

Cascade reactions[1] facilitate complex molecule synthesis through their capacity to generate multiple target-relevant[2] bonds and stereocenters in a single operation.[3] Using thermodynamic control to dictate product formation further streamlines molecular synthesis by minimizing the need to set multiple stereocenters in reaction substrates. Rhenium oxide-mediated allylic alcohol transpositions[4] are useful in the design of thermodynamically-controlled reactions because they adjust regiochemistry[5] and stereochemistry[6] in response to structural influences, as demonstrated by their increasing use in complex molecule synthesis.[7] A schematic of the process that is consistent with experimental observations and computational studies[8] is shown in Scheme 1.
Scheme 1.
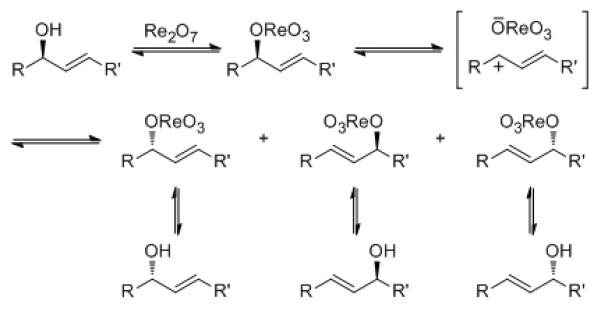
Rhenium oxide-mediated allylic alcohol transposition.
We have begun a program[5c] in which Re2O7-catalyzed allylic alcohol transposition reactions initiate stereoselective ring-forming processes that proceed through trapping the hydroxyl group with a pendent electrophile followed by thermodynamically controlled equilibration. Initial studies employed achiral acetal electrophiles, thereby requiring a stereogenic center in the tether between the nucleophile and the electrophile to control the stereochemical outcome. Trapping transposing alcohols with chiral electrophiles, however, offers significant adavantages for the preparation of enantiomerically pure materials through this sequence. Epoxide groups are particularly attractive for incorporation into these reactions because of their versatile reactivity patterns, whereby they can act as electrophiles to liberate nucleophilic hydroxyl groups upon opening[9] or as nucleophiles to create electrophilic epoxonium ions upon reacting with a cation.[10] Moreover, several methods are commonly employed to prepare epoxides in enantiomerically pure form.[11] These attributes have led to the development of numerous epoxide-opening cascade reactions.[12] In this manuscript we report that epoxides can be used as trapping agents for allylic alcohols in rhenium oxide-mediated transposition reactions. These reactions are used as the basis for a number of cascade processes in which several electrophiles can be used as trapping agents. Ketones are shown to be effective stereochemical conduits, allowing for remote stereoinduction and bidirectional stereogenesis in the synthesis of polycyclic structures. Stereocenters are generated by functionalizing prochiral centers, in contrast to standard epoxide cascade reactions in which an equivalent number of stereocenters are present in the substrates and products. Additionally we show that Re2O7 can be adsorbed on silica gel to provide an easily handled and measured source of the catalyst.
The capacity of epoxides to act as trapping agents was demonstrated (Scheme 2) by exposing 1 to Re2O7 (5 mol%) to produce tetrahydropyrans 2 and 3 in 51% and 38% yields, respectively, after 4 h at rt. Resubjection of the isomers to the reaction conditions resulted in no reaction, thereby establishing that this result arises from kinetic control rather than thermodynamic equilibration. The reaction of secondary alcohol 4 proved to be more complex. After 45 min the reaction produced a mixture of oxepanes 5 and 6 in a nearly 1:1 ratio. Prolonged exposure in the presence of higher catalyst loading (15 mol%) resulted in the formation of tetrahydropyrans 7 and 8, again in a nearly 1:1 ratio. Resubjecting 5 to the reaction conditions predominantly provides 7 and resubjecting 6 to the reaction conditions predominantly provides 8, and 7 and 8 interconvert extremely slowly when resubjected to Re2O7.
Scheme 2.
