

Abstract
Objective
Mice genetically deficient in endothelial nitric oxide synthase (eNOS−/−) are hypertensive with lower circulating nitrite levels, indicating the importance of constitutively produced nitric oxide (NO•) to blood pressure regulation and vascular homeostasis. While the current paradigm holds that this bioactivity derives specifically from expression of eNOS in endothelium, circulating blood cells also express eNOS protein. A functional red cell eNOS that modulates vascular NO• signaling has been proposed.
Approach and Results
To test the hypothesis that blood cells contribute to mammalian blood pressure regulation via eNOS-dependent NO• generation, we cross-transplanted WT and eNOS−/− mice, producing chimeras competent or deficient for eNOS expression in circulating blood cells. Surprisingly, we observed a significant contribution of both endothelial and circulating blood cell eNOS to blood pressure and systemic nitrite levels, the latter being a major component of the circulating NO• reservoir. These effects were abolished by the NOS inhibitor L-NAME and repristinated by the NOS substrate L-Arginine, and were independent of platelet or leukocyte depletion. Mouse erythrocytes were also found to carry an eNOS protein and convert 14C-Arginine into 14C-Citrulline in a NOS-dependent fashion.
Conclusions
These are the first studies to definitively establish a role for a blood borne eNOS, using cross transplant chimera models, that contributes to the regulation of blood pressure and nitrite homeostasis. This work provides evidence suggesting that erythrocyte eNOS may mediate this effect.
Introduction
Hypertension is a complex multifactorial condition associated with cardiovascular disease. Experimental data in mouse models and in human subjects point to a correlation between the production of nitric oxide (NO•) and its oxidative metabolites and hemodynamic parameters, such as nitrite and blood pressure, respectively.1–7
NO• is produced by a nitric oxide synthase (NOS) catalysing the conversion of L-Arginine to equimolar amounts of NO• and citrulline in the presence of oxygen and the cofactors calcium, calmodulin, NADPH and tetrahydrobiopterin.8 There are three NOS isoforms; endothelial NOS (eNOS), inducible NOS (iNOS) and neuronal NOS (nNOS); expressed in multiple cell types such as endothelium, epithelium, leukocytes, platelets, and neurons.8 Whereas iNOS participates in host defence, inflammatory stress and airway epithelial nitric oxide (NO•) formation, the constitutively expressed isoforms, nNOS and eNOS, are important to physiological processes that include neuronal signalling, inhibition of the hemostatic system, vasodilation and blood pressure (BP) control.
In the cardiovascular system, eNOS contributes to the regulation of blood flow and blood pressure, and is an inhibitor of platelet activation and aggregation as well as leukocyte adhesion and migration. Furthermore, endothelial eNOS appears to contribute to the formation of bioactive circulating NO• metabolites, such as the nitrite anion and S-nitrosothiols, that mediate important endocrine activities such as hypoxic vasodilation,9, 10 blood pressure regulation,6 and cytoprotection following myocardial infarction.11, 12 Mice genetically deficient in endothelial nitric oxide synthase (eNOS−/−) are hypertensive with lower circulating nitrite levels, indicating the importance of constitutively produced nitric oxide (NO•) to blood pressure regulation and vascular homeostasis.1–4, 13
Conventional wisdom holds that the pleiotropic effects of eNOS are primarily determined by enzyme expressed in the endothelium. In addition to endothelial cells, most circulating blood cells – including leukocytes,14–18 platelets,14, 19, 20 and red blood cells21, 22 – also carries eNOS transcript and/or protein. In RBC an active red cell eNOS modulates intrinsic erythrocyte deformability, platelet activation and extra-erythrocytic NO• metabolites, such as nitrite.21–24 A physiological in vivo effect for circulating eNOS on blood pressure regulation, or the formation of the circulating NO• metabolite reservoir, has never been evaluated.
The aim of this work was to test in vivo the hypothesis that a functional circulating cell eNOS regulates systemic blood pressure and the formation of the NO• metabolite pool. To limit eNOS functionality to circulating blood cells, we utilized cross-transplantation methodologies with wild type (WT) and eNOS deficient (eNOS−/−) mice. The resulting chimeric mice were characterized for circulating nitrite levels and blood pressures (while anesthetized and awake), as well as eNOS expression and activity by western blotting, real time RT-PCR, flow cytometry, enzymatic NOS activity, chemiluminescence detection and functional wire myography.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
eNOS−/− mice are hypertensive with low circulating nitrite levels
To investigate the hypothesis that circulating blood cell eNOS contributes to the control of blood pressure, systemic blood pressure – systolic and diastolic – was assessed and compared (to 2 decimal places accuracy) in awake mice by radiotelemetry as well as in anesthetized mice via carotid artery cannulation. Mice with global knock out of functional eNOS (eNOS−/−) are hypertensive when compared with normal WT mice (mmHg MAP: 126 ± 3.18 versus 105.4 ± 2.66, p = 0.0016), confirming an important physiological role for eNOS in basal blood pressure regulation (Fig. 1A), as previously reported for this13 and all other eNOS−/− strains.2, 4 They also show lower whole blood (0.72 ± 0.08 μM) (Fig. 1B) and plasma (0.38 ± 0.04 μM) (Fig. 1C) nitrite levels than WT mice (whole blood: 1.08 ± 0.11 μM, and plasma: 0.50 ± 0.54 μM).1 These values indicate the importance of the eNOS enzyme in regulating blood pressure and nitrite homeostasis.
Fig. 1.
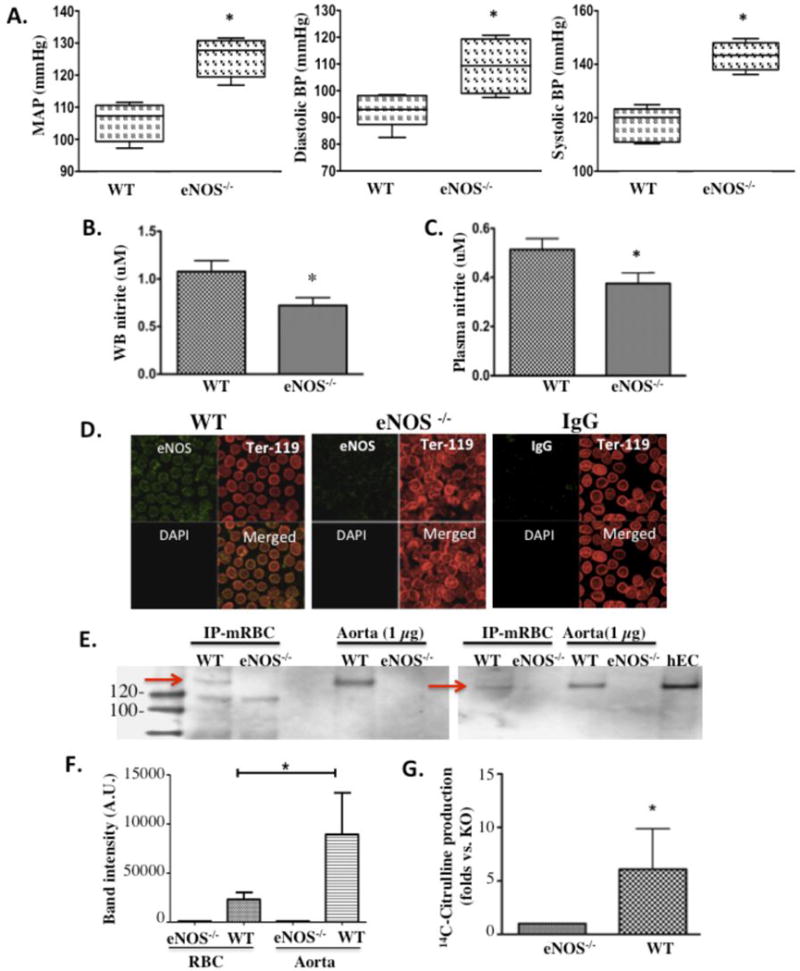
Mouse red blood cells (RBC) contain an active eNOS. In wild type (WT) versus eNOS−/− mice: (A) mean arterial pressure (MAP), systolic BP and diastolic BP (WT: n=5, KO: n=4), (B) whole blood (WB) nitrite (n=5 per group), and (C) plasma nitrite (n=5 per group). (D) Laser scanning microscopic images representative of n=5 independent experiments showing positive staining for eNOS protein (green) in Ter-119pos (red) RBCs freshly isolated from WT (left panel) and Harvard eNOS−/− (center panel) mice. Absence of nuclei in RBCs is shown by negative DAPI (blue) staining. IgG control showing negligible background staining. (E) eNOS expression in RBCs from WT and Düsseldorf eNOS−/− mice was assessed by immunoprecipitation and western blot analysis and compared to eNOS expression in aortic tissue (data from 2 of 6 independent gels are represented). (F) Densitometric assessment of eNOS expression in mouse RBCs as compared to aorta (n=3). (G) Conversion of 14C-Arginine to 14C-Citrulline as a measurement of NOS activity in RBCs from WT and Harvard eNOS−/− mice (n=4 per group). Data are expressed as mean ± SEM. * denotes p<0.05
eNOS−/− mice lack expression and activity of red cell eNOS
Red blood cells make up the largest cellular compartment in blood and are the circulating reservoir for nitrite.25 To define the presence of an active eNOS protein in red blood cells and its deletion in eNOS−/− mice, immunocytochemistry and immunoprecipitation experiments were performed. Using laser scanning confocal microscopy, eNOS staining was found to strongly localize in RBCs (Ter-119 positive cells) from WT mice (Fig. 1D, left panel) but not in RBCs from eNOS−/− mice (Fig. 1D, middle panel). Incubation with IgG was also used to verify that staining was specific for eNOS protein. In these control samples IgG staining was weakly and homogenously distributed across both cellular and noncellular areas and thus, not cell-specific (Fig. 1D, right panel). Mouse red cell eNOS was further characterized by immunoprecipitation and western blot analysis. As shown in Figure 1E, eNOS (~135 KD) is detected in RBCs from WT mice, but not KO mice, demonstrating electrophoretic characteristics similar to mouse endothelial eNOS from immunoprecipitation-enriched samples, crude aortic lysates and human endothelial cells (Fig. 1E right lane). The densitometry of bands corresponding to eNOS in RBC and aorta of three independent gels is shown in Figure 1F.
