

Abstract
Primary triple negative breast cancers (TNBC) represent approximately 16% of all breast cancers1 and are a tumour type defined by exclusion, for which comprehensive landscapes of somatic mutation have not been determined. Here we show in 104 early TNBC cases, that at the time of diagnosis these cancers exhibit a wide and continuous spectrum of genomic evolution, with some exhibiting only a handful of somatic aberrations in a few pathways, whereas others contain hundreds of somatic events and multiple pathways implicated. Integration with matched whole transcriptome sequence data revealed that only ~36% of mutations are expressed. By examining single nucleotide variant (SNV) allelic abundance derived from deep re-sequencing (median >20,000 fold) measurements in 2414 somatic mutations, we determine for the first time in an epithelial tumour, the relative abundance of clonal genotypes among cases in the population. We show that TNBC vary widely and continuously in their clonal frequencies at the time of diagnosis, with basal subtype TNBC2,3 exhibiting more variation than non-basal TNBC. Although p53 and PIK3CA/PTEN somatic mutations appear clonally dominant compared with other pathways, in some tumours their clonal frequencies are incompatible with founder status. Mutations in cytoskeletal and cell shape/motility proteins occurred at lower clonal frequencies, suggesting they occurred later during tumour progression. Taken together our results show that future attempts to dissect the biology and therapeutic responses of TNBC will require the determination of individual tumour clonal genotypes.
To understand the patterns of somatic mutation in TNBC we enumerated genome aberrations at all scales, from 104 cases of primary TNBC (Affymetrix SNP6.0: 104 cases, RNA-seq: 80 cases, genome/exome sequence: 65 cases) (Table S1, Figure S1), annotated with clinical information (Table S2). We re-validated 2414 somatic single nucleotide variants4, 5 (SNVs) (Table S3) including 43 non-coding splice site dinucleotide mutations (Table S4), and 104 genes with 107 indels (Table S5) (Supplemental methods). Strikingly, the distribution of somatic mutation abundance varies in a continuous distribution among tumours (Figure 1a) and appears unrelated to the proportion of the genome altered by copy number alterations (CNAs) (Figure 1b) or tumour cellularity (Figure S2). Although this distribution could be partially explained by a false negative rate in mutation discovery, others have noted similar distributions in epithelial cancers6 suggesting the total mutation content of individual tumours may be shaped by biological processes, or differential exposure to mutagenic influences in the population.
Figure 1.
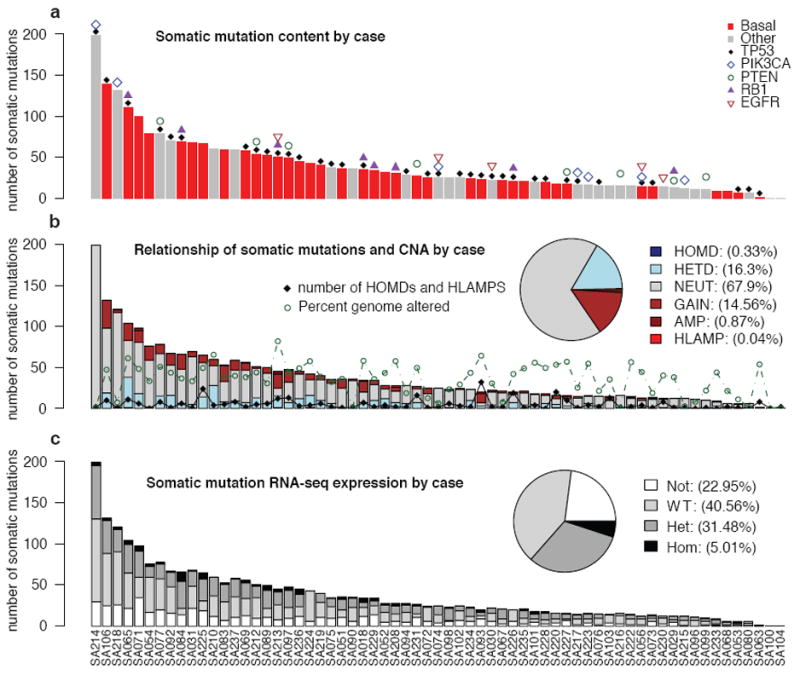
Distribution of number of validated somatic mutations by case over 65 cases. (a) Mutation frequency (Basal (red), Other (gray)). Patients harbouring known driver gene mutations are indicated. (b) Case specific and overall (inset) distributions of mutations in CNA classes: HOMD (homozygous deletion), HETD (hemizygous deletion), NEUT (no copy number change), GAIN (single copy gain), AMP (amplification) and HLAMP (high-level amplification). The number of (HOMD, HLAMP) CNAs (black diamonds) and percentage genome altered (green circles) are indicated. (c) Case specific and overall (inset) distributions of mutations in expression classes: Not (no expression), WT (wildtype expression), Het (mutant and wildtype expression) and Hom (dominant mutant expression).
