

Abstract
Complement factor C5a is a potent proinflammatory mediator that contributes to the pathogenesis of numerous inflammatory diseases. Here, we describe the discovery of NOX-D20, a PEGylated biostable mirror-image mixed (l-)RNA/DNA aptamer (Spiegelmer) that binds to mouse and human C5a with picomolar affinity. In vitro, NOX-D20 inhibited C5a-induced chemotaxis of a CD88-expressing cell line and efficiently antagonized the activation of primary human polymorphonuclear leukocytes (PMN) by C5a. Binding of NOX-D20 to the C5a moiety of human C5 did not interfere with the formation of the terminal membrane attack complex (MAC). In sepsis, for which a specific interventional therapy is currently lacking, complement activation and elevated levels of C5a are suggested to contribute to multiorgan failure and mortality. In the model of polymicrobial sepsis induced by cecal ligation and puncture (CLP), NOX-D20 attenuated inflammation and organ damage, prevented the breakdown of the vascular endothelial barrier, and improved survival. Our study suggests NOX-D20 as a new therapeutic candidate for the treatment of sepsis.
Introduction
The complement system is an integral part of the innate immune defense. Several activation pathways initiate proteolytic cascades that converge in the cleavage and activation of complement components C3 and C5 which results in the elimination of invading bacteria, infected or foreign cells by opsonization and assembly of the membrane attack complex (MAC) and promotes inflammatory processes, mainly by the anaphylatoxins C3a and C5a.1 Despite the essential role of the complement system in host defense, it is well established that certain complement components, most notably the highly potent proinflammatory mediator C5a, contribute to the pathogenesis of a variety of diseases including autoimmune diseases,2 acute inflammatory responses to immune complexes,3 and systemic inflammatory response syndrome caused by ischemia/reperfusion injuries,4 trauma,5 or systemic infection in sepsis.6
Sepsis is one of the major causes of admission and death in intensive care medicine. In the last decade, hospitalization rates for sepsis have continuously increased and despite advances in supportive care mortality is still high.7 Moreover, patients surviving acute sepsis are at high risk for developing long-term disabilities and have reduced life expectancy.8,9 After several anti-sepsis drugs have failed to show clinical efficacy, no specific interventional therapy for severe sepsis is available today.10 Clinical evidence for a role of C5a in sepsis is provided by studies showing that C5a plasma levels are increased in patients with intra-abdominal infection and that high C5a levels are associated with increased mortality in patients with sepsis.11,12 Experimental models established that C5a substantially contributes to overwhelming systemic inflammation, which promotes life-threatening multiorgan failure in sepsis. In the rodent model of polymicrobial sepsis induced by cecal ligation and puncture (CLP), genetic deletion or pharmacological blockade of C5a receptors improves survival.13,14 In agreement with this finding, neutralization of C5a with polyclonal antibodies prevents multiorgan failure and improves survival.15,16 Moreover, C5a inhibition prevents the exhaustion and dysfunction of neutrophils which accounts for impaired bacterial killing during sepsis.15,17 Conversely, C5-deficient mice which are deprived of C5a-mediated effects as well as C5b-dependent MAC formation have no survival advantage over wild-type mice but struggle with strongly increased systemic bacterial load.18 Rats fully depleted from complement by cobra venom factor injection quickly succumb to CLP-induced sepsis.15 Taken together, these results suggest that two opposing aspects of complement activation, the lytic function of the terminal MAC and the proinflammatory function of C5a are both critically involved in sepsis and that the selective blockade of C5a without disturbing the formation of MAC may be a promising new therapeutic approach in human sepsis.
In the present study, we describe the identification and characterization of NOX-D20, a mixed RNA/DNA Spiegelmer (Spiegelmer is a registered trademark of NOXXON Pharma AG) that binds and inhibits human and mouse C5a. Spiegelmers are mirror-image structured oligonucleotides (l-oligonucleotides) that bind and antagonize a pharmacologically relevant target in a manner conceptually similar to monoclonal antibodies. Spiegelmers are generated by first identifying conventional aptamers (d-oligonucleotides) to the mirror-image of the intended target (in the case of a protein-based target a d-polypeptide) from random DNA or RNA libraries by in vitro selection. In a second step, the selected aptamers are synthesized as mirror-image l-oligonucleotides made from l-ribose/l-deoxyribose-containing nucleotides. These so-called Spiegelmers will then bind to the target in its natural configuration (l-polypeptide). The nonnatural chirality makes Spiegelmers resistant to nucleases that are prevalent in biological fluids.19 Spiegelmers generated against a variety of bioactive molecules have shown efficacy in preclinical animal models.20,21,22,23,24,25 Three Spiegelmers are currently in Phase II clinical development and have proven safe, well tolerated, and nonimmunogenic (ref. 26 and data not shown). By showing that NOX-D20 reduces multiorgan failure and improves survival in a rodent model of sepsis, the present study introduces NOX-D20 as a potential candidate for an interventional therapy to prevent sepsis progression and associated, often fatal complications.
Results
Identification of mouse d-C5a-binding aptamers
We had previously identified Spiegelmers that can specifically bind and inhibit human C5a.27 As the preclinical evaluation of these Spiegelmers was hindered by a lack of cross-reactivity to mouse or rat C5a, we sought to generate Spiegelmers targeting mouse C5a as surrogates for the use in animal models. A schematic overview of the discovery process that is described in the following paragraphs is given in Figure 1a.
Figure 1.

Identification of bio-d-mC5a binding aptamers. (a) Schematic overview of the discovery process. (b) Competitive binding assay for aptamer truncation. [32P]-labeled aptamer 274-D5 (83 nt) was incubated with bio-d-mC5a in the presence of unlabeled competitor aptamers 274-D5, 274-D5-001 (48 nt), and 274-D5-002 (44 nt) at indicated concentrations. (c) Secondary structure of 274-D5 as predicted by free energy minimization (ViennaRNA). Primer binding sites are in lower case. (d) Competitive binding assay for sequence optimization. [32P]-labeled aptamer 274-D5-002 was incubated with bio-d-mC5a in the presence of unlabeled competitor aptamers 274-D5-002, 274-C5-002, 274-C8-002, and the composite aptamer 274-C8-002-G14 at indicated concentrations.
After 10 rounds of in vitro selection with continuous enrichment (Supplementary Figure S1), a single family of RNA aptamers binding to biotinylated mirror-image mouse C5a (bio-d-mC5a) was identified (Supplementary Table S1). The most frequently occurring aptamer 274-D5 (83 nt) showed low nanomolar binding affinity to bio-d-mC5a in a competitive binding assay (Figure 1b). Deletion of primer-defined sequences G1–A17 and C66–G83 in 274-D5-001, however, resulted in a substantial loss of binding. A secondary structure prediction suggested a stem structure involving G23–G27 and C62–C66 (Figure 1c). In agreement, truncation of G1–U22 and U67–G83 delivered a 44 nt aptamer, 274-D5-002, that displayed similar binding affinity as the full-length aptamer 274-D5 (Figure 1b). The other aptamers (Supplementary Table S1) were truncated following the same procedure. Two of them, 274-C5-002 and 274-C8-002 with a single (G14) and two (A18 and U26) nucleotide exchanges, respectively, showed better bio-d-mC5a binding than 274-D5-002 (Figure 1d). A combination of these three point mutations resulted in the aptamer 274-C8-002-G14 whose affinity was superior to that of any selected sequence (Figure 1d).
Spiegelmer NOX-D19 binds to mouse and also human (l-)C5a
274-C8-002-G14 was synthesized in its l-configuration (as a Spiegelmer) and designated as NOX-D19001. After coupling of NOX-D19001 to 40 kDa polyethylene glycol (PEG), the resulting molecule was referred to as NOX-D19 (Figure 1a). Surface plasmon resonance (SPR) analysis showed that NOX-D19 not only binds to natural l-mouse C5a (mC5a) with high affinity (Kd = 51 pmol/l) (Figure 2a) but was moreover cross-reactive to human C5a (huC5a) with an affinity of Kd = 1.39 nmol/l (Figure 2b). C5a from rat or monkey (rhesus and cynomolgus) was not bound by NOX-D19 (data not shown). Comparison of C5a amino acid sequences identified two residues, Ser16 and Val28, that are identical in human and mouse C5a but different in rat and monkey C5a suggesting that one or both of these residues may be involved in binding of NOX-D19 (Supplementary Figure S2).