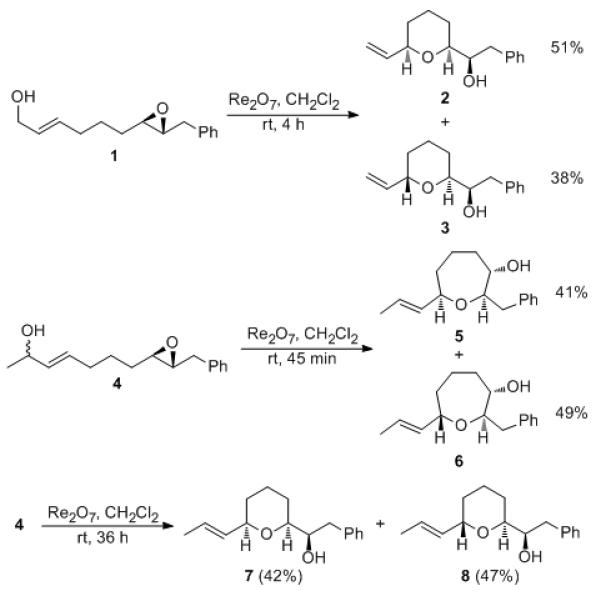
Epoxides as trapping agent in alcohol transposition reactions.
These studies showed that primary and secondary allylic alcohols react through divergent pathways in these processes (Scheme 3). Primary allylic alcohol 1 undergoes transposition to form a mixture of diastereomeric secondary alcohols (9). The Re2O7 or the HOReO3 that forms upon reaction with the hydroxyl group, activates the epoxide group toward nucleophilic attack, thus providing the tetrahydropyran products. Secondary alcohol 4, however, reacts with Re2O7 to form allyl cation 10 due to stabilization from by the additional alkyl group. The epoxide then adds to the cation to form epoxonium ion 11, which reacts with perrhenate through a kinetically preferred[10e] endo-pathway to yield an oxepanyl perrhenate ester that decomposes to yield the observed oxepanyl alcohols as the initial products. Crystallographic[13] and Mosher ester[14] analyses of the products from enantiomerically enriched substrates provided evidence for this pathway by showing that the absolute stereochemistry at the distal carbon of the epoxide (with respect to the allylic alcohol) was retained while the absolute stereochemistry at the proximal carbon was inverted. Re2O7-mediated ionization of the oxepanes yields allyl cations 12 that react with the free hydroxyl groups, predominantly with stereochemical retention, to yield the tetrahydropyrans as thermodynamic products that appear to be inert toward further ionization. Racemization in these processes is minimal, indicating that allylic ethers undergo ionization much faster than the aliphatic ethers.
Scheme 3.
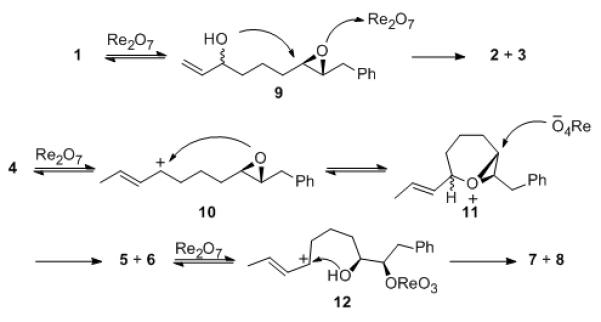
Mechanistic details.
The slow isomerization of 8, in contrast to the isomerizations of 5 and 6, led us to speculate that the exocyclic hydroxyalkyl group suppresses allyl cation formation. This was tested (Scheme 4) by preparing methyl ether 13 as a 1:1 mixture of stereoisomers. This mixture produced the cis-isomer as the nearly exclusive product following exposure to Re2O7 for 5 h at rt, thereby confirming that hydrogen bonding suppresses ionization-based isomerization. A corresponding terminal alkene showed very little isomerization (not shown), confirming the importance of cation stabilization in stereochemical equilibration. These studies indicated that these processes could provide stereochemically pure products if the hydroxyl group that results from epoxide opening were used as a nucleophile in a cascade process.
Scheme 4.
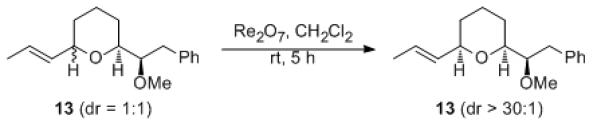
Stereochemical isomerization.
