

Abstract
Proteomics has been successfully used for cell culture on dishes, but more complex cellular systems have proven to be challenging and so far poorly approached with proteomics. Because of the complexity of the angiogenic program, we still do not have a complete understanding of the molecular mechanisms involved in this process, and there have been no in depth quantitative proteomic studies. Plating endothelial cells on matrigel recapitulates aspects of vessel growth, and here we investigate this mechanism by using a spike-in SILAC quantitative proteomic approach. By comparing proteomic changes in primary human endothelial cells morphogenesis on matrigel to general adhesion mechanisms in cells spreading on culture dish, we pinpoint pathways and proteins modulated by endothelial cells. The cell–extracellular matrix adhesion proteome depends on the adhesion substrate, and a detailed proteomic profile of the extracellular matrix secreted by endothelial cells identified CLEC14A as a matrix component, which binds to MMRN2. We verify deregulated levels of these proteins during tumor angiogenesis in models of multistage carcinogenesis. This is the most in depth quantitative proteomic study of endothelial cell morphogenesis, which shows the potential of applying high accuracy quantitative proteomics to in vitro models of vessel growth to shed new light on mechanisms that accompany pathological angiogenesis. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium with the data set identifier PXD000359.
Angiogenesis, the process of new blood vessels growth from existing ones, is crucial to provide oxygen and nutrients to the body during development and in diseases such as cancer, where primary tumors need blood supply to fuel their growth (1). During this process, endothelial cells (ECs)1 sprout, digest the extracellular matrix (ECM), differentiate, migrate, proliferate and change morphology to form a new capillary with a lumen. Finally, the ECs mature cell-cell adhesion structures, secrete new ECM and recruit mural cells to stabilize the newly formed vessel. In pathological conditions these cellular processes are deregulated. For example, tumor blood vessels have abnormal ECM structure and composition (2, 3), which may results in leakiness and chaotic blood flow, and negatively impact conventional anticancer treatments (4). Extensive work is therefore devoted to the characterization of molecular mechanisms regulating ECs in angiogenesis and to discover therapeutic targets with improved efficacy to interfere with this process (5, 6).
In vitro models of angiogenesis have been established in which ECs cultured on or embedded in a thick layer of ECM assemble into tubular structures complete with a lumen-like formation. Particularly, ECs plated on matrigel form a tubular network within hours. With this system some of the cellular processes required in the in vivo situation can be investigated, such as cell-ECM adhesion, ECM remodeling, morphogenesis, and proliferation (7).
Several studies of transcriptome changes associated with in vitro models of angiogenesis, such as matrigel and collagen I assays, have profiled gene regulation and begun to unravel protein functions associated with endothelial morphogenesis (8, 9). However, mRNA levels do not always correlate with protein levels and the cell-ECM interaction is mediated by protein complexes. This makes global MS-based proteomics an attractive approach, but so far this technology has not been explored much in the angiogenesis field (10) and the EC proteome during morphogenesis has been investigated only at a low depth (11). We reasoned that an unbiased and comprehensive proteomic study might uncover novel proteins and cellular processes important for angiogenesis.
In the last decade MS-based technology has improved enormously and it is becoming a versatile tool to investigate proteomes in depth, and to accurately quantify their dynamics. In this context, high resolution mass analyzers used in combination with quantitative strategies, such as SILAC, are becoming invaluable to explore biological systems in vitro and in vivo (12). However, the SILAC-labeling of primary ECs is still challenging and the several passages needed for the full incorporation of the labeled amino acids have so far prevented the use of the cells at early passages, which is critical to avoid interference of culture-induced differentiation of primary cells.
In this study we have employed SILAC in an indirect “spike-in” fashion to accurately quantify a large proportion of the proteome dynamics of early passage human umbilical vein endothelial cells (HUVECs), a cell model that has been largely characterized and used to study angiogenesis in vitro, forming tubules on matrigel or spreading on different ECMs. We provide a detailed portrait of the proteomic changes associated to EC morphogenesis and, in combination with MS-proteomic analyses of protein-protein interaction and profiling of ECM proteins secreted by HUVECs, we discovered CLEC14A and MMRN2 as ECM components highly regulated and functional in EC morphogenesis, and deregulated in specific stage of tumor angiogenesis. This shows the power of combining MS quantitative proteomics to in vitro models of angiogenesis for a deeper understanding of the molecular mechanisms regulating ECs in angiogenesis, and relevant in pathology.
EXPERIMENTAL PROCEDURES
Reagents
Matrigel basement membrane matrix and cell recovery solution were from BD Biosciences. Bovine plasma FN, gelatin, Engelbreth-Holm-Swarm murine sarcoma laminin, sheep IgG, and rabbit anti-MMRN2 used for mouse tissues immunofluorescence were from Sigma; sheep polyclonal anti-CLEC14A (AF4968) and anti-CD31/PECAM-1 (AF806) were from R&D Systems (Minneapolis, MN); goat polyclonal (K-17) anti-MMRN2, used for immunoblotting, and mouse monoclonal (6D365) anti-MMRN2 used for HUVECs immunofluorescence were from Santa Cruz Biotechnology (Santa Cruz, CA); purified rat monoclonal anti-Panendothelial cell antigen (clone Meca32) was from BD Pharmingen; rabbit polyclonal anti-FN1, and rabbit anti-NG2 (chondroitin sulfate proteoglycan polyclonal) were from Chemicon (Temecula, CA); rabbit anti-LAMA4 was kindly provided by Prof. Lydia Sorokin. All secondary antibodies used for immunofluorescence were Alexa Fluor 555 or 488 from Molecular Probes. DAPI nucleic acid stain was from Invitrogen; trypsin, sequencing grade modified was from Promega (Madison, WI).
Endothelial Cell Culture and Transgenic Mouse Models
SILAC HUVECs were grown for four passages in custom M199 (Invitrogen, Carlsbad, CA), without arginine and lysine, and supplemented with 42 mg/l of 13C615N4 l-arginine (that we refer to as heavy arginine), 73 mg/l of 13C615N2 l-lysine (that we refer to as heavy lysine), (Sigma-Aldrich, Cambridge Isotope Laboratories), 100 mg/l heparin (Sigma), 20% 1 kDa dialyzed FBS (dialysis in house, Spectra/Por membrane), and bovine brain extract (kindly provided by Prof. Bussolino's lab). Labeled (95%) HUVECs were lysed with RIPA modified buffer: 150 mm NaCl, 50 mm TrisHCl pH 7.4, 1% igepal, 0.1% sodium deoxycholate, 1 mm EDTA, 10% glycerol and protease mixture inhibitors (Roche). The SILAC lysate was snap frozen and stored at −80 °C until use.
HUVECs for the MS proteomic studies, CLEC14A immunoprecipitation and immunofluorescence staining were isolated from umbilical cords and cultured for 3–4 passages in M199 supplemented with 100 mg/l heparin, 20% FBS (Invitrogen), and bovine brain extract (referred to as M199 complete). For all other experiments, HUVECs were cultured in EGM-2 (Lonza). For the proteome study, three experiments were performed; for replicate 1 (exp1) and 2 (exp2) HUVECs at the second passage (P2) were used; for replicate 3 (exp3) at P3.
The generation of RIP-Tag2 mice has been previously described (13). RIP-Tag2 mice were maintained in the C57Bl/6J background (The Jackson Laboratory, West Grove, PA). From 12 weeks of age, all RIP-Tag2 mice received 50% sugar food (Harlan Teklad) and 5% sugar water to relieve hypoglycemia induced by the insulin-secreting tumors. Generation of K14-HPV16 transgenic mice has been previously reported (14). K14-HPV16 mice were maintained in the FVB/n background (The Jackson Laboratory). One-month-old virgin female transgenic (heterozygous K14-HPV16) and nontransgenic (FVB/n) mice were implanted subcutaneously, in the dorsal back skin, with continuous release pellets that deliver 17-β estradiol (E2) at doses of 0.05 mg over 60 days (Innovative Research of America Inc. Sarasota, FL). Subsequent pellets were administrated at 3 and 5 months of age for a total of 6 months of hormone treatment (15, 16). Mice were housed under the approval and the institutional guidelines governing the care of laboratory mice of the University of Torino Committee on Animal Research and in compliance with National and International laws and policies.
