

Abstract
The ubiquitous C2 domain is a conserved Ca2+-triggered membrane-docking module that targets numerous signaling proteins to membrane surfaces where they regulate diverse processes critical for cell signaling. In this study, we quantitatively compared the equilibrium and kinetic parameters of C2 domains isolated from three functionally distinct signaling proteins: cytosolic phospholipase A2-α (cPLA2-α), protein kinase C-β (PKC-β), and synaptotagmin-IA (Syt-IA). The results show that equilibrium C2 domain docking to mixed phosphatidylcholine and phosphatidylserine membranes occurs at micromolar Ca2+ concentrations for the cPLA2-α C2 domain, but requires 3- and 10-fold higher Ca2+ concentrations for the PKC-β and Syt-IA C2 domains ([Ca2+]1/2 = 4.7, 16,48 μM, respectively). The Ca2+-triggered membrane docking reaction proceeds in at least two steps: rapid Ca2+ binding followed by slow membrane association. The greater Ca2+ sensitivity of the cPLA2-α domain results from its higher intrinsic Ca2+ affinity in the first step compared to the other domains. Assembly and disassembly of the ternary complex in response to rapid Ca2+ addition and removal, respectively, require greater than 400 ms for the cPLA2-α domain, compared to 13 ms for the PKC-β domain and only 6 ms for the Syt-IA domain. Docking of the cPLA2-α domain to zwitterionic lipids is triggered by the binding of two Ca2+ ions and is stabilized via hydrophobic interactions, whereas docking of either the PKC-β or the Syt-IA domain to anionic lipids is triggered by at least three Ca2+ ions and is maintained by electrostatic interactions. Thus, despite their sequence and architectural similarity, C2 domains are functionally specialized modules exhibiting equilibrium and kinetic parameters optimized for distinct Ca2+ signaling applications. This specialization is provided by the carefully tuned structural and electrostatic parameters of their Ca2+- and membrane-binding loops, which yield distinct patterns of Ca2+ coordination and contrasting mechanisms of membrane docking.
During signal transduction in eukaryotic cells, elevation of the second messenger Ca2+ directs diverse fundamental events such as cell division, motility, and contraction, activation of numerous intracellular pathways, secretion of bio-active compounds, and programmed cell death (1). The biochemical events underlying these processes are controlled by the spatial and temporal properties of Ca2+ transients generated as local puffs or sparks, or propagated as global spikes or waves. These transients are highly specialized and differ dramatically between cell types. For instance, at the active presynaptic zones of neuronal cells, brief localized transients exceeding 100 μM Ca2+ direct the release of neurotransmitters via vesicle fusion with the plasma membrane, thus resulting in transmission of signals to postsynaptic neurons on the millisecond time scale (2–4). Smaller Ca2+ oscillations that reach peaks of 1–10 μ M Ca2+ pass through specialized regions of cardiac cells up to 10 times per second, thereby directing the contraction and relaxation phases of the heart beat (5). In nonexcitable cells, micromolar Ca2+ waves of lower frequency and longer duration, lasting from seconds to hours, transmit Ca2+ signals throughout the cytoplasm and nucleus (6). Given the diversity of cellular Ca2+ signals, it is reasonable to postulate that Ca2+-regulated proteins will exhibit highly specialized equilibrium and kinetic Ca2+-binding parameters that are “tuned” to match the levels and time scales of specific Ca2+ transients (7, 8).
The C2 domain is a conserved membrane-targeting motif present in a multitude of Ca2+-regulated signaling proteins (9–11). The C2 domain and the EF-hand motif of the calmodulin superfamily are the two most frequently occurring Ca2+ sensors: for example, the C. elegans, D. melanogaster, and H. sapiens genomes contain at least 61, 57, and 126 C2 domain proteins, respectively, while the same genomes contain at least 58, 92, and 144 EF-hand proteins, respectively (12). Prototypical C2 domains bind Ca2+ during a cytoplasmic transient and trigger protein docking to a specific intracellular membrane (9–11). Subsequently, one or more associated domains modulate signaling by membrane-bound receptors, kinases, GTPases, and regulatory lipids. There are six known functional families of proteins containing C2 domains: (i) kinases that phosphorylate membrane protein targets; (ii) phospholipid-modifying enzymes that generate or inactivate lipid-derived second messengers; (iii) vesicle targetting and fusion proteins; (iv) GTPase-activating proteins; (v) ubiquitination enzymes that target membrane proteins for degradation; and (vi) proteins that form trans-membrane pores (9–11). Whereas prototypical C2 domains bind Ca2+ and dock to phospholipid membranes, other C2 domains dock to membrane protein targets or fail to exhibit Ca2+ regulation (9–11), suggesting a broad range of functional specialization in the C2 domain superfamily. Even prototypical C2 domains have been hypothesized to employ different mechanisms of membrane docking (61).
To elucidate the conserved and specialized features of prototypical C2 domains, it is essential to directly compare the equilibrium and kinetic parameters of C2 domains isolated from functionally distinct signaling proteins. Figure 1A illustrates the conserved β-sandwich architectures of three representative C2 domains of known structure (28, 29, 43) selected for the present study due to their importance as Ca2+ sensors in different pathways and cellular environments. (i) The C2A domain of synaptotagmin-I (Syt-IA)1 is one of the Ca2+ sensors responsible for triggering synaptic vesicle fusion and neurotransmitter release at neuronal synapses (13–15). (ii) The C2 domain of protein kinase C (PKC-β) helps drive the Ca2+-activated docking of this anionic lipid-dependent Ser/Thr kinase to the plasma membrane during regulation of numerous pathways (16–18). (iii) The C2 domain of cytosolic phospholipase A2 (cPLA2-α) binds Ca2+ and targets this protein primarily to nuclear and reticular membranes (19–23), where its enzymatic domain hydrolytically releases arachidonic acid to initiate the inflammatory response blocked by aspirin and ibuprofin (24–26). Previous equilibrium and kinetic studies of these C2 domains have provided much useful information, but have utilized different buffer, ionic, pH, and temperature conditions, contrasting membrane compositions, and in some cases docking site mutations to incorporate spectroscopic probes (30, 47, 61, 64). Such variations prevent quantitative comparisons of C2 domain activation parameters and mechanisms. The present comparison indicates that these three C2 domains differ in their Ca2+ affinity, kinetics, and stoichiometry, as well as in membrane affinity, kinetics, and headgroup selectivity. These results suggest that C2 domains are functionally specialized modules adapted in different cellular contexts to respond to distinct Ca2+ signals.
Figure 1.

Comparison of three representative C2 domains and models for the mechanism of membrane docking. (A) Structures of the cPLA2-α (43), PKC-β (29), and Syt-IA (28) C2 domains illustrating their shared β-sandwich architecture. Yellow spheres denote the crystallographic Ca2+ ion(s) occupying a site formed by three inter-strand loops. (B) Space-filling representation of the three Ca2+-binding loops for each C2 domain, highlighting basic side chains in blue and hydrophobic side chains in green. Yellow spheres illustrate the bound Ca2+ ions of the protein–membrane complex, which are presumed to lie at the same positions observed in the crystal structure of the free domain. [Two of the three Ca2+ ions shown for Syt-IA were modeled into the crystal structure using the homologous PKC-β sites as a guide (29).] The indicated orientation approximates the view from the membrane as the loops approach for docking. (C) Models for the C2 domain–membrane interaction, ranging from specific contacts between the bound Ca2+ ions or the protein surface and phospholipid headgroups (left panels) to nonspecific electrostatic or hydrophobic interactions between the Ca2+–protein complex and the membrane (right panels). Each type of interaction could be regulated by Ca2+ binding, either directly by the presence of bound Ca2+ ions or indirectly by a Ca2+-induced conformational change.
Materials and Methods
Reagents
The lipids used were: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (phosphatidylcholine, PC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (phosphatidylserine, PS), both from Avanti Polar Lipids, and N-(5-dimethylaminonaphthalene-1-sulfonyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (dansyl-PE, dPE) from Molecular Probes. Small unilamellar phospholipid vesicles were prepared by sonication. Solutions and plastic ware were decalcified as described (27).
Purification of C2 Domains
C2 domains of Syt-IA and PKC-β were expressed as glutathione S-transferase (GST) fusion proteins and purified away from GST after cleavage with thrombin as described (28, 29). The cPLA2-α C2 domain was affinity-purified from refolded protein as described (30). Protein concentrations were determined using the tyrosinate method (30).
