

Abstract
NK cells are innate lymphocytes important for host defense against viral infections and malignancy. However, the molecular programs orchestrating NK cell activation are incompletely understood. miR-155 is markedly upregulated following cytokine activation of human and mouse NK cells. Surprisingly, mature human and mouse NK cells transduced to overexpress miR-155, NK cells from mice with NK cell-specific miR-155 overexpression, and miR-155−/− NK cells all secreted more IFN-γ compared to controls. Investigating further, we found that activated NK cells with miR-155 overexpression had increased per cell IFN-γ with normal IFN-γ+ percentages, whereas greater percentages of miR-155−/− NK cells were IFN-γ+. In vivo MCMV-induced IFN-γ expression by NK cells in these miR-155 models recapitulated the in vitro phenotypes. We performed unbiased RISC-Seq on WT and miR-155−/− NK cells, and found that mRNAs targeted by miR-155 were enriched in NK cell activation signaling pathways. Using specific inhibitors, we confirmed these pathways were mechanistically involved in regulating IFN–γ production by miR-155−/− NK cells. These data indicate that miR-155 regulation of NK cell activation is complex, and that miR-155 functions as a dynamic tuner for NK cell activation via both setting the activation threshold as well as controlling the extent of activation in mature NK cells. In summary, miR-155−/− NK cells are more easily activated, through increased expression of proteins in the PI3K, NF-κB, and calcineurin pathways, and miR-155−/− and 155-overexpressing NK cells exhibit increased IFN-γ production through distinct cellular mechanisms.
Keywords: miR-155, NK cell, micro-RNA
INTRODUCTION
Natural killer (NK) cells are innate immune lymphocytes important for host protection against infection and mediate anti-tumor effector responses, especially against hematologic malignancies (1, 2). NK cells develop from the common lymphoid progenitor along with T and B cells (3), but undergo a distinct developmental pathway without DNA rearrangement of a clonal antigen receptor. Mature NK cells integrate activating and inhibitory signals mediated by a wide variety of surface receptors (4). These receptors signal through intracellular or adapter-containing ITAM/ITIM motifs (5). Upon cytokine receptor activation, NK cells acquire enhanced functional competence and also produce NK cell-derived cytokines/chemokines, including IFN-γ, GM-CSF, TNF-α, and MIP1-α (2). When triggered through cell surface receptors, NK cells release cytotoxic granules and kill target cells (6). It is through these primary functions that NK cells provide a rapid response to infected or tumor target cells, as well as participate in complex cross-talk with other immune cell types.
In the physiologic setting of a complex receptor-ligand environment, NK cells continually adapt, highlighted by studies in which transgenic overexpression of activating receptor ligands leads to NK cell functional anergy (7), or transfer of NK cells from an MHC-class I sufficient environment into an MHC-class I deficient environment, where the NK cells rapidly become hypofunctional (8). This ability of NK cells to adapt to their environment by altering their functionality is summarized by a model known as the tuning or ‘rheostat’ model (9), which proposes that the relative strength of the activating and inhibitory signals that an NK cell receives tunes up (arms) or down (disarms) the NK cell responsiveness. This education process both prevents inappropriate NK cell activation, which could lead to autoimmune inflammation and disease, and is responsible for graft-versus-leukemia effects, by setting a threshold for activation (10). While some of the molecular events responsible for regulating NK cell activation and tuning have been defined (11, 12), our understanding of the molecules and intracellular changes that control these processes are incomplete.
microRNAs (miRNAs) are a family of small, non-coding RNAs that mediate downregulation of targeted mRNA transcripts by binding to complementary sites primarily in the 3′UTRs of mRNAs (13). miRNAs have been shown to have a wide variety of roles in cancer (14), inflammation (15), and immune responses (16). In NK cells, miRNAs have been shown to regulate NK cell proliferation, survival, and alter functionality (17–19). Recently, individual miRNAs have been shown to influence NK cell development and function (20–22), including miR-155 (23, 24).
miR-155 is encoded within the BIC non-coding RNA, and its role in T cells, B cells, and macrophages has been characterized (25–28). miR-155 is expressed in resting NK cells (29), and has been shown to be overexpressed in NK cell type lymphoma/leukemia (20). A recent report has identified that miR-155 regulates IFN-γ production by human NK cells partially via repression of SHIP-1, a phosphatase involved in negative regulation of PI3K signaling (23). In addition, mice expressing a Lck-driven miR-155 transgene have NK cell IFN-γ alterations, further linking miR-155 repression of SHIP1 to NK cell activation (24). miR-155 deficient mice were further found to have alterations in murine cytomegalovirus (MCMV) responses (30). However, mice that are deficient in miR-155 (155−/−), or that over-express miR-155 in an NK cell-restricted fashion (155FOE), have not been directly compared and evaluated in depth for alterations in NK cell responses.
Here, we compared the effects of multiple miR-155 alterations, including lentiviral overexpression in mature human and murine NK cells, as well as NK cells from 155−/− and 155FOE mouse models. Unexpectedly, both 155FOE and 155−/− NK cells exhibit an augmented IFN-γ response. We found that 155−/− NK cells had increased IFN-γ secretion due to an increased percentage of IFN-γ+ NK cells without a change in per-cell expression, indicating a novel role for miR-155 in altering the NK cell activation threshold. Conversely, 155FOE NK cells produced more IFN-γ per cell. The cellular mechanism for miR-155’s dual effect on IFN-γ was recapitulated in vivo during MCMV infection of 155−/− and 155FOE mice. Notably, 155−/− mice had decreased MCMV titers, suggesting that the increased functionality of 155−/− NK cells has a biologically significant impact on the early NK-mediated anti-viral cytokine response. Utilizing RISC-Seq analysis of activated NK cells from WT and 155−/− mice, we identified and validated novel miR-155 mRNA targets in NK cells. We further used chemical inhibitors of multiple activation pathways to eliminate IFN-γ production differences, indicating that miR-155 extensively regulates molecules involved in NK cell activation, thereby regulating the NK cell activation threshold.
MATERIALS AND METHODS
Mice
155−/− mice were described previously,(26) and were obtained from The Jackson Laboratory as B6.Cg-Mir155tm1.1Rsky/J. 155FOE mice were generated by crossing Tg(Ncr1-iCre)265Sxl mice previously described (31), with mice containing a miR-155 LoxP-STOP-LoxP expression cassette knocked in to the Rosa locus (26). Rosa26-STOP-eYFP mice were obtained from The Jackson Laboratory as B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J, and has been backcrossed at least 6 generations onto a C57BL/6 background. All other mice were originally generated on a C57BL/6 background. All mice have been bred and maintained in specific pathogen-free housing, and all experiments were conducted in accordance with the guidelines of and with the approval of the Washington University Animal Studies Committee. Mice were used between 8 and 12 weeks of age for all experiments.
