

Abstract
A series of novel HIV-1 protease inhibitors based on two pseudosymmetric dipeptide isosteres have been synthesized and evaluated. The inhibitors were designed by incorporating N-phenyloxazolidinone-5-carboxamides into the hydroxyethylene and (hydroxyethyl)hydrazine dipeptide isosteres as P2 and P2′ ligands. Compounds with (S)-phenyloxazolidinones attached at a position proximal to the central hydroxyl group showed low nM inhibitory activities against wild-type HIV-1 protease. Selected compounds were further evaluated for their inhibitory activities against a panel of multidrug-resistant protease variants and for their antiviral potencies in MT-4 cells. The crystal structures of lopinavir (LPV) and two new inhibitors containing phenyloxazolidinone-based ligands in complex with wild-type HIV-1 protease have been determined. A comparison of the inhibitor–protease structures with the LPV–protease structure provides valuable insight into the binding mode of the new inhibitors to the protease enzyme. Based on the crystal structures and knowledge of structure–activity relationships, new inhibitors can be designed with enhanced enzyme inhibitory and antiviral potencies.
Introduction
Since the discovery of human immunodeficiency virus-1 (HIV-1) as the causative agent of acquired human immunodeficiency syndrome (AIDS), intense efforts have been made to develop antiviral drugs that target various viral proteins to suppress viral replication in host cells. HIV-1 protease has emerged as a promising therapeutic target for antiviral therapy in AIDS patients as it plays a critical role in the virus life cycle by processing the viral Gag and Gag-Pol polyproteins into structural and functional proteins essential for viral maturation. In the past decade, structure-based drug design has led to the discovery of nine FDA-approved protease inhibitors and several others in advanced clinical trials. Currently marketed HIV-1 protease inhibitors, saquinavir (SQV),1 indinavir (IDV),2 ritonavir (RTV),3 nelfinavir (NFV),4 amprenavir (APV),5 lopinavir (LPV),6 atazanavir (ATV),7 tipranavir (TPV),8 and darunavir (DRV)9-12 (Figure 1) are all competitive inhibitors that bind in the active site of the enzyme. Except for the newly approved drug TPV, all drugs contain various noncleavable dipeptide isosteres as core scaffolds to mimic the transition state of the polyprotein substrates of HIV-1 protease. The development and clinical introduction of anti-AIDS HIV-1 protease inhibitors is regarded as a major success of structure-based drug design.13
Figure 1.
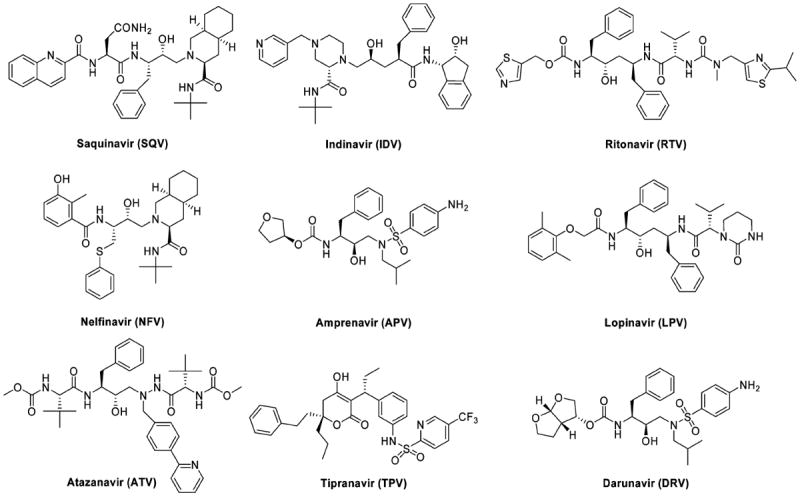
Chemical structures of FDA-approved HIV-1 protease inhibitors.
HIV-1 protease inhibitors are the most potent anti-AIDS drugs reported to date and are essential components of highly active antiretroviral therapy (HAART).14,15 Anti-AIDS chemotherapy based on HIV-1 protease and reverse-transcriptase inhibitors has been remarkably successful in decreasing the mortality rates in HIV-infected patients. However, under the selective pressure of drug therapy, the emergence of many viable multidrug-resistant (MDR) protease variants is posing a great challenge to the efficacy of currently available protease inhibitors.16,17 Therefore, it is increasingly important to discover new classes of protease inhibitors that are less susceptible to drug resistance and show broad spectrum activity against MDR variants. Current efforts toward the development of next generation protease inhibitors have been focused on compounds that retain activity against existing MDR variants, either owing to differences in resistance profiles or with adequate in vivo drug exposure and potency to overcome MDR viruses.18-21
We have been pursuing a structure-based strategy to design and evaluate new classes of HIV-1 protease inhibitors that target ensembles of proteases and exhibit broad spectrum activity against MDR protease variants.22 Recently, we reported the discovery of novel HIV-1 protease inhibitors based on the (hydroxyethylamino)sulfonamide isostere incorporating N-phenyloxazolidinone-5-carboxamides as P2 ligands.23 Preliminary structure–activity relationship (SAR) studies resulted in the identification of several compounds with highly potent inhibitory activities against wild-type and MDR variants of HIV-1 protease. Crystal structures of inhibitors incorporating phenyloxazolidinone-based P2 ligands bound to wild-type HIV-1 protease revealed that the orientation of the oxazolidinone moiety allows it to make a complex network of hydrogen bonds with invariant Asp29 residue of the protease.23 These exciting results prompted us to further explore the utility of phenyloxazolidinones by incorporating the ligands into other transition-state mimetic isostere scaffolds.
In this paper we describe the design, synthesis, and biological evaluation of novel HIV-1 protease inhibitors based on hydroxyethylene and (hydroxyethyl)hydrazine dipeptide isosteres incorporating phenyloxazolidinone-5-carboxamides as P2 and P2′ ligands. The pseudosymmetric dipeptide isosteres, hydroxyethylene and (hydroxyethyl)hydrazine, are the core scaffolds present in approved drugs LPV/RTV and ATV, respectively. In current studies, we have focused our efforts to investigate the stereochemical preference and location of the phenyloxazolidinone-based ligands at both P2 and P2′ positions of the core isosteres. As illustrated in Figure 2, new inhibitors are designed by replacing the P2 and P2′ moieties in the LPV scaffold with (R)- and (S)-enantiomers of N-phenyloxazolidinone-5-carboxamides. Similarly, phenyloxazolidinone moieties are introduced at both P2 and P2′ positions in the ATV scaffold. Herein, the synthesis and inhibitory activities of these designed protease inhibitors against wild-type HIV-1 protease are presented. We also report the inhibitory activities of selected compounds against a panel of MDR protease variants as well as their antiviral activities in cell-based assays. In addition, the crystal structures of LPV and two new inhibitors in complex with wild-type HIV-1 protease are also discussed.
Figure 2.
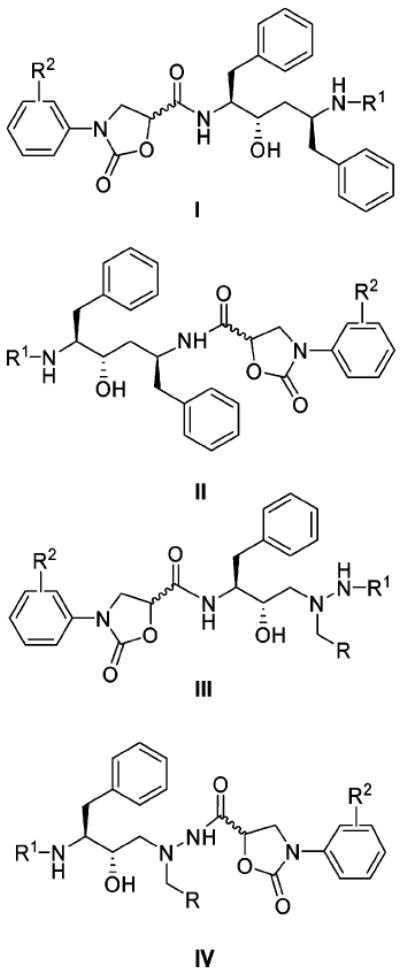
Structures of designed protease inhibitors I–IV based on the hydroxyethylene and (hydroxyethyl)hydrazine isosteres.
Chemistry
The synthesis of hydroxyethylene isostere-based inhibitors I incorporating N-phenyloxazolidinone-5-carboxamides as P2′ ligands was carried out following the synthetic route outlined in Scheme 1. The required Phe–Phe dipeptide isostere, diamino alcohol 1, was prepared in five steps from l-Phe following the reported method.24 The acids A2 and A4, the P2/P2′ ligands in LPV, and acid A3, the P2/P2′ ligand in ATV, were synthesized following reported procedures.25,26 Reaction of the hydroxyethylene core 1 with acids A1–A4 using appropriate conditions provided intermediate compounds 2–5. The coupling reaction of acid A1 with the core fragment 1 was carried out using EDCI/HOBta in a mixture of CH2Cl2 and H2O to avoid epimerization.27 Following the reported procedure, acid fragment A2 was converted to the corresponding acyl chloride and then reacted with the diamino alcohol core 1.25 The coupling reaction of core 1 with acid A3 was carried out using standard peptide coupling conditions (EDCI/HOBt/DIPEA) in a mixture of DMF and CH2Cl2. Acid A4 was treated with SOCl2 in EtOAc, and the resulting acyl chloride was reacted with 1 using the reported procedure.25 Catalytic debenzylation of intermediate compounds 2–5 using Pd/C provided the free amines 6–9 that were subsequently reacted with the activated carboxylic acids 10 or 11a–f to provide the designed inhibitors 12–16. The chiral N-phenyloxazolidinone-5-carboxylic acids 10 and 11a–f were prepared from Cbz-protected aniline derivatives in three steps as previously described.23
Scheme 1.
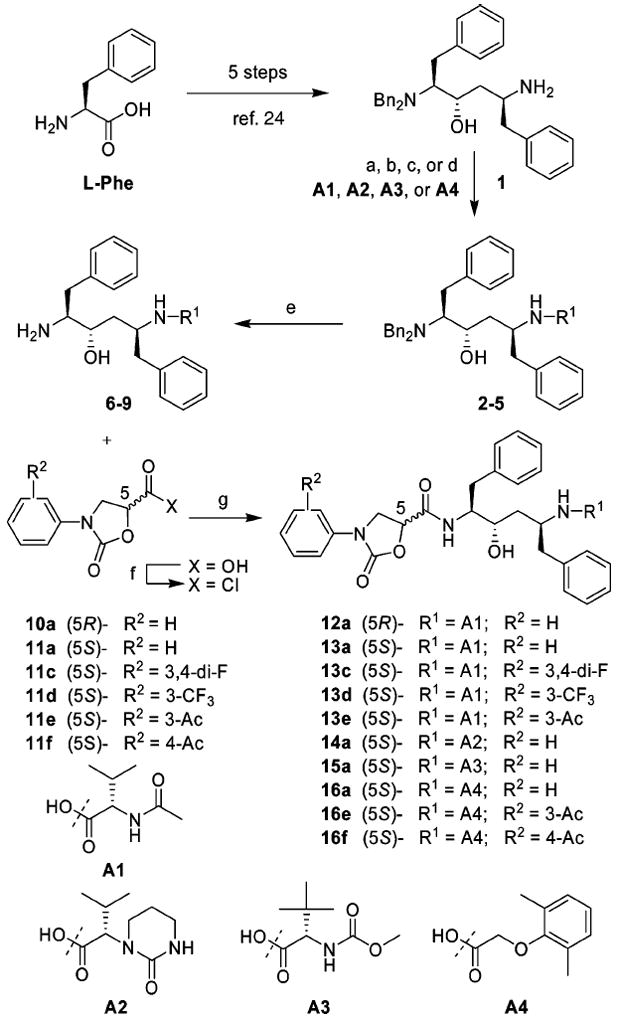
Synthesis of Designed Protease Inhibitors Ia
a Reagents and conditions: (a) acid A1, EDCI, HOBt, H2O–CH2Cl2 (1: 1), 0 °C, 24 h; (b) acid A2, SOCl2, THF, 0 °C to rt, 6 h; coupling: imidazole, EtOAc, acid chloride, 0 °C to rt, overnight; (c) acid A3, EDCI, HOBt, DIPEA, DMF–CH2Cl2 (1:1), 0 °C to rt, 24 h; (d) acid A4, SOCl2, EtOAc, DMF, rt to 50 °C, 6 h; coupling: NaHCO3, EtOAc, H2O, acid chloride, rt, 1 h; (e) Pd/C, HCO2NH4, MeOH, 50 °C, overnight; (f) (COCl)2, rt, overnight; (g) Et3N, THF, 0 °C to rt, 8 h.
The designed inhibitors II incorporating phenyloxazolidinones into hydroxyethylene isostere as P2 ligands were prepared using a slightly different reaction sequence as illustrated in Scheme 2. The hydroxyethylene core 1 was first reacted with activated phenyloxazolidinone-5-carboxylic acids 10 or 11 to provide intermediate compounds 17a and 18a,d that were debenzylated using Pd/C to give free amines 19a and 20a,d. Finally, the amine intermediates 19a and 20a,d were reacted with acyl chlorides prepared from acids A4 and A5 providing inhibitors 21–23. Acylation of amines 19a and 20d with p-nitrophenylcarbonates A6 and A7, prepared from the corresponding alcohols, provided the inhibitors 24a and 25d with carbamate linkage between the core fragment and P2′ ligand.
Scheme 2.
