

Abstract
Aims:
Excess salt intake during pregnancy may alter fetal organ structures and functions leading to increased risks in the development of cardiovascular diseases in later life. The present study determined whether and how the prenatal high-salt (HS) diets affect renin–angiotensin system (RAS) that may mediate cardiac cell death.
Methods and Results:
Angiotensin II receptors, AT1 and AT2, protein expression was increased in the myocardium of the offspring exposed to prenatal HS; apoptotic cells appeared in the myocardium of the adult offspring. Mitochondrion was isolated in cell experiments, and the data showed cardiomyocyte apoptosis requiring cytochrome C release. Pretreating H9C2 cells with AT2 agonist CGP42112A induced cell apoptosis in DNA fragments and activated caspase 3. CGP42112A increased mitochondrion cytochrome C release and apoptosis in the cells.
Conclusion:
Both in vitro and in vivo study demonstrated that cardiomyocyte apoptosis was related to AT2 activation. Prenatal HS diets may reprogram RAS that mediates apoptosis in the offspring myocardium, and AT2 may contribute to cardiomyocyte apoptosis via the cytochrome C release pathway.
Keywords: angiotensin receptors, high salt, apoptosis, offspring heart
Introduction
Both genetic and environmental factors can be involved in the development of cardiovascular diseases. Epidemiological studies showed that cardiovascular diseases could be programed initially in utero.1–4Reprogramming during fetal life occurs in response to adverse fetal environments and results in permanent adaption that may alter organ growth, structure remodeling, and dysfunction, leading to an increased risk in the development of cardiovascular diseases in the offspring.5–8 Previous studies demonstrated that many pregnant women spontaneously prefer salt diets.9 Exposure to excess salt during early developmental periods may cause long-term physiological changes in later life in rats.10 Following in utero exposure to high salt (HS), the offspring showed a suppressed renin–angiotensin system (RAS), indicating HS diet during pregnancy may reprogram RAS.11,12 These may produce offspring with maladaptive phenotypes in a predicted adult life.
Local cardiac RAS plays roles in the maintenance of an appropriate cellular milieu, balancing signaling between inducing and inhibiting cell proliferation as well as mediating adaptive responses to myocardial stress.13 Two major subtypes of angiotensin II receptors (AT1 and AT2) are G-protein-coupled receptors with structural features of 7 transmembrane domains.14 The AT1 and AT2 share limited sequence homology (∼34% amino acid sequence identity).15 The pathophysiological roles and signaling mechanisms of AT2 are less clear.16 The AT2 appears as the dominating subtype in fetuses but decreases rapidly after birth.17 In adults, normal expression of AT2 is largely restricted to certain cell types and certain tissues. An increase in the AT2 expression was observed under pathological conditions, such as myocardial infarction and congestive heart failure.18 The AT2 was found to mediate caspase 3-dependent apoptosis.19 The 3 major transduction mechanisms responsible for AT2 signaling are activation of various protein phosphatases for protein dephosphorylation; activation of the nitric oxide/cyclic guanosine monophosphate system; and stimulation of phospholipase A2.20 Overexpression of AT2 in certain cell lines leads to apoptosis.21
Apoptosis plays a key role in the pathogenesis of cardiac diseases. Cardiomyocyte apoptosis is a rare event in healthy myocardium.22 However, hypertrophied, infarcted, senescent, and failing hearts contain apoptotic cardiomyocytes.23–26 Cardiac apoptosis via AT2 may be a maladaptive response in the stimulation of cellular proliferation and hypertrophy and in controlling cardiac cell populations.27 Limited information is available on prenatally programed pathogenesis of cardiovascular conditions via RAS. Our previous study demonstrated that prenatal high-salt diets induced abnormal heart development in 21-day-old fetus.28 The present study investigated whether prenatal HS diets reprogram offspring’s RAS and whether and how reprogramed local RAS is involved in programed cardiac cell death in the offspring.
Methods
Animals
Female Sprague-Dawley rats (12 weeks old) were obtained from Soochow University Experimental Animal Center. Each rat was caged with a single male rat of verified fertility (history of fertile mating) until successful copulation. Detection of sperm in vaginal smears collected between 9:00 and 11:00 hours was performed in the following morning. Rats were housed in a temperature-controlled 12:12-hour light–dark environment, with food and water ad libitum. The dams were randomized to receive a diet with modified salt content: 0.6% NaCl (normal salt [NS], n = 8) or 8.0% NaCl (HS, n = 8). All diets were based on a standard rodent diet containing 19.3% protein, 39.1% carbohydrates, 3.3% fat, 1.00% calcium, 0.70% phosphate, and 0.68% potassium (Slaccas, Shanghai, China). The salt diet was administered from gestational day 4 until born.29 Male offspring at 2 days (newborn), 1 month, and 5 months after birth were tested.
Rats were anesthetized intraperitoneally with sodium pentobarbital (50-100 mg/kg; Hengrui Medicine, Jiangsu, China). After blood samples were collected from the abdominal aorta into chilled tubes containing 5 μL of heparin (2000 U/mL), the heart was excised and prepared for histological and molecular studies. All procedures and protocols were approved by the Institutional Animal Care and Use Committee and followed the Guidelines for the Care and Use of Laboratory Animals published by US National Institutes of Health (publication no. 85-23).
Immunohistochemistry
Heart tissue was fixed in 10% formalin at room temperature for 8 hours. Immunohistochemical staining using paraffin sections was performed by Soochow University Pathology Laboratory, using polyclonal antibodies raised from rabbit against AT1 and AT2 (Santa Cruz Biotech, Santa Cruz, California) and secondary antibodies (Maixin, Shenzhen, China). 3,3-Diaminobenzidine was used to produce a brown precipitate in immunohistochemical staining. The sections were observed using an image analysis system. Cardiocyte apoptosis was tested using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) detection kit for fragmented DNA in the nucleus (Takara, Kyoto, Japan). Fluorescein-labeled nucleotides in TUNEL assay kit could bind with the DNA 3′-OH ends using recombinant terminal deoxynucleotidyl transferase. The fluorescence was observed with a fluorescence microscope. All analyses were performed in a blind manner. All fluorescence signals were quantified using a UVP Bioimaging system EC3 apparatus (UVP, California).
