

1. Introduction
The substantial level of inherent modularity present in most small molecules suggests that a generalized building block based approach may be applicable to the laboratory synthesis of many of these compounds. In an idealized form of this approach, stable subunits having all of the required functional groups preinstalled in the correct oxidation states and with the desired stereochemical relationships are linked using only one reaction. As a roadmap for creating a general strategy for small molecule synthesis, we aim to identify common substructural motifs that are prevalent in a wide range of natural products and transform them into shelf-stable building blocks that are compatible with iterative assembly.
In this vein, 1,1-disubstituted olefins are prevalent in small molecule pharmaceuticals1 and natural products derived from a wide range of biosynthetic pathways (Figure 1).2 In addition, they are useful synthetic intermediates. 3 Some common strategies for the synthesis of 1,1-disubstituted olefins include the Wittig reaction of methyltriphenylphosphonium bromide and a ketone,4 the Heck reaction of an organohalide or triflate and monosubstituted olefin,5 and other metal catalyzed couplings between a halide or halide surrogate and an organometallic reagent.6
Figure 1.

1,1-Disubstituted olefins are abundant in pharmaceuticals and natural products.
Of particular interest, the 1,1-disubstituted alkenyl boronate motif is an attractive intermediate for the preparation of 1,1-disubstituted olefins because of the versatility of the vinyl boron moiety. 7 While methods for the synthesis of 1,2-disubstituted alkenyl boronates are abundant,8 the methods for the preparation of 1,1-disubstituted alkenyl boronates have been limited. 9 Recent methods for the preparation of these motifs include the copper catalyzed hydroboration of aryl and heteroaryl substituted alkynes.9a Alternatively, aryl substituted vinyl boronates can be prepared from the corresponding vinyl halide via a Miyaura borylation9c or a lithiation/trap sequence.9d,9e However, the synthesis of the requisite vinyl halide can be challenging and often uses conditions that are not compatible with acid-sensitive functionalities. To address these challenges, we envision that a B-protected 1,1-disubstituted halo boronate building block would be extremely useful for the modular synthesis of targets containing a 1,1-disubstituted olefin motif (Scheme 1).
Scheme 1.
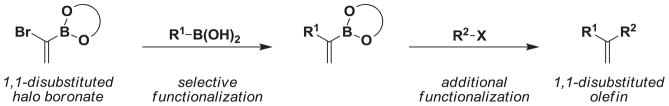
A 1,1-disubstituted halo boronate building block as a useful reagent for the preparation of 1,1-disubstituted olefins.
Along these lines, we are developing a platform of N-methyliminodiacetic acid (MIDA) boronate building blocks.10,11,12,13,14,15,16,17,18,19,20,21,22,23,24 MIDA boronates have many desirable properties that render them exceptionally useful as synthetic intermediates. They are uniformly air-stable, non-toxic, highly crystalline, and monomeric free-flowing solids that are fully compatible with silica gel chromatography. Many methods now exist for preparing MIDA boronates from a wide range of different starting materials, including boronic acids,10,11,12,13,19,25 haloboranes,11,13 boronic esters,16 trialkoxyborate salts,14–17,19 organohalides,19 organolithium reagents,19,23 and Grignard reagents.15,16 The MIDA boronate functional group is inert to anhydrous cross-coupling conditions, yet can be readily transformed into a fully reactive boronic acid or ester using mild conditions.10,14,17 These features enable the simple, efficient, and highly flexible synthesis of a wide range of complex small molecules via iterative cross-coupling of MIDA-protected haloboronic acids.10,11,12,16,17,21,22,24 Finally, a large and growing collection of MIDA boronates are now commercially available.26 Taking advantage of all of these properties, we herein report a 1,1-disubstituted halo MIDA boronate building block, which upon selective functionalization, allows ready access to a variety of 1,1-disubstituted alkenyl boronate motifs with the potential for iterative cross-coupling for the preparation of 1,1-disubstituted olefins.
2. Results
Bromination-elimination reactions provide a convenient method for the synthesis of vinyl halides.27 In particular, 1,1-disubstituted olefins can be prepared from terminal alkenes via this type of transformation.28 With this strategy in mind, vinyl MIDA boronate 1 is an attractive starting point. This building block can easily be prepared on large scale from inexpensive starting materials (Scheme 2), and is now commercially available on both the gram and kilogram scales.29 Bromination of 1 followed by treatment of the resulting vicinal dibromide intermediate with DBU in MeCN provides a very convenient route to (1-bromovinyl)-MIDA boronate 2 in 67% overall yield. Moreover, 2 can be prepared on the decagram scale and isolated in pure form without the use of column chromatography. Building block 2 is also now commercially available.26
Scheme 2.
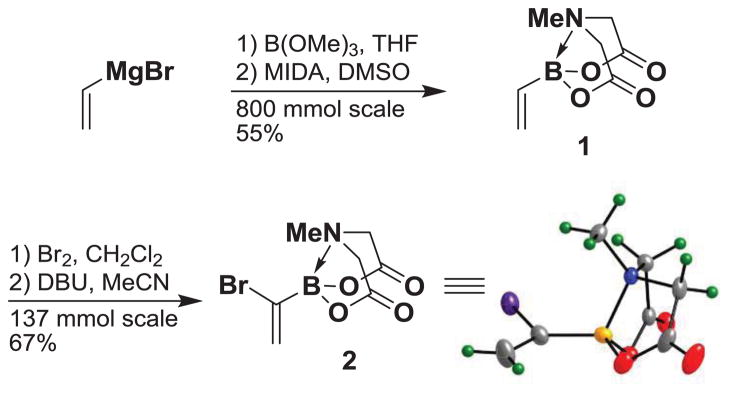
Synthesis of (1-bromovinyl)-MIDA boronate 2.
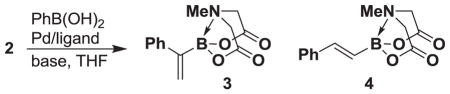
With a readily scalable synthesis of 2 in hand, we have preliminarily explored its utility in the preparation of a range of new 1,1-disubstituted alkenyl boronate building blocks en route to 1,1-disubstituted olefins. In this vein, our initial attempt to couple 2 with phenyl boronic acid under anhydrous cross-coupling conditions surprisingly formed a mixture of products identified as the desired 1,1-disubstituted boronate 3 and the trans-1,2-disubstituted product 4 in a 1:4 ratio (Scheme 3).
Scheme 3.
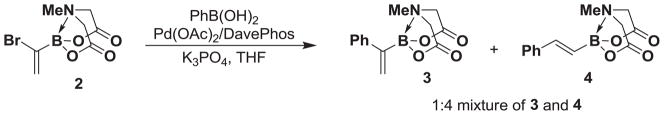
The unoptimized coupling of building block 2 with phenyl boronic acid provides a mixture of products.