The overall pattern (Figure S3a,b) of CNA abundance appears similar (Figure S4) to that seen in a larger, independent series of ~2000 SNP6.0 profiled breast tumours7. Among the most frequently observed events (Table S6) are the tumour suppressor/oncogenes PARK2 (6%), RB1 (5%), PTEN (3%), and EGFR (5%). Here we report intragenic deletions (Figure S5) in the PARK2 tumour suppressor8, 9, specifically linking PARK2 with TNBC for the first time. Consistent with previous reports in breast cancer10, we did not observe frequent recurrent structural rearrangements (Figure S3d, Table S7), although we revalidated many individual fusion events involving known oncogenes/tumour suppressors (e.g. KRAS,RB1,IDH1,ETV6) (Tables S8, S9, S10).
A comparison of the RNA-seq with the genomes/exomes revealed that only 36% of validated somatic SNVs were observed in transcriptome sequence (Table S3, Figure 1c). In a recent lymphoma study, similar proportions were observed (137 of 329 somatic mutations expressed in RNASeq)11. As expected, the proportion of low abundance somatic SNVs observed in RNA is reflected in the distribution of wildtype, heterozygous and homozygous expressed mutations (Figure 1c), consistent with the notion that low abundance alleles may represent rarer clones in the primary tumour. We found 43 splice junction mutations with evidence for an impact on splicing patterns (Table S4), encompassing several known tumour suppressors (p53, PIK3R1, Figure S6) as well as many genes not yet implicated in carcinogenesis. Analysis of 72 somatic mutations in the non-coding space of experimentally determined human regulatory regions12 showed (Table S11) a significant overrepresentation (31.9% vs expected 2.5%, Fisher exact test p=2 × 1019) of mutations within retinoblastoma (Rb) binding sites. Six mutations were predicted to be damaging to Rb binding (Supplemental methods, Figure S7). This is consistent with observations of frequent functional disruption of the Rb regulated cell cycle network13 in TNBC.
We next searched for mutation enrichment patterns in three ways; by single gene mutation frequency over multiple cases; by the mutation frequency over multiple members of a gene family and by correlating mutation status with expression networks. First, similar to other studies14, 15, p53 is the most frequently mutated gene (Table S12) with 62% of basal TNBC (determined by PAM5016 analysis on RNASeq expression profiles) and 43% of non-basal TNBC cases harbouring a validated somatic mutation. We also observed frequent mutations in PIK3CA at 10.2% (7/65), USH2A (Ushers syndrome gene, implicated in actin cytoskeletal functions) at 9.2% (6/65), MYO3A at 9.2%, PTEN and RB1 at 7.7% (5/65) and a further 8 genes (including ATR,UBR5 (EDD1), COL6A3) at 6.2% (4/65) of cases in the cohort (Figure 2a). Considering background mutation rates17, TP53, PIK3CA, RB1, PTEN, MYO3A and GH1 showed evidence of single gene selection (q < 0.1) (Table S13). Additional recurrent mutations of note occurred in the synuclein genes (SYNE1/2, 9.2% 6/65, recently implicated in squamous head and neck cancers18, 19), BRCA2 (3 cases), and several other well-known oncogenes (BRAF, NRAS, ERBB2, and ERBB3) with mutations in 2 cases each. Approximately 20% of cases contained examples of potentially “clinically actionable” somatic aberrations, including BRAF V600E, high level EGFR amplifications and ERBB2/ERBB3 mutations.