Antibodies
Anti-mouse mAbs were obtained from BD Biosciences (San Jose, CA): IFN-γ (XMG1.2), NK1.1 (PK136), NKp46 (29A1.4), CD3 (145-2C11), CD45 (30-F11), CD27 (LG.3A10), CD11b (M1/70), CD19 (1D3), CD212 (114), CD132 (4G3), Ly49A (JR9-318), Ly49C/I (5E6), Ly49G2 (4D11), Ly49D (4E5), Ly49H (3D10); eBioscience (San Diego, CA): CD107a (1D4B), NKG2A (16a11), NKG2ACE (20d5), CD94 (18d3), CD122 (TM-β1); Caltag: GzmB (GB12); BioLegend (San Diego, CA): CD218a (BG/IL18Ra), CD226 (TX42.1); Santa Cruz Biotechnology: SLP-76 (C-20), IKBKE (H-116), SHIP1 (P1C1), and β-actin (C4). Anti-Ly49C (4LO) was kindly provided by W. Yokoyama. Anti-human antibodies were obtained from Beckman Coulter: CD3 (UCHT1), CD56 (N901), CD158a,h (EB6B), CD158b1,b2,j (GL183); BD Biosciences: CD16 (3G8), CD94 (HP-3D9); BioLegend: IFN-γ (B27).
Cell Lines and reagents
Endotoxin-free purified recombinant cytokines (rmIL-12, rmIL-15, rmIL-18, rhIL-12, rhIL-15, rhIL-18) were obtained from Peprotech (Rocky Hill, NJ) and reconstituted in sterile PBS with 0.1% BSA. Anti-NK1.1 (PK136) was purified by the Washington University antibody production core from hybridoma supernatant. Chemical inhibitors (Ly294002, BAY 11-7082, and cyclosporin A) were obtained from EMD Millipore.
IFN-γ ELISA
Sorted GFP+ (LV-GFP, LV-GFP/155), Cre+ (155FOE), or bulk (155−/−) NK cells were plated in duplicate, and cultured for the indicated periods of time in cRPMI + 10% FBS + 10ng/mL each indicated cytokine or immobilized antibody. Cell-free supernatants were collected and frozen at −80, thawed only once, and analyzed by ELISA from eBioscience according to the manufacturer’s instructions.
IFN-γ stimulation assay/Inhibitor Assay
IFN-γ stimulation was performed as previously described (18). Licensing assays were performed as described previously (32). For inhibition assays, IC50 values were obtained from the manufacturer, or in cases in which the inhibitor had been tested in NK cells, were used at the previously demonstrated value (cyclosporin A = 35.7nM (33), BAY 11-7082 = 10μM (manufacturer), Ly294002 = 1.2μM (34)). Conditions with NK cell toxicity were excluded.
Lentiviral Transduction
High titer MND-ΔU3 (35) lentivirus was generated using standard third generation packaging systems, pseudotyped with VSV-G, in ultracentrifuged supernatants from 293T cells. Human NK cells were purified from peripheral blood to >90% purity using RosetteSep (StemCell Technologies, Vancouver, Canada). Mouse NK cells were sorted to >99% purity from RAG−/− spleens. 1×106 human NK cells were cultured in cRPMI + 10% HAB Serum + 100ng/mL rhIL-15, while mouse NK cells were cultured in cRPMI + 10% FBS + 100ng/mL rmIL-15. Lentivirus was then added at an MOI of 20, and the cells were centrifuged (“spinfected”) at 2,000 rpm, 30°C, 45 minutes in a standard swinging bucket table-top centrifuge. Cells were cultured overnight at 37°C in 5% CO2, and spinfection was repeated the next day (day 1). On day 2, media was replaced by removing ½ media, taking care not to disturb the cells, and adding cRPMI + 10%FBS +10ng/mL rmIL-15 or HAB + 10ng/mL rhIL-15 as appropriate. Media was replaced in this manner every other day until harvest. Mouse NK cells were harvested at day 7, while human NK cells were harvested at day 4 or day 9, and then sorted to >99% purity for GFP+ NK cells.
Flow cytometry
All flow cytometry was conducted on a Beckman Coulter Gallios machine, and all cells were sorted using a BD FACSAria II at the Washington University Pathology Flow Cytometry and Fluorescence Activated Cell Sorting Core. Flow cytometry was analyzed using FlowJo (TreeStar, Ashland, OR).
Statistical analysis
Graphical analysis and statistics were performed with GraphPad Prism 5.0. Student t test, one-way ANOVA, and two-way ANOVA were used as appropriate, with p<0.05 considered significant. *p<0.05, **p<0.01, ***p<0.001.
MCMV infection and plaque assays
MCMV infections, stock preparations, and titering were performed as described previously (36, 37). Mice were infected with 10,000 PFU MCMV (Smith Strain), and were injected with BrdU 4 days post-infection. 4 hours post-BrdU injection, spleens were harvested and assessed using standard BrdU intracellular flow cytometry.
RT-qPCR
qPCR was performed on sorted NK cells as indicated by isolating total RNA from Trizol (Invitrogen), according to the manufacturer’s instructions. TaqMan Assays (ABI) were used to create cDNA and RT-qPCR was run on an ABI 7300, according to the manufacturers’ instructions. Relative quantification was determined by the ddCt method, by normalizing either to sno-135 (mouse) or RNU48 (human).
RISC-Seq
1×106 to 3×106 WT or 155−/− NK cells were sorted, and allowed to recover for two days in 100ng/mL IL-15. The NK cells were then activated with 10ng/mL each of IL-12, IL-15, and IL-18 for 24 hours. RISC-Seq was then performed exactly as described (38).
Luciferase Target Validation
Performed as previously described (18) with primers available upon request.
RESULTS
miR-155 expression is markedly upregulated after NK cell activation in vitro and in vivo
miR-155 is moderately expressed in resting mouse NK cells when evaluated by next-generation small RNA sequencing (Fig. 1A).(29) However, miR-155 is markedly upregulated after cytokine activation, especially after stimulation with IL-12, IL-15, and IL-18 (Fig. 1B and (23)). This upregulation was consistent between human and murine NK cells (Fig. 1B). Furthermore, miR-155 is induced in murine NK cells 36 hours after infection with MCMV (Fig. 1C). This corresponds to the peak of IFN-γ production by splenic NK cells (Fig. 1D, (37)). Therefore, miR-155 expression is markedly increased in both mouse and human NK cells after in vitro stimulation and in the context of a physiologic anti-viral response.