Sample Preparation for Proteome Analysis of HUVECs Morphogenesis and Spreading
HUVEC were cultured over-night in M199 10% FBS. Cells were then harvested, re-suspended in M199 10% FBS and seeded at a concentration of 1.2 E6 cells in 10 cm dish. Dishes were previously coated over-night at 4 °C with matrigel diluted 1:1000, 10 μg/ml laminin, 10 μg/ml fibronectin, 4% BSA or 1h at 37 °C with matrigel and matrigel GFR. At the indicated time points, cells were lysed with modified RIPA modified buffer (see above). Cells plated on matrigel were extracted from the matrix by incubating with cell recovery solution for 1h on ice, according to the manufacturer's protocol. Proteins were quantified with Bradford assay (BioRad); 100 μg of each lysate was mixed with 50 μg of SILAC internal standard. The 2:1 ratio non-SILAC:SILAC was used to reduce the noise because of the light signal (5%) contained in the SILAC standard (95% labeled, see above), and improve the accuracy of the protein quantification. Proteins were separated on 4–12% gradient NuPAGE Novex Bis-Tris gel (Invitrogen), two lanes per sample. Each gel lane was cut into 18 slices and the same bands from the two lanes were pooled together. Proteins were in gel digested with trypsin (17) and peptides concentrated and desalted on StageTips (18).
CLEC14A Immunoprecipitation
HUVECs seeded in M199 10% FBS on matrigel overnight were extracted with cell recovery solution and lysed in modified RIPA modified buffer (see above). Lysates were incubated with anti-CLEC14A or sheep IgG cross-linked to the protein G Dynabeads (Invitrogen). Proteins were recovered with Laemmli sample buffer and boiled for 5 min at 95 °C. For Western blot analysis, proteins were separated on 4–12% gradient NuPAGE Novex Bis-Tris gel and transfer on PVDF membrane. For MS analysis, proteins were in-gel digested and peptides loaded on StageTips as described above and analyzed by LC MS/MS (LTQ-Orbitrap XL, see below). Identified proteins were considered significantly enriched in CLEC14A IP compared with IgG with a minimum enrichment of twofold (ratio CLEC14A/IgG) in the two replicates.
Extracellular Matrix and Supernatant Isolation and Analysis
The ECM for MS analysis was isolated according to (19) with minor modifications. Briefly, HUVECs were cultured for 7 days in M199 complete (see above). Cells were then washed with PBS and incubated with a solution 0.5% Triton X-100, 20 mm NH4OH and 1 mm EDTA. Detached cells were aspirated and the remaining ECM thoroughly washed with PBS. The ECM was then solubilized in SDS lysis buffer: 4% SDS, 100 mm DTT and 100 mm TrisHCl pH 7.6, followed by incubation at 95 °C. For MS analysis, proteins from three replicates were in-gel digested and loaded onto StageTips, as described above, and analyzed by LC MS/MS (LTQ-Orbitrap Velos). Only proteins with an intensity measured in the three replicates were considered as components of the HUVEC ECM proteome. See below for more details.
For the analysis of CLEC14A distribution in cells, ECM and supernatant (Fig. 4C), HUVECs were cultured confluent for several days in EGM-2. ECM was prepared as described above; for the supernatant, cells were washed with PBS and incubated in EBM-2 for 6 h. The medium was centrifuged, 12,000 g for 20 min at 4 C. To enrich for secreted proteins, the cleared supernatant was incubated with Strataclean beads (Agilent technologies) according to manufacturer protocol. For the cells, HUVECs were harvested with accutase and lysed in SDS lysis buffer.
Fig. 4.

CLEC14A is a matrixome component and binds ECM proteins with increased abundance in EC morphogenesis. A, Estimated absolute abundance distribution of proteins identified in the ECM produced by HUVECs grown in culture (supplemental Table S6). Ranks were defined according to the median (n = 3) LFQ intensity assigned to each protein. We generated a rank for each order of magnitude (ranges E5 to E10). In red is highlighted the matrixome, which contains 90% of the MS-profiled ECM. The ranks of the matrixome components CLEC14A, MMRN2, HSPG2, and FN1, and the integrins α5 and β1are indicated. Above the plot, the percentage of the ECM represented in the summed ranks is shown. B, Hierarchical clustering, based on Euclidean distance, and heat map of the ratio of 105 matrixome proteins identified during EC morphogenesis (Matr 24h) and spreading (Matr dil and LAM) (supplemental Table S7,S8) (ratio according to supplemental Table S8). The panel highlights the two top clusters of matrixome proteins which increased levels during morphogenesis and that were found more abundant during morphogenesis compared with spreading. These include MMRN2, which was highly enriched in CLEC14A IP (D). Matr/LAM = SILAC ratio Matr 24 h/SILAC ratio LAM; Matr/Matr dil = SILAC ratio Matr 24 h/SILAC ratio Matr dil (see supplemental Table S8). C, Immunoblot analysis for CLEC14A of cell lysate (cells), ECM and supernatant (spr) of HUVECs cultured at confluence in EGM-2 medium. The three lanes are grouped images from different parts of the same gel. D, Scatter plot of proteins enriched (>twofold in two experiments) in CLEC14A MS-based immunoprecipation, which were found quantified in the MS proteomic analysis of HUVEC morphogenesis and spreading. Red dots indicate the proteins significantly up-regulated in Matr 30h (supplemental Table S8), most of which are matrixome proteins (supplemental Table S7). The x axis represents the ratio Matr 30 h/0 h (supplemental Table S2), the y axis the average ratio (CLEC14A/IgG) in two independent CLEC14A immunoprecipatates against IgG control (supplemental Table S8). E, Confocal images of HUVECs plated on matrigel (top panels) or cultured at confluence and stained for CLEC14A, FN1, MMRN2, and LAMA4. Nuclei are stained with DAPI (blue). The square depicts a higher magnification of the boxed area. Images are representative of more than three independent experiments. Scale bar = 25 μm. F, Fully annotated MS/MS spectra (from the Viewer module of MaxQuant) of the CID fragmented peptides DRAEGALLAES(p)PLGSSDA (Score = 138.66). The peptide is phosphorylated at position S483 with a localization probability (56) of 0.999. The list of identified ions and m/z is reported in supplemental Table S9.
MS Data Acquisition and Analysis
Digested peptides were analyzed by EASY-nLC system (Thermo Fisher Scientific) coupled on line to a LTQ-Orbitrap XL (for the EC morphogenesis and the spreading, and immunoprecipitation studies) or Velos (for the ECM study) (Thermo Fisher Scientific) via a nanoelectrospray ion source (Thermo Fisher Scientific). Chromatographic peptide separation was performed in a 15 cm fused silica emitter (Thermo Fisher Scientific) packed in house with reversed-phase Reprosil (Dr. Maisch GmbH) and eluted with a flow of 250 nl/min from 5% to 30% ACN in 0.5% acetic acid, in a 90 min gradient. The full scan MS spectra were acquired with a resolution of 30,000 at m/z 400 in the Orbitrap. The top 5–10 most intense ions were sequentially isolated for fragmentation using collision induced dissociation (CID) (for the EC morphogenesis and spreading, and immunoprecipitation studies) or high-energy collision dissociation (HCD) (for the ECM study), and recorded in the LTQ or Orbitrap, respectively. In the determination of CLEC14A phosphorylation sites, the neutral loss algorithm in the Xcalibur software was enabled for each MS/MS spectrum (20). Data were acquired with Xcalibur software (Thermo Fisher Scientific). The MS files were processed with the MaxQuant software version 1.2.6.20 (21) and searched with Andromeda search engine (22) against the human UniProt database (23) (release-2012 01, 81,213 entries). To search parent mass and fragment ions, an initial mass deviation of 6 ppm and 0.5 Da (CID) or 20 ppm (HCD), respectively, were required. The minimum peptide length was set to 7 amino acids and strict specificity for trypsin cleavage was required, allowing up to two missed cleavage sites. Carbamidomethylation (Cys) was set as fixed modification, whereas oxidation (Met) and N-acetylation were considered as variable modifications. No labeling or double SILAC labeling was defined accordingly. The false discovery rates (FDRs) at the protein and peptide level were set to 1%. Scores were calculated in MaxQuant as described previously (21). The reverse and common contaminants hits (in the ECM proteome analysis, KRT1 and KRT9 were additionally included), were removed from MaxQuant output. Only proteins identified with at least one peptide uniquely assigned to the respective sequence were considered for the analysis (for proteins identified with single peptide ID, see annotated spectra in supplemental Table S9). For SILAC protein quantification, the re-quantification feature was enabled, and the relative quantification of the peptides against their SILAC-labeled counterparts was performed by MaxQuant. Only unique peptides were used for quantification and we required proteins being quantified with at least two ratio counts.
