

Abstract
Site-specific modification of proteins is a major challenge in modern chemical biology due to the large number of reactive functional groups typically present in polypeptides. Because of its importance in biology and medicine, the development of methods for site-specific modification of proteins is an area of intense research. Selective protein modification procedures have been useful for oriented protein immobilization, for studies of naturally-occurring post-translational modifications, for creating antibody-drug conjugates, for the introduction of fluorophores and other small molecules on to proteins, for examining protein structure, folding, dynamics and protein-protein interactions and for the preparation of protein-polymer conjugates. One of the most important approaches for protein labeling is to incorporate bioorthogonal functionalities into proteins at specific sites via enzymatic reactions. The incorporated tags then enable reactions that are chemoselective, whose functional groups are not only inert in biological media, but also do not occur natively in proteins or other macromolecules. This review article summarizes the enzymatic strategies, which enable site-specific functionalization of proteins with a variety of different functional groups. The enzymes covered in this review include formylglycine generating enzyme, sialyltransferases, phosphopantetheinyltransferases, O-GlcNAc post-translational modification, sortagging, transglutaminase, farnesyltransferase, biotin ligase, lipoic acid ligase and N-myristoyl transferase.
Introduction
Site-specific modification of proteins is a challenging problem in modern chemical biology.1–4 With applications in fields including biology, chemistry, and medicine,5 developing methods to site-specifically modify proteins is of great importance. Site-specific protein modifications have been used to create antibody-drug conjugates,6 to study natural post-translational modifications, to introduce fluorophores and other small molecules for biophysical studies, oriented protein immobilization,7 to prepare protein-polymer conjugates,8 and to examine the properties of proteins such as their folding, their dynamics and their interactions with other proteins.9 Protein immobilization is an important first step for many biotechnology applications including the construction of protein microarrays, biosensors, immunoassays and protein conjugates which are used for medical therapies.10, 11 One of the main advantages of site-specific protein modification is that it can be used to create structurally defined covalent linkages between proteins and surfaces, materials or biomolecules.12 This ensures homogeneous coverage and accessibility to the active site of the protein. These are important factors in studying protein expression and localization, to improve bioavailability and pharmacokinetics of protein-based drugs, in structure-function studies, and in the development of biosensors.13
Early methods of protein functionalization exploited the reactivity of either cysteine or lysine residues by reacting the protein with an excess of thiol- or amine-reactive reagents, such as maleimides or N-hydroxysuccinimidyl esters, respectively. In addition to lysine and cysteine modifications, methods have also been developed to chemically modify tyrosine, the N- or C-terminus, and aspartate or glutamate residues.14 Transimination15–17 and periodate oxidation4, 18 are also among common chemical methods for protein modifications which introduce an aldehyde or a ketone moiety at the N-terminus. Although all of these methods are currently in use and have widespread utility, a variety of alternative approaches have been developed to achieve greater reaction yield, higher selectivity and site-specificity, compatibility with complex biological systems, and improved reaction rates.12
One approach for protein labeling is to use reactions that are chemoselective, whose functional groups are both inert in biological media and which do not natively occur in proteins or other biological macromolecules. In many cases, a combined strategy of the described modification methods are used, such as taking advantage of the selectivity of the reactive functional groups on proteins (e.g. lysine of the C-terminus) which is used to introduce a new reactive group that then provides superior reaction kinetics for a second, more complex conjugation reaction. Using this strategy, a large excess of the initial reagent can be used to introduce a reactive handle into a protein, followed by a second reaction using stoichiometric quantities of the biomolecule or probe of interest, which can be more costly to obtain.
The discovery and application of green fluorescent protein (GFP) and its derivatives into chemical biology has had a great impact on exploring protein behavior in living cells and has dramatically increased our knowledge of biological processes.19–21 However, the use of GFP to tag proteins comes with disadvantages; many proteins do not tolerate the fusion with GFP without compromising function or intracellular distribution.22, 23 An alternative to using GFP is to chemically tag proteins with a fluorescent or affinity label. This latter method has the advantage of being relatively simple to perform and usually only uses a small molecule when compared to the size of the protein. One of the major disadvantages is that if done in a nonselective manner, it lacks the precise control of molecular architecture that can be obtained using a genetically encoded tag.