Figure 1. miR-155 is expressed in NK cells and induced after NK cell activation in vitro and in vivo.

miR-155 expression was assessed by deep sequencing (A) or qPCR (B, C) after cytokine stimulation (B) or MCMV infection (C). (A) miR-155 is expressed in resting mouse NK1.1+CD3− splenic NK cells. Data shown are the rank order of all expressed NK cell miRNAs by small RNA sequencing from mouse NK cells (29). Sequencing reads were normalized to total miRNA reads and plotted as log2 (normalized read counts) versus rank order of expression in resting NK cells. The large circle indicates miR-155. (B) Human CD56+CD3− peripheral blood NK cells (left) and mouse NK1.1+CD3− splenic NK cells (right) − were enriched to >95% purity and evaluated for miR-155 expression at rest, or at the indicated time points after cytokine activation. Relative fold change was assessed by miR-155 RT-qPCR. Data summarize 5 mice and 4 donors from 2–3 independent experiments. (C) miR-155 is induced following MCMV infection in NK cells. Ly49H+ and Ly49H− NK1.1+CD3− NK cells were sorted from the spleens of naïve control mice or control mice infected with MCMV (5×104 pfu/mouse) at 1.5 or 7 days p.i. miR-155 mRNA levels were compared by RT-qPCR, and found to be significantly increased in both NK cell subsets at 1.5 days p.i. Data summarize 10 mice from 3 independent experiments. (D) Summary data showing the robust IFN-γ production at day 1.5 post-infection. Data summarize 10 mice from 3 independent experiments.
Overexpression of miR-155 in mature mouse and human NK cells augments activation-induced IFN-γ
Because miR-155 expression increases in activated NK cells, we evaluated the impact of forced lentiviral (LV) over-expression of miR-155 on the IFN-γ response in both mouse and human NK cells. Both control LV-GFP and LV-GFP/155 were able to efficiently transduce human and mouse NK cells (data not shown), and LV-GFP/155 virus resulted in miR-155 overexpression in resting and cytokine-activated mouse and human NK cells (Fig. 2A). Mature NK cells transduced with LV-GFP/155 or LV-GFP control were sorted for GFP+ NK cells, and stimulated to secrete IFN-γ. When miR-155 was over-expressed, we observed enhanced IFN-γ production in human NK cells after stimulation with IL-12 plus IL-18, but not IL-12 plus IL-15 alone, due to a high degree of human donor variability (Fig. 2B, Fig. S2). miR-155 overexpression also increased the MFI of staining with an anti-CD158b1/b2/j mAb (but not other KIRs) on the surface of human NK cells (Fig. S2I). Similar experiments with mature splenic WT NK cells showed that mature mouse NK cells transduced with LV-GFP/155 also produced more IFN-γ protein when stimulated with IL-12 plus IL-18, or with IL-12 plus IL-15 (Fig. 2C). Therefore, forced overexpression of miR-155 results in increased IFN-γ production.
Figure 2. Overexpression of miR-155 in human and mouse mature NK cells results in increased IFN-γ protein secretion.

(A) Relative miR-155 expression measured by qPCR from human and mouse LV-transduced NK cells (mean ± SEM fold change), including both resting and IL-12+IL-18-stimulated NK cells. Data summarize 5–6 donors from 3 independent experiments, and 4–12 mice from 2–3 independent experiments. (B) IFN-γ protein production by human NK cells by ELISA after 12 hours of stimulation with the indicated cytokines. Data summarize 8–10 donors from 2 independent experiments. (C) IFN-γ protein production by mouse NK cells measured by ELISA after 12 hours of stimulation with the indicated cytokines. Data show one experiment of two groups (n=2–3 mice/group), representative of 4 groups of n=2–3 mice, from 3 independent experiments. For (B and C), data are shown as mean ± SEM IFN-γ concentration.
NK cells from 155−/− mice have an intact NK cell compartment and enhanced cytokine-induced IFN-γ production
In order to further investigate the role of miR-155 in regulating NK cells, we examined 155−/− mice (Fig. 3A, Thai et al., 2007). An in depth characterization of the NK cell compartment in 155−/− mice found no changes in NK cell numbers, percentages, development, maturation, survival, or homeostasis from all tissues examined (Fig. S1). NK cells from 155−/− mice have normal expression of most NK cell receptors (Fig. S1), although we observed significantly decreased expression of Ly49G2 and a slight increase in Ly49A expression (Fig. S1D). Therefore, prior to activation, 155−/− NK cells appear similar to WT B6 NK cells. Because NK cells transduced to overexpress miR-155 produced more total IFN-γ, we expected that the 155−/− NK cells would have the opposite phenotype: decreased IFN-γ production after activation. However, we were surprised to find that the 155−/− NK cells secreted more IFN-γ after stimulation with IL-12 plus IL-15 (Fig. 3B) or IL-12 plus IL-18 (Fig. 3C) as measured by ELISA. Furthermore, 155−/− NK cells produce more granzyme B upon stimulation with IL-15 (Fig. S1F), and have increased levels of surface CD107a after NK1.1 ligation (Fig. S1G) suggesting a global enhancement in responses following activation or triggering. We confirmed that Ly49G2 and Ly49A expression was not associated with this IFN-γ phenotype (Fig. S1I), and 155−/− and control NK cells had similar Ly49C-based licensing ratios (Fig. S1J-K). Despite this increase in global activation, the killing of YAC-1 tumors by 155−/− and control NK cells after 48 hours of IL-15 stimulation was not significantly different (Fig. S1H). This may reflect an alteration of threshold following high-dose IL-15 stimulation in vitro. Collectively, these data suggest that 155−/− NK cells are more responsive to activation.
Figure 3. NK cells from 155−/− mice have enhanced IFN-γ production.
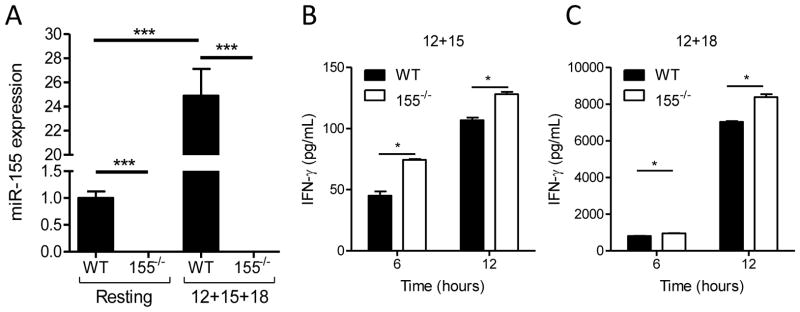
(A) NK cells sorted from 155−/− mice express no detectable miR-155 at rest or following 24 hours of IL-12+IL-15+IL-18 combined activation. Data shown (mean ± SEM fold change) are normalized to WT resting NK cells and summarize 4 pools of 4–5 mice from 3 independent experiments. (B and C) 155−/− NK cells exhibit increased IFN-γ production after 6 or 12 hours of cytokine stimulation. IFN-γ protein was measured by ELISA from cell-free culture supernatants after 6 or 12 hours with IL-12+IL-15 (B) or IL-12+IL-18 (C). Data shown are the mean ± SEM from one experiment (2–3 mice/genotype), representative of 3 independent experiments.