Fluorescence Spectroscopy of Equilibrium Processes
Fluorescence spectroscopy was carried out as described (30) on an SLM 48000S fluorescence spectrometer at 25 °C in standard assay buffer composed of 100 mM KCl, 20 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES), pH 7.4, and 5 mM dithiothreitol (DTT). Excitation and emission slit widths were 4 and 8 nm, respectively. Because of differences in the quantum yields of the C2 domains, the concentrations of the domains necessary to achieve similar fluorescence signals varied, ranging from 0.1 to 1.0 μM. Where necessary, fluorescence signals were corrected for inner filter effects as described (31).
Fluorescence resonance energy transfer (FRET) was employed to measure the Ca2+ dependence of membrane docking as described (30). C2 domain was mixed with target vesicles of PS–PC–dPE (mole percent 47.5%:47.5%:5%; 250 μM total phospholipid), and PC–dPE or PS–dPE (95%: 5%; 100 μM phospholipid). The Ca2+-dependent change in the protein-to-membrane FRET (ΔF) was subjected to nonlinear, least-squares analysis using a modified Hill equation:
| (1) |
where ΔFmax represents the calculated maximal fluorescence change, x is the free Ca2+ concentration, H represents the Hill coefficient, and [Ca2+]1/2 represents the free Ca2+ concentration that induces half-maximal fluorescence change. ΔF values were normalized to ΔFmax to allow comparisons. For the cPLA2-α C2 domain, ΔFmax values from experiments involving PC–dPE vesicles were used to normalize ΔF values from PS–dPE. Likewise, for the PKC-β and Syt-IA domains, ΔFmax values from experiments involving PS-dPE were used for PC–dPE. Fluorescence experiments such as these do not quantitatively determine the Hill coefficient for Ca2+-triggered binding, since the individual binding events in a multistep reaction may yield different microscopic fluorescence changes. Although the Hill coefficient is quantitatively related to the number of bound ligands only in a cooperative system of identical sites (32), for proteins that bind two Ca2+ ions, observed Hill coefficients that exceed 1.4 in fluorescence experiments provide strong evidence for positive cooperativity (33).
Phospholipid titrations were carried out using PS–PC– dPE vesicles (47.5%:47.5%:5%) in the presence of 1 mM excess Ca2+ over ethylenediaminetetraacetic acid (EDTA) in standard buffer as described (30). The Ca2+-dependent change in the protein-to-membrane FRET (ΔF) was analyzed using an equation describing the binding to a single population of independent sites:
| (2) |
Where x is the concentration of phospholipid and KD represents the apparent membrane dissociation constant in the presence of 1 mM free Ca2+.
Intrinsic tryptophan fluorescence was monitored in standard assay buffer as described (30). Emission spectra were recorded from 300 to 400 nm. The Ca2+ dependence of the intrinsic fluorescence change was measured by recording the emission at the fluorescence maximum (325 nm for cPLA2-α and 340 nm for PKC-β), and fluorescence changes were analyzed by eq 1. When present, PS–PC vesicles (50%:50%; 250 μM phospholipid) were used.
The effect of ionic strength on membrane docking was tested by incubating C2 domains with PS–PC–dPE vesicles (47.5%:47.5%:5%; 100 μM phospholipid) in standard assay buffer lacking KCl but containing 1 mM EDTA. All subsequent fluorescence changes were corrected for any small direct effect of NaCl, Ca2+, and EDTA on vesicle fluorescence. After recording the initial dPE emission (F0), 1 mM excess Ca2+ was added, and dPE emission (FCa2+) was recorded. Ionic strength was increased by reciprocal dilution with a like sample prepared in a high concentration of NaCl; dPE emission was recorded (FNaCl) after each increase in NaCl. Finally, excess EDTA was added, and dPE emission was recorded (Fedta). Assuming that the C2 domain–membrane ternary complex is formed from a simple bimolecular interaction between the Ca2+-occupied C2 domain and an excess of phospholipid, then the apparent membrane dissociation constant (Kd) equals the product of the total lipid concentration and the ratio of free to bound C2 domain (34). The concentrations of free and bound C2 domain were approximated by the differences FCa2+ – FNaCl and FNaCl – F0, respectively, to calculate Kd at each concentration of NaCl. Slopes of log [NaCl] plotted against log KD were determined by linear regression to estimate the minimal number of electrostatic interactions involved in the domain–membrane complex based on polyelectrolyte theory (35).
The ability of Na2SO4 to promote membrane docking was carried out essentially as before (36). C2 domains were added to vesicles of PS–PC–dPE (47.5%:47.5%:5%; 100 μM phospholipid) in standard buffer containing 1.9 M Na2SO4 and 5 mM EDTA. The resulting FRET signal was compared to the FRET signal arising from membrane docking induced by Ca2+ in the absence of Na2SO4.
Stopped-Flow Fluorescence Spectroscopy
Stopped flow was carried out essentially as described (30) in standard assay buffer at 25 °C on an Applied Photophysics model 17MV stopped-flow apparatus. The dead-time of the instrument was determined to be 1.5 ms using a control reaction as described (37).
The membrane docking reaction was triggered by stopped-flow mixing a solution containing C2 domain (0.5–2 μM, all concentrations prior to mixing), vesicles of PS–PC–dPE (47.5%:47.5%:5%; 500 μM phospholipid), and trace amounts of EDTA (2 μM) with an equal volume of Ca2+ (400 μM). The resulting protein-to-membrane FRET time course reveals the approach to equilibrium of the docking reaction. Omission of either the C2 domain or Ca2+ eliminated the observed FRET increase upon mixing (data not shown). The dissociation reaction was triggered by stopped-flow mixing a solution of C2 domain (1–8 μM), Ca2+ (100 μM), and vesicles of PS–PC–dPE (47.5%:47.5%:5%; 250 μM) with an equal volume of EDTA (5 mM). The resulting protein-to-membrane FRET time course reveals the irreversible dissociation of C2 domains from membranes. Omission of either the C2 domain or Ca2+ eliminated the observed FRET decrease upon mixing (data not shown). Alternatively, the dissociation reaction was initiated by stopped-flow mixing C2 domain (4 μM), Ca2+ (100 μM), and PS–PC vesicles (50%:50%; 250 μM) with an equal volume of the fluorescent Ca2+ chelator Quin-2 (200 μM). Vesicles were omitted in experiments designed to measure Ca2+ release from the free C2 domain. The Quin-2 fluorescence signal monitored the irreversible release of Ca2+ ions from the C2 domain. Omission of the C2 domain eliminated the Quin-2 fluorescence increase observed after mixing (data not shown). Stoichiometry was determined at pH 7.0 by comparing fluorescence amplitudes (see below) to a standard curve (30). At this pH, all three C2 domains tested were fully membrane-bound at 100 μM Ca2+ (data not shown), which was not the case at pH 7.4 (see Figure 2).
Figure 2.

Ca2+ threshold for membrane docking by C2 domains. Ca2+ was titrated into solutions containing the cPLA2-α (filled circles), PKC-β (open circles), and Syt-IA (open squares) C2 domains and synthetic vesicles composed of PS–PC–dPE (47.5%: 47.5%:5%). Membrane docking was measured by monitoring protein-to-membrane FRET. Solid lines indicate the best-fit of the resulting normalized FRET signals to the modified Hill equation (eq 1). Experimental conditions: 25 °C; 100 mM KCl, 20 mM HEPES, pH 7.4, 5 mM DTT, and 250 μM phospholipid. Results are summarized in Table 1.
The fluorescence signals, F(t), were analyzed using both mono- and biexponential equations:
| (3) |
| (4) |
where k, k1, and k2 are rate constants, ΔF, ΔF1, and ΔF2 are the fluorescence amplitudes, and C is the intrinsic voltage offset of the stopped-flow experiment. F(t) values in all time courses were normalized to ΔF to allow comparison.
Results
The isolated Syt-IA, PKC-β, and cPLA2-α C2 domains were expressed in E. coli and purified to homogeneity. Use of the isolated domains prevented interference by other Ca2+-or membrane-binding motifs present in the full-length proteins, thereby facilitating comparison of the intrinsic C2 domain Ca2+-activation and membrane docking parameters. Moreover, each of the isolated domains has been shown by crystallographic and NMR studies to be monomeric (28, 29, 38–45), obviating the artifactual interdomain cooperativity that could arise in membrane docking experiments involving domains coupled either to multiple sites on affinity beads or to the dimer-forming GST fusion partner (46, 47). To facilitate quantitative comparison of the isolated domains, all experiments were carried out at 25 °C in 100 mM KCl, 20 mM HEPES, pH 7.4, 5 mM DTT unless otherwise noted. Membranes contained an equimolar mixture of the two predominant physiological lipids, phosphatidylcholine (PC) and phosphatidylserine (PS), unless otherwise noted.
