

Abstract
Quantitative proteome analyses suggest that the well-established stain colloidal Coomassie Blue, when used as an infrared dye, may provide sensitive, post-electrophoretic in-gel protein detection that can rival even Sypro Ruby. Considering the central role of two-dimensional gel electrophoresis in top-down proteomic analyses, a more cost effective alternative such as Coomassie Blue could prove an important tool in ongoing refinements of this important analytical technique. To date, no systematic characterization of Coomassie Blue infrared fluorescence detection relative to detection with SR has been reported. Here, seven commercial Coomassie stain reagents and seven stain formulations described in the literature were systematically compared. The selectivity, threshold sensitivity, inter-protein variability, and linear-dynamic range of Coomassie Blue infrared fluorescence detection were assessed in parallel with Sypro Ruby. Notably, several of the Coomassie stain formulations provided infrared fluorescence detection sensitivity to <1 ng of protein in-gel, slightly exceeding the performance of Sypro Ruby. The linear dynamic range of Coomassie Blue infrared fluorescence detection was found to significantly exceed that of Sypro Ruby. However, in two-dimensional gel analyses, because of a blunted fluorescence response, Sypro Ruby was able to detect a few additional protein spots, amounting to 0.6% of the detected proteome. Thus, although both detection methods have their advantages and disadvantages, differences between the two appear to be small. Coomassie Blue infrared fluorescence detection is thus a viable alternative for gel-based proteomics, offering detection comparable to Sypro Ruby, and more reliable quantitative assessments, but at a fraction of the cost.
Gel electrophoresis is an accessible, widely applicable and mature protein resolving technology. As the original top-down approach to proteomic analyses, among its many attributes the high resolution achievable by two dimensional gel-electrophoresis (2DE)1 ensures that it remains an effective analytical technology despite the appearance of alternatives. However, in-gel detection remains a limiting factor for gel-based analyses; available technology generally permits the detection and quantification of only relatively abundant proteins (35). Many critical components in normal physiology and also disease may be several orders of magnitude less abundant and thus below the detection threshold of in-gel stains, or indeed most techniques. Pre- and post-fractionation technologies have been developed to address this central issue in proteomics but these are not without limitations (1–5). Thus improved detection methods for gel-based proteomics continue to be a high priority, and the literature is rich with different in-gel detection methods and innovative improvements (6–34). This history of iterative refinement presents a wealth of choices when selecting a detection strategy for a gel-based proteomic analysis (35).
Perhaps the best known in-gel detection method is the ubiquitous Coomassie Blue (CB) stain; CB has served as a gel stain and protein quantification reagent for over 40 years. Though affordable, robust, easy to use, and compatible with mass spectrometry (MS), CB staining is relatively insensitive. In traditional organic solvent formulations, CB detects ∼ 10 ng of protein in-gel, and some reports suggest poorer sensitivity (27, 29, 36, 37). Sensitivity is hampered by relatively high background staining because of nonspecific retention of dye within the gel matrix (32, 36, 38, 39). The development of colloidal CB (CCB) formulations largely addressed these limitations (12); the concentration of soluble CB was carefully controlled by sequestering the majority of the dye into colloidal particles, mediated by pH, solvent, and the ionic strength of the solution. Minimizing soluble dye concentration and penetration of the gel matrix mitigated background staining, and the introduction of phosphoric acid into the staining reagent enhanced dye-protein interactions (8, 12, 40), contributing to an in-gel staining sensitivity of 5–10 ng protein, with some formulations reportedly yielding sensitivities of 0.1–1 ng (8, 12, 22, 39, 41, 42). Thus CCB achieved higher sensitivity than traditional CB staining, yet maintained all the advantages of the latter, including low cost and compatibility with existing densitometric detection instruments and MS. Although surpassed by newer methods, the practical advantages of CCB ensure that it remains one of the most common gel stains in use.
Fluorescent stains have become the routine and sensitive alternative to visible dyes. Among these, the ruthenium-organometallic family of dyes have been widely applied and the most commercially well-known is Sypro Ruby (SR), which is purported to interact noncovalently with primary amines in proteins (15, 18, 19, 43). Chief among the attributes of these dyes is their high sensitivity. In-gel detection limits of < 1 ng for some proteins have been reported for SR (6, 9, 14, 44, 45). Moreover, SR staining has been reported to yield a greater linear dynamic range (LDR), and reduced interprotein variability (IPV) compared with CCB and silver stains (15, 19, 46–49). SR is easy to use, fully MS compatible, and relatively forgiving of variations in initial conditions (6, 15). The chief consequence of these advances remains high cost; SR and related stains are notoriously expensive, and beyond the budget of many laboratories. Furthermore, despite some small cost advantage relative to SR, none of the available alternatives has been consistently and quantitatively demonstrated to substantially improve on the performance of SR under practical conditions (9, 50).
Notably, there is evidence to suggest that CCB staining is not fundamentally insensitive, but rather that its sensitivity has been limited by traditional densitometric detection (50, 51). When excited in the near IR at ∼650 nm, protein-bound CB in-gel emits light in the range of 700–800 nm. Until recently, the lack of low-cost, widely available and sufficiently sensitive infrared (IR)-capable imaging instruments prevented mainstream adoption of in-gel CB infrared fluorescence detection (IRFD); advances in imaging technology are now making such instruments far more accessible. Initial reports suggested that IRFD of CB-stained gels provided greater sensitivity than traditional densitometric detection (50, 51). Using CB R250, in-gel IRFD was reported to detect as little as 2 ng of protein in-gel, with a LDR of about an order of magnitude (2 to 20 ng, or 10 to 100 ng in separate gels), beyond which the fluorescent response saturated into the μg range (51). Using the G250 dye variant, it was determined that CB-IRFD of 2D gels detected ∼3 times as many proteins as densitometric imaging, and a comparable number of proteins as seen by SR (50). This study also concluded that CB-IRFD yielded a significantly higher signal to background ratio (S/BG) than SR, providing initial evidence that CB-IRFD may be superior to SR in some aspects of stain performance (50).
Despite this initial evidence of the viability of CB-IRF as an in-gel protein detection method, a detailed characterization of this technology has not yet been reported. Here a more thorough, quantitative characterization of CB-IRFD is described, establishing its lowest limit of detection (LLD), IPV, and LDR in comparison to SR. Finally a wealth of modifications and enhancements of CCB formulations have been reported (8, 12, 21, 24, 26, 29, 40, 41, 52–54), and likewise there are many commercially available CCB stain formulations. To date, none of these formulations have been compared quantitatively in terms of their relative performance when detected using IRF. As a general detection method for gel-based proteomics, CB-IRFD was found to provide comparable or even slightly superior performance to SR according to most criteria, including sensitivity and selectivity (50). Furthermore, in terms of LDR, CB-IRFD showed distinct advantages over SR. However, assessing proteomes resolved by 2DE revealed critical distinctions between CB-IRFD and SR in terms of protein quantification versus threshold detection: neither stain could be considered unequivocally superior to the other by all criteria. Nonetheless, IRFD proved the most sensitive method of detecting CB-stained protein in-gel, enabling high sensitivity detection without the need for expensive reagents or even commercial formulations. Overall, CB-IRFD is a viable alternative to SR and other mainstream fluorescent stains, mitigating the high cost of large-scale gel-based proteomic analyses, making high sensitivity gel-based proteomics accessible to all labs. With improvements to CB formulations and/or image acquisition instruments, the performance of this detection technology may be further enhanced.
EXPERIMENTAL PROCEDURES
Materials
All consumables were of electrophoresis grade or higher quality. CHAPS was purchased from Anatrace (Maumee, OH). A range of commercially available, ready-made CB formulations were purchased (Table I). A recombinant protein molecular weight marker from Fermentas (Hanover, MD) was used extensively as a protein standard. Electrophoresis consumables including immobilized pH gradient (IPG) strips, acrylamide, bisacrylamide, low melting agarose, all ampholyte solutions, tris-glycine SDS buffer, and Sypro Ruby total protein stain were from BioRad Laboratories (Hercules, CA). All other consumables, including a series of isolated protein standards, and Coomassie Blue G250 and R250 dyes were purchased from Sigma (St. Louis, MO). The purity of the isolated protein standards was assessed by gel electrophoresis, essentially as previously described (55). The proteins used and their respective purities were: bovine serum albumin (BSA, 67 kDa), 80 ± 1.4%; chicken egg albumin (CEA, 44 kDa), 74 ± 0.71%, bovine erythrocyte carbonic anhydrase (BCA, 29 kDa), 80 ± 2.6%, soybean trypsin inhibitor (STI, 20 kDa), 79 ± 2.4%, and chicken egg lysozyme (CEL, 15 kDa), 87 ± 2.6%. The concentrations of these standard proteins were adjusted according to need.
Table I. In-gel protein staining formulations examined in this study.
| Stain Name | Source | Formulation | Cost per gela | |
|---|---|---|---|---|
| i | SYPRO RUBY | BioRad (Hercules CA) | Not Available | $11.69 |
| ii | BioSafe Coomassie Stain | BioRad (Hercules CA) | Not Available | $3.49 |
| iii | Colloidal Blue Stain | Invitrogen (Carlsbad, CA) | Not Available | $4.76 |
| iv | EZ Blue Gel Staining Reagent | Sigma (St. Louis, MO) | Not Available | $4.66 |
| v | GelCode Blue Stain Reagent | Pierce (Rockford, IL) | Not Available | $8.33 |
| vi | Imperial Protein Stain | Pierce (Rockford, IL) | Not Available | $5.03 |
| vii | PAGE Blue Protein Stain | Fermentas (Hanover, MD) | Not Available | $6.00 |
| viii | Simply Blue Safe Stain | Invitrogen (Carlsbad, CA) | Not Available | $5.49 |
| xiv | Blue Silver | (8) | 0.12% G250, 10% (NH4)2SO4, 10% H3PO4, 20% CH3OH | $0.96 |
| x | Fast Coomassie | (21, 53) | 0.08% G250, 8% H2SO4, 88 mm NaOH, 11.5% TCA | $1.17 |
| xi | Modified Neuhoff G250 | (40) | 0.1% G250, 10% (NH4)2SO4, 3% H3PO4, 20% CH3OH | $0.90 |
| xii | Neuhoff G250 | (12, 32, 39) | 0.08% G250, 8% (NH4)2SO4, 2% H3PO4, 20% CH3OH | $0.76 |
| xiii | Neuhoff R250 | (12, 32, 39) | 0.08% R250, 6% (NH4)2SO4, 2% H3PO4, 20% CH3OH | $0.71 |
| xiv | Sensitive Colloidal | (52) | 0.035% G250, 17% (NH4)2SO4, 3% H3PO4, 34% CH3OH | $1.13 |
| xv | Traditional Coomassie | (41, 54) | 0.1% G250, 10% CH3COOH, 40% CH3OH | $1.27 |
a Cost in Canadian dollars of 50 ml of stain required for a single 9 cm mini-gel, based on list price of reagents without discounts.
Sample Preparation
Mouse tissue samples were collected, handled, and prepared as previously described (56–58). Briefly, whole mouse liver and brain were excised from the sacrificed animal as rapidly as possible and washed in ice cold isotonic PBS solution supplemented with broad spectrum protease, phosphatase, and kinase inhibitors (5, 56, 59). Washed tissues were rapidly dissected into 1 mm cubes, and these were immediately flash frozen in liquid N2 for storage at −80 °C until needed. The frozen tissue samples were homogenized by automated frozen disruption and physically separated into gross soluble and membrane protein fractions as described previously (5, 56, 58, 60).