Figure 2.
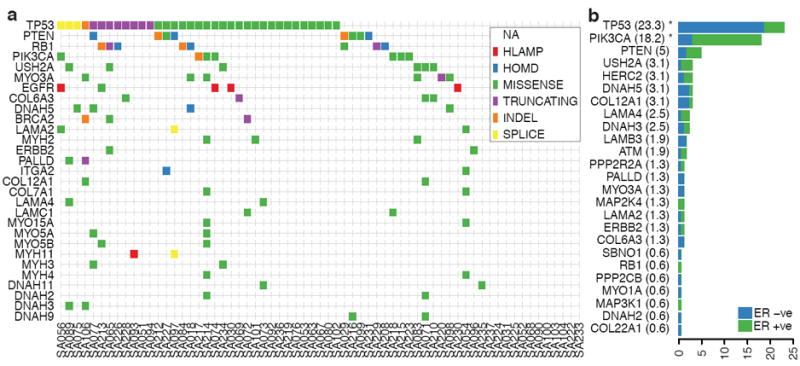
Population patterns of co-occurrence and mutual exclusion of genomic aberrations in TNBC. (a) Case-specific mutations in known driver genes, plus genes from integrin signaling and ECM related proteins (laminins, collagens, integrins, myosins and dynein) derived from all aberration types: high-level amplifications (HLAMP), homozygous deletions (HOMD), missense, truncating, splice site and indel somatic mutations are depicted in genes with at least two aberrations in the population. (b) Distribution of somatic mutations in 25 genes across all exons of 159 additional breast cancers (relative proportion of ER+ cases in green, and ER- in blue), shown as a percentage of cases with one or more mutations.
In the second approach we searched for statistically over represented gene families/protein functions using the Reactome functional protein interaction database20 (Supplemental methods). This analysis quantifies gene family involvement through sparse mutation patterns in functionally connected genes, which would be statistically underrepresented by single gene recurrent mutation analysis. The over-represented pathways (FDR < 0.001) included TP53 related pathways along with chromatin remodeling, PIK3 signaling, ERBB2 signaling, integrin signaling and focal adhesion, WNT/cadherin signaling, growth hormone and nuclear receptor co-activators, ATM/Rb related pathways (Figure 3a, Table S14). We note that the candidate ‘driver’ MYO3A, a cytoskeleton motor protein involved in cell shape/motility, relates to several pathways upstream and downstream of integrin signaling. The mutated genes stretch from extracellular matrix interactions (ECM) (laminins, collagens), ECM receptors (integrins), several proteins regulating actin cytoskeleton dynamics (usherin, palladin, multiple myosins) and microtubule motor proteins (kinesins) (Figure 2a). All of these contribute to cellular processes which have been functionally implicated in cancer progression, however a signature of somatic mutation associated with these proteins has not been previously noted in TNBC. To confirm the mutational spectrum in the general breast cancer population we resequenced all exons of 29 genes in an additional 159 breast cancers (82 ER+ and 77 ER-ve, tumour and matched normal) (Figure 2b) and this confirms that many of the genes found in the discovery cohort were recurrently mutated in an additional population. Whether this pattern of mutation represents the occurrence of disease modifying mutations, or possibly selection from other processes (e.g. transcription related hypermutation) is unknown. Interestingly, the enrichment of cytoskeletal functions in the somatic aberration landscape is also evident from the copy number and alternative splicing landscapes (Figure S8).
Figure 3.