Another alternative strategy to create site-specific protein conjugates is the incorporation of non-natural amino acids into proteins. The incorporation of non-proteogenic residues allows functionality typically not found in proteins to be introduced and exploited. Several systems for expressing proteins with non-natural residues have been described and recently (2009) reviewed.24 Auxotrophic bacterial hosts are commonly used to incorporate non-natural analogues of amino acids, and suppressor tRNA methods have also been developed which enable the incorporation of a variety of modified residues such as p-azidophenylalanine, p-ethynylphenylalanine or p-acetylphenylalanine that contain bioorthogonal functionality.25–28
The development of new methods for site-specific modification of proteins that function under mild conditions is an area of intense research.1, 3, 4, 12 Due to the structural sensitivity of proteins and complex cellular environment in which proteins inhabit, the chemical transformations of proteins need to proceed under mild conditions that are compatible with all functional groups present in the cell, which includes such diverse molecules as proteins, nucleic acids, carbohydrates, and various metabolites. The reaction should occur in water or aqueous solutions at or near physiological pH, have rapid kinetics even with low (sub-millimolar) concentrations of reactants, and occur at physiological temperatures. The search for chemical reactions that satisfy these requirements has revealed the utility of a number of key chemical reactions including the Huisgen [3 + 2] cycloaddition (commonly referred to as the “click” reaction),29–31 the Staudinger ligation,32 a photoinducible reaction of an alkene with tetrazole,33, 34 the Diels-Alder reaction,35–37 an inverse-electron-demand Diels Alder reaction,38, 39 and oxime or hydrazone ligations.40–42
To overcome many of the aforementioned challenges, chemoenzymatic methods that allow site-specific incorporation of labels into proteins have been developed and are an increasingly active area of research.2, 43, 44 Such methods capitalize on the exquisite selectivity of enzymes to accomplish highly selective reactions. Here we review the most recent enzymatic methods for site-specific protein modification including formylglycine generating enzyme, sialyl transferase, phosphopantetheinyl transferase, O-GlcNAc transferase, sortase, transglutaminase, farnesyl transferase, biotin ligase, lipoic acid ligase and N-myristoyl transferase.
Enzymes used for protein modification
Formylglycine generating enzyme (FGE)
Formylglycine generating enzyme (FGE) performs a critical post-translational modification of type I sulfatases by converting a cysteine within the motif CXPXR to the aldehyde-bearing residue formylglycine (FGly).45–48 By introducing this motif into proteins and subsequently reacting it with FGE, an aldehyde group can be site-specifically added to the protein, which can then be used for covalent modification using complimentary aminooxy- or hydrazide-functionalized reagents (Figure 1). Bertozzi and coworkers have refined this strategy by screening FGEs from M. tuberculosis and S. coelicolor against synthetic peptide substrate libraries and identified new peptide sequences that diverge from the canonical FGE-recognized motif.46 Based on their study, E. coli’s FGE activity was found to be similarly promiscuous, which enables the use of novel aldehyde tag sequences for ex vivo modification of recombinant proteins. This finding expanded the range of aldehyde-tag sequences for protein engineering.
Figure 1.

Site-specific modification of proteins containing a genetically encoded aldehyde tag incorporated using formylglycine generating enzyme (FGE). (A) FGE oxidizes a specific cysteine to formylglycine within a 5 residue consensus sequence. (B) Site-specific modification of a protein bearing the genetically encoded aldehyde tag followed by chemoselective bioorthogonal oxime ligation reaction to label the protein of interest.