For the immunoprecipitation and ECM analyses, proteins were quantified according to the MaxQuant label-free algorithm (24); unique and razor (= most likely belonging to the protein group) peptides were used for protein quantification.
The .raw MS files and search/identification files obtained with MaxQuant have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org/cgi/GetDataset) via the PRIDE partner repository (25) with the dataset identifier PXD000359.
MS Data Normalization
For the SILAC data, the H/L normalized ratios in the analyzed MaxQuant output file were inverted and log2 transformed. The distribution of the ratios was further normalized to the median (supplemental Table S2). For quantification, the protein groups were filtered for those with at most one missing quantification value within the three replicates, in each experimental condition (3723 protein groups in supplemental Table S2).
ANOVA Test and One Dimensional Analysis
For the ANOVA test, the three replicates for each adhesion condition were grouped together and the statistical test corrected with permutation based FDR of 0.5% in a similar way as described for t-tests by Tusher and coworkers (26) (supplemental Table S2, “Quantified proteins” datasheet).
The one-dimensional analysis to identify the regulated KEGG categories (27) was performed on the subset of protein ratios that passed the ANOVA test. For each of the KEGG pathways (“KEGG” column in supplemental Table S2) it was tested whether the corresponding ratios (column “Sample/0h”) had a preference to be systematically larger or smaller than the global distribution of protein abundance values. Multiple hypothesis testing was controlled by using a Benjamini-Hochberg FDR threshold of 5%. For categories that passed the test (supplemental Table S5) the median ratio of the proteins belonging to the category was calculated and shown as heat map in Fig. 3. For details see (28).
Fig. 3.

Different pathways are modulated during EC morphogenesis and spreading. A, Heat map showing the average regulation levels of the KEGG categories significantly regulated in ECs morphogenesis (supplemental Table S5). Colors indicate the levels of regulation; blue = down-regulation; red = up-regulation, black = nonsignificantly regulated. Colored bars group categories according to the temporal and adhesion condition regulation. Yellow = Matr 12h, orange = Matr 24h, red = Matr 30h, blue = Spreading 24h. Black bars highlight the categories discussed in the manuscript. ARVC = Arrhythmogenic right ventricular cardiomyopathy. Details of the one-dimensional annotation analysis are provided in the Experimental Procedures. B, Heat map and hierarchical clustering (proteins clustered based on Euclidean correlation distances) of 33 integrin adhesome proteins as in (42). The two highlighted clusters represent proteins with increased abundance in tubule morphogenesis (red) or spreading (blue). GOCC indicates the GO cellular component categories most overrepresented in the cluster. The colors of the heat map are based on the median of the SILAC ratio (sample/0h) as reported in supplemental Table S2.
Immunofluorescence
For the HUVECs, 8.0 E4 cells were seeded on gelatin or matrigel coated glass slides, 11 mm diameter, and grown in M199 complete medium for 5–7 days or in M199 10% FBS for 9h, respectively. Cells were fixed with 3.7% paraformaldehyde and saturated in PBS with 0.01% fish skin gelatin. Staining was performed by incubating cells sequentially with primary antibodies and further with the Alexa-conjugated secondary antibodies diluted in PBS 0.01% fish skin gelatin. Nuclei were stained with DAPI. Images were collected by confocal microscopy (DMIRE2; Leica Microsystems) with 63×/1.4 oil objectives using the Leica Confocal Software (version 2.5, build 1227). Quantification was performed with Imaris software.
For the tissues, frozen sections were air-dried and fixed with Zinc-fixative (calcium acetate 0.5 g, zinc acetate 5.0 g, zinc chloride 5.0 g, 0.1 m Tris buffer for 1000 ml of solution, pH 6.5–7). Samples were blocked with 1% BSA and 5% donkey serum in PBS for 1 h at room temperature. Tissues were incubated with the primary antibodies. After washing with PBS, samples were incubated with Alexa-conjugated secondary antibodies and counterstained with DAPI. Stainings were analyzed by using confocal laser-scanning microscope (TCS SP2 AOBS, Leica Microsystems).
To analyze Clec14a and Mmrn2 protein levels (red channel) during RIP-Tag2 and HPV/E2 tumorigenesis the immunofluorescence image acquisition was performed maintaining the same laser power, gain and offset settings. Ten different fields for each mouse at different stage of tumor progression (n = 5 mice/stage) were considered. In each 400× power picture we selected a region of interest (ROI) close to each blood vessel (green channel). We quantified the mean fluorescence intensity of red and green channels by means of the Leica Confocal Software Histogram Quantification Tool. We calculated the ratio of red to green channel mean fluorescence intensity. Then, the quantification of Clec14a and Mmrn2 abundance was normalized on vessel density and total tumor vascularity.
Three-Dimensional Reconstruction of Mouse Vessel
High-resolution confocal image stacks of ECs, pericytes and Clec14a were reconstructed by isosurface rendering using Imaris software (version 6.4.0, Bitplane, AG, Zurich, Switzerland). Isosurface rendering is a computer generated representation of a specified range of fluorescence intensities in a data set that allow the creation of an artificial solid object of a specific area. Selected confocal images were imported into Imaris to obtain a precise three-dimensional vessel reconstruction and to better point out the colocalization between ECs, pericytes, and Clec14a.
Immunohistochemistry
After deparaffinization slides were incubated in Target Retrieval Solution, Citrate pH6 (Dako). Immunostaining was performed with LSAB+ Kit (Dako) according to the manufacturer's instructions. Slides were counterstained with hematoxylin. Analysis of the samples followed an informed consent approved by the local ethics committee (Department of Pathology, Wroclaw Medical University, Poland).
Matrigel and Three-Dimensional Tubulogenesis
The matrigel assay performed with siRNA HUVECs was carried on in 96 well plate. Briefly, 6.5–10 E3 HUVECs were seeded on 30 μl of solidified matrigel in 100 μl of EGM-2 and let grown at the indicated time.
For the three-dimensional tubulogenesis assay, collagen-coated beads were covered with HUVECs, embedded into fibrin gel and co-cultured with fibroblasts (CAF2, (29)) as previously described (30). Quantifications were performed after 11 days of co-culture.
siRNA
The day before oligofection, HUVECs were seeded in six-well plates at a concentration of 2.0 E4 cells/well. Oligofection of the siRNA duplexes was performed according to the manufacturer's protocol. Briefly, HUVECs were transfected with 375 pmol of nontargeting siRNA (Dharmacon-Thermo), or two different Stealth Select RNAi (Invitrogen). SiCLEC14A#1 GGGAGCGUGAUUUCCAAGUUUAAUU; SiCLEC14A#2: GAGUGAUCCUGAGCCCGCUGCUUUG; siMMRN2#1: GGUGAGCGAGUAUGGUUUGAGUUAA, siMMRN2#2: CACAGCUGCAGUGAUGGAAGCAAAU. After 48 h since the oligofection, HUVECs were used for experiments.