1DE of Isolated Protein Standards and 1DE/2DE of Solubilized Tissue Samples
1DE was carried out essentially according to Laemmli (61) with some minor modifications (62, 63). All 1DE gels were of tris-glycine composition, and 1 mm thickness with 4 mm wide wells. Prefractionated and solubilized mouse liver samples were resolved on standard 12.5% T, 2.6% C gels, with 5% T, 2.6% C stacking gels. For greater resolution of the range of isolated protein standards, these were resolved using 15% T, 2.6% C gels. PAGEruler unstained molecular weight marker from Fermentas (Hanover, MD) was used as a mixed-protein standard as it consisted of 13 recombinant proteins covering a range of molecular weights (10–200 kDa) at a reasonable per-application cost. The supplier graciously provided data regarding the concentration of each protein component. The product was used according to the manufacturer's instructions; no reducing agent was added and the sample was not warmed before gel electrophoresis. Dilutions of the product were resolved in standard 1 mm thick SDS-PAGE gels (composition as above) with 4 mm wide wells.
2DE of solubilized mouse brain soluble and membrane proteins fractions was carried out essentially as previously described. A total of 100 μg protein was loaded per separation, and the second dimensions were standard 9 cm format gels of 12.5% T, 2.6% C tris-glycine composition and 1 mm in thickness (56, 57, 60).
Standardization of CB Staining Parameters
The literature is rich with a variety of mass spectrometry-compatible fixation, washing and staining parameters for CB staining. As it would have been unreasonable and likely superfluous to test every possible combination of these different parameters, the gel fixation, washing, staining, and destaining steps were standardized; reagents and incubations specified for each CB stain formulation were replaced with a single unified staining and destaining protocol. SR was the only exception, being fixed and washed identically to CB-stained gels, but using previously optimized conditions for staining and destaining (56, 57). Thus, in the unified protocol, all gels were (1) fixed according to protocols for SR staining, in 10% methanol : 7% acetic acid (v/v) solution for 4 h, with continuous rocking; (2) washed 10 times with water, for 20 min per wash; and (3) stained, destained, and imaged as described below.
CB Staining
All commercial stains were single reagents requiring no preparation time, except for the Colloidal Blue Stain kit (Invitrogen) which contained two solutions and required some further preparation steps; the stain was prepared according to the manufacturer's instructions for staining tris-glycine polyacrylamide gels. Several stain formulations were evolutions of the original CCB protocol described by Neuhoff and colleagues (Table I) consisting of some combination of methanol, ammonium sulfate, phosphoric acid, and CB dye in aqueous solution (12, 39); here, ammonium sulfate was used throughout rather than the aluminum sulfate originally used in one formulation (40). These stain reagents were prepared by the same method and the required amounts of water and methanol were combined before the addition of ammonium sulfate and phosphoric acid, with the requisite amount of the dye added last. The stain was allowed to mix well for 15 min before use. There were two exceptions. The Traditional Coomassie stain (formulation xv) was a soluble-dye CB formulation (i.e. noncolloidal); this was prepared by combining the components in the order listed in Table I. The Fast Coomassie stain (formulation x) used a fundamentally different chemistry than the Neuhoff-derived stains (21, 53). Briefly, an aqueous 0.1% Coomassie G250 solution was acidified with H2SO4 to a final concentration of 0.5 m, producing a dark red-brown solution which was stirred continuously for 3 h. The stirred solution was vacuum filtered through a regenerated cellulose membrane (22 μm pore size). The filtered solution was sequentially combined with 10 m NaOH, then 100% trichloroacetic acid to achieve the appropriate final concentrations (Table I) and pH, yielding the final stain reagent, a dark green suspension.
Several of the published stain formulations were observed to form precipitates over extended storage. Accordingly, all stains were prepared immediately before use and none were stored for any length of time.
Each fixed, washed gel was incubated in 50 ml of CB staining solution for 4 h at room temperature with continuous rocking. The stain was removed and the gels were destained with 10 washes of distilled water. Five initial rapid washes (5 min each) quickly removed excess stain from the gel and container surfaces. Five additional washes of 20 min each further removed excess dye yielding uniform, clear backgrounds. The only exception was formulation xv, which was a soluble-dye (i.e. non-colloidal) CB formulation; these gels were destained three times for 60 min per wash in a 50% methanol, 10% acetic acid solution, followed by three 60 min washes in water to re-swell the gel.
CB in-gel was detected fluorescently in the IR spectrum using the laser-based Odyssey imager (Licor, Lincoln NB). Gels were excited simultaneously at 685 and 785 nm (default settings), and emission was collected using the 700 channel (i.e. < 750 nm); 16-bit tiffs were natively acquired at 84 μm pixel resolution and medium quality. Image intensities were normalized by adjusting the photomultiplier gain settings to provide consistent sub-saturation maximum signal intensity.
SR Staining and Detection
SR staining was carried out essentially according to the manufacturer's specifications, with minor modifications as previously described (56, 57). Fixed, washed mini gels were stained in 50 ml of new, undiluted SR reagent (BioRad, Hercules, CA) with continuous rocking for 12 h (overnight), in the dark. The stain was removed and the gel washed with fixative solution (2 × 15 min) and then with distilled water (4 × 30 min). SR was detected in the visible spectrum as previously described; excited at 480 nm and emission acquired at 620 nm using the CCD-based ProXpress Proteomic Imaging System (PerkinElmer, Boston MA). All SR gel images were acquired at 100 μm pixel resolution. Image intensities were normalized by altering image exposure time to achieve a consistent, subsaturation maximum peak height (signal intensity). Images were exported as 16-bit tiff files using Profinder Software (PerkinElmer, Boston MA).
Image Analysis
All 16-bit tiff images were analyzed using Progenesis Workstation v2005. The fluorescent signal from bands in 1D gels and spots in 2D gels were quantified as fluorescent volumes (FV) where:
That is, volume was taken as the sum of the volume of all pixels that make up that band or spot. For all 1DE separations, the background was assessed in empty gel areas between sample lanes, immediately adjacent to the protein of interest. For all 2DE separations, background was calculated using the average-on-boundary method, in which the average height of all pixels bordering the spot was used to calculate the background volume for that protein. Only background-subtracted fluorescent volumes are reported. The calculated background volume was additionally used to calculate S/BG and signal squared to background (S2/BG) ratios where indicated.
Evaluation of In-gel Protein Detection Criteria
In addition to S/BG and S2/BG, three additional criteria were quantitatively assessed to provide a detailed analysis of CB-IRFD and SR as in-gel protein detection methods.
1) Detection Sensitivity
A dilution series of the recombinant protein mixture was designed to provide data points above and below ∼1 ng of protein, the accepted detection threshold for SR (6, 9, 15, 19, 44, 64). The lowest limit of detection (LLD) was defined as the smallest quantity of protein that could be statistically distinguished from background and thus consistently detected (t test, p < 0.05). Although not the strictest definition of LLD, it was used here as a practical tool for the routine quantitative comparison of a large number of different staining formulations. Indeed, this is essentially how in-gel detection sensitivity has most commonly been assessed in the literature (65); unfortunately, this is usually only assessed for an arbitrary “standard” such as BSA. However, the possibility that a staining method may be significantly less sensitive for other proteins (see IPV, below) is often overlooked. To account for this, the lowest practical sensitivity (LPS) was derived from the same dilution series but was calculated as the highest quantity of protein in the series that was statistically indistinguishable from background. To better characterize a stain based on this full range of information, an integrated measure of practical sensitivity (IMPS) was adopted that mathematically incorporated LLD and LPS (Fig. 3C). IMPS was defined as:
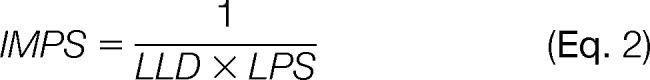 |
Fig. 3.

The sensitivity of fluorescent staining formulations. Derived from Fig. 2. A, Lowest limit of detection (LLD) was taken as the minimal amount of protein that could be discriminated from background. B, Lowest Practical Sensitivity (LPS) was taken as the maximum amount of protein that was indistinguishable from background. C, Integrated Measure of Practical Sensitivity (IMPS) was taken as the inverse product of LLD × LPS. * Indicate significant differences from SR (stain i). † Indicate significant differences from best performing commercial CB formulation by each measure of sensitivity (formulations xiv, xii and xii, in panels A, B, and C, respectively). ‡ Indicate significant differences from the best performing published CB formulation in each experiment (formulations xiv, xii and xii, in panels A, B, and C, respectively). § Indicates no significant differences between the commercial and published CB formulations providing the lowest LLD and LPS (formulations ii and xii in both parts A and B). £ Indicates a significant difference between commercial and published CB formulations providing the highest IMPS. (One-way ANOVA, F = 11.8, 22.7, 24.9 for parts A, B and C, respectively, p < 0.001, Post-hoc Tukey analysis, p < 0.05, n = 3). Error bars indicate S.D.
Thus the IMPS mathematically placed equal weight on both the LLD and the LPS for a given detection method, which was logical, considering both are likely to impact the analysis of gel-based proteomes, and the extent to which one is more important than the other is unclear and likely a matter for debate. Moreover, the IMPS was taken as the reciprocal of these terms to provide an intuitive single-value measurement that could be used for rapid communication and comparison of different stains; the larger the value, the more sensitive the stain under practical conditions, taking both the LLD and LPS into account.
2) Interprotein Variability
An inherent variation in signal strength yielded by the same quantity of different proteins; detection sensitivity is thus somewhat protein-dependent. Two different measures of IPV were calculated. The IPV range was calculated as the maximum extent of fluorescent signal intensities expected based on linear fits of the empirical data for different proteins in the mixture of standards. Because the magnitudes of fluorescent signals varied greatly from stain to stain, the data were normalized as a percentage of the maximum. That is:
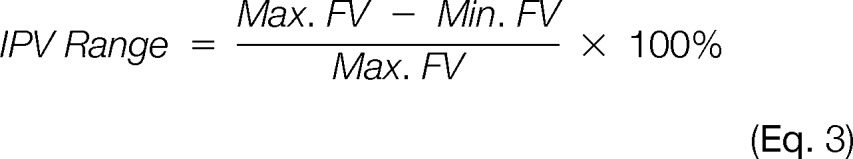 |
The IPV range was thus calculated for each stain at three points (5, 10, and 20 ng protein) in the mid-range of the dilution series. As a second measure of IPV, the distribution of fluorescent responses for the different proteins in the dilution series was assessed. Rather than use any single protein as the basis for comparison, an overall fit of all 13 proteins in the mixture was taken as the basis for comparison. The integral of the absolute difference between this overall fit of all the data, and the linear fit for each individual protein was taken as a physical measure of the divergence of that individual fit from the overall data set.
 |
Where:
Findividual was the linear fit for an individual protein in the mixture of recombinant standards, diluted in series
Foverall was the linear fit of all data points in the dilution series data for a given stain
The limits of the definite integral were taken to be 10 and 20 ng. The lower limit was chosen because none of the stains tested had an LLD significantly worse than 10 ng; to assess the integral IPV below 10 ng would require selective extrapolation for stains with LLD values in this range. The upper limit was chosen to include as much empirical data as possible, without placing undue weight on the small number of fits available for larger quantities of protein (i.e. only two of the 13 proteins in the mixture). Integral values were normalized to the magnitude of the fluorescent signal to account for different signal intensities between stain formulations and summed for all 13 proteins in the mixture, yielding a single numerical value representing the total observed deviation of individual proteins from the dataset for a given stain. Thus the IPV range and integral IPV represented the range and distribution, respectively, of the dilution series data for all 13 proteins in the standard mixture.