Mice with NK cell-specific miR-155 overexpression (155FOE) have a normal NK cell compartment and produce more IFN-γ following activation
We hypothesized that the incongruous phenotype between 155−/− and LV-GFP/155 models could be explained by in vitro culture and/or lentiviral transduction. To further investigate this premise we generated a conditional miR-155 overexpression knock-in model(26) combined with an NK cell-specific Cre (Ncr1-iCre),(31) to allow for specific miR-155 overexpression in NK cells (155FOE). In this model, miR-155 overexpression commences at an early stage of dedicated NK cell development and persists throughout the lifespan of the mature NK cell, with Cre+ NK cells marked by GFP (NK cells routinely ≥85% Cre+). Cre+ NK cells from 155FOE mice exhibit increased miR-155 expression, compared to both WT Cre+ NK cells, or the small number of Cre−NK cells within 155FOE mice (Fig. 4A). Similar to 155−/− mice, resting 155FOE NK percentages, numbers, maturation, surface receptor expression, and ex vivo expansion were normal (Fig. S1D, L-P), with the exception of an increased percentage of Ly49G2+ NK cells (Fig. S1D). We next investigated IFN-γ production by cytokine-activated 155FOE NK cells. Sorted GFP+ 155FOE NK cells or control Cre+ (RosaYFP) NK cells were stimulated with IL-12 plus IL-15 or IL-12 plus IL-18 and analyzed for IFN-γ production by ELISA (Fig. 4B, C). In this model, IFN-γ production was also increased after stimulation, compared to controls. Therefore, forced miR-155 overexpression initiated early in NK development again lead to increased total IFN-γ production in mature mouse NK cells.
Figure 4. NK cells from 155FOE mice have increased levels of miR-155 and increased IFN-γ production.
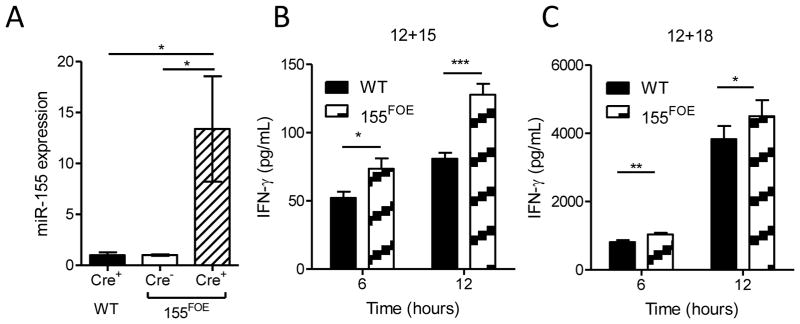
(A) miR-155 expression was measured using RT-qPCR in sorted resting WT GFP+ NK cells, 155FOE GFP− and GFP+ NK cells from the spleen, normalized to WT GFP+ NK cells with sno135 as the calibrator. Data summarize at least 3 mice per group from 3 independent experiments. (B and C) IFN-γ secretion as measured by ELISA of cell-free supernatants by flow sorted GFP+ NK cells after 6 or 12 hours of stimulation with IL-12+IL-15 (B) or IL-12+IL-18 (C). Data shown are mean ± SEM IFN-γ concentration of one experiment of 2–3 mice per group, representative of 2 independent experiments.
Distinct cellular mechanisms are responsible for enhanced IFN-γ production by 155−/− versus 155FOE NK cells
In an effort to better understand the seemingly disparate finding that both 155−/− and 155FOE NK cells produce more IFN-γ than control NK cells, we investigated per-cell IFN-γ production by intracellular flow cytometry. We found that 155−/− NK cells had an increased percentage of IFN-γ+ NK cells following activation with NK1.1 ligation, IL-12 plus IL-15, or IL-12 plus IL-18 (Fig. 5A, C–E), with either no change or a modest reduction of IFN-γ protein per NK cell, determined by median fluorescence intensity (MFI) (Fig. 5A, F–H). In contrast, NK cells from 155FOE mice had increased per NK cell expression of IFN-γ (MFI) following activation with NK1.1 ligation or IL-12 plus IL-15 (Fig. 5B, F-H), without alterations in the percentage of NK cells responding (Fig. 5B, C–E). In the IL-12 plus IL-18 experiments, the percentage positive and MFI for IFN-γ was maximal in control NK cells, thereby providing a minimal dynamic range to detect IFN-γ increases after miR-155 alteration. Since our genetic mouse models of miR-155 loss and overexpression in NK cells revealed different cellular mechanisms of enhanced IFN-γ production, we analyzed per NK cell IFN-γ production in mature human and mouse NK cells following lentiviral overexpression of miR-155. Consistent with 155FOE mice, LV-GFP/155 transduced human (Fig. S2A–C) and mouse (Fig. S2D–F) NK cells demonstrated increased IFN-γ production per NK cell, as well as an increased percentage of IFN-γ+ NK cells in LV-GFP/155 compared to control NK cells (Fig. S2C, F). The net sum of the percentage of IFN-γ+ NK cells and the amount of IFN-γ produced by each cell recapitulates and explains the increased total IFN-γ measured by ELISA in both of these models (Figs. 2–4), suggesting a role for an activation threshold alteration in the 155−/− NK cells. Indeed, α-NK1.1 mAb dilutions revealed a concentration at which 155−/− NK cells produced IFN-γ, but WT NK cells did not (Fig. 5I). Though these differences occur primarily in Ly49C+ cells, which are the primary IFN-γ producers, we observed no alterations in the licensing ratio in the 155−/− NK cells (Fig. S1J-K). Therefore, distinct cellular mechanisms explain the phenomenon of increased total IFN-γ production in all three models.
Figure 5. NK cells from 155−/− and 155FOE mice produce increased IFN-γ by different cellular mechanisms.