Statistical Analysis
For Clec14a and Mmrn2 protein levels in mouse vessels and 3D tubulogenesis p values were calculated using a two-tail Mann Whitney test. For other experiments, unless indicated otherwise, p values were calculated using a two tail unpaired t test. Calculations were performed using GraphPad Prism. * = p < 0.05; ** p < 0.01; *** p < 0.001.
RESULTS
Quantitative Proteomics of HUVECs During Tubulogenesis and Spreading
To follow proteomic changes that contribute to vessel growth, we performed a time-resolved quantitative proteomic study of HUVECs plated on matrigel (Matr) for 0h (cells harvested but not plated on the matrix), 12 h, 24 h, and 30 h and on growth factor reduced (GFR) matrigel. Network structures were clearly evident after only 12 h, and by 30 h there were fully formed tubule. To contrast the morphogenetic process to general adhesion mechanisms, we seeded HUVECs for 24 h on culture dishes coated with ECMs relevant in angiogenesis: diluted matrigel (Matr dil), laminin (LAM), fibronectin (FN), and the nonspecific bovine serum albumin (BSA). On these matrices HUVECs spread without forming tubules (Fig. 1A). No major morphological differences were observed comparing cells on normal and GFR matrigel or comparing cells spreading on different ECMs (not shown).
Fig. 1.

SILAC-based quantitative proteomics identifies proteome changes in HUVECs morphogenesis and spreading. A, The right panel depicts the adhesion conditions analyzed with MS. Phase contrast pictures are representative of HUVECs forming capillary-like network (tubule morphogenesis) and spreading on ECM-coated dish. The workflow shows the main steps of sample preparation and MS analysis. Identified and quantified proteins were obtained from three independent experiments. B, C, Changes in abundance of protein markers for ECs and angiogenesis (B), and vessel maturation (C). Protein levels are represented as average of the Sample/SILAC ratio in the three experiments, normalized by the average protein amount at time 0 (0h/SILAC ratio). Bars indicate the S.D. (n > = 2).
For accurate, in-depth characterization of the proteomes in these different environments we employed SILAC in an indirect “spike-in” format (31). HUVECs in culture were metabolically labeled with heavy arginine and lysine as a reference, and then spiked into the lysate of each of the samples described above. This approach allowed us to profile the proteome of HUVECs at early passage (P), P2-P3. Mixed proteomes were then analyzed by online liquid chromatography mass spectrometry (LC MS/MS). We employed a linear ion trap Orbitrap instrument and processed the MS data in the MaxQuant environment (21) (Fig. 1A).
MS analysis was performed on three independent replicates, which unambiguously identified 6678 proteins. Of those, 3723 were accurately quantified in all adhesive conditions (supplemental Tables S1 and S2). Several known factors required for angiogenesis in vivo, including the vascular endothelial growth factor (VEGF) receptors, VEGFR2/KDR and NRP1, the angiopoietin receptor TIE1 (32–34), the EC markers von Willebrand factor (VWF), ve-cadherin (CDH5) and PECAM1, and the EC tumor markers vimentin (VIM) and CD59 (35), markedly increased levels during EC morphogenesis (Fig. 1B). Additionally, we observed that CDH5, the endothelial nitric oxide synthase (NOS3) and the ECM components fibronectin (FN1) and laminin 411 (LAMA4, LAMB1 and LAMC1), which promote the maturation and stabilization of newly formed vessels (36–38), increased levels in morphogenesis. Conversely, the abundance of the receptor ephrin A2 (EPHA2), which determines cell–cell destabilization (39), decreased abundance over the time course (Fig. 1C).
These data demonstrate that HUVECs grown on matrigel regulate part of the molecular machinery involved in physiological and pathological angiogenesis in vivo, and that the morphogenesis time course analyzed here reflects late aspects of the angiogenic process. We thus show the validity of combining quantitative proteomics and an in vitro model of angiogenesis to investigate vessel growth.
Diverse Proteome Regulation in Tubule-forming and Spreading HUVECs
Statistical test using ANOVA revealed that 1401 of the 3723 accurately quantified proteins were regulated during morphogenesis and spreading (supplemental Table S2) with excellent reproducibility (Spearman rank correlation = 0.90, supplemental Table S3). We then calculated changes in protein abundance over the time as the median SILAC ratio between replicates in the specific adhesion condition divided by the median SILAC ratio measured at the start of the experiment (0h), subsequently referred to as “ratio” (Sample/0 h). To subdivide the regulated proteins in up and downregulated and assign them to the different adhesion conditions, we required a minimum ratio higher than one standard deviation (S.D.) from the mean of the all calculated ratios (mean1,401prot = 0.01; S.D. = 0.534) (supplemental Table S4).
Our analysis revealed that an increasing number of proteins were regulated as tubulogenesis progressed, which was greater than that found during spreading (Fig. 2A). Furthermore, the Venn diagram analysis of the regulated proteins during morphogenesis showed an almost complete overlap between subsequent time points (Fig. 2B), suggesting a progressive differentiation of the cells, as also indicated by increasing levels of several EC markers (Figs. 1B, 1C). Conversely, clear differences were identified between cell morphogenesis and spreading. As representative examples, less than 20% of proteins were commonly regulated between Matr 24 h and Matr dil, or Matr 24 h and LAM (Fig. 2C). This distinct proteome remodeling was confirmed by an unbiased principle component analysis (PCA) of the ratio profiles of the differently cultured HUVECs, which clearly separated them according to their macroscopic appearance (Fig. 2D). Accordingly, a close similarity was detected between cell morphogenesis on normal and GFR matrigel, as well as between cell spreading on different ECMs. Of note, the component number two separated the earlier, Matr 12 h, from the later time points, Matr 24 h and 30 h. This suggests that our time course experiment may contain distinct proteomic signatures for early and late morphogenesis.
Fig. 2.
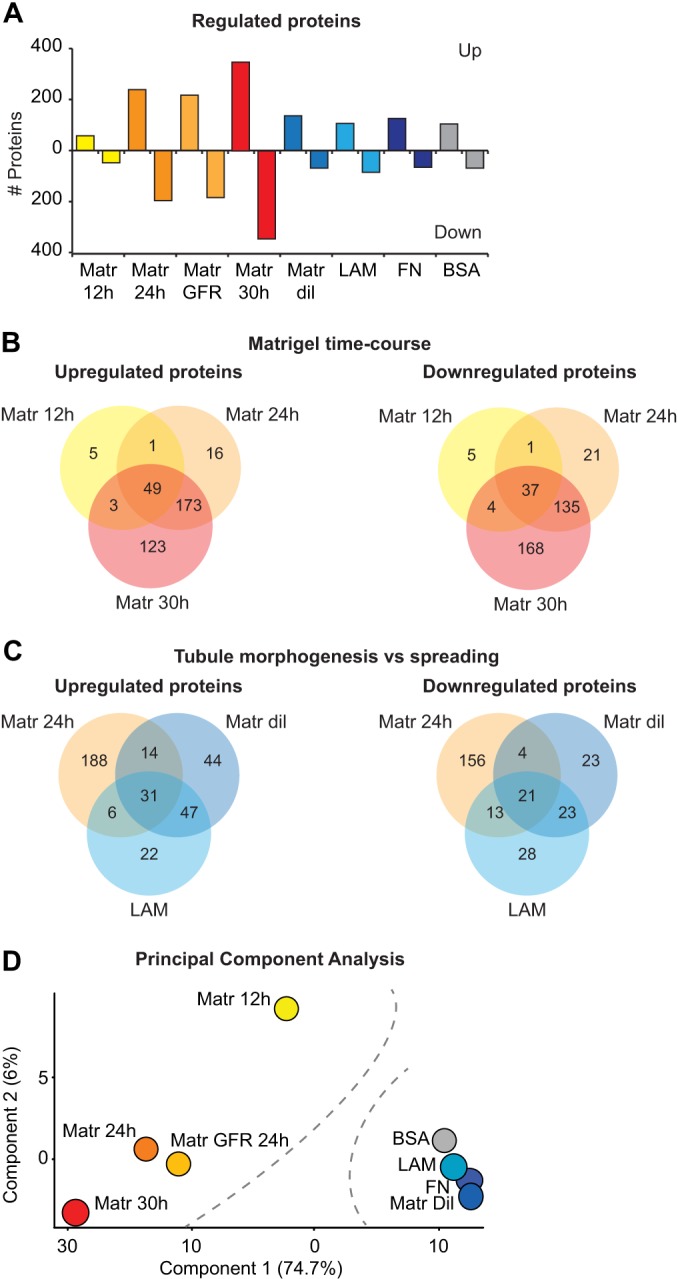
Different proteome changes occur during cell morphogenesis and spreading. A, Number of proteins significantly up and down-regulated in the different adhesion conditions as reported in supplemental Table S4. B, C, Venn diagrams (based on the protein Gene Names) of the proteins up and down-regulated during the EC morphogenesis time course (Matr 12 h, 24 h, and 30 h) (B), and those regulated in EC morphogenesis (24 h Matr) or spreading (Matr dil and LAM) (C). D, Two principal components, which captured 80.7% of the total variance of the HUVEC proteome changes during the different experimental conditions, are shown. Dotted lines highlight the separation between cells forming tubules on matrigel and spreading on ECMs. PCA was performed on the ratios (Sample/0 h) that passed the ANOVA test as in supplemental Table S2.
