed more slowly through an unperturbed mitosis (Online Supplementary Figure S3A) and this delay was even more pronounced in the presence of low doses of nocodazole (Online Supplementary Figure S3A). Importantly, induction of BubR1 in Kasumi-1 cells led to a decrease in the percentage of bipolar divisions in low-dose nocodazole and in accordance with its reported role as a promoter of apoptosis, we observed that the proportion of cells undergoing cell death before anaphase was dramatically increased by more than 3-fold from 20% to over 60% (Figure 5D). Propidium iodide staining and FACS analyses further substantiated this observation. In the presence of low-dose nocodazole the proportion of G2/M cells was increased in asynchronously cycling BubR1-induced Kasumi-1 cells as compared to non-induced controls, indicating enhanced mitotic arrest upon induction of BubR1 (Figure 5C, Online Supplementary Figure S3C).
Although epigenetic silencing of BubR1 through promoter hypermethylation has been described in some tumors, we and others could not detect significant BubR1 up-regulation following demethylating therapy in myeloblastic cells (Online Supplementary Figure S5A).21 Moreover, acetylation has been proposed to cause stabilization of BubR1.22 However, following treatment of Kasumi-1 cells with a deacetylase inhibitor in different conditions, we did not detect stabilization of BubR1 (Online Supplementary Figure S5B).
Inducible expression of cyclin B in myeloblastic cell lines (Kasumi-1 and HL-60) enhances response to spindle poison
We next addressed the question whether, according to its proposed role as an inhibitor of APC/CCdc20, inducible re-expression of BubR1 exerts its effects via stabilization of the APC/C target cyclin B. To this end, we established a system capable of inducible retrovirus-delivered cyclin B expression. In analogy to the inducible expression of BubR1, we noted maximal cyclin B expression levels by day 3 after induction (Figure 5E). We performed live-cell imaging to evaluate the role of forced cyclin B expression on mitoses in Kasumi-1 cells and found that cyclin B induction was associated with a more pronounced mitotic delay in the presence of low doses of nocodazole (Online Supplementary Figure S3B) and enhanced accumulation of Kasumi-1 and HL-60 in G2/M as assessed by flow cytometry (Figure 5E, Online Supplementary Figures S3D and S4A). To test the connection between the stability of cyclin B and the response to antimitotic treatment, we expressed a more stable form of cyclin B with an N-terminal tag in Kasumi-1 cells. As a control we included Kasumi-1 cells expressing a cyclin B derivative with a C-terminal tag that exhibited normal degradation kinetics. The generation and degradation characteristics of both cyclin B derivatives have been described previously.13 Importantly, we provide evidence that a more stable form of cyclin B rendered Kasumi-1 cells more sensitive to spindle poison (Online Supplementary Figure S4B).
Discussion
It is a well-known clinical phenomenon that acute lymphoblastic leukemia and Burkitt’s lymphoma respond better than AML to spindle poison-based therapy.1,2 By using these two well-characterized entities as examples for malignancies that show differential responses to spindle poison, we tested the role of correct SAC function in order to mediate therapeutic response. In this study, we identified that the SAC component BubR1 is repressed in the vast majority of AML cases and demonstrated that re-expression of BubR1 in an AML cell line is capable of enhancing response to spindle poison by reconstituting SAC function. We are thereby the first to demonstrate that BubR1 is commonly down-regulated in AML cells, while Burkitt’s lymphoma cells express high levels of BubR1. The finding that a truncated version of the leukemogenic AML1-ETO fusion protein is a repressor of BubR123 further substantiates that deregulated expression of BubR1 plays an important role in AML. Moreover, in a case of AML the sequence encoding the BubR1-interactor Blinkin was fused to the MLL gene, also supporting the notion that interference with BubR1 function is related to leuke-mogenesis.24,25 However, while it has been reported that BubR1 is affected in a limited number of AML cases, our results provide evidence that down-regulation of BubR1 is a more general feature in AML. This finding suggests that BubR1, due to its frequent repression in AML, is an important factor that contributes to the poor response of AML to spindle poison-based therapy. We could not detect any effect on BubR1 levels following treatment with the demethylating agent decitabine or inhibition of deacetylation by trichostatin A. The molecular basis of the down-regulation of BubR1 during myeloid transformation does, therefore, remain an open issue.