Equilibrium Ca2+ Dependence of Membrane Docking
The Ca2+ dependence of membrane docking by these C2 domains was measured using fluorescence resonance energy transfer (FRET) (Figure 2). This assay detects the transfer of energy from intrinsic tryptophan donors in the C2 domain to trace amounts of the acceptor dansyl-phosphatidylethanolamine (dPE), incorporated into synthetic phospholipid vesicles composed of phosphatidylcholine (PC) and phosphatidylserine (PS), upon formation of the domain–membrane complex. Nonlinear least-squares analysis of the data using the Hill equation (eq 1) yielded the [Ca2+]1/2 values and Hill coefficients (H) summarized in Table 1. These data allow the first direct comparison of equilibrium Ca2+ sensitivities for the docking of monovalent C2 domains to membranes under identical conditions. In the presence of 250 μM total phospholipid (mole percent 47.5% PC, 47.5% PS, 5% dPE), the apparent Hill coefficients all significantly exceeded 1.4, indicating that membrane docking is triggered by the binding of multiple Ca2+ ions with positive cooperativity (see Materials and Methods). The [Ca2+]1/2 values for membrane docking varied 10-fold, ranging from 4.7 μM for the cPLA2-α C2 domain to 16 μM for PKC-β and 48 μM for Syt-IA C2 domains. For the cPLA2-α and PKC-β C2 domains, we obtained similar [Ca2+]1/2 values (4.1 and 10 μM, respectively) in separate experiments monitoring docking-induced changes to intrinsic Trp fluorescence, confirming the FRET results and indicating the dPE acceptor used in the FRET assay was nonperturbing (Figures 3 and 4; Table 1). Thus, the membrane-docking equilibrium of the cPLA2-α C2 domain is 3-fold more sensitive to Ca2+ than that of the PKC-β domain and is 10-fold more sensitive than that of the Syt-IA domain.
Table 1. Equilibrium Parameters of Ca2+ and Membrane Binding to C2 Domainsa.
| Ca2+ dependence of membrane binding | intrinsic Ca2+ binding | membrane binding | ||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||
| protein-to-membrane FRETb | intrinsic fluorescencec | intrinsic fluorescenced | protein-to-membrane FRETe | |||||
|
|
|
|
|
|||||
| C2 domain | [Ca2+]1/2 (μM) | H | [Ca2+]1/2 (μM) | H | [Ca2+] 1/2 (μM) | H | KD (μM) | |
| cPLA2-α | 4.7 ± 0.8 | 1.7 ± 0.2 | 4.1 ± 0.3 | 1.8 ± 0.1 | 14 ± 2 | 1.7 ± 0.2 | 24 ± 7 | |
| PKC-β | 16 ± 4 | 1.6 ± 0.1 | 10 ± 3 | 2.0 ± 0.2 | 39 ± 2 | 1.6 ± 0.1 | 3.4 ± 1.0 | |
| Syt-IA | 48 ± 12 | 2.0 ± 0.2 | ND | ND | ND | ND | 19 ± 6 | |
Results represent the average (±SEM) of 3–4 independent steady-state fluorescence experiments. [Ca2+]1/2 and H represent the best-fit midpoint and apparent Hill coefficient for Ca2+ titrations (eq 1). KD is the best-fit apparent dissociation constant for phospholipid binding to a population of independent binding sites (eq 2). Experimental conditions: 25 °C; 100 mM KCl, 20 mM HEPES, pH 7.4, 5 mM DTT. ND, not determined.
Determined in the presence of PS–PC–dPE vesicles (47.5%:47.5%:5%; 250 μM phospholipid), as in Figure 2.
Determined in the presence of PS–PC vesicles (50%:50%; 250 μM phospholipid), as in Figure 4.
Determined in the absence of vesicles, as in Figure 4.
Determined in the presence of 1 mM free Ca2+ using PS–PC–dPE vesicles (47.5%:47.5%:5%), as in Figure 5.
Figure 3.

Intrinsic fluorescence changes in C2 domains induced by Ca2+ binding and membrane docking. Fluorescence emission spectra of the cPLA2-α (A), PKC-β (B), and Syt-IA (C) C2 domains were recorded in the absence (open symbols) or presence (closed symbols) of PS–PC vesicles (50%:50%, 250 μM phospholipid). Buffer-subtracted emission spectra were recorded before (squares) or after (circles) addition of 1 mM excess Ca2+. Maximum emission intensities were normalized to unity prior to Ca2+ addition: for clarity, spectra recorded in the presence of PS–PC vesicles prior to Ca2+ addition were omitted. Subsequent addition of excess EDTA returned fluorescence signals back to that prior to Ca2+ addition (not shown). Experimental conditions: 25 °C; 100 mM KCl, 20 mM HEPES, pH 7.4, 5 mM DTT, and 1 mM EDTA.
Figure 4.

Intrinsic and apparent Ca2+ affinities of cPLA2-α and PKC-β C2 domains. Ca2+ was titrated into solutions containing the cPLA2-α (A) and PKC-β (B) C2 domains in the absence (open symbols) or presence (filled symbols) of PS–PC vesicles (50%: 50%; 250 μM phospholipid). Intrinsic tryptophan emission was recorded at the emission λmax. Solid lines indicate the best-fit of the resulting normalized fluorescence signals to the modified Hill equation (eq 1). Experimental conditions: 25 °C; 100 mM KCl, 20 mM HEPES, pH 7.4, and 5 mM DTT. Results are summarized in Table 1.
Assuming that a given C2 domain rapidly binds multiple Ca2+ ions before docking to the membrane surface (this assumption is justified below), Ca2+-triggered membrane docking may be depicted by a two-step mechanism:
where n represents the Ca2+ stoichiometry, PL represents phospholipid membrane, and C2•Can•PL represents the C2 domain–membrane ternary complex. Thermodynamic principles require that an observed [Ca2+]1/2 value for assembly of the ternary complex depends on both (i) the intrinsic Ca2+ affinity of the free C2 domain (Step I) and (ii) the membrane affinity of the Ca2+-occupied domain (Step II). To deconvolute these factors, the intrinsic Ca2+ and membrane affinities were determined, allowing the first quantitative comparison of such parameters for native C2 domains.
Equilibrium Binding ofCa2+ to the Free C2 Domain
Ca2+ binding to free C2 domains was measured by monitoring Ca2+-induced changes in the intrinsic tryptophan fluorescence, since tryptophan emission has been used to detect environmental changes in the cPLA2-α C2 domain that are directly coupled to Ca2+ binding (30, 48, 49). The emission spectrum of the cPLA2-α C2 domain was characterized by a blue-shifted emission maximum (λmax ∼ 325 nm) arising from its sole buried tryptophan, Trp71 (Figure 3A). Ca2+ binding to the free cPLA2-α C2 domain increased its fluorescence emission intensity by approximately 15%. By contrast, the emission spectrum of the PKC-β C2 domain was red-shifted (λmax ∼ 340 nm), suggesting that emission from its three solvent-exposed tryptophans, Trp245, Trp247, and Trp274, dominates over that from the one buried tryptophan, Trp223, which occupies the position corresponding to Trp71 in the cPLA2-α C2 domain (Figure 3B). Nevertheless, Ca2+ binding to the free PKC-β C2 domain also affected its fluorescence emission, increasing its intensity by approximately 30% and causing a slight blue shift. The fluorescence emission spectrum of the Syt-IA domain was also characterized by a red-shifted maximum (λmax ∼ 340 nm) due to the presence of its sole, solvent-exposed tryptophan, Trp259 (Figure 3C). In agreement with a previous study (50), its intrinsic fluorescence was insensitive to ligand binding.
