

Abstract
Systemic Candida albicans infection causes high morbidity and mortality and is associated with neutropenia; however, the roles of other innate immune cells in pathogenesis are poorly defined. Here, using a mouse model of systemic candidiasis, we found that resident macrophages accumulated in the kidney, the main target organ of infection, and formed direct contacts with the fungus in vivo mainly within the first few hours after infection. Macrophage accumulation and contact with Candida were both markedly reduced in mice lacking chemokine receptor CX3CR1, which was found almost exclusively on resident macrophages in uninfected kidneys. Infected Cx3cr1–/– mice uniformly succumbed to Candida-induced renal failure, but exhibited clearance of the fungus in all other organs tested. Renal macrophage deficiency in infected Cx3cr1–/– mice was due to reduced macrophage survival, not impaired proliferation, trafficking, or differentiation. In humans, the dysfunctional CX3CR1 allele CX3CR1-M280 was associated with increased risk of systemic candidiasis. Together, these data indicate that CX3CR1-mediated renal resident macrophage survival is a critical innate mechanism of early fungal control that influences host survival in systemic candidiasis.
Introduction
Systemic candidiasis is the fourth leading cause of nosocomial bloodstream infection in intensive care units (1, 2). Its incidence has dramatically risen over the past decades to more than 20 cases/100,000 population, with an annual cost of approximately 2 billion dollars in the US (1–4). Vaccines are not available, and despite antifungal therapy, mortality of infected patients exceeds 30%–40% (1, 2). Strikingly, the risk of systemic candidiasis and its clinical outcome vary significantly even among patients with similar predisposing clinical and microbiological factors (1, 2), indicating that important host-related risk factors remain to be discovered.
A model of systemic candidiasis has been established in which mice develop renal failure and septic shock (5), similar to infected humans (6–8), and in which the kidney is the primary target organ (5, 9). Neutrophils are critical in anti-Candida host defense in this model (10–12), consistent with predisposition to systemic candidiasis in patients with innate immune defects (13), particularly neutropenia (2). Much less is known about the role in pathogenesis of mononuclear phagocytes, which also accumulate in Candida-infected tissues (9, 14) and can directly kill Candida (15). Thus, monocytes/macrophages may contribute to fungal control, but the mechanisms of trafficking, activation, and effector function remain unclear. To interrogate the roles of monocytes/macrophages in systemic candidiasis, we focused on CCR2 and CX3CR1, the “signature” chemokine receptors for inflammatory and resident monocyte/macrophages, respectively (16, 17). Importantly, we found that CX3CR1+ resident macrophages play a critical role for survival and control of fungal proliferation in the mouse model of systemic candidiasis and show that genetic variation at CX3CR1 is a risk factor for the infection in humans.
Results
Cx3cr1 is critical for survival in a mouse model of systemic candidiasis.
To identify monocyte/macrophage-targeted molecular factors that may be important in anti-Candida host defense, we examined the expression of Ccr2 and Cx3cr1 and their ligands Ccl2/Mcp-1 and Cx3cl1/fractalkine, respectively, in WT Candida-infected kidneys. mRNA for Ccr2 and Cx3cr1 and their ligands was significantly and durably induced after infection (Figure 1, A and B). We then assessed receptor expression on kidney monocytes/macrophages. Monocytes were defined as MHCII–F4/80intCD11c–CD11b+ leukocytes with typical monocyte morphology by transmission electron microscopy (TEM) and could be divided into Ly6Chi and Ly6Clo subpopulations. Macrophages were defined as MHCIIhiF4/80hiCD11cloCD11b+ leukocytes and exhibited classic macrophage morphological features such as eccentric nuclei, abundant cytoplasm, and numerous phagocytic vacuoles and lysosomal granules by TEM (Figure 1C). Ccr2 was expressed on all Ly6Chi monocytes, approximately 50%–70% of Ly6Clo monocytes, and approximately 70%–80% of macrophages, whereas Cx3cr1 was expressed at high levels on Ly6Clo monocytes and macrophages and intermediate levels on Ly6Chi monocytes (Figure 1D).
Figure 1. Systemic candidiasis induces the expression of Ccr2, Cx3cr1, and their ligands.

(A) Ccr2 and Cx3cr1 are induced in the kidney of Candida-infected Cx3cr1+/+ mice (n = 8–10; 2 independent experiments). P < 0.0001 for Ccr2 and Cx3cr1 expression on days 3, 6, and 9 versus day 0. (B) Ccl2 and Cx3cl1 are induced in the kidney of Candida-infected Cx3cr1+/+ mice (n = 8–10; 2 independent experiments). P < 0.0001 for Ccl2 and Cx3cl1 expression on days 3, 6, and 9 versus day 0. (C) Phenotypic characterization of monocytes, macrophages, and dendritic cells in the kidney in the mouse model of systemic candidiasis. Representative FACS analysis shows the gating strategy used to define MHCII–F4/80int monocytes, MHCIIhiF4/80hi macrophages, and MHCIIhiF4/80lo dendritic cells after pregating on live CD45+CD3–CD19–NK1.1–Ly6G– cells. Also shown are representative TEM of the 3 populations FACS sorted at day 6 after infection. Scale bars: 2 μm. (D) Representative FACS histograms of Ccr2 and Cx3cr1 staining on kidney Ly6Chi and Ly6Clo monocytes and macrophages at day 6 after infection (n = 6; 2 independent experiments).
Mortality of Ccr2–/– mice following systemic candidiasis was only slightly increased compared with Ccr2+/+ mice (Figure 2A). In contrast, using an LD50 inoculum defined in Cx3cr1+/+ control mice, Candida infection was uniformly fatal in Cx3cr1–/– mice (Figure 2A). This phenotype was reproduced using 2 independent Cx3cr1-deficient mouse lines, 3 different Candida inocula, and 3 different Candida strains (Figure 2A and data not shown), and therefore is robust. Cx3cr1 deficiency caused a more pronounced weight loss (Figure 2B) and resulted in significantly higher kidney fungal burden throughout the course of the infection (Figure 2C). Moreover, while Candida was mostly contained within abscesses in Cx3cr1+/+ kidneys, pseudohyphae inexorably invaded the renal pelvis and frequently formed large fungal balls in Cx3cr1–/– mice (Figure 2, D and E). Cx3cr1–/– mice also exhibited greater fungal burden in the brain and liver but not in the spleen; however, Candida was eventually controlled in these organs (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI71307DS1).
Figure 2. Systemic candidiasis is uniformly fatal in Cx3cr1–/– mice due to uncontrolled fungal proliferation in the kidney and renal failure.

(A) Mortality. Data represent summary results from Ccr2+/+ and Ccr2–/– mice (left; n = 15; 2 independent experiments), Cx3cr1+/+ and Cx3cr1–/– mice (middle; n = 33–35; 3 independent experiments), and Cx3cr1+/+, Cx3cr1gfp/+ and Cx3cr1gfp/gfp mice (right; n = 25–29; 2 independent experiments; P < 0.0001 for Cx3cr1+/+ versus Cx3cr1gfp/gfp, P < 0.0001 for Cx3cr1gfp/+ versus Cx3cr1gfp/gfp mice; P = 0.09 for Cx3cr1+/+ versus Cx3cr1gfp/+ mice). (B) Weight loss. *P < 0.01 (n = 23–24; 2 independent experiments). (C) Fungal burden in the kidney. *P = 0.02; **P < 0.001 (n = 19–23; 3 independent experiments). (D) PAS staining. Cross-sections of Cx3cr1+/+ and Cx3cr1–/– kidneys on day 6 after Candida infection (n = 8; 3 independent experiments). Original magnification, ×200. (E) Candida fungal balls in the Cx3cr1gfp/gfp renal pelvis. Confocal images of Cx3cr1gfp/+ and Cx3cr1gfp/gfp kidney at day 6 after infection with dTomato-expressing Candida (n = 3; 2 independent experiments). Scale bars: 200 μm. Objective, ×2. (F) Anatomic pathology of infected kidneys. Representative kidneys are shown for 2 mice of each genotype sacrificed at day 9 after infection. (G) Renal function. *P < 0.001 (n = 8–10; 2 independent experiments). (H) Histopathology. Representative H&E staining of kidney sections at day 9 after infection. Original magnification, ×20 (top row); ×100 (bottom row) (n = 8; 3 independent experiments). (I) Proinflammatory cytokine induction at day 9 after infection. *P = 0.03. **P < 0.01 (n = 8–10, day 9 after infection; 2 independent experiments). All quantitative data represent mean ± SEM.
Cx3cr1–/– kidneys appeared paler and more swollen than WT kidneys by gross pathology (Figure 2F).
Accordingly, infected Cx3cr1–/– mice developed severe kidney failure after infection (Figure 2G) associated with more extensive inflammatory changes, tubular casts, and papillary necrosis (Figure 2, D and H) and greater induction of proinflammatory cytokines than in kidneys of infected WT mice (Figure 2I). In contrast, Cx3cr1 deficiency did not affect the histology of brain, liver, or spleen of infected mice (Supplemental Figure 1B). Thus, for the remainder of our studies, we focused on the effects of Cx3cr1 in the kidney.
Cx3cr1 on hematopoietic and nonhematopoietic cells confers protection against systemic candidiasis.
Because Cx3cr1 is expressed on both hematopoietic and nonhematopoietic cells (16–20), we asked whether increased susceptibility of Cx3cr1–/– mice to systemic candidiasis was due to Cx3cr1 deficiency in the hematopoietic or nonhematopoietic compartment, or both. Therefore, we generated BM chimeras and infected them with Candida 10 weeks after successful reconstitution (Supplemental Figure 2). Both Cx3cr1+/+ mice reconstituted with Cx3cr1–/– BM and Cx3cr1–/– mice reconstituted with Cx3cr1+/+ BM were susceptible to systemic candidiasis and recapitulated the phenotype observed in Cx3cr1–/– mice and Cx3cr1–/–→Cx3cr1–/– chimeras (Figure 3A), suggesting that Cx3cr1 within both hematopoietic and nonhematopoietic compartments is required for protection.
Figure 3. Cx3cr1-expressing kidney mononuclear phagocytes engulf Candida early after infection in a mouse model of systemic candidiasis.

(A) Cx3cr1 on both hematopoietic and nonhematopoietic cells is critical for survival in systemic candidiasis. Upper right identifies the 4 groups of BM chimeras (donor→recipient). P < 0.001 for WT→WT versus each of the other 3 groups (n = 15–25/group). Data are shown from 1 of 2 independent experiments with similar pattern of results. (B) Cx3cr1-expressing mononuclear phagocytes are present constitutively in uninfected kidney (left; renal cortex) and accumulate after Candida infection (middle, day 6 after infection; corticomedullary junction), surrounding but not invading the renal collecting system (right, day 6 after infection; corticomedullary junction). Images are representative confocal micrographs from Cx3cr1gfp/+ kidneys (n = 3–4; 2 independent experiments). Scale bars: 200 μm (left); 150 μm (middle); 20 μm (right). Objective, ×10 (left and middle), ×60 (right). (C) Representative FACS histogram of Cx3cr1 staining on kidney neutrophils and dendritic cells at day 6 after infection (n = 6; 2 independent experiments). (D) Cx3cl1 is expressed in glomeruli (left) and tubular cells (middle), predominantly at the corticomedullary junction. Images were generated by Cx3cl1 IHC staining of Cx3cr1+/+ kidneys obtained 6 days after infection. Isotype control staining is also seen (right) (n = 6; 2 independent experiments). Original magnification, ×600. (E) Relative mRNA expression of Cx3cl1 in macrophages (day 0 and 6 after infection), endothelial cells (day 0), and tubular cells (day 0) FACS-sorted from Cx3cr1+/+ kidneys (n = 4–6; 2 to 3 independent experiments). Data represent mean ± SEM.