We hypothesized that 4 is formed through a competitive reaction pathway involving oxidative addition of the palladium catalyst to 2 followed by β-hydride elimination, reinsertion of the Pd-H species to form a trans-1,2-disubstituted olefin, transmetalation with the boronic acid, and reductive elimination (Figure 2, pathway 2). Guided by this hypothesis, we explored reaction conditions that could limit the β-hydride elimination pathway (pathway 2) and/or accelerate the rate of the desired transmetalation reaction that leads to pathway 1 (Figure 2).
Figure 2.
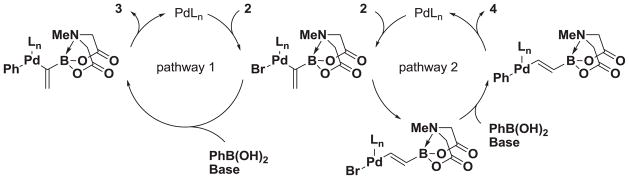
Proposed mechanism for the formation of 1,1-disubstituted product 3 and 1,2-disubstituted product 4.
Along these lines, silver salts have been proposed to increase the rate of transmetalation in the Suzuki-Miyaura cross-coupling reaction.30 To test if this could favor pathway 1 over pathway 2, we employed silver oxide as the base (Table 1, entry 1) in combination with Pd(OAc)2 and tricyclohexylphosphine (Cy3P) and observed 3 as the major product (3:1), although with low conversion (30%). Alternatively, the use of silver carbonate (entry 2) produced 3 in a slightly better ratio (4:1) and with greater conversion (60%). As a second variable, bidentate ligands, such as 1,1′-bis(diphenylphosphino)ferrocene (dppf), have been used to limit β-hydride elimination pathways in cross-coupling reactions.31 Use of PdCl2dppf (entry 3) with silver carbonate provided 3 as the major product (4:1) with greatly improved conversion over previous reaction conditions. A survey of other bidentate phosphine ligands revealed 1,5-bis(diphenylphosphino)pentane (dppp, entry 4) as a ligand that provided 3 in a 10:1 ratio. As a final variable, ancillary ligands were investigated. A survey of various palladium sources revealed that Pd2dba3 combined with 1,6-bis(diphenylphosphino)hexane (dpph, entry 6) provided 3 in a >20:1 ratio and with excellent conversion. Interestingly, during the course of this study, we also found that exclusive formation of the trans product 4 can be achieved in 85% conversion using Pd(OAc)2, SPhos, and K3PO4 in THF (entry 7).
Table 1.
Optimization studies for the coupling of 2.
| ||||
|---|---|---|---|---|
| entry | Pd/ligand | base | % conversiona | 3:4a |
| 1 | Pd(OAc)2/Cy3P | Ag2O | 30 | 3:1 |
| 2 | Pd(OAc)2/Cy3P | Ag2CO3 | 60 | 4:1 |
| 3 | PdCl2dppf | Ag2CO3 | >95 | 4:1 |
| 4 | Pd(OAc)2/dppp | Ag2CO3 | 70 | 10:1 |
| 5 | Pd2dba3/dppp | Ag2CO3 | 70 | 12:1 |
| 6 | Pd2dba3/dpph | Ag2CO3 | >95 | >20:1 |
| 7 | Pd(OAc)2/SPhos | K3PO4 | 85 | 1:20 |
The reaction conversion and ratio of 3:4 was determined based on the 1H NMR of the crude reaction mixture.
These optimized conditions for the formation of 1,1-disubstituted boronate 3 have proven to be scalable. On a 1 mmol scale, cross-coupled product 3 was obtained in 61% isolated yield as a single isomer (Scheme 4). We further explored the utility of 2 as a substrate for iterative cross-coupling (ICC) reactions. The in situ hydrolysis of 3 followed by cross-coupling of the resulting boronic acid with aryl iodide 5 yielded 6 as a single isomer.
Scheme 4.
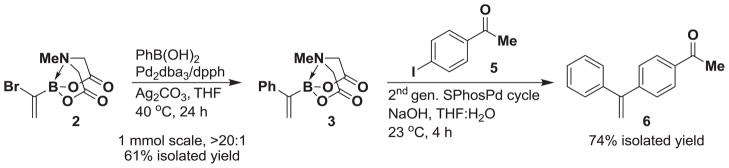
MIDA boronate 2 is a substrate for iterative Suzuki-Miyaura coupling reactions.
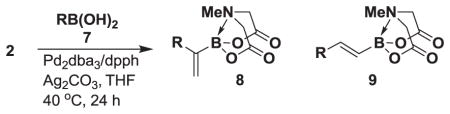
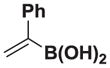
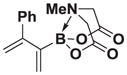
We next tested the generality of the optimized cross-coupling conditions with a variety of aryl, heteroaryl, and vinyl boronic acids (Table 2). Good to moderate conversions were observed in all cases. The coupling of boronic acids with electron neutral (entries 1–3), withdrawing (entries 4–6), and donating (entires 7–8) groups formed the desired product in all cases. Specifically, the coupling of 4-vinylphenylboronic acid 7a and m-tolylboronic acid 7b proceeded with excellent selectivity (>20:1), high conversion (>95%), and a 81% isolated yield for 8a and 90% isolated yield for 8b. Furthermore, boronic acids with ortho substituents, such as 7c, are tolerated, providing the cross-coupled product 8c in >20:1 selectivity and 50% isolated yield. However, the presence of electron withdrawing groups, including 4-methoxycarbonyl, 4-cyano, 4-fluoro, and 4-trifluoromethoxy (entries 4–6), resulted in moderate selectivities (10:1 to 4:1) albeit still with good conversions (70–90%). The electron withdrawing groups on the boronic acid may reduce the rate of the desired transmetalation reaction (Figure 2, pathway 1), therefore making the β-hydride elimination pathway more competitive (pathway 2). In contrast, electron donating groups, including 3,5-dimethoxy and 4-dimethylamino (entries 7–8) provided the cross-coupled products with excellent selectivity (>20:1) and a 66% isolated yield for 8g. Additionally, the coupling of 2-heterocyclic boronic acids, including 2-benzofuranboronic acid 7i and 2-thiopheneboronic acid 7j produced the 1,1-product in >20:1 in both cases and a 82% isolated yield for 8i. Finally, the coupling of vinyl boronic acids also produced the 1,1-product in >20:1 and 76% isolated yield for 8k and 65% isolated yield for 8l.
Table 2.