These results provide clear evidence that major proteomic differences accompany EC morphogenesis and spreading. Using this data set, specific proteins and cellular processes needed by the cells to regulate EC morphogenesis can be pinpointed.
EC Morphogenesis and Spreading Involve Different Remodeling of the Cell–ECM Adhesion Machinery
To gain bioinformatic insights into the processes involved in EC morphogenesis, we applied a ‘one-dimensional annotation distribution’ analysis, which has been recently developed in our group (28), and identified the regulated KEGG pathways during EC morphogenesis and spreading. Fig. 3A (see supplemental Table S5 for details) shows their levels of regulation in the different experimental conditions.
Proliferation-related categories, DNA replication and cell-cycle, were down-regulated in most of the experimental conditions. This may reflect a general decrease in cell proliferation because of the reduced amount of serum in the medium used during the experiment, which was half of the amount used during the culturing of the cells (see Experimental Procedures). Conversely, most of the KEGG categories had distinct regulation during EC morphogenesis and spreading, and we distinguished four major groups: categories regulated at early, medium and late time during EC morphogenesis, and categories regulated only during EC spreading (Fig. 3A and supplemental Table S5 for detailed list of the proteins).
After 12 h on matrigel, HUVECs up-regulated cell-ECM adhesion proteins involved in ECM-receptor interaction (p = 1.5E-5), which include the ECM components and angiogenesis regulators FN1, HSPG2, and laminin 411 (38, 40), and several integrin receptors. This regulation was maintained over the time. Cell–ECM adhesion-related categories were found up-regulated during EC spreading as well, though different ones compared with EC morphogenesis. Among them the category focal adhesion (best p = 3.1E-4), which includes the protein kinases PAK2 and ERK1/2, and the myosin regulators MYL9 and MYL12A (supplemental Table S5). We investigated adhesion mechanism in more detail, and explored the regulation of proteins that have been manually annotated as component of the integrin adhesome (41). This dataset includes two hundreds proteins involved in cell-ECM interaction mediated by integrin receptors (42). Of those, 91 were quantified in our SILAC-based analysis, of which 33 were significantly regulated during EC morphogenesis and spreading (according to ANOVA test, see above). Heat map and hierarchical clustering based on the protein abundance profile of these 33 proteins revealed two main clusters with proteins distinctly regulated in EC morphogenesis and spreading (Fig. 3B). Fisher test-based category enrichment analysis revealed that the major difference between the proteins contained in the two clusters was their subcellular localization: membrane (p = 1.7E-3; Benjamin Hochberg FDR = 0.08) for the cluster of proteins with increased levels in morphogenesis only, whereas intracellular (p = 2.3E-4, Benjamin Hochberg FDR = 0.02) for the cluster of proteins with increased levels during EC spreading and decreased in morphogenesis (Fig. 3B).
Additionally our data revealed that proteins which localize in the lysosome (p24h = 1.7E-19) and peroxisome (p24h = 3.0E-4) compartments, and a substantial portion of metabolic proteins and enzymes, were regulated in morphogenesis, while not changing during EC spreading. At 12 h, these included members of sphingolipids metabolism (p = 2.8E-4), oxidative phosphorylation (p = 4.1E-9), glycine, serine, and threonine (p = 2.7E-3), and purine (p = 8.2E-4) metabolism. Furthermore, at 24 h morphogenesis, also glycosaminoglycan degradation (p = 5.8E-5) and valine, leucine, and isoleucine (p = 3.4E-3) metabolic pathways were regulated, and, at 30 h, increased levels were measured for glycosphingolipid metabolism (p = 5.9E-3).
Finally, some categories were found significantly regulated only during EC spreading. These included proteins involved in nucleotide excision repair (best p = 1.2E-4), RNA degradation (best p = 2.3E-4), and splicing (best p = 1.8E-12).
This proteomic analysis provides a detailed portrait of the processes regulated in HUVECs during morphogenesis and spreading, and revealed the specific categories regulated depending on the adhesion substrate, and in the following we explore these aspects in more depth.
CLEC14A is an Extracellular Protein Component of the HUVEC Matrixome
Endothelial cell–ECM adhesion is a crucial mechanism in angiogenesis, because it regulates multiple steps of blood vessel growth, including vessel maturation. Moreover, a correct composition and structure of the ECM surrounding blood vessel is crucial to maintain its structure and functionality. Our analysis pointed out that ECM-receptor interaction proteins were significantly regulated during ECs morphogenesis, and we observed that among the most up-regulated proteins (the 10% most up-regulated proteins in Matr 30 h are reported in supplemental Table S4) there were ECM components, such as LAMC1, NID1 and LAMB1, which regulate angiogenesis in vivo (43–45). We therefore investigated ECM proteins further. We set up a protocol to purify ECM produced by cells in culture (see Experimental Procedures) and performed an extensive MS profile of the ECM isolated from HUVECs. We identified 1358 proteins, and estimation of the protein absolute abundance revealed that a subset of only 128 proteins made up 90% of the endothelial ECM protein mass, which we term “matrixome” (Fig. 4A, supplemental Table S6). The matrixome included known ECM components, such as FN1 and HSPG2, which were estimated to constitute more than 40% of the total matrixome mass. In contrast membrane proteins, such as the ECM receptors integrin α5 and β1, had low abundance (Fig. 4 supplemental). To identify ECM proteins relevant in angiogenesis in vivo, we searched for matrixome components highly abundant in HUVECs after 24 h morphogenesis on matrigel and whose levels were higher in morphogenesis (Matr 24 h) compared with spreading (adhesion on Matr dil or LAM for 24 h). This pinpointed known secreted proteins (46) (47), such as the ECM component COL12A1, the growth factor TGFB1, the prelamin-A/C (LMNA) and a histone protein (HIST1H4A), but also proteins which have not been previously localized at the ECM (Fig. 4B, supplemental Table S7). Among them we found the c-type lectin domain family XIV member A (CLEC14A), a single pass transmembrane glycoprotein member of the endosialin family (supplemental Fig. S1A). Another endosialin family member, the complement component C1q receptor (CD93), which has been previously shown to undergo shedding in the presence of inflammatory signals (48), was identified as a matrixome component with up-regulation during morphogenesis (Fig. 4B). Western blot analysis of cell, supernatant and ECM fractions of HUVECs in culture confirmed the presence of CLEC14A in the ECM. Additionally, a 40–50kDa band was detected in the supernatant and in the ECM preparation (Fig. 4C), highlighting that different forms of CLEC14A are present in the extracellular fractions.