To better understand the biological function of Cx3cr1, we next examined Cx3cr1 expression on kidney hematopoietic and nonhematopoietic cells using Cx3cr1gfp/+ mice, which are significantly protected from infection compared with Cx3cr1gfp/gfp mice but slightly more susceptible than WT mice (Figure 2A). We did not detect Cx3cr1 on endothelial or epithelial cells by FACS (data not shown) or confocal microscopy (Figure 3B). Within the hematopoietic compartment, Cx3cr1 was expressed on monocytes/macrophages (Figure 1D) but not neutrophils (Figure 3C), the 2 predominant leukocyte subsets in the kidney (Supplemental Table 1), and on a subset of T cells (∼25%), NK cells (∼20%), and dendritic cells (CD11b+CD103– greater than CD103+CD11b–), but not on B cells (Figure 3C and data not shown). Because the monocyte/macrophage is the major Cx3cr1+ cell type in the model and because clodronate administration, which depletes monocytes/macrophages, has been reported to increase mortality in the model (11), we focused our analysis of mechanisms on the effects of Cx3cr1 on mononuclear phagocyte distribution and function in the kidney.
Mononuclear phagocytes interact with Candida early after infection in a Cx3cr1-dependent manner.
We first aimed to characterize the distribution of mononuclear phagocytes and their topographic association with fractalkine and Candida in the kidney. Using Cx3cr1gfp/+ mice, we found that mononuclear phagocytes formed a network in the uninfected kidney and accumulated throughout the renal parenchyma surrounding glomeruli and renal tubules after infection (Figure 3B). Fractalkine was detected in glomerular endothelial cells and in tubular cells (Figure 3D), confirming previous reports (21). Kidney macrophages also expressed fractalkine (Figure 3E), which may act in an autocrine manner on the cells (18, 22).
We then employed confocal microscopy to visualize the interaction of kidney mononuclear phagocytes with the fungus. Kidneys were harvested from Cx3cr1gfp/+ mice infected with dTomato-expressing Candida and imaged immediately. Candida rapidly invaded the kidney and formed 20-μm–long filaments as early as 2 hours after intravenous injection (Figure 4A). At this time point, the majority of Candida yeast and pseudohyphal elements were in contact with mononuclear phagocytes, internalized within or encased by them (Figure 4A), respectively. By day 1 after infection, mononuclear phagocytes were no longer in direct contact with Candida; instead, pseudohyphae were only occasionally surrounded by mononuclear phagocytes in the renal interstitium, and more than 90% of pseudohyphal structures had already extended within renal tubules (Figure 4A and Supplemental Figure 3). Conversely, mononuclear phagocytes surrounded the tubules and were not detected within the renal collecting system at any time-point analyzed (Figure 3B and Figure 4A).
Figure 4. Cx3cr1 deficiency results in decreased steady state and Candida-induced accumulation of kidney macrophages as well as reduced interaction of kidney mononuclear phagocytes with Candida.

(A) Cx3cr1-expressing mononuclear phagocytes directly interact with C. albicans only early in infected mouse kidney. Images depict 3 different types of interaction, as indicated by the labels, in the kidneys of Cx3cr1gfp/+ mice 2 hours (left and middle) and 1 day (right) after infection (n = 3; 3 independent experiments). Scale bars: 5 μm (left, middle); 20 μm (right). Objective, ×60. (B) Decreased frequency of dTomato-Candida in contact with GFP-marked mononuclear phagocytes in the kidney in the absence of Cx3cr1. Left, data are summarized from 3 experiments 2 hours after Candida challenge. *P = 0.002. Right, representative confocal images. Scale bars: 5 μm. Objective, ×60. (C) Cx3cr1 deficiency does not affect binding of EGFP-Candida by BMMs in vitro (n = 9; 3 independent experiments). (D) Cx3cr1 deficiency does not affect phagocytosis of EGFP-Candida by BMMs in vitro (n = 12; 4 independent experiments). (E) Total macrophages in uninfected mouse kidney. Left, summary data obtained by FACS for MHCIIhiF4/80hiCD11b+ cells isolated from kidneys from the indicated mouse strains. *P < 0.001 (n = 9; 3 independent experiments). Right, representative confocal images from the indicated mouse strains (n = 4; 2 independent experiments). Scale bars: 50 μm. Objective, ×10. (F) Number of macrophages extracted from mouse kidney over time after Candida infection. *P = 0.02; **P < 0.01; ***P < 0.001 (n = 9–15/time-point; 3 to 4 independent experiments).
After characterizing the interaction of kidney mononuclear phagocytes with Candida in Cx3cr1gfp/+ mice, we asked whether Cx3cr1 deficiency impedes the ability of mononuclear phagocytes to contact Candida early after infection. Strikingly, while approximately 90% of Candida elements were either internalized or encircled by Cx3cr1gfp/+ mononuclear phagocytes, only approximately 60% of Candida elements were in contact with Cx3cr1gfp/gfp mononuclear phagocytes in the kidney 2 hours after infection (Figure 4B). Hence, our data demonstrate that mononuclear phagocytes interact with Candida almost exclusively very early after infection and this interaction is dependent on functional Cx3cr1. Because resident macrophages are by far the most abundant Cx3cr1+ mononuclear phagocytes compared with resident monocytes and dendritic cells in the kidney at this very early phase of the infection (∼80%–85%; Supplemental Table 1), we next focused on the function of macrophages in the model.
Cx3cr1 deficiency results in significantly decreased macrophage accumulation in the kidney both at steady state and throughout infection.
To gain insight into the Cx3cr1-dependent interaction of macrophages with Candida, we first investigated whether Cx3cr1 deficiency adversely affects Candida recognition, as Cx3cr1 has adhesion properties (23). Cx3cr1+/+ and Cx3cr1–/– BM-derived macrophages (BMMs) had similar capacities for Candida (Figure 4C) or zymosan binding (data not shown), and Candida internalization (Figure 4D). Cx3cr1 deficiency also did not alter dectin-1 or TLR2 expression on macrophages (data not shown).
Based on these findings, we hypothesized that Cx3cr1 deficiency impairs in vivo kidney macrophage:Candida interaction by influencing macrophage accumulation in that organ. Indeed, Cx3cr1 deficiency was associated with approximately 50% reduction in the number of macrophages in Cx3cr1–/– kidneys and decreased macrophage density in Cx3cr1gfp/gfp kidneys at steady state (Figure 4E). Furthermore, Cx3cr1–/– kidneys accumulated approximately 50% fewer macrophages than Cx3cr1+/+ kidneys throughout the entire course of the infection (Figure 4F). In contrast, more neutrophils accumulated (Figure 5, A and B, and Supplemental Figure 4) and induction of neutrophil-targeted chemoattractants was greater in Cx3cr1–/– infected kidneys (Figure 5, C–F).
Figure 5. Cx3cr1 deficiency is associated with increased accumulation of neutrophils in the kidney after Candida infection.

(A) Number of neutrophils extracted from mouse kidney over time after Candida infection. *P < 0.01; **P < 0.001 (n = 9–15; 3 to 4 independent experiments). (B) Neutrophil/macrophage ratio in the kidney after Candida infection (n = 9–15; 3 to 4 independent experiments). (C–F) Kidney induction by Candida infection of the neutrophil-targeted chemokine receptor Cxcr2 and its ligands Cxcl1 and Cxcl2. RNA analysis, *P < 0.05; **P < 0.01 (n = 8–10; 2 independent experiments). Protein analysis, *P < 0.05 (n = 4; 1 independent experiment). All quantitative data represent mean ± SEM.
Cx3cr1 mediates macrophage survival and inhibits caspase-dependent apoptosis associated with Akt activation.
We next aimed to investigate the mechanism(s) underlying the decreased macrophage accumulation in Cx3cr1–/– kidneys, which could be explained by the net effect of decreased (a) in situ macrophage proliferation, and/or (b) monocyte trafficking into the kidney, and/or (c) monocyte differentiation into macrophages within the kidney, and/or (d) macrophage survival in the kidney. Cx3cr1 has been shown to mediate cell proliferation (22, 24); however, the percentage of proliferating macrophages, measured by Ki-67 positivity using FACS, was similar in Cx3cr1+/+ and Cx3cr1–/– kidneys at steady state and throughout infection (Figure 6A). Also, levels of IL-4, which may drive local macrophage proliferation in vivo (25), were not decreased in infected Cx3cr1–/– kidneys (data not shown).
Figure 6. Cx3cr1 deficiency increases apoptosis of kidney macrophages, but does not affect kidney macrophage differentiation or proliferation or monocyte trafficking from blood to kidney.

(A) Kidney macrophage proliferation. Leukocytes were purified from kidneys of the indicated mouse lines, and endogenous MHCIIhiF4/80hiCD11b+ cells were analyzed for Ki67 expression (n = 6–10; 2 independent experiments). (B) Monocyte trafficking from blood to kidney after Candida infection. Adoptive transfer of BM-derived monocytes from the indicated uninfected mouse lines into WT mice 3 days after Candida infection. WT recipient mice were sacrificed 12 hours after cell transfer, and the number of GFP+ monocytes purified from kidneys was measured (n = 6–8; 2 independent experiments). (C) Endogenous monocyte accumulation in kidney after Candida infection (n = 9–15; 3 to 4 independent experiments). (D) Macrophage differentiation. WT recipient mice were sacrificed 3 days after cell transfer, and the percentage of GFP+ cells from the indicated uninfected mouse lines that differentiated into MHCIIhiF4/80hiCD11b+ macrophages in kidneys was determined. (n = 6; 2 independent experiments). (E) Persistence of macrophages in the infected kidney 3 days after monocyte transfer is Cx3cr1 dependent. Left, quantitation. *P = 0.001 (n = 6; 2 independent experiments). Right, representative FACS plots. (F) Endogenous kidney macrophage survival before and after Candida infection. *P < 0.05; **P < 0.01; ***P < 0.001 (n = 6–8; 2 independent experiments). (G) Representative FACS plots for survival marker expression by kidney macrophages of uninfected mice quantified in F. (H) Cx3cr1 modulates caspase-3 cleavage and Akt phosphorylation. Endogenous kidney macrophages were sorted from Candida-infected kidneys 6 days after infection. Left, representative Western blot; middle and right, quantitation of Western blot data. β-actin is shown as loading control. *P = 0.03; **P < 0.01 (n = 4; 2 independent experiments). All quantitative data represent mean ± SEM.
To test the role of Cx3cr1 on monocyte trafficking and differentiation directly, we transferred sorted GFP+CD115+Ly6ChiCD11b+ monocytes from uninfected Cx3cr1gfp/+ and Cx3cr1gfp/gfp BM into WT mice 3 days after infection. Cx3cr1 did not affect monocyte trafficking, as similar numbers of Cx3cr1gfp/+ and Cx3cr1gfp/gfp cells were recruited into the kidney 12 hours after transfer (Figure 6B). Consistent with the dispensable role of Cx3cr1 on monocyte trafficking, monocyte accumulation was similar in Cx3cr1+/+ and Cx3cr1–/– kidneys (Figure 6C). Moreover, Cx3cr1 did not affect macrophage differentiation, as the percentage of Cx3cr1gfp/+ and Cx3cr1gfp/gfp cells that were MHCIIhiF4/80+CD11b+ was similar 3 days after monocyte transfer (Figure 6D).
While monocyte trafficking and differentiation were Cx3cr1 independent, considerably fewer Cx3cr1gfp/gfp than Cx3cr1gfp/+ macrophages persisted in the kidney 3 days after monocyte transfer (Figure 6E), suggesting a survival defect. Indeed, Cx3cr1+/+ kidneys had a higher proportion of annexin V–7-AAD– viable macrophages compared with Cx3cr1–/– kidneys at steady state and throughout infection (Figure 6, F and G). Correspondingly, the proportion of annexin V+7-AAD+ dead and annexin V+7-AAD– apoptotic macrophages was higher in Cx3cr1–/– than Cx3cr1+/+ kidneys before and after infection (Figure 6, F and G). Conversely, kidney neutrophils, which are Cx3cr1 negative (Figure 3C), had similar viability in Cx3cr1+/+ and Cx3cr1–/– mice (data not shown).