To assess eNOS activity in WT RBCs we analyzed conversion of radioactive (14C-labeled) arginine to citrulline. We found that membrane preparations of WT RBCs efficiently converted 14C-Arginine into 14C-Citrulline (0.306 +/− 0.107 fmol/min for WT versus 0.107 +/− 0.061 fmol/min for eNOS−/−), as determined by significantly higher (6-fold) reactive counts in the reaction supernatant versus their eNOS−/− counterparts (Fig. 1G).
Vascular eNOS expression is not conferred by bone marrow transplantation
WT and eNOS−/− mice were cross-transplanted to elucidate the contribution of blood cell eNOS to intravascular nitrite formation and physiologic blood pressure regulation in vivo. Chimeric animals resulting from this cross-transplantation strategy either expressed eNOS only in blood cells (BC) or in the entire rest of the animal, specifically the vascular endothelial cells (EC). These mice are hereafter respectively referred to as BC+/EC− and BC−/EC+ chimeras to simply summarize group identity. Control chimeras globally competent (BC+/EC+) and globally deficient (BC−/EC−) for eNOS (i.e. obtained by transplantation of WT bone marrow into irradiated WT mice, or eNOS−/− bone marrow into irradiated eNOS−/− mice) were created for comparison purposes using the same bone marrow (BM) transplant protocol with WT marrow transplanted into WT mice and eNOS−/− marrow transplanted into eNOS−/− mice (Fig. 2A). At 6–8 weeks post-transplantation, flow cytometric analysis of relative CD45 expression (45.1 versus 45.2, mismatched for BM donors and recipients) by peripheral leukocytes confirmed that the blood cell compartments of cross-transplanted WT and eNOS−/− BM recipients converted greater than 90% to the donor phenotype (Fig. 2B). Leukocyte and platelet counts were within normal ranges for all chimeras and did not significantly differ between groups (Table SI). Western blot analysis (Fig. 2C) demonstrated that eNOS expression was undetectable in aortas of BC+/EC− chimeras, similar to untransplanted eNOS−/− (KO) mice. (These data were confirmed by real time RT-PCR (Figs. 2D,E), indicating that transfer of WT BM into eNOS−/− recipients does not give rise to vascular eNOS expression. This finding is consistent with a recent lineage tracing study that uses GFP to show no significant contribution of transplanted eNOS WT cells to the vascular endothelium of eNOS−/− mice.26 Thus, vessel walls of the chimeras used in the current study retained their pre-transplantation phenotypes while blood took on the phenotype of the BM donor. The possibility of a role for compensatory upregulation of vascular cyclooxygenase (COX-1 or COX-2) in the transplanted chimeras was also ruled out; RT-PCR confirmed similar COX-1 (Fig. 2F) and COX-2 (Fig. 2G) mRNA expression in all 4 chimeric groups.
Fig. 2.
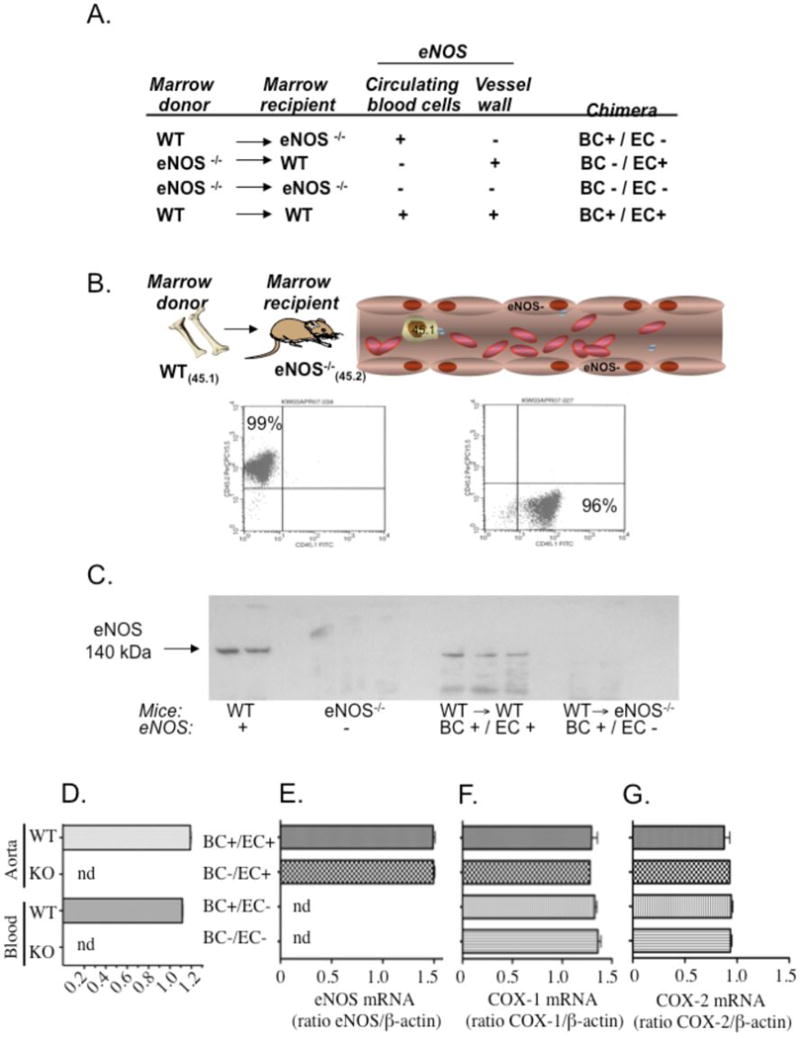
Transplantation of eNOS deficient recipients with bone marrow (BM) from wild type (WT) donors does not confer eNOS expression on vascular endothelium. (A) Table of BM transplanted chimeras used in the present study, outlining mouse BM donors/recipients and sites of eNOS expression at 6–8 weeks post-transplant. (B) Representative flow cytometry data (histograms) evidencing ≥ 90% conversion of BM recipients’ blood (peripheral leukocytes) to the donor phenotype (CD45.1 or CD45.2) prior to use in nitrite and blood pressure experiments (representative experiment of n=340+). (C) Representative western blot of aortic homogenates from WT mice (n=2, lanes 1–2), eNOS−/− mice (n=3, lanes 4–6), WT recipients of WT marrow (WT→WT; n=3, lanes 8–10), and eNOS deficient recipients of WT marrow (WT→eNOS−/−, n=2, lanes 12–13). (D) eNOS mRNA in aortas and blood (all cellular components) of C57Bl/6J (WT) compared to eNOS deficient (eNOS−/−) mice. Ratios of eNOS/β-actin mRNA are given as mean ± SEM for each group (n=2 per group). nd denotes nondetectable at 40 cycles. (E) Cross-transplantation of eNOS−/− mice with WT marrow does not give rise to eNOS mRNA expression in aortas (BC+/EC+: n=2, BC−/EC+: n=3, BC+/EC−: n=5, BC−/EC−: n=3). (F, G) Expression of (F) cyclooxygenase-1 (COX-1) and (G) cyclooxygenase-2 (COX-2) mRNA in aortas of cross-transplanted chimeras is similar between groups (BC+/EC+: n=3, BC−/EC+: n=2, BC+/EC−: n=4, BC−/EC−: n=3). BC+/EC+ (globally competent for eNOS), BC−/EC+ (deficient of blood eNOS), BC+/EC− (deficient of vascular eNOS) and BC−/EC− (globally deficient of eNOS). Ratios of β-actin mRNA are given as mean ± SEM for each group. nd denotes non-detectable eNOS mRNA at 40 cycles.
Vascular reactivity and cGMP levels are not impaired by bone marrow transplantation
To assess the effects of lethal irradiation and bone marrow transplantation on vascular function, wire myography was used to compare vascular reactivity of aortic rings from mice competent for vascular eNOS (WT, BC+/EC+, BC−/EC+). Endothelial-independent (phenylephrine, sodium nitroprusside) (Fig. 3A and 3C) and endothelial-dependent (acetylcholine) (Fig. 3B) relaxation and contraction responses were similar between all 3 groups. Moreover, cGMP levels were measured in the aortas of cross-transplanted Düsseldorf eNOS−/− chimeras (Fig. 3D) as a parameter of eNOS-dependent vascular reactivity. Both BC−/EC+ and BC+/EC+ chimeras, demonstrated cGMP levels similar to WT controls, indicating that any blood pressure differences of chimeric mice lacking blood cell eNOS (BC−/EC+), when compared to WT controls with global competency for eNOS (BC+/EC+), are not a consequence of altered vascular function from the irradiation and transplantation procedure.