To investigate the relationship between CLEC14A and the ECM further we performed a quantitative MS proteomic interaction screen of endogenous CLEC14A immunoprecipitated from HUVECs seeded on matrigel. This identified 53 interaction candidates (Table S8). Of those, LAMA4, LAMC1, VIM, HSPG2, FN1, DBN1, and MMRN2 were up-regulated during EC tubulogenesis on matrigel, similar to CLEC14A (Fig. 4D), and, except for DBN1 and LAMC1, identified as matrixome components. Strikingly, immunofluorescent confocal microscopy showed CLEC14A partial colocalizing with FN1, MMRN2 and LAMA4 (Fig. 4E and supplemental Fig. S1B). MMRN2, an endothelial-specific matricellular glycoprotein (49), was the most enriched matrixome protein in the CLEC14A pull-downs. By immunoprecipitation of endogenous CLEC14A, we verified CLEC14A-MMRN2 interaction in HUVECs in culture (supplemental Fig. S1C). Furthermore, as CLEC14A, MMRN2 was regulated at higher levels during morphogenesis compared with spreading. Altogether, these results prompted us to investigate CLEC14A and MMRN2 in vivo (see below).
Lastly, post-translational modification analysis of immunoprecipitated CLEC14A uncovered a phosphorylation site at Ser483 (Fig. 4F), in proximity of the PDZ domain-binding region at the C terminus of the cytoplasmic tail. A functional role for the C terminus PDZ binding domain was previously shown for CD93 (50), thus suggesting that Ser483 may be of interest for future functional investigations.
Clec14a and Mmrn2 Protein Levels in Blood Vessels Increase During Tumorigenesis in Multistep Mouse Models
We assessed CLEC14A and MMRN2 function in tubule morphogenesis via small interfering RNA (siRNA) knock-down. CLEC14A or MMRN2 protein levels were silenced by siRNA, which compromised the formation of tubule structures when plated on matrigel, compared with control siRNA-transfected HUVECs (Fig. 5A and supplemental Fig. S2A,B). We verified CLEC14A and MMRN2 function with an independent 3D tubulogenesis assay (Fig. 5B and supplemental Fig. S2C, S2D).
Fig. 5.

CLEC14A and MMRN2 are required in EC morphogenesis and are highly expressed in RIP-Tag2 insulinoma and HPV16/E2 invasive carcinoma. A, Quantification of matrigel assay performed with HUVEC transfected with control pool siRNA (siCtl) or two independent siRNA for CLEC14A (red) or MMRN2 (blue). Tubule length is expressed on an arbitrary scale where 1 represents the length in the Ctl cells. Bars indicate the median + S.E. (n = 3). B, Quantification (box plot, minimum maximum value, n>20 cells coated beads) of the 3D tubulogenesis assay performed with HUVEC transfected with siCtl or two independent siRNA for CLEC14A or MMRN2 and co-culture with fibroblasts. C, Immunohistochemistry for CLEC14A and EC marker PECAM1 of consecutive sections of human ductal carcinoma. HE = hematoxylin-eosin staining. Pictures are representative of three patients. D, Representative immunofluorescence for Clec14a and EC marker Meca32 of normal, angiogenic and tumor pancreatic islets of RIP-Tag2 mice. Ac = acinus; n = normal tissue; A = angiogenic tissue; T = tumor tissue. Nuclei are stained with DAPI (blue). (E, F) Mean fluorescence intensity (MFI) (calculated as under Experimental Procedures) of Clec14a and Mmrn2 in blood vessels of normal, angiogenic and tumor islets in RIP-Tag2 mice (E) or normal, CIN3 and invasive SCC in HPV16/E2 model (F). Clec14a and Mmrn2 levels were normalized on vessel density and total tumor vascularity. N/E2 = normal cervix, treated with 17β-estradiol (E2); CIN3 = high grade cervical intraepithelial neoplasia; SCC = squamous cell carcinoma. Bars indicate S.D. (n = 5 mice/stage). G, High resolution confocal images of a RIP-Tag2 insulinoma blood vessel stained for ECs (Meca32), pericytes (NG2), and Clec14a. H, 3D reconstruction by isosurface rendering of high-resolution confocal image stacks in (G, boxed area).
Immunohistochemistry on a panel of human tumor tissues and adjacent normal tissue showed similar staining patterns between CLEC14A and the EC marker PECAM1 in arteries and veins (Fig. 5C and supplemental Fig. S2E). Because tumor vessels strongly stained for CLEC14A, we further investigated whether CLEC14A levels were modulated during tumor progression. We employed two mouse models of spontaneous tumorigenesis that recapitulate the progressive development of human cancer. RIP-Tag2 mice develop pancreatic neuroendocrine tumors, which progress from hyperplastic islets to angiogenic and then tumor islets. HPV16/E2 mice display cervical cancer through progression from low-grade cervical intraepithelial neoplasia, to high-grade dysplasia (CIN-3), and invasive squamous cell carcinoma (SCC). Fluorescence confocal microscopy showed partial colocalization between Clec14a and the EC marker Meca32 in blood vessels of normal, angiogenic and tumor pancreatic islets in RIP-Tag2 mice (Fig. 5D). Accurate fluorescence quantification revealed significantly higher Clec14a levels in blood vessels of tumor compared with normal and angiogenic islets (Fig. 5E). Similarly, higher levels of Clec14a were measured in blood vessels of CIN-3 and SCC of HPV16/E2 mice, compared with nontransgenic E2-treated normal cervix (Fig. 5F). Similar to Clec14a, significantly higher levels of Mmrn2 were measured in tumor compared with normal vessels in RIP-Tag2 and HPV16/E2 mice. However, in blood vessels of RIP-Tag2 tumor angiogenic islets, only Mmrn2 was found elevated and not Clec14a (Figs. 5E, 5F).
Finally, confocal analysis of tumor vessels in RIP-Tag2 and HPV16/E2 mice showed partial colocalization between Clec14a, Mmrn2 and Lama4 (supplemental Fig. S3A, S3B), and 3D reconstruction of confocal images of the RIP-Tag2 tumor vasculature placed Clec14a at the interface between ECs and pericytes (Figs. 5G, 5H), thus supporting the presence of Clec14a in the ECM also in vivo.
Together, these results show that Clec14a and Mmrn2 are required in EC morphogenesis and that they are deregulated at specific stages of cancer progression in mouse models.
DISCUSSION
Our mass spectrometric analysis of HUVECs during tubules morphogenesis in matrigel shows that this process involves significant changes of about 20% of the proteome, which we measured to a depth of more than 3,700 proteins, the most in depth endothelial proteome generated so far. The accuracy of the quantitative approach allowed us to shed new light on EC regulation. This is illustrated by a very reproducible regulation in abundance of a remarkable number of cell adhesion and metabolism-related proteins and enzymes. Furthermore, we provide the first in depth characterization of the HUVEC matrixome, which we proved to be an excellent resource of proteins deregulated in tumor angiogenesis.
Cell–matrix adhesion regulates EC sprouting and migration during angiogenesis, and provides support and stability to the newly formed capillary. Importantly, adhesion mechanisms can be influenced by the environment surrounding the cell, and here we show that ECs differently modulate their adhesion machinery during morphogenesis and spreading (Fig. 3A). An important difference between the matrigel and spreading systems used for this study is the mechanical property of the matrix, where matrigel is soft and other ECMs are rigid. Accordingly, among the regulated cell adhesion proteins the levels of the cell contractility regulators MYL12A and MYL9 decreased in morphogenesis, while they were augmented during spreading. Because increasing ECM rigidity has been shown to support tumor progression (51), we speculate that the differentially regulated adhesion proteins (supplemental Table S5) may be relevant to understanding the role of ECM stiffness on EC behavior in tumors.