Consistent with a survival defect, higher amounts of cleaved caspase-3 were detected in Cx3cr1-deficient macrophages sorted from infected kidneys (Figure 6H). The fractalkine-Cx3cr1 axis has been reported to mediate survival of rat microglia and rat and human smooth muscle cells via different mechanisms, which all involve PI3K-dependent Akt activation (22, 24, 26, 27). In agreement, Akt phosphorylation was reduced in sorted Cx3cr1–/– kidney macrophages (Figure 6H). Similarly, decreased expression of cleaved caspase-3 and increased Akt phosphorylation were detected by FACS in Cx3cr1+/+ compared with Cx3cr1–/– kidney-resident macrophages at steady state (Supplemental Figure 5). These data demonstrate that Cx3cr1 deficiency results in decreased macrophage survival in the kidney associated with increased caspase-dependent apoptosis.
Kidney-resident macrophages inactivate Candida.
Although decreased kidney macrophage accumulation correlated with increased fungal proliferation in the kidney and diminished mouse survival, it is unknown whether renal resident macrophages, which are the principal cells that come in contact with the fungus early after infection, have direct anti-Candida activity. To address this question, we sorted kidney-resident macrophages from uninfected Cx3cr1+/+ mice and incubated them with Candida ex vivo at an effector/target cell ratio of 2:1. Using alamarBlue reduction as a measure of fungal inactivation (28), we found that kidney-resident macrophages inactivated approximately 42% of fungal cells (Figure 7). We then asked whether Cx3cr1 deficiency impairs the ability of kidney-resident macrophages to inactivate Candida. We found that Cx3cr1–/– kidney-resident macrophages exhibited a modest 12% decrease in Candida killing (i.e., ∼37%) compared with Cx3cr1+/+ cells (Figure 7). These data show that renal-resident macrophages exert direct anti-Candida activity, which is slightly decreased in Cx3cr1–/– cells.
Figure 7. Kidney-resident macrophages exhibit Candida-killing capacity, which is slightly decreased in Cx3cr1-deficient cells.

Sorted Cx3cr1+/+ and Cx3cr1–/– kidney-resident macrophages inactivate 42% versus 37% of Candida yeast cells, respectively. *P = 0.028 (n = 6; 3 independent experiments).
The decreased accumulation and effector function of kidney-resident macrophages in Cx3cr1-deficient mice is associated with uncontrolled fungal proliferation very early after infection.
Because Cx3cr1+ kidney-resident macrophages come in contact with Candida in vivo only within the first few hours after infection, and because Cx3cr1 deficiency adversely affects the accumulation, survival, and fungicidal activity of these cells, we reasoned that the very early interactions of tissue-resident macrophages with Candida, when inflammatory monocytes and neutrophils are not yet recruited in the kidney, are critical for controlling the progression of the infection. In line with this, the fungal load in the kidneys of Cx3cr1–/– mice was significantly increased as early as 12 hours after infection (Figure 8).
Figure 8. Cx3cr1 deficiency results in increased fungal proliferation in the kidney very early after Candida infection.

(A) Shown are 12 hours (n = 18–19; 2 independent experiments) and (B) 24 hours after infection (n = 8–9; 2 independent experiments). *P < 0.001.
CX3CR1-M280 is associated with systemic candidiasis in humans.
In humans, the CX3CR1-M280 SNP caused by a cytosine-to-thymidine substitution at nucleotide 839 that changes a threonine at position 280 to methionine results in reduced fractalkine binding and impaired fractalkine-induced cell adhesion and chemotaxis (29). To test the hypothesis that this SNP may influence susceptibility to systemic candidiasis, we genotyped 281 candidemic patients and 384 noninfected controls from 2 patient cohorts from the US and the Netherlands (ref. 30 and Supplemental Tables 2 and 3).
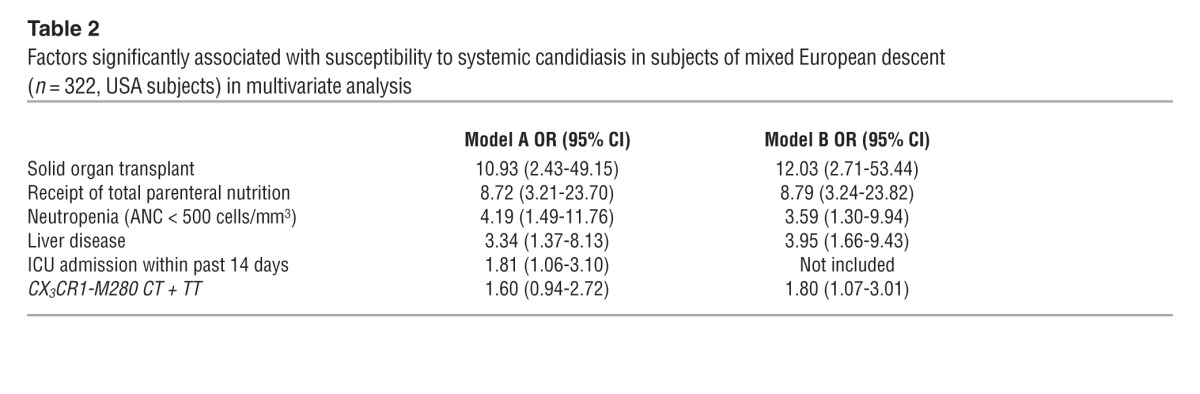
The genotypes were in Hardy-Weinberg equilibrium in both groups. Comparison of frequencies of patients carrying the mutant CX3CR1-M280 allele versus the WT genotype among candidemic and control subjects in the combined groups yielded an odds ratio (OR) of 1.78 for patients of mixed European descent (Table 1; P = 0.0017; 95% CI, 1.24–2.55), but no difference for African-American subjects (Supplemental Table 4). In a multivariate model controlling for other clinical factors associated with systemic candidiasis, including solid organ transplantation, neutropenia, receipt of total parenteral nutrition, and liver disease, the presence of the mutant CX3CR1-M280 allele was significantly associated with candidemia (Table 2; OR, 1.80; P = 0.0262, 95% CI, 1.07–3.01). When intensive care unit admission status was added to the model, the association of CX3CR1-M280 with candidemia decreased slightly (OR, 1.60; P = 0.0685; 95% CI, 0.94–2.72).
Table 1.
Association of the mutant CX3CR1-M280 allele with susceptibility to systemic candidiasis in subjects of mixed European descent from 2 cohorts (Netherlands and USA)
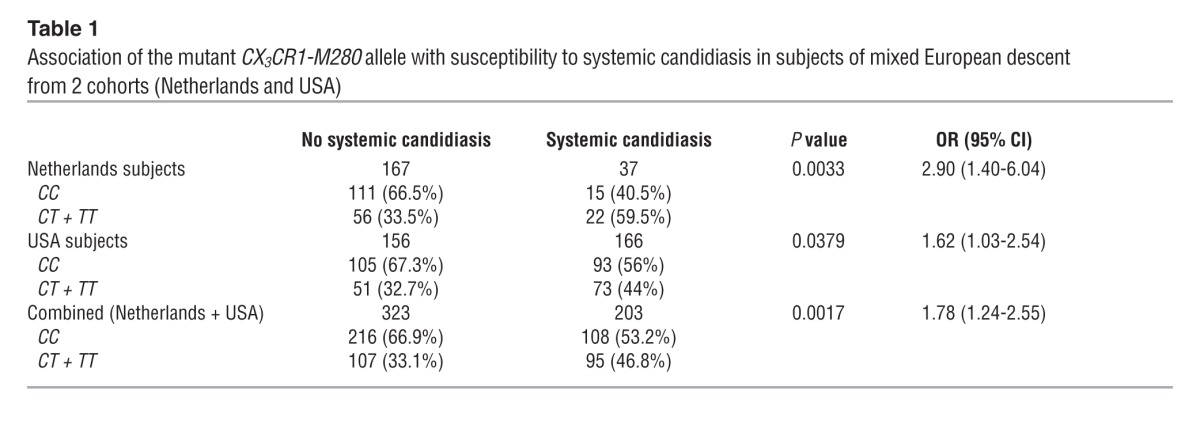
Table 2.
Factors significantly associated with susceptibility to systemic candidiasis in subjects of mixed European descent (n = 322, USA subjects) in multivariate analysis
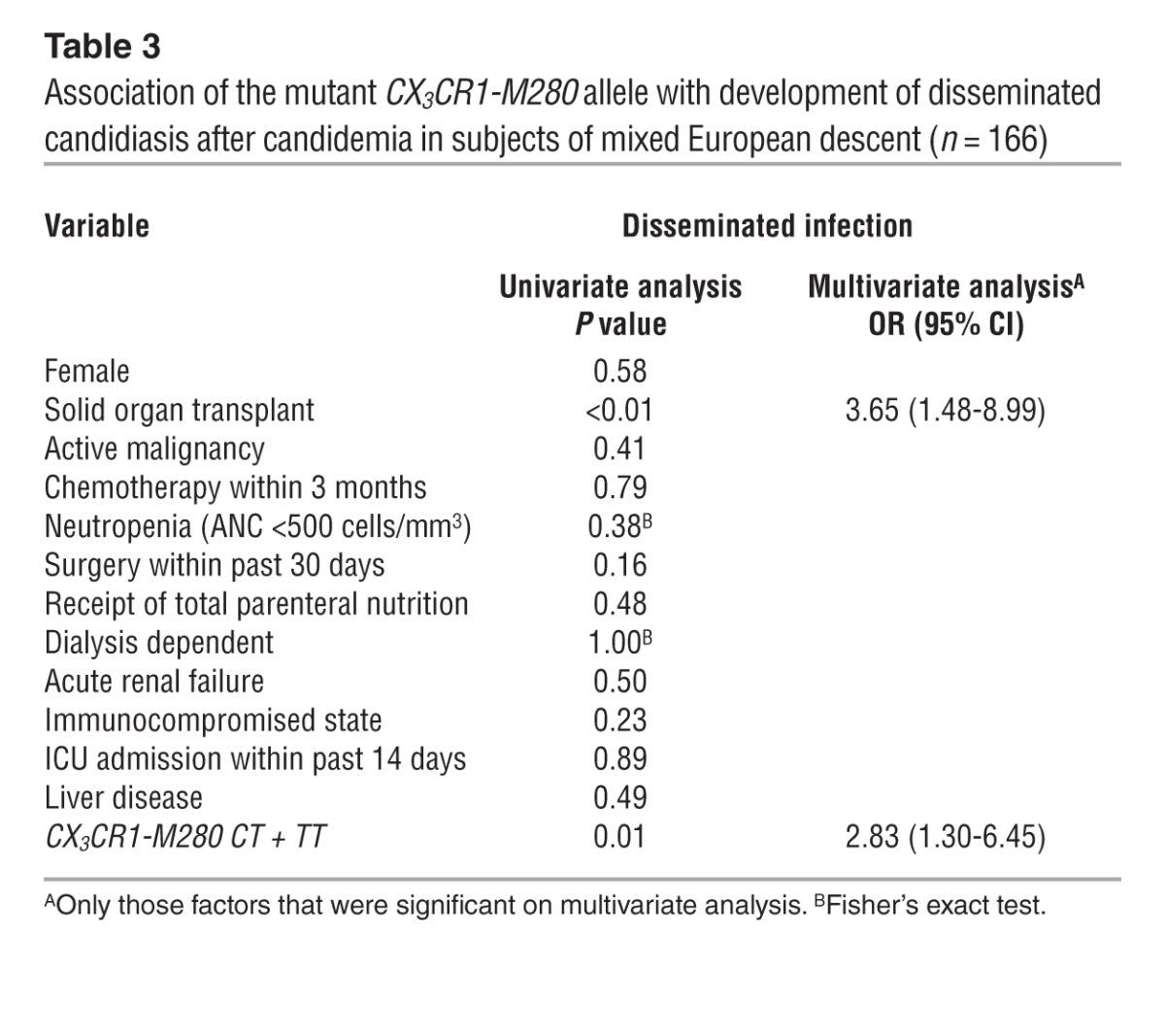
While there was no association of CX3CR1-M280 with persistent fungemia or mortality (data not shown), candidemic patients of mixed European descent carrying the mutant allele were more likely to develop disseminated infection (30.1%) than those lacking this allele (14%) (Table 3; P = 0.01; OR, 2.65; 95% CI, 1.22–5.74). This association remained significant in multivariate analysis when controlling for other clinical factors; in the final model, presence of the CX3CR1-M280 allele (OR, 2.83; P = 0.0104; 95% CI, 1.30–6.45) and solid organ transplantation (OR, 3.65; P = 0.0045; 95% CI, 1.48–8.99) were the only factors independently associated with development of disseminated candidiasis.