Fig. 3.
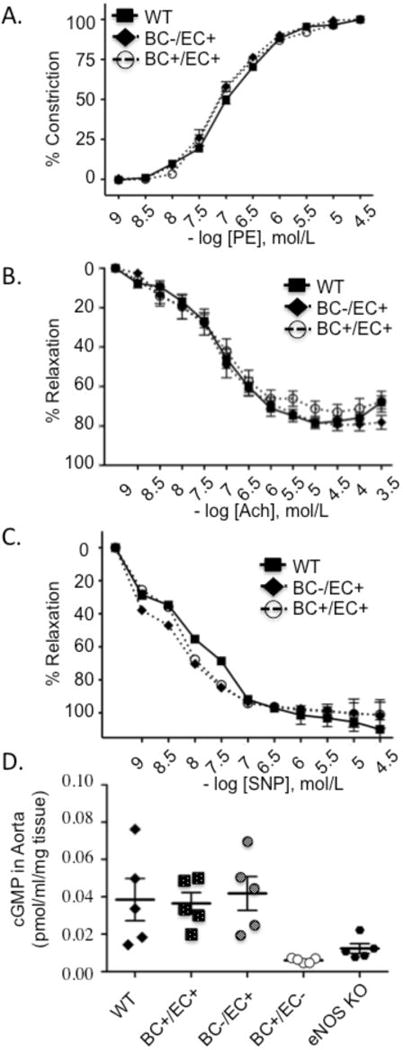
Lethal irradiation, bone marrow (BM) transplantation and blood eNOS deficiency do not impair endothelial and vascular smooth muscle cell function in aortic segments of BC−/EC+ and BC+/EC+ chimeras compared to wild type (WT) controls. (A) Contractions to cumulative concentrations of phenylephrine (PE: 10−9 to 10−4) and relaxations to cumulative concentrations of (B) acetylcholine (Ach: 10−9 to 10−3.5) and (C) sodium nitroprusside (SNP: 10−9 to 10−4.5) were examined in segments preconstricted with PE (10−6). We analyzed 2 rings/mouse (n=5 mice per group). No statistically significant differences in contraction or relaxation responses were noted between groups. (D) Cyclic guanosine monophosphate (cGMP) concentration in aortic tissue of chimeras obtained from Düsseldorf eNOS−/− mice as assessed by enzymatic immunoassay. BC+/EC+ (globally competent for eNOS), BC−/EC+ (deficient of blood eNOS), BC+/EC− (deficient of vascular eNOS), n=5 per group. Data are expressed as mean ± SEM.
Blood cell eNOS rescues eNOS−/− mice from hypertension and low nitrite levels
To gain insight into relative contributions of blood cell versus vessel wall eNOS to circulating nitric oxide derivatives, we measured whole blood (Fig. 4A) and plasma (Fig. 4B) nitrite in anesthetized, cross-transplanted chimeras. Transplantation of WT recipients with marrow from eNOS−/− donors (BC−/EC+) decreased circulating nitrite levels by roughly 1/3 compared to irradiated and transplanted WT controls (BC+/EC+) (plasma: 0.53 ± 0.10 μM versus 0.79 ± 0.07 μM, p<0.05; whole blood: 0.81 ± 0.14 versus 1.19 ± 0.15, p<0.05). Conversely, transplantation of eNOS−/− recipients with marrow from WT donors (BC+/EC−) increased circulating nitrite levels compared to irradiated and transplanted eNOS−/− controls (BC−/EC− (plasma: 0.46 ± 0.07 μM versus 0.32 ± 0.02 μM, p=0.08; whole blood: 0.68 ±0.07 versus 0.49 ± 0.02, p<0.05), indicating that both vascular and blood cell eNOS are important sources of circulating nitrite in vivo.
Fig. 4.
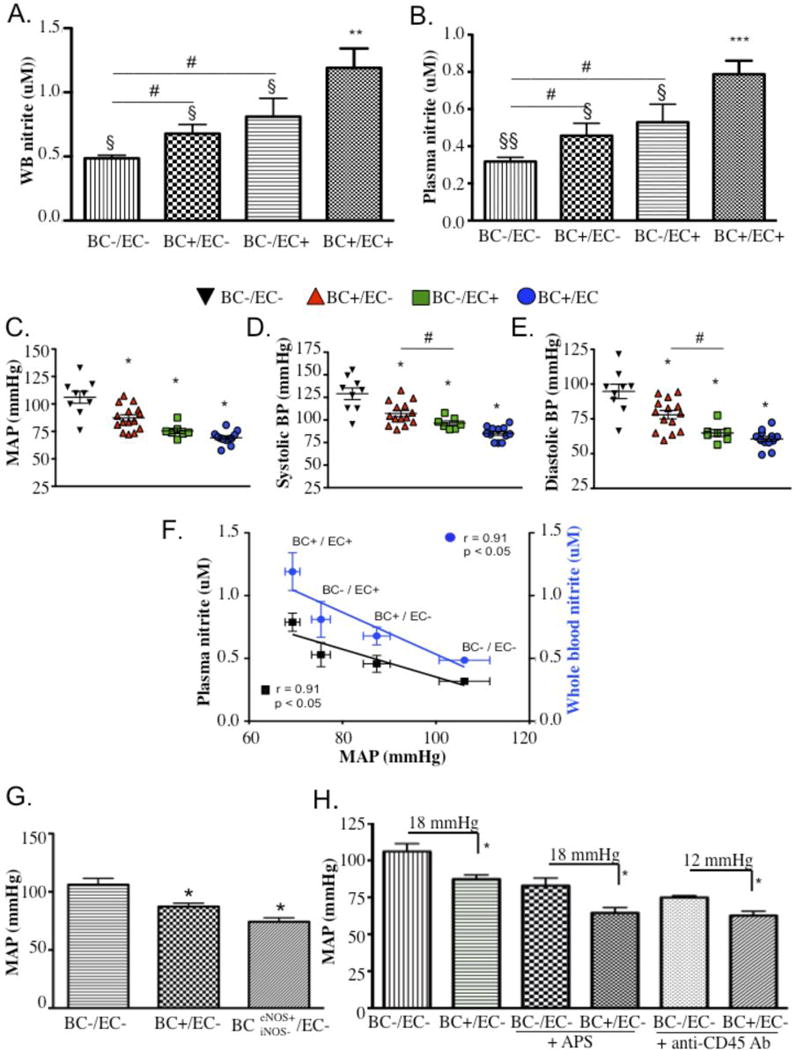
Blood cell eNOS rescues eNOS−/− mice from low circulating nitrite levels and hypertension. Nitrite concentrations in (A) whole blood (BC+/EC+: n=10, BC−/EC+: n=8, BC+/EC−: n=8, BC−/EC−: n=7) and (B) plasma (BC+/EC+: n=10, BC−/EC+: n=5, BC+/EC−: n=8, BC−/EC−: n=7) of cross-transplanted chimeras. BC+/EC+ (globally competent for eNOS), BC−/EC+ (deficient of blood eNOS), BC+/EC− (deficient of vascular eNOS) and BC−/EC− (globally deficient of eNOS). Data are expressed as mean ± SEM versus BC−/EC−: * denotes p<0.05 for plasma and whole blood; ** denotes p<0.005 for whole blood and p<0.0001 for plasma; § denotes p<0.05 and §§ denotes p<0.005 versus BC+/EC+; # denotes p<0.05 by one way t-test; (C, D, E) Blood pressures for eNOS−/− chimeras (BC−/EC+: n=8; BC+/EC+: n=12; BC+/EC−: n=14; BC−/EC−: n=9; (C) Mean arterial pressure (MAP: p=0.0142 for BC−/EC+ versus BC+/EC+, p=0.0014 for BC+/EC− versus BC−/EC−), (D) systolic blood pressure (p=0.0008 for BC−/EC+ versus BC+/EC+, p=0.0018 for BC+/EC− versus BC−/EC−) and (E) diastolic blood pressure (p=0.0001 for BC−/EC+ versus BC+/EC+, p=0.0021 for BC+/EC− versus BC−/EC−) in anesthetized cross-transplanted chimeras. ** denotes p<0.005, *** denotes p<0.0005, and **** denotes p<.0001 versus BC−/EC−. Blood pressure data analyzed by one-way Student’s t-test. (F) Linear regression of MAP averages and nitrite concentrations in plasma (left Y-axis) and whole blood (right Y-axis) across all chimeric groups. Blood pressure data expressed as mean ± SEM (SEM for BC−/EC− MAPs for whole blood and plasma are ±0.023 and 0.022 mmHg, respectively). Correlations of blood pressure and plasma nitrite are expressed as r and p value for plasma (blue type) and whole blood (black type). Blood pressure lowering effects of eNOS competent blood occurs independently of blood iNOS, platelets and leukocytes. (G) Mean arterial pressures (MAP) in anesthetized eNOS deficient chimeras in the absence of blood eNOS (BC−/EC−: n=9), the presence of blood eNOS (BC+/EC−: n=15) and the presence of eNOS, but not iNOS (BCeNOS+iNOS−/EC−, n=4) in blood. * denotes p<0.05 versus BC−/EC−; (H) MAPs in the absence or presence of platelet depletion (+APS; BC+/EC−: n=5 and BC−/EC−: n=7) or leukocyte depletion (+ anti-CD45 Ab; BC+/EC−: n=3 and BC−/EC−: n=4) in anesthetized BC+/EC− and BC−/EC− chimeras are shown. Blood pressure data are expressed as mean ± SEM.