Tumor vessels often display absent or abnormally thick ECM, which impact their function (3). Our MS analyses highlighted significantly increased levels of ECM proteins during HUVECs morphogenesis (supplemental Table S7), and we discovered that CLEC14A is an ECM component. We showed that CLEC14A binds to and colocalizes, in vitro and in vivo, with FN1 and LAMA4, which are master regulators of angiogenesis and previously found deregulated in tumor vessels (2, 52) and with MMRN2 (Fig. 4). In contrast to our results, MMRN2 overexpression has been reported to inhibit angiogenesis by affecting VEGF signaling (53). This suggests that MMRN2 effects on endothelial cells might be context and dose-dependent and that further studies are needed to elucidate its function in angiogenesis. Furthermore, we show that CLEC14A and MMRN2 are EC marker, functional in HUVECs, and that CLEC14A is highly expressed in tumor compared with normal vessels. This confirmed previous literature (54, 55). Additionally, through multiple-stage mouse tumor models, RIP-Tag2 and HPV16/E2, we identified Clec14a and Mmrn2 deregulation at specific stages of tumorigenesis (Figs. 5, supplemental Figs. S2, S3). These mice will therefore be valuable models to further investigate Clec14a and Mmrn2 functions in tumor angiogenesis. Similarly to CLEC14A, other matrixome components contained in our screen (supplemental Table S7) and that were not previously known to be localized at the ECM may be interesting candidates for further investigation of their role in normal and pathological angiogenesis.
Our results show that applying modern proteomic technologies to established in vitro model of angiogenesis is a powerful approach to discover functional regulations in endothelial cells, relevant for the angiogenic process in vivo. Here we have functionally explored ECM proteins, but other processes were also found regulated. For instance, significantly altered abundance was measured for a remarkable number of metabolic proteins, a phenomenon that to our knowledge has not been observed in morphogenesis before (Figs. 3A, supplemental Table S5). In conclusion, we have demonstrated that our proteomic study of ECs morphogenesis and spreading provides valuable information that can help to unravel molecular mechanisms regulating endothelial cells behavior. Our data may contain the basis for further exciting insights to be investigated in angiogenesis in vivo, especially with a view to interfering with this process in pathologies.
Supplementary Material
Acknowledgments
We thank the PRIDE team. Piotr Ziolkowski for providing the human tumor tissues. Simona Pavan and Wolfgang Eiermann kindly provided some of the HUVECs and umbilical cords.
Footnotes
* This work was supported by the Max Planck Society, PROSPECT, a 7th framework program of the European Union (grant agreement HEALTH-F4-2008-201648), and Cancer Research UK. EG and FM were supported by Associazione Italiana per la Ricerca sul Cancro (AIRC) (to E.G., IG # 11600); by AIRC 2010 Special Program in Molecular Clinical Oncology 5 × 1000 Project no. 9970; Fondazione Piemontese per la Ricerca sul Cancro (FPRC), Intramural Grant Program 2010 (5 × 1000 2008); SZ was partially supported by the fellowship “L. Fontana e M. Lionello” granted by Fondazione Italiana per la Ricerca sul Cancro (FIRC) and FM by the fellowship “26 fellowship - FIRC” granted by FIRC.
 This article contains supplemental Figs. S1 to S3 and Tables S1 to S9.
This article contains supplemental Figs. S1 to S3 and Tables S1 to S9.
1 The abbreviations used are:
- EC
- endothelial cell
- SILAC
- stable isotope labeling by amino acids in cell culture
- HUVEC
- human umbilical vein endothelial cells
- ECM
- extracellular matrix
- LTQ
- linear trap quadrupole.
REFERENCES
- 1. Carmeliet P., Jain R. K. (2011) Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 10, 417–427 [DOI] [PubMed] [Google Scholar]
- 2. Baluk P., Morikawa S., Haskell A., Mancuso M., McDonald D. M. (2003) Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. The Am. J. Pathol. 163, 1801–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kalluri R. (2003) Basement membranes: structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 3, 422–433 [DOI] [PubMed] [Google Scholar]
- 4. Goel S., Duda D. G., Xu L., Munn L. L., Boucher Y., Fukumura D., Jain R. K. (2011) Normalization of the vasculature for treatment of cancer and other diseases. Physiolog. Rev. 91, 1071–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Folkman J. (2003) Angiogenesis inhibitors: a new class of drugs. Cancer Biol. Ther. 2, S127–S133 [PubMed] [Google Scholar]
- 6. Ferrara N., Kerbel R. S. (2005) Angiogenesis as a therapeutic target. Nature 438, 967–974 [DOI] [PubMed] [Google Scholar]
- 7. Arnaoutova I., George J., Kleinman H. K., Benton G. (2009) The endothelial cell tube formation assay on basement membrane turns 20: state of the science and the art. Angiogenesis 12, 267–274 [DOI] [PubMed] [Google Scholar]
- 8. van Beijnum J. R., Griffioen A. W. (2005) In silico analysis of angiogenesis associated gene expression identifies angiogenic stage related profiles. Biochim. Biophys. Acta 1755, 121–134 [DOI] [PubMed] [Google Scholar]
- 9. Mellberg S., Dimberg A., Bahram F., Hayashi M., Rennel E., Ameur A., Westholm J. O., Larsson E., Lindahl P., Cross M. J., et al. (2009) Transcriptional profiling reveals a critical role for tyrosine phosphatase VE-PTP in regulation of VEGFR2 activity and endothelial cell morphogenesis. FASEB J. 23, 1490–1502 [DOI] [PubMed] [Google Scholar]
- 10. Hernandez-Fernaud J. R., Reid S. E., Neilson L. J., Zanivan S. (2012) Quantitative mass spectrometry-based proteomics in angiogenesis. Proteomics Clin. Appl. 7, 464–476 [DOI] [PubMed] [Google Scholar]
- 11. Bohman S., Matsumoto T., Suh K., Dimberg A., Jakobsson L., Yuspa S., Claesson-Welsh L. (2005) Proteomic analysis of vascular endothelial growth factor-induced endothelial cell differentiation reveals a role for chloride intracellular channel 4 (CLIC4) in tubular morphogenesis. J. Biol. Chem. 280, 42397–42404 [DOI] [PubMed] [Google Scholar]
- 12. Lamond A. I., Uhlen M., Horning S., Makarov A., Robinson C. V., Serrano L., Hartl F. U., Baumeister W., Werenskiold A. K., Andersen J. S., Vorm O., Linial M., Aebersold R., Mann M. (2012) Advancing cell biology through proteomics in space and time (PROSPECTS). Mol Cell Proteomics 11, O112 017731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hanahan D. (1985) Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 315, 115–122 [DOI] [PubMed] [Google Scholar]
- 14. Coussens L. M., Hanahan D., Arbeit J. M. (1996) Genetic predisposition and parameters of malignant progression in K14-HPV16 transgenic mice. Am. J. Pathol. 149, 1899–1917 [PMC free article] [PubMed] [Google Scholar]
- 15. Arbeit J. M., Howley P. M., Hanahan D. (1996) Chronic estrogen-induced cervical and vaginal squamous carcinogenesis in human papillomavirus type 16 transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 93, 2930–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Giraudo E., Inoue M., Hanahan D. (2004) An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Invest. 114, 623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protocols 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 18. Rappsilber J., Ishihama Y., Mann M. (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 [DOI] [PubMed] [Google Scholar]
- 19. Vlodavsky I. (2001). Preparation of extracellular matrices produced by cultured corneal endothelial and PF-HR9 endodermal cells. Current protocols in cell biology/editorial board, Juan S. Bonifacino. [et al Chapter10, Unit 10 14] [DOI] [PubMed] [Google Scholar]