Representative intracellular IFN-γ flow cytometry histograms of 155−/− (A, light gray) and 155FOE (B, dark gray) compared to WT NK cells (A, black) or GFP+ control NK cells (B, black), after stimulation with IL-12+IL-15 for 6 hours. (C–E) Normalized summary data for percent IFN-γ positive NK cells (left) demonstrates a significantly higher percentage of 155−/− NK cells are positive for IFN-γ, while 155FOE NK cells are unchanged, compared to control NK cells. (F–H) The IFN-γ expression on a per cell basis (MFI) of IFN-γ+ NK cells (right) was unaffected or modestly decreased within 155−/− NK cells, while 155FOE NK cells exhibited a significant increase in the expression of IFN-γ on a per cell basis, in all conditions except IL-12+IL-18. (I) 155−/− NK cells are activated to produce IFN-γ at concentrations that do not activate WT NK cells, indicating an activation threshold defect. Data in (C–H) summarize 5–20 mice per group from 2–7 independent experiments. Data in (I) are from 3 mice per group in one experiment, representative of three independent experiments.
MCMV infection recapitulates in vitro IFN-γ responses of 155−/− and 155FOE NK cells, and leads to decreased viral titers in 155−/− mice
To determine whether or not the increased activation of 155−/− NK cells was relevant in vivo, we infected 155−/− and B6 WT mice, or 155FOE and RosaYFP control mice, with MCMV. At 36 hours postinfection (p.i.), an increased percentage of 155−/− NK cells responded to MCMV infection by producing IFN-γ (Fig. 6A–C), compared to control NK cells. In contrast, 155FOE NK cells had increased IFN-γ MFI with no alteration in the IFN-γ+ NK cell percentage (Fig. 6D–F). Therefore, these in vivo results recapitulated the in vitro IFN-γ phenotype. We next evaluated whether the increased NK cell-derived IFN-γ at 1.5 days p.i. affected the course of MCMV infection by measuring MCMV viral titers of 155−/− mice at 3 days p.i. (Fig. 6G). 155−/− mice had decreased MCMV titers at 3 days p.i., consistent with the observed enhanced IFN-γ response. Thus, NK cells from 155−/− mice exhibit enhanced early IFN-γ production during the cytokine-dependent phase, and miR-155 disrupts the anti-MCMV NK cell response.
Figure 6. 155−/− and 155FOE NK cells produce more IFN-γ during MCMV infection in vivo.

(A–C).Age and gender-matched 155−/− or B6 mice were infected with 5×104 PFU MCMV, and splenic NK cells were assessed for intracellular IFN-γ production by flow cytometry after 36 hours. Representative flow plots (A) and summary data (B–C) showing increased mean ± SEM IFN-γ+ percentage (B) and MFI (normalized to WT in each experiment) (C) of NK cells in 155−/− mice, compared to WT controls at 36 hours p.i. Data summarize 6–7 mice per group from 2 independent experiments. (D–F) Representative flow plot (D) and summary data (E–F) showing increased mean ± SEM IFN-γ percentage (E) and MFI (F) of NK cells in 155FOE mice, compared to Cre+ controls at 36 hours p.i. Data summarize 4–6 mice per group from 2 independent experiments. (G) Groups of MCMV-infected WT and 155−/− mice were assessed for splenic viral titers 3 days p.i. (mean ± SEM viral titer). Dotted line represents the limit of detection for the assay (0.92). Data summarize 7–8 mice/group from 2 independent experiments.
RISC-Seq identifies novel miR-155 targets in multiple NK cell activation pathways
In order to determine relevant targets of miR-155 involved in threshold and IFN-γ production, WT and 155−/− NK cells were activated with IL-12, IL-15, and IL-18 for 24 hours to induce mRNA targets that are likely regulated by miR-155 at its peak expression level in NK cells. We then sequenced (RISC-Seq) paired total RNA and RNA from immunoprecipitated RNA-Induced Silencing Complex (RISC), which contains miRNAs and their targets from both WT and 155−/− NK cells (Fig. S3A).(38, 39) An mRNA transcript that is a target of miR-155 is expected to be enriched in the RISC of WT NK cells compared to 155−/− NK cells (i.e., a RISC incorporation ratio, RIR, > 1) (Fig. 7A). To improve the signal/noise ratio of this analysis, we filtered by predicted miR-155 targets in TargetScan (Fig. 7B).(40) This resulted in marked enrichment of targets with a RIR > 1.2, with an almost complete loss of targets with a RIR < 0.83. This filter approach did not produce any mRNA target enrichment when performed with an irrelevant miRNA target prediction (Fig. S3 B-C). We next biochemically validated miR-155 targets identified using miR-155 overexpression coupled with a luciferase sensor plasmid containing the 3′UTR of the putative target mRNAs (Fehniger et al., 2010; Sullivan et al., 2012) (Fig. 7C) and Western Blot of 155−/− and WT NK cells for SHIP1, SLP-76, and IKBKE (Fig. 7D). Notably, all candidates identified by RISC-Seq that were tested by either luciferase assay or Western blot were confirmed to be true miR-155 targets by these validation methods. Together, these data indicate that miR-155 targets multiple molecules in NK cell activation pathways.
Figure 7. RISC-Seq identifies miR-155 targets in NK cells.

(A) Unfiltered results of RISC-Seq, displayed as WT total RNA expression versus RISC Incorporation Ratio (WT/155−/−). Graph depicted is limited to targets with a fold change between 0.5 and 2 to align with (B). 116 transcripts lie outside of these depicted boundaries, but none of these were targets predicted by TargetScan. Targets validated in (C) are shown as predicted (open circle) or not predicted (open square) by TargetScan, and the open diamond represents SHIP1, a previously validated target. Numbers above graph represent the percentage of total mRNA transcripts within each RISC Incorporation Ratio (RIR) quartile: Group 1: RI > 1.2, Group 2: 1 < RI < 1.2, Group 3: 0.83 < RI < 1, Group 4: RI < 0.83. Data represent 2 biological replicates for groups of WT and 155−/− NK cells following IL-12, IL-15, and IL-18 activation. (B) Dataset in (A) filtered by cross-referencing with miR-155 predicted targets (TargetScan). (C) Luciferase validation in 293T cells of RISC-Seq targets, as indicated in (A) and (B). Data summarize two independent experiments of duplicate wells. (D) Western Blot analysis of miR-155 target expression in WT and 155−/− NK cells. Relative overexpression for 155−/− NK cells is indicated.
mRNA transcripts with a RIR > 1 standard deviation from the mean were analyzed using DAVID pathway analysis, and the KEGG lymphocyte (T cell) activation pathway was found to be significantly overrepresented. We hypothesized that the decreased activation threshold of 155−/− NK cells was due to an increase in the protein levels of the miR-155-targeted members of this pathway, and that treatment with specific inhibitors would elucidate the proteins critical for miR-155 threshold effects. In order to directly test these confirmed targets in the acquisition of a decreased threshold of activation in the 155−/− NK cells, we utilized chemical inhibitors of NK cell activation pathways; specifically, PI3K (Ly294002), calcineurin (cyclosporin A), and NF-κB (BAY 11-7082). We utilized these inhibitors in an NK1.1 ligation assay, since this stimulus demonstrated the greatest threshold effect. We found that inhibition of either calcineurin (Fig. 8A) or PI3K (Fig. 8B) led to a dose-responsive decrease in the differential between 155−/− and WT NK cell IFN-γ production, while inhibition of NF-κB led to a differential response only at concentrations above its IC50 (Fig. 8C), and an inhibitor of p38 Kinase, SB203580, had no effect on IFN-γ production (Fig. 8D). These data indicate that the PI3K and calcineurin pathways contribute to the increased production of IFN-γ in 155−/− NK cells, and collectively, form the basis of the decreased threshold of activation in these cells (Fig. 8E).