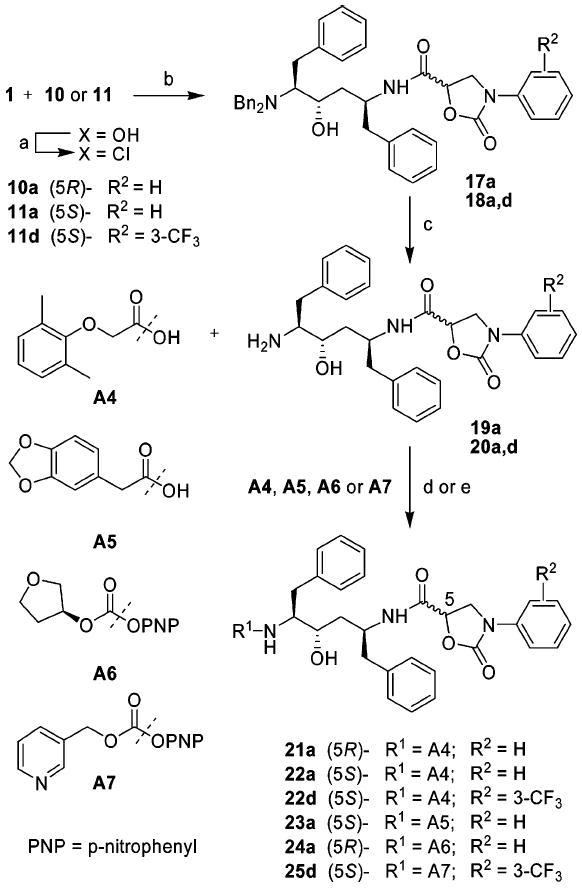
Synthesis of Designed Protease Inhibitors IIa
a Reagents and conditions: (a) (COCl)2, rt, overnight; (b) Et3N, THF, 0 °C to rt, 8 h; (c) Pd/C, HCO2NH4, MeOH, 50 °C, overnight; (d) Acid A4 or A5, SOCl2, EtOAc, DMF, rt to 50 °C, 6 h; coupling: NaHCO3, EtOAc, H2O, acid chloride, rt, 1h; (e) A6 or A7, DIPEA, DMF–CH2Cl2 (1:1), 0 °C to rt, 12 h.
The (hydroxyethyl)hydrazine isostere-based inhibitors III and IV were prepared from commercially available chiral epoxide, (1S,2R)-(1-oxiranyl-2-phenylethyl)carbamic acid tert-butyl ester 26, via a modification of published sequence; the general synthetic route is outlined in Scheme 3.26,28 Ring opening of chiral epoxide 26 with Cbz-isobutylcarbazate 27 provided the orthogonally-protected dipeptide isostere 28. The carbazate 27 can be prepared in two steps by the reaction of Cbz-carbazate with isobutyraldehyde followed by selective reduction of the resulting imine using NaCNBH3.28 Removal of the Cbz protection in (hydroxyethyl)hydrazine core 28 by hydrogenation provided the intermediate amine 29 that was reacted with N-methoxycarbonyl-l-tert-leucine A3 under standard peptide coupling conditions to furnish the intermediate compound 30. Deprotection of Boc and reaction of the resulting free amine 31 with activated N-phenyl-oxazolidinone-5-carboxylic acid 11a provided the designed inhibitor 32. The synthesis of designed inhibitor IV incorporating the phenyloxazolidinone moiety as P2′ ligands was carried out using a similar strategy. Reaction of the free amine 29 with activated N-phenyloxazolidinone-5-carboxylic acid 11a yielded the intermediate 33. Removal of Boc protection and subsequent reaction of the free amine 34 with N-methoxycarbonyl-l-tert-leucine A3 using standard peptide coupling conditions provided target compound 35.
Scheme 3.
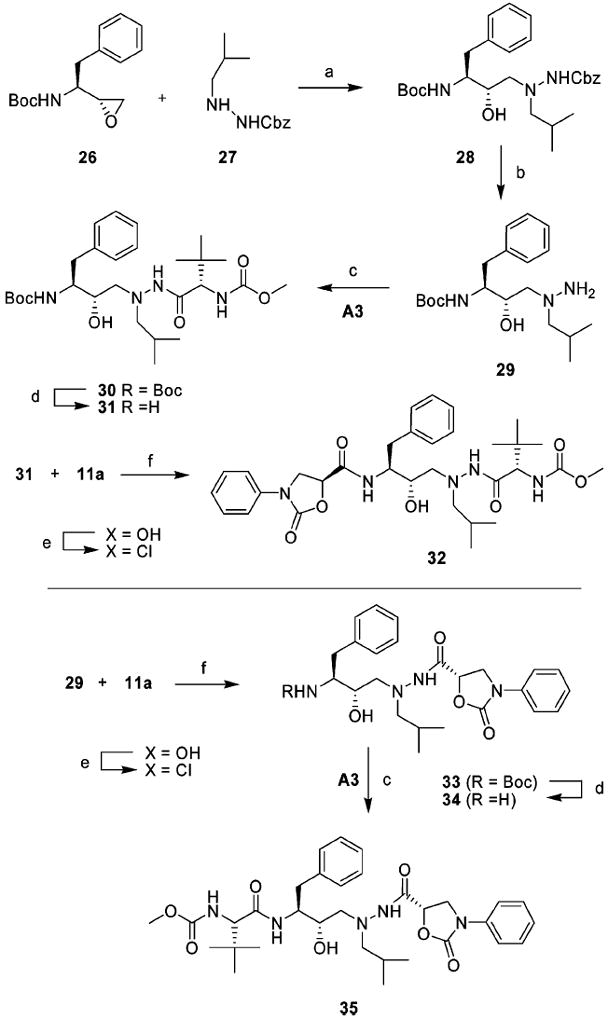
Synthesis of Designed Protease Inhibitors III and IVa
a Reagents and conditions: (a) i-PrOH, 80 °C, 24 h; (b) Pd/C, MeOH, rt, overnight; (c) acid A3, EDCI, HOBt, DIPEA, DMF–CH2Cl2 (1:1), 0 °C to rt, 12 h (d) TFA, CH2Cl2, 1 h; (e) acid 11a, (COCl)2, rt, overnight; (f) Et3N, THF, 0 °C to rt, 8 h.
The symmetrically acylated protease inhibitors 39 and 40 incorporating phenyloxazolidinones at both P2 and P2′ positions were prepared from the bis-Boc-protected (hydroxyethyl)-hydrazine isostere 37, as shown in Scheme 4. The symmetrically protected compound 37 is a key intermediate in the synthesis of approved drug ATV and was prepared from (1S,2R)-epoxide 26 and carbazate 36 following a reported procedure.29 Simultaneous removal of both the Boc groups in 37 using 4 N HCl in dioxane provided the free bis-amine intermediate 38 that was coupled with activated phenyloxazolidinone-5-carboxylic acid 11a or 11e to provide the target compounds 39 and 40.
Scheme 4.
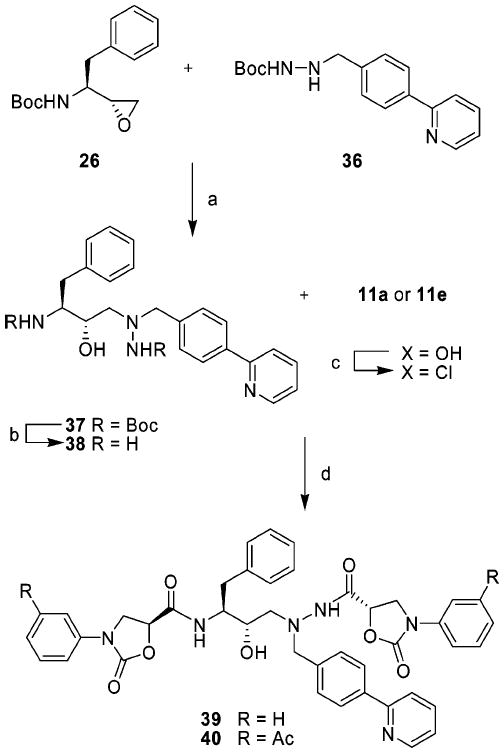
Synthesis of Symmetrically Acylated Protease Inhibitorsa
a Reagents and conditions: (a) i-PrOH, 80 °C, 24 h; (b) 4 N HCl–dioxane, 50 °C, 4 h; (c) acid 11a or 11e, (COCl)2, rt, overnight; (d) Et3N, THF, 0 °C to rt, 8 h.
Results and Discussion
Our previous studies of the (hydroxyethylamino)sulfonamide isostere-based protease inhibitors indicate that compounds incorporating (5S)-enantiomer of N-phenyloxazolidinone-5-carboxamides are significantly more potent inhibitors of HIV-1 protease than the corresponding (5R)-analogues.23 As hydroxyethylene and (hydroxyethyl)hydrazine isosteres are pseudosymmetric, it was important to investigate the stereochemical preference and optimal location of the phenyloxazolidinonebased ligands at both P2 and P2′ positions. In addition, both isosteres have opposite (S)-configuration at the transition-state hydroxyl group compared to the (R)-configuration in hydroxyethylamine isostere. Therefore, we initially focused our efforts to determine the optimal stereochemistry and position of the phenyloxazolidinone moieties in these core isosteres. A series of inhibitors were prepared incorporating either (R)- or (S)-enantiomer of phenyloxazolidinones into the hydroxyethylene and (hydroxyethyl)hydrazine isosteres as P2 as well as P2′ ligands. All the compounds were evaluated for their inhibitory activities against wild-type HIV-1 protease (Q7K) using a fluorescence resonance energy transfer (FRET)-based assay; Ki values are presented in Tables 1 and 2.
Table 1.
Inhibitory Activities of Hydroxyethylene Isostere-Based Compounds against Wild-Type HIV-1 Protease
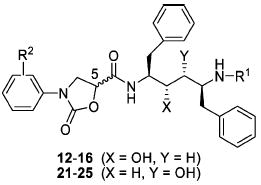
| |||
|---|---|---|---|
| Compd | R1 | R2 | Ki (nM) |
| 12a |
|
H (5R) | 131.4 |
| 13a |
|
H (5S) | 0.85 |
| 13c |
|
3,4-di-F (5S) | 2.13 |
| 13d |
|
3-CF3 (5S) | 2.62 |
| 13e |
|
3-Ac (5S) | 0.98 |
| 14a |
|
H (5S) | 2.04 |
| 15a |
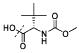
|
H (5S) | 3.51 |
| 16a |
|
H (5S) | 8.05 |
| 16e |
|
3-Ac (5S) | 35.11 |
| 16f |
|
4-Ac (5S) | 39.87 |
| 21a |
|
H (5R) | 4731 |
| 22a |
|
H (5S) | 7.28 |
| 22d |
|
3-CF3 (5S) | 21.63 |
| 23a |
|
H (5S) | 657 |
| 24a |
|
H (5R) | 1800 |
| 25d |
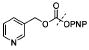
|
3-CF3 (5S) | 41.09 |
Table 2.
Inhibitory Activities of (Hydroxyethyl)hydrazine Isostere-Based Compounds against Wild-Type HIV-1 Protease
| Compd | Structure | Ki (nM) |
|---|---|---|
| 32 |
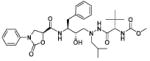
|
7.0 |
| 35 |
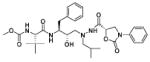
|
10.6 |
| 39 |
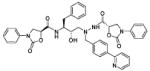
|
300 |
| 40 |
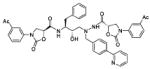
|
1090 |
In the hydroxyethylene isostere-based inhibitor series, we first investigated the stereochemical preference of N-phenyloxazolidinone-5-carboxamides attached at P2′ position, proximal to the central hydroxyl group, replacing the 2,6-dimethylphenoxyacetyl moiety in LPV scaffold. Two compounds with either (R)- or (S)-enantiomer of N-phenyloxazolidinone-5-carboxylic acid (10a or 11a) attached to the hydroxyethylene core 1 at the P2′ position and acid A1, a close mimic of P2–P3 moiety in LPV, attached at the P2 position were prepared and tested. Interestingly, compound 13a with (S)-N-phenyloxazolidinone was more potent (Ki = 0.85 nM) compared to compound 12a, with the corresponding (R)-enantiomer (Ki = 131.4 nM). These results are similar to those observed for (hydroxyethylamino)sulfonamide isostere-based inhibitors and clearly indicate that the compounds with (5S)-N-phenyloxazolidinone-5-carbaxamides as P2′ ligands are more potent than the corresponding (5R)-analogues.
Several inhibitors were prepared using various (S)-N-phenyloxazolidinone-5-carboxylic acids 11a–f attached to the core isostere 1 at P2′ position and one of the four acids A1–A4 attached at P2 position. Compared to inhibitor 13a, the inhibitors with the substituted phenyl ring of the oxazolidinone moiety 13c–e were less potent; only 13e was equipotent to 13a, exhibiting a Ki value of 0.98 nM. To increase the potency, the N-acetylvaline group A1 was replaced with the LPV cyclic urea moiety A2, but with no improvement in inhibitory activity (compound 14a, Ki = 2.04 nM). Similarly, compound 15a containing the N-methoxycarbonyl-l-tert-leucine moiety, the P2/P2′ ligand in ATV, as P2 ligand displayed low nanomolar inhibitory activity (Ki = 3.51 nM), but this modification also did not improve the potency. Introduction of a more hydrophobic moiety, 2,6-dimethylphenoxyacetyl A4, as the P2 ligand further reduced the inhibitory activity (compound 16a, Ki = 8.05 nM); inhibitors 16e,f containing substituted phenyloxazolidinones were less potent compared to the unsubstituted analogue 16a.