Table 3.
Association of the mutant CX3CR1-M280 allele with development of disseminated candidiasis after candidemia in subjects of mixed European descent (n = 166)
Discussion
In the present study we reveal a role of Cx3cr1-dependent tissue-resident mononuclear phagocytes in host defense against systemic fungal infection and show that the dysfunctional CX3CR1-M280 SNP is associated with susceptibility to human systemic candidiasis. The receptor appears to act directly by promoting macrophage survival and accumulation in tissue, which facilitates effective interaction between tissue-resident macrophages and Candida early after infection. Our conclusions are based on detailed analysis of differences in clinical, microbiological, pathological, immunological, imaging, biochemical and molecular parameters between WT and Cx3cr1-deficient mice. To the best of our knowledge, our study is the first to reveal the direct interaction of mononuclear phagocytes with the fungus at the site of infection and the first to identify a monocyte/macrophage-specific molecular factor that protects against systemic candidiasis in mice and humans.
Our results are consistent with a single report published in 1994 that had not been followed up showing that clodronate-induced monocyte/macrophage depletion in mice results in increased fungal burden and mortality after systemic candidiasis (11). No information was previously available on mechanisms by which monocytes/macrophages might promote antifungal protection or on whether anti-Candida host defense is mediated by resident monocytes/macrophages, recruited inflammatory monocytes/macrophages, or both. Our investigation of the roles of chemokine receptors CCR2 and CX3CR1 in anti-Candida host defense could elucidate this latter point because they are “signature” molecules for mouse and human inflammatory and resident monocytes/macrophages, respectively (16, 17). Specifically, mouse Ly6Chi inflammatory monocytes/macrophages correlate with human classical CD14++CD16– cells, which both express CCR2 and CX3CR1, whereas mouse Ly6Clo resident monocytes/macrophages correlate with human nonclassical CD14+CD16+ cells, which both express CX3CR1, but not CCR2 (17). While the role of CCR2+ inflammatory monocytes/macrophages has been investigated in several infection models, the contribution of CX3CR1+ resident monocytes/macrophages in host defense remains less well defined (31).
Our study strongly supports a predominant role of resident macrophages over recruited inflammatory monocytes/macrophages in anti-Candida innate host defense. First, Ccr2–/– mice, which are monocytopenic and unable to mobilize inflammatory monocytes into tissue after infection (31), did not display significantly enhanced susceptibility to systemic candidiasis. In contrast, Cx3cr1–/– mice were highly susceptible to infection despite the redundant role of Cx3cr1 in monocyte accumulation in the kidney. Also, our confocal imaging data provide evidence that Cx3cr1+ macrophages in the infected kidney interact with Candida within the first hours after infection, when only resident macrophages but not recruited inflammatory monocytes/macrophages are present in tissue.
Our study identifies Cx3cr1 as a protective molecular factor in systemic candidiasis, which appears to operate by promoting macrophage accumulation in the kidney. The receptor has previously been shown to mediate macrophage accumulation in lamina propria, liver, and lung, but not spleen (32). The resultant approximately 50% decrease of tissue-resident macrophages in Cx3cr1–/– kidneys is associated with a significant reduction in Candida-macrophage contact in the kidney. Specifically, the percentage of Candida elements that are not in contact with kidney tissue–resident macrophages is approximately 4-fold greater in Cx3cr1–/– than Cx3cr1+/+ kidneys (∼40% versus ∼10%, respectively; Figure 4B) during the first 2 hours after infection. The ineffective macrophage-Candida contact in Cx3cr1–/– kidneys early after infection, when inflammatory monocytes and neutrophils are not yet recruited, appears detrimental for arresting Candida proliferation, as fungal burden in Cx3cr1–/– kidneys is significantly increased as early as 12 hours after infection (Figure 8). Of interest, while Cx3cr1 is important for control of Candida proliferation in kidney, liver, and brain, it is not in the spleen, the only organ among those examined in which resident macrophages are Cx3cr1-negative (33) and do not depend on Cx3cr1 for tissue accumulation (32). Of note, although the capacity of kidney-resident macrophages to kill Candida was slightly decreased in Cx3cr1–/– cells, it appears that the effects of Cx3cr1 on macrophage accumulation predominate over the effects of the receptor on macrophage-killing activity in accounting for the large differences in kidney fungal burden and mouse survival between Cx3cr1+/+ and Cx3cr1–/– mice. Nonetheless, future studies will be required to determine the mechanism by which Cx3cr1 mediates macrophage candidacidal activity.
The mechanism of Cx3cr1-dependent macrophage accumulation in the kidney does not relate to monocyte trafficking or differentiation. This differs from the previously reported roles of Cx3cr1 on monocyte recruitment into inflamed tissue during splenic listeriosis and renal ischemia-reperfusion (34, 35) and on monocyte differentiation in liver fibrosis (36). Conversely, our data are consistent with the redundant role of Cx3cr1 on monocyte trafficking and differentiation in the peritoneum (20). Instead, we show that Cx3cr1 mediates macrophage accumulation by providing survival signals both at steady state and throughout infection, which protect from apoptosis. These data reveal suggest a novel role for Cx3cr1-dependent macrophage survival in acute fungal control and are consistent with the reported Cx3cr1-dependent antiapoptotic effects on macrophages in aorta and liver (36, 37). Additional work is required to define the molecular mechanisms that mediate Cx3cr1-dependent inhibition of macrophage caspase-dependent apoptosis downstream of Akt activation. Furthermore, future studies will be needed to determine which nonhematopoietic Cx3cr1+ cell or cells confer protection against systemic candidiasis and by which mechanism or mechanisms and to define the role of Cx3cr1+ renal resident dendritic cells in early innate immune responses against the fungus.
Two striking findings arise from our confocal studies. First, Candida promptly invades the kidney and forms 20- to 25-μm–long filaments within just 2 hours after intravenous injection. Conversely, Candida filamentation, a key virulence factor (38), is not seen in liver or spleen (9). The rapidity of kidney-specific filamentation is remarkable in light of the requirement of approximately 2–3 hours for formation of approximately 5-μm germ-tubes in vitro using “filamentation-priming” conditions (39) and underscores the importance for future research aiming to identify the kidney-specific factors that are permissive for accelerated filamentation.
Second, Candida is almost exclusively found within renal tubules by 24 hours after infection, where it penetrates from the renal interstitium. The universal invasion of Candida into renal tubules by day 1 after infection appears to act as an immune-evading mechanism because (a) monocytes/macrophages do not enter into the renal collecting system,and (b) neutrophils, which are recruited into renal tubules and colocalize with Candida (9), are unable to confer protection beyond the first 24 hours after infection (10); in fact, neutrophils within tubules mediate immunopathology in the late phase of infection (10, 40). Furthermore, the localization of Candida outside renal tubules during only the first hours after infection highlights the role of tissue-resident kidney macrophages as an innate “barrier” to the rapid spread of Candida from the renal interstitium into the tubules. Future studies using dynamic intravital imaging will be required to better define the in vivo interaction of tissue-resident macrophages with Candida in the infected kidney early after infection before inexorable fungal growth ensues within the renal collecting system.
The dysfunctional CX3CR1-M280 SNP, which has been associated with protection from cardiovascular disease (29) in agreement with protection against atherosclerosis in Cx3cr1–/– mice (41), provided a unique opportunity to extend our mouse findings to humans. Hence, consistent with our data in Cx3cr1–/– mice, we found that the CX3CR1-M280 allele is associated with an increased susceptibility to candidemia and disseminated disease in patients of mixed European descent. The lack of such association in African-American individuals may reflect race-specific differences in the pathogenesis of the infection or could be due to the small sample size. Because mortality of candidemic patients exceeds 30%–40% despite antifungal therapy, identification of genetic factors such as CX3CR1-M280 that may predispose to systemic candidiasis could lead to individualized risk stratification and antifungal prophylaxis strategies in patients. Moreover, because the clinical outcome of systemic candidiasis varies substantially among patients with similar clinical and microbiological risk factors, discovery of genetic factors such as CX3CR1-M280 that predispose to disseminated disease could result in individualized prognostication strategies and in identification of candidemic patients in which intensified diagnostic and therapeutic interventions may be warranted. Like any genetic association study, it is possible that the association reported here is due to population stratification or linkage with another as-yet-unknown SNP; thus, this finding requires validation in future cohort studies. However, the fact that Cx3cr1–/– mice have heightened susceptibility to systemic candidiasis suggests that the association is direct.
In conclusion, our study demonstrates that CX3CR1 is critical for innate host defense against the most common human fungal pathogen, acting by promoting macrophage survival and accumulation in tissue, which is associated with early efficient contact of tissue-resident macrophages with the fungus and control of Candida growth and immune evasion. In agreement, we identify the dysfunctional CX3CR1-M280 SNP as a potential novel factor for risk assessment and prognostication in systemic candidiasis in humans.
Methods
Mice.
Female Cx3cr1+/+ WT and Cx3cr1–/– C57BL/6 mice, and CD45.1+ congenic B6.SJL mice were obtained from Taconic Farms. The Cx3cr1–/– mice were generated as previously described (41) and were backcrossed onto the C57BL/6 background for 10 generations. Female Cx3cr1+/+ WT and Cx3cr1gfp/gfp C57BL/6 mice were obtained from the Jackson Laboratories. The Cx3cr1gfp/gfp mice were generated as described previously (20) and were backcrossed onto the C57BL/6 background for 10 generations. Cx3cr1gfp/+ mice were obtained by crossing Cx3cr1gfp/gfp with Cx3cr1+/+ WT mice. Female Ccr2–/– C57BL/6 mice were obtained from the Jackson Laboratories and were backcrossed onto the C57BL/6 background for 9 generations. Mice were maintained under specific pathogen–free housing conditions and were used at 8 to 12 weeks of age unless otherwise stated.
Fungal strains and mouse model of systemic candidiasis.
The Candida albicans strains SC5314, CAF2-1 (gifts of John Bennett, National Institute of Allergy and Infectious Diseases [NIAID]), and UC820 were used in the present study. SC5314-EGFP, a prototrophic derivative of SC5314, was previously described (42). CAF2-1-dTomato, a derivative of the prototrophic CAF2 strain (43), was created by transformation with a custom synthetic construct containing the PENO1 promoter, a C. albicans codon–optimized dTomato gene, the TTEF terminator, and a nourseothricin resistance marker (synthesized by Genscript Inc.). Transformants were selected on 100 μg/ml nourseothricin and screened both for correct integration at the ENO1 locus by PCR and for high fluorescence by flow cytometry.
Candida was grown in yeast extract, peptone, and dextrose medium containing penicillin and streptomycin (Mediatech Inc.) in a shaking incubator at 30°C.
Cells were centrifuged, washed in PBS, counted using a hemocytometer, and injected into mice via the lateral tail vein. 105 Candida yeast cells were injected per mouse unless stated otherwise.
Fungal burden determination.