3) Linear Dynamic Range
The overall detection capacity of a stain in terms of the linear relationship between the absolute quantity of a given protein and the fluorescent signal measured from bound stain. A large linear range enables simultaneous detection, quantification and meaningful comparison of higher and lower abundance proteins in biological samples. Beyond the LDR, the relationship between signal and the quantity of protein is uncertain, and it is unacceptable to assume a simple correlation. Thus, because of the IPV of the stain response, a given detection method may yield substantially different LDRs for different proteins. Currently, all that can be done is to assess the LDR for several standard proteins and take this as an indication of how a given stain can handle the complexity of biological protein mixtures. For thoroughness, it was anticipated that the actual LDR might extend beyond the microgram range, and thus quantities of protein well beyond this expected maximum had to be included on the same gel. Accordingly, five commonly available isolated standard proteins with a range of molecular weights were used. Dilution series of the isolated protein standards were designed to extend below the LLD and beyond the documented LDR of SR, and to fit within a limited number of wells on a mini-gel for convenience. Images were acquired so as to yield consistent sub-saturation signal intensity for the tallest pixel in the band corresponding to the largest quantity of protein in the dilution series. The fluorescent signal volumes for each band were determined by quantitative image analysis enabling determination of the LLD for these proteins as was done for the initial recombinant standards. From the LLD, the range of linearity for each protein was determined by successive regression analyses; LDR was defined as the range of protein quantity for which linear fits of the data had R2 ≥ 0.995. Above the LDR, fluorescence characteristically plateaued and R2 values of linear fits incorporating these data points rapidly declined.
Analysis of 2D-Gel Data
2D gels were used as a final point of comparison between the different detection methods. Four replicates of each gel, prepared in parallel, were subjected to either SR or CB staining, and imaged accordingly as described above. All gels were warped and matched using automated digital image analysis.
Several analytical criteria were used to compare the efficacy of the different stains for detecting 2D-gel proteomic maps. The total number of spots detected in each gel was quantified as previously described (50, 56). As means of spot filtering, spots that were detected and positionally matched with 100% consistency between replicate gels for a given stain were considered more valid and more reliable than spots which were detected in some replicates, but not others. Irreproducible spots could, in principal, be aberrations, or contamination such as dust spots, stain crystals, etc. Thus, focusing on the so-called ‘100%-reproducible’ spots seemed a rational course of action for subsequent quantitative analysis and comparison. Correspondingly, the number of 100%-reproducible spots, that is, spots that were consistently detected and positionally matched in each replicate gel for a given condition, was quantified.
The inter-replicate variability of the different detection methods was assessed for 2D gel spots that were detected and positionally matched in all four replicates for each condition. For such spots, the coefficient of variation (CV) of the fluorescent volume was determined as the ratio of the standard deviation/mean. The distributions of the CV of FVs for all reproducibly detected spots with each detection method were overlaid for comparison.
Comparative analysis of the 100%-reproducible spots between stains was carried out to determine if there were spots that some stains failed to detect. Additionally, a quantitative analysis was carried out for proteins detected by both detection methods, to determine if CB-IRFD reported different relative quantities of proteins, compared with the benchmark, SR. The S2/BG ratio for 2D gels was additionally determined, essentially as described for 1D gels (above).
Finally, the absolute quantity of protein in a given spot resolved in 2D gels was estimated with the following equation
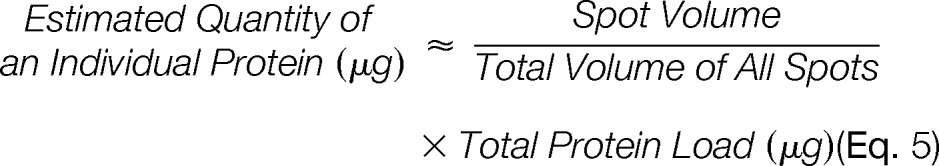 |
There were necessarily a number of assumptions that must be made to accept this estimation: (1) a linear correlation existed between fluorescence and protein quantity across the entire range of protein quantities present in the gel; (2) a consistent correlation existed between fluorescence and protein quantity that did not vary from protein to protein (i.e. zero IPV); (3) the quantity of total protein loaded, based on a total protein assay, against a single protein standard, had real meaning; (4) the 2D gel electrophoresis processes were 100% efficient, and all the protein loaded entered both 1st and 2nd dimension gels and then was fully resolved; and (5) proteins below detection threshold collectively amounted to an insignificantly small proportion of the total protein load, and thus their overall contribution was not significant compared with the contributions of detectable proteins in the resolved proteome. These were not insignificant assumptions. The first two assumptions were clearly invalid and the final three assumptions were also likely problematic. However, momentarily accepting these assumptions for the sake of estimation, the quantity of protein resolved in a given spot could thus be derived according to Equation 5 (above).
Statistics
In quantitative and differential image analyses of 2D gels, only 100% reproducible spots (spots that were detected and positionally matched) across the entire dataset (n = 4 gels) were considered. Protein spots that were inconsistently detected in replicate gels were eliminated from all calculations. Quantitative comparisons of resolved proteomes were tested for statistical significance using the integrated statistics package of Progenesis Work station. All comparisons of 2D gels involved two groups (i.e. experimental versus control) and thus t-tests were employed to determine the significance of individual differences. Only differences that were significant (p < 0.05) are reported. For experiments involving a large number of groups, such as screening the different CB stains, a one-way ANOVA (p < 0.05) with Tukey's HSD post-hoc analyses was used to test for differences between groups.
RESULTS AND DISCUSSION
Here the characteristics of in-gel protein detection using CB-IRF were explored in comparison with SR. The goal was to determine which CB formulations were most effective with IRFD and if this might be an effective alternative to SR for proteomic analyses. Several CB formulations outperformed SR in certain detection parameters, but important practical limitations were identified that must be considered when using either stain for gel-based proteomics.
Normalization Of Gel Staining Parameters
Only a few CB formulations have been characterized in terms of their IRF properties (50, 51). To meaningfully compare the efficacies of the different CB stains in a timely and efficient manner, only the stain formulation per se was assessed and a single, unified fixation, washing, staining, and destaining protocol was adopted (see Methods). Although this might mean that some stains may not have performed optimally, according to their original design, the unified protocol was rationally designed to minimize this possibility.
Assessing the Selectivity of Detection Methods
The S/BG ratio was taken as a straightforward measure of the selectivity of a stain for protein relative to the gel matrix. All 14 CB formulations were compared with SR for staining of two different protein mixtures resolved by 1DE: (1) a commercially available mixture of recombinant protein standards; and (2) membrane proteins extracted from mouse liver (Fig. 1 and supplemental Fig. S1). S/BG ratios for all bands in a given lane of the 1D gels were aggregated; essentially, selectivity was initially assessed as previously described (50). However, by this criterion, certain CB stains were unable to detect more than a handful of proteins, while yielding S/BG ratios as high as those stains that detected many more proteins and visibly performed much more comparably to SR (data not shown). For example, staining with CB formulations vi, xiii and xv yielded very weak IRFD signal intensity (Fig. 2 and supplemental Fig. S2); low signal intensity was accompanied by correspondingly low background, yielding S/BG ratios indistinguishable from SR (data not shown). Thus, the S/BG ratio was not an effective indicator of stain performance (Fig. 2 and supplemental Fig. S2). In practice, stains that produce untenably low signal intensity (i.e. below the optimal parameters of the detection instrument) will ultimately compromise sensitivity; no amount of signal integration or photomultiplier gain manipulation can reasonably be expected to rescue data from such low intensity signals.
Fig. 1.

Assessing selectivity of SR and CB staining. A, Mouse liver membrane samples were separated by 1DE (10 μg of total protein per lane). B, A commercially available mixture of 13 recombinant protein standards was separated by 1DE (total protein load was 2.5 μg per lane, and the quantity of individual protein species ranged from 80 to 480 ng). Gels were stained with SR (Stain i; imaged using standard fluorescence settings) or with one of 14 different CB stain formulations and imaged using IRF (formulations ii and xii shown here; all other CB formulations shown in supplemental Fig. S1). Data from quantitative image analysis of mouse liver membrane proteins (C) and resolved protein standards (D) was used to calculate S2/BG of detected proteins. * Indicate significant differences from SR (i.e. stain i). † Indicate significant differences from best performing commercial CB formulation in this experiment (i.e. stain v). ‡ Indicate significant differences from best performing published CB formulation in this experiment (i.e. stain x). § Indicate no significant difference between the commercial and published stain formulations with the highest S2/N. (One-way ANOVA, F = 68.9 and 65.7 for parts C and D, respectively, p < 0.001, Post-hoc Tukey Analysis, p < 0.05, n = 3). Error bars indicate S.D. Refer to supplemental Fig. S1 in Supplementary Data for the complete series of gel images.
Fig. 2.

Assessing sensitivity of SR and CB. A dilution series of a commercially available mixture of 13 recombinant protein standards was resolved by 1DE and detected with either SR (A) or stained with one of 14 different CB formulations and detected using IRF. Only CB formulations ii (B) and xii (C) are shown here; all other formulations are shown in supplemental Fig. S2. The fluorescent volume in each band detected in images A–C are plotted in the corresponding right hand panels D–F, respectively. Trend lines indicate overall linear fits of the entire dataset. Error bars represent S.D., n = 3. Refer to supplemental Fig. S2 in Supplementary Data for the complete series of gel images.
As an alternative measure of selectivity, the S2/BG ratio better emphasized signal intensity as an important criterion while still accounting for background. All assessments of detection selectivity reported henceforth are thus given as the S2/BG ratio, and these two terms are used interchangeably. Considering the mouse liver membrane proteins, several CB formulations showed significantly better selectivity than SR (Fig. 1A and supplemental Fig. S1A); this was consistent with previous work indicating potential performance advantages of CB over SR (50). The selectivity of all 14 CB formulations was additionally assessed using recombinant protein standards and SR yielded among the highest S2/BG ratios; several CB formulations matched the performance of SR, but none exceeded it (Fig. 1B and supplemental Fig. S1B).
Assessing Detection Sensitivity
Detection sensitivity has been the primary driving force behind the development of fluorescent stains such as SR, and no alternative will gain traction in the field unless it performs comparably. As such, the threshold sensitivity of each of the 14 CB stains was assessed (Fig. 2 and supplemental Fig. S2). In the resolved dilution series, the background subtracted fluorescent volume contributed by each band was individually determined; thus, as the quantity of protein decreased in a dilution series, the fluorescent volumes approached zero (Fig. 2 and supplemental Fig. S2). To simplify presentation, data points that were statistically indistinguishable from background were omitted. Several different mathematical representations of the threshold sensitivity were explored. Linear plots of the empirical data could have been used to extrapolate a theoretical maximum sensitivity for each stain as the point at which the plots of protein quantity intercept background; by such analyses, SR and several CB formulations were theoretically capable of detecting sub-nanogram protein levels (data not shown). Alternatively, the LLD is a robust, established and widely accepted criterion of sensitivity. Several CB stains performed indistinguishably from SR, achieving detection of nanogram to sub-nanogram quantities of protein (Fig. 3A). These data were consistent with indications that CB-IRFD was as sensitive or more so than SR (Fig. 1) (50).
However, because of IPV of the stain response (discussed in the following section) the threshold detection sensitivity was somewhat protein-dependent. Here it was observed that as little as 500 pg of some proteins in the mixture of recombinant standards provided sufficient signal intensity to be distinguished from background, whereas others were only detected at several nanograms (Fig. 2 and supplemental Fig. S2). Thus, LPS was considered as an additional tool for assessing stains. Whereas the LLD identified those proteins in a mixture with the highest stain affinity and thus the greatest fluorescent signal, the LPS was defined by those proteins in the mixture that had the poorest stain affinity, yielding the weakest fluorescent signals. In practical terms, the LPS established the quantity of protein below which some proteins in a mixture would be undetectable (Fig. 3B). Notably, CB formulations with the poorest LLD also had among the poorest LPS (formulations vi, xiii and xv, Fig. 3). These stains additionally had the poorest selectivity of all the formulations tested (see previous section; Figs. 1 and 3). Thus by multiple measures, these particular formulations appeared to be less effective choices for in-gel IRFD of proteins. However, it should be noted that formulation vi in particular was easily the equal of the other CCB formulations when scanned densitometrically (data not shown).