In another study, they introduced the peptide sequence recognized by the endoplasmic reticulum (ER)-resident FGE into heterologous proteins expressed in mammalian cells. Applying the FGE technique, they were able to site-specifically modify monoclonal antibodies as well as membrane-associated and cytosolic proteins expressed in mammalian systems.47
Plasma membrane-associated proteins, similar to secreted proteins, traffic through the secretory pathway and thus can potentially be substrates for the ER-resident FGE. They explored this idea by introducing a 13 residue aldehyde tagging sequence onto the N-terminus of the platelet-derived growth factor receptor (PDGFR) transmembrane (TM) domain. This protein, along with human FGE, was expressed for 48 h in CHO cells. Next, the cells were reacted with biotin-hydrazide followed by AlexaFluor488-streptavidin and subjected to subsequent analysis by flow cytometry. Their results showed that CHO cells expressing aldehyde tagged PDGFR-TM manifested a clear increase in fluorescence compared with cells expressing the unmodified PDGFR-TM protein.47
In additional work, a bacterial FGE homolog derived from S. coelicolor was introduced into a mammalian expression vector. A GFP was genetically encoded with a 13 residue aldehyde tag at its N-terminus denoted as Ald13-GFP. Both proteins were expressed in human embryonic kidney (HEK)293T cells. After cell lysis and purification, the fusion protein was reacted with biotin-hydrazide and analyzed by non-reducing PAGE and Western blotting. Their results showed that the Ald13-GFP was efficiently labeled with biotin-hydrazide.47
Overall, the aldehyde tag offers a versatile method for site-specific modification of membrane-associated and cytosolic proteins in mammalian cells. It only requires a peptide sequence 6–13 residues in length that can be varied for the protein modification applications. The 6 residue tag is smaller but the 13 residue tag provides higher levels of conversion of Cys to FGly presumably due the greater accessibility to the enzyme active site provided by the longer flanking sequences. Because of its site-selective nature, the aldehyde tag is promising for the development of new protein-conjugates for research and therapeutic purposes.
Sialylation
The surfaces of many cells, including both prokaryotes and eukaryotes, are decorated with glycan chains.49 These glycans play important roles in a wide range of biological processes such as cell–cell interaction, protein recognition and small molecule–cell recognition.49 One important type of glycan modification is the addition of sialic acid to proteins (Figure 2).50 Sialic acid is a monosaccharide with a nine-carbon backbone and is a generic term for the O- or N-substituted derivatives of neuraminic acid. Sialic acid refers to a wide family of related sugar acids that occur prominently at the terminal position of many eukaryotic surface-exposed glycoconjugates, where they confer important properties upon the resulting cell surface.50, 51 One of the most abundant and best-studied sialic acid members is N-acetylneuraminic acid (Neu5Ac). Besides Neu5Ac, there are numerous naturally occurring variations of sialic acid, including substitutions at the C-5 position, or modifications of different hydroxyl groups. Sialic acids are found widely distributed in animal tissues and, to a lesser extent, in other species, ranging from plants and fungi to yeasts and bacteria, predominantly in glycoproteins and gangliosides.
Figure 2.
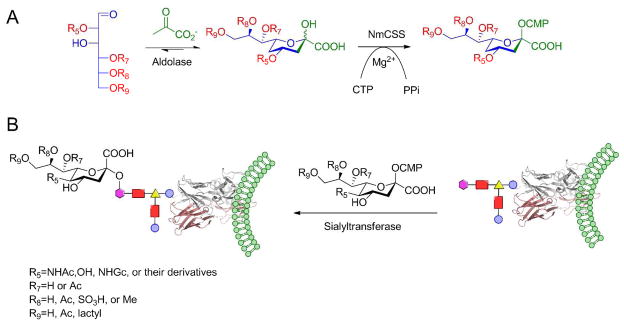
(A) Synthesis of sialosides containing sialic acid modifications via a chemoenzymatic approach. (B) Application of sialyltransferase for cell surface labeling using sialic acid derivatives. Rx (X=5–9) represent groups that can contain an azide, an alkyne or other bioorthogonal functional groups.
Sialic acid-containing structures play important roles in cellular recognition and communication.51 Accordingly, many pathogenic bacteria have evolved the molecular machinery to decorate their cell surfaces with sialic acid to mimic that of their host. This results in the ability to resist the host’s innate immune response and also allows them to interact specifically with different host-cell surfaces.52 Therefore understanding sialylation will enable a better understanding of how certain pathogens function and could be used to create more effective drugs.