For concomitant measurement of both circulating nitrite levels and corresponding blood pressures, mice were analyzed under anesthetized conditions and blood pressure was measured by carotid artery cannulation (Figs. 4C–E). Transplantation of WT bone marrow into eNOS−/− background (BC+/EC−) significantly decreased blood pressures relative to BC−/EC− chimeras (MAP: 87.33 ± 2.88 mmHg versus 106.1 ± 5.43 mmHg, p=0.0014; Fig. 4C). Similar results were obtained by radiotelemetry measurement of BP in awake BC+/EC− chimeras versus their BC−/EC− counterparts (Fig. 5B; diastolic: p=0.0036, systolic: p= 0.0112; MAP: p=0.0056). While the BP-lowering effect afforded by blood cell eNOS in otherwise eNOS-deficient mice was low (5 mmHg), radiotelemetry experiments demonstrated its reproducibility in two investigated eNOS−/− strains (Harvard and UNC; Figs. 5C,D). Additionally, transplantation of eNOS−/− bone marrow into WT background significantly increased blood pressures relative to BC+/EC+ chimeras under awake conditions (Fig. 5B, systolic: p=0.0171, diastolic: p<0.0001, MAP: p=0.0011), again supporting a role for blood cell eNOS in blood pressure regulation. Differences in the baseline blood pressures obtained in the anesthetized versus awake BC−/EC+ chimeras suggest an effect for the anesthesia (Fig. 4C–E and Fig. 5B). The data also demonstrate an effect for eNOS in the vascular wall, apparent in anesthetized and awake BC+/EC− chimeras versus their BC+/EC+ counterparts (MAP (mmHg) of 75.39 ± 2.0 versus 106.1 ± 5.43, p<0.05, Fig. 4C; and 36 hr MAP (AUC) of 4100 versus 3764, p=0.0003, Fig. 5A,B). In sum, our findings support a role for blood eNOS, in addition to vascular eNOS, in blood pressure regulation.
Fig. 5.
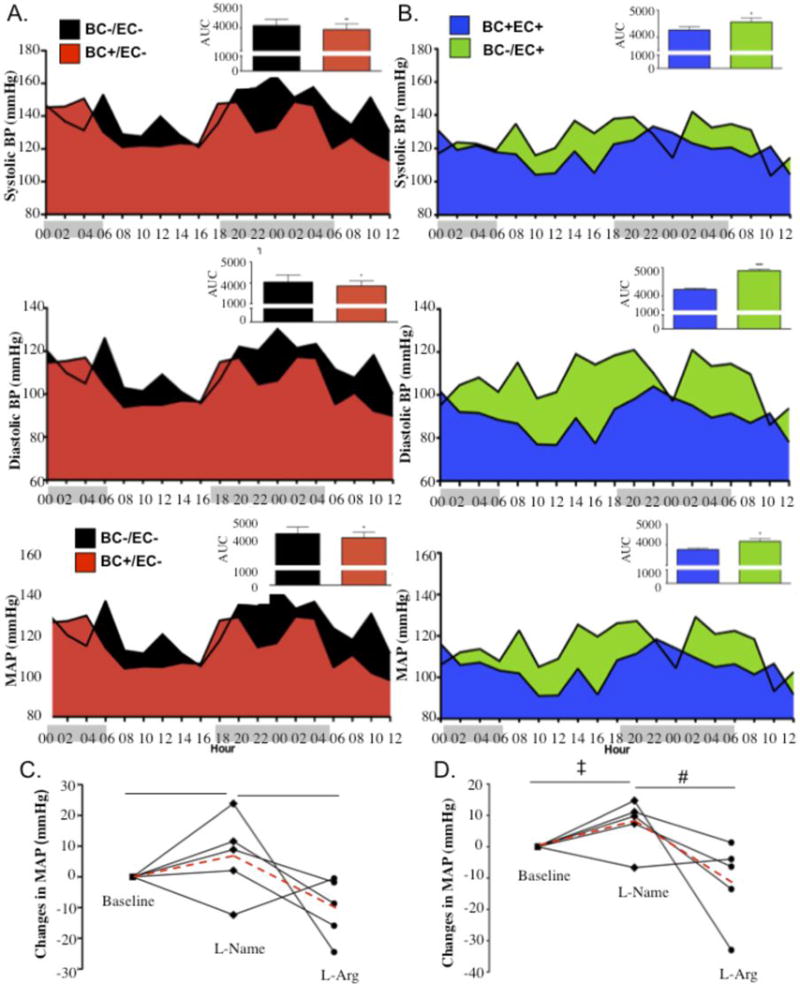
Blood pressure lowering effects of eNOS competent blood are responsive to L-NAME and L-Arginine (L-Arg) treatments. Thirty-six hour radiotelemetry-detected baseline BPs in (A) BC−/EC+ (n=3) versus BC+/EC+ (n=4) (systolic: p=0.0171, diastolic: p<0.0001, MAP: p=0.0011) and (B) BC+/EC− (n=3,) versus BC−/EC− (n=4) (systolic: p=0.0112, diastolic: p=0.0036, MAP: p=0.0056), with group averages calculated for each 2 hour interval and area under the curve (AUC) by 2-way ANOVA. Comparisons of moving averages, assessed by 2-way ANOVA, were also statistically significant (p<.0001). X-axis depicts hours of darkness (gray shade) versus hours of light (white shade) (C, D) Change in radiotelemetry-detected MAP (mmHg) in (C) Harvard BC+/EC− chimeras (n=5) and (D) UNC BC+/EC− chimeras (n=5) after oral treatment with L-NAME or L-Arginine. Blood pressure data for individual animals shown as solid lines; group averages shown as dashed red line. ‡ denotes p<0.05 for L-NAME versus baseline. # denotes p<0.05 for L-Arginine versus L-NAME. * denotes p<0.05 using Student’s t-test.
Given our previous observations of a contribution from both vascular and blood eNOS to intravascular nitrite, and the proposed contribution of nitrite to blood pressure regulation,6, 9, 27 we also explored the relationship between mean plasma and whole blood nitrite levels versus mean arterial pressures (MAP) and found an inverse correlation between these parameters across all chimeric groups (plasma: r = −0.91, p<0.05; whole blood: r = −0.91, p=0.05) (Fig. 4F). Together, these data support an interrelated role for blood cell eNOS in nitrite formation and blood pressure regulation. However, the cause and effect remain to be established.
Blood eNOS lowers BP independently of blood iNOS, platelets and leukocytes
Constitutive expression of eNOS in immune cells has been shown to serve overlapping physiological and pathophysiological roles, most notably in activating the proinflammatory profile (iNOS and NFkB gene expression) of monocytes/macrophages (human and rodent) in a manner consistent with nanomolar-level NO• production that is Ca2+/CAM and cGMP-dependent.28 The possibility that restoration of immune cell eNOS in an otherwise eNOS-deficient setting, such as the BC+/EC− chimera, could regulate blood pressure was addressed in eNOS−/− mice cross-transplanted with BM from iNOS−/− donors (BCeNOS+iNOS−/EC−). The resulting chimeric group, possessing blood eNOS but not blood iNOS, maintained a lower blood pressure than the BC−/EC− group, indicating that blood iNOS was not participating in physiological blood pressure control in these mice (Fig. 4G).
We further investigated the blood cell type contributing to blood pressure regulation by depleting BC+/BC− chimeras of their platelets or leukocytes. Platelet eNOS is a well documented antagonist of platelet activation and aggregation.29 Most leukocytes also express eNOS,15–18 although its function in these cells has yet to be clearly defined. To determine whether platelets are the blood cell source of eNOS involved in physiological blood pressure regulation, BC+/EC− chimeras and their hypertensive counterparts (BC−/EC−) were pharmacologically depleted of platelets (> 90%) using an anti-thrombocyte serum (Table SII). While platelet depletion lowered MAPs equally in both groups, the blood pressure lowering effect of red cell eNOS persisted between treated groups (18.45 ± 2.54 mmHg) to a similar extent as between untreated (platelet-competent) groups (18.80 ± 3.60 mmHg) (Fig. 4H)
Leukocytes were also depleted using an anti-CD45 antibody that reacts with the pan-leukocyte antigen CD45 (Ly5), causing clearance of leukocytes from the circulation. Treatment of BC+/EC− and BC−/EC− chimeras with this leuko-depleting antibody substantially decreased their circulating leukocyte counts by ≥ 75% (Table SII). Similar to thrombocytopenia, leukopenia decreased blood pressure in both treated groups but did not eradicate the significant blood pressure lowering effect of red cell eNOS (Fig. 4H).
The persistence of lower arterial blood pressures in BC+/EC− chimeric mice, relative to eNOS−/− controls (BC−/EC−), after platelet or leukocyte depleting treatments indicates that platelet- and leukocyte-derived eNOS are not responsible for the blood pressure-modulating effect of circulating blood eNOS, and supports a role for a functional red cell eNOS in blood pressure control.