Measurement of Plasma Renin Activity, AT1, AT2, and Angiotensin Converting Enzyme
Blood samples in tubes with heparin lithium were centrifuged (800g at 4°C for 10 minutes), and then the plasma was collected. Plasma samples were used for measuring the renin activity, AT1, AT2, and angiotensin converting enzyme (ACE) levels by Sino-UK Institute of Biological Technology28 using radioimmunoassay methods.
Mitochondrion and Cytosolic Protein Isolation
Mitochondrion was separated from cytoplasm in the myocardium from either offspring’s heart tissue or H9C2 cells (H9C2 cell line is a subclone of the original clonal cell line derived from the embryonic rat heart) using a mitochondria-extraction kit according to the manufacturer’s instructions (Solarbio, Beijing, China).
Western Blotting
Mitochondrion and cytosolic protein from both offspring’s heart tissue and H9C2 cells were prepared as described previously. Homogenate of heart tissue was processed for Western blotting as reported previously.28 Samples were randomly chosen 8 animals (1 per litter) each group. The protein concentration (1.5-2.5 mg/mL) was determined using the Bradford and standard bovine serum albumin (DC Protein Assay; Bio-Rad, California). Equal amounts of total protein (30 μg) were loaded on polyacylamide gels and then transferred to nitrocellulose membranes and blocked with 5% dry milk in Tris-buffered saline/Tween 20. The primary antibodies were diluted (AT1, 1:500; AT2, 1:1000; Bcl-2-associated X [Bax], 1:500; cyclooxygenase 4 [Cox-4], 1:500; cytochrome C, 1:300; activated-caspase 3, 1:500; Santa Cruz Biotech). Blots were then probed with a horseradish peroxidase-coupled goat anti-rabbit secondary antibody (1:5000), and β-actin antibody (1:5000) was probed with rabbit anti-mouse secondary antibody. Blots were developed using enhanced chemiluminescence Western blotting detection reagents (Thermo, Illinois), and specific bands were quantified using a UVP Bioimaging system EC3 apparatus (UVP).
Cells and Experimental Treatments
Cell line H9C2, obtained from Shanghai Institutes for Biological Sciences, was cultured in Dulbecco modified Eagle medium (Invitrogen, California) with 15% fetal bovine serum (Hyclone, Utah) and 1% penicillin/streptomycin. The cells were grown in 5% CO2 at 37°C in a humidified atmosphere culture incubator. The AT2 (Sigma, MO) or AT2 agonist CGP42211A (Sigma) was added to the medium at 0, 0.1, 1, and 10 μmol/L, pretreated with either AT2 antagonist PD123319 (10 μmol/L) or AT1 antagonist (losartan, 10 μmol/L; Sigma), and then apoptosis was examined.
Flow Cytometry
H9C2 was added to AT II (0, 0.1, 1, or 10 μmol/L) or CGP42112A (0, 0.1, 1, or 10 μmol/L) combined with pretreatment of either AT1 receptor antagonist losartan (10 μmol/L ) or AT2 receptor antagonist PD123319 (10 μmol/L). Apoptosis was determined using annexin V conjugates for apoptosis detection kit (Invitrogen) with flow cytometry. After treatment with the probes from the kit, apoptotic cells show green fluorescence, dead cells show red and green fluorescence, and live cells show little or no fluorescence. Apoptotic cells were analyzed using a Beckman FC500 machine. All experiments were repeated for at least 4 times.
DNA Extraction and Gel Electrophoresis
Myocardium (from 5-month-old male offspring) and H9C2 cells were washed twice with phosphate-buffered saline and lysed in lysis buffer containing 10 nmol/L Tris-HCl (pH 7.5), 1 mmol/L EDTA, and 0.5% Triton X-100. After lysing, the debris was removed by centrifugation at 12 000g for 20 minutes. Ribonuclease A (RNase A; Sigma) was added at a final concentration of 40 μg/mL to the lysates, which were incubated for 1 hour at 37°C with gentle agitation. Proteinase K (Sigma) was added to the RNase A-treated lysates (40 μg/mL). The lysates were further incubated for 1 hour at 37°C with gentle agitation. DNA in the supernatant was precipitated and dissolved in Tris-EDTA (TE) buffer (10 nmol/L Tris, 1 mmol/L EDTA, pH 8.0). The extracted DNA and molecular marker (100 bp DNA ladder; Takara) were subjected to 2.0% agarose gel (containing 0.5 μg/mL ethidium bromide) electrophoresis with the electrophoretic apparatus (Bio-Rad). DNA ladder formation was visualized under a UV transilluminator (UVP).
Statistical Analysis
Data were expressed as mean ± standard deviation or median. Student t test and 1-way analysis of variance were used. For Western blots, the NS group served as reference, and the mean value of individual measurements was set as 100%. The results were considered significant when probability of error (P) was <.05.
Results
There was no difference in litter size (NS: 11.5 ± 1.2 vs HS: 10.9 ± 1.2, P = .32, NS: n = 8; HS: n = 8) and body weight in the newborn (male pup birth weight [NS: 6.47 ± 0.14 g vs HS: 6.62 ± 0.18 g, P = .09, NS: n = 42; HS: n = 39]; female pup birth weight [NS: 6.26 ± 0.08 g vs HS: 6.33 ± 0.07 g, P = .13, NS: n = 46; HS: n = 41]). There was also no difference in the ratio for female-to-male offspring (NS: 0.92 ± 0.12 vs HS: 0.9 ± 0.05, P = .59, NS: n = 8; HS: n = 8).