Figure 2.

Characterization and postselection optimization of C5a-binding Spiegelmer. SPR measurement of NOX-D19 binding to (a) mouse and (b) human C5a. Kinetic rate constants ka and kd are shown as mean ± SEM. Data are representative for at least 3 individual measurements. (c) Kinetic rate constants of 2′-deoxyribonucleotide-modified NOX-D19001 variants binding to huC5a were determined by SPR measurement. For unmodified NOX-D19001 (black diamond; dotted lines) mean ± SD of 5 injections is shown. Six modified variants of NOX-D19001 (D09, D16, D17, D30, D32, and D40) with increased overall affinity (Kd = kd/ka) were chosen and combined to the six-times modified Spiegelmer NOX-D19001-6xDNA (black square). (d) Inhibition by NOX-D19 (black diamonds) and NOX-D20 (black squares) of CD88+ BA/F3 cell chemotaxis stimulated with 0.1 nmol/l huC5a. Mean ± SD of triplicate measurements is shown. Data are representative for four independent experiments. RU, response units.
Affinity improvement by backbone modification
To evaluate the possibility of improving the affinity of the C5a-binding Spiegelmer through sugar backbone modifications, ribonucleotides were replaced by the corresponding 2′-deoxyribonucleotides at individual positions of the NOX-D19001 sequence and the resulting Spiegelmers were assessed for their binding to huC5a (Figure 2c). At few selected positions, ribo- to deoxyribonucleotide exchange led to an improved overall affinity (Kd = kd/ka) with the highest affinity improvement being observed for uridine to deoxyuridine exchange at position 9 (D09, ka = 1.82 × 106 mol/l−1 s−1; kd = 1.29 × 10−3 s−1; Kd = 0.71 nmol/l). After detailed kinetic analyses, six positions (D09, D16, D17, D30, D32, and D40) were chosen and combined in one molecule. The resulting Spiegelmer, NOX-D19001-6xDNA, had an equilibrium dissociation constant Kd = 0.36 nmol/l (ka = 1.83 × 106 mol/l−1 s−1; kd = 6.61 × 10−4 s−1) which corresponds to an approximately fourfold affinity increase driven by both faster association and slower dissociation compared with the unmodified Spiegelmer NOX-D19001 (Kd = 1.36 nmol/l; ka = 1.08 × 106 mol/l−1 s−1; kd = 1.47 × 10−3 s−1) (Figures 1a and 2c). In subsequent experiments, the assumed terminal helix of NOX-D19001-6xDNA was truncated by two more base pairs without compromising the affinity giving rise to the 40 mer NOX-D20001 (Supplementary Table S2 and Figure 1a). When conjugated to 40 kDa PEG, this Spiegelmer was designated NOX-D20 (Figure 1a).
Spiegelmer NOX-D20 shows improved binding and inhibition
NOX-D20 inhibited huC5a-stimulated chemotaxis of CD88+ BA/F3 cells with an inhibitory constant (IC50) of 0.28 ± 0.09 nmol/l compared with 1.9 ± 0.14 nmol/l (mean ± SD) for NOX-D19 which confirmed that higher binding affinity translates into an improved inhibitory potency (Figure 2d). Control Spiegelmers of the reverse NOX-D19 and NOX-D20 sequence did not inhibit huC5a-induced chemotaxis at concentrations up to 1 µmol/l (Supplementary Figure S3). The binding affinity to mouse C5a was improved from 51 pmol/l for NOX-D19 to 19 pmol/l for NOX-D20 (Figure 2a and Supplementary Figure S4). Chemotaxis induced by mouse C5a was inhibited with similar IC50 of 0.21 ± 0.1 nmol/l and 0.14 ± 0.05 nmol/l (mean ± SD) by NOX-D19 and NOX-D20, respectively, due to the stoichiometric limitation of the assay (Supplementary Figure S5).
NOX-D20 binds to C5 but does not inhibit C5 cleavage
Using SPR, we found that NOX-D20 not only binds to C5a (Figure 3a) but with similar affinity also to its metabolite C5a(desArg) (Figure 3b) and also to C5 (Figure 3c). Whether binding of NOX-D20 to C5 would interfere with C5 cleavage and C5b-dependent terminal MAC formation was evaluated in an in vitro hemolysis assay using sheep erythrocytes. In contrast to the anti-C5 aptamer C5C628 that dose-dependently inhibited erythrocyte lysis, no inhibition was observed for NOX-D20 at concentrations up to 10 µmol/l (Figure 3d). This shows that binding of NOX-D20 to the C5a moiety of C5 does not interfere with the cleavage of C5 and complement-mediated cell lysis.
Figure 3.
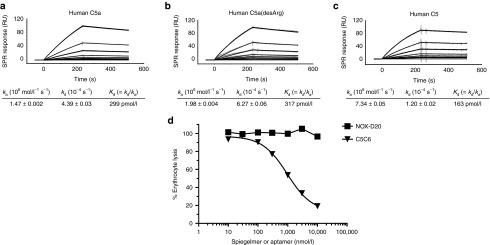
NOX-D20 binds to C5 but does not inhibit complement-mediated hemolysis. SPR measurement of NOX-D20 binding to human (a) C5a, (b) C5a(desArg), and (c) C5. Kinetic rate constants ka and kd are shown as mean ± SEM. Data are representative for at least three individual measurements. (d) Human serum pretreated with NOX-D20 (black squares) or C5-binding aptamer C5C6 (black triangles) was incubated with opsonized sheep erythrocytes. Hemolysis was quantified by photometric measurement of hemoglobin in the supernatant at 405 nm. Normalized data representative for three independent experiments is shown. RU, response units.
NOX-D20 prolongs survival and attenuates multiorgan failure in CLP-induced sepsis
In vivo efficacy of NOX-D20 was tested in CLP-induced polymicrobial sepsis, a widely used rodent model resembling important aspects of clinical sepsis.29 Vehicle-treated mice subjected to CLP surgery had a median survival of 3 days (Figure 4a). Daily treatment with 1 mg/kg NOX-D20 significantly prolonged median survival to 7 days. An increase of the dose to 3 mg/kg NOX-D20 had no additional protective effect (median survival 6.5 days). Notably, a single dose of 1 mg/kg NOX-D20 after CLP surgery followed by daily vehicle injections was as effective as daily NOX-D20 treatment (median survival of 6.5 days) (Figure 4a). As expected, no mortality occurred in sham operated mice. Statistical significance of increased survival in all three treatments groups over vehicle was confirmed by the log-rank test.
Figure 4.
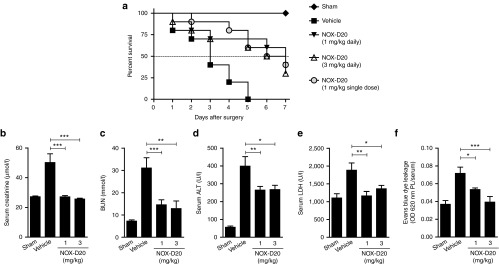
NOX-D20 improves survival in CLP-induced polymicrobial sepsis. Mice (n = 9–10 per group) were treated with daily i.p. injections of vehicle (black squares), 1 mg/kg NOX-D20 (black triangles) or 3 mg/kg NOX-D20 (open triangles) for 7 days. One group of mice received a single i.p. dose of 1 mg/kg NOX-D20 after surgery followed by daily vehicle injections (gray circles). Sham operated mice (n = 5) receiving daily vehicle injections were used as controls (black diamonds). (a) Survival was monitored daily and log-rank test was performed for statistical analysis. At day 1 (b) serum creatinine, (c) blood urea nitrogen (BUN), and (d) serum alanine aminotransferase (ALT) levels were determined (n = 9–10 for vehicle and 3 mg/kg NOX-D20; n = 20 for 1 mg/kg NOX-D20; n = 5 for sham). (e) Serum lactate dehydrogenase (LDH) levels were determined 18 hours after CLP surgery and vehicle or NOX-D20 injection (n = 7–10 per group). (f) Mice were i.v. injected with 0.25% w/v Evans blue after CLP surgery and vehicle or NOX-D20 injection (n = 8–10 per group). Ratio of Evans blue in peritoneal lavage (PL) and serum after 18 hours is shown as a measure for capillary leakage. Means ± SEM are shown. For statistical analysis, one-way analysis of variance and Dunnett's comparison was performed (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001).