Substrate scope under the optimized reaction conditions.
| ||||
|---|---|---|---|---|
| entry | 7 | 8 | 8:9 | % conversiona (isolated yield) |
| 1 |
 7a |
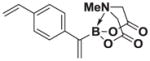 8a |
>20:1 | >95 (81%) |
| 2 |
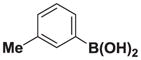 7b |
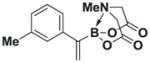 8b |
>20:1 | >95 (90%) |
| 3 |
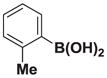 7c |
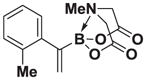 8c |
>20:1 | >95 (50%) |
| 4 |
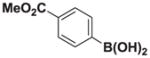 7d |
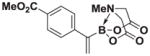 8d |
10:1 | 85 (45%)b |
| 5 |
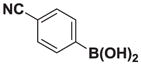 7e |
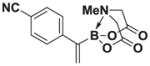 8e |
8:1 | 70 (39%)b |
| 6 |
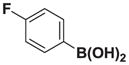 7f |
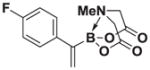 8f |
4:1 | 90 (43%)b |
| 7 |
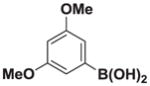 7g |
 8g |
>20:1 | 85 (66%) |
| 8 |
 7h |
 8h |
>20:1 | 50 (28%) |
| 9 |
 7i |
 8i |
>20:1 | 95 (82%) |
| 10 |
 7j |
 8j |
>20:1 | 50 (24%) |
| 11 |
7k |
 8k |
>20:1 | 90 (76%) |
| 12 |
 7l |
 8l |
>20:1 | 80 (65%) |
The reaction conversion and ratio of 8:9 was determined based on the 1H NMR of the crude reaction mixture.
Isolated yield as an inseparable mixture of isomers.
3. Summary and conclusions
Access to a wide range of bifunctional building blocks representing motifs that commonly appear in small molecules stands to greatly increase the efficiency and flexibility of the synthesis of this broadly important class of compounds. The 1,1-disubstituted olefin represents a very important substructure that appears in a wide range of pharmaceuticals and natural products. We herein described a very practical and scalable synthesis of the bifunctional building block 2 and demonstrated its utility for the synthesis of a wide range of 1,1-disubstituted boronates. Collectively, these findings expand the utility of ICC with MIDA boronates as a simple and flexible platform for the efficient synthesis of a wide range of functional small molecules.
4. Experimental
4.1. Materials
Commercial reagents were purchased from Sigma-Aldrich, Fisher Scientific, Alfa Aesar, TCI America, Strem Chemicals Inc., or Frontier Scientific and were used without further purification unless otherwise noted. Solvents were purified via passage through packed columns as described by Pangborn and coworkers32 (THF, Et2O, CH3CN, CH2Cl2: dry neutral alumina; hexane, benzene, and toluene: dry neutral alumina and Q5 reactant; DMSO, DMF: activated molecular sieves). All water was deionized prior to use.
4.2. General experimental procedures
Unless noted, all reactions were performed in flame-dried round bottom or modified Schlenk flasks fitted with rubber septa under a positive pressure of argon or nitrogen. Organic solutions were concentrated via rotary evaporation under reduced pressure with a bath temperature of 35 – 40 °C. Reactions were monitored by analytical thin layer chromatography (TLC) performed using the indicated solvent on E. Merck silica gel 60 F254 plates (0.25mm). Compounds were visualized by exposure to a UV lamp (λ = 254 nm) and/or a solution of basic KMnO4 followed by brief heating using a Varitemp heat gun. MIDA boronates are compatible with standard silica gel chromatography, including standard loading techniques. Column chromatography was performed using standard methods33 or on a Teledyne-Isco CombiFlash Rf purification system using Merck silica gel grade 9385 (60Å, 230–400 mesh). For loading, compounds were adsorbed onto non acid-washed Celite in vacuo from an acetone solution. Specifically, for a 1 g mixture of crude material the sample is dissolved in reagent grade acetone (25 to 50 mL) and to the flask is added Celite 545 Filter Aid (5 to 15 g). The mixture is then concentrated in vacuo to afford a powder, which is then loaded on top of a silica gel column. The procedure is typically repeated with a small amount of acetone (5 mL) and Celite (2 g) to ensure quantitative transfer.
4.3. Structural analysis
1H NMR spectra were recorded at 23 °C on one of the following instruments: Varian Unity 400, Varian Unity 500, Varian Unity Inova 500NB. Chemical shifts (δ) are reported inparts per million (ppm) downfield from tetramethylsilane and referenced to residual protium in the NMR solvent (CHCl3, δ = 7.26; CD2HCN, δ = 1.94, center line; d6-acetone, δ = 2.05, center line) or to added tetramethylsilane (δ = 0.00). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, sept = septet, m = multiplet, b = broad, app = apparent), coupling constant (J) in Hertz (Hz), and integration. 13C NMR spectra were recorded at 23 °C on a Varian Unity 400 or Varian Unity 500. Chemical shifts (δ) are reported in ppm downfield from tetramethylsilane and referenced to carbon resonances in the NMR solvent (CDCl3, δ = 77.0, center line; CD3CN, δ = 1.30, center line; d6-acetone, δ = 29.80, center line) or to added tetramethylsilane (δ = 0.00). Carbons bearing boron substituents were not observed (quadrupolar relaxation). High resolution mass spectra (HRMS) were performed by Furong Sun, Haijun Yao, and Beth Eves at the University of Illinois School of Chemical Sciences Mass Spectrometry Laboratory. X-ray crystallographic analyses were carried out by Dr. Danielle Gray at the University of Illinois George L. Clark X-Ray facility.
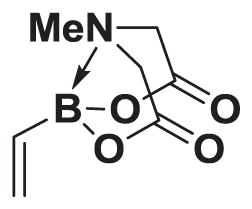
4.4. MIDA boronate 1
To a 3 L 3-neck round bottom flask equipped with a stir bar was added B(OMe)3 (94 mL, 840 mmol) and THF (600 mL). The solution was cooled to −78 °C. Vinylmagnesium bromide (1.0 M in THF, 800 mL, 800 mmol) was added dropwise via cannula over 2 h 45 min. The resulting solution was stirred at −78°C for 15 min, followed by stirring at 23 °C for 2 h 30 min. In a separate 2 L 3-neck round bottom flask equipped with a stir bar, internal thermometer, and distillation apparatus was added dry MIDA (235.0 g, 1.6 mol) and DMSO (600 mL). The solution was heated with an oil bath to an internal temperature of 110 – 115 °C. The borate suspension was added dropwise to the hot MIDA solution via a Teflon cannula dropwise over 2 h 10 min, keeping the internal temperature between 105 and 115 °C. After full addition of the borate solution, the reaction solution was cooled to 23 °C. The resulting solution was transferred to a separatory funnel containing H2O (1 L), brine (1 L), EtOAc (1.5 L) and acetone (1 L). The mixture was shaken and the aqueous layer was removed and extracted with EtOAc:acetone (2:1, 2 × 600 mL). The combined organic layers were washed with H2O (2 × 500 mL). The combined water washes were back extracted with EtOAc:acetone (2:1, 2 × 300 mL). The combined organic phases were dried over MgSO4, filtered, and concentrated in vacuo. The resulting solid was suspended in 300 mL acetone and 4 L Et2O was added to precipitate the product. The resulting solid was collected by vacuum filtration to yield vinyl MIDA boronate 1 as a white solid (81.2 g, 55%). Spectral data for 1 were consistent with those previously reported from our laboratories.13
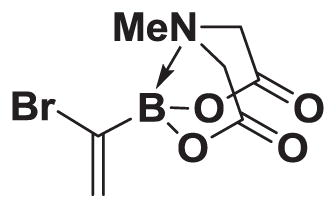
4.5. MIDA boronate 2
To a 2 L round bottom flask equipped with a stir bar and charged with vinyl MIDA boronate 1 (25.0 g, 137 mmol) was added CH2Cl2 (1.2 L). The resulting clear, colorless solution was cooled to 0 °C in an ice bath. Neat bromine (12.5 mL, 239 mmol) was added dropwise over 5 min to give a cloudy orange solution. The solution was warmed to 23 °C over 30 min. The resulting orange solution was concentrated in vacuo to give a yellow solid. Residual bromine was removed by azeotroping with CH2Cl2 (2 × 100 mL). The resulting solid was suspended in MeCN (1 L). 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU, 51.0 mL, 342 mmol) was added in one portion. The resulting mixture stirred at 23 °C for 1 h. The solution was poured into 1 M aq. HCl (1 L) and diluted with EtOAc:acetone (3:1, 1 L). After shaking, the layers were separated. The organic layer was washed with saturated aqueous sodium bisulfite:brine (3:2, 1 × 500 mL) and brine (1 × 250 mL). The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The resulting residue was passed through a plug of silica, eluting with acetone. The resulting solid was suspended in THF (50 mL) onto which Et2O (1.8 L) was layered to precipitate the product. The product 2 was collected by vacuum filtration (24.0 g, 67%).