The Ca2+ dependences of these fluorescence increases were measured in order to directly compare the intrinsic Ca2+ affinities of the cPLA2-α and PKC-β C2 domains (Figure 4). The resulting [Ca2+]1/2 values (Table 1) indicate that the free cPLA2-α C2 domain cooperatively binds Ca2+ with higher affinity ([Ca2+]1/2 = 14 μM) than the PKC-β domain ([Ca2+]1/2 = 39 μM), where each of these [Ca2+]1/2 values represents an average affinity for the binding of multiple Ca2+ ions. For comparison, NMR and optical measurements have shown that the free Syt-IA C2 domain binds Ca2+ with multiple apparent dissociation constants ranging from 60 μM to >1 mM (38, 40, 41, 47). Thus, both the free cPLA2-α and PKC-β C2 domains bind Ca2+ with considerably higher affinity than the free Syt-IA domain.
Equilibrium Docking of the Ca2+-Occupied C2 Domain to Membrane
The membrane affinity of each Ca2+-occupied C2 domain was measured by the FRET assay (Figure 5), yielding the apparent phospholipid dissociation constants (Kd) in the presence of 1 mM Ca2+ (Table 1). These KD values varied approximately 7-fold, ranging from 3.4 μM for PKC-β to 24 and 19 μM for the cPLA2-α and Syt-IA C2 domains, respectively. Therefore, the greater Ca2+ sensitivity of the cPLA2-α C2 domain relative to PKC-β in membrane docking is due to the higher intrinsic affinity of the free cPLA2-α C2 domain for Ca2+, whereas the lesser Ca2+ sensitivity of the Syt-IA domain is due to a combination of both its reduced intrinsic Ca2+ affinity and its lower affinity for phospholipid membranes. The latter findings are surprising, especially given the similar structures, headgroup selectivities, and docking mechanisms exhibited by the PKC-β and Syt-IA membrane docking loops (see below).
Figure 5.

Apparent membrane-binding affinities of C2 domains. Vesicles of PS–PC–dPE (47.5%:47.5%:5%) were titrated into solutions containing the cPLA2-α (filled circles), PKC-β (open circles), and Syt-IA (open squares) C2 domains and 1 mM free Ca2+. Membrane docking was measured by monitoring protein-tomembrane FRET. Solid lines indicate the best-fit of the resulting normalized FRET signals to a single, independent-site equation (eq 2). Experimental conditions: 25 °C; 100 mM KCl,20 mM HEPES, pH 7.4, 5 mM DTT, 2 mM CaCl2, and 1 mM EDTA. Results are summarized in Table 1.
Kinetics of Ca2+ Binding and Membrane Docking
The kinetics of the Ca2+-triggered membrane docking reaction were analyzed in vitro by stopped-flow fluorescence spectroscopy. To probe the kinetics of ternary complex formation, Ca2+ was mixed with a solution of C2 domain and phospholipid vesicles while monitoring membrane docking via protein-to-membrane FRET. This experiment measures a complex, multistep approach to equilibrium involving the binding of multiple Ca2+ ions and docking to the membrane surface. The resulting time courses exhibited simple, monoexponential behavior (Figure 6, eq 3). Such simple behavior for a multistep approach to equilibrium is consistent with the existence of a reversible, rate-determining step that controls the kinetics of ternary complex formation and dissociation. Nonlinear best-fit analysis yielded the apparent, pseudo-first-order rate constants of ternary complex formation (kform) summarized in Table 2. These data allow the first quantitative comparison of rate constants for the reactions of native C2 domains with membranes. In the presence of Ca2+ (200 μM) and phospholipid (250 μM; mole percent 47.5% PS, 47.5% PC, 5% dPE), equilibration of ternary complex formation was slowest for cPLA2-α, intermediate for PKC-β, and fastest for the Syt-IA domain. Different relative rates (PKC-β < cPLA2-α < Syt-IA) were reported in an independent study where membrane binding was monitored by tryptophan emission (47), which also detects the rapid binding of Ca2+ to the free cPLA2-α C2 domain and thus may artifactually increase the apparent rate of ternary complex formation (30). Our results indicate that the apparent time constants for ternary complex formation (τform = 1/kform) varied nearly 10-fold, ranging from 4.3 ms for Syt-IA and 5.6 ms for PKC-β to 38 ms for the cPLA2-α domain. Notably, at physiological Ca2+ concentrations, the membrane docking equilibrium is reached considerably more quickly for the Syt-IA and PKC-β C2 domains than for the cPLA2-α domain.
Figure 6.

Kinetics of C2 domain activation. Ca2+-triggered docking of C2 domains to membranes was initiated by stopped-flow mixing a solution containing the indicated C2 domain, PS–PC–dPE vesicles (47.5%:47.5%:5%; 500 μM phospholipid), and trace amounts of EDTA (2 μM) with an equal volume of Ca2+ (400 μM). Membrane docking was measured by monitoring protein-tomembrane FRET. Solid lines represent the best-fit of the resulting normalized FRET signals to a monoexponential equation (eq 3). Omission of either the C2 domain or Ca2+ eliminated the observed FRET increase upon mixing (data not shown). Experimental conditions: 25 °C; 100 mM KCl, 20 mM HEPES, pH 7.4, and 5 mM DTT. Results are summarized in Table 2.
Table 2. Kinetic Parameters of C2 Domain Activation and Deactivation Time Coursesa.
| membrane dissociationc | Ca2+ dissociationd | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||||
| membrane associationb | kdiss (s−1) | kdiss (s−1) | ||||||||
|
|
|
|
||||||||
| C2 domain | kform (s−1) | (n) | kdiss1(s−1) | kdiss2 (s−1) | (n) | kdiss1(s−1) | kdiss2 (s−1) | (n) | stoichiometry (mol/mol)e | (n) |
| cPLA2-α | 26 ± 1 | (6) | 14 ± 1 | 2.5 ± 0.1 | (10) | 24 ± 8 | 2.9 ± 0.4 | (9) | 2.0 ± 0.2 | (5) |
| PKC-β | 180 ± 10 | (6) | 132 ± 1 | (12) | 67 ± 3 | (10) | 3.0 ± 0.1 | (10) | ||
| Syt-IA | 230 ± 20 | (7) | 520 ± 20 | (12) | 590 ± 30 | (11) | 3.1 ± 0.3 | (9) | ||
Results represent the average (±SEM) of the indicated n independent stopped-flow fluorescence experiments. Experimental conditions: 25 °C; 100 mM KCl, 20 mM HEPES, pH 7.4, 5 mM DTT.
The observed rate constant for equilibration of the membrane association reaction, initiated by rapidly mixing a solution of C2 domain and PS–PC–dPE vesicles (47.5%:47.5%:5%, 250 μM phospholipid final) with a solution of Ca2+ (200 μM final), as in Figure 6. The resulting time course was best fit by a monoexponential equation (eq 3) to yield kform as defined in the text.
The rate constant for C2 domain dissociation from membranes, measured by mixing a solution of C2 domain, Ca2+, and phospholipid vesicles (PS– PC–dPE, 47.5%:47.5%:5%) with a solution of EDTA, as in Figure 7A. The dissociation rate constants kdiss1 and kdiss2 were determined by best fitting the data with a biexponential equation (eq 4) representing a two-step mechanism (cPLA2-α) or a monoexponential equation (eq 3) representing a one-step mechanism (PKC-β, Syt-IA).
The rate constant for Ca2+ dissociation from the membrane-bound domain, measured by rapidly mixing a solution of C2 domain, Ca2+, and phospholipid vesicles (PS–PC, 50%:50%) with a solution of the fluorescent Ca2+ chelator Quin-2, as in Figure 7B. The dissociation rate constants kdiss1 and kdiss2 were extracted as in the previous footnote.
Ca2+ stoichiometry (mol of Ca2+ bound/mol of C2 domain) was determined at pH 7.0.
In principle, because the reaction is an approach to equilibrium, the observed kinetics of ternary complex formation depend on the association and dissociation rate constants for each of the reversible steps in the binding of multiple Ca2+ ions and membrane docking (51). Two kinetic observations strongly suggest that this complicated multistep reaction can be simplified to a reversible, two-step scheme in which a rapid Ca2+-binding equilibrium is coupled to a slow, rate-determining membrane docking equilibrium (Scheme 1) (51). First, the rate constant of Ca2+ dissociation for each free C2 domain is considerably larger than the apparent rate constants of ternary complex assembly and disassembly (see below), indicating that Ca2+ binding to the free C2 domain achieves equilibrium on a much shorter time scale than membrane docking. Second, kform for formation of the ternary complex varies hyperbolically with the concentration of Ca2+ at a fixed, saturating concentration of phospholipid vesicles but varies linearly with the concentration of phospholipid vesicles at a fixed, saturating concentration of Ca2+ [Nalefski and Falke, unpublished; also see ref (47)]. Such results indicate that Ca2+ occupancy precedes the membrane association event detected by protein-to-membrane FRET, as depicted in Scheme 1.