High mortality in sepsis mainly arises from the damage and acute failure of vital organs. Kidney failure is indicated by the significant elevation of serum creatinine and blood urea nitrogen levels in vehicle-treated compared with sham mice one day after CLP surgery (Figure 4b,c). NOX-D20 treatment prevented any increase in serum creatinine and suppressed the elevation of blood urea nitrogen implying a protective effect of NOX-D20 on renal function (Figure 4b,c). Furthermore, NOX-D20–treated mice displayed significantly reduced serum levels of alanine aminotransferase, a marker for liver injury and hepatocellular dysfunction, compared with vehicle-treated mice (Figure 4d). Finally, elevated serum levels of lactate dehydrogenase which occur after tissue injury and may therefore be regarded as an indicator for multiorgan failure were effectively blocked by NOX-D20 (Figure 4e). For all parameters tested, 1 mg/kg NOX-D20 was sufficient to significantly reduce the sepsis-induced elevation of serum levels.
In addition to multiorgan failure, breakdown of the endothelial barrier and edema formation is a common fatal event in sepsis. By using Evans blue, a twofold increase in relative plasma protein extravasation into the peritoneal cavity was observed upon the induction of sepsis (Figure 4f). This increase in protein extravasation could be dose-dependently blocked by NOX-D20, whereby a dose of 3 mg/kg completely prevented sepsis-associated vascular leakage (Figure 4f).
NOX-D20 suppresses peritonitis
Activation of resident mononuclear cells in the peritoneal cavity leading to a fulminant release of proinflammatory and chemotactic mediators is a critical event in the pathogenesis of sepsis. We evaluated potential effects of C5a blockade by NOX-D20 on this initial inflammatory response. After previous experiments had shown that a dose of 1 mg/kg NOX-D20 was sufficient for in vivo efficacy, an additional group treated with a reduced dose of 0.1 mg/kg NOX-D20 was included. As expected, CLP-induced peritonitis was associated with a strong local and systemic upregulation of the proinflammatory cytokines interleukin-6 (IL-6) and tumor necrosis factor α (TNF-α) (Figure 5). Treatment of mice with 1 mg/kg and 3 mg/kg NOX-D20 significantly reduced TNF-α and IL-6 concentrations in the peritoneal lavage (PL) (Figure 5a) and in serum (Figure 5b). 0.1 mg/kg NOX-D20 was sufficient to significantly suppress both TNF-α levels in PL and IL-6 levels in serum.
Figure 5.
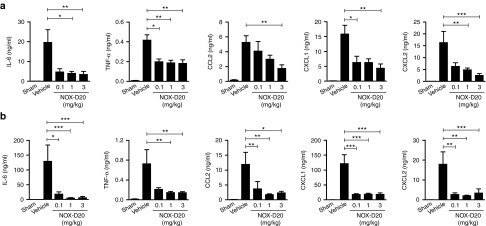
NOX-D20 suppresses proinflammatory mediator release in CLP-induced sepsis. Cytokine and chemokine concentrations in (a) peritoneal lavage (PL) and (b) serum were determined 18 hours after CLP surgery and NOX-D20 or vehicle injection. Mean ± SEM for n = 7–10 mice per group is shown. For statistical analysis, one-way analysis of variance and Dunnett's comparison was performed on log-transformed values (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001).
In mice, the recruitment of phagocytic cells, namely neutrophils, into the peritoneal cavity critically depends on the chemokines CCL2 (monocyte chemotactic protein-1), CXCL1 (keratinocyte chemoattractant), and CXCL2 (macrophage inflammatory protein-2). All three chemokines were significantly increased in PL and serum of vehicle-treated septic mice (Figure 5a,b). C5a inhibition by NOX-D20 largely blocked this upregulation. In PL, doses of 0.1 mg/kg and 1 mg/kg NOX-D20 significantly reduced CXCL1 and CXCL2 levels, respectively, whereas the highest dose of 3 mg/kg NOX-D20 was required to significantly suppress CCL2 levels (Figure 5a). In serum, the lowest dose of NOX-D20 was sufficient to significantly reduce the increase of CCL2, CXCL1, and CXCL2 (Figure 5b).
Eighteen hours after CLP surgery, a massive infiltration of polymorphonuclear leukocytes (PMN) into the peritoneal cavity was observed in vehicle-treated mice (Figure 6a). Monocytes accumulated at significant numbers as well (Figure 6b). No infiltration of macrophages or lymphocytes was observed at this time point. Treatment with 0.1 mg/kg NOX-D20 was sufficient to significantly decrease the numbers of both PMN and monocytes accumulating in the peritoneal cavity of septic mice (Figure 6a,b).
Figure 6.

NOX-D20 suppresses PMN recruitment and activation. Cell numbers of (a) PMN and (b) monocytes were determined in the peritoneal lavage by cytospin 18 hours after CLP surgery and NOX-D20 or vehicle injection. Mean ± SEM of n = 8–10 mice is shown. For statistical analysis, one-way analysis of variance and Dunnett's comparison was performed. (c,d) Primary human PMN were stimulated in vitro with huC5a. Inhibition of (c) PMN chemotaxis and (d) elastase release by NOX-D20 is shown. Mean ± SEM of triplicate measurement of PMN from three individual donors is shown. For statistical analysis, one-way analysis of variance and Dunnett's comparison was performed on log-transformed values (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). PMN, polymorphonuclear leukocytes.
NOX-D20 inhibits activation of primary human PMN
In agreement with the results from mice above, NOX-D20 also efficiently blocked huC5a-stimulated chemotaxis of primary human peripheral blood PMN in vitro (Figure 6c). The activation of neutrophils by C5a may directly contribute to organ injury through stimulating the release of cytotoxic proteases such as neutrophil elastase. In vitro, NOX-D20 efficiently inhibited the release of elastase from primary human neutrophils (Figure 6d). Of note, in this assay format cells were stimulated with 30 nmol/l huC5a. Thus, for stoichiometric reasons, half-maximal elastase release in the presence of 30 nmol/l NOX-D20 indicates a highly efficient neutralization of C5a.
Pharmacokinetics of NOX-D20 in mice
A pharmacokinetic study with single intraperitoneal doses of 1 mg/kg and 10 mg/kg NOX-D20 was carried out in mice. Plasma levels of NOX-D20 were determined over 4 weeks using an SPR-based assay with a complementary l-DNA hybridization probe. Three hours after dosing, the plasma concentrations (Cmax) of NOX-D20 peaked at 1.8 ± 0.02 µmol/l for the 1 mg/kg and 12.0 ± 0.7 µmol/l for the 10 mg/kg dose (means ± SEM, n = 4) (Figure 7). The plasma elimination of both doses was biphasic with half-lives (t1/2) in the initial elimination phase (3–48 hours) of 7.2 hours (95% CI: 5.8–9.5 hours) for 1 mg/kg and 7.9 hours (95% CI: 6.4–10.3 hours) for 10 mg/kg NOX-D20. The calculated area under the curve for 1 mg/kg was 11.95% of that for 10 mg/kg NOX-D20 indicating a linear dose–exposition relationship. In the later phase (4–28 days after dosing), an attenuated elimination of NOX-D20 from plasma with t1/2 > 7 days for both dosing groups was observed. The transition from initial into late phase elimination occurred within the range or slightly below measured plasma C5 concentrations (130 ± 26 nmol/l; mean ± SEM of n = 8) providing evidence for a target-driven slowing-down of late phase elimination. After 28 days, plasma levels were still at 8.5–12 nmol/l in both dosing groups (Figure 7).
Figure 7.

Pharmacokinetics study. NMRI mice were i.p. injected with a single dose of 1 mg/kg or 10 mg/kg NOX-D20. Blood was drawn at indicated time points and NOX-D20 concentrations were determined by SPR measurement. Mean ± SEM of four mice per group and time point is shown. Linear regression of log-transformed plasma concentrations at 3–48 hours and 4–28 days is shown. Lower limit of quantification was 0.79 nmol/l NOX-D20. Plasma C5 levels were determined by ELISA (duplicate measurement) in plasma of mice (n = 4) treated with vehicle 10 minutes and 24 hours before blood sampling. Mean C5 plasma concentration is shown as solid line. Gray area indicates 95% CI.