1H NMR (500 MHz, d6-acetone) δ 6.39 (bs, 1H), 6.19 (bs, 1H), 4.37 (d, J = 17.0 Hz, 2H), 4.16 (d, J = 17.0 Hz, 2H), 3.16 (s, 3H). 13C NMR (125 MHz, d6-acetone) δ 168.4, 129.5, 63.4, 47.2. HRMS (ESI+) calculated for C7H10BBrNO4 (M+H)+: 261.9886. Found: 261.9873. X-ray quality crystals were grown by vapor diffusion of Et2O into a dissolved solution of 2 in acetone.34 MIDA boronate 2 is commercially available.26
4.6. General procedure for Table 1
In a glovebox, to a 7 mL vial containing a stir bar and silver salt (0.4 mmol, 4.0 eq) was added a 0.25 mL aliquot of each of the following three stock solutions: 1) 262 mg MIDA boronate 2 in 2.5 mL THF (0.1 mmol, 1.0 eq of 2 added to each reaction); 2) 183 mg phenylboronic acid in 2.5 mL THF (0.15 mmol, 1.5 eq of boronic acid added to each reaction); 3) A solution of 0.05 mmol palladium salt and 0.2 mmol ligand in 2.5 mL THF was prepared and stirred at 23 °C for 30 min. before adding an aliquot to the reaction vials (0.005 mmol, 0.05 eq of palladium added to each reaction and 0.02 mmol, 0.2 eq of ligand added to each reaction). An additional 0.25 mL of THF was added to each reaction. The vials were capped, removed from the glovebox, and placed in a 40 °C heating block with stirring for 24 h. The crude reaction mixtures were filtered through a plug of Celite, concentrated in vacuo, and analyzed by 1H NMR.
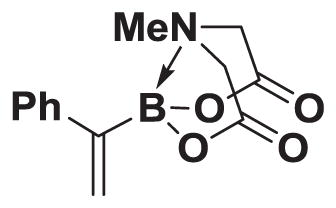
4.7. MIDA boronate 3
Preparation of catalyst solution
In a glovebox, to a 7 mL vial charged with dpph (90.9 mg, 0.20 mmol) and Pd2dba3 (45.8 mg, 0.05 mmol) was added THF (5.0 mL). The solution was stirred at 23 °C for 30 min.
The freshly prepared catalyst solution was used in the following reaction
In a glovebox, to a 20 mL vial with stir bar, charged with MIDA boronate 2 (261.9 mg, 1.0 mmol) was added Ag2CO3 (1.10 g, 4.0 mmol). Phenylboronic acid (182.8 mg, 1.5 mmol) was added as a solution in THF (5.0 mL). The prepared catalyst solution was added in one portion. The vial was sealed with a septum cap and removed from the glovebox. The solution was stirred at 40 °C for 24 h. The reaction mixture was filtered through a pad of Celite, concentrated in vacuo, dry loaded onto Celite from an acetone solution, and purified via flash chromatography on silica (Et2O:MeCN 100:0 → 80:20) to afford MIDA boronate 3 (158.0 mg, 61%).
1H NMR (400 MHz, d6-acetone) δ 7.42–7.38 (m, 2H), 7.33–7.28 (m, 2H), 7.26–7.21 (m, 1H), 5.73 (bd, J = 3.0 Hz, 1H), 5.69 (bs, 1H), 4.23 (d, J = 17.0 Hz, 2H), 3.85 (d, J = 17.0 Hz, 2H), 2.82 (s, 3H). 13 C NMR (125 MHz, d6-acetone) δ 168.9, 145.2, 129.1, 128.3, 128.1, 127.3, 62.6, 47.6. HRMS (ESI+) calculated for C13H15BNO4 (M+H)+: 260.1094. Found: 260.1094. TLC (Et2O:MeCN 6:1) Rf = 0.44, visualized by short wave UV. MIDA boronate 3 is commercially available.26
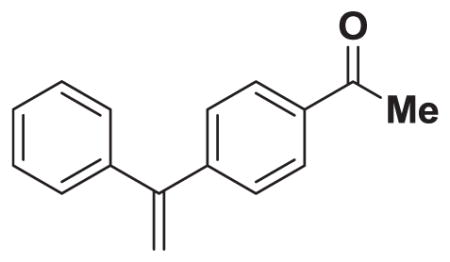
4.8. 1,1-disubstituted olefin 6
In a glovebox, to a 20 mL vial with stir bar, charged with MIDA boronate 3 (100.0 mg, 0.39 mmol, 1.0 eq) was added 4-iodoacetephenone (113.9 mg, 0.46 mmol, 1.2 eq), 2nd gen. SPhosPd cycle (20.9 mg, 0.029 mmol, 0.075 eq), and THF (3.5 mL). The vial was sealed with a septum cap and removed from the glovebox. To the reaction solution was added 1 M aq. NaOH via syringe (2.9 mL, 2.9 mmol). The solution was stirred at 23 °C for 4 h. The reaction mixture was poured into a separatory funnel containing aqueous sodium phosphate buffer (0.5 M, pH 7.0, 10 mL) and diluted with Et2O (10 mL). The mixture was shaken and the layers were separated. The aqueous phase was extracted with Et2O (2 × 10 mL). The combined organics were dried over MgSO4, filtered, and then concentrated in vacuo. The crude material was dry loaded onto Celite from an acetone solution, and purified via flash chromatography on silica (hexane:EtOAc 100:0 → 90:10) to afford 1,1-disubstituted olefin 6 (63.5 mg, 74%). Spectral data for 6 were consistent with those previously reported.35
4.9. General procedure for Table 2
Preparation of catalyst solution
In a glovebox, to a 20 mL vial charged with dpph (136.4 mg, 0.30 mmol) and Pd2dba3 (68.7 mg, 0.075 mmol) was added THF (7.5 mL). The solution was stirred at 23 °C for 30 min.