Blood cell eNOS effects on BP are abolished by NOS inhibition in conscious mice
To further assess the role of blood eNOS availability and function in blood pressure regulation, conscious blood pressure responses to NOS inhibition with L-NAME or repristination with L-Arginine were measured in BC+/EC− chimeras via radiotelemetry. Baseline blood pressure (pretreatment) was allowed to stabilize for 10 days following blood pressure sensor implantation. The average blood pressure for the BC+/EC− group was significantly lower than that of its BC−/EC− counterpart (systolic: p=0.0112, diastolic: p=0.0036, MAP: p=0.0056), strongly supporting a role for blood eNOS in physiological blood pressure regulation (Fig. 5A). Mice were then treated with L-NAME in the drinking water for 4 days (days 11–14) followed by L-Arginine in the drinking water for 3 days (days 15–17). Blood pressure averages were calculated for the hours 22:00–04:00 on day 10 (baseline), day 14 (L-NAME) and day 17 (L-Arginine). Even in the absence of eNOS in the vascular wall and its exclusive presence in blood (BC+/EC−), blood pressure responses to oral L-NAME and oral L-Arginine followed a classical pattern (Fig. 5C): L-NAME consumption increased MAP compared to baseline (3.16 +/− 4.77 mmHg, NS), while L-Arginine consumption decreased MAP compared to L-NAME (−7.43 +/− 4.43 mmHg, p<0.05). These experiments, performed in chimeras generated from the Harvard eNOS−/− mouse (our usual eNOS deficient mouse line used for all eNOS−/− experiments except where noted), were repeated in a second group of BC+/EC− chimeras generated from a different, commercially available eNOS−/− mouse (UNC).4 The UNC BC+/EC− chimeras demonstrated similar significant MAP responses to L-NAME (+7.25 ± 3.68 mmHg, p<0.05) and L-Arginine (−11.09 ± 5.97 mmHg, p<0.05) (Fig. 5D). These classical blood pressure responses to the NO• synthase inhibitor and substrate, noted in separate BC+/EC− groups created from different eNOS−/− strains (Figs. 5C,D) and absent in the UNC-eNOS−/− mice (as shown previously)30 further support the existence of a blood cell eNOS that participates in physiological blood pressure regulation. BC−/EC− obtained from the Harvard strain mounted a paradoxical (decreased BP) response to L-NAME (Fig. SI), as previously shown for Harvard eNOS−/−,13 however, the differences were not statistically significant. Taken together, these results point to a role for NOS in the decreased BPs observed in the chimeras expressing eNOS only in blood. Until now, conventional wisdom has held that eNOS-mediated control of blood pressure is primarily dependent on eNOS enzyme expressed in the endothelium. These data indicate that blood cell eNOS also contributes to the regulation of blood pressure.
Discussion
The role of eNOS in vascular endothelium has been shown both in vitro and in vivo to participate in autocrine and paracrine NO• signaling and the control of basal blood pressure.8 Most circulating blood cells – including leukocytes, platelets and red blood cells (RBCs) – have been shown to contain eNOS.15–18, 21, 22, 29, 31 It has been suggested that red cell eNOS21–24 is capable of producing NO• under normoxic conditions. Data from our laboratory and others have demonstrated NOS-derived effects exerted by RBCs, including inhibition of ADP-induced platelet aggregation;22, 32 protection of isolated, perfused hearts from ischemia/reperfusion injury;33 as well as decreased RBC-released NO• metabolite (nitrite, nitrate, nitros(yl)ated species) levels in response to NOS inhibition.22, 24 Circulating angiogenic cells also contain a functional eNOS, but their number in peripheral blood is lower than 0.01% of peripheral blood mononuclear cell numbers (0.0067±0.0097 per 100).34–36 On the other hand, RBCs are the most abundant cells in blood and transport hemoglobin and contain relatively high levels of nitrite.25 Hence, it is reasonable to consider a role for RBCs as potential regulators of the circulating NO• pool and blood pressure responses. However, a role for red cell eNOS in NO• signaling remains controversial, and a specific role for blood cell eNOS in blood pressure regulation has not been investigated previously.
In the present study, cross-transplanted chimeric mice genetically competent or deficient for eNOS in circulating blood cells are used to demonstrate a previously unrecognized role for blood cell eNOS in blood pressure regulation and in vivo nitrite homeostasis. Our data support a persistent blood-derived eNOS effect on blood pressure after in vivo platelet- and leukocyte-depletion, as well as RBC catalysis of arginine-to citrulline and generation of intracellular NO• metabolites. While these studies provide strong evidence for circulating blood cells in NOS-dependent control of blood pressure and nitrite production, definitive clarification of the role of the erythrocyte in this process will require the use of erythroid cell specific knock-out approaches in future studies.
We performed numerous control experiments to test the hypothesis that red cell eNOS regulates blood pressure. We verified that endothelial function/integrity was not affected by our experimental setting, as demonstrated by an absence of change in endothelial-dependent and independent vasodilatation of aortic rings, or aortic cGMP levels relative to the appropriate control group or wild type mice. The BP-lowering effects of circulating blood cell eNOS were not due to incorporation of bone marrow cells into vascular endothelium, as demonstrated here by the absence of eNOS in western blot analyzed BC+/EC− aortas. Consistent with our observation, a recent study involving transplantation of GFPpos (eNOS competent) BM into sublethally irradiated eNOS−/− mice confirmed that BM-derived endothelial progenitor cells (EPCs) do not incorporate into vascular endothelium.26 Furthermore, we performed control experiments to rule out the contribution of other NOS isoforms to observed changes in BP. We did not detect a compensatory upregulation of other NOS isoforms or the cyclo-oxygenase 2 gene in the vasculature of the Harvard eNOS−/− mice, a finding that has been previously reported but remains controversial due to reported disparities between different strains of eNOS−/− mice.13, 30, 37–40 If other NOS isoforms are responsible for the blood pressure effects we observed in the BC+/EC− chimeras, then a general NOS inhibitor such as L-NAME should have reversed those effects. We instead noted that the BC−/EC− chimera mounted a paradoxical hypotensive response to L-NAME treatment (shown in supplemental Fig. S1), a result that has been previously described in the Harvard strain by different groups,13, 30 while the BC+/EC− chimera mounted the classical increased blood pressure response. Paul Huang and others have reported an upregulation of neuronal NOS (nNOS) in the Harvard eNOS KO mouse13, 41, 42 and have speculated on its possible role in the mutant’s hypotensive BP response to L-Name treatment.13 In the presence of a nonspecific NOS inhibitor such as L-NAME, a compensatory role for nNOS is unlikely to account for our observation. Furthermore, while Paul Kubes’ group has shown that nNOS is upregulated in brain and skeletal muscle of the Harvard eNOS KO mice, it was not sufficient to compensate for eNOS deficiency during H2O2-stimulated leukocyte infiltration in postcapillary venules.43 H2O2 has been proposed by several groups as an important endothelium derived hyperpolarizing factor (EDHF), but in a NOS-dependent manner.38,44 Additionally, nonspecific effects of L-NAME treatment have been reported: antagonism of muscarinic acetylcholine receptors45 and inhibition of cytochrome c reduction in vitro.46 Indeed, we observed normal endothelial function in wild type mice with elevated BP due to blood cell deficiency of eNOS (BC−/EC+ chimeras), an unexpected finding given the general association of elevated BP and endothelial dysfunction. A study by Suda et al.30 found that coronary vascular lesions develop in wild type mice after long term (8 week) treatment with L-NAME alone or with coadministration of L-NAME and the antihypertensive drug hydralazine, calling into question L-Name’s nonspecific and BP-independent effects on vascular function.
With regard to a specific role for the red blood cell in these observations, we did not observe an effect of leukocytes or platelets on eNOS-dependent blood pressure regulation. Reductions in blood pressure in our BC+/EC− mice were not abrogated by platelet-, leukocyte- or iNOS-depleting interventions, arguing against a significant role for any of these factors in physiological blood pressure regulation. While these interventions lowered blood pressure in all groups, the difference in blood pressure observed between the BC+/EC− and BC−/EC− chimeras remained significant and equivalent. The intervention-associated hypotensive responses could be attributable to deficiency of platelet- or neutrophil-derived thromboxane A(2),47 reactive oxygen species, and/or augmented prostacyclin levels.48 Our anti-leukocyte intervention targeted circulating lymphocytes and neutrophils alike (≥ 75% depletion). This may be important given the antihypertensive effects previously shown to be elicited in rats and mice by thymocyte-49 or neutrophil-depleting50 interventions. It is unlikely that these hypotensive effects could have masked an augmentation in blood pressure due to platelet or leukocyte eNOS deficiency, because the absolute decreases in blood pressure in the BC+/EC− chimera remained constant with and without depletion of leukocytes and platelets.
A recent study evaluating similar chimera experiments reported results that differed from ours. That study observed no blood pressure-reducing effect for GFPpos WT BM when transplanted into eNOS−/− mice.26 This discrepancy in blood pressure results is likely due to differences in study design. Whole body irradiation and BM transplantation are not without potentially confounding effects, thus the WT and eNOS−/− controls used in the current study were also irradiated and BM transplanted, whereas the controls used in the other study were not.