Discussion
There is emerging evidence for a pathogenic role of the complement factor C5a in a number of diseases that are associated with complement activation and inflammation.2,3,4,5,6 In sepsis, C5a contributes to the exceeding inflammatory response and life-threatening organ failure. Blockade of C5a by polyclonal antibodies or inactivation of its receptors has been shown to improve the outcome of experimental sepsis.13,14,15,30 Here, we report the identification and characterization of NOX-D20, a biostable mirror-image mixed (l-)RNA/DNA aptamer (Spiegelmer) that binds to mouse as well as human C5a with picomolar affinities that are in the range or even below those described for C5a binding to its receptors.31,32 Consistently, NOX-D20 efficiently competed with C5a receptor binding and blocked C5a-induced cellular responses of both a CD88-expressing cell line and primary human PMN in vitro.
The generation of NOX-D20 involved an approach to improve binding affinity by the deletion of oxygen atoms in the 2′-position of ribose at selected positions through ribo- to deoxyribonucleotide exchanges. So far, chemical modifications (primarily ribose modifications such as 2′-O-methyl, 2′-amino, or 2′-fluoro) in aptamers and other oligonucleotide modalities have mostly been used to reduce nuclease susceptibility thereby increasing in vivo stability.33 In this context, it was observed that such modifications can also affect the affinity of aptamers.34 Spiegelmers are inherently stable against nucleolytic attacks due to their mirror-image configuration which enabled us to use ribose modifications solely for the systematic improvement of the target binding affinity. The replacement of ribonucleotides by the corresponding deoxyribonucleotides at six defined positions delivered a Spiegelmer (NOX-D19001-6xDNA) with substantially improved affinity and inhibitory constant. We observed that truncation of the predicted terminal helix without loss of affinity was possible for this deoxy-modified Spiegelmer (resulting in NOX-D20001) but not for all-RNA NOX-D19001 (data not shown). Therefore, we speculate that stabilization of the Spiegelmer's secondary structure contributes to affinity improvement. In addition, intermolecular interactions between the Spiegelmer and its target may be enhanced by substitution of selected 2′-hydroxyl groups by hydrogen, possibly involving nucleotides located within potential loop regions such as the most effective single exchange, U-to-dU at position 9. In analogy to NOX-D20, we have found affinity improvement for a DNA-Spiegelmer by introduction of ribonucleotides at selected positions, i.e., the addition, not the deletion, of oxygen atoms.25 This shows the potential of sugar backbone modifications for affinity improvement and highlights a unique advantage of chemically synthesized oligonucleotide-based drugs like Spiegelmers.
Various pathways link C5a and systemic inflammation to the pathogenesis of sepsis and are the basis of the protective effects of C5a antagonism in preclinical models of sepsis.10 Neutrophils are widely considered the first responders to an infection and can be observed in the peritoneal cavity within a few hours after induction of bacterial peritonitis.35 The pivotal role of C5a for neutrophil recruitment and activation has been demonstrated for immune complex-induced lung injury where genetic deletion or pharmacological blockade of C5a receptor markedly decreased PMN recruitment into the lung and strongly reduced proinflammatory mediator release.3,36 Activation of neutrophils moreover results in the release of cytotoxic substances such as reactive oxygen species and lysosomal enzymes (e.g., elastase) that directly contribute to organ damage.10 Eventually, excessive stimulation of phagocytic innate immune cells by C5a promotes the development of immune dysfunction associated with an increased susceptibility to infections.11,13,15,17 C5a-mediated activation of the coagulation/fibrinolytic system may furthermore result in enhanced clotting, diffuse fibrin deposition on the microvasculature, and tissue hypoxia.37,38 Altogether, these events result in the damage and failure of vital organs which is a reason for the still high mortality rate in patients with sepsis. NOX-D20, the novel C5a-neutralizing Spiegelmer described here, prevented C5a-induced release of elastase from PMN in vitro, limited the infiltration of neutrophils into the peritoneal cavity of septic mice and attenuated the upregulation of proinflammatory cytokines and chemokines in vivo. The suppression of serum parameters for liver and general tissue injury (alanine aminotransferase and lactate dehydrogenase) as well as renal glomerular filtration rate (creatinine, blood urea nitrogen) indicates that NOX-D20 prevents the progression of sepsis and the development of multiorgan dysfunction thereby improving survival. In addition, systemic inflammation also affects the cardiovascular system leading to increased cardiac output, systemic vasodilation and a breakdown of the capillary barrier function resulting in plasma extravasation and edema formation.10,39 Accordingly, edema formation in the lungs and acute lung injury is among the most frequent complications in patients with severe sepsis.6 In both CLP-induced sepsis and immune complex-induced lung injury polyclonal anti-C5a antibodies improved respiratory function.16 Similarly, NOX-D20 suppressed plasma protein extravasation into the peritoneum and NOX-D19 has recently been shown to limit microvascular leakiness and to prevent airway ischemia in C3-deficient tracheal transplant recipients.40
An alternative strategy to interfere with C5a-mediated signaling that has been pursued for many years is the blockade of the C5a receptor (CD88). A new generation of small-molecule CD88 antagonists as well as a CD88-targeting monoclonal antibody have recently progressed into clinical trials for rheumatoid arthritis and antineutrophil cytoplasmic autoantibody–associated renal vasculitis.41,42 Before this, PMX-53, a cyclic peptide CD88 antagonist, and analogs thereof had been successfully used in different preclinical models, including CLP-induced sepsis in mice13 but clinical development was discontinued following repeated trial failures.42 For other clinical stage CD88 blockers, data from sepsis models have not been published, most probably due to a lack of rodent CD88 cross-reactivity.42 There is experimental evidence that in sepsis CD88 and also C5L2, a second receptor for C5a and C5a(desArg), synergistically contribute to pathogenesis.14 NOX-D20 will most likely block signals mediated by both receptors. In chronic indications, oral availability of small-molecule CD88 blockers is certainly an advantage over injectable macromolecules like Spiegelmers and antibodies. However, this does not apply to intensive care settings where parenteral administration is the route of choice.
In contrast to C5a-specific interventions, inhibition of the complement cascade by blockade of C5 is not considered to be indicated in sepsis since C5b-dependent lysis is important for controlling the bacterial load.30 In CLP-induced sepsis, C5-deficient mice do not have any survival advantage over wild-type but struggle with strongly increased systemic bacterial load,18 whereas animals in which C5a is selectively antagonized are protected.13,14,15 In agreement with this observation, an anti-C5 antibody which inhibits C5 cleavage prevented potentially harmful C5a-mediated granulocyte activation but simultaneously disabled C5b-mediated bacterial clearance in a human whole blood in vitro model of meningococcal sepsis.43 In blood cultures treated with a C5a-specific antibody, however, granulocyte activation was effectively suppressed without precluding efficient bacterial clearance. Finally, clinical data show that long-term inhibition of C5 cleavage is associated with an increased risk of meningococcal infections.30 NOX-D20, although binding to C5, does not interfere with complement-mediated hemolysis thus offering a C5a-specific treatment option.