The second series of inhibitors were prepared by transposing the cyclic urea P2–P3 moiety in LPV with various phenyloxazolidinone ligands attached at the P2 position, distal to the central hydroxyl group, of the core isostere. Utilizing the 2,6-dimethylphenoxyacetyl moiety A4 at P2′ position, a pair of inhibitors were prepared with (R)- and (S)-enantiomer of phenyloxazolidinones. Again, compound 22a containing the (S)-enantiomer of the ligand was significantly more potent (Ki = 7.28 nM) than compound 21a containing the corresponding (R)-enantiomer of the ligand (Ki = 4731 nM). Introducing the 3-CF3 group on the phenyl ring of oxazolidinone moiety in compound 22d (Ki = 21.63 nM) decreased the potency compared to the unsubstituted compound 22a. Replacement of the 2,6-dimethylphenoxyacetyl moiety A4 with 1,3-(benzo)dioxolane group A5 resulted in a significant loss of activity in compound 23a (Ki = 657 nM). Compounds 24a and 25d containing P2′ ligands A6 and A7 attached to the core isostere via carbamate linkage also showed poor inhibitory activities.
From the above two series, compounds 16a and 22a, alternately, contain the same phenyloxazolidinone moiety at the P2′ and P2 positions, proximal and distal to the central hydroxyl group, and the 2,6-dimethylphenoxyacetyl group on the other end of the core isostere. Both compounds show similar inhibitory activities, suggesting that the protease inhibition activity is not particularly sensitive to the position of the phenyloxazolidinone moiety relative to the central hydroxyl group. This trend has also been observed in previous SAR studies of hydroxyethylene isostere-based inhibitors containing hexahydrofurofuranyloxy (bis-tetrahydrofuran) and 2,6-dimethylphenoxyacetyl moieties as P2/P2′ ligands.30
The strong preference observed for the (S)-phenyloxazolidinones at both P2 and P2′ positions in the hydroxyethylene isostere-based inhibitors lead us to focus only on the (S)-enantiomer of the ligand in (hydroxyethyl)hydrazine isosterebased inhibitors. The inhibitor 32 with the phenyloxazolidinone moiety attached at the P2 position, proximal to central hydroxyl group, and N-methoxycarbonyl-l-tert-leucine moiety A3 at the P2′ position showed a moderate inhibitory activity against HIV-1 protease, with a Ki value of 7.0 nM (Table 2). Exchanging the position of phenyloxazolidinone moiety with N-methoxycarbonyl-l-tert-leucine A3 from P2 to P2′ position, distal to the central hydroxyl group, in compound 35, slightly decreased the inhibitory activity (Ki = 10.6 nM). These results indicate that in (hydroxyethyl)hydrazine isostere-based inhibitors the phenyloxazolidinone moiety can be attached at both proximal and distal positions to the central hydroxyl group without significantly affecting protease inhibition activity. The symmetrically acylated compounds 39 and 40 incorporating phenyloxazolidinones at both P2 and P2′ positions showed very weak inhibitory activity against HIV-1 protease.
Selected hydroxyethylene and (hydroxyethyl)hydrazine isostere-based inhibitors were further evaluated for their inhibitory activities against a panel of MDR protease variants each representing a different pattern of drug resistance. The mutant variants were selected by examining the Stanford HIV-1 Drug Resistance Database (http://hivdb.stanford.edu), which contains sequences of viral isolates from HIV-1-infected patients. The selected protease variants represent the pattern of resistance mutations that occur under the selective pressure of three or more currently prescribed protease inhibitors.31 These MDR variants are L10I, G48V, I54V, L63P, V82A (M1), D30N, L63P, N88D (M2), and L10I, L63P, A71V, G73S, I84V, L90M (M3). The Ki values of selected inhibitors against M1–M3 MDR variants of protease are presented in Table 3. For comparison, several currently marketed protease inhibitors, SQV, RTV, APV, LPV, and ATV, were also studied. The data shows that all protease inhibitors lose affinity against mutant variants compared to the wild-type protease, however, the relative loss of activity is different in different inhibitors. The first generation drugs, SQV and RTV, lose significant activity against all three mutant variants. The relative loss of activity for APV is not that significant against all mutants but its Ki value for wild-type is also relatively high. LPV significantly loses activity against protease variants M1 and M3 but still retains high affinity against M2 variant. ATV loses moderately against variants M1 and M3 but still retains high affinity against the M2 variant. The most potent inhibitor in the current series, 13a, was also a potent inhibitor of the M2 variant but significantly loses activity against M1 and M3 variants; the analogues 13c and 13e also lose activities against M1 and M3 variants but retain low nM inhibitory activities against the M2 variant. Most other inhibitors in this series have very similar activities against wild-type and M2 variant, but have decreased affinities against M1 and M3 variant proteases.
Table 3.
Inhibitory Activities of Selected Compounds against MDR Variants of HIV-1 Protease and Their Antiviral Potenciesa
| cmpd |
Ki (nM)
|
EC50b (μM) | |||
|---|---|---|---|---|---|
| Wt | M1 | M2 | M3 | ||
| 13a | 0.85 | 51.2 | 3.1 | 112 | 0.79 |
| 13c | 2.13 | 168 | 3.9 | 282 | 0.36 |
| 13e | 0.98 | 82.3 | 1.67 | 122 | 0.62 |
| 14a | 2.04 | 175 | 13.9 | 185 | 1.21 |
| 15a | 3.51 | 131 | 14.0 | 392 | 1.03 |
| 22a | 7.28 | 364 | 18.3 | 447 | ND |
| 32 | 7.0 | 231 | 39.2 | 549 | ND |
| SQV | 0.065 | 89.53 | 1.03 | 78.44 | 0.01 |
| RTV | 0.055 | 3.025 | 0.46 | 2.81 | ND |
| APV | 0.10 | 0.15 | 0.21 | 1.40 | 0.04 |
| LPV | 0.005 | 6.10 | 0.04 | 0.90 | 0.01 |
| ATV | 0.046 | 0.33 | 0.009 | 0.49 | ND |
Wt: Q7K; M1: L10I, G48V, I54V, L63P, V82A; M2: D30N, L63P, N88D; M3: L10I, L63P, A71V, G73S, I84V, L90M.
Antiviral activities were determined in MT-4 cells.
We also determined the antiviral activities of selected compounds in MT-4 cells; results are summarized in Table 3. All tested compounds showed only moderate antiviral activity compared to the drugs SQV, APV, and LPV. Compound 13c with a 3,4-difluorophenyl-oxazolidinone group at the P2′ position and N-acetylvaline at the P2 position was the most potent in this series, with an EC50 value of 0.36 μM. Two close analogues, 13a and 13e, also exhibit submicromolar antiviral potency, with EC50 values of 0.79 and 0.62 μM, respectively.
Crystal Structures of HIV-1 Protease Complexes
We have determined the crystal structures of two new inhibitors incorporating phenyloxazolidinone-based ligands, 13e and 14a, in complex with wild-type HIV-1 protease to a resolution of 1.85 and 1.9 Å, respectively. Both compounds have the same hydroxyethylene dipeptide isostere central core as that of LPV, but different substituent groups at P2 and P2′ positions (Figure 3). We also determined the crystal structure of LPV complexed with wild-type HIV-1 protease, as the structure available in the PDB, 1MUI,32 is of low resolution. In the crystal structure of 14a, there is some excess density near the oxazolidinone ring that could not be modeled. Otherwise, in all the three crystal structures, the inhibitor has unambiguous density and has one orientation. The crystallographic data and the refinement statistics for all the three structures are presented in Table 4.
Figure 3.
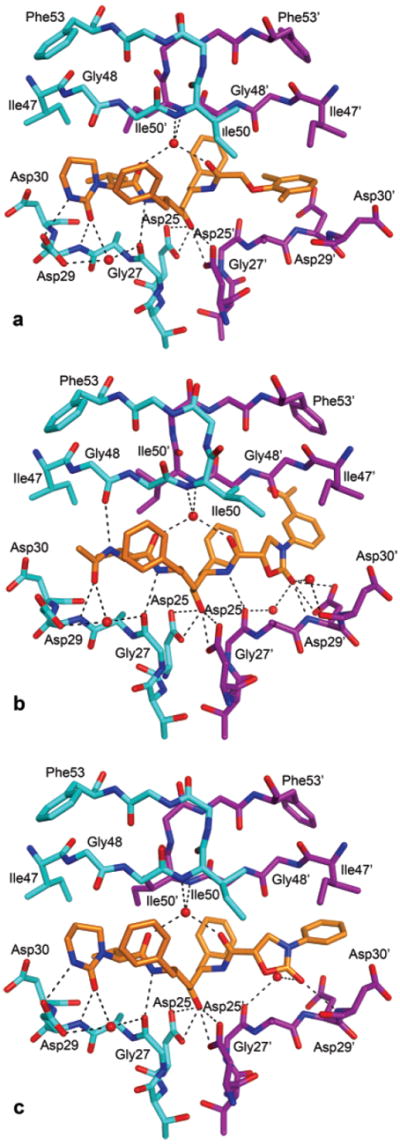
Hydrogen bonds, shown as dotted black lines, observed in the protease complexes of (a) LPV, (b) 13e, and (c) 14a. The two monomers and the inhibitor are shown in cyan, magenta, and orange, respectively. Nitrogen and oxygen atoms are in blue and red, respectively.
Table 4.
Crystallographic Data Collection and Refinement Statistics for Complexes of LPV, 13e, and 14a with the Wild-Type HIV-1 Protease
| LPV | 13e | 14a | |
|---|---|---|---|
| resolution (Å) | 1.95 | 1.85 | 1.90 |
| temperature (°C) | −80 | −80 | −80 |
| space group | P212121 | P212121 | P212121 |
| a (Å) | 51.16 | 50.76 | 50.84 |
| b (Å) | 57.98 | 58.17 | 58.53 |
| c (Å) | 61.12 | 61.44 | 61.87 |
| Z | 4 | 4 | 4 |
| Rmerge (%) | 4.4 | 3.8 | 5.4 |
| completeness (%) | 97.0 | 95.5 | 99.7 |
| total no. of reflections | 45 575 | 102 303 | 95 174 |
| No. of unique reflections | 13 461 | 15 398 | 15 088 |
| Rfree (%) | 23.6 | 22.4 | 23.5 |
| Rfactor (%) | 19.2 | 17.4 | 17.5 |
| rmsda in: | |||
| bond length (Å) | 0.008 | 0.008 | 0.009 |
| bond angle (°) | 1.195 | 1.207 | 1.292 |
| No. of crystallographic waters | 108 | 135 | 136 |
| PDBb code | 2Q5K | 2Q54 | 2Q55 |
rmsd, root-mean-square deviation.
PDB, protein data bank.
In inhibitor 13e, the P2–P3 cyclic urea moiety and the P2′ 2,6-dimethylphenoxyacetyl group of LPV are substituted with N-acetylvaline and phenyloxazolidinone groups, respectively, whereas in 14a only the P2′ 2,6-dimethylphenoxyacetyl group of LPV is replaced with phenyloxazolidinone group. When the crystal structures of 13e and 14a are superimposed onto LPV, the backbone of 14a superimposes well with LPV compared to 13e with LPV. The backbone region of Gly48 in LPV and 14a rotates away to accommodate the trimethylene portion of the cyclic urea moiety as previously observed in the LPV–protease structure.32 Consequently, the flap region of monomer A in 14a resembles more of LPV while it is very different in 13e. The flap regions of the monomer B in the crystal structures of 13e and 14a superimpose well onto each other than with LPV. The flap in monomer B of LPV is about 1 Å closer to the inhibitor compared to 13e and 14a. This may be due to the presence of the phenyloxazolidinone ring in 13e and 14a, which makes interactions with the active site of the protease.
The difference in the protease backbone among the three structures, especially among the flap regions, is also evident from the double difference plots.33 The double difference plot elucidates the differences of the domain-to-domain distances of the protease for various structures irrespective of the inhibitor present in the structures. Two complexes can be compared using these plots without actually superimposing them, thus reducing the bias as to which residues are chosen for superimposition. The double difference plots of LPV-13e and 14a–13e (Figure 4a,c) show that the flap region in monomer A of LPV and 14a is greater than 0.5 Å apart from the other Cα atoms of the protease compared to that in 13e. The LPV-14a plot (Figure 4b) shows no variation in the monomer A of 14a compared to LPV, while the double difference plot of 14a–13e shows very little variation in the monomer B of 14a compared to that in 13e. Thus, the backbone CR of the monomer A of the protease in 14a resembles the monomer A of LPV, where they have a common P2 group in the inhibitor, and the monomer B of 14a resembles the monomer B of 13e, where they have phenyloxazolidinone ring as the P2′ group. The presence of different groups on the LPV core reflects on the packing of the protease.
Figure 4.
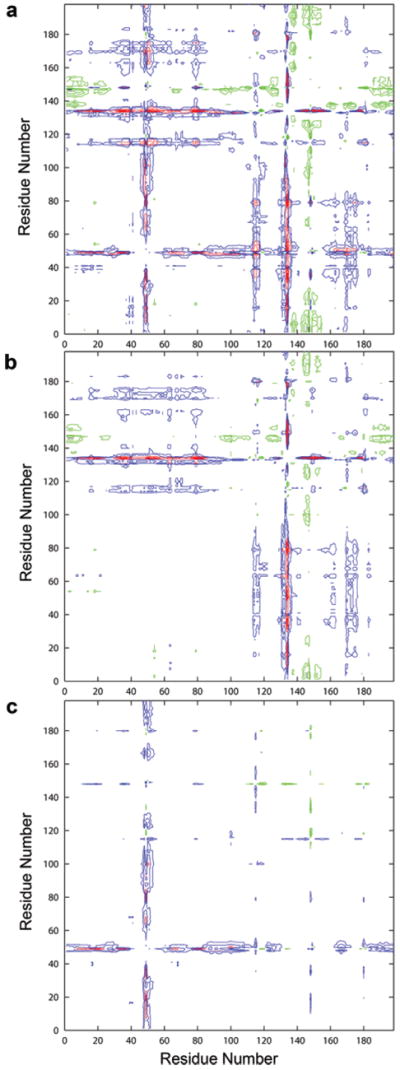
Double difference plots showing the relative shifts in the wild-type HIV-1 protease complexes of (a) 13e in reference to LPV, (b) 14a in reference to LPV, and (c) 14a in reference to 13e. Double difference plots contour differences in internal Cα–Cα distances between two complexes. Contours in the plots show whether the respective distances in the two complexes being compared are closer or further apart. Black indicates a difference of < −1 Å, green indicates a difference of between −0.99 and −0.5 Å, blue indicates a difference of between 0.5 and 0.99 Å, and red indicates a difference of > 1 Å.