Infected mice were euthanized on days 3, 6, and 9 after infection to determine the tissue fungal burden in the kidney, brain, liver, and spleen. The organs were aseptically removed, weighed, homogenized using the Omni Tissue Homogenizer (Omni International), serially diluted, and plated on yeast extract, peptone, and dextrose agar plates containing penicillin and streptomycin. CFUs were determined after 48 hours of incubation at 37°C and results were expressed as CFUs/g of tissue. From 2 to 3 independent experiments were performed using a combined total of 22 to 23 Cx3cr1+/+ and 19 to 22 Cx3cr1–/– mice per time-point.
Mouse histology and immunohistochemistry.
Mice were euthanized on days 3, 6, and 9 after infection and the kidney, brain, spleen, and liver were removed, fixed with 10% formalin, and embedded in paraffin. Tissue sections were prepared for H&E, PAS staining, or immunohistochemistry (IHC). For IHC staining, tissue sections were placed on poly-l-lysine–coated glass slides, deparaffinized in xylene, and rehydrated in a graded series of alcohol. After antigen retrieval for 20 minutes at 85°C, endogenous peroxidase blocking was carried out in alcohol solution containing 0.3% hydrogen peroxide for 10 minutes at room temperature. The slides were then incubated in 5% BSA (Sigma-Aldrich) solution for 20 minutes to block nonspecific protein binding and washed with Tris-buffered saline containing 0.05% Tween 20 (TBST). Then the primary goat polyclonal anti-rat Cx3cl1 (1:20; AF537; R&D Systems) or normal goat IgG isotype control (1:20; AB-108-C; R&D Systems) was added in 1% BSA and slides were incubated overnight at room temperature. The slides were washed twice with TBST, and the secondary biotinylated anti-goat IgG antibody (1:500; BA-5000; Vector) was added. The slides were incubated for 30 minutes at room temperature and, following a TBST wash, detection of immunoreaction was achieved using the streptavidin–horseradish peroxidase (SA–horseradish peroxidase) system (1:400; Dako) containing SA-peroxidase complex after 30 minutes incubation at room temperature. Color was developed with 3,3′-diaminobenzidine (Sigma-Aldrich) and hydrogen peroxide (Sigma-Aldrich), and slides were subsequently counterstained with hematoxylin, dehydrated in graded ethanol, and mounted with Permount (Fisher Scientific). At least 2 different sections of each organ per time point were tested per experiment in 3 independent experiments using a combined total of 8 Cx3cr1+/+ and 8 Cx3cr1–/– mice.
Confocal microscopy.
Confocal microscopy was performed on freshly excised, unfixed kidneys from Cx3cr1gfp/+ and Cx3cr1gfp/gfp mice at 2 hours, day 1, and day 6 after intravenous Candida challenge. The animals were euthanized by carbon dioxide asphyxiation, and both kidneys were rapidly excised, sliced longitudinally, and placed in PBS solution on ice. The sliced tissue was then placed into a custom-made dish fitted with 40 mm round glass coverslip no. 1.5. Imaging was carried out within 30 minutes after excision to ensure preserved morphology of the fresh tissue using an Olympus IX81 inverted microscope equipped with a Fluoview 1000 confocal scanning head (Olympus America). GFP was excited with a 488-nm laser, whereas dTomato was excited with a 561-nm laser. The emission was collected using appropriate filter sets. Cx3cr1+/+ animals and animals injected with Candida not expressing dTomato were used as controls to determine the proper acquisition settings for GFP-expressing cells and dTomato-expressing fungus, respectively. The images were acquired using the following objectives: ×2, ×10, and UPLSAPO ×60 numerical aperture (NA) 1.2 water immersion objective (Olympus America). The focal plane for lower magnification objectives was set at approximately 10 μm from the surface of the tissue, and then single plane snapshots were acquired. Some of the images using the ×60 objective were acquired as 43-slice Z-stacks. Pixel width was set to 414 nm, and the Z-step was set to 0.79 μm to achieve minimal overlap between the adjacent slices. Images were prepared in Imaris Bitplant Scientific software (version 7.5.2) by taking snapshots of maximal intensity projections of the Z-stacks.
To compare the interaction of dTomato-Candida with Cx3cr1gfp/+ versus Cx3cr1gfp/gfp kidney mononuclear phagocytes in vivo, both kidneys from Cx3cr1gfp/+ and Cx3cr1gfp/gfp mice infected 2 hours prior to euthanization were processed as mentioned above. The percentage of Candida in contact with mononuclear phagocytes was quantified by evaluating 11–28 randomly selected dTomato-Candida structures on the surface of each kidney and determining whether they were in contact with GFP-expressing mononuclear phagocytes. Three independent experiments were performed using a combined total of 3 Cx3cr1gfp/+ and 3 Cx3cr1gfp/gfp mice per time point.
Determination of serum blood urea nitrogen and creatinine.
Blood urea nitrogen (BUN) and creatinine concentrations were measured in the serum of Cx3cr1+/+ and Cx3cr1–/– mice at day 9 after infection as markers of renal function. Blood was collected by cardiac puncture and stored at –80°C until analysis of BUN and creatinine at the NIH Clinical Chemistry Laboratory. Two independent experiments were performed using a combined total of 8–10 Cx3cr1+/+ and 8–10 Cx3cr1–/– mice.
Gene expression analysis by real-time quantitative PCR.
Control uninfected and Candida-infected Cx3cr1+/+ and Cx3cr1–/– mice were euthanized on days 3, 6, and 9 after infection (n = 4–5 mice per time point), and mRNA was extracted from the kidney using Trizol (Invitrogen) and the RNeasy kit (QIAGEN) according to the manufacturer’s instructions. cDNA was generated using the qScript cDNA SuperMix kit (Quanta BioSciences) with oligodT and random primers, and quantitative PCR (qPCR) was performed by SYBR Green (PerfeCTa SYBR Green FastMix ROX; Quanta BioSciences) or TaqMan detection (PerfeCTa qPCR FastMix ROX; Quanta BioSciences) with the 7900HT Fast Real-Time PCR System (Applied Biosystems) in a total reaction volume of 22 μl using 2 μl cDNA, 11 μl 2× SYBR Green or TaqMan PCR Master mix, and 1.1 μl of forward primer/1.1 μl of reverse primer (SYBR Green) or 1.1 μl primer–probe mix (TaqMan). All qPCR assays were performed in duplicate and the relative gene expression of each molecule compared with uninfected mice was determined after normalization with GAPDH transcript levels using the ΔΔCT method, displayed as mRNA fold change. The primers using SYBR Green detection were designed from GenBank sequences using software Primer3 ( http://frodo.wi.mit.edu/primer3/) and purchased from Invitrogen (sequences shown in Supplemental Table 5), whereas TaqMan primers/probes were predesigned by Applied Biosystems. Two independent experiments were performed using a combined total of 8–10 Cx3cr1+/+ and 8–10 Cx3cr1–/– mice per time point.
Chemokine protein quantification.
Kidneys from Cx3cr1+/+ and Cx3cr1–/– mice were harvested before infection and at days 3, 6, and 9 after infection and were homogenized with an Omni Tissue Homogenizer into PBS with 0.5% Tween 20 (Bio-Rad) and a protease inhibitor cocktail (11873580001; Roche Applied Science) and centrifuged at 15,682 g for 10 minutes at 4°C. The supernatants were clarified, aliquoted, and frozen at –80°C until use. Mouse Cxcl1 and Cxcl2 were quantified using a Luminex Magnetic bead array Milliplex MAP Kit (MCYTOMAG-70K; Millipore). One experiment was performed using a total of 4 Cx3cr1+/+ and 4 Cx3cr1–/– mice per time point.
Single-cell suspension from mouse kidney.
Mice were euthanized before infection and at days 1, 3, 6, and 9 after infection (n = 3–5 mice/time point). Mice were anesthetized using ketamine/xylazine and perfused with 10 ml of normal saline before kidney harvesting. Single-cell suspensions from kidney were prepared using previously described methods (9). In brief, kidneys were finely minced and digested at 37°C in digestion solution (RPMI 1640 with 20 mM HEPES [Mediatech] without serum) containing liberase TL (05401020001; Roche) and grade II DNAse I (10104159001; Roche) for 20 minutes with shaking. Digested tissue was passed through a 70-μm filter and washed; the remaining red cells were lysed with ACK lysis buffer (Lonza). Then,the cells were passed through a 40-μm filter, washed, and suspended in 40% Percoll (GE Healthcare). The suspension was overlaid on 70% Percoll and centrifuged at 836 g for 30 minutes at room temperature. The leukocytes and nonhematopoietic cells at the interphase were isolated, washed 3 times in FACS buffer (PBS plus 0.5% BSA plus 0.01% NaN3), suspended in PBS, and passed though a 40-μm filter.
Flow cytometry.
The single-cell suspensions were first stained with a LIVE/DEAD fluorescent dye (L-23105; Invitrogen) for 10 minutes (1:500) in PBS at 4°C and then incubated with rat anti-mouse CD16/32 (2.4G2; BD Biosciences) for 10 minutes (1:100) in FACS buffer at 4°C to block Fc receptors. For staining of surface antigens, cells were incubated with fluorochrome-conjugated (FITC, PE, PE-Cy7, allophycocyanin [APC], APC-Cy7, APC-eFluor 780, Alexa Fluor 700, eFluor 450, eFluor 605 NC, or PerCP-Cy5.5) antibodies against mouse CD45 (Ly-5), CD45.1 (A20), CD45.2 (clone 104) (eBioscience); Ly6C (AL-21), Ly6G (1A8) (BD Biosciences); F4/80 (BM8), CD11b (M1/70), MHC II (M5/114.15.2), CD11c (N418), CD103 (2E7), TCR-β (H57-597), CD3e (145-2C11), CD4 (GK1.5), CD8 (53-6.7), CD19 (1D3), NK1.1 (PK136), CD117/c-Kit (2B8), TLR2/CD282 (6C2) (eBioscience); dectin-1 (GE2) (AbD Serotec); CD29/Integrin-β1 (HMb1-1), CD31/PECAM-1 (clone 390), CD102/ICAM-2 (3C4), and CD324/E-cadherin (DECMA-1) (eBioscience). After 3 washes with FACS buffer, stained cells were analyzed on a 5-laser LSRII (BD Biosciences), and the data were analyzed using FACS Diva (BD Biosciences) and FlowJo software (Treestar). Only single cells were analyzed, and cell numbers were quantified using PE-conjugated fluorescent counting beads (Spherotech).
FACS analysis of macrophages and neutrophils for annexin V and 7-AAD expression.
To determine whether Cx3cr1 deficiency affects macrophage and neutrophil survival before and after Candida infection, we performed FACS analysis on kidney leukocytes from Cx3cr1+/+ and Cx3cr1–/– mice on days 0, 1, 3, 6, and 9 after infection. Cells were initially incubated for 10 minutes with rat anti-mouse CD16/32 and then stained for 30 minutes with the combination of fluorochrome-conjugated Ly6G (BD Biosciences), CD45, MHCII, F4/80, CD11b, CD3, CD19, and NK1.1 (eBioscience). Then the cells were washed twice with PBS, suspended in 1× annexin V binding buffer (BD Biosciences), and incubated for 15 minutes with 5 μl each of FITC-conjugated annexin V and 7-AAD according to the manufacturer’s instructions (BD Biosciences). The cells were washed and FACS was performed on a 5-laser LSRII. Two independent experiments were performed using a total of 6 to 8 Cx3cr1+/+ and 6 to 8 Cx3cr1–/– mice per time point.
FACS analysis of macrophages for Ki-67 expression.