The ability of NOX-D20 to bind to C5 may even positively impact on its pharmacodynamic and pharmacokinetic profile. In contrast to compounds that would bind to C5a only after it was cleaved from C5, NOX-D20's mode of action is likely to improve in vivo efficacy as it will prevent the release of active C5a by ongoing complement activation. We measured mean C5 plasma levels of 130 nmol/l in mice whereas plasma levels of C5a are much lower.40 A single i.p. injection of 1 mg/kg NOX-D20 resulting in a maximal plasma concentration of 1.8 µmol/l Spiegelmer could therefore be sufficient to occupy most C5 present in circulation. Pharmacokinetic analysis showed that in the initial elimination phase, i.e., between 3 and 48 hours after administration, NOX-D20 had a plasma half-life of 7–8 hours thereby showing a substantially longer presence in circulation than peptidic or nonpeptidic small-molecule complement inhibitors.30,44,45 Plasma half-lives of 6–8 hours are routinely observed for Spiegelmers conjugated to 40 kDa PEG whereas unPEGylated Spiegelmers are rapidly cleared from circulation with t1/2 < 30 minutes (ref. 46 and data not shown). In a second phase, elimination of NOX-D20 was attenuated with t1/2 > 7 days and at day 28 Spiegelmer levels of about 10 nmol/l were still present in circulation. Transition from early into late phase elimination occurred 48–96 hours after dosing in a concentration range similar to the levels of C5 measured in plasma. This provides first evidence that by binding to the large protein C5, the half-life of NOX-D20 is prolonged. Similar biphasic elimination profiles have been described for other inhibitors that bind to soluble complement components, such as a single-chain anti-C5 antibody fragment47 and C3 inhibitor compstatin analogs.48 For OmCI, a tick-derived C5 inhibitor, the decelerated elimination has been shown to be dependent on the presence of C5.44 Taken together, this suggests a NOX-D20 PK model with an early PEG-driven and a late target-driven attenuation of Spiegelmer elimination. Clinical data from the C5-blocking antibody eculizumab show that long-term C5 inhibition primarily involves safety issues that are related to the blockade of C5b-mediated bacterial lysis.30 For this reason, long-term inhibition of C5a is not expected to create serious adverse events.
In recent years, numerous approaches to improve the outcome of sepsis by targeting selected pathways failed to show efficacy in clinical trials. Examples are the blockade of proinflammatory mediators (TNF-α or IL-1) and the inhibition of coagulation by activated protein C.10 C5a is involved in a number of pathways that contribute to the pathogenesis of sepsis and there is conclusive experimental evidence that anti-C5a treatment can provide a new therapeutic option for patients with sepsis.30 Spiegelmers are a novel class of chemically synthesized, biostable, and nonimmunogenic agents that efficiently inhibit protein–protein interactions. In Phase I clinical studies, three different Spiegelmer-based compounds were demonstrated to be safe and well tolerated (ref. 26 and data not shown). Here, we have described NOX-D20, a Spiegelmer that specifically antagonizes C5a and does not interfere with C5b-mediated MAC formation. In a preclinical in vivo model of sepsis NOX-D20 suppressed local and systemic inflammation, prevented organ failure, and improved survival. Cross-reactivity of NOX-D20 to mouse and human C5a will facilitate the translation of preclinical results into the clinical setting making NOX-D20 a promising candidate for interventional therapies in sepsis and potentially other diseases associated with excessive complement activation and inflammation.
Materials and Methods
Peptides and nucleic acids. Mouse C5a (1–77) (UniProt ID: P06684) in the all-d-configuration with biotin covalently coupled to its amino terminus by a di-aminoethyloxyethyloxyacetyl linker (bio-d-mC5a) was synthesized at ALMAC Sciences (Edinburgh, UK). Recombinant human and mouse C5a was from R&D Systems (Wiesbaden, Germany), recombinant human C5a(desArg) from Hycult Biotech (Beutelsbach, Germany), and human C5 purified from serum from Sigma-Aldrich (Taufkirchen, Germany). Recombinant rat and cynomolgus/rhesus monkey C5a were prepared at NOXXON Pharma AG (Berlin, Germany). Oligonucleotides were synthesized at NOXXON Pharma AG using standard solid-phase phosphoramidite chemistry as previously described.49 To increase the plasma residence time, the 5′-end of Spiegelmers was covalently linked to a Y-shaped branched 40 kDa PEG moiety (JenKem, Allen, TX) using an aminohexyl linker (American International Chemical, Framingham, MA). Concentrations and doses always refer to the oligonucleotide part as anhydrous free acid. The oligonucleotide sequence of NOX-D20 is GCGAUG(dU)GGUGGU(dG)(dA)AGGGUUGUUGGG(dU)G(dU)CGACGCA(dC)GC.
In vitro selection. An RNA library was generated from a synthetic single-stranded DNA library with 34 random positions and flanking primer binding sites for the forward primer carrying a T7-promotor (5′-TCTAATACGACTCACTATAGGAGCTCAGACCGTACACC-3′) and the reverse primer (5′-CTGGAACCGACACTGCAGCC-3′). Double-stranded DNA (dsDNA) was generated by single-step fill-in reaction using Vent (exo-) polymerase (New England BioLabs, Frankfurt am Main, Germany) and the forward primer followed by RNA transcription with T7 RNA polymerase (Stratagene, Waldbronn, Germany) to yield the RNA library: GGAGCUCAGACCGUACACCUGUGC-N34-GCACAGGCUGCAGUGUCGGUUCCAG. In the first round of in vitro selection, 4 nmol RNA library (= 2.4 × 1015 individual molecules) 5′-labeled with [−32P]-ATP by T4 polynucleotide kinase (Invitrogen, Karlsruhe, Germany) were incubated with equimolar bio-d-mC5a in selection buffer (20 mmol/l Tris, pH 7.4; 150 mmol/l NaCl; 5 mmol/l KCl; 1 mmol/l MgCl2; 1 mmol/l CaCl2; 0.1% Tween 20; 50 µg/ml bovine serum albumin (BSA); 10 µg/ml unspecific l-RNA; 4 U/ml RNaseOut, Invitrogen) at 37 °C for 16 hours. Bio-d-mC5a/RNA complexes were immobilized on streptavidin or neutravidin-coupled agarose or ultralink plus beads (Pierce, Thermo Scientific, Nidderau, Germany). The percentage of target-bound RNA molecules was measured in a scintillation counter (LS6500; Beckman Coulter, Fullerton, CA). After washing with selection buffer, bio-d-mC5a–bound RNA molecules were reverse transcribed (SCRIPT reverse transcriptase, Jena Bioscience, Jena, Germany), PCR amplified (Vent (exo-) polymerase; New England BioLabs), and transcribed to single-stranded RNA. In subsequent rounds, the stringency of in vitro selection was continuously increased by reducing incubation times and the concentrations of both bio-d-mC5a and the enriched RNA library. Preselection steps with beads in the absence of target were performed before each selection round to avoid enrichment of bead-binding aptamers. dsDNA obtained from round 10 was cloned and sequenced (LGC Genomics, Berlin, Germany). ViennaRNAfold was used for minimum free energy secondary structure prediction.50
Competitive aptamer binding assay. 5′-[32P]-labeled aptamers were incubated in selection buffer with bio-d-mC5a at concentrations enabling binding of 5–10% aptamer. Unlabeled aptamer was added at indicated concentrations to compete with labeled aptamer for binding to bio-d-mC5a. After 3–4 hours at 37 °C bio-d-mC5a/aptamer complexes were immobilized on neutravidin agarose beads, washed with selection buffer, and bead-bound radioactivity was measured. The amount of bound labeled aptamer decreases with increasing concentrations and/or affinity of the competitor aptamer. Data were normalized to the amount of bead-bound [32P]-aptamer in the absence of competitor.
Determination of selectivity and specificity by SPR. SPR measurements were performed with a Biacore 2000 instrument (GE Healthcare, Munich, Germany). Proteins were immobilized by amine coupling to an NHS/EDC-activated carboxy-dextran–coated CM5 sensor chip (GE Healthcare). To adjust the molecular weight difference, 300–600 response units (RU) of C5a and C5a(desArg) and 6,500–12,500 RU of C5 were immobilized. Spiegelmers were injected in twofold dilution series ranging from 1 µmol/l to 120 pmol/l at a flow of 30 µl/minutes. Measurements were conducted at 37 °C in 20 mmol/l Tris–HCl pH 7.4, 150 mmol/l NaCl, 5 mmol/l KCl, 1 mmol/l MgCl2 and 1 mmol/l CaCl2. Regeneration was performed by injection of 60 μl 5 mol/l NaCl. Samples were referenced to a NHS/EDC-activated and ethanolamine-blocked control flow cell. Data analysis and calculation of equilibrium dissociation constants Kd was done with the BIAevaluation 3.1.1 software (BIACORE AB, Uppsala, Sweden) using a Langmuir 1:1 stoichiometric fitting algorithm with a fixed refractive index correction value RI = 0, a defined mass transfer coefficient kt = 1 × 107 (RU mol/l−1 s−1) and a local fitting of the maximal response (Rmax). Data fitting was performed using the injected Spiegelmer concentrations from 120 pmol/l up to 7.8 nmol/l. For kd values <1 × 10−4 (s−1), long-term dissociation studies (1,400 s) were performed to determine exact off-rate values. Each measurement was done at least three times. Kinetic rate constants are shown as mean ± SEM.