Scheme 1.
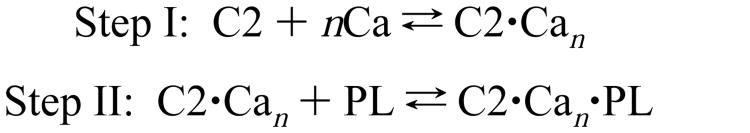
Kinetics of Membrane Dissociation and Ca2+ Release
To probe the kinetics of ternary complex dissociation after a Ca2+ signal ends, a rapidly decaying Ca2+ signal was simulated in vitro by stopped-flow mixing EDTA with the preformed ternary complex consisting of Ca2+, membrane, and C2 domain. The experiment follows the loss of protein-to-membrane FRET as the C2 domain is released from the membrane surface. This reaction is essentially irreversible, since EDTA rapidly binds free Ca2+ within the dead-time of the instrument (∼1.5 ms) and thereby prevents re-docking of the released apo protein. Overall, the reaction provides the first quantitative comparison of rate constants for the dissociation of native C2 domains from membranes. The time courses observed for the Syt-IA and PKC-β domains were monoexponential (Figure 7A, eq 3), indicating the existence of a rate-determining step during the multistep disassembly of the ternary complex. It is not yet clear whether this rate-determining step is the release of Ca2+ from the membrane-bound ternary complex, or the release of the Ca2+-occupied C2 domain from the membrane. To acknowledge this ambiguity, the dissociation reaction is written as a single kinetically resolvable step:
Figure 7.

Kinetics of C2 domain inactivation. (A) C2 domain dissociation from membranes triggered by Ca2+ removal was initiated by stopped-flow mixing a solution containing the indicated C2 domain, Ca2+ (100 μM), and PS–PC–dPE vesicles (47.5%: 47.5%:5%; 250 μM phospholipid) with an equal volume of EDTA (5 mM). Membrane docking was measured by monitoring protein-to-membrane FRET. Solid lines represent the best-fit of the resulting normalized FRET signals to a monoexponential equation (eq 3) for the PKC-β and Syt-IA domains or a biexponential equation (eq 4) for the cPLA2-α domain. Omission of either the C2 domain or Ca2+ eliminated the observed FRET decrease upon mixing (data not shown). (B) Ca2+ release from C2 domain–membrane complexes triggered by Ca2+ removal was initiated by stopped-flow mixing a solution containing the indicated C2 domain (4 μM), Ca2+ (100 μM), and PS–PC vesicles (50%:50%; 250 μM phospholipid) with an equal volume of the fluorescent Ca2+ chelator Quin-2 (200 μM). The release of Ca2+ ions was measured by monitoring the increase in Quin-2 fluorescence emission. Solid lines represent the best-fit of the resulting normalized Quin-2 signal to a monoexponential equation (eq 3) for the PKC-β and Syt-IA C2 domains or a biexponential equation (eq 4) for the cPLA2-α domain. Omission of the C2 domain eliminated the observed Quin-2 fluorescence increase after mixing (data not shown). Unlike Ca2+ release from the free cPLA2-α C2 domain, Ca2+ release from the free PKC-β and Syt-IA domains was too rapid to be observed in the absence of membranes (data not shown). Experimental conditions: 25 °C; 100 mM KCl, 20 mM HEPES, pH 7.4, and 5 mM DTT. Results are summarized in Table 2.
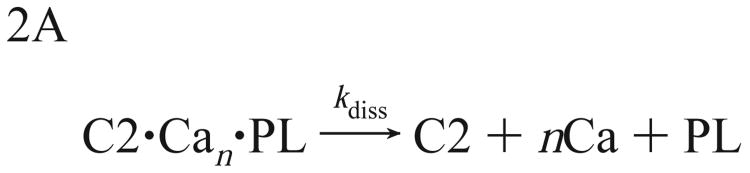
where kdiss is the measured first-order rate constant. Membrane dissociation was most rapid for the Syt-IA C2 domain (k diss = 520 s−1) and was considerably slower for the PKC-β domain (kdiss= 132 s−1) (Table 2). In contrast to the monoexponential kinetics observed for the Syt-IA and PKC-β domains (Figure 7A), the time course for the cPLA2-α domain was optimally fit with a biexponential equation (eq 4) as we have shown previously (30). These more complex kinetics imply a two-step reaction involving: (i) dissociation of the first Ca2+ ion from the domain, which perturbs the intrinsic tryptophan of the membrane-bound domain (kdiss1 = 14 s−1; see below); and (ii) dissociation of the domain from the membrane concomitantly with the loss of FRET and dissociation of the second Ca2+ ion (kdiss2 = 2.5 s−1). The resulting sequential two-step process is depicted by
Thus, during the slow decay of the cPLA2-α ternary complex, one of the Ca2+ ions dissociates before the domain is released from the membrane. Overall, the results indicate that the effective lifetimes of the membrane-bound state following removal of free Ca2+ vary considerably, ranging from 1.9 and 7.6 ms for the Syt-IA and PKC-β C2 domains, respectively, to 400 ms for the cPLA2-α domain. Since this experiment measures the average time for dissociation of the domain from the membrane after removal of the Ca2+ signal, it measures the duration that the C2 domain “remembers” the Ca2+ signal. Thus, the duration of Ca2+ memories for C2 domains from different signaling proteins varies over several hundred milliseconds: the Syt-IA and PKC-β C2 domains have considerably shorter Ca2+ memories than the cPLA2-α domain.
The time course of Ca2+ release from C2 domains was measured in order to determine the number of Ca2+ ions in the domain–membrane ternary complex and the degree to which their release is correlated with disassembly of the complex. Recent evidence suggests that the Ca2+-binding loops of the C2 domain directly contact the membrane surface (42, 44, 45, 47, 49, 50, 52, 57). As a result, bound Ca2+ ions could become trapped within the protein– membrane interface until the ternary complex dissociates (30). To monitor Ca2+ release from the free or membranebound C2 domain, the fluorescent Ca2+ chelator Quin-2 was mixed by stopped flow with the Ca2+-occupied C2 domain; then the appearance of free Ca2+ was detected as an increase of the Quin-2 fluorescence (Figure 7B). Quin-2 binds free Ca2+ within the instrument dead-time (58) and does not add a lag time to the kinetics. The number of Ca2+ ions released per domain was determined by comparing the fluorescence signal to a standard curve. While the dissociation of two Ca2+ ions from the free cPLA2-α C2 domain is slow enough to be quantified [k = 110 s−1, ref (30)], the dissociation of Ca2+ from the free C2 domains of SytI-A and PKC-β is too rapid to be measured by stopped-flow (k » 700 s−1) (data not shown). By contrast, Ca2+ release from all three premade C2 domain–membrane complexes was readily detected, thereby enabling the first measurement of Ca2+ stoichiometries for membrane-bound SytI-A and PKC-β C2 domains, and of their rate constants for Ca2+ release. Two Ca2+ ions were released from the cPLA2-α ternary complex, whereas three ions were released from the PKC-β and Syt-IA complexes (Table 2). In each case, there was relatively close agreement between the rate constants for the release of Ca2+ and dissociation of C2 domains from membranes (Table 2), indicating that membrane dissociation is kinetically linked to Ca2+ dissociation. Since Ca2+ dissociation from each ternary complex is at least 1 order of magnitude slower than dissociation from the corresponding free C2 domain, these findings support the proposal that the membrane surface occludes the Ca2+-binding sites in all three ternary complexes.