Figure 8. miR-155 targets NK cell activation pathways.

(A–D) NK cells were activated via NK1.1 ligation using plate bound α-NK1.1 in the presence of an inhibitor of calcineurin (A), PI3K (B), NF-κB (C), or p38 Kinase (D) to demonstrate loss of threshold differentials after including inhibitors. Solid lines represent previously determined IC50 values (see Materials and Methods), while dashed lines represent IC50 values obtained by our experiments. The data shown are mean ± SEM and are representative of three independent experiments. (E) Model/summary of miR-155’s role in targeting genes involved in the acquisition of NK cell threshold. RIR Incorporation values are included with each gene. RISC-Seq-identified (RIR>1.2) miR-155 targets (dark box) and inhibitor targets (shaded box) are indicated.
DISCUSSION
In this study, we assessed the impact of miR-155 alterations in human and mouse NK cells using lentiviral overexpression, as well as genetic mouse models of global miR-155 deficiency and NK-specific miR-155 overexpression. We demonstrate that miR-155 is induced upon cytokine activation of both mouse and human NK cells in vitro, as well as in vivo during an ongoing NK cell response to MCMV. We confirmed results of a previous study (23), showing that miR-155 overexpression in mature human NK cells leads to increased IFN-γ production following NK cell activation. Furthermore, we found identical results in mature mouse NK cells, demonstrating consistent miR-155 regulation of NK cell activation in mice and humans in this context. However, we unexpectedly discovered that 155−/− NK cells also produced more IFN-γ upon cytokine activation. How can miR-155 overexpression and deficiency lead to similar IFN-γ phenotypes in NK cells? To address this, we generated a novel 155FOE model that specifically overexpresses miR-155 in developing and mature NK cells, finding that similar to lentivirus-transduced NK cells that overexpress miR-155, 155FOE NK cells also secreted increased amounts of IFN-γ upon activation. We hypothesized that miR-155 loss or overexpression had distinct mechanisms that lead to similar apparent IFN-γ phenotypes. Indeed, a higher proportion of NK cells from 155−/− mice produced IFN-γ following activation, whereas NK cells from 155FOE mice exhibited increased per NK cell expression of IFN-γ following activation. These findings, in multiple complementary experimental systems, suggest a dual role for miR-155 by regulating NK cell activation and cytokine production. When miR-155 is deficient during NK cell development and maturation (e.g., 155−/−) the NK cell threshold for future activation is lowered, allowing more NK cells to express IFN-γ when stimulated. In contrast, forced overexpression of miR-155 in NK cells, either in vitro (lentiviral) in mature NK cells, or in vivo (155FOE) in developing NK cells, allows an NK cell, once it crosses the activation threshold, to have an amplified IFN-γ response (Fig. S4). This is likely due to the previously postulated mechanism of miR-155 negatively regulating SHIP-1, a negative regulator of IFN-γ production. While the Ly49C+/Ly49C− ratio of IFN-γ production (the licensing ratio) is not altered per se (Fig. S1K), we do note that the vast majority of IFN-γ producing cells in these mice are Ly49C+, and thus the licensed subset of NK cells is the major subset affected by miR-155 loss. It remains possible, therefore, that miR-155 may have a role in NK cell licensing. Licensing alterations in both the 155−/− and the 155FOE NK cells are an area of ongoing study. Furthermore, since LV-GFP/155 NK cells develop in a context of normal levels of miR-155, while 155FOE NK cells develop with increased levels of miR-155, the increased percentage of activated NK cells in the LV-GFP/155 further supports a developmental/maturation role for miR-155 in suppressing future activation via threshold modulation. Thus, the combined use of both human and murine mature NK cells in concert with genetic mouse models provides a comprehensive picture of the complex role of miR-155 in directing NK cell activation and IFN-γ production.
The findings presented here that both mature human and mouse NK cells that overexpress miR-155 exhibit enhanced IFN-γ production upon activation are in agreement with a recent study focused primarily on human NK cells (23). Furthermore, the enhanced NK cell IFN-γ phenotype demonstrated in 155FOE mice was also observed in a recent report in an Lck-miR-155 transgenic mouse model (24). However, the Lck-miR-155 transgenic mice were reported to have increased NK cell percentages, numbers, and proliferation, while the 155FOE mice had a normal complement of NK cells in all tissues examined. The level of overexpression of miR-155 was similar in both of these mouse models, suggesting that disparate miR-155 overexpression was not the explanation. The 155FOE is NK cell-specific, tracked using a Cre-reporter allele, and was analyzed on a B6 background. We suspect that cell specificity of miR-155 overexpression, strain of the mice used in the different models, or integration effects may play a role in the different phenotypes observed in these models.
What are the downstream targets that are responsible for miR-155’s effects in mature NK cells? miR-155-target mRNAs have been shown to be cell-context specific (41), and thus we demonstrate here the first miR-155 target set in primary NK cells. Here we report a RISC-Seq approach that identified >20 high probability candidate mRNA targets, and validated many using luciferase sensor assays and Western blot. Notably, this included SHIP-1 (Inpp5d), which has been previously identified and validated as a target of miR-155 (23, 28). Indeed, specific knockdown of SHIP-1 has been shown to phenocopy miR-155 deletion in macrophages. SHIP-1 functions as a negative regulator of IFN-γ production, and thus miR-155 inhibition of SHIP-1 in activated NK cells should result indirectly in greater IFN-γ production. Our data fit with this model of miR-155 directly targeting SHIP-1, with increased IFN-γ MFI in LV-GFP/155 and 155FOE NK cells, and a modestly decreased IFN-γ MFI in some activating conditions in our 155−/− NK cells. Further, a recent study has shown that SHIP-1 deficiency in developing NK cells, contrary to what would be expected for deletion of a negative regulator of activation, actually leads to a decreased percentage of mature cells becoming IFN-γ positive after stimulation (42). These findings are provocative and consistent with our data, because miR-155 deletion would lead to increased SHIP-1 levels in development, and therefore may alter the NK cell activation threshold, leading to increased IFN-γ percentages in mature NK cells. This concept is similar to the known TCR signal amplitude tuning by miR-181a targeting of multiple phosphatases, identified in T cells (43). Thus, SHIP-1 represents one validated miR-155 target that may contribute to the NK cell phenotype. However, previous studies examining NK cell SHIP-1 gain-of-function resulted in only a partial restoration of the NK cell activation phenotype (23), suggesting that targets other than SHIP1 play a role in this phenotype.