The initial set of cascade reactions proceeded through the incorporation of an electrophile or pro-electrophile at the substrate terminus to trap the hydroxyl group following epoxide opening (Scheme 5). Exposing epoxy ester 14 to Re2O7 provided lactone 15 as a single stereoisomer in 71% yield, though MeOH removal proved to be essential for equilibration. Acetal 16 was prepared to study the viability of using the Lewis acidity of the rhenium oxide to promote the formation of an oxocarbenium ion[5b-c,15] as a trapping group. Exposing 16 to Re2O7 at rt led to decomposition, and lowering the reaction temperature provided a low yield of 17. Switching to the more soluble catalyst Ph3SiOReO3,[16] however, allowed the initial phase of the reaction to be conducted at −25 °C. After warming to rt to effect the stereochemical equilibration, 17 was isolated as a single stereoisomer with respect to the tetrahydropyran and as a 5:2 mixture at the anomeric site. This improved efficiency could be attrbibuted to the absence of HOReO3 formation when Ph3SiOReO3 is used as the catalyst. Shorter reaction times resulted in the observation of more stereoisomeric products, confirming that equilibration follows the initial cyclization. Cascade reactions with enone electrophiles provided tetrahydropyranyl ketones, as shown through the conversion of 18 to 19. This reaction provided a bis-tetrahydropyran within 12 h, but required 3.5 d at rt to provide the product as a single stereoisomer. The reaction with an enone electrophile is significant in that it demonstrates bidirectional stereogenesis, in which the introduction of new stereogeneic centers from distal prochiral units are directed by the stereogenic centers in the epoxide group. Notably, this transformation also proceeds with perfect atom economy.[17]
Scheme 5.
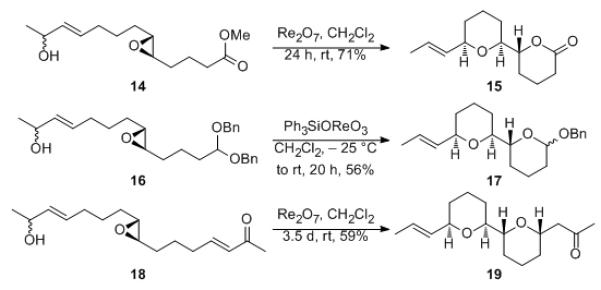
Cascade reactions.
Utilizing ketones as stereochemical conduits provides an alternate strategy to chirality transfer from the epoxide groups to remote sites. This approach uses a ketone group as a nucleophile to open the epoxide, thereby generating a chiral oxocarbenium ion[18] that acts as an electrophilic trap for the transposing alcohol. This process allows for the stereochemical editing phase of the reaction to proceed through acetal ionization and allylic alcohol transposition rather than allylic ether ionization, thus facilitating the process and providing a pathway for stereochemical editing in products that contain terminal alkenes. This is demonstrated (Scheme 6) by the reaction of 20 with Re2O7, which proceeds within 20 h at rt to form spiroacetal 21 as a single stereoisomer in 93% yield. A plausible mechanism for this transformation proceeds through the Lewis acid-mediated opening of the epoxide by the oxygen of the ketone to yield oxocarbenium ion 22. The allylic alcohol reversibly transposes to form a mixture of secondary alcohols (23) that add to the oxocarbenium ion to form 21 and 24. The thermodynamically less stable 24 can revert to a single diastereomer of 23, which undergoes stereochemical editing through allylic alcohol transposition. Eventually this process leads to thermodynamically preferred spiroacetal 21 as the sole product of this transformation. An alternative mechanism in which the transposing alcohols add into the ketone to form a mixture of hemiacetals that add into the epoxide to form 21 and 24 can also be envisioned. Secondary alcohol substrates decomposed under these reaction conditions, presumably due to the myriad pathways that are possible upon forming an allylic cation intermediate. Succcess was achieved, however, by utilizing Ph3SiOReO3 and conducting the initial phases of the reaction at −78 °C followed by a slow and controlled warming. This protocol resulted in the conversion of 25 to 26 in 71% yield. A minor amount (4%) of a diastereomer was isolated, but the overall yield of 26 was compromised when the reaction was run to complete equilibration.
Scheme 6.
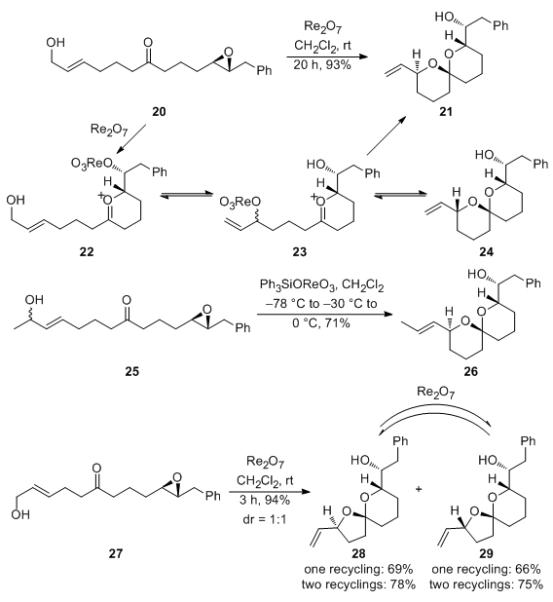
Ketones as stereochemical conduits.