Heart Weight
Heart weight (5-month-old offspring) in the HS group was not statistically different from that of the NS (NS: 303 ± 22 mg vs HS: 330 ± 26mg, P > .05). Body weight in HS offspring was also not statistically different from that of the NS (NS: 336 ± 20 g vs HS: 316 ± 23 g, P > .05). However, the ratio of heart/body weight of the HS offspring was significantly higher than that of the NS (NS: 0.89 ± 0.08 vs HS: 1.05 ± 0.1, P = .002; Table 1).
Table 1.
Body and Heart Weight and the Ratio.
| Group | n | BW, g | HW, mg | HW/BW, mg/g |
|---|---|---|---|---|
| NS | 32 | 336 ± 20 | 302 ± 22 | 0.899 ± 0.08 |
| HS | 34 | 314 ± 23 | 330 ± 26 | 1.051 ± 0.1a |
| t test | P > .05 | P > .05 | P = .002 |
Abbreviations: BW, body weight; HW, heart weight; NS, normal salt; HS, high salt.
a P < .01 versus NS.
Plasma Renin, Angiotensin Enzyme, and Peptides
Plasma angiotensin II and ACE levels were significantly reduced in the newborn HS group compared with the NS. Although plasma 2angiotensin II and ACE seemed lower in the 1-month-old and 5-month-old HS offsprings than that of the NS, there was no statistical difference. For the newborn, 1-month-old, and 5-month-old HS offsprings, there was no significant difference in plasma AT I and plasma renin activity (PRA) levels between the groups (Table 2).
Table 2.
Plasma AT I, AT II, ACE, and PRA in the New Born, 1-Month-Old, and 5-Month-Old Offspring.
| Serum | New Born 2 Days | 1-Month-Old Male | 5-Month-Old Male | |||
|---|---|---|---|---|---|---|
| NS n = 8 | HS n = 8 | NS n = 8 | HS n = 8 | NS n = 8 | HS n = 8 | |
| AT I, ng/mL | 1.93 ± 0.05 | 1.88 ± 0.07 | 1.95 ± 0.13 | 1.84 ± 0.186 | 1.79 ± 0.07 | 1.67 ± 0.20 |
| AT II, pg/mL | 107.44 ± 3.01 | 95.32 ± 3.06a | 116.22 ± 5.55 | 111.26 ± 2.89 | 114.90 ± 5.34 | 107.56 ± 7.45 |
| ACE (μm/mL) | 56.17 ± 2.66 | 32.76 ± 5.12a | 52.34 ± 7.51 | 43.96 ± 4.16 | 49.29 ± 6.01 | 42.07 ± 4.91 |
| PRA, ng/mL | 2.15 ± 0.40 | 2.49 ± 0.41 | 2.50 ± 0.22 | 2.32 ± 0.11 | 2.51 ± 0.09 | 2.02 ± 0.14 |
Abbreviations: NS, normal salt; HS, high salt; PRA, plasma renin activity; ACE, angiotensin converting enzyme; AT, angiotensin receptor.
a P < .05 versus NS.
Expression of AT1 and AT2
Positive immunostaining was observed for both AT1 and AT2 in the myocardium of the newborn, 1-month-old, and 5-month-old HS offsprings (Figure 1). Western blot analysis showed that AT1 was significantly increased in the newborn, 1-month-old, and 5-month-old offsprings exposed to HS. The expression of AT2 was significantly increased in the 5-month-old HS offspring (Figure 2).
Figure 1.

Immunostaining of AT1 and AT2 in the myocardium of the newborn, 1-month-old, and 5-month-old offspring. Positive immunostaining was observed for both AT1 and AT2 in the myocardium of the newborn, 1-month-old, and 5-month-old high-salt (HS) offspring. Positive brown immunostaining was marked with arrows. AT indicates angiotensin receptor.
Figure 2.

Western blot analysis showed that AT1 was significantly increased in the newborn, 1-month-old, and 5-month-old offspring exposed to HS. The expression of AT2 significantly increased only in the 5-month-old HS offspring. **, P < .01 versus NS (NS: n = 8; HS: n = 8). AT indicates angiotensin receptor; HS, high salt; NS, normal salt.
Apoptosis in the Myocardium
The proportion of positive TUNEL staining was significantly higher in the myocardium of the 5-month-old HS offspring than that of the NS (Figure 3). Typical apoptotic DNA laddering was visualized in agarose gel with ethidium bromide staining for DNA extraction from the myocardium of the 5-month-old HS offspring (Figure 4).
Figure 3.
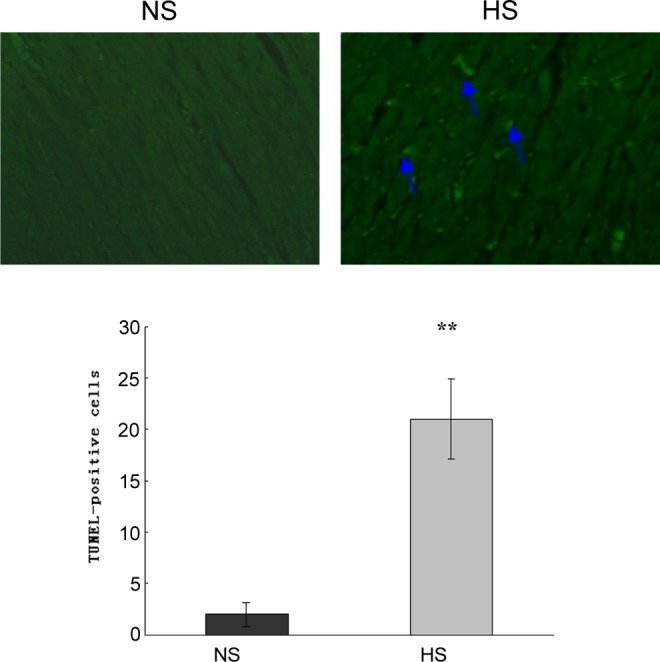
TUNEL assay showed apoptotic cells in the myocardium of the 5-month-old HS offspring. TUNEL-positive nuclei (blue arrows) are marked with green fluorescence. The proportion of positive TUNEL staining was significantly higher in the myocardium of 5-month-old HS offspring compared with the NS. **, P < .01 versus NS (NS: n = 8; HS: n = 8). HS indicates high salt; NS, normal salt; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Figure 4.