The freshly prepared catalyst solution was used in the following reactions
In a glovebox, to a 7 mL vial with stir bar, charged with MIDA boronate 2 (26.2 mg, 0.1 mmol) was added Ag2CO3 (110.0 mg, 0.4 mmol). Boronic acid (0.15 mmol) was added, followed by THF (0.50 mL). An aliquot of the prepared catalyst solution (0.50 mL) was added in one portion. The vial was sealed with a septum cap and removed from the glovebox. The solution was stirred at 40 °C for 24 h. The reaction mixture was filtered through a pad of Celite, concentrated in vacuo, and analyzed by 1H NMR. Select samples were dry loaded onto Celite from an acetone solution, and purified via flash chromatography on silica (Et2O:MeCN 100:0 → 80:20) to afford the MIDA boronate 8.
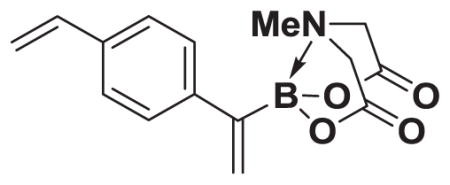
4.10. MIDA boronate 8a
Based on the 1H NMR of the crude reaction mixture, the reaction proceeded to >95% conversion and a >20:1 of 8:9 was obtained. 8a was isolated by column chromatography to provide 57.8 mg (0.25 mmol reaction), 81% yield.
1H NMR (400 MHz, d6-acetone) δ 7.71–7.33 (m, 4H), 6.85–6.68 (m, 2H), 5.90–5.68 (m, 2H), 5.29–5.17 (m, 1H), 4.25 (d, J = 17.0 Hz, 2H), 3.88 (d, J = 17.0 Hz, 2H), 2.81 (s, 3H). 13C NMR (125 MHz, d6-acetone) δ 168.9, 137.4, 136.7, 135.2, 128.5, 128.0, 127.0, 113.7, 62.6, 47.6. HRMS (ESI+) calculated for C15H17BNO4 (M+H)+: 286.1251. Found: 286.1248. TLC (Et2O:MeCN 6:1) Rf = 0.44, visualized by short wave UV.
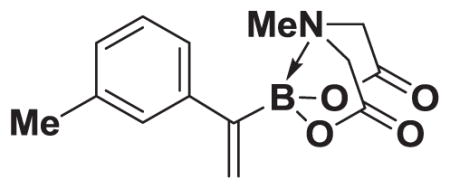
4.11. MIDA boronate 8b
Based on the 1H NMR of the crude reaction mixture, the reaction proceeded to >95% conversion and a >20:1 of 8:9 was obtained. 8b was isolated by column chromatography to provide 61.1 mg (0.25 mmol reaction), 90% yield.
1H NMR (500 MHz, d6-acetone) δ 7.24 (m, 1H), 7.21–7.18 (m, 2H), 7.10–7.05 (m, 1H), 5.73 (bd, J = 3.0 Hz, 1H), 5.68 (bs, 1H), 4.23 (d, J = 17.0 Hz, 2H), 3.83 (d, J = 17.0 Hz, 2H), 2.83 (s, 3H), 2.31 (s, 3H). 13C NMR (125 MHz, d6-acetone) δ 168.7, 145.2, 138.3, 128.9, 128.8, 127.9, 127.6, 125.3, 62.6, 47.4, 21.4. 11B NMR (128 MHz, d6-acetone) δ 11.4. HRMS (ESI+) calculated for C14H17BNO4 (M+H)+: 274.1251. Found: 274.1245. TLC (Et2O:MeCN 6:1) Rf = 0.44, visualized by short wave UV.
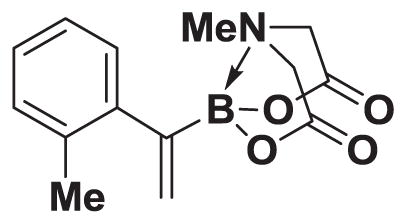
4.12. MIDA boronate 8c
Based on the 1H NMR of the crude reaction mixture, the reaction proceeded to >95% conversion and a >20:1 of 8:9 was obtained. 8c was isolated by column chromatography to provide 68.6 mg (0.50 mmol reaction), 50% yield.
1H NMR (400 MHz, d6-acetone) δ 7.18–7.04 (m, 4H), 5.84 (d, J = 3.5 Hz, 1H), 5.52 (bs), 4.19 (d, J = 17.0 Hz, 2H), 3.88 (d, J = 17.0 Hz, 2H), 3.03 (s, 3H), 2.29 (s, 3H). 13C NMR (125 MHz, d6-acetone) δ 168.7, 144.9, 136.2, 130.9, 128.5, 128.3, 127.0, 126.1, 62.6, 47.4, 20.6. HRMS (ESI+) calculated for C14H17BNO4 (M+H)+: 274.1251. Found: 274.1252. TLC (Et2O:MeCN 6:1) Rf = 0.44, visualized by short wave UV.
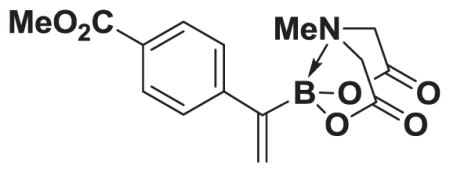
4.13. MIDA boronate 8d
Based on the 1H NMR of the crude reaction mixture, the reaction proceeded to 85% conversion and a 10:1 of 8:9 was obtained. 8d and 9d were isolated by column chromatography as an inseparable mixture of isomers to provide 60.6 mg (0.50 mmol reaction), 45% yield.
1H NMR (400 MHz, d6-acetone) δ 7.70–7.36 (m, 4H), 5.77 (bd, J = 3.0 Hz, 1H), 5.73 (bs, 1H), 4.25 (d, J = 17.0 Hz, 2H), 3.91 (d, J = 17.0 Hz, 2H), 3.85 (s, 3H), 2.81 (s, 3H). 13C NMR (125 MHz, d6-acetone) δ 168.8, 167.1, 134.9, 130.2, 129.2, 129.0, 128.5, 62.7, 52.2, 47.7. HRMS (ESI+) calculated for C15H17BNO6 (M+H)+: 318.1149. Found: 318.1143. TLC (Et2O:MeCN 6:1) Rf = 0.32, visualized by short wave UV.