Tight control of mitosis and mitotic regulators is very important in both benign and malignant hematologic conditions.26–30 Accurate distribution of the chromosomes onto the two developing daughter cells depends on precise surveillance of APC/C-dependent proteolysis by the SAC. We observed a less stringent mitotic control in AML cells than in Burkitt’s lymphoma cells. This became apparent through the finding that the SAC in AML cells required higher doses of spindle poison in order to affect cell cycle kinetics. Moreover, we observed that the APC/C was active and drove mitotic exit in the presence of severe structural aberrations caused by low-dose nocodazole. The serum levels of commonly administered spindle poisons, such as vincristine, vindesine, vinblastine, vinorelbine and taxol, are higher only within a short period of time after administration.20,31–33 Prolonged exposure to rather low serum levels may lead to induction of apoptosis only in those tumors that are highly sensitive to spindle poisons. However, we noted that AML cells are not able to undergo a sustained mitotic block even in the presence of high doses of spindle poison as evidenced in its extreme form by the finding of sister-chromatid separation in the presence of spindle disruption.
Our data indicate that chromatid separation is a consequence of continuous proteasome-dependent proteolysis of cyclin B and securin, and we propose that this triggers erroneous mitotic exit in individual cells. Importantly, we observed that re-expression of BubR1 in AML cells (with low BubR1 expression levels) reinforced a barrier to limit the number of (incorrect) cell divisions in the presence of low-dose nocodazole by stabilizing cyclin B expression and enhancing cell death in mitosis. In rare cases, in which AML cells express higher levels of BubR1, such as in the HL-60 cell line, we noted that this barrier can be reinforced downstream of BubR1 by up-regulation of cyclin B. Importantly, this finding suggests that in the less common instances of higher BubR1 expression in AML, different and so far unknown lesions are of relevance and may collaborate with coexisting alterations to weaken the accuracy of mitotic exit control by reducing the stability of cyclin B. The extent to which the anti-mitotic barrier is reinforced is reflected best by the increase in the number of cells that undergo cell death before anaphase in the presence of low-dose nocodazole, as detected by live-cell imaging. Since BubR1 and cyclin B are known to support apoptosis of cells that are arrested in mitosis, our cell cycle histograms might underestimate the effect of BubR1 and cyclin B reconstitution because dead cells do not add to the G2/M-fraction of cells; this highlights the value of complementary live-cell imaging. Our findings support the view that a weakened SAC per se may predispose to a higher number of inaccurate cell divisions. Interference with microtubule kinetics by spindle poisons may further enhance the probability of such events and increase the level of genetic instability resulting from mitotic inaccuracies. This enhanced genetic instability can either increase lethality resulting in a therapeutic effect in some cells or provide a proliferation advantage in other cells.
A prolonged mitotic arrest is linked to enhanced cell death4,34 as a result of pro-apoptotic signals that accumulate during the time of arrest.7 Interestingly, BubR1 has been implicated in the induction of cell death in mitosis in cancer cells15 and its down-regulation, for instance by promoter hypermethylation in some cancers, has been shown to be one mechanism to evade mitotic arrest and cell death.15,21 Stabilization of cyclin B is a major barrier against premature mitotic exit35,36 and recent studies demonstrated the potential value of targeting APC/C-dependent proteolysis and mitotic exit in the treatment of cancer.7,37,38 BubR1 controls mitotic exit by stabilization of cyclin B and lower expression levels of BubR1 were associated with a reduced response to spindle poison.39 Since re-expression of BubR1 in AML led to a more potent interference with mitotic exit, we provide evidence that the downstream cascade of the SAC is functional in these cells. This important finding establishes cyclin B and the downstream mitotic exit cascade as a target in AML. By probing the response of myeloblastic Kasumi-1 cells expressing a more stable cyclin B variant with reduced degradation kinetics, we provide additional evidence that slowing down the degradation of cyclin B sensitizes cells to spindle poison (Online Supplementary Figure S3C). Stabilization of cyclin B appears to be a promising rationale, since high expression of cyclin B strengthens mitotic arrest and enhances eradication of arrested cells by induction of cell death in mitosis. This is achieved by cyclin B-Cdk1-dependent phosphorylation of the anti-apoptotic regulator Mcl-1 and subsequent destruction, which initiates apoptosis after a prolonged mitotic arrest.9 Since we found that Mcl-1 levels are already significantly decreased in AML cell lines, AML might respond well to therapeutic approaches that keep cyclin B levels stable, such as proteasome inhibitors, and hence enforce a powerful barrier at the metaphase to anaphase transition.
Since diverse tumors respond differently to spindle poisons40 it will be interesting to analyze, in future experiments, whether levels of BubR1 expression could also predict response to antimitotic therapy in these entities.
In summary, while our finding of low expression of the highly important SAC protein BubR1 in the vast majority of AML is striking, there are also additional so far unknown proteins involved to explain why for instance HL-60 with higher BubR1 expression also responds poorly to spindle poisons.