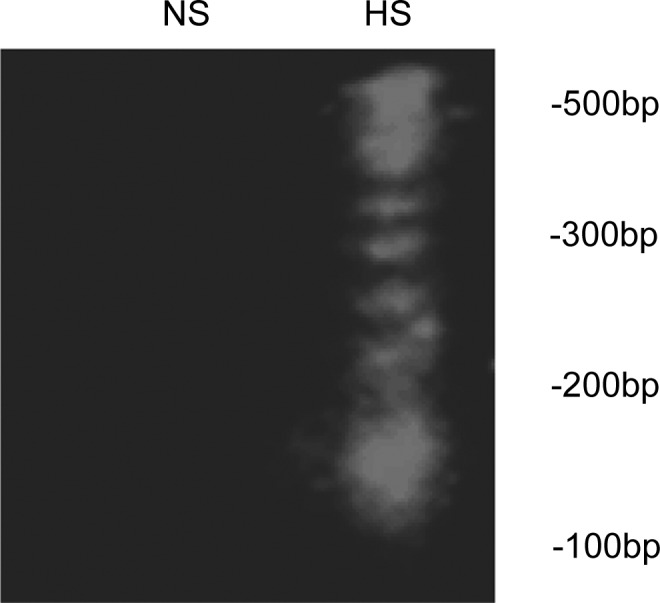
Gel electrophoresis of DNA fragments in the myocardium of the 5-month-old offspring. Apoptotic DNA laddering was visualized in an agarose gel by ethidium bromide staining. DNA was extracted from the 5-month-old myocardium. Right lane: high-salt (HS) 5-month-old myocardium. Left lane: normal-salt (NS) 5-month-old myocardium.
Cytochrome C Pathway-Mediated Myocardium Apoptosis
Mitochondrion was separated from cytoplasm of the myocardium. The Cox-4 was shown as a marker of mitochondrion in the separated mitochondrion samples, and not in the separated cytoplasm. The Bax was significantly increased in the mitochondrion, while cytochrome C was significantly increased in the cytoplasm and decreased in the mitochondrion. Activated caspase 3 was also significantly increased in the cytoplasm from the myocardium of the 5-month-old HS offspring (Figure 5).
Figure 5.

The expression of cytochrome C and activated caspase 3, Bax, and Cox-4 in the mitochondrion and cytoplasm of the 5-month-old offspring’s myocardium. The Cox-4: was a marker for mitochondrion. The Bax was significantly accumulated in the mitochondrion of the 5-month-old HS offspring, and cytochrome C was significantly increased in the mitochondrion and significantly decreased in the cytoplasm. Activated caspase 3 was significantly increased in the cytoplasm. **, P < .01 versus NS; *, P < .05 versus NS (NS: n = 8; HS: n = 8). BAX indicates Bcl-2-associated X; COX-4, cyclooxygenase 4; HS, high salt; NS, normal salt.
AT2 Receptor-Mediated Apoptosis
In flow cytometry assay, apoptosis rate in H9C2 was at the highest level when the cells were treated with 10 μmol/L angiotensin II or 1 μmol/L CGP42112A. PD123319 (10 μmol/L), not losartan (10 μmol/L), inhibited AT2- or CGP42112A-induced apoptosis (Figure 6). In DNA ladder analysis, following pretreatment of CGP42112A (1 or 10 μmol/L), characteristic low-molecular-weight DNA fragments were shown in H9C2 cells. On the other hand, pretreatment of H9C2 with PD123319 decreased CGP42112A-induced DNA fragments (Figure 7). Analysis showed that CGP42112A (1 or 10 μmol/L) significantly induced caspase 3 activation, which was blocked by PD123319. CGP42112A (1 or 10 μmol/L) significantly reduced cytochrome C in H9C2 mitochondrion and significantly increased cytoplasmic cytochrome C in H9C2 (Figure 8).
Figure 6.

Apoptotic cells were measured using flow cytometry. A, H9C2 treated with angiotensin (AT) II at different concentrations, and pretreated with AT1 antagonist losartan or AT2 antagonist PD123319. *, P < .05 versus AT II 0 μmol/L, •, P < .05 versus AT II 0.1 μmol/L; ▴, P < .05 versus AT II 1 μmol/L; ♦, P < .05 versus AT II 10 μmol/L. B, H9C2 treated with CGP42112A at different concentrations and pretreated with losartan or PD123319. Los: losartan; PD: PD123319; CGP: CGP42112A, AT2 agonist; *, P < .05 versus CGP 0 μmol/L; •, P < .05 versus CGP 0.1 μmol/L; ▴, P < .05 versus CGP 1 μmol/L; ♦, P < .05 versus CGP 10 μmol/L. AT indicates angiotensin receptor.
Figure 7.