Important protease–inhibitor hydrogen bonds in the crystal structures of LPV, 13e, and 14a, including those mediated by water molecules, are shown in Figure 3. The hydrogen-bonding distances observed in the three crystal structures are presented in Table 5. In all three complexes, a central water molecule present in the flap region makes hydrogen bonds similar to what has been observed for most HIV-1 protease inhibitors. In all three inhibitors, the central hydroxyl group forms hydrogen bonds with the two catalytic residues Asp25 and Asp25′ in the floor of the active site. The amide nitrogen of the inhibitor attached to the valine group forms a hydrogen bond with the carbonyl group of the Gly27 in all three complexes. The carbonyl group of the N-acetylvaline moiety in 13e forms similar hydrogen bonds as carbonyl groups of the cyclic urea moiety in LPV and 14a, as shown in Figure 3. These include a direct hydrogen bond of the carbonyl group in all three structures, with the main chain nitrogen of the Asp29 and a water-mediated interaction between the carbonyl of Gly27 with the carbonyl group of the cyclic urea in LPV and 14a and the carbonyl of N-acetylvaline moiety in 13e. These are the common hydrogen bonds that are observed in all three structures.
Table 5.
Hydrogen-Bonding Distances between Protease and Inhibitor Atoms in Crystal Structures of LPV, 13e, and 14a
| protease atom | inhibitor atom | distance (Å) for
|
||
|---|---|---|---|---|
| LPV | 13e | 14a | ||
| Asp 29 N | O30 | 2.8 | ||
| Asp 29 OD1 | O30 | 3.2 | ||
| Asp 29 OD2 | O30 | 3.5 | ||
| water | O30 | 3.3 (W69)a | ||
| water | O30 | 3.1 (W12) | 3.3 (W23) | |
| Asp 30 N | O30 | 3.0 | ||
| water | N31 | 3.3 (W23) | ||
| water | O28 | 2.9 (W69) | 3.5 (W23) | |
| Gly 27 O | N4 | 3.3 | ||
| water | O5 | 2.6 (W36) | 2.7 (W6) | 2.7 (W6) |
| Asp 25 OD1 | O4 | 3.0 | 2.7 | 2.8 |
| Asp 25 OD2 | O4 | 2.8 | 2.8 | 2.7 |
| Asp 25′ OD1 | O4 | 2.7 | 3.0 | 2.8 |
| Asp 25′ OD2 | O4 | 2.7 | 2.6 | 3.0 |
| Gly 27′ O | N3 | 3.0 | 3.0 | 3.0 |
| water | O2 | 2.6 (W36) | 2.7 (W6) | 2.7 (W6) |
| water | O1 | 3.2 (W26) | 3.2 (W67) | 3.1 (W3) |
| Asp 29′ N | O1 | 2.7 | 2.9 | 2.8 |
| Asp 29′ OD2 | N2 | 3.0 | 3.1 | |
| Arg 8 NH2 | N2 | 3.5 | ||
| Gly 48′ O | N1 | 3.1 | ||
The corresponding water number is in parentheses.
The P2′ 2,6-dimethylphenoxyacetyl group in LPV does not form any hydrogen bonds to the protease, whereas phenyloxazolidinone moieties in 13e and 14a form hydrogen bonds between the oxazolidinone ring and the protease. The orientation of the oxazolidinone ring in the active site is different in 13e and 14a. Consequently, they form different van der Waals (vdW) and hydrogen bond interactions with the protease. The carbonyl group of the oxazolidinone ring in 13e forms three hydrogen bonds with the main chain nitrogen atom and side chain atoms of Asp29′, whereas it forms one direct hydrogen bond with the nitrogen atom of Asp30′ in 14a. The oxazolidinone ring is also involved in two water-mediated hydrogen bonds in 13e and one water-mediated hydrogen bond in 14a. Inhibitor 13e also forms two hydrogen bonds to the main chain oxygen atoms of Gly27′ and Gly48. The hydrogen bonds formed by the oxazolidinone ring in 13e resemble the pattern of hydrogen bonds formed by the oxazolidinone moiety in the (R)-(hydroxyethylamino)-sulfonamide isostere-based (APV scaffold) inhibitors recently reported by our group.23 Incorporation of the phenyloxazolidinone moiety as the P2 ligand into the APV scaffold improved the binding affinity with respect to APV, but its incorporation as the P2 or P2′ ligand into the LPV scaffold did not improve the binding affinity with respect to LPV. Compared to the LPV–protease structure, the presence of the phenyloxazolidinone moiety at the P2′ position of the LPV scaffold expands the active site of the protease in both 13e– and 14a–protease structures. In contrast, the backbone of the protease changes a little in the structures of APV scaffold-based inhibitors carrying phenyloxazolidinones as the P2 ligands (2I0A and 2I0D)23 compared to the APV–protease structure. The active site in the 13e–protease complex with an N-acetylvaline group at P2 is relatively closely packed than in the 14a–protease complex with P2–P3 cyclic urea moiety. The incorporation of the phenyloxazolidinone moiety as the P2′ ligand into the hydroxyethylene isostere may not alter the backbone of the protease if the P2 ligand is optimized by SAR studies; these compounds may exhibit improved binding affinities compared to 13e.
The loss of activity of these inhibitors with the mutant proteases may be explained by the observed protease–inhibitor interactions in the crystal structures. LPV, 13e, and 14a significantly lose activity against the mutant variants M1 and M3 and are not severely effected by the mutant variant M2. The mutants M1 and M3 are multidrug-resistant variants, while the mutant M2 with D30N as active site mutation is specific to nelfinavir (NFV) drug resistance. As none of the three inhibitors form ionic interactions with the side chain of D30 in the wildtype structures, the mutation D30N has minimal effect on the observed Ki. The mutant M1 has G48V and V82A as active site mutations and M3 has active site mutation I84V. Modeling of these active site mutations into the wild-type crystal structures show that G48V could potentially cause steric clashes with the acetyl group of 13e while a potential loss of van der Waals contacts with V82A and I84V may occur in LPV, 13e, and 14a. Thus, the observed loss of binding affinity for the resistant variants M1 and M3 are likely due to changes in protease–inhibitor interactions, while the M2 protease variant is unlikely to strongly impact the interactions of these inhibitors with the enzyme.
Conclusions
In summary, we have designed, synthesized, and evaluated novel HIV-1 protease inhibitors incorporating N-phenyloxazolidinone-5-carboxamides into hydroxyethylene and (hydroxyethyl) hydrazine isosteres as P2 and P2′ ligands. In each series, compounds with a (5S)-enantiomer of the phenyloxazolidinone moiety are significantly more potent inhibitors of HIV-1 protease than the corresponding (5R)-analogues. The position of the phenyloxazolidinone moiety, proximal or distal relative to the central hydroxyl group, has little effect on the protease inhibition activity. Variations at the P2 and P2′ side chains of the inhibitors resulted in compounds with Ki values in the low nM range. Selected inhibitors showing low nM inhibitory activities against wild-type HIV-1 protease were further evaluated against a panel of MDR protease variants. The antiviral activities of selected compounds were determined in MT-4 cells; three compounds exhibited EC50 values in submicromolar range. The crystal structures of LPV and two new inhibitors (13e and 14a) in complex with wild-type HIV-1 protease have been determined; both 13e and 14a contain phenyloxazolidinone moieties attached to the hydroxyethylene isostere. Analysis and comparison of 13e– and 14a–protease crystal structures with the LPV–protease structure provides insight into the binding mode of these inhibitors to the protease. The information from the current investigations and structural analyses can be utilized to design new protease inhibitors with enhanced enzyme inhibitory and antiviral activities.
Experimental Section
General
1H and 13C NMR spectra were recorded on a Varian 400 MHz NMR spectrometer, operating at 400 MHz for 1H and 100 MHz for 13C. Chemical shifts are reported in ppm relative to the solvent signal, and coupling constant (J) values are reported in Hertz (Hz). Thin-layer chromatography (TLC) was performed on E. Merck silica gel 60-F-254 plates, and spots were visualized with UV light. Flash column chromatography was performed using 230–400 mesh silica gels (E. Merck). High-resolution mass spectra (HRMS) were recorded on Waters Q-TOF Premier mass spectrometer by direct infusion of solutions of each compound using electrospray ionization (ESI) in positive mode. Tetrahydrofuran (THF) was distilled from sodium/benzophenone. Anhydrous dichloromethane, N,N-dimethylformamide (DMF), benzene, and toluene were purchased from Aldrich and used as such. All reagents and chemicals were purchased from commercial vendors and used as received. Analytical reversed-phase high performance liquid chromatography (HPLC) was performed on a Waters-Alliance Separation Module 2695 system equipped with an auto sampler and a Waters 996 photodiode array detector. Purity of the final compounds was determined using two different chromatographic systems. First system: column, Waters XTerra RP-C18 (3.5 μm, 4.6 mm × 150 mm); mobile phase A, 10 mM ammonium acetate in water; mobile phase B, acetonitrile. Using a flow rate of 0.8 mL/min, gradient elution was performed from 30% B to 90% B over 10 min. Second system: column, Zorbax 300SB-C8 (5 μm, 4.6 mm × 250 mm); mobile phase A, 0.1% trifluoroacetic acid in water; mobile phase B, 0.1% trifluoroacetic acid in acetonitrile. Gradient elution was performed from 40% B to 90% B over 10 min at a flow rate of 1 mL/min. A table containing the retention time and purity of each final compound is in Supporting Information.
Synthesis of Protease Inhibitors
The Phe–Phe hydroxyethylene dipeptide isostere 1 was prepared following the reported method.24 The acids A2–A4 were synthesized following reported procedures.25,26 The chiral N-phenyloxazolidinone-5-carboxylic acids 10a and 11a–f were prepared from Cbz-protected aniline derivatives as previously described.23 General procedures for the coupling reactions of acids and core amines are provided in the Supporting Information. The orthogonally and symmetrically protected (hydroxyethyl)hydrazine dipeptide isosteres 28 and 37, respectively, were prepared from chiral epoxide, (1S,2R)-(1-oxiranyl-2-phenylethyl)carbamic acid tert-butyl ester 26, following or adapting reported procedures.26,28,29 Enantiomerically pure epoxide 26 was generously provided by Kaneka, U.S.A.
(5R)-N-[(1S,2S,4S)-4-[[(2S)-2-(Acetylamino)-3-methyl-1-oxobutyl]amino]-2-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]-2-oxo-3-phenyloxazolidine-5-carboxamide (12a)
1H NMR (400 MHz, CDCl3) δ 7.53–7.50 (m, 2H), 7.42–7.38 (m, 2H), 7.24–7.13 (m, 8H), 7.09–6.99 (m, 3H), 6.54 (d, J = 7.2 Hz, 1H), 6.16 (d, J = 8.4 Hz, 1H), 4.77 (dd, J = 10.0, 6.4 Hz, 1H), 4.18 (t, J = 9.6 Hz, 1H), 4.12–4.04 (m, 4H), 3.68 (m, 1H), 3.56 (br s, 1H), 2.89 (d, J = 7.6 Hz, 2H), 2.76 (dd, J = 13.2, 6.4 Hz, 1H), 2.59 (dd, J = 13.6, 7.2 Hz, 1H), 2.0 (m, 1H), 1.98 (s, 3H), 1.68–1.60 (m, 2H), 0.89 (d, J =6.8 Hz, 3H), 0.87 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.58, 170.87, 168.82, 153.58, 138.12, 137.98, 137.66, 129.68 (2C), 129.49 (2C), 129.46 (2C), 128.71 (2C), 128.57 (2C), 126.79, 126.68, 125.01, 118.68 (2C), 70.51, 69.53, 59.18, 55.44, 49.99, 49.17, 41.49, 39.26, 38.25, 31.08, 23.49, 19.55, 18.41. HRMS (ESI) m/z calcd for C35H43N4O6 [M + H]+, 615.3183; found, 615.3153.
(5S)-N-[(1S,2S,4S)-4-[[(2S)-2-(Acetylamino)-3-methyl-1-oxobutyl]amino]-2-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]-2-oxo-3-phenyloxazolidine-5-carboxamide (13a)
1H NMR (400 MHz, CDCl3) δ 7.46–7.39 (m, 4H), 7.24–7.18 (m, 3H), 7.15–7.09 (m, 5H), 7.01 (t, J = 7.2 Hz, 2H), 6.98–6.89 (m, 3H), 6.55 (d, J = 8.8 Hz, 1H), 4.73 (dd, J = 10.0, 6.4 Hz, 1H), 4.35–4.24 (m, 3H), 4.18 (t, J = 8.0 Hz, 1H), 4.07 (t, J = 10.0 Hz, 1H), 3.82 (m, 1H), 3.42 (dd, J = 9.2, 6.4 Hz, 1H), 2.93–2.84 (m, 2 H), 2.83–2.73 (m, 2H), 2.10 (m, 1H), 2.0 (s, 3H), 1.72–1.63 (m, 2H), 0.91 (t, J = 6.4 Hz, 6H); 13C NMR (100 MHz, CDCl3 δ 171.79, 170.89, 168.96, 153.47, 137.99, 137.69, 137.54, 129.63 (2C), 129.55 (2C), 129.38 (2C), 128.64 (2C), 128.49 (2C), 126.72, 126.59, 124.88, 118.50 (2C), 70.47, 69.97, 59.33, 54.66, 48.94, 48.50, 41.28, 39.91, 38.75, 30.51, 23.38, 19.68, 18.46. HRMS (ESI) m/z calcd for C35H43N4O6 [M + H]+, 615.3183; found, 615.3150.