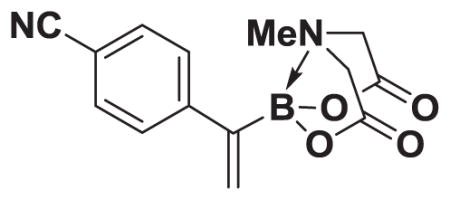
4.14. MIDA boronate 8e
Based on the 1H NMR of the crude reaction mixture, the reaction proceeded to 70% conversion and a 8:1 of 8:9 was obtained. 8e and 9e were isolated by column chromatography as an inseparable mixture of isomers to provide 57.6 mg (0.50 mmol reaction), 39% yield.
1H NMR (400 MHz, d6-acetone) δ 7.79–7.52 (m, 4H), 5.82 (bd, J = 3.0 Hz, 1H), 5.79 (bs, 1H), 4.27 (d, J = 17.0 Hz, 2H), 3.95 (d, J = 17.0 Hz, 2H), 2.82 (s, 3H). 13C NMR (125 MHz, d6-acetone) δ 168.8, 134.2, 132.9. 130.4, 129.3, 119.4, 111.0, 62.6, 4d7.7. HRMS (ESI+) calculated for C14H14BN2O4 (M+H)+: 285.1047. Found: 285.1038. TLC (Et2O:MeCN 6:1) Rf = 0.32, visualized by short wave UV.
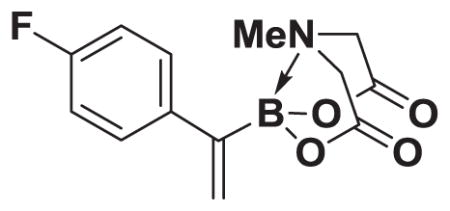
4.15. MIDA boronate 8f
Based on the 1H NMR of the crude reaction mixture, the reaction proceeded to 90% conversion and a 4:1 of 8:9 was obtained. 8f and 9f were isolated by column chromatography as an inseparable mixture of isomers to provide 59.1 mg (0.50 mmol reaction), 43% yield.
1H NMR (400 MHz, d6-acetone) δ 7.49–7.00 (m, 4H), 5.70 (bd, J = 3.0 Hz, 1H), 5.67 (bs, 1H), 4.23 (d, J = 17.0 Hz, 2H), 3.88 (d, J= 17.0 Hz, 2H), 2.80 (s, 3H). 13 C NMR (125 MHz, d6-acetone) δ 163.6, 130.2, 130.1, 129.2, 128.4, 115.8, 62.6, 47.6. HRMS (ESI+) calculated for C13H14BFNO4 (M+H)+: 278.1000. Found: 278.0994. TLC (Et2O:MeCN 6:1) Rf = 0.37, visualized by short wave UV.
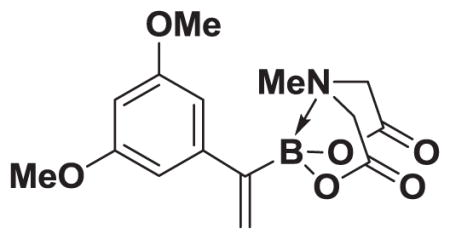
4.16. MIDA boronate 8g
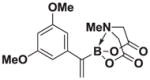
Based on the 1H NMR of the crude reaction mixture, the reaction proceeded to 85% conversion and a >20:1 of 8:9 was obtained. 8g was isolated by column chromatography to provide 21.1 mg (0.1 mmol reaction), 66% yield.
1H NMR (500 MHz, d6-acetone) δ 6.59 (d, J = 2.5 Hz, 2H), 6.38 (t, J = 2.5 Hz, 1H), 5.75 (m, 2H), 4.24 (d, J = 17.0 Hz, 2H), 3.84 (d, J = 17.0 Hz, 2H), 3.77 (s, 6H), 2.86 (s, 3H). 13C NMR (125 MHz, d6-acetone) δ 169.0, 161.7, 147.4, 127.8, 106.3, 99.7, 62.9, 55.5, 47.6. 11B NMR (128 MHz, d6-acetone) δ 11.3. HRMS (ESI+) calculated for C15H19BNO6 (M+H)+: 320.1305. Found: 320.1295. TLC (Et2O:MeCN 6:1) Rf = 0.31, visualized by short wave UV.
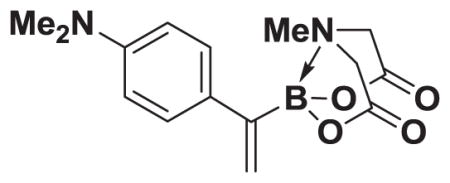
4.17. MIDA boronate 8h
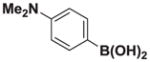
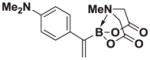
Based on the 1H NMR of the crude reaction mixture, the reaction proceeded to 50% conversion and a >20:1 of 8:9 was obtained. 8h was isolated by column chromatography to provide 40.4 mg (0.50 mmol reaction), 28% yield.
1H NMR (400 MHz, d6-acetone) δ 7.35–7.00 (m, 4H), 5.58 (bs, 1H), 5.55 (bd, J = 3.0 Hz, 1H), 4.18 (d, J = 17.0 Hz, 2H), 3.79 (d, J = 17.0 Hz, 2H), 3.12 (s, 6H), 2.75 (s, 3H). 13C NMR (125 MHz, d6-acetone) δ 168.3, 129.5, 128.9, 128.4, 125.2, 113.2, 62.7, 47.5, 40.5. HRMS (ESI+) calculated for C15H20BN2O4 (M+H)+: 303.1516. Found: 303.1524. TLC (Et2O:MeCN 6:1) Rf = 0.31, visualized by short wave UV.
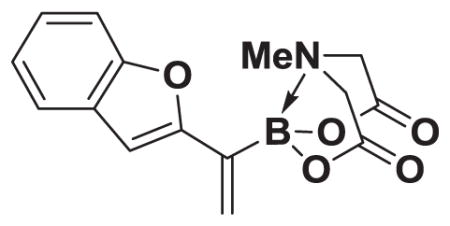
4.18. MIDA boronate 8i
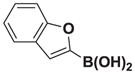
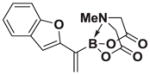
Based on the 1H NMR of the crude reaction mixture, the reaction proceeded to 95% conversion and a >20:1 of 8:9 was obtained. 8i was isolated by column chromatography to provide 24.6 mg (0.1 mmol reaction), 82% yield.
1H NMR (500 MHz, d6-acetone) δ 7.59 (d, J = 8.0 Hz, 1H), 7.47 (d, J = 8.0 Hz, 1H), 7.29 (t, J = 7.0 Hz, 1H), 7.21 (t, J = 7.0 Hz, 1H), 6.92 (s, 1H), 6.45 (s, 1H), 5.80 (d, J = 2.5 Hz, 1H), 4.41 (d, J = 17.0 Hz, 2H), 4.23 (d, J = 17.0 Hz, 2H), 3.03 (s, 3H). 13C NMR (125 MHz, d6-acetone) δ 168.9, 158.4, 154.8, 129.8, 126.5, 125.0, 123.3, 121.7, 111.2, 104.5, 62.9, 47.7. 11B NMR (128 MHz, d6-acetone) δ 11.3. HRMS (ESI+) calculated for C15H15BNO5 (M+H)+: 300.1043. Found: 300.1049.