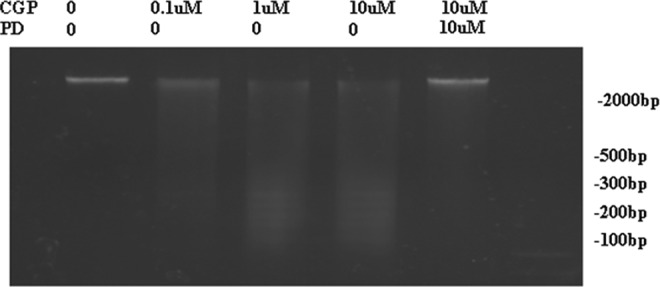
Gel electrophoresis for DNA fragment detection in H9C2 cells. CGP42112A (1 and 10 μmol/L) induced characteristic low-molecular-weight DNA fragments in H9C2 cells, and pretreatment of H9C2 with PD123319 inhibited CGP42112A-induced DNA fragments. PD: PD123319; CGP: CGP42112A, AT2 agonist. AT indicates angiotensin receptor.
Figure 8.

The expression of AT2, activated-caspase 3, and cytochrome C in H9C2 mitochondrion and cytoplasm after treating with CGP42112A or pretreating with PD123319. The 1 or 10 μmol/L CGP42112A significantly increased AT2 expression in H9C2 cells, mitochondrion cytochrome C was significantly reduced, cytoplasm activated caspase 3, and cytochrome C was significantly increased compared to that of 0 μmol/L CGP42112A. These results could be reversed by pretreatment with 10 μmol/L PD123319. PD: PD123319, AT2 antagonist; CGP: CGP42112A, AT2 agonist. **P < .01 versus NS, *P < .05 versus NS, t test. AT indicates angiotensin receptor; NS, normal salt.
Discussion
In the present study, the tested subjects included newborn, adolescents (1-month-old), and adult (5-month-old) male offspring rats, in the determination of the influence of HS intake during pregnancy on the heart of the offspring. Prenatal HS increased myocardium apoptosis in the 5-month-old offspring, and the induction of myocardium apoptosis was related to the elevated expression of AT2 receptors in the heart. An important hypothesis for a relationship between poor fetal development and cardiovascular diseases in adulthood was proposed 20 years ago.30,31 Subsequent investigations provided evidence that underdeveloped babies at birth were associated with an increased risk of heart diseases and hypertension,6,20,32,33 indicating that the prenatal environments could cause permanent changes in the body and increase risks of diseases later in life. Previous studies reported that protein restriction during pregnancy suppressed the intrarenal RAS in the newborn rats and leads to a reduced number of glomeruli, glomerular enlargement, and hypertension in the adults.34–36 Our recent study has demonstrated that fetal heart weight at birth was increased following maternal HS intake during pregnency.28 In the present study, the prenatal treatment with HS diets was the same as that reported28; heart weights of the HS offspring at 5 months was not statically different from that of the NS. Notably, in the fetus exposed to the exactly same HS intake during pregnancy, their heart weight was significantly increased.28 The difference in heart weight between the fetus and the 5-month-old offspring suggests that the affected heart weight by HS diets could be reversed somewhat after birth when feeding returned to normal situation. However, such a kind reverse was limited, since the ratio of heart weight to body weight was still significantly higher in the HS offspring in the present study, indicating a possible chronic influence from prenatal HS intake on cardiac structures.
In pathological increase in heart weight, cellular fibrillation is a common mechanism, and apoptosis is closely linked to the pathological changes.28 In light of this, we determined apoptosis in the cardiac tissue of the offspring heart and found increased positive TUNEL staining in the heart tissue, providing new histological evidence that apoptosis occurred in the heart of the male offspring exposed to prenatal HS. The subsequent experiments added further information with characteristic low-molecular-weight DNA fragments in the myocardium of the 5-month-old HS offspring. Both TUNEL staining and DNA fragments are positive indicators commonly used in the determination of apoptosis.37,38 Together, these data demonstrated that apoptosis occurred in the myocardium of the HS offspring associated with an increased ratio of heart/body weight.
Based on the above findings, 2 questions were thought important. Why and by what pathways HS intake may induce cellular apoptosis in the heart? What happened inside the apoptosis cells? It is well known that HS or high sodium in the body would affect cardiovascular systems via RAS pathways.39,40 Thus, we measured key components of the systemic RAS in the offspring exposed to prenatal HS. The data showed that plasma angiotensin II and ACE levels were reduced only in the newborn exposed to prenatal HS; however, this reduction was back to normal in the 1-month old or 5-month old. Notably, in our previous report, fetal plasma AT II levels were also decreased in the maternal HS-diet group.28 There was no significant difference in AT I and PRA levels between the HS and the NS in the newborn and older offspring. This indicates that cardiac apoptosis and the altered ratio of heart/body weight in the adult offspring were not primarily via systemic RAS pathways.
Local RAS was expressed in the heart during in utero development,41,42 playing a role in growth and differentiation in the heart.43 Han et al reported that unbalanced maternal diets influenced the fetal cardiac RAS.44 It is rational that the heart could exhibit abnormalities if RAS was affected during fetal periods. Our previous fetal study has demonstrated an altered expression of the angiotensin II receptors in the fetal heart exposed to the same HS diets.28 In the present study, protein analysis in the HS offspring showed an increased AT1 in the newborn heart that persisted into adulthood. It is interesting to note that AT2 expression in Western blot was not significantly altered between the HS and the NS offspring at new born and 1-month old. However, local cardiac AT2 presented a significant higher expression in the HS offspring at 5-month old. This may be related to the long-term changes in AT1 in the same tissue. Additional postnatal chronic influence from the abnormal expression of AT1 in the same tissue, AT2 expression could be eventually changed in the 5-month-old HS offspring. This possibility offers a new and interesting clue for future investigation.