Taken together, the findings from this study point consistently toward a functional circulating red blood cell eNOS that is active in physiological vasorelaxation and nitrite homeostasis, and may make contributions to other as yet unidentified biological processes. If eNOS in blood can contribute to blood pressure regulation and nitrite production, then it is likely to have effects under reparative conditions, such as following myocardial ischemia or stroke. Indeed, circulating BM-derived EPCs have been shown to participate in the neovascularization of jeopardized tissue.51 A prior study by Ii and colleagues may have relevance to our observations.52 That study evaluated similar cross-transplantation experiments with BM recipient mice subjected to experimental myocardial infarction. Cardioprotection was observed in the eNOS−/− mice receiving WT BM, which at the time was ascribed to NO• released by EPCs that had incorporated into ischemic myocardium. However, in that study only a small percentage of the incorporated EPCs and the cardiomyocytes in ischemic myocardium expressed eNOS, suggesting that the cardioprotection may have derived from circulating blood cell-derived eNOS activity or nitrite. As a final component of these NO•-related regenerative pathways, numerous groups have demonstrated that circulating nitrite can be reduced in the blood to form NO•, regulate hypoxic vasodilation, cytoprotection following ischemia/reperfusion events, and vascular angiogenesis.9–11, 53–55 It is likely that both direct NO• formation and signaling, as well as indirect NO• oxidation to nitrite, contribute to the therapeutic effects of red cell eNOS in angiogenesis, cytoprotection and blood pressure control.
In summary, the present study provides strong evidence that a circulating blood eNOS participates in nitrite homeostasis and blood pressure regulation under physiological conditions (Fig. 6). These findings provide novel insight into mechanisms of blood pressure control that challenge our conventional perspectives on nitric oxide and nitrite signaling in blood, suggesting a more holistic regulation of vascular function, with both the circulating red blood cells and the endothelium contributing to vascular homeostasis. The existence of a functional red blood cell eNOS opens the door to studies addressing the function and dysfunction of blood eNOS in health and disease, such as a role in red blood cell enzymopathies, hemoglobinopathies and membranopathies, and in infectious diseases such as malaria.
Fig. 6.
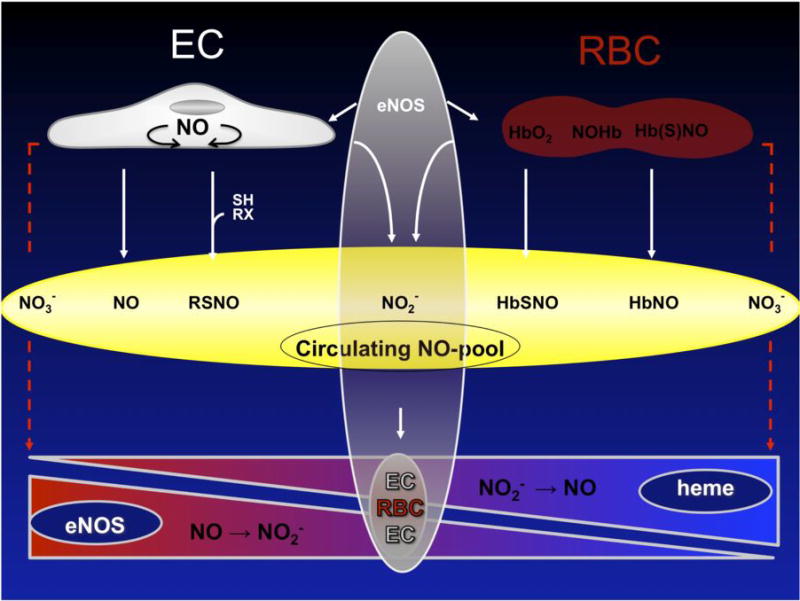
Contribution of blood cell eNOS to regulation of the circulating nitric oxide (NO•) pool and blood pressure under normoxic and hypoxic conditions. The circulating NO• pool consists of various NO• related species that transport bioactive NO• in the mammalian circulation. These oxygenated and nitros(yl)ated NO• species are interconvertible. NO• bioactivity within this pool is regulated by the surrounding redox conditions at the site of delivery. Under hypoxic conditions NO• is formed in blood by the nitrite reductase activity of heme-containing proteins, including deoxyhemoglobin. Under normoxic conditions NO• can be produced by eNOS in endothelial cells (EC) and scavenged by oxyhemoglobin in red blood cells (RBC). An eNOS-derived NO• production in blood cells may offset oxyhemoglobin-dependent NO• scavenging, thereby safeguarding vasodilatory activity in adjacent normoxic vessels. Furthermore, eNOS activity in RBCs might contribute to their intracellular storage pool of “NO• equivalents”, such as nitrite. Thus, a fruitful cycle of continuous NO• formation in blood might arise from blood cell eNOS under normoxic conditions and deoxyhemoglobin-mediated reduction of nitrite under hypoxic conditions. RSH, thiols; RX, other reactive groups; RSNO, nitrosated thiols; HbO2, oxyhemoglobin; NO-Hb, nitrosylated hemoglobin; HbSNO, s-nitrosated hemoglobin; EC, endothelial cells; RBC, red blood cells.
Clinical Perspective
Hypertension is a complex multifactorial condition associated with cardiovascular disease. Accumulating evidence points to a correlation between blood pressure and circulating NO• metabolites, like nitrite. The data presented here demonstrate a role for circulating blood cell eNOS in nitrite homeostasis and blood pressure regulation under physiological conditions. A fruitful cycle of continuous NO• formation in blood might arise from blood cell eNOS under normoxic conditions and deoxyhemoglobin-mediated reduction of nitrite under hypoxic conditions. This cycle of NO• formation within blood and the vasculature may be impaired in cardiovascular disease, where a generalized eNOS dysfunction is often marked by eNOS uncoupling or protein deficiency. Indeed, recent years have seen anemia and RBC dysfunction identified as independent risk factors for cardiovascular disease. There is a growing recognition that intravascular hemolysis represents a fundamental mechanism for human disease through the release of cell-free plasma hemoglobin that inhibits NO• signaling in the subendothelial layer. It is common to hemoglobinopathies and hemolytic anemia, as well as the “red blood cell storage lesion” of aged blood used in transfusion therapy, and may be driven by red cell eNOS-linked enzymopathies and membranopathies.56–62 Thus, our findings may illuminate novel avenues for assaying the general status of eNOS activity and cardiovascular health in the human body.
Supplementary Material
Significance.
These are the first studies, using cross-transplant chimera models, to definitively identify a mouse circulating blood eNOS that regulates intravascular nitrite homeostasis, cGMP signaling, and the control of systemic blood pressure under physiological conditions. Our findings extend the current paradigm for endothelial generation of NO• and nitrite in the regulation of blood pressure, suggesting a contribution from circulating red blood cells to NO• and nitrite synthesis and vascular homeostasis. Hypertension is a complex multifactorial condition associated with cardiovascular disease. Accumulating evidence points to a correlation between blood pressure and circulating NO• metabolites, like nitrite. The existence of a functional red blood cell eNOS invites studies addressing the function and dysfunction of blood eNOS in cardiovascular health and disease. Our findings may illuminate novel avenues for assaying the general status of eNOS activity and cardiovascular health in the human circulation.
Acknowledgments
We thank Paul Huang and Axel Gödecke for providing the Harvard and Düsseldorf eNOS−/− mice, respectively. We thank the staff of the Murine Phenotyping Core and the Laboratory of Animal Medicine and Surgery facility for assistance with the radiotelemetry-monitored blood pressure experiments, and Sivatharsini Sivarajah for the western blot analysis of mouse RBC, and Simone Zander for providing the Chimeras from Düsseldorf eNOS−/− mice. We acknowledge the professional skills and advice of Dr. Christian Heiß, Dr. Christian A. Combs and Dr. Daniela Malide (Light Microscopy Core Facility, NHLBI, NIH) and Prof. Dr. Dieter Häussinger for use of the FACS Canto II flow cytometer.
Sources of Funding: This work was supported in part by NIH R01HL098032, RO1HL096973, RC1DK085852, the Institute for Transfusion Medicine, Hemophilia Center of Western Pennsylvania (MTG), the Division of Intramural Research of the National Heart, Lung and Blood Institute, NIH (GJK), the Deutsche Forschungsgemeinschaft (DFG 405/5-1 and FOR809TP7 Me1821/3-1 to MK) and the Forschungskommission of the Medical Faculty of the Heinrich-Heine-University of Düsseldorf (to MCK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: Dr. Gladwin is a co-inventor on an NIH government patent application on the use of nitrite salts for cardiovascular diseases.