Although the LPS of several CB stains were comparable to that of SR, several performed significantly worse, being unable to detect relatively large amounts of some proteins (Fig. 3B). Moreover, of the stains that had LLD values very close to SR, several had significantly poorer LPS values. Thus, several CB formulations were as sensitive as SR for certain proteins, but markedly less sensitive for others (Figs. 3A, 3B). Taking both LLD and LPS into consideration seemed the most reasonable practice given that the problem of IPV of the stain response seems unlikely to be solved. Thus, the IMPS (Equation 2) was derived to provide a convenient and intuitive means for comparing the threshold sensitivity of different detection methods for a range of different proteins; the higher the IMPS, the better the overall sensitivity of the stain. Although several CB stains had an IMPS that matched that of SR, two formulations, ii and xii, had significantly higher values (Fig. 3C).
Assessing Inter-Protein Variability of Detection Methods
For every staining formulation tested, different proteins yielded different linear correlations between quantity and fluorescent signal volume (Fig. 2 and supplemental Fig. S2). This IPV of the stain response appeared to be an inherent limitation in all protein detection technologies. To characterize in-gel CB-IRFD as fully as possible, IPV was assessed for each stain formulation in parallel with SR (Fig. 2 and supplemental Fig. S2, summarized in Fig. 4).
Fig. 4.

The interprotein variability (IPV) of fluorescent staining formulations. Derived from Fig. 2. Two parameters of IPV are reported: A, The IPV range was calculated as the maximum range of IPV for 13 protein standards at 3 points in the mid-range of the dilution series. B, The integrated IPV was calculated as the integral sum of the deviation of each individual linear fit, from an average fit of all 13 proteins. * Indicate significant differences from SR (i.e. stain i). † Indicate significant differences from best performing commercial CB formulation in each experiment (formulation ix in panel A, formulation xii in panel B). ‡ Indicate significant differences from best performing published CB formulation in each experiment: formulation ix in panel A, formulation xii in panel B. § Indicate no significant difference between the commercial and published formulations providing the lowest IPV. (One-way ANOVA, F = 13.9, 28.5 for parts A and B, respectively, p < 0.001, Post-hoc Tukey analysis, p < 0.05, n = 3). Error bars indicate S.D.
SR provided among the lowest IPV of all stains tested, by both IPV measures; in terms of both IPV range (Equation 3) and integral IPV (Equation 4), several CB formulations were indistinguishable from SR, but none were superior. (Fig. 4) Relative to SR, there were only a few CB formulations for which the trend in IPV range dramatically differed from that of the integral IPV. In particular, CB formulation v yielded an IPV range that was not significantly better than SR, yet the integral IPV was significantly worse. Notably, the difference between poorly performing CB stains and SR was much greater in terms of IPV range than integral IPV. That is, if the IPV range were the only available assessment, one might conclude that SR had about 20% less IPV than the worst performing CB stains; although not an insubstantial difference, the integral IPV revealed a somewhat different picture, indicating that the distribution of data within the IPV range is roughly two- to threefold smaller with SR and the leading CB formulations relative to the poorest performing CB formulations (Fig. 4).
Overall, it was clear that IPV was a concern for all of the stains tested. Detection methods with excessively large IPV were considered less than ideal. However, the extent to which IPV of the stain response is actually a problem for quantitative proteomics is not certain. Most proteomic analyses compare the in-gel signal intensities of the same protein spots between experimental groups, IPV is less of a concern than in attempts at absolute quantification, for which IPV of the stain response might lead to substantial errors. Although IPV is an informative characteristic of in-gel staining, the sensitivity and selectivity are likely more critical. High IPV could be accepted if a stain was sufficiently sensitive, and would be preferable to a stain with low IPV but poor sensitivity. Thus, high IPV may not be a sufficient reason to abandon an otherwise highly sensitive CB stain. In contrast, IPV provided an additional criterion with which to discriminate between otherwise similarly performing stain formulations. For instance, whereas CB formulations iv and vii were among the most sensitive (Fig. 3), there were several other formulations with comparable sensitivity, but simultaneously significantly lower IPV (Fig. 4). Although likely inherent to all proteomic detection methods, eliminating IPV seems unlikely given the diversity of the proteome. Nonetheless, in terms of being aware of limitations and thus practical effects on protein assessments, quantitative evaluation of the IPV characteristics of a chosen detection method is prudent.
Selecting the Optimal CB Formulations for Infrared Fluorescence Detection
Having quantitatively compared 14 CB formulations to determine their relative efficacy as reagents for IRFD of proteins, additional analyses of all of these formulations was deemed unnecessary. Selectivity, sensitivity, and IPV were thus collectively considered as criteria for the identification of the most suitable CB formulations for further comparisons with SR. Individually, none of the criteria were especially convincing support for adopting one stain formulation over others; stains that demonstrated superior performance across criteria were of the most interest. Several CB stain formulations performed strongly in all the criteria assessed. In particular, commercial formulations ii and iii, and published formulations ix, x, xii, and xiv all provided results comparable to SR, and slightly better in some respects (Table II). Notably, formulations ii and xii had the highest IMPS values of any of the stains tested, and were significantly more sensitive than SR by this criterion, although their LLDs were indistinguishable from SR (Fig. 3). Conversely, formulations vi, xiii and xv consistently yielded among the poorest performances. The traditional, non-colloidal CB formulation xv was less selective and sensitive than the CCB formulations tested.
Table II. Summary of the relative performances of the 14 different CB formulations, imaged with IRF, compared with SR. (+) Indicate performance significantly superior to SR. (−) Indicate performance significantly inferior to SR. (e) Indicate performance indistinguishable from SR.
| Selectivity–Mouse Liver Sample | Selectivity–Recombinant Sample | LLD | IMPS | IPV Range | Integral IPV | |
|---|---|---|---|---|---|---|
| Stain Formulation | Fig. 1C | Fig. 1D | Fig. 3A | Fig. 3C | Fig. 4A | Fig. 4B |
| ii | + | e | e | + | e | e |
| iii | + | − | e | e | e | e |
| iv | + | − | e | e | − | |
| v | + | e | e | e | e | − |
| vi | − | − | − | e | − | − |
| vii | + | − | e | e | − | − |
| viii | + | − | e | e | e | − |
| ix | e | e | e | e | e | e |
| x | e | e | e | e | e | e |
| xi | e | − | e | e | e | e |
| xii | + | e | e | + | e | e |
| xiii | − | − | − | e | e | − |
| xiv | e | e | e | e | e | e |
| xv | e | − | e | e | − | e |
Of all the CB formulations, only two used the R250 dye variant (i.e. vi and xiii) and both were among the poorest performing IRFD reagents (Table I). Acquiring images using the alternative 800 channel of the Odyssey scanner did not improve the detection of R250-based CB stains (data not shown). Nonetheless, it should be noted that R250-based CB stain reagents in general, and formulation vi in particular qualitatively appeared to be excellent visible dyes (data not shown). Thus it appeared to be the differing spectral properties of the G250 and R250 variants of CB, and not the protein binding properties of these dyes, that was responsible for their differing performance as IRFD reagents.
Among commercial CB reagents tested, formulations ii and iii performed competitively throughout the battery of tests, proving comparable to SR in terms of selectivity, sensitivity, and IPV (Table II). Commercial formulations iv, v and vii were as sensitive and selective (or nearly so) as SR but yielded significantly higher IPV. The relatively poor overall performance of commercial CB formulations relative to the published formulations (Table II) may be related to their long term stability. Unlike published formulations, no precipitates were observed in the commercial formulations despite their prolonged storage. The commercial formulations may have altered dye concentrations, or contain agents to mitigate the formation of dye precipitates, enhancing shelf life, that may have contributed to some quantitative differences in stain performance with IRFD. Nonetheless, the convenience of commercial stains cannot be overlooked; for maximum reproducibility and quality control, the advantages afforded by a commercial product are considerable, despite the higher cost (Table I). Ultimately the decision of the user will be based on a number of practical factors, not only stain performance. Thus, to focus subsequent analyses on the “optimal” CCB formulations in comparison with SR, it was fitting to include both ii and xii in further tests, thus providing additional detail for one commercial and one published CB stain formulation.
Assessing the Linear Dynamic Range of Staining Methods
Simultaneously generating quantitative data from both large and small quantities of a variety of different proteins in a given sample is one of the substantive remaining challenges in proteomics and a major driving force for the continuing development of pre- and postfractionation techniques. A large linear range enables simultaneous detection, quantification and meaningful comparison of higher and lower abundance proteins in biological samples. Thus as an additional parameter with which to characterize CB-IRFD, and for comparison between CB and SR, the LDRs for these detection methods were compared.
Based on detailed assessments across a wide range of protein concentrations, SR yielded a LDR of 2–3 orders of magnitude for four of the five proteins tested (Figs. 5, 6 and supplemental Fig. S3), consistent with previous observations (6, 15, 45). Notably, both CB formulations ii and xii provided significantly greater LDR than SR for four of the five proteins tested (CEA, BCA, STI, and CEL), and for the fifth (BSA) the LDR for CB and SR were indistinguishable (Figs. 5, 6 and supplemental Fig. S3). Notably SR had a greater LDR for BSA than for any of the other standards tested, and this was the only standard for which its LDR approached that of the two CB formulations (Figs. 5, 6 and supplemental Fig. S3). Moreover, SR exhibited significantly greater signal attenuation than CB for the highest quantities of proteins in these determinations (Fig. 5 and supplemental Fig. S3). That is, SR consistently diverged from the linear relationship between fluorescent signal and protein quantity both earlier and more profoundly than did CB (Fig. 5 and supplemental Fig. S3).
Fig. 5.

Assessing the linear dynamic range of SR and CB. A dilution series of isolated protein standards was resolved by 1DE and stained with either SR, or one of the two most sensitive CB formulations, ii and xii, and imaged with IRF. Pictured here in the left hand panels, are images of parallel BSA (A) and STI (B) dilution series. Quantitative image analysis was used to determine the LLD and the LDR for each stain/protein. Data near threshold are expanded in the center panels (C and D), and the full range of data are plotted in the right hand panels (E and F). The LDR was determined by regression analysis. Solid lines indicate the LDR for each stain and standard. Dotted lines indicate projection of the linear fit beyond the LDR. Gel images and quantitative LLD/LDR determinations for CEA, BCA, and CEL are shown in supplemental Fig. S3. ** Indicate CB IRF volumes that were significantly greater than SR. (One-way ANOVA. p < 0.001, Post-hoc Tukey analysis, p < 0.05, n = 3). Error bars indicate S.D. Refer to supplemental Fig. S3 in Supplementary Data for the complete series of gel images.
Fig. 6.