Acknowledgments
We thank D. Wider and M. Zlei for critical discussions and R. Mertelsmann for continuous support. We are grateful to J. Schuringa for sharing information about unpublished results and to A. Kohlmann for advice concerning the choice of appropriate microarray datasets. We are also grateful to S. Taylor and D. Gerlich for providing plasmids and J. Finke and M. Lübbert for providing cell lines.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
RW is supported by grants from the Deutsche Forschungsgemeinschaft (WA 1363/3-1) and Jose-Carreras Leukemia Foundation (DJCLS R 10/14).
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Wagner HP, Swidzinska PM, Hirt A. Variable duration of vincristine-induced metaphase block in leukemic and nornal bone marrow cells of children. Med Pediatr Oncol. 1977;3(1): 75–83 [DOI] [PubMed] [Google Scholar]
- 2.Landini I, Bartolozzi B, Banchelli I, Degli Innocenti A, Nocentini O, Bernabei PA. In vitro activity of vinorelbine on human leukemia cells. J Chemother. 2001;13 (3): 309–15 [DOI] [PubMed] [Google Scholar]
- 3.Stanton RA, Gernert KM, Nettles JH, Aneja R. Drugs that target dynamic microtubules: a new molecular perspective. Med Res Rev. 2011;31(3): 443–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: the mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005;8(1): 7–12 [DOI] [PubMed] [Google Scholar]
- 5.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8(5): 379–93 [DOI] [PubMed] [Google Scholar]
- 6.Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell. 2004;7(5): 637–51 [DOI] [PubMed] [Google Scholar]
- 7.Huang HC, Shi J, Orth JD, Mitchison TJ. Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell. 2009;16(4): 347–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14(2): 111–22 [DOI] [PubMed] [Google Scholar]
- 9.Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010;29(14): 2407–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471 (7336): 110–4 [DOI] [PubMed] [Google Scholar]
- 11.Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471(7336): 104–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol. 2006;16(12): 1194–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schnerch D, Follo M, Krohs J, Felthaus J, Engelhardt M, Wäsch R. Monitoring APC/C activity in the presence of chromosomal misalignment in unperturbed cell populations. Cell Cycle. 2012;11(2): 310–21 [DOI] [PubMed] [Google Scholar]
- 14.Suijkerbuijk SJ, van Osch MH, Bos FL, Hanks S, Rahman N, Kops GJ. Molecular causes for BUBR1 dysfunction in the human cancer predisposition syndrome mosaic variegated aneuploidy. Cancer Res. 2010;70 (12): 4891–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shin HJ, Baek KH, Jeon AH, Park MT, Lee SJ, Kang CM, et al. Dual roles of human BubR1, a mitotic checkpoint kinase, in the monitoring of chromosomal instability. Cancer Cell. 2003;4(6): 483–97 [DOI] [PubMed] [Google Scholar]
- 16.Bohers E, Sarafan-Vasseur N, Drouet A, Paresy M, Latouche JB, Flaman JM, et al. Gradual reduction of BUBR1 protein levels results in premature sister-chromatid separation then in aneuploidy. Hum Genet. 2008;124(5): 473–8 [DOI] [PubMed] [Google Scholar]
- 17.Rio Frio T, Lavoie J, Hamel N, Geyer FC, Kushner YB, Novak DJ, et al. Homozygous BUB1B mutation and susceptibility to gastrointestinal neoplasia. N Engl J Med. 2010;363(27): 2628–37 [DOI] [PubMed] [Google Scholar]
- 18.de Jonge HJ, Woolthuis CM, Vos AZ, Mulder A, van den Berg E, Kluin PM, et al. Gene expression profiling in the leukemic stem cell-enriched CD34(+) fraction identifies target genes that predict prognosis in normal karyotype AML. Leukemia. 2011;25(12): 1825–33 [DOI] [PubMed] [Google Scholar]
- 19.Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010;28(15): 2529–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sethi VS, Jackson DV, Jr, White DR, Richards F, 2nd, Stuart JJ, Muss HB, et al. Pharmacokinetics of vincristine sulfate in adult cancer patients. Cancer Res. 1981;41(9 Pt 1): 3551–5 [PubMed] [Google Scholar]
- 21.Park HY, Jeon YK, Shin HJ, Kim IJ, Kang HC, Jeong SJ, et al. Differential promoter methylation may be a key molecular mechanism in regulating BubR1 expression in cancer cells. Exp Mol Med. 2007;39(2): 195–204 [DOI] [PubMed] [Google Scholar]