References
- 1.Kleinbongard P, Dejam A, Lauer T, Rassaf T, Schindler A, Picker O, Scheeren T, Godecke A, Schrader J, Schulz R, Heusch G, Schaub GA, Bryan NS, Feelisch M, Kelm M. Plasma nitrite reflects constitutive nitric oxide synthase activity in mammals. Free Radic Biol Med. 2003;35:790–796. doi: 10.1016/s0891-5849(03)00406-4. [DOI] [PubMed] [Google Scholar]
- 2.Godecke A, Decking UK, Ding Z, Hirchenhain J, Bidmon HJ, Godecke S, Schrader J. Coronary hemodynamics in endothelial no synthase knockout mice. Circ Res. 1998;82:186–194. doi: 10.1161/01.res.82.2.186. [DOI] [PubMed] [Google Scholar]
- 3.Gregg AR, Schauer A, Shi O, Liu Z, Lee CG, O’Brien WE. Limb reduction defects in endothelial nitric oxide synthase-deficient mice. Am J Physiol. 1998;275:H2319–2324. doi: 10.1152/ajpheart.1998.275.6.H2319. [DOI] [PubMed] [Google Scholar]
- 4.Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sobko T, Marcus C, Govoni M, Kamiya S. Dietary nitrate in japanese traditional foods lowers diastolic blood pressure in healthy volunteers. Nitric Oxide. 2010;22:136–140. doi: 10.1016/j.niox.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 6.Webb AJ, Patel N, Loukogeorgakis S, Okorie M, Aboud Z, Misra S, Rashid R, Miall P, Deanfield J, Benjamin N, MacAllister R, Hobbs AJ, Ahluwalia A. Acute blood pressure lowering, vasoprotective, and antiplatelet properties of dietary nitrate via bioconversion to nitrite. Hypertension. 2008;51:784–790. doi: 10.1161/HYPERTENSIONAHA.107.103523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kapil V, Milsom AB, Okorie M, Maleki-Toyserkani S, Akram F, Rehman F, Arghandawi S, Pearl V, Benjamin N, Loukogeorgakis S, Macallister R, Hobbs AJ, Webb AJ, Ahluwalia A. Inorganic nitrate supplementation lowers blood pressure in humans: Role for nitrite-derived no. Hypertension. 2010;56:274–281. doi: 10.1161/HYPERTENSIONAHA.110.153536. [DOI] [PubMed] [Google Scholar]
- 8.Balligand JL, Feron O, Dessy C. Enos activation by physical forces: From short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol Rev. 2009;89:481–534. doi: 10.1152/physrev.00042.2007. [DOI] [PubMed] [Google Scholar]
- 9.Cosby K, Partovi KS, Crawford JH, et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 10.Maher A, M AB, Gunaruwan P, Abozgula K, Ahmed I, Weaver R, Thomas P, Ashrafian H, B GV, James P, Frenneaux M. Hypoxic modulation of exogenous nitrite-induced vasodilation in humans. Circulation. 2008;117:670–677. doi: 10.1161/CIRCULATIONAHA.107.719591. [DOI] [PubMed] [Google Scholar]
- 11.Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langsotn W, Patel RP, Yet SF, Wang X, Kevil CG, Gladwin MT, Lefer DJ. Cytoprotective effects of nitirite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest. 2005;115:1232–1240. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez FM, Shiva S, Vincent PS, Ringwood LA, Hsu LY, Hon YY, Aletras AH, Cannon RO, 3rd, Gladwin MT, Arai AE. Nitrite anion provides potent cytoprotective and antiapoptotic effects as adjunctive therapy to reperfusion for acute myocardial infarction. Circulation. 2008;117:2986–2994. doi: 10.1161/CIRCULATIONAHA.107.748814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 14.Chen LY, Mehta JL. Variable effects of l-arginine analogs on l-arginine-nitric oxide pathway in human neutrophils and platelets may relate to different nitric oxide synthase isoforms. J Pharmacol Exp Ther. 1996;276:253–257. [PubMed] [Google Scholar]
- 15.Saluja R, Jyoti A, Chatterjee M, Habib S, Verma A, Mitra K, Barthwal MK, Bajpai VK, Dikshit M. Molecular and biochemical characterization of nitric oxide synthase isoforms and their intracellular distribution in human peripheral blood mononuclear cells. Biochim Biophys Acta. 2011;1813:1700–1707. doi: 10.1016/j.bbamcr.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 16.Muhl H, Pfeilschifter J. Endothelial nitric oxide synthase: A determinant of tnfalpha production by human monocytes/macrophages. Biochem Biophys Res Commun. 2003;310:677–680. doi: 10.1016/j.bbrc.2003.09.039. [DOI] [PubMed] [Google Scholar]
- 17.Aubry JP, Dugas N, Lecoanet-Henchoz S, Ouaaz F, Zhao H, Delfraissy JF, Graber P, Kolb JP, Dugas B, Bonnefoy JY. The 25-kda soluble cd23 activates type iii constitutive nitric oxide-synthase activity via cd11b and cd11c expressed by human monocytes. J Immunol. 1997;159:614–622. [PubMed] [Google Scholar]
- 18.de F, Sanchez de Miguel L, Farre J, Gomez J, Romero J, Marcos-Alberca P, Nunez A, Rico L, Lopez-Farre A. Expression of an endothelial-type nitric oxide synthase isoform in human neutrophils: Modification by tumor necrosis factor-alpha and during acute myocardial infarction. J Am Coll Cardiol. 2001;37:800–807. doi: 10.1016/s0735-1097(00)01185-2. [DOI] [PubMed] [Google Scholar]
- 19.Sase K, Michel T. Expression of constitutive endothelial nitric oxide synthase in human blood platelets. Life Sci. 1995;57:2049–2055. doi: 10.1016/0024-3205(95)02191-k. [DOI] [PubMed] [Google Scholar]
- 20.Radomski MW, Palmer RM, Moncada S. An l-arginine/nitric oxide pathway present in human platelets regulates aggregation. Proc Natl Acad Sci U S A. 1990;87:5193–5197. doi: 10.1073/pnas.87.13.5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cortese-Krott MM, Rodriguez-Mateos A, Sansone R, Kuhnle GG, Thasian-Sivarajah S, Krenz T, Horn P, Krisp C, Wolters D, Heiss C, Kroncke KD, Hogg N, Feelisch M, Kelm M. Human red blood cells at work: Identification and visualization of erythrocytic enos activity in health and disease. Blood. 2012;120:4229–4237. doi: 10.1182/blood-2012-07-442277. [DOI] [PubMed] [Google Scholar]
- 22.Kleinbongard P, Schulz R, Rassaf T, et al. Red blood cells express a functional endothelial nitric oxide synthase. Blood. 2006;107:2943–2951. doi: 10.1182/blood-2005-10-3992. [DOI] [PubMed] [Google Scholar]
- 23.Mihov D, Vogel J, Gassmann M, Bogdanova A. Erythropoietin activates nitric oxide synthase in murine erythrocytes. Am J Physiol Cell Physiol. 2009;297:C378–388. doi: 10.1152/ajpcell.00543.2008. [DOI] [PubMed] [Google Scholar]
- 24.Ulker P, Sati L, Celik-Ozenci C, Meiselman HJ, Baskurt OK. Mechanical stimulation of nitric oxide synthesizing mechanisms in erythrocytes. Biorheology. 2009;46:121–132. doi: 10.3233/BIR-2009-0532. [DOI] [PubMed] [Google Scholar]
- 25.Dejam A, Hunter CJ, Pelletier MM, Hsu LL, Machado RF, Shiva S, Power GG, Kelm M, Gladwin MT, Schechter AN. Erythrocytes are the major intravascular storage sites of nitrite in human blood. Blood. 2005;106:734–739. doi: 10.1182/blood-2005-02-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perry TE, Song M, Despres DJ, Kim SM, San H, Yu ZX, Raghavachari N, Schnermann J, Cannon RO, 3rd, Orlic D. Bone marrow-derived cells do not repair endothelium in a mouse model of chronic endothelial cell dysfunction. Cardiovasc Res. 2009;84:317–325. doi: 10.1093/cvr/cvp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bailey SJ, Winyard P, Vanhatalo A, Blackwell JR, Dimenna FJ, Wilkerson DP, Tarr J, Benjamin N, Jones AM. Dietary nitrate supplementation reduces the o2 cost of low-intensity exercise and enhances tolerance to high-intensity exercise in humans. J Appl Physiol. 2009;107:1144–1155. doi: 10.1152/japplphysiol.00722.2009. [DOI] [PubMed] [Google Scholar]
- 28.Conelly L, Jacobs AT, Palacios-Callender M, Moncada S, Hobbs AJ. Macrophage endothelial nitric-oxide synthase autoregulates cellular activation and proinflammatory protein expression. J Biol Chem. 2003;278:26480–26487. doi: 10.1074/jbc.M302238200. [DOI] [PubMed] [Google Scholar]
- 29.Randriamboavonjy V, Fleming I. Endothelial nitric oxide synthase (enos) in platelets: How is it regulated and what is it doing there? Pharmacol Rep. 2005;57(Suppl):59–65. [PubMed] [Google Scholar]
- 30.Suda O, Tsutsui M, Morishita T, Tanimoto A, Horiuchi M, Tasaki H, Huang PL, Sasaguri Y, Yanagihara N, Nakashima Y. Long-term treatment with n(omega)-nitro-l-arginine methyl ester causes arteriosclerotic coronary lesions in endothelial nitric oxide synthase-deficient mice. Circulation. 2002;106:1729–1735. doi: 10.1161/01.cir.0000029749.16101.44. [DOI] [PubMed] [Google Scholar]
- 31.Dudzinski DM, Michel T. Life history of enos: Partners and pathways. Cardiovasc Res. 2007;75:247–260. doi: 10.1016/j.cardiores.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen LY, Mehta JL. Evidence for the presence of l-arginine-nitric oxide pathway in human red blood cells: Relevance in the effects of red blood cells on platelet function. J Cardiovasc Pharmacol. 1998;32:57–61. doi: 10.1097/00005344-199807000-00009. [DOI] [PubMed] [Google Scholar]