Hemolytic assay. Human complement serum (Sigma-Aldrich) was preincubated with Spiegelmer in 96-well plates (Nunc-Immuno Plate, Thermo Scientific) in HEPES buffer (KBR (CFTB) buffer; Virion\Serion, Wuerzburg, Germany) at 37 °C for 60 minutes. As a positive control, the C5-binding and cleavage inhibiting d-RNA aptamer C5C6 (CG*CCGCG*G*UCUCA*G*G*CGCUG*A*G*UCUG*A*G*UUUACCUG*CG*; C is 2′-fluoro-cytidin, U is 2′-fluoro-uridine, G* is 2′-methoxy-guanosine, A* is 2′-methoxy-adensoine; synthesized at NOXXON Pharma) was used.28 Sheep erythrocytes opsonized with rabbit anti-sheep erythrocyte antibodies (“hemolytic system”, Virion\Serion) were added and hemolysis was quantified after 30 minutes incubation at 37 °C by colorimetric measurement of the supernatant at 405 nm (PolarStar Galaxy plate reader, BMG Labtech, Ortenberg, Germany).
Chemotaxis of CD88-expressing cell line. The BA/F3 mouse pro-B cell line was stably transfected using a plasmid coding for the human C5a receptor CD88 (NCBI accession NM_001736 in pcDNA3.1+). Transfected cells were selected by treatment with geneticin and tested for expression of CD88 with RT-PCR. Functionality was tested with a chemotaxis assay. For this purpose, recombinant human (0.1 nmol/l) or mouse (0.3 nmol/l) C5a was preincubated with Spiegelmers at indicated concentrations in HBH buffer (Hanks balanced salt solution (HBSS) + 1 mg/ml BSA + 20 mmol/l HEPES) in the lower compartments of a 96-well Corning Transwell plate with 5 µm pores (Costar Corning, NY) at 37 °C for 20–30 minutes. 1 × 105 CD88+ BA/F3 cells in HBH buffer were added to the upper compartments and were allowed to migrate at 37 °C for 3 hours. After removal of the upper compartments 50 µmol/l resazurin (Sigma-Aldrich) in phosphate-buffered saline was added to the lower compartments and incubated at 37 °C for 2.5 hours. Fluorescence was measured at 590 nm (excitation wavelength 544 nm). Background-corrected and normalized fluorescence values were plotted against Spiegelmer concentration. IC50 values were determined with nonlinear regression (4 parameter fit) using Prism 5 software (GraphPad Software, San Diego, CA).
Chemotaxis and elastase release of primary human PMN. Peripheral blood from healthy donors was collected in acid citrate dextrose–containing blood collection tubes (Sarstedt, Leicester, UK). PMN were isolated using a discontinuous dextran/ficoll gradient centrifugation method. For chemotaxis assays, human C5a (1 nmol/l) was preincubated with indicated concentrations of the Spiegelmer in HBSS + 0.01% BSA + 25 mmol/l HEPES in the lower chamber of a 96-well chemotaxis plate. 1.7 × 105 PMN were added to the upper chambers and chemotaxis was performed at 37 °C for 25 minutes. Following incubation the upper chamber was transferred to a white luminescence plate containing Accutase (Thermo Scientific) to harvest cells bound to the underside of the chemotaxis mesh. Glo reagent (Promega, Southampton, UK) was added and equilibrated for 10 minutes. Luminescence was measured using a Biotek Synergy 2 plate reader (Potton, UK). For elastase release assays, human PMN were primed with TNF-α (10 ng/ml) and cytochalasin B (5 μg/ml) at 37 °C for 30 minutes. Cells were then stimulated for 45 minutes with human C5a (30 nmol/l) which had been preincubated with Spiegelmer at indicated concentrations. Following incubation, 25 μl of the supernatant were incubated with elastase substrate (Calbiochem, Nottingham, UK) in Tris–HCl 0.1 mol/l pH 7.4 at 37 °C for 1 hours with absorbance at 405 nm being measured every 5 minutes. The kinetic data were analyzed to determine the vmax for each sample. The mean percentage elastase activity relative to control was calculated for each sample (background not subtracted). Experiments were performed by GlycoMar (Dunbeg, Oban Argyll, Scotland, UK). Prism 5 software (GraphPad Software) was used for data processing and statistical evaluation.
CLP model. Peritonitis was surgically induced under light isoflurane anesthesia in 10- to 12-week-old, male C57BL/6 mice (Charles River Laboratories, Germany). Incisions were made into the left upper quadrant of the peritoneal cavity, the cecum was exposed and a tight ligature was placed around the cecum with sutures distal to the insertion of the small bowel. One puncture wound was made with a 24-gauge needle into the cecum and small amounts of cecal contents were expressed through the wound. The cecum was replaced into the peritoneal cavity and the laparotomy site was closed. 500 µl saline was given s.c. as fluid replacement. Sham animals underwent the same procedure except for ligation and puncture of the cecum. Vehicle (0.9% NaCl) or Spiegelmer was administered by intraperitoneal injection starting immediately after CLP surgery. Survival was followed for 7 days. Kaplan–Meier survival curves were generated and a log-rank test was performed for statistical analysis using Prism 5 software (GraphPad Software). Blood samples were obtained under light ether anesthesia from the cavernous sinus at the indicated time points. Serum markers of acute kidney failure (creatinine and blood urea nitrogen), acute liver injury (alanine aminotransferase) and endothelial injury (lactate dehydrogenase) were measured using an Olympus analyzer (AU400). Prism 5 software (GraphPad Software) was used for data processing and statistical evaluation. All procedures were carried out at Phenos GmbH (Hannover, Germany) according to the guidelines of the German Society for Animal Science (Gesellschaft fuer Versuchstierkunde; GV-SOLAS) and were approved by the local authorities.
Determination of cytokine and chemokine concentrations in PL and serum. Eighteen hours after CLP surgery, mice were anesthetized with isofluorane for blood sampling. Subsequently, animals were killed and PL was performed using 3 ml phosphate-buffered saline. Total cell count was assessed using a hemocytometer (Omnilab, Gehrden, Germany). Differential cell count was performed on cytospins (cytospin4, Thermo Scientific) after hematoxylin and eosin staining. PL and serum levels of TNF-α, IL-6, and CCL2 were quantified by bead-based flow cytometry assay (CBA Kit; BD Biosciences, Heidelberg, Germany) according to the manufacturer's instructions. For statistical analysis, values below the lower limit of quantification were set to lower limit of quantification. The concentrations of CXCL1 and CXCL2 were determined by ELISA (R&D Systems, Wiesbaden, Germany). Prism 5 software (GraphPad Software) was used for data processing and statistical evaluation.
Capillary leakage. Immediately after CLP surgery, 0.25% w/v Evans blue (200 μl) was injected intravenously. After 18 hours, mice were killed and PL was performed. Optical densities at 620 nm (A620) were measured in PL and serum. Absorption in serum was corrected for contamination with heme pigments (corrected A620 = A620 − (A405 × 0.014). Capillary leakage was determined as the ratio of A620 in PL and corrected A620 in serum. Prism 5 software (GraphPad Software) was used for data processing and statistical evaluation.