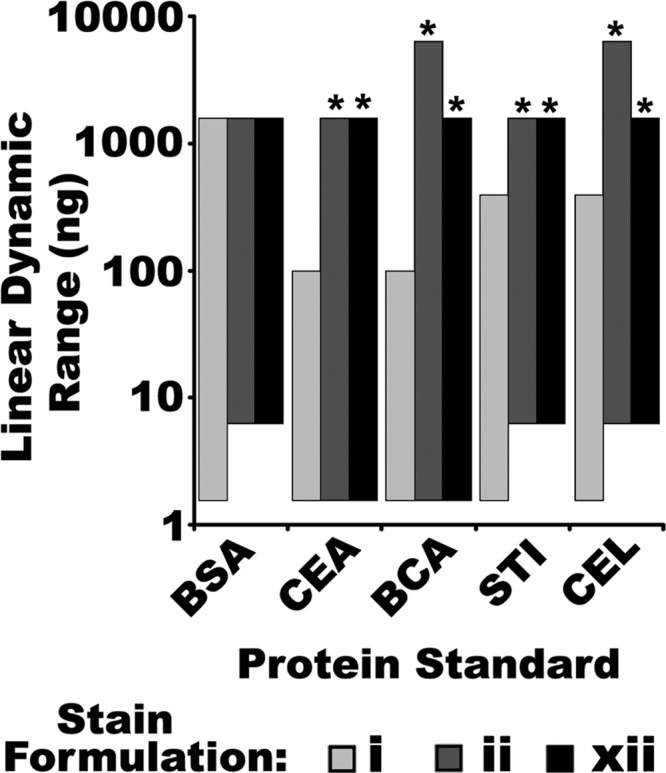
Summary of the LDR from the LLD of SR (stain i) and the two most sensitive CB formulations, ii and xii, imaged with IRF, for five different isolated protein standards. Derived from Fig. 5 and supplemental Fig. S5. * Indicate significant differences from SR (One-way ANOVA, F = 12.2, p < 0.001, Post-hoc Tukey analysis, p < 0.05, n = 3).
In contrast to earlier LLD determinations in which CB was determined to perform essentially equally to (or marginally better than) SR, in these assessments of isolated proteins the CB formulations showed a trend toward poorer threshold sensitivity (Figs. 3 and 6). In assessments of recombinant standards, CB formulations ii and xii were able to detect slightly less than 1 ng of protein (Figs. 2 and 3), but with the isolated proteins used for LDR assessment, neither CB formulation was capable of detecting the 1.5 ng band for 3 out of 5 proteins tested (Figs. 5 and 6); SR detected the 1.5 ng band for all five proteins (Fig. 6). Thus, in assessments of LDR, in which a greater range of protein loads was detected in the same gel, SR was slightly more sensitive than CB (Figs. 5 and 6). This discrepancy may be attributed to IPV of the stain response: by necessity, different samples were used in LDR and LLD determinations. There may additionally have been some fundamental differences between the samples used: in experiments designed to determine the LLD with the greatest possible precision, recombinant protein standards were used, whereas in those designed to determine the LDR, isolated proteins were used. The amino acid sequences and make-up of the recombinant standards were proprietary; these might be substantially modified and thus have different dye binding properties than proteins from a biological source, to maximize resolution, band clarity/sharpness, shelf life, and so forth. Nonetheless, it seemed somewhat unlikely that this alone could fully explain the discrepancy between CB and SR in LDR and LLD assessments.
The most plausible explanation for the observed lower sensitivity of CB relative to SR in the LDR determinations concerns the greater fluorescent signal attenuation seen with SR relative to CB at increasing protein concentrations (Fig. 5 and supplemental Fig. S3). For each protein tested, SR exhibited significantly more pronounced flattening of the fluorescent signal for increasing quantities of protein than did CB (Fig. 5 and supplemental Fig. S3). It might be that the greater degree of fluorescent signal attenuation inherent to SR detection allowed for greater signal integration without saturating the detector. Conversely, CB had a larger LDR for most proteins and less pronounced signal attenuation at increasing protein quantities. It is plausible that this greater linearity of the fluorescent signal limited the degree of signal integration possible below detector saturation. Thus, CB appeared to saturate the detector before SR. If this were the case, SR would have some practical advantages over CB in the detection of low abundance proteins in gels that simultaneously contain very high abundance proteins. This would have to be weighed against the fact that CB-IRFD had a greater LDR than SR, and thus would provide for more comprehensively accurate quantitative proteomic analyses. Thus, LDR assessments yielded the first indication of a critical difference between CB-IRFD and SR.
Assessing the Efficacy of CB for Analyzing Gel-resolved Proteomes
As a final assessment CB and SR were directly compared in the detection of proteomes resolved by 2DE. Total mouse brain soluble and membrane protein fractions were resolved using a refined 2DE protocol (56–58, 60). Replicate gels were stained with either SR or with CB formulation ii or xii and comparative image analysis was used to assess the efficacy of the detection methods for the documentation and quantitative assessment of these proteome maps.
Images of the proteomes detected by CB and SR were comparable on visual inspection, with both staining and detection methods yielding gel images of acceptable quality (Fig. 7). This qualitative assessment was further explored by examining the variability between replicate 2D gel images acquired with each detection method (Fig. 8). Distributions of the CVs of fluorescent volumes of the 100% reproducibly detected and matched spots imaged with each detection method were overlaid and compared (Fig. 8). Qualitatively, the distributions yielded by SR and CB formulations ii and xii were grossly similar and there were no dramatic shifts in the distributions as would be expected if one detection method had significantly more inherent variability relative to another. Moreover, this suggests that most proteins responded relatively consistently to the different staining techniques: given that SR is considered one of the most reproducible fluorescent stains currently available, there appeared not to be a large number of proteins in this diverse biological sample that were inherently irreproducibly stained with CB. On the whole, the variability of CB-IRFD of 2D gels seemed to be comparable to SR. There was a trend for slightly more spots with the smallest CV (< 0.05) when staining with SR relative to CB, and correspondingly, CB had a few more spots with higher CV than SR (Figure 8). This difference amounted to 1.2–1.5% of the 100% reproducible spots detected. Consequently, the mean CV for the two CB stains tested were slightly higher than the mean CV for SR, and this effect was reasonably consistent between the two different samples tested (Fig. 8). However, it should be noted that this comparative assessment of inter-replicate variability simultaneously incorporated all aspects of inter-gel variation typical of 2DE separations, in addition to the variation that occurred during staining, thus making discreet conclusions about staining variability difficult. Nonetheless, although SR may possibly have somewhat less inter-replicate variability for a few spots in 2D gels, and thus ostensibly some quantitative advantage over CB, the difference between SR and CB appeared to be quite small overall, given the considerable overall similarity of the distributions (Fig. 8). This was consistent with quantitative assessments of the selectivity, sensitivity and LDR of SR and CB in 1D gels reported in previous sections: within each experiment conducted and presented here, SR and CB yielded comparable CV, suggesting that the variability between the staining methods was roughly consistent (Figs. 1, 3, 4, and 6, supplemental Figs. S2 and S3).
Fig. 7.
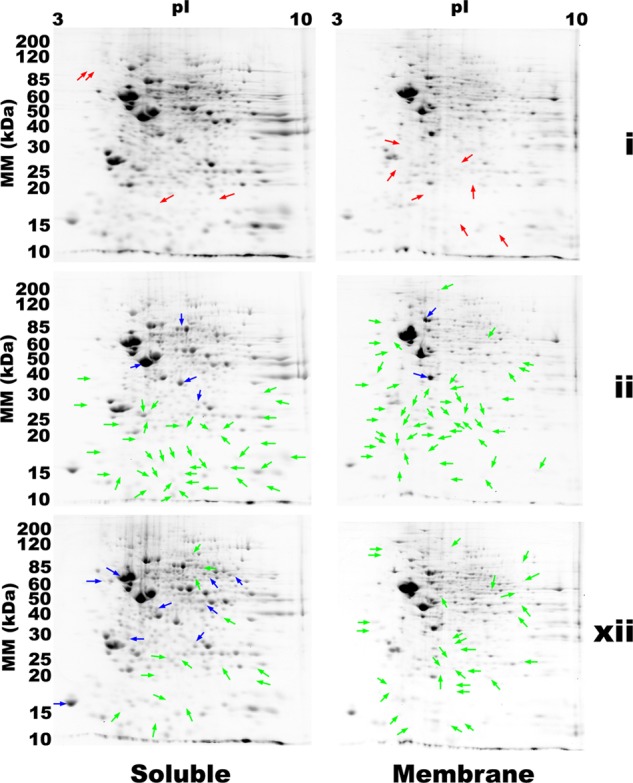
Quantitative differential comparison of mouse brain proteomes resolved by 2DE and detected with either SR (stain i) or one of the two most sensitive CB formulations, ii and xii, imaged with IRF. RED Arrows indicate proteins that were detected with SR, but not by either CB formulation. BLUE and GREEN arrows indicate 1.5-fold (or larger) increases and decreases in relative abundance, respectively, in specific proteins detected with CB-IRF relative to SR (t test, p < 0.05, n = 4). All differences, including those of less than 1.5-fold are summarized in supplemental Tables S1 and S2.
Fig. 8.
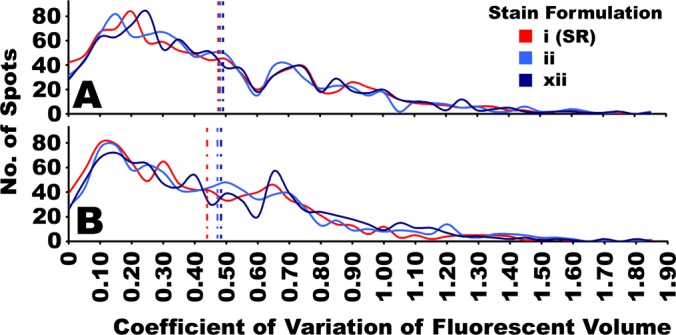
Inter-replicate variability of fluorescently imaged 2D gel proteomes stained with SR or CB. Mouse brain protein fractions; A, soluble fraction; B, membrane fraction, were resolved in a series of 2D gels (Fig. 7) and stained with either SR (stain i), or one of two CB formulations (stains ii and xii). Quantitative image analysis eliminated spots that were not detected in all four replicate gels in each condition, and only spots that were reproducibly detected and positionally matched in all replicates were considered (n = 4). The inter-replicate variability was then assessed as the coefficient of variation (ratio of standard deviation/mean) of the fluorescent volume for each spot. A distribution of the coefficients of variation for all of the reproducible spots detected by each stain tested were overlaid for comparison between stains, indicated with solid lines. Vertical dotted lines indicate the mean value in each distribution. (n = 4).
When overlaid and compared by automated image analysis, CB and SR images were found to be > 99% consistent based on positional matches of protein spots. Differential image analysis indicated a few mouse brain proteins (four soluble and seven membrane) detected with SR but not with either CB formulation (Red Arrows, Fig. 7). Neither CB formulation detected any proteins that were undetectable using SR. Thus, although all three staining methods were capable of detecting comparable numbers of proteins in a given 2DE analysis, SR detected more spots, amounting to 0.6% of the proteome (Figs. 7 and 9). Quantitative image analysis also identified several specific differences between the CB and SR detection methods (Fig. 7). A total of 200 proteins that were successfully detected using both CB and SR, nonetheless differed significantly in their apparent relative abundance between the staining methods (Fig. 7 and supplemental Table S1); that is, the CB and SR signal intensities differed significantly for these specific proteins, despite identical protein loads. Most of these quantitative differences were diminished signals in CB relative to SR; in mouse brain proteomes stained with ii, 48 soluble and 58 membrane proteins, yielded significantly less fluorescent signal with CB than with SR; with xii, 30 soluble and 34 membrane proteins yielded significantly less signal with CB (Fig. 7, supplemental Tables S1 and S2). Many of these apparently lower signals appeared in the low molecular weight range of the gels. There were fewer instances of the opposite effect, wherein certain proteins yielded greater fluorescent signal when detected with CB rather than SR: for ii this amounted to nine soluble and two membrane proteins, and for xii, 14 soluble and 5 membrane proteins (Fig. 7, supplemental Tables S1 and S2). Notably, many of these proteins appeared to be of mid- to high-molecular weight and -abundance (Fig. 7). Overall, it seemed that in these particular assessments, CB was slightly less sensitive than SR, a result inconsistent with the independent assessments of selectivity and sensitivity in which the performance of CB-IRFD was indistinguishable from, and in some cases exceeded that of SR (see above). Other than the fact that samples separated by 1D gel electrophoresis were heated before they were resolved, it was unlikely that there was anything fundamentally different about protein detection in the 1D and 2D gels as the second dimension was compositionally identical, as were staining conditions, and resolved mouse brain proteins were from parallel aliquots of the same homogenized tissue sample. In an effort to connect data derived by 1DE and 2DE assessments, the selectivity for protein in the 2D gels was calculated; notably CB gave significantly higher S2/BG ratios than SR (Fig. 9C), consistent with the 1D gel data on mouse liver membrane proteins (Fig. 1B) and with an initial comparison of SR and CB (50).