To determine whether Cx3cr1 deficiency affects in situ proliferation of macrophages in the kidney before and after Candida infection, we performed FACS analysis on kidney macrophages from Cx3cr1+/+ and Cx3cr1–/– mice on days 0, 3, 6, and 9 after infection. Cells were initially incubated for 10 minutes with a LIVE/DEAD fluorescent dye, followed by a 10-minute incubation with rat anti-mouse CD16/32 and were then stained for 30 minutes with the combination of fluorochrome-conjugated CD45, MHCII, F4/80, CD11b, CD3, CD19, NK1.1, and Ly6G (eBioscience and BD Biosciences). Then the cells were washed twice with PBS, fixed, and permeabilized for 20 minutes with the BD Cytofix/Cytoperm solution (BD Biosciences) and incubated for 30 minutes with PE-conjugated mouse monoclonal anti-human Ki-67 (51-36525X, clone B56; BD Biosciences) or PE-conjugated mouse IgG1 (51-35405X, clone MOPC-21; BD Biosciences) in BD Perm/Wash buffer according to the manufacturer’s instructions (BD Biosciences). After intracellular staining with Ki-67 or isotype control, the cells were washed and FACS was performed on a 5-laser LSRII. Two independent experiments were performed using a combined total of 6 to 8 Cx3cr1+/+ and 6 to 8 Cx3cr1–/– mice per time point.
FACS analysis of macrophages for pAKT and cleaved caspase-3 expression.
To determine whether Cx3cr1 deficiency affects Akt phosphorylation and caspase-3 cleavage in kidney-resident macrophages, we performed FACS analysis on kidney macrophages from uninfected Cx3cr1+/+ and Cx3cr1–/– mice. Cells were initially incubated for 10 minutes with a LIVE/DEAD fluorescent dye, followed by a 10-minute incubation with rat anti-mouse CD16/32, and were then stained for 30 minutes with the combination of fluorochrome-conjugated CD45, MHCII, F4/80, and CD11b (eBioscience).
For cleaved caspase-3 intracellular staining, the cells were washed twice with PBS, fixed, and permeabilized for 20 minutes with the BD Cytofix/Cytoperm solution (BD Biosciences), and incubated for 30 minutes with or without PE-conjugated rabbit anti-cleaved caspase-3 (550821; BD Biosciences) in BD Perm/Wash buffer according to the manufacturer’s instructions (BD Biosciences). The cells were then washed and FACS was performed on a 5-laser LSRII. Three independent experiments were performed using a combined total of 9 Cx3cr1+/+ and 9 Cx3cr1–/– mice.
For pAKT intracellular staining, the cells were washed twice with PBS, fixed with 4% formaldehyde for 10 minutes at room temperature, and permealized with an overnight incubation with ice-cold 90% methanol. The cells were then washed and incubated for 30 minutes with PE-conjugated mouse monoclonal anti-AKT (pT308) (558275, clone J1-223.371; BD Biosciences) or PE-conjugated mouse IgG1, k isotype control (556620, clone MOPC-21; BD Biosciences) in FACS buffer. The cells were then washed and FACS was performed on a 5-laser LSRII. Two independent experiments were performed using a combined total of 6 Cx3cr1+/+ and 6 Cx3cr1–/– mice.
FACS analysis of hematopoietic and nonhematopoietic cells for Ccr2 and Cx3cr1 expression.
To determine the expression of Ccr2 and Cx3cr1 on hematopoietic and nonhematopoietic cells in the kidney, we performed FACS analysis on kidney cells from Cx3cr1+/+ and Cx3cr1gfp/+ mice on days 0, 1, 3, and 6 after infection. Single-cell suspensions were prepared as described above, and cells were incubated for 10 minutes with a LIVE/DEAD fluorescent dye, followed by staining with rat anti-mouse CD16/32 for 10 minutes. Then the cells were stained for 30 minutes with a combination of fluorochrome-conjugated antibodies for labeling CD45+ hematopoietic (MHCII, F4/80, CD11c, Ly6C, Ly6G, CD11b, CD103, CD3, CD19, NK1.1; eBioscience and BD Biosciences) and CD45– nonhematopoietic cells (CD29, CD31, CD102, CD324; eBioscience) and PE-conjugated rat monoclonal anti-mouse Ccr2 (FAB5986P, clone 643854; R&D Systems) or PE-conjugated rat IgG2B (IC013P, clone 141945; R&D Systems). The cells were then washed and FACS was performed on a 5-laser LSRII. Two independent experiments were performed using a combined total of 6 Cx3cr1+/+ and 6 Cx3cr1gfp/+ mice per time point.
Isolation of kidney monocytes, macrophages, and DCs for TEM.
MHCII–F4/80intCD11b+ monocytes, MHCIIhiF4/80hiCD11b+ macrophages, and MHCIIhiF4/80loCD11chi dendritic cells were FACS sorted from Candida-infected Cx3cr1+/+ mice at day 6 after infection for TEM analysis. Single-cell suspensions from kidneys were prepared as described above and, following Fc blockade, cells were stained with FITC-conjugated CD45, eFluor 450–conjugated MHCII, PE-Cy7–conjugated F4/80, PerCP-Cy5.5–conjugated CD3, CD19 and NK1.1, APC-conjugated CD11c, APC-eFluor 780–conjugated CD11b (eBioscience), Alexa Fluor 700–conjugated Ly6C, and PerCP-Cy5.5–conjugated Ly6G (BD Biosciences). The 3 myeloid cell populations were sorted using a FACSAria instrument (BD Biosciences). Purity of sorted monocytes, macrophages, and dendritic cells was greater than 96%. FACS-sorted monocytes, macrophages, and dendritic cells were then centrifuged and cell pellets were fixed overnight at 4°C with 2.5% glutaraldehyde/4% paraformaldehyde in 0.1 M sodium cacodylate buffer (15960; Electron Microscopy Sciences) and then post-fixed for 30 minutes with 0.5% osmium tetroxide/0.8% potassium ferricyanide, 1 hour with 1% tannic acid, and overnight with 1% uranyl acetate at 4°C. Samples were dehydrated with a graded ethanol series and embedded in Spurr’s resin. Thin sections were cut with an RMC MT-7000 Ultramicrotome (Ventana) and stained with 1% uranyl acetate and Reynold’s lead citrate before viewing at 80 kV on a transmission electron microscope (H-7500; Hitachi). Digital images were acquired with a Hamamatsu XR-100 bottom-mount charge-coupled device system (Advanced Microscopy Techniques).
Gene expression of Cx3cl1 on sorted kidney cells.
Cx3cr1+/+ mice were used to extract mRNA from FACS-sorted kidney MHCIIhiF4/80hiCD11b+ macrophages (purity, >92%) at steady state and day 6 after infection, from kidney CD45–CD102+ endothelial cells (purity, >90%) at steady state, and from kidney CD45–E-cadherin+ tubular cells (purity, >86%) at steady state using Trizol per the manufacturer’s protocol. cDNA was generated, and qPCR was performed for Cx3cl1 as described above. Relative gene expression was determined after normalization with GAPDH transcript levels using the ΔΔCT method and displayed as gene amplicons. Two to three independent experiments were performed using a combined total of 4 to 6 Cx3cr1+/+ mice.
Western blot analysis on sorted kidney macrophages.
MHCIIhiF4/80hiCD11b+ macrophages were FACS sorted (purity, >96%) from Cx3cr1+/+ and Cx3cr1–/– kidneys at day 6 after infection for Western blot analysis of cleaved caspase-3 and phosphorylated Akt. Sorted cells were washed once with ice-cold PBS and lysed in buffer containing 50 mM HEPES, 50 mM NaCl, 10% glycerol, 0.5% Nonidet P-40, 2 mM EDTA, and a Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific). Cell lysates were centrifuged at 13,200 g for 10 minutes at 4°C, and equal protein amounts from the supernatant were resuspended in SDS loading buffer with 5% β-mercaptoethanol for downstream Western blot analysis. Proteins were analyzed by SDS-PAGE and transferred to Immobilon P membranes, as previously described (44). Blocking was performed by incubating the membranes with TBST containing 5% nonfat dry milk. Membranes were then incubated with rabbit monoclonal antibodies against cleaved caspase-3 (9661 and 9664; Cell Signaling Technology) or phospho-Akt (Thr308) (2965; Cell Signaling Technology), and polyclonal antibody against β-actin (4967; Cell Signaling Technology) for 2 hours at room temperature under continuous agitation, washed 3 times with TBST, and incubated with horseradish peroxidase–conjugated anti-rabbit secondary antibody (Southern Biotech) for 1 hour at room temperature. Membranes were subsequently washed, and detection of immunoreactive bands was performed using the SuperSignal West Fempto Maximum Sensitivity Substrate chemiluminescence (ECL) detection kit according to the manufacturer’s instructions (Pierce Biotechnology). The protein levels that corresponded to the immunoreactive bands were quantified using ImagePC image analysis software (Scion Corp.). Two independent experiments were performed using a combined total of 16 Cx3cr1+/+ and 16 Cx3cr1–/– kidneys, which were pooled in 4 groups, each consisting of 4 kidneys from 2 mice.
Generation of BM chimeras.
For BM cell transfers, femurs and tibias from donor 6- to 8-week-old Cx3cr1+/+ (CD45.1+) and Cx3cr1–/– (CD45.2+) mice were removed aseptically and BM was flushed using cold PBS supplemented with 2 mM EDTA. Six- to eight-week-old recipient Cx3cr1+/+ (CD45.1+) and Cx3cr1–/– (CD45.2+) mice were irradiated with 9 Gy and were reconstituted 6 hours after irradiation with 5 × 106 Cx3cr1+/+ (WT→WT and WT→Cx3cr1–/– mice) or Cx3cr1–/– (Cx3cr1–/–→Cx3cr1–/– and Cx3cr1–/–→WT mice) cells by lateral tail-vein injection. Mice were given trimethoprim/sulfamethoxazole in the drinking water for the first 4 weeks of reconstitution before being switched to antibiotic-free water. Chimeras were infected with Candida 10 weeks after transplantation. Prior to infection, we confirmed that mice reconstituted with congenic BM stem cells had achieved a satisfactory level of chimerism by assessing the number of CD45.1+ (Cx3cr1+/+) and CD45.2+ (Cx3cr1–/–) leukocytes in the blood and kidney, using flow cytometry. In the blood, the proportion of donor-derived monocytes, neutrophils, and B cells exceeded 95%, whereas that of T cells exceeded 85% (Supplemental Figure 2). In the kidney, the proportion of donor-derived macrophages exceeded 90% in Cx3cr1–/–→WT mice and 96% in WT→Cx3cr1–/– mice (Supplemental Figure 2). Two independent experiments were performed using a combined total of 15 WT→WT, 15 Cx3cr1–/–→Cx3cr1–/–, 25 WT→Cx3cr1–/–, and 25 Cx3cr1–/–→WT mice.
Isolation and adoptive transfer of BM monocytes.
3 × 106 Fms-like tyrosine kinase 3 ligand–expressing (FLT3L-expressing) B16 cells (gift of Brian Kelsall, NIAID) were injected subcutaneously in Cx3cr1gfp/+ and Cx3cr1gfp/gfp donor mice 12–14 days before monocyte isolation to increase their total BM monocyte numbers (45). BM was then flushed from the tibias and femurs of donor mice as described above. After red blood cell lysis using ACK lysis buffer (Lonza), CD115+ cells were enriched using the AutoMACS automatic magnetic cell-sorting system on an AutoMACS separator (Miltenyi Biotec) using a biotinylated CD115 antibody (AFS98; eBioscience) followed by anti-biotin microbeads (130-090-485; Miltenyi Biotec) according to the manufacturer’s instructions. CD115-enriched cells were then stained for Alexa Fluor 700–conjugated Ly6C (AL-21), APC-eFluor 780–conjugated CD11b (M1/70), SA-PE, and a mixture of APC-conjugated lineage antibodies (TCR-β [H57-597], CD4 [GK1.5], CD8 [53-6.7], CD19 [1D3], NK1.1 [PK136], CD11c [N418], CD117/c-Kit [2B8] and Ly6G [1A8]). Monocytes were sorted as Lin–CD115biotin/SA+GFP(Cx3cr1)+Ly6ChiCD11b+ cells using a FACSAria (BD Biosciences) (45). Monocyte purity after sorting was greater than 98%. Monocytes were resuspended in sterile PBS at a final concentration of 20 × 106 cells/ml, and a total of 4 × 106 (200 μl) Cx3cr1gfp/+ or Cx3cr1gfp/gfp monocytes were injected intravenously per animal in Cx3cr1+/+ Candida-infected mice on day 3 after infection.