Pharmacokinetics of NOX-D20 in mice. Plasma levels of NOX-D20 were determined over 4 weeks in NMRI mice after a single intraperitoneal injection of 1 mg/kg or 10 mg/kg NOX-D20 (16 mice per group, 4 mice per time point). Blood samples were taken 10 minutes, 1 hours, 3 hours, 8 hours, 24 hours, 48 hours, 3 days, 7 days, 14 days, 21 days, and 28 days after dosing and lithium-heparin plasma was prepared. Dosing and blood sampling were performed by Heidelberg Pharma (Heidelberg, Germany). All animal procedures were approved by the local ethical committee and performed in accordance with the national guidelines for the care and use of animals in biomedical research. NOX-D20 plasma levels were quantified by SPR measurement. First, neutravidin (Thermo Fisher) was immobilized (20,000–30,000 RU) by amine coupling to an NHS/EDC-activated carboxy-dextran–coated CM5 sensor chip (GE Healthcare). A biotinylated l-DNA hybridization probe (biotin-2x hexaethyleneglycol-5′-ACACCCAACAACCCTTCACCAC-3′) complementary to a central stretch of NOX-D20 was then immobilized to 1,500–1,600 RU. An NHS/EDC-activated and ethanolamine-blocked flow cell and a neutravidin-coupled biotin-blocked flow cell were used as controls. Regeneration was performed by injection 15 µl of Glycine-HCl pH 1.5 (GE Healthcare) and 15 µl of 50 mmol/l NaOH (GE Healthcare). Association and dissociation of the binding event was recorded under optimized buffer conditions at 37 °C for 120 seconds each at a flow of 20 µl/minutes. The increase in response correlates with the concentration of NOX-D20 and was determined by report points at 0 and 140 seconds of injection. Data analysis was performed with the BIA evaluation 3.1.1 software. Data processing was performed using Prism 5 software (GraphPad Software). A standard curve was used to calculate the concentrations of NOX-D20 in unknown samples. The lower limit of quantification was 0.79 nmol/l NOX-D20. Plasma half-lives (t1/2) were calculated from a linear regression of log-transformed plasma concentrations of NOX-D20 at 3–48 hours and 4–28 days after dosing with t1/2 = (−log 2/m) and m = slope of the regression line. C5 levels were determined by ELISA in lithium-heparin plasma of control mice (n = 4) treated with vehicle 10 minutes and 24 hours before blood sampling according to manufacturer's instructions (UscnLife Science, Hoelzel Diagonistic, Koeln, Germany).
SUPPLEMENTARY MATERIAL Figure S1. Enrichment of bio-d-mC5a binding aptamers from a random RNA library. Figure S2. C5a sequence alignment. Figure S3. Reverse control Spiegelmers do not inhibit huC5a-induced chemotaxis. Figure S4. NOX-D20 binds to mouse C5a. Figure S5. NOX-D19 and NOX-D20 inhibit mouse C5a-induced chemotaxis. Table S1. Sequences and frequencies of RNA aptamers identified after 10 rounds of in vitro selection. Table S2. Truncation of NOX-D19001-6xDNA.
Acknowledgments
The authors thank Lucas Bethge, Gabriele Anlauf, and Tino Struck for providing aptamers and Spiegelmers; Lisa Bauer and Katrin Schindele for technical assistance in the in vitro pharmacology group; Dirk Zboralski for supportive experiments; and Christian Mihm for editing of the figures. K.H., C.M., K.B., W.P., A.V., and S.K. are employees of NOXXON Pharma AG, a company that has filed patents on the C5a-inhibiting Spiegelmers and has a commercial interest in Spiegelmers. The work was done in Berlin and Hannover, Germany. The other authors declare no conflict of interest.
Supplementary Material
References
- Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, et al. Arthritis critically dependent on innate immune system players. Immunity. 2002;16:157–168. doi: 10.1016/s1074-7613(02)00275-3. [DOI] [PubMed] [Google Scholar]
- Shushakova N, Skokowa J, Schulman J, Baumann U, Zwirner J, Schmidt RE, et al. C5a anaphylatoxin is a major regulator of activating versus inhibitory FcgammaRs in immune complex-induced lung disease. J Clin Invest. 2002;110:1823–1830. doi: 10.1172/JCI200216577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banz Y, Rieben R. Role of complement and perspectives for intervention in ischemia-reperfusion damage. Ann Med. 2012;44:205–217. doi: 10.3109/07853890.2010.535556. [DOI] [PubMed] [Google Scholar]
- Burk AM, Martin M, Flierl MA, Rittirsch D, Helm M, Lampl L, et al. Early complementopathy after multiple injuries in humans. Shock. 2012;37:348–354. doi: 10.1097/SHK.0b013e3182471795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosmann M, Ward PA. Role of C3, C5 and anaphylatoxin receptors in acute lung injury and in sepsis. Adv Exp Med Biol. 2012;946:147–159. doi: 10.1007/978-1-4614-0106-3_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar G, Kumar N, Taneja A, Kaleekal T, Tarima S, McGinley E, Milwaukee Initiative in Critical Care Outcomes Research Group of Investigators et al. Nationwide trends of severe sepsis in the 21st century (2000-2007). Chest. 2011;140:1223–1231. doi: 10.1378/chest.11-0352. [DOI] [PubMed] [Google Scholar]
- Winters BD, Eberlein M, Leung J, Needham DM, Pronovost PJ, Sevransky JE. Long-term mortality and quality of life in sepsis: a systematic review. Crit Care Med. 2010;38:1276–1283. doi: 10.1097/CCM.0b013e3181d8cc1d. [DOI] [PubMed] [Google Scholar]
- Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. 2010;304:1787–1794. doi: 10.1001/jama.2010.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry DE. Sepsis, systemic inflammatory response, and multiple organ dysfunction: the mystery continues. Am Surg. 2012;78:1–8. [PubMed] [Google Scholar]
- Solomkin JS, Jenkins MK, Nelson RD, Chenoweth D, Simmons RL. Neutrophil dysfunction in sepsis. II. Evidence for the role of complement activation products in cellular deactivation. Surgery. 1981;90:319–327. [PubMed] [Google Scholar]
- Gressner OA, Koch A, Sanson E, Trautwein C, Tacke F. High C5a levels are associated with increased mortality in sepsis patients–no enhancing effect by actin-free Gc-globulin. Clin Biochem. 2008;41:974–980. doi: 10.1016/j.clinbiochem.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Huber-Lang MS, Riedeman NC, Sarma JV, Younkin EM, McGuire SR, Laudes IJ, et al. Protection of innate immunity by C5aR antagonist in septic mice. FASEB J. 2002;16:1567–1574. doi: 10.1096/fj.02-0209com. [DOI] [PubMed] [Google Scholar]
- Rittirsch D, Flierl MA, Nadeau BA, Day DE, Huber-Lang M, Mackay CR, et al. Functional roles for C5a receptors in sepsis. Nat Med. 2008;14:551–557. doi: 10.1038/nm1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czermak BJ, Sarma V, Pierson CL, Warner RL, Huber-Lang M, Bless NM, et al. Protective effects of C5a blockade in sepsis. Nat Med. 1999;5:788–792. doi: 10.1038/10512. [DOI] [PubMed] [Google Scholar]
- Huber-Lang M, Sarma VJ, Lu KT, McGuire SR, Padgaonkar VA, Guo RF, et al. Role of C5a in multiorgan failure during sepsis. J Immunol. 2001;166:1193–1199. doi: 10.4049/jimmunol.166.2.1193. [DOI] [PubMed] [Google Scholar]
- Huber-Lang MS, Younkin EM, Sarma JV, McGuire SR, Lu KT, Guo RF, et al. Complement-induced impairment of innate immunity during sepsis. J Immunol. 2002;169:3223–3231. doi: 10.4049/jimmunol.169.6.3223. [DOI] [PubMed] [Google Scholar]
- Flierl MA, Rittirsch D, Nadeau BA, Day DE, Zetoune FS, Sarma JV, et al. Functions of the complement components C3 and C5 during sepsis. FASEB J. 2008;22:3483–3490. doi: 10.1096/fj.08-110595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klussmann S, Nolte A, Bald R, Erdmann VA, Fürste JP. Mirror-image RNA that binds D-adenosine. Nat Biotechnol. 1996;14:1112–1115. doi: 10.1038/nbt0996-1112. [DOI] [PubMed] [Google Scholar]