Fig. 9.
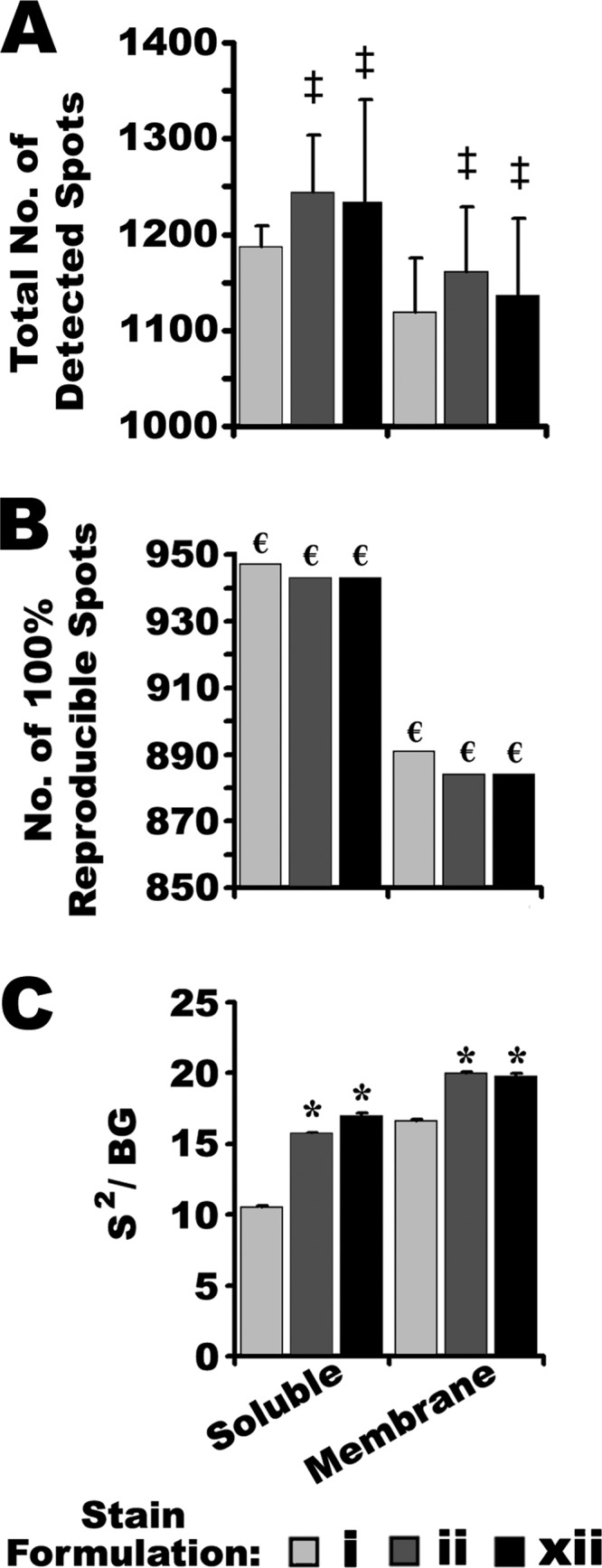
Summary of quantitative image analysis of resolved mouse brain membrane and soluble proteomes detected with either SR (stain i) or one of the two most sensitive CB formulations (ii and xii), imaged using IRF, and derived from Fig. 7. A, The total number of proteins in 2D gels, detected by each staining method. Error bars indicate S.D. of the mean. ‡ Indicates no significant difference from SR (One-way ANOVA: p = 0.533, F = 0.698, n = 4 for soluble proteins; p = 0.693, F = 0.382, n = 4 for membrane proteins). B, The number of proteins that were 100% reproducibly detected and positionally matched in each set of replicate 2D gels (n = 4 for each condition). € Indicates data that are invariant by definition. C, The S2/BG ratio of proteins detected in 2D gels. Error bars indicate S.D. of the mean. * Indicates significant differences from SR (One-way ANOVA: p < 0.001, F = 19.3; Post-hoc Tukey Analysis: p < 0.05, n = 4).
The simplest remaining explanation for this apparent performance discrepancy originates with the differing LDR performance of CB relative to SR. The detection of electrophoretically resolved tissue proteomes in 2D gels was more like assessing LDR (Figs. 5, 6 and supplemental Fig. S3) than threshold sensitivity (Fig. 2 and supplemental Fig. S2); biological extracts simultaneously contain proteins with an enormous range of relative abundances. Thus the differing LDRs of CB and SR did not influence the quantification of relatively small amounts of proteins, as in determinations of maximal dye sensitivity, as even the highest quantities of protein in these gels were well within the LDR of the two detection methods (Figs. 2, 3, and 6). However, when imaging gels that simultaneously contained very large and very small quantities of protein, as did the mouse brain proteomes resolved by 2DE (Fig. 7), and 1D gels used for LDR assessments (Fig. 5 and supplemental Fig. S3), many resolved proteins were beyond the LDR of the detection method. To assess this possibility the quantities of individual proteins resolved by 2DE were estimated (Table III). Notably, these estimates were likely to be conservative considering the degree of fluorescent signal attenuation observed with SR for large protein quantities (Fig. 5 and supplemental Fig. S3). Although crude, these estimates illustrate an important point: in proteomes resolved by 2DE, without depletion of highly abundant proteins, some proteins are present in quantities in excess of the LDR for standard proteins assessed in isolation (Fig. 6).
Table III. Summary of estimated absolute quantities of individual proteins resolved in 2D gels of mouse brain soluble and membrane samples, followed by staining with SR (stain i) or the two most sensitive CB stains (formulations ii and xii). Values were derived from gel images presented in Fig. 7, using Equation 5.
| Stain Formulation | Soluble |
Membrane |
||||
|---|---|---|---|---|---|---|
| i | ii | xii | i | ii | xii | |
| No. of proteins with estimated abundance: | ||||||
| >1000 ng | 6 | 11 | 8 | 6 | 10 | 6 |
| 750–1000 ng | 6 | 7 | 5 | 1 | 6 | 3 |
| 500–750 ng | 18 | 16 | 14 | 16 | 6 | 10 |
| Average estimated quantity of the most abundant protein detected (μg ± S.D.) | 2.4 ± 0.78 | 5.7 ± 0.37 | 4.0 ± 0.13 | 2.5 ± 0.58 | 7.7 ± 0.48 | 4.0 ± 0.89 |
With the exception of BSA, for every standard protein tested the LDR of CB was superior to that of SR (Fig. 6). Moreover, for every standard tested in LDR assessments, the highest quantities of protein loaded yielded significantly lower fluorescent volumes when detected by SR (Fig. 5 and supplemental Fig. S3); the depression of fluorescent signal for high quantities of protein was greater for SR than CB for most proteins tested (Fig. 5 and supplemental Fig. S3), and thus presumably many other proteins as well. Accepting this to also be true in the analysis of biological samples by 2DE, then if the fluorescent signals for the highest abundance proteins resolved were more attenuated when detected by SR, it would be expected that these gels could be integrated slightly longer during imaging before saturation of these signals. Thus it appeared SR had some practical advantage over CB in the detection of low abundance proteins (i.e. amounting to 0.6% of the total detected proteome).
To elaborate, it is standard practice to adjust the parameters of the detector to produce consistent signal intensity between multiple images. Unless thus normalized, small variations in staining could compromise comparative analyses. For the CCD-based instrument this normalization was achieved by adjusting signal integration time to achieve a consistent maximum peak height; for the scanner-based instrument the photomultiplier gain was adjusted. In each case the goal was to ensure normalization of a series of gels processed in parallel to provide images of consistent pixel intensity; minimally this ensures that the most intense peak does not saturate the detector.
Thus it appears in so normalizing images detected by SR and CB, SR was advantaged. Because large quantities of SR stained-protein have been demonstrated to give a particularly “flattened” or attenuated fluorescent signal response (Fig. 5 and supplemental Fig. S3), SR-stained gels can be integrated slightly longer without saturating these signals. Thus, signals from low abundance proteins at or slightly below detection threshold are enhanced by virtue of this increased integration. Conversely, CB-IRFD was demonstrated to have a higher LDR and less pronounced attenuation of fluorescent signals with increasing protein abundance (Fig. 5 and supplemental Fig. S3). Under these circumstances, signal integration of CB-IRF to the same extent as for SR gels was impossible: the gain of the detector could not be increased to a comparable extent without saturation of the most abundant spots. Thus in CB stained gels, the most highly abundant signals saturated the detector earlier than in parallel SR stained gels, limiting the detection of faint signals from a few lower abundance proteins. This confirms the previous observation that with increasing quantities of protein, SR experienced greater signal attenuation than did CB (Fig. 5 and supplemental Fig. S3); the same effects observed in direct assessments of LDR using isolated proteins were also at work in assessments of proteomes resolved in 2D gels.
A central objective of proteomics is to provide an accurate representation of the native biological complexity of a sample. In this regard, with greater LDR, CB would appear to have provided the more accurate quantitative data over a larger range of protein quantities in-gel; the quantitative range of SR was comparatively limited, thus presenting a relatively skewed representation of the abundances of proteins in 2D gels. In other words, the difference in signal intensity between lowest and highest abundance proteins was artificially narrowed to some degree by SR staining. This apparent property of SR presents a compromise of sorts: relative to CB, SR detects a few more proteins in 2D gels but with a more limited LDR.
Thus, from a strictly quantitative standpoint, it might be considered beneficial to limit the total protein load in an analysis such that even the most highly abundant proteins are within the LDR of the detection method. Given some assumptions, even with a reasonable amount of total protein (i.e. 100 μg), proteomes resolved by 2DE likely contain many proteins in quantities beyond the LDR of current detection methods (Table III); substantially larger total protein loads are possible, and commonplace in the literature. Most efforts to resolve extracts that are not fractionated are therefore apparently “overloaded” with many proteins present in amounts substantially exceeding the LDR of current detection methods. For such proteins, quantitative data derived under false assumption of a linear relationship between protein quantity and fluorescent signal, are thus suspect. One approach to address this might be to load less total protein on 2D gels, and take steps to ensure that the quantities of proteins in the analyte do not exceed the LDR of the detection method. However, barring exhaustive assessment of the LDRs of all known proteins in a given sample, this optimal total protein load could at best only be estimated based on the LDR of one or a few standard proteins, and for a given stain. Moreover, it seems highly unlikely that such a practice would be accepted with much enthusiasm as one of the explicit goals of proteomics is the simultaneous detection and analysis of as much of the proteome as possible. Thus it is common practice to apply as much sample as is possible without detriment to resolution, for the purpose of achieving improved detection of relatively low abundance proteins, and thus increased overall coverage of the proteome. Here the indications are that by overloading gels, the accuracy of subsequent quantitative assessments is placed in question. If quantitative assessments are to be made as part of credible comparative analyses, these potential sources of error must be better addressed.