- 20. Schroeder M. J., Shabanowitz J., Schwartz J. C., Hunt D. F., Coon J. J. (2004) A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal. Chem. 76, 3590–3598 [DOI] [PubMed] [Google Scholar]
- 21. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 22. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011). Andromeda - a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 23. Consortium. U. (2010) The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 38, D142–D148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luber C. A., Cox J., Lauterbach H., Fancke B., Selbach M., Tschopp J., Akira S., Wiegand M., Hochrein H., O'Keeffe M., Mann M. (2010) Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity 32, 279–289 [DOI] [PubMed] [Google Scholar]
- 25. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Pérez-Riverol Y., Reisinger F., Ríos D., Wang R., Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tusher V. G., Tibshirani R., Chu G. (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U. S. A. 98, 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kanehisa M., Goto S., Sato Y., Furumichi M., Tanabe M. (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cox J., Mann M. (2012) 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinformatics 13, S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Orimo A., Gupta P. B., Sgroi D. C., Arenzana-Seisdedos F., Delaunay T., Naeem R., Carey V. J., Richardson A. L., Weinberg R. A. (2005) Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121, 335–348 [DOI] [PubMed] [Google Scholar]
- 30. Nakatsu M. N., Hughes C. C. (2008) An optimized three-dimensional in vitro model for the analysis of angiogenesis. Methods Enzymol. 443, 65–82 [DOI] [PubMed] [Google Scholar]
- 31. Geiger T., Wisniewski J. R., Cox J., Zanivan S., Kruger M., Ishihama Y., Mann M. (2011) Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics. Nat. Protocols 6, 147–157 [DOI] [PubMed] [Google Scholar]
- 32. Carmeliet P., Mackman N., Moons L., Luther T., Gressens P., Van Vlaenderen I., Demunck H., Kasper M., Breier G., Evrard P., Müller M., Risau W., Edgington T., Collen D. (1996) Role of tissue factor in embryonic blood vessel development. Nature 383, 73–75 [DOI] [PubMed] [Google Scholar]
- 33. Sato T. N., Tozawa Y., Deutsch U., Wolburg-Buchholz K., Fujiwara Y., Gendron-Maguire M., Gridley T., Wolburg H., Risau W., Qin Y. (1995) Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature 376, 70–74 [DOI] [PubMed] [Google Scholar]
- 34. Kawasaki T., Kitsukawa T., Bekku Y., Matsuda Y., Sanbo M., Yagi T., Fujisawa H. (1999) A requirement for neuropilin-1 in embryonic vessel formation. Development 126, 4895–4902 [DOI] [PubMed] [Google Scholar]
- 35. van Beijnum J. R., Dings R. P., van der Linden E., Zwaans B. M., Ramaekers F. C., Mayo K. H., Griffioen A. W. (2006) Gene expression of tumor angiogenesis dissected: specific targeting of colon cancer angiogenic vasculature. Blood 108, 2339–2348 [DOI] [PubMed] [Google Scholar]
- 36. Dejana E., Tournier-Lasserve E., Weinstein B. M. (2009) The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Developmental Cell 16, 209–221 [DOI] [PubMed] [Google Scholar]
- 37. Kashiwagi S., Tsukada K., Xu L., Miyazaki J., Kozin S. V., Tyrrell J. A., Sessa W. C., Gerweck L. E., Jain R. K., Fukumura D. (2008) Perivascular nitric oxide gradients normalize tumor vasculature. Nat. Med. 14, 255–257 [DOI] [PubMed] [Google Scholar]
- 38. Grant D. S., Kleinman H. K. (1997) Regulation of capillary formation by laminin and other components of the extracellular matrix. EXS 79, 317–333 [DOI] [PubMed] [Google Scholar]
- 39. Zhou N., Zhao W. D., Liu D. X., Liang Y., Fang W.G., Li B., Chen Y. H. (2011) Inactivation of EphA2 promotes tight junction formation and impairs angiogenesis in brain endothelial cells. Microvasc. Res. 82, 113–121 [DOI] [PubMed] [Google Scholar]
- 40. Iozzo R. V. (2005) Basement membrane proteoglycans: from cellar to ceiling. Nat. Rev. 6, 646–656 [DOI] [PubMed] [Google Scholar]
- 41. Zaidel-Bar R. (2009) Evolution of complexity in the integrin adhesome. J. Cell Biol. 186, 317–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zanivan S., Meves A., Behrendt K., Schoof E. M., Neilson L. J., Cox J., Tang H. R., Kalna G., van Ree J. H., van Deursen J. M., Trempus C. S., Machesky L. M., Linding R., Wickström S. A., Fässler R., Mann M. (2013) In vivo SILAC-based proteomics reveals phosphoproteome changes during mouse skin carcinogenesis. Cell Rep. 3, 552–566 [DOI] [PubMed] [Google Scholar]
- 43. Bagri A., Tessier-Lavigne M., Watts R. J. (2009) Neuropilins in tumor biology. Clin. Cancer Res. 15, 1860–1864 [DOI] [PubMed] [Google Scholar]
- 44. Dulak J., Deshane J., Jozkowicz A., Agarwal A. (2008) Heme oxygenase-1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation 117, 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hallmann R., Horn N., Selg M., Wendler O., Pausch F., Sorokin L. M. (2005) Expression and function of laminins in the embryonic and mature vasculature. Physiolog. Rev. 85, 979–1000 [DOI] [PubMed] [Google Scholar]
- 46. de la Cuesta F., Barderas M. G., Calvo E., Zubiri I., Maroto A. S., Darde V. M., Martin-Rojas T., Gil-Dones F., Posada-Ayala M., Tejerina T., Lopez J. A., Vivanco F., Alvarez-Llamas G. (2012) Secretome analysis of atherosclerotic and nonatherosclerotic arteries reveals dynamic extracellular remodeling during pathogenesis. J. Proteomics 75, 2960–2971 [DOI] [PubMed] [Google Scholar]
- 47. Greco T. M., Seeholzer S. H., Mak A., Spruce L., Ischiropoulos H. (2010) Quantitative mass spectrometry-based proteomics reveals the dynamic range of primary mouse astrocyte protein secretion. J. Proteome Res 9, 2764–2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Greenlee M. C., Sullivan S. A., Bohlson S. S. (2008) CD93 and related family members: their role in innate immunity. Curr. Drug Targets 9, 130–138 [DOI] [PubMed] [Google Scholar]
- 49. Sanz-Moncasi M. P., Garin-Chesa P., Stockert E., Jaffe E. A., Old L. J., Rettig W. J. (1994) Identification of a high molecular weight endothelial cell surface glycoprotein, endoGlyx-1, in normal and tumor blood vessels. Lab. Invest. 71, 366–373 [PubMed] [Google Scholar]
- 50. Bohlson S. S., Silva R., Fonseca M. I., Tenner A. J. (2005) CD93 is rapidly shed from the surface of human myeloid cells and the soluble form is detected in human plasma. J. Immunol. 175, 1239–1247 [DOI] [PubMed] [Google Scholar]
- 51. Butcher D. T., Alliston T., Weaver V. M. (2009) A tense situation: forcing tumour progression. Nat. Rev. Cancer 9, 108–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ljubimova J. Y., Fugita M., Khazenzon N. M., Das A., Pikul B. B., Newman D., Sekiguchi K., Sorokin L. M., Sasaki T., Black K. L. (2004) Association between laminin-8 and glial tumor grade, recurrence, and patient survival. Cancer 101, 604–612 [DOI] [PubMed] [Google Scholar]
- 53. Lorenzon E., Colladel R., Andreuzzi E., Marastoni S., Todaro F., Schiappacassi M., Ligresti G., Colombatti A., Mongiat M. (2012) MULTIMERIN2 impairs tumor angiogenesis and growth by interfering with VEGF A/VEGFR2 pathway. Oncogene 31, 3136–3147 [DOI] [PubMed] [Google Scholar]
- 54. Mura M., Swain R. K., Zhuang X., Vorschmitt H., Reynolds G., Durant S., Beesley J. F., Herbert J. M., Sheldon H., Andre M., Sanderson S., Glen K., Luu N. T., McGettrick H. M., Antczak P., Falciani F., Nash G. B., Nagy Z. S., Bicknell R. (2012) Identification and angiogenic role of the novel tumor endothelial marker CLEC14A. Oncogene 31, 293–305 [DOI] [PubMed] [Google Scholar]
- 55. Rho S. S., Choi H. J., Min J. K., Lee H. W., Park H., Kim Y. M., Kwon Y. G. (2011) Clec14a is specifically expressed in endothelial cells and mediates cell to cell adhesion. Biochem. Biophys. Res. Commun. 404, 103–108 [DOI] [PubMed] [Google Scholar]
- 56. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.