(5S)-N-[(1S,2S,4S)-4-[[(2S)-2-(Acetylamino)-3-methyl-1-oxobutyl]amino]-2-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]-3-(3,4-difluorophenyl)-2-oxo-oxazolidine-5-carboxamide (13c)
1H NMR (400 MHz, CDCl3) δ 7.55–7.49 (m, 1H), 7.22 (m, 2H), 7.18-7.10 (m, 6H), 7.02 (m, 3H), 6.94–6.86 (m, 2H), 6.71 (d, J = 8.4 Hz), 6.27 (d, J = 8.4 Hz, 1H), 4.74 (dd, J = 10.0, 6.0 Hz, 1H), 4.27 (m, 2H), 4.12 (t, J = 7.6 Hz, 1H), 4.08 (m, 1H), 4.02 (t, J = 10.0 Hz, 1H), 3.78 (m, 1H), 3.39 (dd, J = 9.6, 6.4 Hz, 1H), 2.88–2.74 (m, 5H), 2.08 (m, 1H), 1.98 (s, 3H), 1.67 (m, 2H), 0.90 (d, J = 6.4 Hz, 3H), 0.88 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.79, 170.90, 168.64, 153.25, 150.52 (dd, J = 246.1, 13.2 Hz), 147.46 (dd, J = 245.1, 12.5 Hz), 138.04, 137.75, 134.08 (m), 129.67, 129.62 (2C), 129.57 (2C), 128.63 (2C), 128.45 (2C), 126.72, 126.53, 117.79 (d, J = 16.9 Hz), 113.94 (m), 108.52 (d, J = 22.7 Hz), 70.47, 69.86, 59.39, 54.66, 48.95, 48.48, 41.23, 39.95, 38.69, 30.52, 23.35, 19.65, 18.52. HRMS (ESI) m/z calcd for C35H41F2N4O6 [M + H]+, 651.2994; found, 651.3009.
(5S)-N-[(1S,2S,4S)-4-[[(2S)-2-(Acetylamino)-3-methyl-1-oxobutyl]amino]-2-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]-2-oxo-3-[(3-trifluoromethyl)phenyl]oxazolidine-5-carboxamide (13d)
1H NMR (400 MHz, CDCl3) δ 7.68 (t, J = 8.0 Hz, 2H), 7.54 (t, J = 8.0 Hz, 1H), 7.46 (d, J = 8.0 Hz, 1H), 7.21 (d, J = 7.2 Hz, 2H), 7.15 (t, J = 8.0 Hz, 3H), 7.09 (d, J = 7.2 Hz, 2H), 6.99–6.93 (m, 3H), 6.86 (t, J = 7.2 Hz, 1H), 6.48 (d, J = 8.4 Hz, 1H), 4.77 (dd, J = 10.4, 6.4 Hz, 1H), 4.30 (m, 2H), 4.15 (t, J = 7.6 Hz, 1H), 4.08 (t, J = 9.2 Hz, 1H), 3.82 (m, 1H), 3.40 (dd, J = 8.8, 6.0 Hz, 1H), 2.90–2.76 (m, 4H), 2.09 (m, 1H), 2.0 (s, 3H), 1.78–1.65 (m, 2H), 0.91 (d, J = 6.8 Hz, 3H), 0.89 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.79, 170.93, 168.67, 153.25, 138.14, 137.95, 137.69, 131.66 (d, J = 32.9 Hz), 130.01, 129.60 (2C), 129.57 (2C), 128.66 (2C), 128.45 (2C), 126.76, 126.51, 121.41, 121.30 (m), 115.05 (m), 70.57, 69.99, 59.40, 54.64, 49.05, 48.31, 41.29, 40.08, 38.72, 30.44, 23.36, 19.65, 18.46. HRMS (ESI) m/z calcd for C36H42F3N4O6 [M + H]+, 683.3056; found, 683.3018.
(5S)-N-[(1S,2S,4S)-4-[[(2S)-2-(Acetylamino)-3-methyl-1-oxobutyl]amino]-2-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]-3-(3-acetylphenyl)-2-oxo-oxazolidine-5-carboxamide (13e)
1H NMR (400 MHz, CDCl3) δ 7.93 (br s, 1H), 7.83–7.76 (m, 2H), 7.53 (t, J = 8.0 Hz, 2H), 7.22 (d, J = 7.2 Hz, 2H), 7.17–7.10 (m, 5H), 7.0 (t, J = 7.6 Hz, 2H), 6.94–6.87 (m, 2H), 6.77 (d, J = 7.6 Hz, 1H), 6.35 (d, J = 8.0 Hz, 1H), 4.77 (dd, J = 9.6, 6.0 Hz, 1H), 4.28 (m, 2H), 4.17–4.08 (m, 2H), 3.80 (m, 1H), 3.47 (dd, J = 9.2, 6.4 Hz, 1H), 2.88 (m, 2H), 2.80 (m, 2H), 2.64 (s, 3H), 2.09 (m, 1H), 2.0 (s, 3H), 1.76–1.65 (m, 2H), 0.91 (d, J = 6.4 Hz, 3 H), 0.89 (d, J = 7.2 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 197.64, 171.78, 170.92, 168.73, 153.48, 138.13, 138.10, 138.02, 137.75, 129.74, 129.65 (2C), 129.59 (2C), 128.62 (2C), 128.47 (2C), 126.71, 126.56, 124.76, 122.99, 117.63, 70.44, 70.07, 59.36, 54.62, 48.93, 48.43, 41.25, 39.93, 38.72, 30.57, 26.94, 23.36, 19.66, 18.51. HRMS (ESI) m/z calcd for C37H45N4O7 [M + H]+, 657.3288; found, 657.3283.
(5S)-N-[(1S,2S,4S)-2-Hydroxy-4-[[(2S)-3-methyl-1-oxo-2-(tetrahydro-2-oxo-1(2H)-pyrimidinyl)butyl]amino]-5-phenyl-1-(phenylmethyl)pentyl]-2-oxo-3-phenyloxazolidine-5-carboxamide (14a)
1H NMR (400 MHz, CDCl3) δ 7.74 (br d, J = 8.8 Hz, 1H), 7.46–7.35 (m, 4H), 7.27 (m, 2H), 7.22–7.12 (m, 5H), 6.99 (t, J = 7.6 Hz, 2H), 6.80 (t, J = 7.6 Hz, 1H), 5.96 (br s, 1H), 5.37 (br s, 1H), 4.79 (dd, J = 10.4, 6.4 Hz, 1H), 4.58 (m, 1H), 4.37 (m, 1H), 4.22 (d, J = 10.8 Hz, 1H), 4.06 (t, J = 9.6 Hz, 1H), 3.89 (m, 1H), 3.37 (dd, J = 9.2, 6.0 Hz, 1H), 3.30–3.23 (m, 2H), 3.16–3.01 (m, 3H), 2.87 (dd, J = 13.6, 5.6 Hz, 1H), 2.78 (dd, J = 13.2, 5.2 Hz, 1H), 2.70 (dd, J = 13.2, 8.0 Hz, 1H), 2.26 (m, 1H), 1.81–1.76 (m, 3H), 1.60 (m, 2H), 0.92 (d, J = 6.8 Hz, 3H), 0.86 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.27, 168.98, 156.99, 154.05, 138.48, 138.42, 137.54, 129.82 (2C), 129.62 (2C), 129.33 (2C), 128.44 (2C), 128.40 (2C), 126.38, 126.18, 124.84, 118.60 (2C), 71.23, 70.04, 63.53, 53.60, 48.75, 48.36, 42.06, 40.88, 40.28, 38.53, 31.81, 25.90, 21.81, 20.01, 18.97. HRMS (ESI) m/z calcd for C37H46N5O6 [M + H]+, 656.3448; found, 656.3435.
(5S)-N-[(1S,2S,4S)-4-[[(2S)-3,3-Dimethyl-2-N-(methoxycarbonyl)-1-oxo-butyl]amino]-2-hydroxy-5-phenyl-1-(phenylmethyl)-pentyl]-2-oxo-3-phenyloxazolidine-5-carboxamide (15a)
1H NMR (400 MHz, CDCl3) δ 7.47–7.38 (m, 4H), 7.28–7.24 (m, 2H), 7.20–7.17 (m, 2 H), 7.14–7.10 (m, 4 H), 7.03 (t, J = 8.0 Hz, 2 H), 6.93 (m, 1H), 6.75 (d, J = 12.0 Hz, 1H), 6.02 (d, J = 8.0 Hz, 1H), 5.30 (br d, J = 8.0 Hz, 1H), 4.77 (dd, J = 12.0, 8.0 Hz, 1H), 4.25–4.16 (m, 2H), 4.08 (t, J = 9.6 Hz, 1H), 3.78–3.73 (m, 2H), 3.69 (s, 3H), 3.48 (dd, J = 9.2, 6.0 Hz, 1H), 2.88–2.78 (m, 4H), 1.73 (m, 1H), 1.60 (m 1H), 0.93 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 171.19, 168.97, 157.33, 153.27, 137.88, 137.61, 137.32, 129.51 (2C), 129.48 (2C), 129.36 (2C), 128.83 (2C), 128.56 (2C), 126.97, 126.64, 124.83, 118.53 (2C), 70.69, 69.95, 63.62, 54.91, 52.77, 49.33, 48.45, 41.66, 40.41, 38.69, 34.42, 26.78 (3C). HRMS (ESI) m/z calcd for C36H45N4O7 [M + H]+, 645.3288; found, 645.3295.
(5S)-N-[(1S,2S,4S)-4-[(2,6-Dimethylphenoxyacetyl)]amino]-3-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]-2-oxo-3-phenyloxazolidine-5-carboxamide (16a)
1H NMR (400 MHz, CDCl3) δ 7.46 (m, 2H), 7.41 (m, 2H), 7.29 (m, 2H), 7.24–7.19 (m, 4H), 7.17–7.14 (m, 2H), 7.05 (t, J = 7.2 Hz, 2H), 7.01–6.93 (m, 5H), 6.82 (d, J = 9.6 Hz, 1H), 4.83 (dd, J = 10.0, 6.0 Hz, 1H), 4.39 (m, 1H), 4.27 (m, 1H), 4.27 (d, J = 15.2 Hz, 1H, overlapping signal), 4.12–4.07 (m, 2H), 3.86 (m, 1H), 3.73 (d, J = 4.4 Hz, 1H), 3.50 (dd, J = 9.6, 6.4 Hz, 1H), 3.01–2.83 (m, 4H), 2.13 (s, 6H), 1.85–1.75 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 169.54, 168.99, 154.22, 153.23, 137.93, 137.64, 137.22, 130.67, 129.57 (2C), 129.43 (2C), 129.36 (2C), 129.34 (2C), 128.95 (2C), 128.57 (2C), 127.13, 126.63, 125.04, 124.79, 118.49 (2C), 70.79, 70.28, 69.97, 54.85, 48.95, 48.46, 41.83, 41.39, 38.84, 16.39 (2C). HRMS (ESI) m/z calcd for C38H42N3O6 [M + H]+, 636.3074; found, 636.3094.
(5S)-3-(3-Acetylphenyl)-N-[(1S,2S,4S)-4-[(2,6-Dimethylphenoxyacetyl)]amino]-3-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]-2-oxo-oxazolidine-5-carboxamide (16e)
1H NMR (400 MHz, CDCl3) δ 7.92 (t, J = 2.4 Hz, 1H), 7.84 (m, 1H), 7.76 (m, 1H), 7.52 (d, J = 7.6 Hz, 1H), 7.31–7.27 (m, 2H), 7.24–7.19 (m, 3H), 7.15 (dd, J = 8.8, 1.6 Hz, 2H), 7.04 (t, J = 7.6 Hz, 2H), 7.01–6.90 (m, 5H), 6.81 (d, J = 10.0 Hz, 1H), 4.86 (dd, J = 9.6, 6.4 Hz, 1H), 4.39 (m, 1H), 4.31 (m, 1H), 4.27 (d, J = 15.2 Hz, 1H, overlapping singal), 4.15–4.08 (m, 2H), 3.89–3.82 (m, 2H), 3.53 (dd, J = 9.6, 6.4 Hz, 1H), 3.02–2.83 (m, 4H), 2.65 (s, 3H), 2.13 (s, 6H), 1.85–1.75 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 197.67, 169.59, 168.74, 154.21, 153.20, 138.22, 138.08, 137.94, 137.20, 130.66, 129.70, 129.60 (2C), 129.42 (2C), 129.34 (2C), 128.97 (2C), 128.55 (2C), 127.15, 126.62, 125.06, 124.66, 123.01, 117.59 70.76, 70.27, 70.04, 54.81, 48.95, 48.37, 41.77, 41.43, 38.84, 26.98, 16.39 (2C). HRMS (ESI) m/z calcd for C40H44N3O7 [M + H]+, 678.3179; found, 678.3204.
(5S)-3-(4-Acetylphenyl)-N-[(1S,2S,4S)-4-[(2,6-Dimethylphenoxyacetyl)]amino]-3-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]-2-oxo-oxazolidine-5-carboxamide (16f)
1H NMR (400 MHz, CDCl3) δ 8.01 (m, 2H), 7.57 (m, 2H), 7.29 (m, 2H), 7.24–7.19 (m, 3H), 7.14 (dd, J = 8.0, 1.2 Hz, 2H), 7.03–6.94 (m, 6H), 6.88 (dt, J = 8.4, 1.2 Hz, 1H), 6.79 (d, J = 10.0 Hz, 1H), 4.85 (dd, J = 9.6, 6.0 Hz, 1H), 4.39 (m, 1H), 4.30 (m, 1H), 4.27 (d, J = 15.2 Hz, 1H), 4.14–4.09 (m, 2H), 3.38 (m, 1H), 3.84 (br s, 1H), 3.49 (dd, J = 9.2, 6.4 Hz, 1H), 3.02–2.83 (m, 4H), 2.62 (s, 3H), 2.13 (s, 6H), 1.86–1.75 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 197.07, 169.59, 168.63, 154.21, 152.86, 141.69, 137.93, 137.19, 133.16, 130.65, 129.88 (2C), 129.59 (2C), 129.42 (2C), 129.36 (2C), 128.98 (2C), 128.50 (2C), 127.16, 126.61, 125.07, 117.61 (2C), 70.78, 70.27, 69.95, 54.84, 48.91, 48.16, 41.75, 41.43, 38.86, 26.75, 16.39 (2C). HRMS (ESI) m/z calcd for C40H44N3O7 [M + H]+, 678.3179; found, 678.3206.