Spot Picking from Coomassie-Stained Gels
With a substantially more sensitive method of documenting Coomassie-stained gels, it has become possible to detect some Coomassie-stained protein spots in-gel that are invisible to the naked eye. Generally speaking, it would not be possible to accurately or reliably excise such proteins from the gel manually. Moreover, current automated spot picking systems are not equipped with sensors capable of IRFD of CB-stained gels. Nonetheless, very faint CB-stained spots can be excised from gels routinely by a triangulation method. That is, given three fixed landmarks around the spot of interest (either higher abundance protein spots, or small holes cut in the gel matrix) the absolute position of the spot of interest can be calculated in two-dimensional space by triangulation with sufficient accuracy to excise that spot with confidence. Once picked, the gel can be re-imaged using a high sensitivity imaging instrument (restaining the gel, if necessary), and compared with the original image to ensure that the spot of interest has been excised. Automated spot-picking instruments with appropriate triangulation software are widely available, and greatly streamline this method. Thus it is expected that accurately excising low abundance CB-stained proteins for downstream mass spectrometry or other analyses would not be materially different from excising SR- or other fluorescently labeled proteins that happen to fall below the detection limit of the spot picking instrument.
Concluding Remarks
Here the use of CB as a reagent for IRFD of gel-resolved proteomes was characterized systematically, and compared with SR as a potential alternative for gel-based proteomic analysis. Of a selection of CB formulations, those providing the greatest IRFD performance were identified based on four critical detection parameters: selectivity, sensitivity, IPV, and LDR; within these parameters, CB is capable of matching or exceeding the performance of SR. Taking into consideration that staining protocols for specific CB formulations may not have been optimal, even more substantial differences between CB and SR are possible. It is expected that further optimization of CB-IRFD, either by refining CB staining formulations or protocols, or improvements in IR gel imaging instruments, could well contribute to even greater CB-IRFD performance.
However, based only on these data, it cannot be unequivocally determined if CB-IRFD is superior to SR for in-gel proteomic analysis. Each method has practical advantages and disadvantages. One advantage of SR may be its relatively limited LDR, and consequently slightly better sensitivity for lower abundance proteins. To keep this in perspective, the enhanced detection with SR here amounted to 0.6% of all the spots detected in the mouse brain proteome (Fig. 9). Should a potentially important protein alteration be found within this small subset of the proteome, this narrow advantage would nonetheless prove critical, but may simultaneously place the quantitative relevance of the findings in question. CB appears to provide the more accurate representation of protein quantities in a given proteome, as a result of its wider LDR. CB and SR were shown to have comparable inter-replicate variability: it could be argued that SR had some advantage in terms of the numbers of proteins detected in 2D gels with very low CV, but this difference was small compared with the overall similarity of distributions of variation in 2D gels. Although the relative merits SR and CB could be endlessly debated, it should be noted that fluorescence detection of CB met or exceeded the performance of SR in some encouraging aspects: any gap between these methods appeared to be quite narrow and could potentially be reduced as additional optimizations to CB-IRFD are implemented. Overall, though further comparisons may prove necessary for specific applications and with specific imaging equipment, here it has been quantitatively shown that CB-IRFD is a competitive in-gel fluorescence detection technology for top-down proteomics. A variety of imagers capable of IRFD are currently available: those dedicated to IR, such as the Licor Odyssey family of imagers are cost-effective, and alternatives such as the FLA family of laser-based imagers developed by Fuji serve a range of imaging demands as might be expected of any modern research facility. The initial investment in such instruments is, to some extent, offset by savings in reagents: certainly in terms of consumable expenses, CB is superior to SR and related stains. Moreover, to be fair, it should be noted that for the best possible SR performance, a specialized imager is likewise required. Thus in terms of quantitative performance, accessibility, and breadth of potential users, CB staining has clear advantages, and will likely continue to have a place in the field for years to come.
Supplementary Material
Acknowledgments
We thank Dr. V. Wee Yong and colleagues for their generous contribution of instrument time that was necessary for the completion of this work.
Footnotes
* JRC was supported by the Canadian Institutes of Health Research, the Natural Sciences and Engineering Research Council of Canada, the Alberta Heritage Foundation for Medical Research, and the University of Western Sydney. RHB held a Studentship from the Alberta Heritage Foundation at the time this research was carried out. This work formed part of RHB's PhD dissertation.
 This article contains supplemental Figs. S1 to S3 and Tables S1 and S2.
This article contains supplemental Figs. S1 to S3 and Tables S1 and S2.
1 The abbreviations used are:
- 2DE
- two dimensional gel electrophoresis
- 1DE
- one dimensional gel electrophoresis
- BCA
- bovine carbonic anhydrase from erythrocytes
- BSA
- bovine serum albumin
- CB
- Coomassie Blue
- CCB
- colloidal Coomassie Blue
- CEA
- chicken egg albumin
- CEL
- chicken egg lysozyme
- CV
- coefficient of variation
- FV
- fluorescent volume
- IMPS
- integrated measure of practical sensitivity
- IPG
- immobilized pH gradient
- IPV
- inter-protein variability (of fluorescent signal intensity)
- IR
- infrared
- IRF
- infrared fluorescence
- IRFD
- infrared fluorescence detection
- LDR
- linear dynamic range
- LLD
- lowest limit of detection
- LPS
- lowest practical sensitivity
- S/BG
- signal/background ratio
- S2/BG
- signal squared/background ratio
- SR
- sypro ruby
- STI
- soybean trypsin inhibitor.
REFERENCES
- 1. Zuo X., Speicher D. W. (2000) A method for global analysis of complex proteomes using sample prefractionation by solution isoelectrofocusing prior to two-dimensional electrophoresis. Anal. Biochem. 284, 266–278 [DOI] [PubMed] [Google Scholar]
- 2. Issaq H. J., Conrads T. P., Janini G. M., Veenstra T. D. (2002) Methods for fractionation, separation and profiling of proteins and peptides. Electrophoresis 23, 3048–3061 [DOI] [PubMed] [Google Scholar]
- 3. Righetti P. G., Castagna A., Herbert B., Candiano G. (2005) How to bring the “unseen” proteome to the limelight via electrophoretic pre-fractionation techniques. Biosci. Rep. 25, 3–17 [DOI] [PubMed] [Google Scholar]
- 4. Wang H., Qian W. J., Chin M. H., Petyuk V. A., Barry R. C., Liu T., Gritsenko M. A., Mottaz H. M., Moore R. J., Camp Ii D. G., Khan A. H., Smith D. J., Smith R. D. (2006) Characterization of the mouse brain proteome using global proteomic analysis complemented with cysteinyl-peptide enrichment. J. Proteome. Res. 5, 361–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Butt R. H., Coorssen J. R. (2005) Postfractionation for enhanced proteomic analyses: routine electrophoretic methods increase the resolution of standard 2D-PAGE. J. Proteome Res. 4, 982–991 [DOI] [PubMed] [Google Scholar]
- 6. Berggren K. N., Schulenberg B., Lopez M. F., Steinberg T. H., Bogdanova A., Smejkal G., Wang A., Patton W. F. (2002) An improved formulation of SYPRO Ruby protein gel stain: comparison with the original formulation and with a ruthenium II tris (bathophenanthroline disulfonate) formulation. Proteomics 2, 486–498 [DOI] [PubMed] [Google Scholar]
- 7. Campbell K. P., MacLennan D. H., Jorgensen A. O. (1983) Staining of the Ca2+-binding proteins, calsequestrin, calmodulin, troponin C, and S-100, with the cationic carbocyanine dye “Stains-all”. J. Biol. Chem. 258, 11267–11273 [PubMed] [Google Scholar]
- 8. Candiano G., Bruschi M., Musante L., Santucci L., Ghiggeri G. M., Carnemolla B., Orecchia P., Zardi L., Righetti P. G. (2004) Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis 25, 1327–1333 [DOI] [PubMed] [Google Scholar]
- 9. Chevalier F., Rofidal V., Vanova P., Bergoin A., Rossignol M. (2004) Proteomic capacity of recent fluorescent dyes for protein staining. Phytochemistry 65, 1499–1506 [DOI] [PubMed] [Google Scholar]
- 10. Fernandez-Patron C., Castellanos-Serra L., Rodriguez P. (1992) Reverse staining of sodium dodecyl sulfate polyacrylamide gels by imidazole-zinc salts: sensitive detection of unmodified proteins. BioTechniques 12, 564–573 [PubMed] [Google Scholar]
- 11. Mackintosh J. A., Choi H. Y., Bae S. H., Veal D. A., Bell P. J., Ferrari B. C., Van Dyk D. D., Verrills N. M., Paik Y. K., Karuso P. (2003) A fluorescent natural product for ultra sensitive detection of proteins in one-dimensional and two-dimensional gel electrophoresis. Proteomics 3, 2273–2288 [DOI] [PubMed] [Google Scholar]
- 12. Neuhoff V., Arold N., Taube D., Ehrhardt W. (1988) Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 9, 255–262 [DOI] [PubMed] [Google Scholar]
- 13. Porro M., Viti S., Antoni G., Saletti M. (1982) Ultrasensitive silver-stain method for the detection of proteins in polyacrylamide gels and immunoprecipitates on agarose gels. Anal. Biochem. 127, 316–321 [DOI] [PubMed] [Google Scholar]
- 14. Smejkal G. B., Robinson M. H., Lazarev A. (2004) Comparison of fluorescent stains: relative photostability and differential staining of proteins in two-dimensional gels. Electrophoresis 25, 2511–2519 [DOI] [PubMed] [Google Scholar]
- 15. Steinberg T. H., Jones L. J., Haugland R. P., Singer V. L. (1996) SYPRO orange and SYPRO red protein gel stains: one-step fluorescent staining of denaturing gels for detection of nanogram levels of protein. Anal. Biochem. 239, 223–237 [DOI] [PubMed] [Google Scholar]
- 16. Ahnert N., Patton W. F., Schulenberg B. (2004) Optimized conditions for diluting and reusing a fluorescent protein gel stain. Electrophoresis 25, 2506–2510 [DOI] [PubMed] [Google Scholar]
- 17. Bell P. J., Karuso P. (2003) Epicocconone, a novel fluorescent compound from the fungus epicoccumnigrum. J. Am. Chem. Soc. 125, 9304–9305 [DOI] [PubMed] [Google Scholar]
- 18. Berggren K., Steinberg T. H., Lauber W. M., Carroll J. A., Lopez M. F., Chernokalskaya E., Zieske L., Diwu Z., Haugland R. P., Patton W. F. (1999) A luminescent ruthenium complex for ultrasensitive detection of proteins immobilized on membrane supports. Anal. Biochem. 276, 129–143 [DOI] [PubMed] [Google Scholar]