The AT II contributes to apoptosis in vitro and in vivo.45–47 The angiotensin II-induced apoptosis in primary and transformed epithelial cells and coronary artery endothelial cells were mediated by AT1 receptors.48,49 By contrast, AT2 receptor activation contributed to apoptosis in fibroblasts, smooth muscle cells, human umbilical vein endothelial cells, primary lung endothelial cells, and PC12W cells (rat pheochromocytoma cell line).19,50–52 The angiotensin II-induced signaling has been demonstrated to be cell type dependent, and the signaling specificity is related to the biological responses.19 In the present study, positive TUNEL staining was observed in the 5-month-old HS offspring in association with an alteration in the AT1 and AT2 expression in the heart, demonstrating that the local RAS or its receptors in the heart, not the systemic RAS in circulation, may be responsible for apoptosis in the cardiac tissue of the HS offspring.
H9C2 is a subclone of the original clonal cell line derived from embryonic rat heart tissue. In the determination of whether AT1 and AT2 pathways are involved in cellular apoptosis in the heart, our in vitro study showed that angiotensin II induced cardiocyte apoptosis in H9C2 cells, indicating angiotensin II may cause cardiac apoptosis either via AT1, or AT2, or both. The subsequent experiments using AT2 agonist CGP42112A demonstrated a similar pattern of cellular apoptosis, providing a new evidence of the role of AT2 in cardiac apoptosis. In addition, CGP42112A-mediated apoptosis in the cells was dose dependent (from 0.1 to 1 μmol/L) in increasing activated caspase 3 and DNA fragment in the present study. In flow cytometry analysis, PD123319 (AT2 antagonist) not losartan (AT1 antagonist) inhibited AT II- or CGP42112A-induced apoptosis. At the same time, pretreatment of the cells with PD123319, not losartan, inhibited CGP42112A-induced DNA fragments. Further experiments in the present study demonstrated that CGP42112A-induced caspase 3 activation could be blocked by PD123319. Together, the data provide new information that AT2 may play a key role in apoptosis in cardiac cells. Notably, although the cells used in our in vitro studies were also cardiac cells, the experimental conditions were different from the cardiac cells in the heart tissue of the HS offspring. Both AT1 and AT2 expression were increased in the HS offspring’s heart in association with cardiac apoptosis, while only AT2 pathway was shown to be mediated with cellular death in vitro. This can be considered a major contribution of AT2 pathway; however, an involvement of AT1 in such case should not be simply excluded in the HS offspring.
To determine pathways inside the cardiac cells during apoptosis, mitochondrion was separated from cytoplasm of the myocardium in the present study. The Cox-4, as a marker of mitochondrion, was shown in the separated mitochondrion samples and not in the separated cytoplasm. Then, we determined possible apoptosis pathway involved inside the cells. The data showed that activated caspase 3 and cytochrome C were increased, and Bax decreased in the cardiocyte cytoplasm. Previous studies showed that cytochrome C plays a critical role in apoptosis.53 On the other hand, Bax was increased in the mitochondrion, indicating Bax translocated to mitochondrion. The Bax proteins regulate apoptosis, in part, by controlling the formation of the mitochondrion apoptosis-induced channel (MAC), which is a cytochrome C release channel induced in an intrinsic apoptotic pathway. The MAC appeared when Bax translocated to the mitochondrion, and cytochrome C was released in dying cells due to apoptosis.54 Previous studies have also demonstrated that caspase 3 activation in response to cytochrome C release from mitochondrion led to apoptosis.55 The present study demonstrated that cardiocyte apoptosis required cytochrome C release from the mitochondrion, providing new information on molecular pathways inside the apoptosis cells in the HS offspring’s heart.
Finally, the present study also showed that CGP42112A reduced cytochrome C in H9C2 mitochondrion and increased cytochrome C in cytoplasm, suggesting that AT2 may mediate cardiocyte apoptosis via the pathway in release of cytochrome C.
In conclusion, the present study demonstrated that apoptosis in the myocardium of the offspring exposed to prenatal HS intake, indicating an increase in the risk of development of cardiac diseases. The data showed that altered expression of the angiotensin II receptors in the local RAS in the heart may play a critical role in cell death and provide new evidence that the pathway is important in RAS-mediated cell apoptosis in release of cytochrome C in the heart of the male offspring exposed to prenatal HS, adding new information for further understanding the long-term influence of prenatal conditions on cardiovascular health and diseases in fetal origins.
Footnotes
Authors’ Note: Juanxiu lv and Peiwen Zhang contributed equally to this work.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported by 2013BAI04B05 and National Natural Science Foundation (81100431, 81100448, 81030006, 0973211, and 81070540) as well as Jiangsu Province’s Key Discipline/Laboratory of Medicine and “ChuangXin TuanDui” grants.