Here, we identified a number of these additional miR-155 mRNA targets directly in primary NK cells in our RISC-Seq experiments. A number of miR-155 NK cell mRNA targets are important members of signaling cascades central to NK cell activation, providing an alternative explanation for how miR-155 may tune the activation threshold of NK cells (Fig 8). Additionally, we found that novel targets identified in the PI3K, NF-κB, and calcineurin pathways are able to contribute to the increased IFN-γ production in NK cells, as chemical inhibition of these pathways results in a loss of the enhanced IFN-γ production phenotype, though to a far lesser extent for NF-κB, indicating that this pathway is less important for miR-155’s impact on IFN-γ. Thus, these additional miR-155 targets in the NK cell activation pathway contribute to NK cell activation phenotypes. The individual and collective contribution of these targets to tuning the NK cell threshold will be evaluated further in future studies.
We also demonstrate a regulatory role for miR-155 during early anti-viral NK cell responses in vivo, with effects on early cytokine-dependent IFN-γ production. IFN-γ production by 155−/− NK cells was increased in vivo at day 1.5 post-MCMV infection, which correlated with a decreased viral titer at 3 days, and was consistent with our in vitro cytokine-dependent IFN-γ phenotype. MCMV studies of 155FOE mice are ongoing. These findings may have powerful implications in the human NK cell response to MCMV, and extending the study of miR-155 in this setting is of exceptional interest. A recently published study (30) identified SOCS1 and Noxa as targets of miR-155, and demonstrated a blunted Ly49H+ expansion at later time points. Future studies in a miR-155 floxed knock-out model that selectively lacks miR-155 in NK cells will be used to investigate the NK cell intrinsic role of miR-155.
In this study we have defined a novel dual role for miR-155 in the regulation of both mature NK cell IFN–γ production and setting a threshold of activation during NK cell development and maturation. This regulation of activation by miR-155 is evident both in vitro and in vivo during an NK cell response to viral infection. While SHIP-1 represents a validated target of miR-155 that is likely important in these processes, novel mRNA targets in activation signaling pathways identified here contribute to setting the NK cell activation threshold. miR-155 also regulates the NK cell IFN-γ response during MCMV infection, indicating that the observed miR-155-mediated changes in IFN-γ influence host defense against a viral pathogen. These findings further support the role for miR-155, and miRNAs in general, as ‘tuners’ of immune cell activation. Future studies of miR-155, especially in the context of cell-specific deletion or overexpression, will undoubtedly uncover more of miR-155’s diverse functions in NK cells.
Supplementary Material
Acknowledgments
This work was supported by NIH T32 HL708836 (RPS), NIH AI078994 (ARF), and K08HL093299 from the NIH, ASH Scholar Award from the ASH foundation, Physician-Scientist Early Career Award from HHMI (TAF). The authors thank the Siteman Cancer Center Flow Cytometry Core (NIH P30CA091842) and the Washington University Pathology & Immunology Flow Cytometry Core, as well as the Washington University Facility of Rheumatic Diseases Core Center (NIH P30AR048335).
The authors thank Drs. Wayne Yokoyama, Megan Cooper, Daniel Link, Marco Colonna, and Thad Stappenbeck for insightful discussion.
Abbreviations
- miRNA
microRNA
- MCMV
murine cytomegalovirus
- RISC
RNA-Induced Silencing Complex
- RIR
RISC Incorporation Ratio
References
- 1.Yokoyama WM, Kim S, French AR. The dynamic life of natural killer cells. Annu Rev Immunol. 2004;22:405–429. doi: 10.1146/annurev.immunol.22.012703.104711. [DOI] [PubMed] [Google Scholar]
- 2.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 3.Di Santo JP. Natural killer cell developmental pathways: a question of balance. Annu Rev Immunol. 2006;24:257–286. doi: 10.1146/annurev.immunol.24.021605.090700. [DOI] [PubMed] [Google Scholar]
- 4.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 5.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orange JS. Formation and function of the lytic NK-cell immunological synapse. Nat Rev Immunol. 2008;8:713–725. doi: 10.1038/nri2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tripathy SK, Keyel PA, Yang L, Pingel JT, Cheng TP, Schneeberger A, Yokoyama WM. Continuous engagement of a self-specific activation receptor induces NK cell tolerance. J Exp Med. 2008;205:1829–1841. doi: 10.1084/jem.20072446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elliott JM, Wahle JA, Yokoyama WM. MHC class I-deficient natural killer cells acquire a licensed phenotype after transfer into an MHC class I-sufficient environment. J Exp Med. 2010;207:2073–2079. doi: 10.1084/jem.20100986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Höglund P, Brodin P. Current perspectives of natural killer cell education by MHC class I molecules. Nat Rev Immunol. 2010;10:724–734. doi: 10.1038/nri2835. [DOI] [PubMed] [Google Scholar]
- 10.Long EO, Sik Kim H, Liu D, Peterson ME, Rajagopalan S. Controlling Natural Killer Cell Responses: Integration of Signals for Activation and Inhibition. Annu Rev Immunol. 2013;31:227–258. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hesslein DGT, Lanier LL. Transcriptional control of natural killer cell development and function. Adv Immunol. 2011;109:45–85. doi: 10.1016/B978-0-12-387664-5.00002-9. [DOI] [PubMed] [Google Scholar]
- 12.Ramirez K, Kee BL. Transcriptional regulation of natural killer cell development. Curr Opin Immunol. 2010;22:193–198. doi: 10.1016/j.coi.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 14.Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med. 2009;60:167–179. doi: 10.1146/annurev.med.59.053006.104707. [DOI] [PubMed] [Google Scholar]
- 15.O’Connell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295–312. doi: 10.1146/annurev-immunol-020711-075013. [DOI] [PubMed] [Google Scholar]