- 22.Choi E, Choe H, Min J, Choi JY, Kim J, Lee H. BubR1 acetylation at prometaphase is required for modulating APC/C activity and timing of mitosis. EMBO J. 2009;28(14): 2077–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyapati A, Yan M, Peterson LF, Biggs JR, Le Beau MM, Zhang DE. A leukemia fusion protein attenuates the spindle checkpoint and promotes aneuploidy. Blood. 2007;109 (9): 3963–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayette S, Tigaud I, Vanier A, Martel S, Corbo L, Charrin C, et al. AF15q14, a novel partner gene fused to the MLL gene in an acute myeloid leukaemia with a t(11;15)(q23;q14). Oncogene. 2000;19(38): 4446–50 [DOI] [PubMed] [Google Scholar]
- 25.Kiyomitsu T, Obuse C, Yanagida M. Human Blinkin/AF15q14 is required for chromosome alignment and the mitotic checkpoint through direct interaction with Bub1 and BubR1. Dev Cell. 2007;13(5): 663–76 [DOI] [PubMed] [Google Scholar]
- 26.Nolte-'t Hoen EN, Almeida CR, Cohen NR, Nedvetzki S, Yarwood H, Davis DM. Increased surveillance of cells in mitosis by human NK cells suggests a novel strategy for limiting tumor growth and viral replication. Blood. 2007;109(2): 670–3 [DOI] [PubMed] [Google Scholar]
- 27.Vitrat N, Cohen-Solal K, Pique C, Le Couedic JP, Norol F, Larsen AK, et al. Endomitosis of human megakaryocytes are due to abortive mitosis. Blood. 1998;91(10): 3711–23 [PubMed] [Google Scholar]
- 28.Sanchez-Aguilera A, Montalban C, de la Cueva P, Sanchez-Verde L, Morente MM, Garcia-Cosio M, et al. Tumor microenvironment and mitotic checkpoint are key factors in the outcome of classic Hodgkin lymphoma. Blood. 2006;108(2): 662–8 [DOI] [PubMed] [Google Scholar]
- 29.Staal FJ, de Ridder D, Szczepanski T, Schonewille T, van der Linden EC, van Wering ER, et al. Genome-wide expression analysis of paired diagnosis-relapse samples in ALL indicates involvement of pathways related to DNA replication, cell cycle and DNA repair, independent of immune phenotype. Leukemia. 2010;24(3): 491–9 [DOI] [PubMed] [Google Scholar]
- 30.Lin SF, Lin PM, Yang MC, Liu TC, Chang JG, Sue YC, et al. Expression of hBUB1 in acute myeloid leukemia. Leuk Lymphoma. 2002;43(2): 385–91 [DOI] [PubMed] [Google Scholar]
- 31.Owellen RJ, Root MA, Hains FO. Pharmacokinetics of vindesine and vincristine in humans. Cancer Res. 1977;37(8 Pt 1): 2603–7 [PubMed] [Google Scholar]
- 32.Gebbia V, Puozzo C. Oral versus intravenous vinorelbine: clinical safety profile. Expert Opin Drug Saf. 2005;4(5): 915–28 [DOI] [PubMed] [Google Scholar]
- 33.Rizzo J, Riley C, von Hoff D, Kuhn J, Phillips J, Brown T. Analysis of anticancer drugs in biological fluids: determination of taxol with application to clinical pharmacokinetics. J Pharm Biomed Anal. 1990;8(2): 159–64 [DOI] [PubMed] [Google Scholar]
- 34.Tao W, South VJ, Zhang Y, Davide JP, Farrell L, Kohl NE, et al. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer Cell. 2005;8(1): 49–59 [DOI] [PubMed] [Google Scholar]
- 35.Wolf F, Wandke C, Isenberg N, Geley S. Dose-dependent effects of stable cyclin B1 on progression through mitosis in human cells. EMBO J. 2006;25(12): 2802–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wäsch R, Cross FR. APC-dependent proteolysis of the mitotic cyclin Clb2 is essential for mitotic exit. Nature. 2002;418(6897): 556–62 [DOI] [PubMed] [Google Scholar]
- 37.Manchado E, Guillamot M, de Carcer G, Eguren M, Trickey M, Garcia-Higuera I, et al. Targeting mitotic exit leads to tumor regression in vivo: modulation by Cdk1, Mastl, and the PP2A/B55alpha, delta phosphatase. Cancer Cell. 2010;18(6): 641–54 [DOI] [PubMed] [Google Scholar]
- 38.Wäsch R. Targeting mitotic exit for cancer treatment. Expert Opin Ther Targets. 2011;15(7): 785–8 [DOI] [PubMed] [Google Scholar]
- 39.Greene LM, Campiani G, Lawler M, Williams DC, Zisterer DM. BubR1 is required for a sustained mitotic spindle checkpoint arrest in human cancer cells treated with tubulin-targeting pyrrolo-1,5-benzox-azepines. Mol Pharmacol. 2008;73 (2): 419–30 [DOI] [PubMed] [Google Scholar]
- 40.Matson DR, Stukenberg PT. Spindle poisons and cell fate: a tale of two pathways. Mol Interv. 2011;11(2): 141–50 [DOI] [PMC free article] [PubMed] [Google Scholar]