References
- 1. Huxley RR, Shiell AW, Law CM. The role of size at birth and postnatal catch-up growth in determining systolic blood pressure: a systematic review of the literature. J Hypertens. 2000;18(7):815–831. [DOI] [PubMed] [Google Scholar]
- 2. Eriksson J, Forsen T, Tuomilehto J, Osmond C, Barker D. Fetal and childhood growth and hypertension in adult life. Hypertension. 2000;36(5):790–794. [DOI] [PubMed] [Google Scholar]
- 3. Bertram C, Trowern AR, Copin N, Jackson AA, Whorwood CB. The maternal diet during pregnancy programs altered expression of the glucocorticoid receptor and type 2 11β-hydroxysteroid dehydrogenase: potential molecular mechanisms underlying the programming of hypertension in utero. Endocrinology. 2001;142(7):2841–2853. [DOI] [PubMed] [Google Scholar]
- 4. Boguszewski MC, Johannsson G, Fortes LC, Sverrisdottir YB. Low birth size and final height predict high sympathetic nerve activity in adulthood. J Hypertens. 2004;22(6):1157–1163. [DOI] [PubMed] [Google Scholar]
- 5. Bauer R, Walter B, Bauer K, Klupsch R, Patt S, Zwiener U. Intrauterine growth restriction reduces nephron number and renal excretory function in newborn piglets. Acta Physiol Scand. 2002;176(2):83–90. [DOI] [PubMed] [Google Scholar]
- 6. Manning J, Vehaskari M. Postnatal modulation of prenatally programmed hypertension by dietary Na and ACE inhibition. Am J Physiol Regul Integr Comp Physiol. 2005;288(1):R80–R84. [DOI] [PubMed] [Google Scholar]
- 7. Leeson CP, Kattenhorn M, Morley R, Lucas A, Dean field JE. Impact of low birth weight and cardiovascular risk factors on endothelial function in early adult life. Circulation. 2001;103(9):1264–1268. [DOI] [PubMed] [Google Scholar]
- 8. Law CM, Shiell AW, Newsome CA, Syddall HE, Shinebourne EA. Is blood pressure inversely related to birth weight? The strength of evidence from systematic review of the literature. J Hypertens. 1996;14(8):935–941. [PubMed] [Google Scholar]
- 9. Morris MJ, Na ES, Johnson AK. Salt craving: the psychobiology of pathogenic sodium intake. Physiol Behav. 2008;94(5):709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McBride SM, Culver B, Flynn FW. Dietary sodium manipulation during critical periods in development sensitize adult offspring to amphetamines. Am J Physiol Regul Integr Comp Physiol. 2008;295(3):899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bayorh MA, Ganafa AA, Emmett N, Socci RR, Eatman D, Fridie IL. Alterations in aldosterone and angiotensin II levels in salt-induced hypertension. Clin Exp Hypertens. 2005;27(4):355–367. [PubMed] [Google Scholar]
- 12. Ramos DR, Costa NL, Jang KL, et al. Maternal high-sodium intake alters the responsiveness of the renin–angiotensin system in adult offspring. Life Sci. 2012;90(19-20):785–792. [DOI] [PubMed] [Google Scholar]
- 13. De Mello WC, Danser AHJ. Angiotensin II and the heart. On the intracrine renin angiotensin system. Hypertension. 2000;35(6):1183–1188. [DOI] [PubMed] [Google Scholar]
- 14. Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415(6868):206–212. [DOI] [PubMed] [Google Scholar]
- 15. Iwai N, Inagami T. Identification of two subtypes in the rat type I angiotensin II receptor. FEBS Lett. 1992;298(2-3):257–260. [DOI] [PubMed] [Google Scholar]
- 16. De Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International Union of Pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52(3):415–472. [PubMed] [Google Scholar]
- 17. Grady EF, Sechi LA, Griffin CA, Schambelan M, Kalinyak JE. Expression of AT2 receptors in the developing rat fetus. J Clin Invest. 1991;88(3):921–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Verdecchia1 P, Angeli F, Gattobigio R, Reboldi GP. Do angiotensin II receptor blockers increase the risk of myocardial infarction? Eur Heart J. 2005;26(22):2381–2386. [DOI] [PubMed] [Google Scholar]
- 19. Lee YH, Mungunsukh O, Tutino RL, Marquez AP, Day RM. Angiotensin II-induced apoptosis requires regulation of nucleoli and Bcl-xL by SHP-2 in primary lung endothelial cells. J Cell Sci. 2010;123(pt 10):1634–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stanner SA, Bulmer K, Andres C, et al. Does malnutrition in utero determine diabetes and coronary heart disease in adulthood? Results from the Leningrad siege study, a cross sectional study. BMJ. 1997;315(7119):1342–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miura S, Karnik SS. Ligand-independent signals from angiotensin II type 2 receptor induce apoptosis. EMBO J. 2000;19(15):4026–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Buja LM, Vela D. Cardiomyocyte death and renewal in the normal and diseased heart. Cardiovasc Pathol. 2008;17(6):349–374. [DOI] [PubMed] [Google Scholar]
- 23. Bing OHL. Hypothesis: apoptosis may be a mechanism for the transition to heart failure in rats with chronic pressure overload. J Mol Cell Cardiol. 1994;26(8):943–948. [DOI] [PubMed] [Google Scholar]
- 24. Rodriguez M, Lucchesi BR, Schaper J. Apoptosis in myocardial infarction. Ann Med. 2002;34(6):470–479. [DOI] [PubMed] [Google Scholar]
- 25. Nitahara JA, Cheng W, Liu Y, et al. Intracellular calcium, DNAse activity and myocyte apoptosis in aging Fischer 344 rats. J Mol Cell Cardiol. 1998;30(3):519–535. [DOI] [PubMed] [Google Scholar]
- 26. Freude B, Master TN, Kostin S, Robicsek F, Schaper J. Cardiomyocyte apoptosis in acute and chronic conditions. Basic Res Cardiol. 1998;93(2):85–89. [DOI] [PubMed] [Google Scholar]