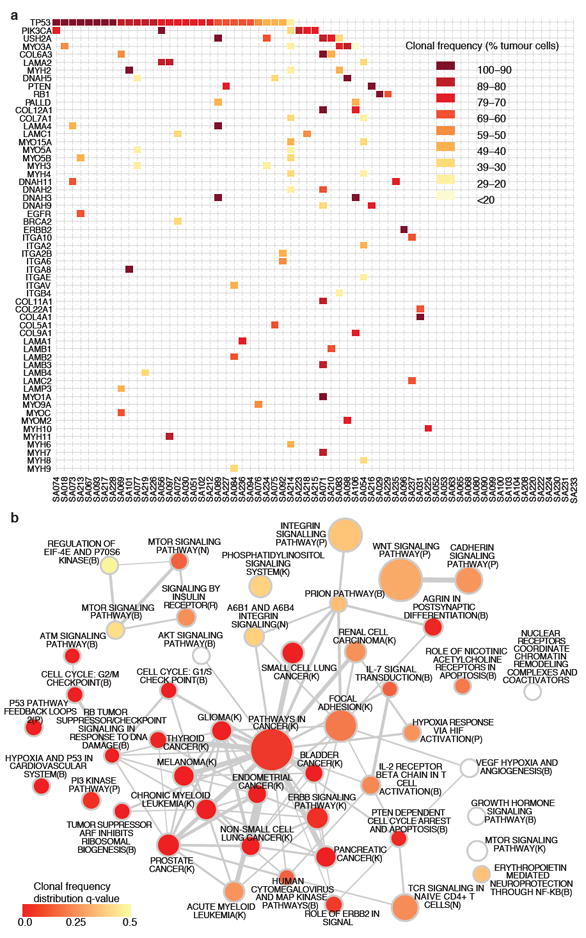
Network analysis of recurrently mutated genes by somatic point mutations and indels (254 genes). (a) Significantly over-represented pathways (FDR < 0.001) from recurrently mutated genes (see Supplemental methods). Node shading encodes the adjusted p-value (q-value) of the comparison of the distribution of clonal frequencies of mutations in a given pathway to the overall distribution of clonal frequencies. A spectrum of higher (red) and lower (yellow) clonal frequencies is evident. (b) Case-specific mutations shaded according to clonal frequencies in known driver genes, plus genes from integrin signaling and ECM related proteins (laminins, collagens, integrins, myosins and dyneins).
Third, we integrated both the CNA and mutation data with expression to reveal genomic events associated with extreme changes in transcription of interacting genes20 (Table 1), using a bipartite graph-based method (driverNet, Supplemental methods). The somatic aberrations showing statistically significant association with extreme expression in this analysis (p<0.05) (Table 1, S15) implicates well known oncogenes and tumour suppressors (TP53, PIK3CA, NRAS, EGFR, RB1) and suggests several new genes of interest, including PRPS2 (a nucleotide biosynthesis enzyme, rank 7), harboring homozygous deletions in 3 cases, NRC31 (a glucocorticoid receptor, rank 10) with SNVs in 3 cases, four PKC related genes PRKCZ, PRKCQ, PRKG1 and PRKCE (1 case with a mutation, 2 cases with mutations, 2 cases with homozygous deletions and 1 case with homozygous deletion, respectively) and ATM (rank 30, 2 cases with mutations). The gene networks show a partial overlap with driverNet applied to the TCGA ovarian high grade serous data21 (Table S16).
Table 1.
Analysis of the top somatically aberrated genes (by node degree) connected (by Reactome gene sets) to genes that exhibited outlying expression from their population level distributions as computed by driverNet.