- 19. Berggren K., Chernokalskaya E., Steinberg T. H., Kemper C., Lopez M. F., Diwu Z., Haugland R. P., Patton W. F. (2000) Background-free, high sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gels using a luminescent ruthenium complex. Electrophoresis 21, 2509–2521 [DOI] [PubMed] [Google Scholar]
- 20. Berggren K. N., Chernokalskaya E., Lopez M. F., Beechem J. M., Patton W. F. (2001) Comparison of three different fluorescent visualization strategies for detecting Escherichia coli ATP synthase subunits after sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteomics 1, 54–65 [DOI] [PubMed] [Google Scholar]
- 21. Blakesley R. W., Boezi J. A. (1977) A new staining technique for proteins in polyacrylamide gels using coomassie brilliant blue G250. Anal. Biochem. 82, 580–582 [DOI] [PubMed] [Google Scholar]
- 22. Choi J. K., Yoon S. H., Hong H. Y., Choi D. K., Yoo G. S. (1996) A modified Coomassie blue staining of proteins in polyacrylamide gels with Bismark brown R. Anal. Biochem. 236, 82–84 [DOI] [PubMed] [Google Scholar]
- 23. Choi J. K., Tak K. H., Jin L. T., Hwang S. Y., Kwon T. I., Yoo G. S. (2002) Background-free, fast protein staining in sodium dodecyl sulfate polyacrylamide gel using counterion dyes, zincon and ethyl violet. Electrophoresis 23, 4053–4059 [DOI] [PubMed] [Google Scholar]
- 24. Choi J. K., Chae H. Z., Hwang S. Y., Choi H. I., Jin L. T., Yoo G. S. (2004) Fast visible dye staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gels compatible with matrix assisted laser desorption/ionization-mass spectrometry. Electrophoresis 25, 1136–1141 [DOI] [PubMed] [Google Scholar]
- 25. Coghlan D. R., Mackintosh J. A., Karuso P. (2005) Mechanism of reversible fluorescent staining of protein with epicocconone. Org. Lett. 7, 2401–2404 [DOI] [PubMed] [Google Scholar]
- 26. de Moreno M. R., Smith J. F., Smith R. V. (1985) Silver staining of proteins in polyacrylamide gels: increased sensitivity through a combined Coomassie blue-silver stain procedure. Anal. Biochem. 151, 466–470 [DOI] [PubMed] [Google Scholar]
- 27. de Moreno M. R., Smith J. F., Smith R. V. (1986) Mechanism studies of coomassie blue and silver staining of proteins. J. Pharm. Sci. 75, 907–911 [DOI] [PubMed] [Google Scholar]
- 28. Flory M. R., Griffin T. J., Martin D., Aebersold R. (2002) Advances in quantitative proteomics using stable isotope tags. Trends Biotechnol. 20, S23–S29 [DOI] [PubMed] [Google Scholar]
- 29. Irie S., Sezaki M., Kato Y. (1982) A faithful double stain of proteins in the polyacrylamide gels with Coomassie blue and silver. Anal. Biochem. 126, 350–354 [DOI] [PubMed] [Google Scholar]
- 30. Kang C., Kim H. J., Kang D., Jung D. Y., Suh M. (2003) Highly sensitive and simple fluorescence staining of proteins in sodium dodecyl sulfate-polyacrylamide-based gels by using hydrophobic tail-mediated enhancement of fluorescein luminescence. Electrophoresis 24, 3297–3304 [DOI] [PubMed] [Google Scholar]
- 31. Lamanda A., Zahn A., Röder D., Langen H. (2004) Improved Ruthenium II tris (bathophenantroline disulfonate) staining and destaining protocol for a better signal-to-background ratio and improved baseline resolution. Proteomics 4, 599–608 [DOI] [PubMed] [Google Scholar]
- 32. Neuhoff V., Stamm R., Pardowitz I., Arold N., Ehrhardt W., Taube D. (1990) Essential problems in quantification of proteins following colloidal staining with coomassie brilliant blue dyes in polyacrylamide gels, and their solution. Electrophoresis 11, 101–117 [DOI] [PubMed] [Google Scholar]
- 33. Ohsawa K., Ebata N. (1983) Silver stain for detecting 10-femtogram quantities of protein after polyacrylamide gel electrophoresis. Anal. Biochem. 135, 409–415 [DOI] [PubMed] [Google Scholar]
- 34. Patton W. F. (2000) A thousand points of light: the application of fluorescence detection technologies to two-dimensional gel electrophoresis and proteomics. Electrophoresis 21, 1123–1144 [DOI] [PubMed] [Google Scholar]
- 35. Gauci V. J., Wright E. P., Coorssen J. R. (2011) Quantitative proteomics: assessing the spectrum of in-gel protein detection methods. J. Chem. Biol. 4, 3–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Molnar A., Kulcsar P., Prestwich G. D. (1990) Effective removal of excess Coomassie blue from polyacrylamide gels. BioTechniques 9, 534, 536 [PubMed] [Google Scholar]
- 37. Zehr B. D., Savin T. J., Hall R. E. (1989) A one-step, low background coomassie staining procedure for polyacrylamide gels. Anal. Biochem. 182, 157–159 [DOI] [PubMed] [Google Scholar]
- 38. Duhamel R. C., Brendel K. (1983) Coomassie Blue R-250 metachromatic staining of histone 5 from goose and chicken erythrocytes. Comp Biochem. Physiol B. 75, 133–135 [DOI] [PubMed] [Google Scholar]
- 39. Neuhoff V., Stamm R., Eibl H. (1985) Clear background and highly sensitive protein staining with Coomassie Blue dyes in polyacrylamide gels: A systematic analysis. Electrophoresis 6, 427–448 [Google Scholar]
- 40. Kang D., Gho Y. S. S. M., Kang C. (2002) Highly sensitive and fast protein detection with Coomassie Brilliant Blue in sodium dodecyl sulfate - polyacrylamide gel electrophoresis. Bull. Korean Chem. Soc. 23, 1511–1512 [Google Scholar]
- 41. Winkler C., Denker K., Wortelkamp S., Sickmann A. (2007) Silver- and Coomassie-staining protocols: detection limits and compatibility with ESI MS. Electrophoresis 28, 2095–2099 [DOI] [PubMed] [Google Scholar]
- 42. Sasse J., Gallagher S. R. (2003) Staining proteins in gels. Curr. Protoc. Mol. Biol. Chapter 10, Unit [DOI] [PubMed] [Google Scholar]
- 43. Terpetschnig E., Szmacinski H., Malak H., Lakowicz J. R. (1995) Metal-ligand complexes as a new class of long-lived fluorophores for protein hydrodynamics. Biophys. J. 68, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rabilloud T., Strub J. M., Luche S., Van D. A., Lunardi J. (2001) A comparison between Sypro Ruby and ruthenium II tris (bathophenanthroline disulfonate) as fluorescent stains for protein detection in gels. Proteomics 1, 699–704 [DOI] [PubMed] [Google Scholar]
- 45. Steinberg T. H., Chernokalskaya E., Berggren K., Lopez M. F., Diwu Z., Haugland R. P., Patton W. F. (2000) Ultrasensitive fluorescence protein detection in isoelectric focusing gels using a ruthenium metal chelate stain. Electrophoresis 21, 486–496 [DOI] [PubMed] [Google Scholar]
- 46. Lanne B., Panfilov O. (2005) Protein staining influences the quality of mass spectra obtained by peptide mass fingerprinting after separation on 2-d gels. A comparison of staining with coomassie brilliant blue and sypro ruby. J. Proteome. Res. 4, 175–179 [DOI] [PubMed] [Google Scholar]
- 47. Lin J. F., Chen Q. X., Tian H. Y., Gao X., Yu M. L., Xu G. J., Zhao F. K. (2008) Stain efficiency and MALDI-TOF MS compatibility of seven visible staining procedures. Anal. Bioanal. Chem. 390, 1765–1773 [DOI] [PubMed] [Google Scholar]
- 48. Lopez M. F., Berggren K., Chernokalskaya E., Lazarev A., Robinson M., Patton W. F. (2000) A comparison of silver stain and SYPRO Ruby Protein Gel Stain with respect to protein detection in two-dimensional gels and identification by peptide mass profiling. Electrophoresis 21, 3673–3683 [DOI] [PubMed] [Google Scholar]
- 49. Tannu N. S., Sanchez-Brambila G., Kirby P., Andacht T. M. (2006) Effect of staining reagent on peptide mass fingerprinting from in-gel trypsin digestions: a comparison of SyproRuby and DeepPurple. Electrophoresis 27, 3136–3143 [DOI] [PubMed] [Google Scholar]
- 50. Harris L. R., Churchward M. A., Butt R. H., Coorssen J. R. (2007) Assessing detection methods for gel-based proteomic analyses. J. Proteome. Res. 6, 1418–1425 [DOI] [PubMed] [Google Scholar]
- 51. Luo S., Wehr N. B., Levine R. L. (2006) Quantitation of protein on gels and blots by infrared fluorescence of Coomassie blue and Fast Green. Anal. Biochem. 350, 233–238 [DOI] [PubMed] [Google Scholar]
- 52. Anderson N. L., Esquer-Blasco R., Hofmann J. P., Meheus L., Raymackers J., Steiner S., Witzmann F., Anderson N. G. (1995) An updated two-dimensional gel database of rat liver proteins useful in gene regulation and drug effect studies. Electrophoresis 16, 1977–1981 [DOI] [PubMed] [Google Scholar]
- 53. Diezel W., Kopperschläger G., Hofmann E. (1972) An improved procedure for protein staining in polyacrylamide gels with a new type of Coomassie Brilliant Blue. Anal. Biochem. 48, 617–620 [DOI] [PubMed] [Google Scholar]
- 54. Weber K., Osborn M. (1969) The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis. J. Biol. Chem. 244, 4406–4412 [PubMed] [Google Scholar]
- 55. Coorssen J. R., Blank P. S., Albertorio F., Bezrukov L., Kolosova I., Chen X., Backlund P. S., Jr., Zimmerberg J. (2003) Regulated secretion: SNARE density, vesicle fusion and calcium dependence. J. Cell Sci. 116, 2087–2097 [DOI] [PubMed] [Google Scholar]
- 56. Butt R. H., Coorssen J. R. (2006) Pre-extraction sample handling by automated frozen disruption significantly improves subsequent proteomic analyses. J Proteome. Res. 5, 437–448 [DOI] [PubMed] [Google Scholar]
- 57. Butt R. H., Pfeifer T. A., Delaney A., Grigliatti T. A., Tetzlaff W. G., Coorssen J. R. (2007) Enabling coupled quantitative genomics and proteomics analyses from rat spinal cord samples. Mol. Cell Proteomics 6, 1574–1588 [DOI] [PubMed] [Google Scholar]
- 58. Churchward M. A., Butt R. H., Lang J. C., Hsu K. K., Coorssen J. R. (2005) Enhanced detergent extraction for analysis of membrane proteomes by two-dimensional gel electrophoresis. Proteome. Sci. 3, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Coorssen J. R., Blank P. S., Tahara M., Zimmerberg J. (1998) Biochemical and functional studies of cortical vesicle fusion: the SNARE complex and Ca2+ sensitivity. J. Cell Biol. 143, 1845–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Butt R. H., Lee M. W., Pirshahid S. A., Backlund P. S., Wood S., Coorssen J. R. (2006) An initial proteomic analysis of human preterm labor: placental membranes. J Proteome. Res. 5, 3161–3172 [DOI] [PubMed] [Google Scholar]
- 61. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 62. Cannon-Carlson S., Tang J. (1997) Modification of the Laemmli sodium dodecyl sulfate-polyacrylamide gel electrophoresis procedure to eliminate artifacts on reducing and nonreducing gels. Anal. Biochem. 246, 146–148 [DOI] [PubMed] [Google Scholar]
- 63. Coorssen J. R., Blank P. S., Albertorio F., Bezrukov L., Kolosova I., Backlund P. S., Jr., Zimmerberg J. (2002) Quantitative femto- to attomole immunodetection of regulated secretory vesicle proteins critical to exocytosis. Anal. Biochem. 307, 54–62 [DOI] [PubMed] [Google Scholar]
- 64. Smejkal G. B., Li C., Robinson M. H., Lazarev A. V., Lawrence N. P., Chernokalskaya E. (2006) Simultaneous reduction and alkylation of protein disulfides in a centrifugal ultrafiltration device prior to two-dimensional gel electrophoresis. J. Proteome. Res. 5, 983–987 [DOI] [PubMed] [Google Scholar]
- 65. Gauci V. J., Wright E. P., Coorssen J. R. (2011) Quantitative proteomics: assessing the spectrum of in-gel protein detection methods. J. Chem. Biol. 4, 3–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.