- Denekas T, Tröltzsch M, Vater A, Klussmann S, Messlinger K. Inhibition of stimulated meningeal blood flow by a calcitonin gene-related peptide binding mirror-image RNA oligonucleotide. Br J Pharmacol. 2006;148:536–543. doi: 10.1038/sj.bjp.0706742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maasch C, Vater A, Buchner K, Purschke WG, Eulberg D, Vonhoff S, et al. Polyetheylenimine-polyplexes of Spiegelmer NOX-A50 directed against intracellular high mobility group protein A1 (HMGA1) reduce tumor growth in vivo. J Biol Chem. 2010;285:40012–40018. doi: 10.1074/jbc.M110.178533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purschke WG, Eulberg D, Buchner K, Vonhoff S, Klussmann S. An L-RNA-based aquaretic agent that inhibits vasopressin in vivo. Proc Natl Acad Sci USA. 2006;103:5173–5178. doi: 10.1073/pnas.0509663103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2012;61:416–426. doi: 10.1136/gutjnl-2011-300304. [DOI] [PubMed] [Google Scholar]
- Schwoebel F, van Eijk LT, Zboralski D, Sell S, Buchner K, Maasch C, et al. The effects of the anti-hepcidin Spiegelmer NOX-H94 on inflammation-induced anemia in cynomolgus monkeys. Blood. 2013;121:2311–2315. doi: 10.1182/blood-2012-09-456756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vater A, Sell S, Kaczmarek P, Maasch C, Buchner K, Pruszynska-Oszmalek E, et al. A Mixed Mirror-image DNA/RNA Aptamer Inhibits Glucagon and Acutely Improves Glucose Tolerance in Models of Type 1 and Type 2 Diabetes. J Biol Chem. 2013;288:21136–21147. doi: 10.1074/jbc.M112.444414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vater A, Sahlmann J, Kroger N, Zollner S, Lioznov M, Maasch C, et al. Hematopoietic stem and progenitor cell mobilization in mouse and man by a first-in-class mirror-image oligonucleotide inhibitor of CXCL12. Clin Pharmacol Ther. 2013;94:150–157. doi: 10.1038/clpt.2013.58. [DOI] [PubMed] [Google Scholar]
- Vater A, Jarosch F, Purschke W, Reutter R, Maasch C, Buchner K, et al. Identification and characterisation of C5a-inhibiting biostable aptamers. Mol Immunol. 2010;47:2290. [Google Scholar]
- Biesecker G, Dihel L, Enney K, Bendele RA. Derivation of RNA aptamer inhibitors of human complement C5. Immunopharmacology. 1999;42:219–230. doi: 10.1016/s0162-3109(99)00020-x. [DOI] [PubMed] [Google Scholar]
- Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PA, Guo RF, Riedemann NC. Manipulation of the complement system for benefit in sepsis. Crit Care Res Pract. 2012;2012:427607. doi: 10.1155/2012/427607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain SA, Monk PN. The orphan receptor C5L2 has high affinity binding sites for complement fragments C5a and C5a des-Arg(74). J Biol Chem. 2002;277:7165–7169. doi: 10.1074/jbc.C100714200. [DOI] [PubMed] [Google Scholar]
- Gerard NP, Gerard C. The chemotactic receptor for human C5a anaphylatoxin. Nature. 1991;349:614–617. doi: 10.1038/349614a0. [DOI] [PubMed] [Google Scholar]
- Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green LS, Jellinek D, Bell C, Beebe LA, Feistner BD, Gill SC, et al. Nuclease-resistant nucleic acid ligands to vascular permeability factor/vascular endothelial growth factor. Chem Biol. 1995;2:683–695. doi: 10.1016/1074-5521(95)90032-2. [DOI] [PubMed] [Google Scholar]
- Leendertse M, Willems RJ, Giebelen IA, Roelofs JJ, van Rooijen N, Bonten MJ, et al. Peritoneal macrophages are important for the early containment of Enterococcus faecium peritonitis in mice. Innate Immun. 2009;15:3–12. doi: 10.1177/1753425908100238. [DOI] [PubMed] [Google Scholar]
- Skokowa J, Ali SR, Felda O, Kumar V, Konrad S, Shushakova N, et al. Macrophages induce the inflammatory response in the pulmonary Arthus reaction through G alpha i2 activation that controls C5aR and Fc receptor cooperation. J Immunol. 2005;174:3041–3050. doi: 10.4049/jimmunol.174.5.3041. [DOI] [PubMed] [Google Scholar]
- Laudes IJ, Chu JC, Sikranth S, Huber-Lang M, Guo RF, Riedemann N, et al. Anti-c5a ameliorates coagulation/fibrinolytic protein changes in a rat model of sepsis. Am J Pathol. 2002;160:1867–1875. doi: 10.1016/S0002-9440(10)61133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambas K, Markiewski MM, Pneumatikos IA, Rafail SS, Theodorou V, Konstantonis D, et al. C5a and TNF-alpha up-regulate the expression of tissue factor in intra-alveolar neutrophils of patients with the acute respiratory distress syndrome. J Immunol. 2008;180:7368–7375. doi: 10.4049/jimmunol.180.11.7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumpers P, Gueler F, David S, Slyke PV, Dumont DJ, Park JK, et al. The synthetic tie2 agonist peptide vasculotide protects against vascular leakage and reduces mortality in murine abdominal sepsis. Crit Care. 2011;15:R261. doi: 10.1186/cc10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Maasch C, Vater A, Klussmann S, Morser J, Leung LL, et al. Targeting complement component 5a promotes vascular integrity and limits airway remodeling. Proc Natl Acad Sci USA. 2013;110:6061–6066. doi: 10.1073/pnas.1217991110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff TM, Nandakumar KS, Tedesco F. Inhibiting the C5-C5a receptor axis. Mol Immunol. 2011;48:1631–1642. doi: 10.1016/j.molimm.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Klos A, Wende E, Wareham KJ, Monk PN. International Union of Pharmacology. LXXXVII. Complement peptide C5a, C4a, and C3a receptors. Pharmacol Rev. 2013;65:500–543. doi: 10.1124/pr.111.005223. [DOI] [PubMed] [Google Scholar]
- Sprong T, Brandtzaeg P, Fung M, Pharo AM, Høiby EA, Michaelsen TE, et al. Inhibition of C5a-induced inflammation with preserved C5b-9-mediated bactericidal activity in a human whole blood model of meningococcal sepsis. Blood. 2003;102:3702–3710. doi: 10.1182/blood-2003-03-0703. [DOI] [PubMed] [Google Scholar]
- Hepburn NJ, Williams AS, Nunn MA, Chamberlain-Banoub JC, Hamer J, Morgan BP, et al. In vivo characterization and therapeutic efficacy of a C5-specific inhibitor from the soft tick Ornithodoros moubata. J Biol Chem. 2007;282:8292–8299. doi: 10.1074/jbc.M609858200. [DOI] [PubMed] [Google Scholar]
- Strachan AJ, Shiels IA, Reid RC, Fairlie DP, Taylor SM. Inhibition of immune-complex mediated dermal inflammation in rats following either oral or topical administration of a small molecule C5a receptor antagonist. Br J Pharmacol. 2001;134:1778–1786. doi: 10.1038/sj.bjp.0704417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisgard R, Kuhnast B, Vonhoff S, Younes C, Hinnen F, Verbavatz JM, et al. In vivo biodistribution and pharmacokinetics of 18F-labelled Spiegelmers: a new class of oligonucleotidic radiopharmaceuticals. Eur J Nucl Med Mol Imaging. 2005;32:470–477. doi: 10.1007/s00259-004-1669-8. [DOI] [PubMed] [Google Scholar]
- Evans MJ, Rollins SA, Wolff DW, Rother RP, Norin AJ, Therrien DM, et al. In vitro and in vivo inhibition of complement activity by a single-chain Fv fragment recognizing human C5. Mol Immunol. 1995;32:1183–1195. doi: 10.1016/0161-5890(95)00099-2. [DOI] [PubMed] [Google Scholar]
- Qu H, Ricklin D, Bai H, Chen H, Reis ES, Maciejewski M, et al. New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology. 2013;218:496–505. doi: 10.1016/j.imbio.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann S, Hoos J, Klussmann S, Vonhoff S. RNA aptamers and spiegelmers: synthesis, purification, and post-synthetic PEG conjugation. Curr Protoc Nucleic Acid Chem. 2011;Chapter 4:Unit 4.46.1–Unit 4.4630. doi: 10.1002/0471142700.nc0446s46. [DOI] [PubMed] [Google Scholar]
- Gruber AR, Lorenz R, Bernhart SH, Neuböck R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res. 2008;36 Web Server issue:W70–W74. doi: 10.1093/nar/gkn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.