Lower homologs are also suitable substrates for the process, though product stereocontrol is lost in these transformations due to the greater conformational freedom for tetrahydrofurans in comparison to tetrahydropyrans. Exposing 27 to Re2O7 provided a 1:1 mixture of spirocycles 28 and 29 in 94% overall yield after 3 h. While thermodynamic control fails to deliver a major product, either isomer is available in useful quantities through a simple recycling protocol. Thus, 28 can be obtained in 69% yield by resubjecting 29 to the reaction conditions and in 78% yield after two recyclings. Alternatively 29 can be isolated in 66% yield after subjecting 28 to one recycling and in 75% yield after two recyclings.
The use of ketones as stereochemical conduits can be merged with the inclusion of enones as terminal electrophiles to provide impressive leaps in molecular complexity through bidirectional stereogenesis. This is demonstrated (Scheme 7) by the conversion of 30 to 31. This atom economical transformation, in which a compound with one ring and two stereogenic centers is converted to a structure with three rings and five stereogenic centers, proceeded in 8 h at rt to form the product with complete stereocontrol in 84% yield.
Scheme 7.
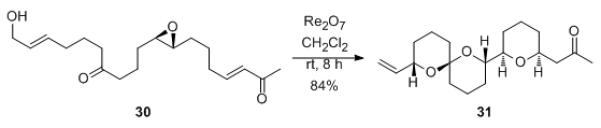
Bidirectional stereogenesis with a ketone conduit.
The difficulty associated with delivering small quantities of Re2O7 with precision led us to explore alternative reagent preparations. We discovered that a supported catalyst can be prepared through a convenient protocol in which a slurry of Re2O7 and silica gel in Et2O is stirred for several hours followed by drying under vacuum.[19] We prepared a 10% (w/w) mixture of Re2O7 on SiO2 through this protocol and showed that it is a competent catalyst for the transformations that we have previously developed. Yields and reaction times were similar when the immobilized and free catalysts were employed, with the conversion of 20 to 21 proceeding in 93% yield after 12 h and the conversion of 30 to 31 proceeding in 76% yield after 24 h. Notably, the immobilized catalyst could be used for the conversion of 16 to 17 in 60% yield, indicating that this easily accessed material could be an alternative to the much more expensive Ph3SiOReO3. The freely flowing immobilized catalyst is easily weighed, even in humid environments that cause Re2O7 to liquefy. Reactions with the immobilized catalyst do not require the induction periods that are often observed for the free catalyst because of superior dispersion. Filtering the catalyst from reactions provided a reagent that showed diminished activity, indicating that partial catalyst leaching occurs during the reaction. Thus this preparation primarily serves to facilitate the transformations from an operational perspective, particularly for reactions that utilize high molecular weight substrates.
We have demonstrated that epoxide cascade reactions can be initiated by rhenium oxide-mediated allylic alcohol transposition reactions. These transformations are possible because rhenium oxide acts in dual roles as a transposition catalyst and as an acid that enhances electrophilicity of the epoxide group and promotes product ionization, thereby allowing thermodynamically controlled stereochemical equilibration. Substrate syntheses are facilitated by relying on the functionalization of prochiral carbons rather than the generation of stereochemically defined nucleophiles for epoxide opening. Reactions proceed either by direct addition of the transposing alcohol into the activated epoxide or by using ketones as conduits that relay the stereochemical information from the epoxide to distal regions of the molecule. Maximal complexity increases are observed when an electrophile is present to trap the hydroxyl group that is liberated upon epoxide opening. Enones can be used in this capacity to provide atom economical transformations that exhibit bidirectional stereogenesis, in which the stereogenic centers in the epoxide guide the formation of stereocenters on opposite ends of the structure.