- 27. Diwan A, Wansapura J, Syed FM, Matkovich SJ, Lorenz JN, Dorn GW II. Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation. Circulation. 2008;117(3):396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ding Y, Lv J, Mao C, et al. High-salt diet during pregnancy and angiotensin-related cardiac changes. J Hypertens. 2010;28(6):1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dubin NH, Baros NA, Cox RT, King TM. Implantation and fetal survival in the rat as affected by intrauterine injection of normal sterile saline. Biol Reprod. 1979;21(1):47–52. [DOI] [PubMed] [Google Scholar]
- 30. Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2(8663):577–580. [DOI] [PubMed] [Google Scholar]
- 31. Curhan GC, Chertow GM, Willett WC, et al. Birth weight and adult hypertension and obesity in women. Circulation. 1996;94(6):1310–1315. [DOI] [PubMed] [Google Scholar]
- 32. Bagby SP, Ogden BE, LeBard LS, Woods LL, Corless CL, Luo Z. Maternal protein restriction during nephrogenesis in microswine causes asymmetric intrauterine growth retardation (IUGR) in neonates and hypertension (HTN) with body weight excess in adults [Abstract]. J Am Soc Nephrol. 2001;12:461A. [Google Scholar]
- 33. Woods LL, Ingelfinger JR, Nyengaard JR, Rasch R. Maternal protein restriction suppresses the newborn renin–angiotensin system and programs adult hypertension in rats. Pediatric Res. 2001;49(4):460–467. [DOI] [PubMed] [Google Scholar]
- 34. Woods LL. Fetal origins of adult hypertension: a renal mechanism? Curr Opin Nephrol Hypertens. 2000;9(4):419–425. [DOI] [PubMed] [Google Scholar]
- 35. Woods LL, Weeks DA, Rasch R. Programming of adult blood pressure by maternal protein restriction: role of nephrogenesis. Kidney Int. 2004;65(4):1339–1348. [DOI] [PubMed] [Google Scholar]
- 36. Ingelfinger JR, Woods LL. Perinatal programming, renal development, and adult renal function. Am J Hypertens. 2002;15(2 pt 2):46S–49S. [DOI] [PubMed] [Google Scholar]
- 37. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Henery S, George T, Hall B, Basiji D, Ortyn W, Morrissey P. Quantitative image based apoptotic index measurement using multispectral imaging flow cytometry: a comparison with standard photometric methods. Apoptosis. 2008;13(8):1054–1063. [DOI] [PubMed] [Google Scholar]
- 39. Rabkin SW. Apoptosis in human acute myocardial infarction: the rationale for clinical trials of apoptosis inhibition in acute myocardial infarction. Scholarly Res Exc 2009;2009:10. [Google Scholar]
- 40. Xu F, Mao C, Hu Y, Rui C, Xu Z, Zhang L. Cardiovascular effects of losartan and its relevant clinical application. Curr Med Chem. 2009;16(29):3841–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fitzsimons JT. Angiotensin, thirst, and sodium appetite. Physiol Rev. 1998;78(3):583–686. [DOI] [PubMed] [Google Scholar]
- 42. Vicky AC, Leigh JE. Minireview: natriuretic peptides during development of the fetal heart and circulation. Endocrinology. 2003;144(6):2191–2194. [DOI] [PubMed] [Google Scholar]
- 43. Schütz S, Le Moullec JM, Corvol P, Gasc JM. Early expression of all the components of the renin–angiotensin-system in human development. Am J Pathol. 1996;149(6):2067–2079. [PMC free article] [PubMed] [Google Scholar]
- 44. Kawamura M, Itoh H, Yura S, et al. Undernutrition in utero augments systolic blood pressure and cardiac remodeling in adult mouse offspring: possible involvement of local cardiac angiotensin system in developmental origins of cardiovascular disease. Endocrinology. 2007;148(3):1218–1225. [DOI] [PubMed] [Google Scholar]
- 45. Han HC, Austin KJ, Nathanielsz PW, Ford SP, Nijland MJ, Hansen TR. Maternal nutrient restriction alters gene expression in the ovine fetal heart. J Physiol. 2004;588(pt 1):111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Akishita M, Nagai K, Xi H, et al. Renin–angiotensin system modulates oxidative stress–induced endothelial cell apoptosis in rats. Hypertension. 2005;45(6):1188–1193. [DOI] [PubMed] [Google Scholar]
- 47. Lukkarinen HP, Laine J, Aho H, Zagariya A, Vidyasagar D, Kääpä PO. Angiotensin II receptor inhibition prevents pneumocyte apoptosis in surfactant-depleted rat lungs. Pediatr Pulmonol. 2005;39(4):349–358. [DOI] [PubMed] [Google Scholar]
- 48. Singh VP, Le B, Khode R, Baker KM, Kumar R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes. 2008;57(12):3297–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li D, Yang B, Philips MI, Mehta JL. Proapoptotic effects of Ang II in human coronary artery endothelial cells: role of AT1 receptor and PKC activation. Am J Physiol. 1999;276(3 pt 2):786–792. [DOI] [PubMed] [Google Scholar]
- 50. Papp M, Li X, Zhuang J, Wang R, Uhal BD. Angiotensin receptor subtype AT1 mediates alveolar epithelial cell apoptosis in response to ANG II. Am J Physiol Lung Cell Mol Physiol. 2002;282(4):713–718. [DOI] [PubMed] [Google Scholar]
- 51. Cui T, Nakagami H, Iwai M, et al. Pivotal role of tyrosine phosphatase SHP-1 in AT2 receptor mediated apoptosis in rat fetal vascular smooth muscle cell. Cardiovasc Res. 2001;49(4):863–871. [DOI] [PubMed] [Google Scholar]
- 52. Horiuchi M, Hayashida W, Kambe T, Yamada T, Dzau VJ. Angiotensin type 2 receptor dephosphorylates Bcl-2 by activating mitogen-activated protein kinase phosphatase-1 and induces apoptosis. J Biol Chem. 1997;272(30):19022–19026. [DOI] [PubMed] [Google Scholar]
- 53. Horiuchi M, Akishita M, Dzau VJ. Molecular and cellular mechanism of angiotensin II-mediated apoptosis. Endocr Res. 1998;24(3-4):307–314. [DOI] [PubMed] [Google Scholar]
- 54. Goldstein JC, Muñoz-Pinedo C, Ricci JE, et al. Cytochrome C is released in a single step during apoptosis. Cell Death Differ. 2005;12(5):453–462. [DOI] [PubMed] [Google Scholar]
- 55. Dejean LM, Martinez-Caballero S, Guo L, et al. Oligomeric bax is a component of the putative cytochrome C releases channel MAC, mitochondrial apoptosis-induced channel. Mol Biol Cell. 2005;16(5):2424–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]