To determine the role of Cx3cr1 on trafficking of monocytes from the blood into the kidney, mice were euthanized 12 hours after monocyte transfer, and to determine the role of Cx3cr1 on differentiation of monocytes into kidney macrophages and cell persistence into the kidney, mice were euthanized 3 days after monocyte transfer. The kidneys were harvested and single-cell suspensions were prepared as described above. After LIVE/DEAD staining and Fc blockade, leukocytes were stained with eFluor 605 NC–conjugated CD45, eFluor 450-conjugated MHCII, PE-Cy7–conjugated F4/80, APC-conjugated CD11c, Alexa Fluor 700–conjugated Ly6C, and APC-eFluor 780–conjugated CD11b. Monocyte migration from the blood into the kidney was calculated based on the number of GFP+ Cx3cr1gfp/+ versus Cx3cr1gfp/gfp monocytes detected in the kidney at 12 hours after transfer. Two independent experiments were performed using a combined total of 14 Cx3cr1+/+ mice, 8 injected with Cx3cr1gfp/+ monocytes and 6 injected with Cx3cr1gfp/gfp monocytes. Monocyte differentiation into kidney macrophages was calculated based on the percentage of GFP+ Cx3cr1gfp/+ versus Cx3cr1gfp/gfp cells that had become MHCIIhiF4/80hiCD11b+ macrophages 3 days after transfer. Cell persistence in the kidney was calculated based on the number of GFP+ Cx3cr1gfp/+ versus Cx3cr1gfp/gfp cells detected in the kidney at 3 days after transfer. Two independent experiments were performed using a combined total of 12 Cx3cr1+/+ mice, 6 each injected with Cx3cr1gfp/+ or Cx3cr1gfp/gfp monocytes.
Generation of BMMs and FACS analysis for Cx3cr1 expression.
BMMs were generated by culturing BM cells obtained as described above from Cx3cr1+/+ and Cx3cr1–/– mice in RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamine (Mediatech), 20 mM HEPES, 1% penicillin/streptomycin and 40 ng/ml M-CSF (Peprotech Inc.) for 5 to 7 days, at which time point more than 95% of cells were viable and expressed F4/80 and CD11b (data not shown). BMMs were Cx3cr1+ as determined by FACS using a combined total of 6 Cx3cr1+/+ and 6 Cx3cr1gfp/+ mice in 2 independent experiments (data not shown).
Expression of dectin-1 and TLR2 on BMMs.
106 Cx3cr1+/+ or Cx3cr1–/– BMMs were incubated for 10 minutes with a LIVE/DEAD fluorescent dye, followed by a 10-minute incubation with rat anti-mouse CD16/32 to block Fc receptors. Then the cells were stained for 30 minutes with FITC-conjugated rat monoclonal anti-mouse dectin-1 (2A11; AbD Serotec), PE-conjugated rat monoclonal anti-mouse TLR2/CD282 (6C2; eBioscience), or rat IgG2B conjugated with FITC or PE (eB149/10H5; eBioscience). The cells were then washed and FACS was performed on a 5-laser LSRII. Two independent experiments were performed using a combined total of 4 Cx3cr1+/+ and 5 Cx3cr1–/– mice.
Binding of Candida and zymosan by BMMs.
To determine whether Cx3cr1 deficiency affects the capacity of macrophages to bind Candida or zymosan, 2.5 × 105 Cx3cr1+/+ or Cx3cr1–/– BMMs were plated in 24-well tissue culture plates (Corning Inc.) and were incubated with EGFP-Candida yeast cells (or FITC-labeled zymosan particles [Z2841; Invitrogen]) at a ratio of 25 yeast cells (or 25 zymosan particles) per macrophage as previously described (46). The yeast cells (or zymosan particles) were allowed to bind to BMMs for 30 minutes on ice, and unbound yeast cells (or zymosan particles) were washed 3 times with RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamine, and 1% penicillin/streptomycin. Then the BMMs were lysed with 3% Triton X-100 (Sigma-Aldrich) and transferred to black 96-well plates (Greiner Bio-One Inc.). Fluorescence was measured using a fluorescent plate reader with excitation wavelength of 484 nm (or 490 nm for zymosan particles) and emission wavelength of 510 nm (or 514 nm for zymosan particles). From 2 to 3 independent experiments were performed using a combined total of 6 to 9 Cx3cr1+/+ and 6 to 9 Cx3cr1–/– mice.
Candida phagocytosis by BMMs.
To determine whether Cx3cr1 deficiency affects the capacity of macrophages to internalize Candida, 106 Cx3cr1+/+ or Cx3cr1–/– BMMs were incubated for 30 minutes at 37°C with opsonized EGFP-Candida yeast cells at a Candida/BMM ratio of 1:1 or 5:1. After washing, BMMs were incubated with a biotinylated anti–C. albicans antibody (ab53892; Abcam) for 30 minutes at 4°C followed by a 20-minute incubation with APC-conjugated SA (eBioscience). The percentage of Candida phagocytosis by BMMs was then determined using FACS by initially gating on FITC+ macrophages and then by determining the percentage of FITC+ macrophages that had internalized Candida (FITC+APC–) versus those that had Candida bound on the cell surface (FITC+APC+). Four independent experiments were performed using a combined total of 12 Cx3cr1+/+ and 12 Cx3cr1–/– mice.
Candida killing by kidney-resident macrophages.
To determine whether kidney-resident macrophages have Candida-killing capacity and to assess whether Cx3cr1 deficiency affects the capacity of these cells to kill Candida, we used the alamarBlue-based fluorescence assay, as previously described (28). In brief, 5 × 104 FACS-sorted kidney MHCIIhiF4/80hiCD11b+ macrophages (purity, >97%) from uninfected Cx3cr1+/+ or Cx3cr1–/– mice were incubated in flat-bottom 96-well plates (Corning Inc.) with opsonized Candida yeast cells at a macrophage/Candida ratio of 2:1 in duplicate for 3 hours at 37°C in a 5% CO2 incubator. Then the wells were treated with 0.02% Triton X-100 in water for 5 minutes to lyse macrophages, washed twice with PBS, and incubated with alamarBlue (Invitrogen) for 18 hours at 37°C. Fluorescence was measured using a POLARstar OPTIMA plate reader (BMG LABTECH). Killing was calculated by comparing the fluorescence of Candida incubated with macrophages with that of Candida incubated without macrophages. Three independent experiments were performed using a combined total of 6 Cx3cr1+/+ and 6 Cx3cr1–/– mice.
Human subjects.
Patients were enrolled between January 2003 and January 2009 at the Duke University Hospital (DUMC) and Radboud University Nijmegen Medical Center (RUNMC) (Supplemental Table 2). The clinical characteristics of the Candida-infected and noninfected control patients are summarized in Supplemental Table 3.
CX3CR1 genotype and susceptibility to systemic candidiasis.
To be included in the analysis of susceptibility to systemic candidiasis, infected subjects from both DUMC and RUNMC must have had 1 or more positive blood cultures for a Candida species while hospitalized at the participating center; infected subjects were enrolled consecutively. Noninfected controls from DUMC must have been hospitalized with no history or evidence of candidemia/systemic candidiasis or any other systemic fungal infection. None of the control patients were on antifungal therapy/prophylaxis. Although it was impracticable to match controls on specific risk factors, noninfected control individuals were recruited consecutively from the same hospital wards/services as Candida-infected patients during the study period, with a similar balance of medical, surgical, and oncology patients in Candida-infected and noninfected control groups. Subjects were excluded from the study if insufficient volume of blood or clinical data were available to allow for analysis. In addition, allogeneic hematopoietic stem cell transplant patients were excluded. Immunocompromised status was defined as any of the following conditions: neutropenia (absolute neutrophil count <500/mm3), CD4+ lymphopenia (absolute CD4 count <250/mm3 associated with or without HIV), chronic steroid use or receipt of immunosuppressive chemotherapy/immunosuppressant medications within 30 days prior to baseline, diabetes mellitus, and end-stage renal disease requiring hemodialysis. Acute renal failure was defined based on clinical diagnosis, including doubling of serum creatinine and/or requirement for acute dialysis (intermittent hemodialysis or continuous veno-venous hemofiltration dialysis). Noninfected controls from RUNMC were healthy volunteers from the Netherlands enrolled between 2009 and 2011 without history of infection or underlying chronic disease. None of the control patients from RUNMC were on antifungal therapy/prophylaxis. Intergroup comparisons between the 2 groups of noninfected subjects and between the 2 groups of Candida-infected subjects (at DUMC and RUNMC) were performed regarding similarity in genetic distribution of the studied CX3CR1 SNP prior to further statistical analysis of Candida-infected versus noninfected subjects.
CX3CR1 genotype and association with clinical outcomes after systemic candidiasis.
Due to the lack of detailed clinical data on the Dutch candidemia patients recruited at RUNMC, the correlations of the CX3CR1 genetic data with clinical outcome were performed only for the patients from DUMC (n = 244). Infected subjects at DUMC were followed prospectively following diagnosis of candidemia to determine clinical outcome. Mortality data, including date of death, was ascertained from hospital records and/or the National Social Security Death Index. Disseminated infection was defined as the presence of Candida species at sterile body sites outside the bloodstream with the exception of urine. The most frequent manifestations of disseminated infection were abdominal abscess/peritonitis (55%), hepatosplenic candidiasis (13%), and endocarditis/endovascular infection (11%). Persistent fungemia was defined as 5 days or more of persistently positive Candida blood cultures.
CX3CR1-M280 genotyping.
A 50-μl PCR reaction was performed to genotype this site using 200 ng of genomic DNA isolated from whole blood using standard procedures and 15 pmoles of each of the following primers: 5′-AGAATCATCCAGACGCTGTTTTCC-3′ and 5′-CACAGGACAGCCAGGCATTTCC-3′. Other conditions were as follows: 2.0 mM MgCl2, 175 μM each of dNTP, 1.5 U Taq polymerase, and 1X buffer (Life Technologies). Amplification reactions were performed using 35 cycles of 95°C, 69°C, and 72°C for 30 seconds each, preceded by a single cycle of 95°C for 3 minutes and followed by a single cycle of 72°C for 10 minutes. This produced an amplicon of 311 bp. In order to type the alleles at codon 280, 5 μl of the resulting PCR reaction mix was digested with 1.5 U of Bst4CI (New England BioLabs) in a 20-μl reaction incubated overnight at 65°C according to the manufacturer’s instructions. This resulted in 107- and 204-bp fragments when a C was present at nucleotide 839 of the open reading frame, while the amplicon remained uncut if a T was present. The genotype was determined by electrophoresis on a 2% agarose gel stained with Gelstar (FMC Bioproducts) according to the manufacturer’s instructions.
Statistics.
The mouse experimental data were analyzed using the 2-tailed unpaired t-test or the Mann-Whitney test where appropriate with Prism 6.0 software (GraphPad Software) and are presented as the mean ± SEM. The cutoff for statistical significance was defined as P < 0.05.
For the analysis of the impact of the CX3CR1 genotype on susceptibility to candidemia, statistical comparisons of frequencies were made between Candida-infected versus noninfected subjects using the χ2 test. Data were analyzed for individuals of mixed European descent separately from African-Americans, as allelic frequencies were expected to differ between these 2 populations; indeed, examination of race-stratified genotype frequencies for the tested SNP confirmed this expectation (Table 1 and Supplemental Table 4). Univariate correlations were performed by use of the Pearson correlation coefficient or Fisher’s exact test as appropriate. Differences in genotypic frequencies for the CX3CR1-M280 allele were examined for significance using the Pearson χ2 test or Fisher’s exact test. Standard summary statistics (OR and 95% CI) were reported for these tests of association. Baseline demographic and clinical categorical variables in the case and control groups were compared by χ2 analysis or Fisher’s exact test (as appropriate), and variables with P < 0.1 were analyzed as confounders of the CX3CR1-M280 SNP using logistic regression. SAS software version 9.2 (SAS Corporation) was used for all analyses.