(5R)-N-[(1S,3S,4S)-4-[(2,6-Dimethylphenoxyacetyl)]amino]-3-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]-2-oxo-3-phenyloxazolidine-5-carboxamide (21a)
1H NMR (400 MHz, CDCl3) δ 7.50–7.48 (m, 2H), 7.39–7.35 (m, 2H), 7.28–7.23 (m, 2H), 7.21–7.17 (m, 6H), 7.16–7.11 (m, 4H), 6.97–6.92 (m, 3H) 6.87 (d, J = 8.4 Hz, 1H), 4.83 (dd, J = 9.6, 5.6 Hz, 1H), 4.22 (m, 1H), 4.20–4.15 (m, 2H), 4.18 (s, overlapping signal, 2H), 4.08 (dd, J = 9.2, 5.6 Hz, 1H), 3.69 (m, 1H), 3.19 (d, J = 5.2 Hz, 1H), 2.91 (dd, J = 7.6, 2.0 Hz, 2H), 2.86 (t, J = 6.0 Hz, 2H), 2.13 (s, 6H), 1.76 (m, 1H), 1.69 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 169.38, 168.94, 154.43, 153.11, 138.06, 137.53, 137.18, 130.53, 129.45 (4C), 129.30 (2C), 128.86 (2C), 128.81 (2C), 127.09, 126.83, 125.05, 124.92, 118.26 (2C), 70.45, 70.27, 70.06, 54.84, 49.98, 48.35, 41.57, 39.61, 38.23, 16.42 (2C). HRMS (ESI) m/z calcd for C38H42N3O6 [M + H]+, 636.3074; found, 636.3081.
(5S)-N-[(1S,3S,4S)-4-[(2,6-Dimethylphenoxyacetyl)]amino]-3-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]-2-oxo-3-phenyloxazolidine-5-carboxamide (22a)
1H NMR (400 MHz, CDCl3) δ 7.46–7.38 (m, 4H), 7.32–7.24 (m, 4H), 7.22–7.17 (m, 2H), 7.06 (d, J = 4.0 Hz, 4H), 7.01–6.92 (m, 4H), 6.61 (d, J = 9.2 Hz, 1H), 4.76 (dd, J = 10.4, 6.0 Hz, 1H), 4.34 (m, 1H), 4.28 (m, 1H), 4.26 (AB d, J = 15.2 Hz, 1H), 4.19 (AB d, J = 15.2 Hz, 1H), 4.12 (t, J = 9.6 Hz, 1H), 3.82 (m, 1H), 3.63 (dd, J = 9.6, 6.0 Hz, 1H), 3.27 (d, J = 5.2 Hz, 1H), 3.01 (d, J = 8.0 Hz, 2H), 2.91 (dd, J = 13.6, 5.6 1H), 2.67 (dd, J = 13.6, 8.4 Hz, 1H), 2.18 (s, 6H), 1.84–1.70 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 169.38, 168.94, 154.43, 153.11, 138.06, 137.53, 137.18, 130.61, 129.52 (2C), 129.38 (4C), 129.35 (2C), 128.84 (2C), 128.73 (2C), 126.92, 126.85, 124.98, 124.91, 118.56 (2C), 70.47, 70.08, 69.93, 54.67, 49.33, 48.42, 41.87, 40.23, 38.28, 16.47 (2C). HRMS (ESI) m/z calcd for C38H42N3O6 [M + H]+, 636.3074; found, 636.3120.
(5S)-N-[(1S,3S,4S)-4-[(2,6-Dimethylphenoxyacetyl)]amino]-3-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]-2-oxo-3-[(3-trifluoromethyl)phenyl]oxazolidine-5-carboxamide (22d)
1H NMR (400 MHz, CDCl3) δ 7.69 (m, 1H), 7.66 (d, J = 8.4 Hz, 1H), 7.52 (t, J = 7.2 Hz, 1H), 7.44 (d, J = 7.6 Hz, 1H), 7.32–7.20 (m, 5H) 7.08–6.93 (m, 8H), 6.89 (t, J = 7.2 Hz, 1H), 6.73 (d, J = 8.8 Hz, 1H), 4.78 (dd, J = 10.0, 5.6 Hz, 1H), 4.40 (m, 1H), 4.30 (m, 1H), 4.26 (AB d, J = 14.8 Hz, 1H), 4.19 (AB d, J = 14.8 Hz, 1H), 4.10 (t, J = 9.6 Hz, 1H), 3.85 (m, 1H), 3.59 (dd, J = 9.6, 6.0 Hz, 1H), 3.01 (d, J = 8.0 Hz, 2H), 2.93 (dd, J = 13.6, 5.6 Hz, 1H), 2.67 (dd, J = 13.6, 9.6 Hz, 1H), 2.16 (s, 6H), 1.86–1.74 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 169.47, 168.59, 154.40, 152.96, 138.51, 138.07, 137.35, 131.86 (m), 130.60, 129.97, 129.52 (2C), 129.40 (2C), 129.35 (2C), 128.83 (2C), 128.64 (2C), 126.86, 126.78, 125.0, 121.38, 121.34 (m), 115.53 (m), 70.45, 70.04, 70.0, 54.72, 49.24, 48.23, 41.76, 40.17, 38.24, 16.45 (2C). HRMS (ESI) m/z calcd for C39H41F3N3O6 [M + H]+, 704.2947; found, 704.2948.
(5S)-N-[(1S,3S,4S)-4-[[2-(Benzo[1,3]-dioxol-5-yl)]acetyl]amino]-3-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]-2-oxo-3-phenyloxazolidine-5-carboxamide (23a)
1H NMR (400 MHz, CDCl3) δ 7.47–7.43 (m, 3H), 7.42–7.37 (m, 2H), 7.24–7.20 (m, 4H), 7.17–7.11 (m, 3H), 7.05 (t, J = 7.2 Hz, 2H), 6.94 (m, 3H), 6.90 (d, J = 10.0 Hz, 1H), 6.74 (d, J = 8.0 Hz, 1H), 6.53 (s, 1H), 5.97 (dd, J = 4.8, 1.6 Hz, 2H), 5.54 (d, J = 8.4 Hz, 1H), 4.81 (dd, J = 9.6, 6.0 Hz, 1H), 4.24–4.17 (m, 2H), 4.08 (m, 2H), 3.78 (m, 1H), 3.50 (dd, J = 9.2, 6.4 Hz, 1H), 3.39 (s, 2H), 2.90–2.79 (m, 3H), 1.62 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 172.08, 168.98, 153.31, 148.33, 147.23, 138.05, 137.66, 137.09, 129.60 (2C), 129.39 (2C), 129.36 (2C), 128.82 (2C), 128.54 (2C), 128.0, 126.98, 126.60, 124.79, 123.02, 118.52 (2C), 110.11, 108.96, 101.41, 70.46, 70.02, 54.96, 49.15, 48.48, 43.52, 41.22, 40.91, 38.78. HRMS (ESI) m/z calcd for C37H38N3O7 [M + H]+, 636.2710; found, 636.2743.
[(1S,2S,4S)-2-Hydroxy-4-[[(5R)-(2-oxo-3-phenyloxazolidine)-5-carbonyl]amino]-5-phenyl-1-(phenylmethyl)pentyl]carbamic Acid (3S)-Tetrahydro-3-furanyl Ester (24a)
1H NMR (400 MHz, CDCl3) δ 7.52–7.48 (m, 2H), 7.40 (t, J = 7.6 Hz, 2H), 7.28–7.24 (m, 2H), 7.22–7.16 (m, 5H), 7.14–7.10 (m, 4H), 7.02 (d, J = 6.8 Hz, 1H), 6.91 (t, J = 9.2 Hz, 1H), 5.13 (m, 1H), 5.0 (d, J = 9.6 Hz, 1H), 4.81 (dd, J = 9.6, 6.0 Hz, 1H), 4.24–4.17 (m, 2H), 4.13–4.05 (m, 2H), 3.86–3.66 (m, 7H), 3.57 (br s, 1H), 2.91 (d, J = 8.0 Hz, 1H), 2.83 (t, J = 5.6 Hz, 1H), 2.67 (m, 1H), 2.07 (m, 1H), 1.68 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 168.91, 156.35, 153.43, 137.96, 137.56, 137.22, 129.47 (2C), 129.45 (2C), 129.42 (2C), 128.79 (2C), 128.76 (2), 126.92, 126.74, 125.08, 118.64 (2C), 75.50, 73.56, 70.47, 69.80, 67.18, 55.34, 49.91, 48.32, 41.45, 39.28, 38.18, 32.98; HRMS (ESI) m/z calcd for C33H38N3O7 [M + H]+, 588.2710; found, 588.2690.
[(1S,2S,4S)-2-Hydroxy-4-[[(5S)-[(2-oxo-3-[(3-trifluoromethyl)-phenyl]oxazolidine)]-5-carbonyl]amino]-5-phenyl-1-(phenylmethyl) pentyl]carbamic Acid (Pyridine-3-yl)-methyl Ester (25d)
1H NMR (400 MHz, CDCl3) δ 8.56 (m, 2H), 7.71–7.61 (m, 3H), 7.52 (m, 1H), 7.45 (d, J = 7.6 Hz, 1H), 7.30–7.26 (m, 3H), 7.21 (t, J = 6.4 Hz, 2H), 7.13 (t, J 7.2 = Hz, 2H), 7.04–6.98 (m, 4H), 6.90 (d, J = 7.6 Hz, 1H), 6.61 (d, J = 8.8 Hz, 1H), 5.08 (s, 2H), 4.99 (d, J = 8.4 Hz, 1H), 4.77 (dd, J = 9.6, 5.2 Hz, 1H), 4.29 (m, 1H), 4.14–4.03 (m, 2H), 3.80 (br s, 1H), 3.47 (dd, J = 9.2, 6.4 Hz, 1H), 2.90–2.80 (m, 4H), 1.76–1.72 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 168.72, 156.49, 153.0, 149.53, 138.21, 138.06, 137.76, 137.20, 136.07, 132.34, 131.69, 129.97, 129.51 (2C), 129.34 (2C), 128.80 (2C), 128.54 (2C), 126.99, 129.78, 123.69, 121.41, 121.29, 115.12, 70.69, 69.97, 64.39, 54.59, 49.23, 48.21, 41.76, 40.17, 38.69. HRMS (ESI) m/z calcd for C36H36F3N4O6 [M + H]+, 677.2587; found, 677.2561.
(6S,7S)-2-Methyl-4-[[N-(methoxycarbonyl)-L-tert-leucinyl]-amino]-6-hydroxy-7-[[(5S)-(2-oxo-3-phenyloxazolidine)-5-carbonyl] amino]-2-azaoctane (32)
1H NMR (400 MHz, CDCl3) δ 7.45–7.37 (m, 4H), 7.19–7.15 (m, 1H), 7.12 (d, J = 7.6 Hz, 2H),7.01–6.94 (m, 2H), 6.88 (t, J = 7.2, 1H), 6.80 (br s, 1H), 5.43 (d, J = 9.2, 1H), 4.80 (dd, J = 10.0, 6.4 1H), 4.71 (br s, 1H), 4.17 (m, 1H), 4.03 (t, J = 9.2 Hz, 1H), 3.74 (d, J = 9.2 Hz, 1H), 3.66 (s, 3H), 3.63 (d, J = 7.2 Hz, 1H), 3.36 (t, J = 6.8 Hz, 1H), 2.91–2.86 (m, 2H), 2.63 (d, J = 6.8 Hz, 2H), 2.57 (dd, J = 12.0, 6.0 Hz, 1H), 2.47 (dd, J = 12.0, 8.4 Hz, 1H), 2.01 (br s, 1H), 1.70 (m, 1H), 1.0 (s, 9H), 0.96 (d, J = 6.8 Hz, 3H), 0.89 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.20, 168.95, 157.36, 153.22, 137.98, 137.67, 129.60 (2C), 129.32 (2C), 128.45, 126.55 (2C), 124.72, 118.48 (2C), 69.94, 68.34, 66.94, 63.17, 61.77, 52.73, 52.01, 48.38, 39.24, 34.47, 26.81, 26.75 (3C), 20.75, 20.59. HRMS (ESI) m/z calcd for C32H46N5O7 [M + H]+, 612.3397; found, 612.3330.