Mechanism of Ca2+-Triggered Membrane Docking
Given that the three C2 domains exhibit significantly different Ca2+ thresholds for membrane docking, different kinetics of membrane association and dissociation, and different Ca2+ stoichiometries, it is possible that the fundamental forces used by these domains to bind membranes could differ as well. A recent study hypothesized that different C2 domains may utilize contrasting mechanisms of membrane docking (61). To further test this idea, the phospholipid headgroup selectivities of the cPLA2-α, PKC-β, and Syt-IA C2 domains were probed in the FRET assay by replacing the equimolar PS—PC mixture in vesicles with pure PS or PC (Figure 8A,B). Ca2+ binding triggered the docking of both the PKC-β and Syt-IA C2 domains to anionic PS but not zwitterionic PC, whereas the cPLA2-α C2 domain docked to PC but not PS. Similarly, the PKC-β and Syt-IA C2 domains exhibited Ca2+-triggered docking to other anionic phospholipids, including phosphatidylglycerol, phosphatidic acid, and phosphatidylinositol (data not shown), in agreement with previous studies on the PKC-α (55) and Syt-IA (57, 59, 60) C2 domains. By contrast, the cPLA2-α C2 domain failed to dock to these anionic membranes [data not shown and ref (36)]. The effect of ionic strength on membrane docking by these C2 domains was tested to determine the importance of electrostatic interactions (50, 57, 61). Ca2+-dependent docking of the PKC-β and Syt-IA domains, but not the cPLA2-α domain, to membranes containing a PS−PC mixture was inhibited by increasing ionic strength (Figure 9). Furthermore, where Kd again represents the apparent dissociation constant for phospholipid binding (see Materials and Methods), plots of log KD vs log [NaCl] for PKC-β and Syt-IA were linear between 150 and 700 mM NaCl (data not shown). The resulting slopes allow calculation of the effective number of charges involved (35) and indicate a minimum of three to four ionic interactions between the PKC-β and Syt-IA C2 domains and the anionic lipid surface.
Figure 8.

Phospholipid headgroup selectivity of C2 domains. Ca2+ was titrated into solutions containing the cPLA2-α (filled circles), PKC-β (open circles), and Syt-IA (open squares) C2 domains and vesicles of PC–dPE (95%:5%) in (A) or PS–dPE (95%:5%) in (B). Membrane docking was measured by protein-to-membrane FRET. Solid lines represent the best-fit of the resulting normalized FRET signals to the modified Hill equation (eq 1) where applicable. Experimental conditions: 25 °C; 100 mM KCl, 20 mM HEPES, pH 7.4, 5 mM DTT, and 100 μM phospholipid.
Figure 9.

Role of electrostatic interactions in membrane docking by C2 domains. 1 mM free Ca2+ was added to solutions containing the cPLA2-α (filled circles), PKC-β (open circles), and Syt-IA (open squares) C2 domains and vesicles of PS–PC–dPE (47.5%:47.5%: 5%) in assay buffer lacking added KCl. Membrane docking was measured by monitoring protein-to-membrane FRET; all subsequent FRET signals were corrected for small effects of NaCl on dPE emission. The NaCl concentration was raised incrementally, and the resulting FRET signal was normalized to the initial value. Following the NaCl additions, excess EDTA was added (dashed line) to demonstrate the reversibility of cPLA2-α C2 domain binding to membranes. Experimental conditions: 25 °C; 20 mM HEPES, pH 7.4, 5 mM DTT, less than 10 mM KCl, 1 mM EDTA, and 250 μM phospholipid.
The ability of Na2SO4 to induce membrane docking was determined in order to directly test the importance of hydrophobic interactions in membrane docking. Na2SO4, which strengthens hydrophobic interactions (36), promoted Ca2+-independent docking of the cPLA2-α C2 domain, but not the PKC-β and Syt-IA domains, to PS–PC vesicles (Figure 10). These results support the hypothesis that membrane docking by the cPLA2-α C2 domain is stabilized mainly through hydrophobic interactions with the membrane, whereas the docking of the Syt-IA or PKC-β C2 domain is stabilized primarily by electrostatic interactions between anionic lipid headgroups and the Ca2+–protein complex (36, 40, 41, 49, 50, 53–55, 57, 61).
Figure 10.

Role of hydrophobic interactions in membrane docking by C2 domains. The indicated C2 domains and vesicles of PS –PC–dPE (47.5%:47.5%:5%) were incubated in standard assay buffer containing 1.9 M Na2SO4 and 5 mM EDTA (filled bars) or 2 mM CaCl2 and 1 mM EDTA (open bars). Membrane docking was measured by monitoring protein-to-membrane FRET. The resulting FRET signals were normalized to the Ca2+-triggered FRET signals. Error bars represent SEM of 4 or more experiments. Experimental conditions: 25 °C; 100 mM KCl, 20 mM HEPES, pH 7.4, 5 mM DTT, and 100 μM phospholipid.
Discussion
Our results demonstrate that the C2 domains of cPLA2-α, PKC-β, and Syt-IA can be selectively triggered to dock to membranes by Ca2+ concentrations that vary over a 10-fold range, from approximately 5 to 50 μM Ca2+. These in vitro [Ca2+]1/2 values likely represent upper limits to the values that would be obtained in vivo, where intracellular membranes also contain a mixture of PS and PC but are present in much higher local concentrations. Most importantly, the results predict that these three C2 domains will exhibit significantly different Ca2+ activation thresholds in vivo, such that the relative Ca2+ concentrations needed for intracellular membrane docking are lowest for cPLA2-α, intermediate for PKC-β, and highest for the Syt-IA domain. The relatively low Ca2+ threshold of the cPLA2-α C2 domain suggests that it is well adapted for low-amplitude Ca2+ signals and does not require significant assistance in membrane binding from other domains. By contrast, the higher Ca2+ threshold of the PKC-β C2 domain is consistent with the proposal that activation of the full-length enzyme requires assistance from other domains, including the docking of its regulatory C1 domain to membrane-partitioned diacylglycerol and phosphatidylserine (62, 63). The high Ca2+ threshold of the Syt-IA domain, on the other hand, suggests that it is best adapted for high-amplitude Ca2+ signals such as those generated at presynaptic plasma membranes (3, 4).
Figure 1A illustrates the similar architectures of the cPLA2-α, PKC-β, and Syt-IA C2 domains, each of which is a sandwich of two four-stranded, antiparallel β-sheets. The three Ca2+-binding loops lie at one end of the β-sandwich where they also serve as the primary membrane docking surface (42, 44, 45, 47, 49, 50, 52, 57). Figure 1B summarizes the observed Ca2+ stoichiometries of the membrane-bound C2 domains. Notably, these loops bind different numbers of Ca2+ ions: the cPLA2-α C2 domain binds two Ca2+ ions in solution, in crystals, and while bound to membranes (30, 43–45), whereas the PKC-β C2 domain binds two to three Ca2+ ions in solution and in crystals (29, 38), and the Syt-IA C2 domain binds from one to three Ca2+ ions in crystals and in solution (28, 38, 41). Our results provide the first direct evidence that the latter domains bind three or more Ca2+ ions when docked to target membranes. At least in part, the differing Ca2+ sensitivities of the three C2 domains are proposed to arise from specialized modifications of the four potential Ca2+-binding sites (I-IV) on their Ca2+-binding loops. For example, the higher Ca2+ affinity of the free cPLA2-α domain could stem from the fact that it binds only two Ca2+ ions in sites I and II, while the Syt-IA and PKC-β C2 domains bind three Ca2+ ions in sites II, III, and IV (29). Alternatively, the lower Ca2+ affinity of the Syt-IA and PKC-β domains could stem from the higher density of positive charges on their Ca2+-binding loops, which may destabilize the Ca2+ –protein complex by repulsion between like charges.
The Ca2+-binding loops are also specialized to generate different types of protein–membrane interactions. Figures 1B and 1C provide a close-up view of the Ca2+-binding loops and summarize four different mechanisms that could contribute to membrane docking, respectively. Figure 1C presents four major interactions that could stabilize membrane docking: (i) direct inner sphere coordination of the bound Ca2+ ions by anionic phospholipid headgroup oxygens, (ii) other types of electrostatic interactions between the Ca2+ –protein complex and the membrane surface, (iii) recognition of one or more headgroups by a specific binding site, and (iv) nonspecific hydrophobic interactions between protein side chains and the membrane interior. Figure 1B indicates that the Ca2+-binding loops of the cPLA2-α domain possess numerous solvent-exposed hydrophobic side chains that account for the hydrophobic nature of membrane docking by this C2 domain, whereas the Ca2+-binding loops of the Syt-IA and PKC-β C2 domains appear to utilize multiple basic side chains and a third Ca2+ ion to drive electrostatic membrane docking. Our results emphasize the importance of hydrophobic interactions in the docking of cPLA2-α C2 domain to its preferred zwitterionic lipids and electrostatic interactions in the docking of PKC-β and Syt-IA C2 domains to their preferred anionic lipids (36, 40, 41, 49, 50, 53–55, 57, 61). The different lipid preferences of C2 domains may allow their activation parameters, including Ca2+ affinity, to be tuned by the varying lipid compositions of membranes in different regions of the cell.