- 33.Yang BC, Nichols WW, Mehta JL. Cardioprotective effects of red blood cells on ischemia and reperfusion injury in isolated rat heart: Release of nitric oxide as a potential mechanism. J Cardiovasc Pharmacol Ther. 1996;1:297–306. doi: 10.1177/107424849600100405. [DOI] [PubMed] [Google Scholar]
- 34.Furstenberger G, von Moos R, Senn HJ, Boneberg EM. Real-time pcr of cd146 mrna in peripheral blood enables the relative quantification of circulating endothelial cells and is an indicator of angiogenesis. Br J Cancer. 2005;93:793–798. doi: 10.1038/sj.bjc.6602782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mariucci S, Rovati B, Bencardino K, Manzoni M, Danova M. Flow cytometric detection of circulating endothelial cells and endothelial progenitor cells in healthy subjects. Int J Lab Hematol. 32:e40–48. doi: 10.1111/j.1751-553X.2008.01105.x. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt-Lucke C, Fichtlscherer S, Aicher A, Tschope C, Schultheiss HP, Zeiher AM, Dimmeler S. Quantification of circulating endothelial progenitor cells using the modified ishage protocol. PLoS One. 2010;5:e13790. doi: 10.1371/journal.pone.0013790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kojda G, Laursen JB, Ramasamy S, Kent JD, Kurz S, Burchfield J, Shesely EG, Harrison DG. Protein expression, vascular reactivity and soluble guanylate cyclase activity in mice lacking the endothelial cell nitric oxide synthase: Contributions of nos isoforms to blood pressure and heart rate control. Cardiovasc Res. 1999;42:206–213. doi: 10.1016/s0008-6363(98)00315-0. [DOI] [PubMed] [Google Scholar]
- 38.Takaki A, Morikawa K, Tsutsui M, Murayama Y, Tekes E, Yamagishi H, Ohashi J, Yada T, Yanagihara N, Shimokawa H. Crucial role of nitric oxide synthases system in endothelium-dependent hyperpolarization in mice. J Exp Med. 2008;205:2053–2063. doi: 10.1084/jem.20080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao X, Chen YR, He G, Zhang A, Druhan LJ, Strauch AR, Zweier JL. Endothelial nitric oxide synthase (nos3) knockout decreases nos2 induction, limiting hyperoxygenation and conferring protection in the postischemic heart. Am J Physiol Heart Circ Physiol. 2007;292:H1541–1550. doi: 10.1152/ajpheart.00264.2006. [DOI] [PubMed] [Google Scholar]
- 40.Sharp BR, Jones SP, Rimmer DM, Lefer DJ. Differential response to myocardial reperfusion injury in enos-deficient mice. Am J Physiol Heart Circ Physiol. 2002;282:H2422–2426. doi: 10.1152/ajpheart.00855.2001. [DOI] [PubMed] [Google Scholar]
- 41.Meng W, Ayata C, Waeber C, Huang PL, Moskowitz MA. Neuronal nos-cgmp-dependent ach-induced relaxation in pial arterioles of endothelial nos knockout mice. Am J Physiol. 1998;274:H411–415. doi: 10.1152/ajpheart.1998.274.2.H411. [DOI] [PubMed] [Google Scholar]
- 42.Meng W, Ma J, Ayata C, Hara H, Huang PL, Fishman MC, Moskowitz MA. Ach dilates pial arterioles in endothelial and neuronal nos knockout mice by no-dependent mechanisms. Am J Physiol. 1996;271:H1145–1150. doi: 10.1152/ajpheart.1996.271.3.H1145. [DOI] [PubMed] [Google Scholar]
- 43.Sanz MJ, Hickey MJ, Johnston B, McCafferty DM, Raharjo E, Huang PL, Kubes P. Neuronal nitric oxide synthase (nos) regulates leukocyte-endothelial cell interactions in endothelial nos deficient mice. Br J Pharmacol. 2001;134:305–312. doi: 10.1038/sj.bjp.0704234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takaki A, Morikawa K, Murayama Y, Yamagishi H, Hosoya M, Ohashi J, Shimokawa H. Roles of endothelial oxidases in endothelium-derived hyperpolarizing factor responses in mice. J Cardiovasc Pharmacol. 2008;52:510–517. doi: 10.1097/FJC.0b013e318190358b. [DOI] [PubMed] [Google Scholar]
- 45.Buxton IL, Cheek DJ, Eckman D, Westfall DP, Sanders KM, Keef KD. Ng-nitro l-arginine methyl ester and other alkyl esters of arginine are muscarinic receptor antagonists. Circ Res. 1993;72:387–395. doi: 10.1161/01.res.72.2.387. [DOI] [PubMed] [Google Scholar]
- 46.Peterson DA, Peterson DC, Archer S, Weir EK. The non specificity of specific nitric oxide synthase inhibitors. Biochem Biophys Res Commun. 1992;187:797–801. doi: 10.1016/0006-291x(92)91266-s. [DOI] [PubMed] [Google Scholar]
- 47.Vanhoutte PM. Cox-1 and vascular disease. Clin Pharmacol Ther. 2009;86:212–215. doi: 10.1038/clpt.2009.108. [DOI] [PubMed] [Google Scholar]
- 48.Vanhoutte PM, Tang EH. Endothelium-dependent contractions: When a good guy turns bad! J Physiol. 2008;586:5295–5304. doi: 10.1113/jphysiol.2008.161430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khraibi AA. Association between disturbances in the immune system and hypertension. Am J Hypertens. 1991;4:635–641. doi: 10.1093/ajh/4.7.635. [DOI] [PubMed] [Google Scholar]
- 50.Morton J, Coles B, Wright K, Gallimore A, Morrow JD, Terry ES, Anning PB, Morgan BP, Dioszeghy V, Kuhn H, Chaitidis P, Hobbs AJ, Jones SA, O’Donnell VB. Circulating neutrophils maintain physiological blood pressure by suppressing bacteria and ifngamma-dependent inos expression in the vasculature of healthy mice. Blood. 2008;111:5187–5194. doi: 10.1182/blood-2007-10-117283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jujo K, Ii M, Losordo D. Endothelial progenitor cells in neovascularization of infarcted myocardium. J Mol Cell Cardiol. 2008;45:530–544. doi: 10.1016/j.yjmcc.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ii M, Nishimura H, Iwakura A, Wecker A, Eaton E, Asahara T, Losordo DW. Endothelial progenitor cells are rapidly recruited to myocardium and mediate protective effect of ischemic preconditioning via “imported” nitric oxide synthase activity. Circulation. 2005;111:1114–1120. doi: 10.1161/01.CIR.0000157144.24888.7E. [DOI] [PubMed] [Google Scholar]
- 53.Dezfulian C, Raat N, Shiva S, Gladwin MT. Role of the anion nitrite in ischemia-reperfusion cytorprotection and therapeutics. Cardiovasc Res. 2007;75:327–338. doi: 10.1016/j.cardiores.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frerart F, Lobysheva I, Gallez B, Dessy C, Feron O. Vascular caveolin deficiency supports the angiogenic effects of nitrite, a major end product of nitric oxide metabolism in tumors. Mol Cancer Res. 2009;7:1056–1063. doi: 10.1158/1541-7786.MCR-08-0388. [DOI] [PubMed] [Google Scholar]
- 55.Raat NJ, Noguchi AC, Liu VB, Raghavachari N, Liu D, Xu X, Shiva S, Munson PJ, Gladwin MT. Dietary nitrate and nitrite modulate blood and organ nitrite and the cellular ischemic stress response. Free Radic Biol Med. 2009;47:510–517. doi: 10.1016/j.freeradbiomed.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gladwin MT, Kanias T, Kim-Shapiro DB. Hemolysis and cell-free hemoglobin drive an intrinsic mechanism for human disease. J Clin Invest. 2012;122:1205–1208. doi: 10.1172/JCI62972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim-Shapiro DB, Lee J, Gladwin MT. Storage lesion: Role of red blood cell breakdown. Transfusion. 2011;51:844–851. doi: 10.1111/j.1537-2995.2011.03100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Donadee C, Raat NJ, Kanias T, Tejero J, Lee JS, Kelley EE, Zhao X, Liu C, Reynolds H, Azarov I, Frizzell S, Meyer EM, Donnenberg AD, Qu L, Triulzi D, Kim-Shapiro DB, Gladwin MT. Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion. Circulation. 2011;124:465–476. doi: 10.1161/CIRCULATIONAHA.110.008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee JS, Gladwin MT. Bad blood: The risks of red cell storage. Nat Med. 2010;16:381–382. doi: 10.1038/nm0410-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Minneci PC, Deans KJ, Zhi H, Yuen PS, Star RA, Banks SM, Schechter AN, Natanson C, Gladwin MT, Solomon SB. Hemolysis-associated endothelial dysfunction mediated by accelerated no inactivation by decompartmentalized oxyhemoglobin. J Clin Invest. 2005;115:3409–3417. doi: 10.1172/JCI25040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hod EA, Zhang N, Sokol SA, Wojczyk BS, Francis RO, Ansaldi D, Francis KP, Della-Latta P, Whittier S, Sheth S, Hendrickson JE, Zimring JC, Brittenham GM, Spitalnik SL. Transfusion of red blood cells after prolonged storage produces harmful effects that are mediated by iron and inflammation. Blood. 2010;115:4284–4292. doi: 10.1182/blood-2009-10-245001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baek JH, D’Agnillo F, Vallelian F, Pereira CP, Williams MC, Jia Y, Schaer DJ, Buehler PW. Hemoglobin-driven pathophysiology is an in vivo consequence of the red blood cell storage lesion that can be attenuated in guinea pigs by haptoglobin therapy. J Clin Invest. 2012;122:1444–1458. doi: 10.1172/JCI59770. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.