| rank | gene | gband | SNV Indel | HLAMP | HOMD | events | p-value |
|---|---|---|---|---|---|---|---|
| 1 | TP53 | 17p13.1 | 35 | 0 | 0 | 2242 | 0 |
| 2 | PIK3CA | 3q26.32 | 7 | 0 | 0 | 441 | 1.00E-04 |
| 3 | NRAS | 1p13.2 | 2 | 0 | 0 | 271 | 4.00E-04 |
| 4 | EGFR | 7p11.2 | 1 | 5 | 0 | 220 | 4.00E-04 |
| 5 | RB1 | 13q14.2 | 5 | 0 | 5 | 184 | 5.00E-04 |
| 6 | PGM2 | 4p14 | 1 | 0 | 1 | 172 | 5.00E-04 |
| 7 | PRPS2 | 23p22.2 | 0 | 0 | 3 | 171 | 5.00E-04 |
| 8 | PTEN | 10q23.31 | 5 | 0 | 3 | 150 | 5.00E-04 |
| 9 | PRKCE | 2p21 | 0 | 0 | 1 | 136 | 7.00E-04 |
| 10 | NR3C1 | 5q31.3 | 3 | 0 | 0 | 130 | 7.00E-04 |
| 11 | CREBBP | 16p13.3 | 1 | 0 | 1 | 119 | 8.00E-04 |
| 12 | CS | 12q13.2 | 1 | 0 | 0 | 108 | 0.0011 |
| 13 | MAN2A2 | 15q26.1 | 2 | 0 | 1 | 104 | 0.0012 |
| 14 | HMGCS2 | 1p12 | 1 | 2 | 0 | 100 | 0.0013 |
| 15 | HEXA | 15q24.1 | 2 | 1 | 0 | 97 | 0.0013 |
| 16 | ADCY9 | 16p13.3 | 2 | 1 | 0 | 91 | 0.0017 |
| 17 | OR4N4 | 15q11.2 | 0 | 0 | 5 | 90 | 0.0017 |
| 18 | LCLAT1 | 2p23.1 | 0 | 0 | 1 | 85 | 0.002 |
| 19 | DGKI | 7q33 | 2 | 0 | 0 | 82 | 0.0022 |
| 20 | CYP2A6 | 19q13.2 | 1 | 0 | 0 | 80 | 0.0024 |
| 21 | JAK1 | 1p31.3 | 1 | 0 | 0 | 78 | 0.0026 |
| 22 | POLR1A | 2p11.2 | 2 | 0 | 0 | 78 | 0.0026 |
| 23 | PLD1 | 3q26.31 | 1 | 0 | 0 | 69 | 0.0038 |
| 24 | IDH3B | 20p13 | 1 | 0 | 1 | 68 | 0.004 |
| 25 | PAPSS2 | 10q23.2 | 0 | 0 | 3 | 67 | 0.0041 |
| 26 | PRKX | 23p22.33 | 0 | 0 | 2 | 65 | 0.0046 |
| 27 | TPH2 | 12q21.1 | 1 | 0 | 0 | 65 | 0.0046 |
| 28 | UGT2B17 | 4q13.2 | 0 | 0 | 1 | 63 | 0.0053 |
| 29 | RRM2 | 2p25.1 | 1 | 0 | 0 | 57 | 0.0072 |
| 30 | ATM | 11q22.3 | 1 | 0 | 0 | 55 | 0.0084 |
| 31 | CLCA1 | 1p22.3 | 2 | 0 | 0 | 54 | 0.009 |
| 32 | PRKCZ | 1p36.33 | 1 | 0 | 0 | 53 | 0.0095 |
Columns: Rank: by driverNet algorithm (Supplemental methods); Gene: somatically aberrated gene; gband: chromosomal band containing gene; SNV/Indel: number of cases harbouring an SNV or indel in the gene; HLAMP: number of cases harbouring a predicted high level amplification; HOMD: number of cases harbouring a predicted homozygous deletion; \textbf{SNV/Indel, HLAMP, HOMD}: number of cases harbouring a SNV/Indel, highlevel amplification and homozygous deletion respectively; events: number of gene expression outliers (see Supplemental methods) coincident with a genomic aberration and where the outlying gene is connected to the aberrated gene; p-value: statistical significance based on a randomly generated background distribution (Supplemental methods).