Overall, the present results indicate that membrane docking triggered by Ca2+ progresses via two distinct steps (Scheme 1): the C2 domain first loads rapidly with Ca2+ ions, thereby triggering the slower membrane docking step. Rapid Ca2+ removal results in disassembly of the C2 domain ternary complexes, whereupon bound Ca2+ ions trapped at the protein−membrane interface are released as the domain dissociates from the membrane (Schemes 2A and 2B). A recent crystal structure of the ternary complex formed by the PKC-α C2 domain, Ca2+ ions, and phosphatidylserine (56) directly supports this model. These findings further support the proposal that the Ca2+-binding loops interact directly with the membrane surface, as well as the hypothesis that one or more Ca2+ ions are directly coordinated both by protein and by headgroup oxygens in the ternary complex.
Scheme 2.
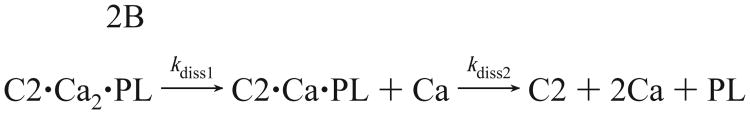
Importantly, the kinetics of membrane docking by C2 domains are tuned to different time scales, resulting in domain activation−inactivation cycles that vary in duration. This cycle is initiated by rapid Ca2+ addition that triggers membrane association and is terminated by rapid Ca2+ removal. A full cycle (1/kform + 1/kdiss) requires approximately 6 and 13 ms for the Syt-IA and PKC-β C2 domains, respectively, but at least 400 ms for the cPLA2-α C2 domain. The comparatively short cycle times of the Syt-IA and PKC-β C2 domains are consistent with their roles as rapid on/off switches in the Ca2+ triggering of synaptic events and membrane protein phosphorylation, respectively. The longer cycle time of the cPLA2-α domain provides a longer memory after a Ca2+ signal ends, thereby ensuring a minimal number of enzyme turnovers for each activation event. Additionally, the longer time constant could be used to damp out fluctuating Ca2+ signals, whereas the short time constants of the Syt-IA and PKC-β C2 domains would ensure that these domains faithfully track Ca2+ transients. It follows that rapid changes in the local cytoplasmic Ca2+ concentration will cause different C2 domains to dock to membranes on time scales that differ by tens of milliseconds or more, which could result in stepwise activation of signaling components.
We further propose that the different lifetimes observed for C2 domains arise directly from their contrasting membrane-binding mechanisms. For example, the rapid formation and dissociation kinetics of the Syt-IA and PKC-β ternary complexes (C2·Ca3·PL) are consistent with C2 domain docking to the headgroup layer of the membrane surface via multiple nonspecific electrostatic interactions that are rapidly formed and broken. By contrast, the slow formation and dissociation of the cPLA2 ternary complex (C2·Ca3·PL) is consistent with the hypothesis that this C2 domain may undergo more extensive hydrophobic penetration into the membrane (43, 49, 52–54, 61), which raises the transition state barrier for the docking reaction and slows both membrane docking and release. Thus, there is considerable heterogeneity in the mechanism of membrane docking, indicating that each C2 domain uses a specialized combination of the components summarized in Figure 1C to stabilize the docking interaction. Finally, the role played by Ca2+ in initiating membrane docking is not fully understood. In addition to its established role in triggering electrostatic changes in the membrane binding surface of the Syt-IA C2 domain (40, 41, 57), Ca2+ may provide strong energetic contributions via direct interactions with phospholipid head-groups (56) or by triggering changes to protein conformation or dynamics.
In summary, the activation parameters of C2 domains are highly specialized for their biological context. The equilibrium and kinetics of Ca2+ activation, membrane docking, and inactivation are carefully optimized so that the C2 domain will act in concert with other signaling protein domains, thereby yielding a sensor that is activated by the appropriate Ca2+ threshold concentration and that exhibits the optimal activation and inactivation response times. The observed diversity of Ca2+ activation parameters enables C2 domains to function in different intraprotein and intracellular environments. Such diversity also suggests that pathways utilizing multiple C2 domains could exhibit stepwise activation and inactivation of components, thereby triggering complex sequences of events in a carefully controlled manner. Finally, the present findings illustrate that comparative studies of related proteins provide important insights that complement and extend the information provided by genomic analysis of protein families.
Footnotes
Support provided by NIH Grants F32 GM-18383 (E.A.N.) and R01 GM-48203 (J.J.F.)
Abbreviations: cPLA2-α, cytosolic phospholipase A2-α; PKC-β, protein kinase C-β; Syt-I, synaptotagmin-I; PC, phosphatidylcholine; PS, phosphatidylserine; dansyl-PE, N-(5-dimethylaminonaphthalene-1-sulfonyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine; GST, glutathione S-transferase; FRET, fluorescence resonance energy transfer; DTT, dithiothreitol; HEPES, N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid; EDTA, ethylenediaminetetraacetic acid.
References
- 1.Berridge MJ, Bootman MD, Lipp P. Nature. 1998;395:645. doi: 10.1038/27094. [DOI] [PubMed] [Google Scholar]
- 2.Llinás R, Steinberg IZ, Walton K. Biophys J. 1981;33:323. doi: 10.1016/S0006-3495(81)84899-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Llinás R, Sugimori M, Silver RB. Science. 1992;256:677. doi: 10.1126/science.1350109. [DOI] [PubMed] [Google Scholar]
- 4.Heidelberger R, Heinemann C, Neher E, Matthews G. Nature. 1994;371:513. doi: 10.1038/371513a0. [DOI] [PubMed] [Google Scholar]
- 5.Cheng H, Lederer MR, Xiao RP, Gomez AM, Zhou Y, Ziman B, Spurgeon H, Lakatta EG, Lederer WJ. Cell Calcium. 1996;20:129. doi: 10.1016/s0143-4160(96)90102-5. [DOI] [PubMed] [Google Scholar]
- 6.Thomas AP, Bird G, St J, Hajnoóczky G, Robb-Gaspers LD, Putney JW. FASEB J. 1996;10:1505. [PubMed] [Google Scholar]
- 7.Falke JJ, Drake SK, Hazard AL, Peersen OB. Q Rev Biophys. 1994;27:219. doi: 10.1017/s0033583500003012. [DOI] [PubMed] [Google Scholar]
- 8.Linse S, Forsén S. Adv Second Messenger Phosphoprotein Res. 1995;30:89. doi: 10.1016/s1040-7952(05)80005-9. [DOI] [PubMed] [Google Scholar]
- 9.Ponting CP, Parker PJ. Protein Sci. 1996;5:162. doi: 10.1002/pro.5560050120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nalefski EA, Falke JJ. Protein Sci. 1996;5:2375. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rizo J, Südhof TC. J Biol Chem. 1998;273:15879. doi: 10.1074/jbc.273.26.15879. [DOI] [PubMed] [Google Scholar]
- 12.Schultz J, Milpetz F, Bork P, Ponting CP. Proc Natl Acad Sci U S A. 1998;95:5857. doi: 10.1073/pnas.95.11.5857. see http://smart.embl-heidelberg.de/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brose N, Petrenko AG, Südhof TC, Jahn R. Science. 1992;256:1021. doi: 10.1126/science.1589771. [DOI] [PubMed] [Google Scholar]