Supplementary Material
Acknowledgments
[**] This work was supported by grants from the National Institutes of Health (P50-GM06082) and the National Science Foundation (CHE-1151979). We thank Dr. Steve Geib for crystallographic analyses, and Dr. Damodaran Krishnan and Ms. Sage Bowser for assistance with NMR experiments.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- [1] a).Anderson EA. Org. Biomol. Chem. 2011;9:3997. doi: 10.1039/c1ob05212h. [DOI] [PubMed] [Google Scholar]; b) Grondal C, Jeanty M, Enders D. Nature Chem. 2010;2:167. doi: 10.1038/nchem.539. [DOI] [PubMed] [Google Scholar]; c) Nicolaou KC, Chen JS. Chem. Soc. Rev. 2009;38:2993. doi: 10.1039/b903290h. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Nicolaou KC, Edmonds DJ, Bulger PG. Angew. Chem. 2006;118:7292. doi: 10.1002/anie.200601872. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2006;45:7134. [Google Scholar]; e) Tietze LF. Chem. Rev. 1996;96:115. doi: 10.1021/cr950027e. [DOI] [PubMed] [Google Scholar]
- [2].Hendrickson JB. J. Am. Chem. Soc. 1975;97:5784. [Google Scholar]
- [3] a).Wender PA, Miller BL. Nature. 2009;460:197. doi: 10.1038/460197a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wender PA, Miller BL. In: Connectivity Analysis and Multibond-forming Processes in Organic Synthesis: Theory and Applications. Hudlicky T, editor. JAI Press; 1993. pp. 27–66. [Google Scholar]
- [4].For a review of rhenium oxide-mediated transformations, see Bellemin-Laponnaz S. ChemCatChem. 2009;1:357.; for examples of allylic alcohol transposition, see: Narasaka K, Kusama H, Hiyashi Y. Tetrahedron. 1992;48:2059.; Bellemin-Laponnaz S, Gisie H, LeNy JP, Osborn JA. Angew. Chem. 1997;109:1011.; Angew. Chem. Int. Ed. 1997;36:976.
- [5] a).Morrill C, Grubbs RH. J. Am. Chem. Soc. 2005;127:2842. doi: 10.1021/ja044054a. [DOI] [PubMed] [Google Scholar]; b) Morrill C, Beutner GL, Grubbs RH. J. Org. Chem. 2006;71:7813. doi: 10.1021/jo061436l. [DOI] [PubMed] [Google Scholar]; c) Hansen EC, Lee D. J. Am. Chem. Soc. 2006;128:8142. doi: 10.1021/ja0620639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6] a).Jung HH, Seiders JR, II, Floreancig PE. Angew. Chem. 2007;119:8616. doi: 10.1002/anie.200702999. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:8464. [Google Scholar]; b) Herrmann AT, Saito T, Stivala CE, Tom J, Zakarian A. J. Am. Chem. Soc. 2010;132:5962. doi: 10.1021/ja101673v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xie Y, Floreancig PE. Chem. Sci. 2011;2:2423. [Google Scholar]
- [7] a).Trost BM, Toste FD. J. Am. Chem. Soc. 2000;122:11262. [Google Scholar]; b) Hutchison JM, Lindsay HA, Dormi SS, Jones GD, Vivic DA, McIntosh MC. Org. Lett. 2006;8:3663. doi: 10.1021/ol061072j. [DOI] [PubMed] [Google Scholar]; c) Yun SY, Hansen EC, Volchkov I, Lo WY, Lee D. Angew. Chem. 2010;122:4357. doi: 10.1002/anie.201001681. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2010;49:4261. [Google Scholar]; d) Trost BM, Amans D, Seganish WM, Chung CK. Chem. Eur. J. 2012;18:2961. doi: 10.1002/chem.201102899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bellemin-Laponnaz S, LeNy JP, Dedieu A. Chem. Eur. J. 1999;5:57. [Google Scholar]
- [9].For recent examples, see: Simpson GL, Heffron TP, Merino E, Jamison TF. J. Am. Chem. Soc. 2006;128:1056. doi: 10.1021/ja057973p.; Marshall JA, Mikowski AM. Org. Lett. 2006;8:4375. doi: 10.1021/ol061826u.; Morimoto Y, Okita T, Takaishi M, Tanaka T. Angew. Chem. 2007;119:1150. doi: 10.1002/anie.200603806.; Angew. Chem. Int. Ed. 2007;46:1132.; Vilotijevic I, Jamison TF. Science. 2007;317:1189. doi: 10.1126/science.1146421.; Morimoto Y, Yata H, Nishikawa Y. Angew. Chem. 2007;119:6601. doi: 10.1002/anie.200701737.; Angew. Chem. Int. Ed. 2007;46:6481.; Marshall JA, Hann RK. J. Org. Chem. 2008;73:6753. doi: 10.1021/jo801188w.; Xiong Z, Busch R, Corey EJ. Org. Lett. 2010;12:1512. doi: 10.1021/ol100213e.; Morten CJ, Byers JA, Jamison TF. J. Am. Chem. Soc. 2011;133:1902. doi: 10.1021/ja1088748.