Within the infected DUMC cohort, allelic frequencies were further assessed in association with 4 prespecified clinical outcomes: (a) disseminated disease, (b) persistent fungemia, (c) all-cause mortality at 14 days, and (d) all-cause mortality at 30 days. Variables with P < 0.1 were further assessed in a multivariable logistic regression model using backward elimination. Variables with P < 0.05 were retained in the final predictive model. ORs and 95% CIs were reported for variables that remained significant in the final multivariable model.
Study approval.
All mice were maintained at an American Association for the Accreditation of Laboratory Animal Care–accredited animal facility at the NIAID and housed in accordance with the procedures outlined in the Guide for the Care and Use of Laboratory Animals under the auspices of a protocol approved by the Animal Care and Use Committee of the NIAID.
The study patients were enrolled after informed consent (or waiver as approved by the Institutional Review Board) at DUMC and RUNMC. The study was approved by the Institutional Review Boards at each study center and was performed in accordance with the Declaration of Helsinki.
Supplementary Material
Acknowledgments
This work was supported by the Division of Intramural Research (DIR), NIAID, NIH, and in part by the Transition Program in Clinical Research (TPCR) of the DIR, NIAID, NIH. M.G. Netea was supported by an ERC Consolidator Grant (no. 310372). The funding source had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors thank Elizabeth Fischer of the Research Technologies Branch, NIAID, for performing the TEM, and the NIAID FACS sorting facility and the Comparative Medicine Branch animal facilities for technical assistance and animal handling. This work was presented in part at the 52nd Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, California, September 9–12, 2012, and at the 2nd Gordon Research Conference on Immunology of Fungal Infections, Galveston, Texas, January 13–18, 2013.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Note regarding evaluation of this manuscript: Manuscripts authored by scientists associated with Duke University, The University of North Carolina at Chapel Hill, Duke-NUS, and the Sanford-Burnham Medical Research Institute are handled not by members of the editorial board but rather by the science editors, who consult with selected external editors and reviewers.
Citation for this article: J Clin Invest. 2013;123(12):5035–5051. doi:10.1172/JCI71307.
Michail S. Lionakis’s present address is: Fungal Pathogenesis Unit, Laboratory of Clinical Infectious Diseases, National Institute of Allergy and Infectious Diseases, NIH, Bethesda, Maryland, USA.
References
- 1.Zaoutis TE, Argon J, Chu J, Berlin JA, Walsh TJ, Feudtner C. The epidemiology and attributable outcomes of candidemia in adults and children hospitalized in the United States: a propensity analysis. Clin Infect Dis. 2005;41(9):1232–1239. doi: 10.1086/496922. [DOI] [PubMed] [Google Scholar]
- 2.Horn DL, et al. Epidemiology and outcomes of candidemia in 2019 patients: data from the prospective antifungal therapy alliance registry. Clin Infect Dis. 2009;48(12):1695–1703. doi: 10.1086/599039. [DOI] [PubMed] [Google Scholar]
- 3.Miller LG, Hajjeh RA, Edwards JE., Jr Estimating the cost of nosocomial candidemia in the united states. Clin Infect Dis. 2001;32(7):1110. doi: 10.1086/319613. [DOI] [PubMed] [Google Scholar]
- 4.Wilson LS, Reyes CM, Stolpman M, Speckman J, Allen K, Beney J. The direct cost and incidence of systemic fungal infections. Value Health. 2002;5(1):26–34. doi: 10.1046/j.1524-4733.2002.51108.x. [DOI] [PubMed] [Google Scholar]
- 5.Spellberg B, Ibrahim AS, Edwards JE, Jr, Filler SG. Mice with disseminated candidiasis die of progressive sepsis. J Infect Dis. 2005;192(2):336–343. doi: 10.1086/430952. [DOI] [PubMed] [Google Scholar]
- 6.Louria DB, Stiff DP, Bennett B. Disseminated moniliasis in the adult. Medicine. 1962;41:307–338. [Google Scholar]
- 7.Sabesin SM. Renal failure and disseminated candidiasis. Arch Intern Med. 1962;110:526–534. doi: 10.1001/archinte.1962.03620220118020. [DOI] [PubMed] [Google Scholar]
- 8.Pappas PG. Invasive candidiasis. Infect Dis Clin North Am. 2006;20(3):485–506. doi: 10.1016/j.idc.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 9.Lionakis MS, Lim JK, Lee CC, Murphy PM. Organ-specific innate immune responses in a mouse model of invasive candidiasis. J Innate Immun. 2011;3(2):180–199. doi: 10.1159/000321157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romani L, Mencacci A, Cenci E, Del Sero G, Bistoni F, Puccetti P. An immunoregulatory role for neutrophils in CD4+ T helper subset selection in mice with candidiasis. J Immunol. 1997;158(5):2356–2362. [PubMed] [Google Scholar]
- 11.Qian Q, Jutila MA, Van Rooijen N, Cutler JE. Elimination of mouse splenic macrophages correlates with increased susceptibility to experimental disseminated candidiasis. J Immunol. 1994;152(10):5000–5008. [PubMed] [Google Scholar]
- 12.Lionakis MS, Netea MG. Candida and host determinants of susceptibility to invasive candidiasis. . PLoS Pathog. 2013;9(1):e1003079. doi: 10.1371/journal.ppat.1003079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lionakis MS. Genetic susceptibility to fungal infections in humans. Curr Fungal Infect Rep. 2012;6(1):11–22. doi: 10.1007/s12281-011-0076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Legrand F, et al. Adjuvant corticosteroid therapy for chronic disseminated candidiasis. Clin Infect Dis. 2008;46(5):696–702. doi: 10.1086/527390. [DOI] [PubMed] [Google Scholar]
- 15.Brown GD. Innate antifungal immunity: the key role of phagocytes. Annu Rev Immunol. 2011;29:1–21. doi: 10.1146/annurev-immunol-030409-101229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murphy PM, et al. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52(1):145–176. [PubMed] [Google Scholar]
- 17.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71–82. doi: 10.1016/S1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 18. Yang XP, et al. Fractalkine upregulates intercellular adhesion molecule-1 in endothelial cells through CX3CR1 and the Jak Stat5 pathway. Circ Res. 2007. 101 10 1001 1008 [DOI] [PubMed] [Google Scholar]
- 19.Jamieson WL, Shimizu S, D’Ambrosio JA, Meucci O, Fatatis A. CX3CR1 is expressed by prostate epithelial cells and androgens regulate the levels of CX3CL1/fractalkine in the bone marrow: potential role in prostate cancer bone tropism. Cancer Res. 2008;68(6):1715–1722. doi: 10.1158/0008-5472.CAN-07-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jung S, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20(11):4106–4114. doi: 10.1128/MCB.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim KW, et al. In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood. 2011;118(22):e156–e167. doi: 10.1182/blood-2011-04-348946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandrasekar B, et al. Fractalkine (CX3CL1) stimulated by nuclear factor kappaB (NF-κB)-dependent inflammatory signals induces aortic smooth muscle cell proliferation through an autocrine pathway. Biochem J. 2003;373(pt 2):547–558. doi: 10.1042/BJ20030207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fong AM, et al. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J Exp Med. 1998;188(8):1413–1419. doi: 10.1084/jem.188.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.White GE, Tan TC, John AE, Whatling C, McPheat WL, Greaves DR. Fractalkine has anti-apoptotic and proliferative effects on human vascular smooth muscle cells via epidermal growth factor receptor signalling. Cardiovasc Res. 2010;85(4):825–835. doi: 10.1093/cvr/cvp341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jenkins SJ, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332(6035):1284–1288. doi: 10.1126/science.1204351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boehme SA, Lio FM, Maciejewski-Lenoir D, Bacon KB, Conlon PJ. The chemokine fractalkine inhibits Fas-mediated cell death of brain microglia. J Immunol. 2000;165(1):397–403. doi: 10.4049/jimmunol.165.1.397. [DOI] [PubMed] [Google Scholar]
- 27.White GE, Greaves DR. Fractalkine: a survivor’s guide: chemokines as antiapoptotic mediators. Arterioscler Thromb Vasc Biol. 2012;32(3):589–594. doi: 10.1161/ATVBAHA.111.237412. [DOI] [PubMed] [Google Scholar]
- 28.Johnnidis JB, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451(7182):1125–1129. doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- 29.McDermott DH, et al. Chemokine receptor mutant CX3CR1-M280 has impaired adhesive function and correlates with protection from cardiovascular disease in humans. J Clin Invest. 2003;111(8):1241–1250. doi: 10.1172/JCI16790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Plantinga TS, et al. Toll-like receptor 1 polymorphisms increase susceptibility to candidemia. J Infect Dis. 2012;205(6):934–943. doi: 10.1093/infdis/jir867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Medina-Contreras O, et al. CX3CR1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J Clin Invest. 2011;121(12):4787–4795. doi: 10.1172/JCI59150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gautier EL, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13(11):1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Auffray C, et al. CX3CR1+ CD115+ CD135+ common macrophage/DC precursors and the role of CX3CR1 in their response to inflammation. J Exp Med. 2009;206(3):595–606. doi: 10.1084/jem.20081385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li L, et al. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int. 2008;74(12):1526–1537. doi: 10.1038/ki.2008.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karlmark KR, et al. The fractalkine receptor CX3CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology. 2010;52(5):1769–1782. doi: 10.1002/hep.23894. [DOI] [PubMed] [Google Scholar]
- 37.Landsman L, et al. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood. 2009;113(4):963–972. doi: 10.1182/blood-2008-07-170787. [DOI] [PubMed] [Google Scholar]
- 38.Lo HJ, Köhler JR, DiDomenico B, Loebenberg D, Cacciapuoti A, Fink GR. Nonfilamentous C. albicans mutants are avirulent. . Cell. 1997;90(5):939–949. doi: 10.1016/S0092-8674(00)80358-X. [DOI] [PubMed] [Google Scholar]
- 39.Gow NA. Germ tube growth in Candida albicans. . Curr Top Med Mycol. 1997;8(1–2):43–55. [PubMed] [Google Scholar]
- 40.Lionakis MS, et al. Chemokine receptor Ccr1 drives neutrophil-mediated kidney immunopathology and mortality in invasive candidiasis. PLoS Pathog. 2012;8(8):e1002865. doi: 10.1371/journal.ppat.1002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Combadière C, et al. Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation. 2003;107(7):1009–1016. doi: 10.1161/01.CIR.0000057548.68243.42. [DOI] [PubMed] [Google Scholar]
- 42.Wheeler RT, Kombe D, Agarwala SD, Fink GR. Dynamic, morphotype-specific Candida albicans beta-glucan exposure during infection and drug treatment. . PLoS Pathog. 2008;4(12):e1000227. doi: 10.1371/journal.ppat.1000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fonzi WA, Irwin MY. Isogenic strain construction and gene mapping in Candida albicans. . Genetics. 1993;134(3):717–728. doi: 10.1093/genetics/134.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mikelis C, Sfaelou E, Koutsioumpa M, Kieffer N, Papadimitriou E. Integrin α(v)β(3) is a pleiotrophin receptor required for pleiotrophin-induced endothelial cell migration through receptor protein tyrosine phosphatase beta/zeta. FASEB J. 2009;23(5):1459–1469. doi: 10.1096/fj.08-117564. [DOI] [PubMed] [Google Scholar]
- 45.Rivollier A, He J, Kole A, Valatas V, Kelsall BL. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med. 2012;209(1):139–155. doi: 10.1084/jem.20101387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kerrigan AM, de Sousa Mda G, Brown GD. Simple assays for measuring innate interactions with fungi. Methods Mol Biol. 2012;845:303–317. doi: 10.1007/978-1-61779-539-8_20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.