Having identified candidate driver genes and significantly over-represented pathways, we asked how these are distributed among individual tumours by clustering a pathway-patient mutation matrix (Figure S9). The abundance of implicated pathways can be seen to be only partially related to the total number of mutations in a case, groups 1 and 2 having on average fewer mutations per case. Frequent involvement of pathways with p53, PTEN, PI3K as members, is noted (Figure S9 and legend) however, the case groupings also vary by the progressive inclusion of additional pathways (e.g. WNT signaling, integrin signaling, ERBB signaling, hypoxia and PI3-kinase). More than two thirds of cases contained one or more mutations in the actin/cytoskeletal functions group of genes (Figure S9). Some 12% of cases did not contain somatic aberrations in any of the frequent drivers or cytoskeletal genes (Table S12). This suggests that primary TNBC are mutationally heterogenous from the outset, with some patients’ tumours having a small number of implied pathways and few mutations, whereas other patients present with tumours containing extensive mutation burdens and multiple pathway involvement.
Stimulated by the observation that early primary TNBC display a wide variation of mutation content, we asked whether the clonal composition of these primary cancers is similarly varied. We and others have shown22, 23 how deep frequency measurements of allelic abundance can be used to study tumour clonal evolution. Clonal mutation frequency (Figure 4a) can be estimated from allele abundance, once the influence of copy number states, regional loss of heterozygosity (LOH state) and tumour cellularity have been considered (although we note approximately 68% of SNVs in this study are in diploid, neutral regions). To extend allelic abundance measurements to estimation of clonal frequencies, we implemented a Dirichlet process clustering model (pyclone, Supplemental methods, Figure S10) that simultaneously estimates the genotype and clonal frequency given a list of deeply sequenced mutations and their local copy number and heterozygosity contexts.
Figure 4.
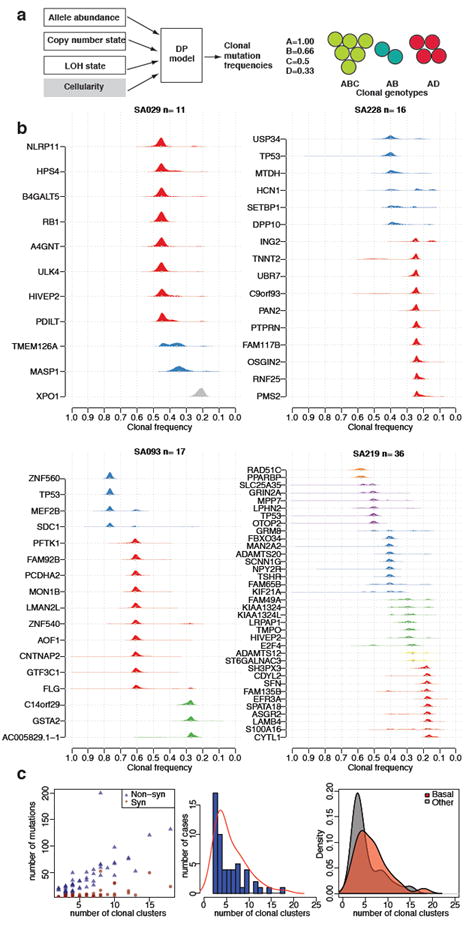
Clonal evolution in TNBC. (a) Schematic representation of integration of CNA, LOH, allelic abundance measurements and normal cell contamination for clonal frequency estimation (left). Example of a mixture of three clonal genotypes and their resulting clonal frequencies. (b) Estimated clonal frequencies for four cases are shown as the distribution of posterior probabilities from the pyclone model(Supplemental methods). Clonal frequency distributions are coloured by their frequency group membership. (c) (left) Relationship of mutation abundance (synonymous and non-synonymous) and the inferred number of clonal clusters. (middle) Distribution and kernel density (red line) of the number of inferred clonal clusters over 54 TNBCs. (right) Kernel density distribution of clonal clusters for basal (red) and non-basal (grey) tumours.