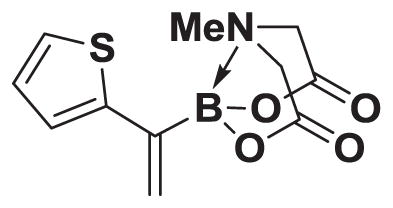
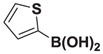
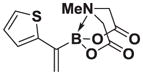
4.19. MIDA boronate 8j
Based on the 1H NMR of the crude reaction mixture, the reaction proceeded to 50% conversion and a >20:1 of 8:9 was obtained. 8j was isolated by column chromatography to provide 31.3 mg (0.50 mmol reaction), 24% yield.
1H NMR (400 MHz, d6-acetone) δ 7.80–7.00 (m, 3H), 5.83 (bs, 1H), 5.56 (bs, 1H), 4.30 (d, J = 17.0 Hz, 2H), 4.00 (d, J = 17.0 Hz, 2H), 3.13 (s, 3H). 13 C NMR (125 MHz, d6-acetone) δ 168.3, 129.5, 128.5, 126.1, 125.7, 125.3, 62.9, 47.8. HRMS (ESI+) calculated for C11H13BNO4S (M+H)+: 266.0658. Found: 266.0647. TLC (Et2O:MeCN 6:1) Rf = 0.39, visualized by short wave UV.
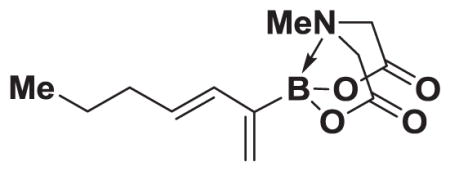
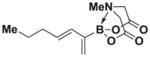
4.20. MIDA boronate 8k
Based on the 1H NMR of the crude reaction mixture, the reaction proceeded to 90% conversion and a >20:1 of 8:9 was obtained. 8k was isolated by column chromatography to provide 47.7 mg (0.25 mmol reaction), 76% yield.
1H NMR (500 MHz, d6-acetone) δ 6.20 (d, J = 15.0 Hz, 1H), 5.90 (dt, J = 15.0 Hz, 6.5 Hz, 1H), 5.61 (bs, 1H), 5.34 (bd, J = 3.5 Hz, 1H), 4.25 (d, J = 17.0 Hz, 2H), 4.01 (d, J = 17.0 Hz, 2H), 2.97 (s, 3H), 2.06 (m, 2H), 1.41 (s, J = 7.0 Hz, 2H), 0.89 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 167.3, 132.9, 132.8, 125.9, 61.9, 47.0, 35.4, 22.5, 13.7. 11B NMR (128 MHz, CDCl3) δ 10.7. HRMS (ESI+) calculated for C12H19BNO4 (M+H)+: 252.1407. Found: 252.1405. TLC (Et2O:MeCN 6:1) Rf = 0.48, visualized by short wave UV.
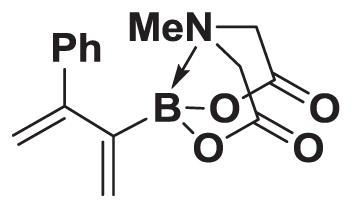
4.21. MIDA boronate 8l
Based on the 1H NMR of the crude reaction mixture, the reaction proceeded to 80% conversion and a >20:1 of 8:9 was obtained. 8l was isolated by column chromatography to provide 46.3 mg (0.25 mmol reaction), 65% yield.
1H NMR (400 MHz, d6-acetone) δ 7.47–7.19 (m, 5H), 5.58 (d, J = 2.0 Hz, 1H), 5.31 (d, J = 2.0 Hz, 1H), 5.25 (d, J = 2.0 Hz, 1H), 5.23 (d, J = 2.0 Hz, 1H), 4.16 (d, J = 17.0 Hz, 2H), 3.91 (d, J = 17.0 Hz, 2H), 3.05 (s, 3H). 13 C NMR (125 MHz, d6-acetone) δ 168.6, 154.5, 142.3, 129.1, 128.8, 128.2, 128.1, 113.9, 62.7, 47.5. HRMS (ESI+) calculated for C15H17BNO4 (M+H)+: 286.1251. Found: 286.1244. TLC (Et2O:MeCN 6:1) Rf = 0.36, visualized by short wave UV.
Acknowledgments
We gratefully acknowledge the NIH for funding (R01 GM080436). E.M.W. is an NSF Graduate Fellow and M.D.B. is an Early Career Scientist of the Howard Hughes Medical Institute.
Footnotes
Dedicated to Professor Paul Wender with deepest admiration on his receipt of the 2012 Tetrahedron Prize for Creativity in Organic Chemistry.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Boehm MF, Zhang L, Badea BA, White SK, Mais DE, Suto CM, Goldman ME, Heyman RA. J Med Chem. 1994;37:2930–2941. doi: 10.1021/jm00044a014. [DOI] [PubMed] [Google Scholar]; (b) Carney JR, Krenisky JM, Williamson RT, Luo J. J Nat Prod. 2002;65:203–205. doi: 10.1021/np010374l. [DOI] [PubMed] [Google Scholar]; (c) Borris RP, Cordell GA. J Nat Prod. 1984;47:270–278. doi: 10.1021/np50032a006. [DOI] [PubMed] [Google Scholar]; (d) Jarboe CH, Porter LA, Buckler RT. J Med Chem. 1968;11:729–731. doi: 10.1021/jm00310a020. [DOI] [PubMed] [Google Scholar]
- 2.(a) Arnone A, Dimodugno V, Nasini G, Depava OV. Gazz Chim Ital. 1990;120:397. [Google Scholar]; (b) Corey EJ, Roberts BE. J Am Chem Soc. 1997;119:12425–12431. [Google Scholar]; (c) Oppolzer W, Thirring K. J Am Chem Soc. 1982;104:4978–4979. [Google Scholar]; (d) Ghosh AK, Wang Y. J Am Chem Soc. 2000;122:11027–11028. doi: 10.1021/ja0027416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Organ MG, Bratovanov S. Tetrahedron Lett. 2000;41:6945–6949. [Google Scholar]; (b) Taber DF, Christos TE, Neubert TD, Batra D. J Org Chem. 1999;64:9673–9678. [Google Scholar]; (c) Tiefenbacher K, Mulzer J. Angew Chem, Int Ed. 2008;47:2548–2555. doi: 10.1002/anie.200705303. [DOI] [PubMed] [Google Scholar]
- 4.Hamze A, Giraud A, Messaoudi S, Provot O, Peyrat JF, Bignon J, Liu JM, Wdzieczak-Bakala J, Thoret S, Dubois J, Brion JD, Alami M. ChemMedChem. 2009;4:1912. doi: 10.1002/cmdc.200900290. [DOI] [PubMed] [Google Scholar]
- 5.(a) Dounay AB, Overman LE. Chem Rev. 2003;103:2945–2963. doi: 10.1021/cr020039h. [DOI] [PubMed] [Google Scholar]; (b) Cabri W, Candiani I, DeBernardinis S, Francalanci F, Penco S. J Org Chem. 1991;56:5796–5800. [Google Scholar]
- 6.(a) Sabarre A, Love J. Org Lett. 2008;10:3941–3944. doi: 10.1021/ol8012843. [DOI] [PubMed] [Google Scholar]; (b) Hamze A, Brion JD, Alami M. Org Lett. 2012;14:2782–2785. doi: 10.1021/ol3010112. [DOI] [PubMed] [Google Scholar]; (c) Bhilare SV, Darvatkar NB, Deorukhkar AR, Raut DG, Trivedi GK, Salunkhe MM. Tetrahedron Lett. 2009;50:893. [Google Scholar]; (d) Hashimoto T, Kutubi S, Izumi T, Rahman A, Kitamura TJ. Organomet Chem. 2011;696:99. [Google Scholar]; (e) Li R, Wang SR, Lu W. Org Lett. 2007;9:2219. doi: 10.1021/ol070737u. [DOI] [PubMed] [Google Scholar]; (f) Reetz MT, Sommer K. Eur J Org Chem. 2003:3485. [Google Scholar]