- [10] a).Hayashi N, Fujiwara K, Murai A. Tetrahedron. 1997;53:12425. [Google Scholar]; b) Zakarian A, Batch A, Holton RA. J. Am. Chem. Soc. 2003;125:7822. doi: 10.1021/ja029225v. [DOI] [PubMed] [Google Scholar]; c) Valentine JC, McDonald FE, Neiwert WA, Hardcastle KI. J. Am. Chem. Soc. 2005;127:4586. doi: 10.1021/ja050013i. [DOI] [PubMed] [Google Scholar]; d) Tong R, Valentine JC, McDonald FE, Cao R, Fang X, Hardcastle KI. J. Am. Chem. Soc. 2007;129:1050. doi: 10.1021/ja068826+. [DOI] [PubMed] [Google Scholar]; e) Wan S, Gunaydin H, Houk KN, Floreancig PE. J. Am. Chem. Soc. 2007;129:7915. doi: 10.1021/ja0709674. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Tanuwidjala J, Ng S-S, Jamison TF. J. Am. Chem. Soc. 2009;131:12084. doi: 10.1021/ja9052366. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Clausen DJ, Wan S, Floreancig PE. Angew. Chem. 2011;123:5284. doi: 10.1002/anie.201007757. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2011;50:5178. [Google Scholar]
- [11].For representative examples, see: Katsuki T, Sharpless KB. J. Am. Chem. Soc. 1980;102:5974.; Wang Z-X, Tu Y, Frohn M, Zhang J-R, Shi Y. J. Am. Chem. Soc. 1997;119:11224.; Schaus SE, Brandes BD, Larrow JF, Tokunga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J. Am. Chem. Soc. 2002;124:1307. doi: 10.1021/ja016737l.; Kakei H, Tsuji R, Ohshima T, Shibasaki M. J. Am. Chem. Soc. 2005;127:8962. doi: 10.1021/ja052466t.; Wang X, List B. Angew. Chem. 2008;120:1135.; Angew. Chem. Int. Ed. 2008;47:1119.; Li Z, Yamamoto H. J. Am. Chem. Soc. 2010;132:7878. doi: 10.1021/ja100951u.
- [12].For reviews, see: Vilotijevic I, Jamison TF. Angew. Chem. 2009;121:5352. doi: 10.1002/anie.200900600.; Angew. Chem. Int. Ed. 2009;48:5250.; Vilotijevic I, Jamison TF. Marine Drugs. 2010;8:763. doi: 10.3390/md8030763.
- [13].Please see the Supporting Information. CCDC 903395, 903396, and 903397 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- [14] a).Dale JA, Dull DL, Mosher HS. J. Org. Chem. 1969;34:2543. [Google Scholar]; b) Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J. Am. Chem. Soc. 1991;113:4092. [Google Scholar]
- [15].Tadpetch K, Rychnovsky SD. Org. Lett. 2008;10:4839. doi: 10.1021/ol8019204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schoop T, Roesky HW, Noltemeyer M, Schmidt H-G. Organometallics. 1993;12:571. [Google Scholar]
- [17].Trost BM. Angew. Chem. 1995;107:285. [Google Scholar]; Angew. Chem., Int. Ed. 1995;34:259. [Google Scholar]
- [18] a).Mulholland RL, Jr., Chamberlin AR. J. Org. Chem. 1988;53:1082. [Google Scholar]; b) Fotsch CH, Chamberlin AR. J. Org. Chem. 1991;56:4141. [Google Scholar]; c) Rychnovsky SD, Dahanukar VH. Tetrahedron Lett. 1996;37:339. [Google Scholar]
- [19].For a more rigorous preparation of Re2O7•SiO2, see: Scott SL, Basset J-M. J. Am. Chem. Soc. 1994;116:12069.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.