(6S,7S)-2-Methyl-4-[[(5S)-(2-oxo-3-phenyloxazolidine)-5-carbonyl]amino]-6-hydroxy-7-[[N-(methoxycarbonyl)-L-tert-leucinyl]amino]-8-phenyl-2-azaoctane (35)
1H NMR (400 MHz, CDCl3) δ 7.51–7.48 (m, 2H), 7.45–7.41 (m, 2H), 7.26–7.20 (m, 1H), 7.14 (t, J = 9.2 Hz, 3H), 7.06 (t, J = 7.6 Hz, 2H), 6.87 (t, J = 7.2 Hz, 1H), 6.38 (d, J = 8.4 Hz, 1H), 5.21 (d, J = 8.4 Hz, 1H), 4.99 (dd, J = 9.6, 6.8 Hz, 1H), 4.49 (br s, 1H), 4.28 (t, J = 9.2 Hz, 1H), 4.02 (q, J = 8.4 Hz, 1H), 3.93 (t, J = 8.0 Hz, 1H), 3.79 (d, J = 8.8 Hz, 1H), 3.64 (s, 3H), 3.42 (d, J = 9.2 Hz, 1H), 2.90 (d, J = 7.6 Hz, 2H), 2.72 (t, J = 10.8 Hz, 1H), 2.58–2.51 (m, 2H), 2.44 (dd, J = 12.4, 8.8 Hz, 1H), 1.66 (m, 1H), 1.52 (m, 1H), 0.95 (d, J = 6.8 Hz, 3H), 0.93 (s, 9H), 0.89 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.92, 168.09, 157.06, 153.11, 138.16, 137.39, 129.50 (2C), 129.46 (2C), 128.48 (2C), 126.59, 125.24, 118.86 (2C), 70.01, 66.94, 66.80, 63.74, 62.53, 52.55, 52.18, 48.59, 38.44, 34.52, 26.83 (3C), 26.64, 20.72, 20.58. HRMS (ESI) m/z calcd for C32H46N5O7 [M + H]+, 612.3397; found, 612.3369.
(4S,5S)-1-[4-Pyridin-2-yl)phenyl]-2,5-bis-[[(5S)-(2-oxo-3-phenyloxazolidine)-5-carbonyl]amino]-4-hydroxy-6-phenyl-2-azahexane (39)
1H NMR (400 MHz, CDCl3) δ 8.67 (m, 1H), 7.91 (d, J = 8.8 Hz, 2H), 7.73 (m, 1H), 7.63 (d, J = 8.4 Hz, 1H), 7.43–7.34 (m, 8H), 7.28–7.22 (m, 3H), 7.19–7.14 (m, 4H), 7.08 (t, J = 7.6 Hz, 2H), 6.87 (t, J = 7.2 Hz, 1H), 6.82 (d, J = 10.0 Hz, 1H), 4.79–4.73 (m, 2H), 4.63 (br s, 1H), 4.20–4.15 (m, 2H), 4.10–4.02 (m, 3H), 3.84 (dd, J = 9.2, 6.8 Hz, 1H), 3.68 (br d, J = 8.4 Hz, 1H), 3.50 (dd, J = 9.2, 6.8 Hz, 1H), 2.94 (d, J = 7.6 Hz, 2H), 2.79–2.69 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 168.56, 168.20, 156.94, 153.13, 152.82, 149.91, 139.56, 137.24, 137.02 (2C), 136.91, 135.70, 129.58 (2C), 129.48 (2C), 129.44 (2C), 129.40, 128.73 (2C), 127.61, 127.39 (2C), 127.08, 125.20, 125.17, 122.51, 120.76, 118.69 (2C), 69.97, 69.86, 68.07, 62.56, 61.12, 52.65, 48.30, 48.23, 38.39. HRMS (ESI) m/z calcd for C42H41N6O7 [M + H]+, 741.3037; found, 740.3023.
(4S,5S)-1-[4-Pyridin-2-yl)phenyl]-2,5-bis-[[(5S)-[3-(3-acetylphenyl)-2-oxo-oxazolidine]-5-carbonyl]amino]-4-hydroxy-6-phenyl-2-azahexane (40)
1H NMR (400 MHz, CDCl3) δ 8.62 (m, 1H), 7.94–7.88 (m, 4H), 7.84 (m, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.72–7.64 (m, 4H), 7.59 (d, J = 7.6 Hz, 1H), 7.51 (t, J = 8.0 Hz, 1H), 7.44 (d, J = 8.4 Hz, 2H), 7.39 (t, J = 7.6 Hz, 1H), 7.20 (m, 1H), 7.12 (d, J = 7.2 Hz, 2H), 6.97–6.91 (m, 3H), 6.77 (t, J = 7.6 Hz, 1H), 4.87–4.81 (m, 2H), 4.53 (br s, 1H), 4.27–4.19 (m, 2H), 4.13–4.04 (m, 3H), 3.90 (dd, J = 9.2, 6.4 Hz, 1H), 3.73 (br d, J = 7.6 Hz, 1H), 3.53 (dd, J = 9.2, 6.4 Hz, 1H), 2.93–2.82 (m, 4H), 2.64 (s, 3H), 2.57 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 197.67, 197.47, 168.63, 168.28, 156.85, 153.24, 152.98, 149.85, 139.31, 138.21, 138.07 (2C), 137.90, 137.75, 137.0, 136.27, 129.72 (2C), 129.69 (2C), 129.60 (2C), 128.47, 127.26, 126.62, 124.79, 124.68, 123.06, 122.90, 122.49, 120.68, 117.64, 117.62, 70.08, 69.89, 68.29, 62.34, 61.50, 51.97, 48.31, 48.24, 38.92, 26.98, 26.92. HRMS (ESI) m/z calcd for C46H45N6O9 [M + H]+, 825.3248; found, 825.3234.
HIV-1 Protease Inhibition Assays
The HIV-1 protease inhibitory activities of all new designed inhibitors were determined by FRET method.23,34 Protease substrate, (Arg-Glu(EDANS)-Ser-Gln-Asn-Tyr-Pro-Ile-Val-Gln-Lys(DABCYL)-Arg) was purchased from Molecular Probe. The energy transfer donor (EDANS) and acceptor (DABCYL) dyes are labeled at two ends of the peptide, respectively, to perform FRET. Fluorescence measurements were carried out on a fluorescence spectrophotometer (Photon Technology International) at 30 °C. Excitation and emission wavelengths were set at 340 and 490 nm, respectively. Each reaction was recorded for about 10 min. Wild-type HIV-1 protease (Q7K) and its MDR variants M1 (L10I, G48V, I54V, L63P, V82A), M2 (D30N, L63P, N88D), and M3 (L10I, L63P, A71V, G73S, I84V, L90M) were desalted through PD-10 columns (Amersham Biosciences). Sodium acetate (20 mM, pH 5) was used as elution buffer. Apparent protease concentrations were around 50 nM estimated by UV spectrophotometry at 280 nm. All inhibitors were dissolved in dimethylsulfoxide (DMSO) and diluted to appropriate concentrations. Protease (2 μL) and inhibitor (2 μL) or DMSO were mixed and incubated for 20–30 min at room temperature before initializing substrate cleavage reaction. For all experiments, 150 μL of 1 μM substrate were used in substrate buffer [0.1 M sodium acetate, 1 M sodium chloride, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM dithiothreitol (DTT), 2% DMSO, and 1 mg/mL bovine serum albumin (BSA), with an adjusted pH of 4.7]. Inhibitor binding dissociation constant (Ki) values were obtained by nonlinear regression fitting (GraFit 5, Erithacus software) to the plot of initial velocity as a function of inhibitor concentrations based on the Morrison equation.35 The initial velocities were derived from the linear range of reaction curves.
Cell-Based Antiviral Assay
The antiviral activity of compounds against wild-type HIV-1 virus (HIV-14–3) was determined by the 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay method.36 Briefly, exponentially growing MT-4 cells were infected with 100 CCID50 HIV-1, washed in 1 × PBS solution, and seeded in 96 well plates at 4 × 104 cells per well in a total volume of 200 μL RPMI media (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum. Various concentrations of protease inhibitors were added to each well of the 96 well plates in triplicate at 0.5% DMSO final concentration, mixed well, and incubated for 5 days at 37 °C, 5% CO2. Mock infections were carried out in parallel with all protease inhibitor challenges as negative controls and to examine possible drug toxicity. At the end of incubation, 20 μL of an MTT (Sigma-Aldrich, St Louis, MO) solution (7.5 mg/mL in PBS) was added to all wells and incubated for an additional 4 h, 37 °C, 5% CO2. Minimal volumes of media were removed from each well and solubilization of formazan crystals achieved by the addition of 100 μL 10% Triton X-100 in acidified isopropanol followed by vigorous mixing. The absorbencies were read on a Safire plate reader (Tecan) at 540 and 690 nm (for nonspecific absorption). The percent protection achieved by the given concentration of each compound in HIV-infected cells was calculated, and the dose achieving 50% protection defined as the 50% effective dose (EC50) according to the formula was calculated.36 Grafit software was used for all curve fitting and calculation of subsequent EC50 values.
Protein Crystallography
Protein expression, isolation, and purification were carried out as previously described.33 The protein used for crystallizing LPV, 13e, and 14a was further purified using a Pharmacia Superdex 75 FPLC column. Crystals were set up with a 3-fold molar excess of inhibitors to protease of 1.2–1.8 mg/mL concentration. The hanging drop vapor diffusion method was used for crystallization as previously described.37 The reservoir solution consisted of 126 mM phosphate buffer at pH 6.2, 63 mM sodium citrate and ammonium sulfate in a range of 24 to 29%. Intensity data were collected on an in-house Rigaku X-ray generator equipped with an R-axis IV image plate system. Data were collected at −80 °C and the data processing was carried out using the programs DENZO and ScalePack,38,39 respectively. Data collection statistics are listed in Table 4.
The CCP4i interface to the CCP4 suite40 was used to refine the structures. Structure solution was obtained with the molecular replacement package AMoRe,41 with 1F7A42 as the starting model. The molecular replacement phases were further improved using ARP/wARP43 by building solvent molecules into the unaccounted regions of electron density. Model building was performed using the interactive graphics programs O44 and Coot.45 Conjugate gradient refinement using Refmac546 was performed by incorporating Schomaker and Trueblood tensor formulation of TLS (translation, libration, screw-rotation) parameters.47-49 The working R (Rfactor) and its cross validation (Rfree) were monitored throughout the refinement. The refinement statistics are also shown in Table 4. The graphics were generated using the program PyMOL.50
Structural Analysis
For comparison of two structures, complexes were superimposed using the α-carbon backbone of the relatively immobile terminal domain51 (residues 1–9 and 86–99 of both the monomers) of the protease structure.
Double Difference Plots
The double difference plots are constructed to assess intramolecular shifts in response to ligand binding. Initially, Cαi–Cαj distances ([dij]A) were computed between all of the Cα atoms within a protease dimer of a complex, A. This is repeated for a second complex, B, with which the first structure is compared. The difference in these distances ([Dij] = |[dij]A − [dij]B|) was plotted as a two-dimensional contour plot as a function of residue numbers (i and j) by using “gnuplot” (http://www.gnuplot.info).
Supplementary Material
Acknowledgments
We thank Ellen A. Nalivaika for the expression and purification of wild-type and MDR variants of HIV-1 protease. We also thank Drs. Bruce Tidor, Michael K. Gilson, Ronald Swanstrom, Robert Shafer, and members of the Rana and Schiffer labs for helpful discussions. A generous gift of chiral epoxides from Kaneka U.S.A. is gratefully acknowledged. Some FDA-approved protease inhibitors were obtained through the AIDS Research and Reference Reagent Program, NIAID, NIH. This work was supported in part by a National Institutes of Health grant (GM66524).
Footnotes
Abbreviations: EDCI, 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide hydrochloride; HOBt, N-hydroxybenzotriazole hydrate; DIPEA, diisopropylethylamine; FRET, fluorescence resonance energy transfer.