- 7.(a) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–2483. [Google Scholar]; (b) Hall DG. Boronic Acids. Wiley-VCH; Weinheim, Germany: 2005. [Google Scholar]
- 8.(a) Zweifel G, Brown HC. Org React. 1963;13:1. [Google Scholar]; (b) Miyaura N, Maruoka K. Synthesis of Organometallic Compounds. Wiley; Chichester, UK: 1997. [Google Scholar]
- 9.(a) Jang H, Zhugralin AR, Lee Y, Hoveyda AH. J Am Chem Soc. 2011;133:7859–7871. doi: 10.1021/ja2007643. [DOI] [PubMed] [Google Scholar]; (b) Gao F, Hoveyda AH. J Am Chem Soc. 2010;132:10961–10963. doi: 10.1021/ja104896b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Takagi J, Takahashi K, Ishiyama T, Miyaura N. J Am Chem Soc. 2002;124:8001–8006. doi: 10.1021/ja0202255. [DOI] [PubMed] [Google Scholar]; (d) Morrill C, Funk TW, Grubbs RH. Tetrahedron Lett. 2004;45:7733–7736. [Google Scholar]; (e) Moran WJ, Morken JP. Org Lett. 2006;8:2413–2415. doi: 10.1021/ol060735u. [DOI] [PubMed] [Google Scholar]; (f) Takahashi K, Ishiyama T, Miyaura N. J Organomet Chem. 2001;625:47–53. [Google Scholar]
- 10.Gillis EP, Burke MD. J Am Chem Soc. 2007;129:6716–6717. doi: 10.1021/ja0716204. [DOI] [PubMed] [Google Scholar]
- 11.Lee SJ, Gray KC, Paek JS, Burke MD. J Am Chem Soc. 2008;130:466–468. doi: 10.1021/ja078129x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gillis EP, Burke MD. J Am Chem Soc. 2008;130:14084–14085. doi: 10.1021/ja8063759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uno BE, Gillis EP, Burke MD. Tetrahedron. 2009;65:3130–3138. [Google Scholar]
- 14.Knapp DM, Gillis EP, Burke MD. J Am Chem Soc. 2009;131:6961–6963. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Struble JR, Lee SJ, Burke MD. Tetrahedron. 2010;66:4710–4718. [Google Scholar]
- 16.Woerly EM, Cherney AH, Davis EK, Burke MD. J Am Chem Soc. 2010;132:6941–6943. doi: 10.1021/ja102721p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee SJ, Anderson TM, Burke MD. Angew Chem Int Ed. 2010;49:8860–8863. doi: 10.1002/anie.201004911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ballmer SG, Gillis EP, Burke MD. Org Syn. 2009;86:344–359. [Google Scholar]
- 19.Dick GR, Knapp DM, Gillis EP, Burke MD. Org Lett. 2010;12:2314–2317. doi: 10.1021/ol100671v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woerly EM, Struble JR, Palyam N, O’Hara SP, Burke MD. Tetrahedron. 2011;67:4333–4343. doi: 10.1016/j.tet.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujii S, Chang SY, Burke MD. Angew Chem Int Ed. 2011;50:7862–7864. doi: 10.1002/anie.201102688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J, Burke MD. J Am Chem Soc. 2011;133:13774–13777. doi: 10.1021/ja205912y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dick GR, Woerly EM, Burke MD. Angew Chem Int Ed. 2012;51:2667–2672. doi: 10.1002/anie.201108608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gray KC, Palacios DS, Dailey I, Endo MM, Uno BE, Wilcock BC, Burke MD. Proc Natl Acad Sci USA. 2012;109:2234–2239. doi: 10.1073/pnas.1117280109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mancilla T, Contreras R, Wrackmeyer B. J Organomet Chem. 1986;307:1–6. [Google Scholar]
- 26.www.aldrich.com/mida
- 27.(a) see references 13 and 17Brown HC, Subrahmanyam C, Hamaoka T, Ravindran N, Bowman DH, Misumi S, Unni MK, Somayaji V, Bhat NG. J Org Chem. 1989;54:6068–6075.Morrill C, Grubbs RH. J Org Chem. 2003;68:6031–6034. doi: 10.1021/jo0345345.
- 28.For examples see Chauvelier J. Ann Chim. 1948;12:393–444.Mitchell RH. Can J Chem. 1977;55:210–219.(c) reference 7.
- 29.Sigma Aldrich: 704415; AllyChem: AC111044; BoroPharm
- 30.(a) Uenishi Y, Beau JM, Armstrong RW, Kishi Y. J Am Chem Soc. 1987;109:4756–4758. [Google Scholar]; (b) Weibel JM, Blanc A, Pale P. Chem Rev. 2008;108:3149–3173. doi: 10.1021/cr078365q. [DOI] [PubMed] [Google Scholar]
- 31.(a) Driver MS, Hartwig JF. J Am Chem Soc. 1996;118:7217–7218. [Google Scholar]; (b) Alcazar-Roman LM, Hartwig JF, Rheingold AL, Liable-Sands LM, Guzei IA. J Am Chem Soc. 2000;122:4618–4630. [Google Scholar]; (c) Birkholz MN, Freixa Z, van Leeuwen PWNM. Chem Soc Rev. 2009;38:1099–1118. doi: 10.1039/b806211k. [DOI] [PubMed] [Google Scholar]
- 32.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518. [Google Scholar]
- 33.Still WC, Kahn M, Mitra A. J Org Chem. 1978;43:2923. [Google Scholar]
- 34.CCDC 937425 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.cdcc.cam.ac.uk/data_request.cif
- 35.Xu ZH, McArthur CR, Leznoff CC. Can J Chem. 1983;61:1405–1409. [Google Scholar]