- 14.Südhof TC, Rizo J. Neuron. 1996;17:379. doi: 10.1016/s0896-6273(00)80171-3. [DOI] [PubMed] [Google Scholar]
- 15.Fukuda M, Mikoshiba K. Bioessays. 1997;19:593. doi: 10.1002/bies.950190710. [DOI] [PubMed] [Google Scholar]
- 16.Nishizuka Y. Nature. 1988;334:661. doi: 10.1038/334661a0. [DOI] [PubMed] [Google Scholar]
- 17.Dekker LV, Parker PJ. Trends Biochem Sci. 1994;19:73. doi: 10.1016/0968-0004(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 18.Newton AC. Curr Opin Cell Biol. 1997;9:161. doi: 10.1016/s0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- 19.Clark JD, Lin LL, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, Knopf JL. Cell. 1991;65:1043. doi: 10.1016/0092-8674(91)90556-e. [DOI] [PubMed] [Google Scholar]
- 20.Nalefski EA, Sultzman LA, Martin DM, Kriz RW, Towler P, Knopf J, Clark JD. J Biol Chem. 1994;269:18239. [PubMed] [Google Scholar]
- 21.Glover S, de Carvalho M, Bayburt T, Jonas M, Chi E, Leslie CC, Gelb MH. J Biol Chem. 1995;270:15359. doi: 10.1074/jbc.270.25.15359. [DOI] [PubMed] [Google Scholar]
- 22.Schievella A, Regier M, Smith WL, Lin LL. J Biol Chem. 1995;270:30749. doi: 10.1074/jbc.270.51.30749. [DOI] [PubMed] [Google Scholar]
- 23.Gijón MA, Spencer DM, Kaiser AL, Leslie CC. J Cell Biol. 1999;145:1219. doi: 10.1083/jcb.145.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clark JD, Schievella AR, Nalefski EA, Lin LL. J Lipid Mediators Cell Signalling. 1995;12:83. doi: 10.1016/0929-7855(95)00012-f. [DOI] [PubMed] [Google Scholar]
- 25.Kramer RM, Sharp JD. FEBS Lett. 1997;410:49. doi: 10.1016/s0014-5793(97)00322-0. [DOI] [PubMed] [Google Scholar]
- 26.Leslie CC. J Biol Chem. 1997;272:16709. doi: 10.1074/jbc.272.27.16709. [DOI] [PubMed] [Google Scholar]
- 27.Needham JV, Chen TY, Falke JJ. Biochemistry. 1993;32:3363. doi: 10.1021/bi00064a020. [DOI] [PubMed] [Google Scholar]
- 28.Sutton RB, Davletov BA, Berghuis AM, Südhof TC, Sprang SR. Cell. 1995;80:929. doi: 10.1016/0092-8674(95)90296-1. [DOI] [PubMed] [Google Scholar]
- 29.Sutton RB, Sprang SR. Structure. 1998;6:1395. doi: 10.1016/s0969-2126(98)00139-7. [DOI] [PubMed] [Google Scholar]
- 30.Nalefski EA, Slazas MM, Falke JJ. Biochemistry. 1997;36:12011. doi: 10.1021/bi9717340. [DOI] [PubMed] [Google Scholar]
- 31.Lakowicz JR. Principles of Fluorescence Spectroscopy. Kluwer/Plenum Publishers; New York: 1999. [Google Scholar]
- 32.Wyman J, Gill SJ. Binding and Linkage. University Science Books; Mill Valley, CA: 1990. [Google Scholar]
- 33.Grabarek Z, Gergely J. J Biol Chem. 1983;258:14103. [PubMed] [Google Scholar]
- 34.Rebecci M, Peterson A, McLaughlin S. Biochemistry. 1992;31:12742. doi: 10.1021/bi00166a005. [DOI] [PubMed] [Google Scholar]
- 35.Olson ST, Halvorson HR, Björk I. J Biol Chem. 1991;266:6342. [PubMed] [Google Scholar]
- 36.Nalefski EA, McDonagh T, Somers W, Seehra J, Falke JJ, Clark JD. J Biol Chem. 1998;273:1365. doi: 10.1074/jbc.273.3.1365. [DOI] [PubMed] [Google Scholar]
- 37.Tonomura B, Nakatani H, Ohnishi M, Yamaguchi-Ito J, Hiromi K. Anal Biochem. 1978;84:370. doi: 10.1016/0003-2697(78)90054-4. [DOI] [PubMed] [Google Scholar]
- 38.Shao X, Davletov BA, Sutton RB, Südhof TC, Rizo J. Science. 1996;273:248. doi: 10.1126/science.273.5272.248. [DOI] [PubMed] [Google Scholar]
- 39.Shao X, Fernandez I, Südhof TC, Rizo J. Biochemistry. 1998;37:16106. doi: 10.1021/bi981789h. [DOI] [PubMed] [Google Scholar]
- 40.Shao X, Li C, Fernandez I, Zhang X, Südhof TC, Rizo J. Neuron. 1998;18:133. doi: 10.1016/s0896-6273(01)80052-0. [DOI] [PubMed] [Google Scholar]
- 41.Ubach J, Zhang X, Shao X, Südhof TC, Rizo J. EMBO J. 1998;17:3921. doi: 10.1093/emboj/17.14.3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chae YK, Abildgaard F, Chapman ER, Markley JL. J Biol Chem. 1998;273:25659. doi: 10.1074/jbc.273.40.25659. [DOI] [PubMed] [Google Scholar]
- 43.Perisic O, Fong S, Lynch DE, Bycroft M, Williams RL. J Biol Chem. 1998;273:1596. doi: 10.1074/jbc.273.3.1596. [DOI] [PubMed] [Google Scholar]
- 44.Xu GY, McDonagh T, Yu HA, Nalefski EA, Clark JD, Cumming DA. J Mol Biol. 1998;280:485. doi: 10.1006/jmbi.1998.1874. [DOI] [PubMed] [Google Scholar]
- 45.Dessen A, Tang J, Schmidt H, Stahl M, Clark JD, Seehra J, Somers WS. Cell. 1999;97:349. doi: 10.1016/s0092-8674(00)80744-8. [DOI] [PubMed] [Google Scholar]
- 46.Tudyka T, Skerra A. Protein Sci. 1997;6:2180. doi: 10.1002/pro.5560061012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis AF, Baj J, Fasshauer D, Wolowick MJ, Lewis JL, Chapman ER. Neuron. 1999;24:363. doi: 10.1016/s0896-6273(00)80850-8. [DOI] [PubMed] [Google Scholar]
- 48.Hixon MS, Ball A, Gelb MH. Biochemistry. 1998;37:8516. doi: 10.1021/bi980416d. [DOI] [PubMed] [Google Scholar]
- 49.Nalefski EA, Falke JJ. Biochemistry. 1998;37:17642. doi: 10.1021/bi982372e. [DOI] [PubMed] [Google Scholar]
- 50.Chapman ER, Davis AF. J Biol Chem. 1998;273:13995. doi: 10.1074/jbc.273.22.13995. [DOI] [PubMed] [Google Scholar]
- 51.Johnson KA. In: The Enzymes. Sigman DS, editor. Academic Press; San Diego, CA: 1992. pp. 1–61. [Google Scholar]
- 52.Ball A, Nielsen R, Gelb MH, Robinson BH. Proc Natl Acad Sci U S A. 1999;96:6637. doi: 10.1073/pnas.96.12.6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bittova L, Summandea M, Cho W. J Biol Chem. 1999;274:9665. doi: 10.1074/jbc.274.14.9665. [DOI] [PubMed] [Google Scholar]
- 54.Perisic O, Paterson HF, Mosedale G, Lara-Gonzalez S, Williams RL. J Biol Chem. 1999;274:14979. doi: 10.1074/jbc.274.21.14979. [DOI] [PubMed] [Google Scholar]
- 55.Medkova M, Cho W. J Biol Chem. 1999;274:19852. doi: 10.1074/jbc.274.28.19852. [DOI] [PubMed] [Google Scholar]
- 56.Verdaguer N, Corbalan-Garcia S, Ochoa WF, Fita I, Gómez-Fernàndez JC. EMBO J. 1999;18:6329. doi: 10.1093/emboj/18.22.6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang X, Rizo J, Südhof TC. Biochemistry. 1998;37:12395. doi: 10.1021/bi9807512. [DOI] [PubMed] [Google Scholar]
- 58.Bayley P, Ahlström P, Martin SR, Forsén S. Biochem Biophys Res Commun. 1984;120:185. doi: 10.1016/0006-291x(84)91431-1. [DOI] [PubMed] [Google Scholar]
- 59.Davletov B, Südhof TC. J Biol Chem. 1993;268:26386. [PubMed] [Google Scholar]
- 60.Chapman ER, Jahn R. J Biol Chem. 1994;269:5735. [PubMed] [Google Scholar]
- 61.Davletov B, Perisic O, Williams RL. J Biol Chem. 1998;273:19093. doi: 10.1074/jbc.273.30.19093. [DOI] [PubMed] [Google Scholar]
- 62.Johnson JE, Zimmerman ML, Daleke DL, Newton AC. Biochemistry. 1998;37:12020. doi: 10.1021/bi981107q. [DOI] [PubMed] [Google Scholar]
- 63.Newton AC, Johnson JE. Biochim Biophys Acta. 1998;1376:155. doi: 10.1016/s0304-4157(98)00003-3. [DOI] [PubMed] [Google Scholar]
- 64.Johnson JE, Giorgione J, Newton AC. Biochemistry. 2000;39:11360–11369. doi: 10.1021/bi000902c. [DOI] [PubMed] [Google Scholar]