Supporting Information Available: Detailed procedures for the coupling reactions and results of HPLC analysis of final compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Roberts NA, Martin JA, Kinchington D, Broadhurst AV, Craig JC, Duncan IB, Galpin SA, Handa BK, Kay J, Krohn A, Lambert RW, Merrett JH, Mills JS, Parkes KEB, Redshaw S, Ritchie AJ, Taylor DL, Thomas GJ, Machin PJ. Rational design of peptide-based HIV proteinase inhibitors. Science. 1990;248:358–361. doi: 10.1126/science.2183354. [DOI] [PubMed] [Google Scholar]
- 2.Dorsey BD, Levin RB, McDaniel SL, Vacca JP, Guare JP, Darke PL, Zugay JA, Emini EA, Schleif WA, Quintero JC, Lin JH, Chen I-W, Holloway MK, Fitzgerald PMD, Axel MG, Ostovic D, Anderson PS, Huff JR. L-735,524: The design of a potent and orally bioavailable HIV protease inhibitor. J Med Chem. 1994;37:3443–3451. doi: 10.1021/jm00047a001. [DOI] [PubMed] [Google Scholar]
- 3.Kempf DJ, Marsh KC, Denissen JF, McDonald E, Vasavanonda S, Flentge CA, Green BE, Fino L, Park CH, Kong XP, Wideburg NE, Saldivar A, Ruiz L, Kati WM, Sham HL, Robins T, Stewart KD, Hsu A, Plattner JJ, Leonard JM, Norbeck DW. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc Natl Acad Sci U S A. 1995;92:2484–2488. doi: 10.1073/pnas.92.7.2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaldor SW, Kalish VJ, Davies JF, II, Shetty BV, Fritz JE, Appelt K, Burgess JA, Campanale KM, Chirgadze NY, Clawson DK, Dressman BA, Hatch SD, Khalil DA, Kosa MB, Lubbehusen PP, Muesing MA, Patick AK, Reich SH, Su KS, Tatlock JH. Viracept (nelfinavir mesylate, AG1343): A potent, orally bioavailable inhibitor of HIV-1 protease. J Med Chem. 1997;40:3979–3985. doi: 10.1021/jm9704098. [DOI] [PubMed] [Google Scholar]
- 5.Kim EE, Baker CT, Dwyer MD, Murcko MA, Rao BG, Tung RD, Navia MA. Crystal structure of HIV-1 protease in complex with VX-478, a potent and orally bioavailable inhibitor of the enzyme. J Am Chem Soc. 1995;117:1181–1182. [Google Scholar]
- 6.Sham HL, Kempf DJ, Molla A, Marsh KC, Kumar GN, Chen CM, Kati W, Stewart K, Lal R, Hsu A, Betebenner D, Korneyeva M, Vasavanonda S, McDonald E, Saldivar A, Wideburg N, Chen X, Niu P, Park C, Jayanti V, Grabowski B, Granneman GR, Sun E, Japour AJ, Leonard JM, Plattner JJ, Norbeck DW. ABT-378, a highly potent inhibitor of the human immunodeficiency virus protease. Antimicrob Agents Chemother. 1998;42:3218–3224. doi: 10.1128/aac.42.12.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robinson BS, Riccardi KA, Gong YF, Guo Q, Stock DA, Blair WS, Terry BJ, Deminie CA, Djang F, Colonno RJ, Lin PF. BMS-232632, a highly potent human immunodeficiency virus protease inhibitor that can be used in combination with other available antiretroviral agents. Antimicrob Agents Chemother. 2000;44:2093–2099. doi: 10.1128/aac.44.8.2093-2099.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turner SR, Strohbach JW, Tommasi RA, Aristoff PA, Johnson PD, Skulnick HI, Dolak LA, Seest EP, Tomich PK, Bohanon MJ, Horng MM, Lynn JC, Chong KT, Hinshaw RR, Watenpaugh KD, Janakiraman MN, Thaisrivongs S. Tipranavir (PNU-140690): A potent, orally bioavailable nonpeptidic HIV protease inhibitor of the 5,6-dihydro-4-hydroxy-2-pyrone sulfonamide class. J Med Chem. 1998;41:3467–3476. doi: 10.1021/jm9802158. [DOI] [PubMed] [Google Scholar]
- 9.Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King NM, Prabu-Jeyabalan M, Nalivaika EA, Wigerinck P, de Bethune MP, Schiffer CA. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J Virol. 2004;78:12012–12021. doi: 10.1128/JVI.78.21.12012-12021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Surleraux DL, Tahri A, Verschueren WG, Pille GM, de Kock HA, Jonckers TH, Peeters A, De Meyer S, Azijn H, Pauwels R, de Bethune MP, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PB. Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J Med Chem. 2005;48:1813–1822. doi: 10.1021/jm049560p. [DOI] [PubMed] [Google Scholar]
- 12.De Meyer S, Azijn H, Surleraux D, Jochmans D, Tahri A, Pauwels R, Wigerinck P, de Bethune MP. TMC114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates. Antimicrob Agents Chemother. 2005;49:2314–2321. doi: 10.1128/AAC.49.6.2314-2321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wlodawer A, Vondrasek J. Inhibitors of HIV-1 protease: A major success of structure-assisted drug design. Annu Rev Biophys Biomol Struct. 1998;27:249–284. doi: 10.1146/annurev.biophys.27.1.249. [DOI] [PubMed] [Google Scholar]
- 14.Bartlett JA, DeMasi R, Quinn J, Moxham C, Rousseau F. Overview of the effectiveness of triple combination therapy in antiretroviral-naive HIV-1 infected adults. AIDS. 2001;15:1369–1377. doi: 10.1097/00002030-200107270-00006. [DOI] [PubMed] [Google Scholar]
- 15.Gulick RM, Mellors JW, Havlir D, Eron JJ, Meibohm A, Condra JH, Valentine FT, McMahon D, Gonzalez C, Jonas L, Emini EA, Chodakewitz JA, Isaacs R, Richman DD. 3-Year suppression of HIV viremia with indinavir, zidovudine, and lamivudine. Ann Intern Med. 2000;133:35–39. doi: 10.7326/0003-4819-133-1-200007040-00007. [DOI] [PubMed] [Google Scholar]
- 16.Condra JH, Schleif WA, Blahy OM, Gabryelski LJ, Graham DJ, Quintero JC, Rhodes A, Robbins HL, Roth E, Shivaprakash M, Titus D, Yang T, Tepplert H, Squires KE, Deutsch PJ, Emini EA. In vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature. 1995;374:569–571. doi: 10.1038/374569a0. [DOI] [PubMed] [Google Scholar]
- 17.Clavel F, Hance AJ. HIV drug resistance. Structural and thermodynamic basis of resistance to HIV-1 protease inhibition: Implications for inhibitor design. N Engl J Med. 2004;350:1023–1035. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- 18.Miller JF, Andrews CW, Brieger M, Furfine ES, Hale MR, Hanlon MH, Hazen RJ, Kaldor I, McLean EW, Reynolds D. Ultra-potent P1 modified arylsulfonamide HIV protease inhibitors: The discovery of GW0385. Bioorg Med Chem Lett. 2006;16:1788–1794. doi: 10.1016/j.bmcl.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 19.Surleraux DL, de Kock HA, Verschueren WG, Pille GM, Maes LJ, Peeters A, Vendeville S, De Meyer S, Azijn H, Pauwels R, de Bethune MP, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PB. Design of HIV-1 protease inhibitors active on multidrug-resistant virus. J Med Chem. 2005;48:1965–1973. doi: 10.1021/jm049454n. [DOI] [PubMed] [Google Scholar]
- 20.Randolph JT, Huang PP, Flosi WJ, DeGoey D, Klein LL, Yeung CM, Flentge C, Sun M, Zhao C, Dekhtyar T. Synthesis, antiviral activity, and pharmacokinetic evaluation of P3 pyridylmethyl analogs of oximinoarylsulfonyl HIV-1 protease inhibitors. Bioorg Med Chem. 2006;14:4035–4046. doi: 10.1016/j.bmc.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. Structure-based design of novel HIV-1 protease inhibitors to combat drug resistance. J Med Chem. 2006;49:5252–5261. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 22.King NM, Prabu-Jeyabalan M, Nalivaika EA, Schiffer CA. Combating susceptibility to drug resistance: Lessons from HIV-1 protease. Chem Biol. 2004;11:1333–1338. doi: 10.1016/j.chembiol.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 23.Ali A, Reddy GSKK, Cao H, Anjum SG, Nalam MNL, Schiffer CA, Rana TM. Discovery of HIV-1 protease inhibitors with picomolar affinities incorporating N-aryl-oxazolidinone-5-carboxamides as novel P2 ligands. J Med Chem. 2006;49:7342–7356. doi: 10.1021/jm060666p. [DOI] [PubMed] [Google Scholar]
- 24.Haight AR, Stuk TL, Allen MS, Bhagavatula L, Fitzgerald M, Hannick SM, Kerdesky FAJ, Menzia JA, Parekh SI, Robbins TA, Scarpetti D, Tien JHJ. Reduction of an enaminone: Synthesis of the diamino alcohol core of ritonavir. Org Process Res Dev. 1999;3:94–100. [Google Scholar]
- 25.Stoner EJ, Cooper AJ, Dickman DA, Kolaczkowski L, Lallaman JE, Liu JH, Oliver-Shaffer PA, Patel KM, Paterson JB, Plata DJ, Riley DA, Sham HL, Stengel PJ, Tien JHJ. Synthesis of HIV Protease Inhibitor ABT-378 (Lopinavir) Org Process Res Dev. 2000;4:264–269. [Google Scholar]
- 26.Bold G, Fässler A, Capraro HG, Cozens R, Klimkait T, Lazdins J, Mestan J, Poncioni B, Rosel J, Stover D, Tintelnot-Blomley M, Acemoglu F, Beck W, Boss E, Eschbach M, Hurlimann T, Masso E, Roussel S, Ucci-Stoll K, Wyss D, Lang M. New aza-dipeptide analogues as potent and orally absorbed HIV-1 protease inhibitors: Candidates for clinical development. J Med Chem. 1998;41:3387–3401. doi: 10.1021/jm970873c. [DOI] [PubMed] [Google Scholar]
- 27.Ho G-J, Emerson KM, Mathre DJ, Shuman RF, Grabowski EJJ. Carbodiimide-mediated amide formation in a two-phase system. A high-yield and low-racemization procedure for peptide synthesis. J Org Chem. 1995;60:3569–3570. [Google Scholar]
- 28.Fässler A, Bold G, Capraro HG, Cozens R, Mestan J, Poncioni B, Rosel J, Tintelnot-Blomley M, Lang M. Aza-peptide analogs as potent human immunodeficiency virus type-1 protease inhibitors with oral bioavailability. J Med Chem. 1996;39:3203–3216. doi: 10.1021/jm960022p. [DOI] [PubMed] [Google Scholar]
- 29.Xu Z, Singh J, Schwinden MD, Zheng B, Kissick TP, Patel B, Humora MJ, Quiroz F, Dong L, Hsieh DM, Heikes JE, Pudipeddi M, Lindrud MD, Srivastava SK, Kronenthal DR, Mueller RH. Process research and development for an efficient synthesis of the HIV protease inhibitor BMS-232632. Org Process Res Dev. 2002;6:323–328. [Google Scholar]
- 30.Chen X, Kempf DJ, Li L, Sham HL, Vasavanonda S, Wideburg NE, Saldivar A, Marsh KC, McDonald E, Norbeck DW. Synthesis and SAR studies of potent HIV protease inhibitors containing novel dimethylphenoxyl acetates as P2 ligands. Bioorg Med Chem Lett. 2003;13:3657–3660. doi: 10.1016/j.bmcl.2003.08.043. [DOI] [PubMed] [Google Scholar]
- 31.Wu TD, Schiffer CA, Gonzales MJ, Taylor J, Kantor R, Chou S, Israelski D, Zolopa AR, Fessel WJ, Shafer RW. Mutation patterns and structural correlates in human immunodeficiency virus type 1 protease following different protease inhibitor treatments. J Virol. 2003;77:4836–4847. doi: 10.1128/JVI.77.8.4836-4847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stoll V, Qin W, Stewart KD, Jakob C, Park C, Walter K, Simmer RL, Helfrich R, Bussiere D, Kao J, Kempf D, Sham HL, Norbeck DW. X-ray crystallographic structure of ABT-378 (Lopinavir) bound to HIV-1 protease. Bioorg Med Chem. 2002;10:2803–2806. doi: 10.1016/s0968-0896(02)00051-2. [DOI] [PubMed] [Google Scholar]
- 33.King NM, Melnick L, Prabu-Jeyabalan M, Nalivaika EA, Yang SS, Gao Y, Nie X, Zepp C, Heefner DL, Schiffer CA. Lack of synergy for inhibitors targeting a multi-drug-resistant HIV-1 protease. Protein Sci. 2002;11:418–429. doi: 10.1110/ps.25502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matayoshi ED, Wang GT, Krafft GA, Erickson J. Novel fluorogenic substrates for assaying retroviral proteases by resonance energy transfer. Science. 1990;247:954–958. doi: 10.1126/science.2106161. [DOI] [PubMed] [Google Scholar]
- 35.Greco WR, Hakala MT. Evaluation of methods for estimating the dissociation constant of tight binding enzyme inhibitors. J Biol Chem. 1979;254:12104–12109. [PubMed] [Google Scholar]
- 36.Pauwels R, Balzarini J, Baba M, Snoeck R, Schols D, Herdewijn P, Desmyter J, De Clercq E, Pauwels R, Balzarini J, Baba M, Snoeck R, Schols D, Herdewijn P, Desmyter J, De Clercq E. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J Virol Methods. 1988;20:309–321. doi: 10.1016/0166-0934(88)90134-6. [DOI] [PubMed] [Google Scholar]
- 37.Prabu-Jeyabalan M, Nalivaika EA, King NM, Schiffer CA. Viability of a drug-resistant human immunodeficiency virus type 1 protease variant: Structural insights for better antiviral therapy. J Virol. 2003;77:1306–1315. doi: 10.1128/JVI.77.2.1306-1315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Minor W. XdisplayF. Purdue University; West Lafayette, Indiana: 1993. [Google Scholar]
- 39.Otwinowski Z, Minor W, Charles W Carter., Jr . Methods Enzymol. Vol. 276. Academic Press; New York: 1997. Processing of X-ray diffraction data collected in oscillation mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 40.Collaborative Computational Project, Number 4. The CCP4suite: Programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. [Google Scholar]
- 41.Navaza J. AMoRe: An automated package for molecular replacement. Acta Crystallogr. 1994;A50:157–163. [Google Scholar]
- 42.Prabu-Jeyabalan M, Nalivaika E, Schiffer CA. How does a symmetric dimer recognize an asymmetric substrate? A substrate complex of HIV-1 protease. J Mol Biol. 2000;301:1207–1220. doi: 10.1006/jmbi.2000.4018. [DOI] [PubMed] [Google Scholar]
- 43.Morris RJ, Perrakis A, Lamzin VS. ARP/wARP’s model-building algorithms. I. The main chain. Acta Crystallogr. 2002;D58:968–975. doi: 10.1107/s0907444902005462. [DOI] [PubMed] [Google Scholar]
- 44.Jones TA, Bergdoll M, Kjeldgaard MO. Crystallographic and Modeling Methods in Molecular Design. Springer-Verlag Press; Berlin: 1990. A macromolecular modeling environment; pp. 189–195. [Google Scholar]
- 45.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 46.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 47.Schomaker V, Trueblood KN. On the rigid-body motion of molecules in crystals. Acta Crystallogr. 1968;B24:63–76. [Google Scholar]
- 48.Kuriyan J, Weis WI. Rigid protein motion as a model for crystallographic temperature factors. Proc Natl Acad Sci U S A. 1991;88:2773–2777. doi: 10.1073/pnas.88.7.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tickle IJ, Moss DS. IUCr99 Computing School. IUCr; London: 1999. Modelling rigid-body thermal motion in macromolecular crystal structure refinement. [Google Scholar]
- 50.DeLano WL. The PyMOL User’s Manual. DeLano Scientific; San Carlos, CA: 2002. [Google Scholar]
- 51.Rose RB, Craik CS, Stroud RM. Domain flexibility in retroviral proteases: Structural implications for drug resistant mutations. Biochemistry. 1998;37:2607–2621. doi: 10.1021/bi9716074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.