Using the set of deeply sequenced (median 20, 000x), validated SNVs, our analysis revealed (Figure 4b) that groups of mutations within individual cases exhibit different clonal frequencies, indicative of distinct clonal genotypes. Remarkably, the tumours exhibit a wide spectrum of modes over clonal frequencies (Figure 4b and Figure S11), with some cases showing only one or two frequency modes (Figure 4b, top 2 panels), indicating a smaller number of clonal genotypes, whereas other tumours exhibit multiple clonal frequency modes, indicating more extensive clonal evolution. Consistent with early ”driver gene” status, mutations in known tumour suppressors such as p53 tend to occur in the highest clonal frequency group in most tumours. However in some cases (e.g. SA219, SA236 Figure 4b, Figure S11) p53 resides in lower abundance clonal frequency groups (Figure S12, Figure 3a) suggesting it was not the founding event. Although the number of clonal frequency modes tends to increase with the number of mutations, the relationship is not strictly linear (Figure 4c). To determine whether basal and non-basal cancers differ in their clonality, we compared the distribution of clonal modes (clusters) by case, and as an overall distribution and note that basal TNBC have more clonal frequency modes than non-basal TNBC (Figure 4c). Both of these distributions emphasize a key observation, namely, that at the time of diagnosis TNBC already display a widely varying clonal evolution that mirrors the variation in mutational evolution.
Finally, we asked where significant pathways appear in the distribution of clonal frequency groups. We examined the clonal frequency of genes in each pathway and ascertained if there was a deviation away from the distribution of clonal frequency for all mutations. As expected, pathways harbouring p53 and PIK3CA showed significantly skewed distributions (Wilcoxon, q<0.01, Figure 3b: red nodes, Figure S12) towards higher clonal frequencies, consistent with their roles in early tumourigenesis (Figure 3a, Table S17). Intriguingly, pathways with cytoskeletal genes such as myosins, laminins, collagens and integrins tend to have lower median clonal frequencies suggesting that somatic mutations in these genes are acquired much later (Figure 3b lighter nodes). Notably, the median clonal frequency for Reactome pathway ”p53 pathway feedback loops” including 46 mutations in ATM, ATR, NRAS, PIK3CA, PTEN, SIAH1, and TP53 was 73% (Wilcoxon, q=0.0007) whereas ”Integrin cell surface interactions” including 23 mutations in integrin, laminin and collagen genes had a median clonal frequency of 42% (Wilcoxon, q=0.9569).
Primary triple negative breast cancers are still treated as if they were a single disease entity, yet it is clear they do not behave as a single entity in response to current therapies. Here we show for the first time using next generation sequencing mutational profiling methods, that treatment naive TNBC display a complete spectrum of mutational and clonal evolution, with some patients tumours showing only a few somatic coding sequence point mutations and a limited number of molecular pathways implicated, whereas other patients tumours exhibit significant additional mutational involvement. Moreover, the clonal heterogeneity of these cancers is also a continuum, with some patients presenting with low clonality cancers and other cases exhibiting more extensive clonal evolution at diagnosis. In this respect the basal expression subtype TNBC also tend to exhibit higher clonality at diagnosis, although the relationship is not exact.
In clonally evolving tumours identification of genes by single gene mutation frequency measurements will likely only implicate early driver genes, because the subsequent involvement of multiple additional pathways during tumour progression is unlikely to be observed as a frequent single gene mutation. The clonality analysis emphasizes this point: known drivers such as p53 and PIK3CA/PTEN, appear among the highest clonal frequencies, whereas cell shape/motility and ECM signaling genes appear in the lower clonal frequency groups, distributed over many genes. Although p53 somatic mutations are clearly early events, the clonal frequencies observed in some TNBC suggest they are not always the first event, raising a question about what drives early clonal expansion in some of these cancers. Our findings suggest that each TNBC at the time of primary diagnosis may be at a very different phase of molecular progression, with possible implications for approaches to the biology of ’low clonality’ vs ’high clonality’ primary tumours.
Methods
All Methods and the associated references are available in the Supplemental Information.