Unfortunately, the study of sialylation is difficult due to the complex structure of sialic acid, which presents numerous synthetic challenges. This complexity is due to the hindered tertiary anomeric center and the lack of a neighboring participating group in sialic acids. Using enzymatic methods to create and conjugate sialic acid derivatives is a much more promising method to study sialylation. Thus, a highly active sialyltransferase with broad substrate functionality would be particularly useful in this field.
Chen and coworkers reported the discovery of a water-soluble and highly active multifunctional sialyltransferase from P. multocida.53 They used a one-pot, three-enzyme system for the efficient synthesis of a diverse set of sialoside libraries (Figure 2).54 In this system, using a recombinant E. coli K12 sialic acid aldolase and a recombinant N. meningitidis cytidine monophosphate (CMP)-Sia-synthetase, CMP-Sia derivatives were generated in situ from sialic acid precursors. They were able to perform these reactions under mild conditions at 37 °C, pH 8.5, over the course of only a few hours; additionally, they did not observe significant sialidase or α-2,6-sialyltransferase (SiaT) activity. More recently (2012), using the established one-pot three-enzyme strategy and starting from C6-modified mannose derivatives, Chen and coworkers chemoenzymatically synthesized a library of thirty α-2-3 and α-2-6-linked sialyl galactosides containing C9-modified sialic acids.55 They used these sialosides to evaluate substrate specificity of various sialidases. A high-throughput sialidase substrate specificity assay was used to elucidate the importance of the C9-OH group in sialidase recognition. Their results showed that different sialidases have various promiscuities towards the different synthetic substrates, with V. cholerae sialidase having the highest substrate promiscuity.
Overall, the newly discovered SiaT has broad substrate specificity, substantial expression levels, and high water-solubility. Thus, the new SiaT is a powerful tool for synthesizing structurally diverse sialosides and can be used to further understand their important biological functions. In a broader sense, SiaT may be useful for engineering novel glycosylated proteins for a variety of applications in biotechnology and medicine.
Phosphopantetheinyl transferase (PPTase)
Peptide carrier protein (PCP), and acyl carrier protein (ACP) are small folding domains (80-100 residues in size), which can either be embedded or stand alone as key parts of the biosynthetic machinery of nonribosomal peptide synthetases (NRPSs), fatty acid synthases (FASs) and polyketide synthases (PKSs).56–59 All PCP and ACP domains must be posttranslationally modified by PPTases in order for these enzymatically synthesized assemblies to be active. PPTases modify PCP and ACP by an installation of a phosphopantetheinyl (Ppant) prosthetic group 20 Å in length through a phosphodiester bond to the hydroxyl group of a conserved serine residue.60 The Ppant group is a fragment derived from coenzyme A (CoASH), which is the native substrate of PPTases. It functions as a swinging arm to provide successive anchoring points for the attachment of the growing peptide, fatty acyl chains, or polyketide as they elongate down the NRPS, FAS, or PKS enzymatic assembly lines.
Walsh and coworkers developed a strategy for the site-specific modification of proteins with small molecules using phosphopantetheinyltransferase, Sfp, a post-translational modification enzyme. Recently, a number of groups have exploited the utility of Sfp to transfer a wide range of molecules, conjugated to the thiol moiety of Coenzyme A (CoA), onto different carrier proteins (Figure 3). Sfp and related PPTases have also been used to label proteins, containing the PPTase recognition sequence (peptide carrier protein, PCP, or acyl carrier protein ACP), with biotin61 or fluorophores,62 the former of which has been used to non-covalently immobilize arrays of protein onto avidin-coated slides.
Figure 3.

(A) Sfp-catalyzed modification of PCP, ACP or ybbR tag at a specific Ser residue by CoA-SR conjugates. (B) Using Sfp or related PPTases for direct site-specific protein immobilization via surface immobilized CoA as a substrate and a protein of interest containing a genetically encoded carrier protein or a ybbR tag. (C) Labeling cell surface proteins containing ACP, PCP or a ybbR tag using PPTase.