- 16.Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;9:839–845. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 17.Bezman NA, Cedars E, Steiner DF, Blelloch R, Hesslein DGT, Lanier LL. Distinct Requirements of MicroRNAs in NK Cell Activation, Survival, and Function. J Immunol. 2010;185:3835–3846. doi: 10.4049/jimmunol.1000980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sullivan RP, Leong JW, Schneider SE, Keppel CKR, Germino E, French AR, Fehniger TA. MicroRNA deficient NK cells exhibit decreased survival but enhanced function. J Immunol. 2012;188:3019–3030. doi: 10.4049/jimmunol.1102294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas MF, Abdul-Wajid S, Panduro M, Babiarz JE, Rajaram M, Woodruff P, Lanier LL, Heissmeyer V, Ansel KM. Eri1 regulates microRNA homeostasis and mouse lymphocyte development and anti-viral function. Blood. 2012;120:130–142. doi: 10.1182/blood-2011-11-394072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leong J, Sullivan R, Fehniger T. Natural Killer Cell Regulation by MicroRNAs in Health and Disease. J Biomed Biotechnol. 2012;2012(63232):1–12. doi: 10.1155/2012/632329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sullivan RP, Leong JW, Fehniger TA. MicroRNA regulation of natural killer cells. Front Immunol. 2013;4(44):1–12. doi: 10.3389/fimmu.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beaulieu AM, Bezman NA, Lee JE, Matloubian M, Sun JC, Lanier LL. MicroRNA function in NK-cell biology. Immunol Rev. 2013;253:40–52. doi: 10.1111/imr.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trotta R, Chen L, Ciarlariello D, Josyula S, Mao C, Costinean S, Yu L, Butchar JP, Tridandapani S, Croce CM, Caligiuri MA. MiR-155 regulates IFN-γ production in natural killer cells. Blood. 2012;119:3478–3485. doi: 10.1182/blood-2011-12-398099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trotta R, Chen L, Costinean S, Josyula S, Mundy-Bosse BL, Ciarlariello D, Mao C, Briercheck EL, McConnell KK, Mishra A, Yu L, Croce CM, Caligiuri MA. Overexpression of miR-155 causes expansion, arrest in terminal differentiation and functional activation of mouse natural killer cells. Blood. 2013;121:3126–3134. doi: 10.1182/blood-2012-12-467597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, Vetrie D, Okkenhaug K, Enright AJ, Dougan G, Turner M, Bradley A. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, Schmidt-Supprian M, Rajewsky N, Yancopoulos G, Rao A, Rajewsky K. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 27.O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci USA. 2007;104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci USA. 2009;106:7113–7118. doi: 10.1073/pnas.0902636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fehniger TA, Wylie T, Germino E, Leong JW, Magrini VJ, Koul S, Keppel CR, Schneider SE, Koboldt DC, Sullivan RP, Heinz ME, Crosby SD, Nagarajan R, Ramsingh G, Link DC, Ley TJ, Mardis ER. Next-generation sequencing identifies the natural killer cell microRNA transcriptome. Genome Res. 2010;20:1590–1604. doi: 10.1101/gr.107995.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zawislak CL, Beaulieu AM, Loeb GB, Karo J, Canner D, Bezman NA, Lanier LL, Rudensky AY, Sun JC. Stage-specific regulation of natural killer cell homeostasis and response against viral infection by microRNA-155. Proc Natl Acad Sci USA. 2013;110(17):6967–6972. doi: 10.1073/pnas.1304410110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T, Rülicke T, Mueller M, Casanova E, Sexl V. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood. 2011;117:1565–1573. doi: 10.1182/blood-2010-06-291633. [DOI] [PubMed] [Google Scholar]
- 32.Jonsson aH, Yang L, Kim S, Taffner SM, Yokoyama WM. Effects of MHC class I alleles on licensing of Ly49A+ NK cells. J Immunol. 2010;184:3424–3432. doi: 10.4049/jimmunol.0904057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rossum HH, van FPHTM, Romijn, Sellar KJ, Smit NPM, van der Boog PJM, de Fijter JW, van Pelt J. Variation in leukocyte subset concentrations affects calcineurin activity measurement: implications for pharmacodynamic monitoring strategies. Clin Chem. 2008;54:517–524. doi: 10.1373/clinchem.2007.097253. [DOI] [PubMed] [Google Scholar]
- 34.Ortaldo JR, Bere EW, Hodge D, Young HA. Activating Ly-49 NK receptors: central role in cytokine and chemokine production. J Immunol. 2001;166:4994–4999. doi: 10.4049/jimmunol.166.8.4994. [DOI] [PubMed] [Google Scholar]
- 35.Bahner I, Sumiyoshi T, Kagoda M, Swartout R, Peterson D, Pepper K, Dorey F, Reiser J, Kohn DB. Lentiviral vector transduction of a dominant-negative Rev gene into human CD34+ hematopoietic progenitor cells potently inhibits human immunodeficiency virus-1 replication. Mol Ther. 2007;15:76–85. doi: 10.1038/sj.mt.6300025. [DOI] [PubMed] [Google Scholar]
- 36.Brown MG, Dokun AO, Heusel JW, Smith HR, Beckman DL, Blattenberger EA, Dubbelde CE, Stone LR, Scalzo AA, Yokoyama WM. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science. 2001;292:934–937. doi: 10.1126/science.1060042. [DOI] [PubMed] [Google Scholar]
- 37.Fogel LA, Sun MM, Geurs TL, Carayannopoulos LN, French AR. Markers of Nonselective and Specific NK Cell Activation. J Immunol. 2013;190(12):6269–6276. doi: 10.4049/jimmunol.1202533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matkovich SJ, Van Booven DJ, Eschenbacher WH, Dorn GW. RISC RNA sequencing for context-specific identification of in vivo microRNA targets. Circ Res. 2011;108:18–26. doi: 10.1161/CIRCRESAHA.110.233528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karginov FV, Conaco C, Xuan Z, Schmidt BH, Parker JS, Mandel G, Hannon GJ. A biochemical approach to identifying microRNA targets. Proc Natl Acad Sci USA. 2007;104:19291–19296. doi: 10.1073/pnas.0709971104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Cell Biol. 2011;18:1139–1146. doi: 10.1038/nsmb.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loeb GB, Khan AA, Canner D, Hiatt JB, Shendure J, Darnell RB, Leslie CS, Rudensky AY. Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Mol Cell. 2012;48:760–770. doi: 10.1016/j.molcel.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Banh C, Miah SMS, Kerr WG, Brossay L. Mouse natural killer cell development and maturation are differentially regulated by SHIP-1. Blood. 2012;120(23):4583–4590. doi: 10.1182/blood-2012-04-425009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li QJ, Chau J, Ebert PJR, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P, Klein LO, Davis MM, Chen CZ. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.