Supplementary Material
Acknowledgments
General acknowledgements. The support of the BC Cancer Agency Tumour Bank (TTR), Alberta CBCF Breast Tumour Bank and the Addenbrookes (Cambridge UK) Tumour bank is acknowledged. Technical support is acknowledged from the Centre for Translational Genomics (CTAG), the Michael Smith Genome Sciences Centre technical group, the BCCA Flow Cytometry Core Facility in the Terry Fox Laboratory. Supported by the BC Cancer Foundation, US Department of Defense CDMRP progam, Candian Breast Cancer Foundation (BC Yukon) (SA, SS), Michael Smith Foundation for Health Research (SS), US National Institutes of Health (NIH) Roadmap Epigenomics Program, NIH grant 5U01ES017154-02 (to M.H, M.A.M, J.C and T.T), Cancer Research UK (CC, PDP, CC) and the National Institute of General Medical Sciences (R01GM084875 to WWW), the Canadian Breast Cancer Research Alliance and the Canadian Cancer Society (to CE). We thank Boris Reva, Yevgeniy Antipin and Chris Sander (Memorial Sloan Kettering Cancer Center) for assistance with MutationAssessor and Guanming Wu (Ontario Institute for Cancer Research) for assistance with Reactome.
Footnotes
Author Contributions SA, SPS, CC, MAM designed and implemented the research plan and wrote the manuscript. SPS, AR, RG, GaH, JD, GH, AlB, AMP, KS, AC, RyG, AHM, JR, DL, IB, RV, SC, MG, IMM SJ, ChC, OR, PP conducted bioinformatic analyses of the data and/or advice on analytic methodology. GT conducted histopathologic review and immunohistohemistry. AO, YZ, GT, KT, LP, JK, AB, DY, AT, ND, TZ, KM, MH conducted sequencing or experimental validation of somatic aberrations. DY, AM, SWGC, GBM conducted proteome validation of splicing. PW, KG, StC, SFC, GT, JM, PDP, DH: collection and interpretation of clinical data. SD, JC, TT, MS, PG, CE: contributed materials/reagents. KH, VT, TH, MH, MAM: sequence data generation.
Supplementary Information All methods and supplementary results are available with this submission.
Competing Interests The authors declare that they have no competing financial interests.
Aligned exome/genome sequence data, RNASeq data and Affymetrix SNP6.0 datasets are available at the European Genome phenome archive: http://www.ebi.ac.uk/ega/ under study accession number EGAS00001000132.
References
- 1.Blows FM, et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: a collaborative analysis of data for 10,159 cases from 12 studies. PLoS medicine. 2010;7:e1000279. doi: 10.1371/journal.pmed.1000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 3.Sørlie T. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roth A, et al. JointSNVmix : A probabilistic model for accurate detection of somatic mutations in normal/tumour paired next generation sequencing data. Bioinformatics. 2012 doi: 10.1093/bioinformatics/bts053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding J, et al. Feature-based classifiers for somatic mutation detection in tumour-normal paired sequencing data. Bioinformatics. 2012;28:167–175. doi: 10.1093/bioinformatics/btr629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding L, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012 doi: 10.1038/nature10983. accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poulogiannis G, et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proceedings of the National Academy of Sciences. 2010;107:15145–15150. doi: 10.1073/pnas.1009941107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bignell GR, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stephens PJ, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morin RD, et al. Frequent mutation of histone-modifying genes in non-hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chicas A, et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell. 2010;17:376–387. doi: 10.1016/j.ccr.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herschkowitz JI, He X, Fan C, Perou CM. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast cancer research : BCR. 2008;10:R75. doi: 10.1186/bcr2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langerød A, et al. TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast cancer research : BCR. 2007;9:R30. doi: 10.1186/bcr1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Børresen-Dale A-L. TP53 and breast cancer. Human mutation. 2003;21:292–300. doi: 10.1002/humu.10174. [DOI] [PubMed] [Google Scholar]
- 16.Parker JS, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Youn A, Simon R. Identifying cancer driver genes in tumor genome sequencing studies. Bioinformatics. 2011;27:175–181. doi: 10.1093/bioinformatics/btq630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agrawal N, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science (New York, NY) 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stransky N, et al. The mutational landscape of head and neck squamous cell carcinoma. Science (New York, NY) 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu G, Feng X, Stein L. A human functional protein interaction network and its application to cancer data analysis. Genome Biology. 2010;11:R53. doi: 10.1186/gb-2010-11-5-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shah SP, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 23.Ding L, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.