In the early work reported by Walsh and coworkers, the target proteins were expressed as fusions to PCP (80-100 amino acids), which were excised from a nonribosomal peptide synthetase.61 Next, Sfp was used to catalyze the covalent attachment of the small molecule phosphopantetheine to a specific serine residue within the PCP. The labeling reaction proceeded with high specificity and efficiency, targeting the PCP-fusion proteins in the cell lysate. The PCP tag was shown to be compatible with various proteins because its fusion onto them did not compromise the functional properties of the parent protein. Additionally, Sfp was shown to be promiscuous with respect to the small molecule probes linked to CoA. This work highlighted the potential of the PCP tag for site-specific protein labeling with small molecules. Moreover, they employed a PCP tag for biotin labeling of proteins and then demonstrated that the resulting biotinylated proteins could be used to create a protein microarray for enzymatic screening. Overall, the relatively small size of the PCP domain (80-100 amino acids compared to 238 residues for GFP fusion proteins), the portability of PCP to various target proteins, the high efficiency of Sfp-catalyzed PCP post-translational modification and the promiscuity of Sfp with various small molecule probes makes this approach an attractive alternative for protein labeling.
Having access to promiscuous metabolic pathways can provide an interesting alternative for site-specific protein labeling ex vivo. Burkart and coworkers used the PPTase strategy for protein labeling by harnessing an intrinsic bacterial metabolic pathway.63, 64 They treated E. coli cells with a cell-permeable, nonhydrolyzable, fluorescent pantetheine analogue (coumarin derivative), which was used by the CoA biosynthetic pathway to synthesize CoA-analogues ex vivo. It is likely that E. coli internalizes the pantetheine analogue via a sodium-dependent symporter where it undergoes phosphorylation by CoAA. Further processing by CoAD and CoAE results in stepwise adenylation and further phosphorylation yields the final CoA analogue. This compound is then retained within the cell and is available for downstream transfer to a carrier protein domain. This work showed how cell-penetrating analogues of metabolic precursors could be used ex vivo to site-specifically label proteins. The key feature of this approach is the availability of a viable uptake mechanism. Using this strategy, CoA-bound reporter molecules containing coumarin-derivatives were successfully generated for post-translational protein modification ex vivo. This method may be applicable to natural product pathway manipulation as well as applications in conventional molecular and cellular biology. The significant tolerance to structural modification manifested by the enzymes in this pathway should allow delivery of a wide variety of chemical moieties to ex vivo processes. In very recent work, Burkart and coworkers used PPTase and acyl carrier proteins (ACP-fusion proteins) for visualization and functionalization studies and demonstrated the reversibility of this labeling approach.65 Treatment of acyl carrier proteins with Sfp and coumarin-CoA generated fluorescently labeled proteins. They then showed that treatment with ACP-hydrolase can reverse this reaction. Thus, subsequent treatment of coumarin-labeled lysate with recombinant ACP-hydrolase uniformly removed the coumarin-4′-phosphopantetheinyl (PPant) from ACP. This iterative enzymatic methodology was then used to reversibly label acyl carrier protein variants in vitro and was subsequently employed for NMR structural studies of protein-substrate interactions.
In related work, Johnsson and coworkers used the PPTase strategy to site-specifically label cell surface proteins in eukaryotes and prokaryotes with a fluorescent probe.66 They expressed a-agglutinin receptor Aga2p as a C-terminal fusion with ACP (Aga2p-ACP) in S. cerevisiae. Aga2p is known to interact with Aga1p through two disulfide bonds, which cause its localization to the cell surface.67 Incubation of yeast cells that coexpressed Aga1p and Aga2p-ACP with CoA-Cy3 (a fluorescent analogue of the natural substrate) and PPTase led to a clear fluorescent signal at the cell membrane. Aga2p-ACP was also modified with a biotinylated CoA analogue, and subsequently incubated with commercially available CdSe quantum dots conjugated to streptavidin. The resulting bright fluorescence of yeast displaying Aga2p-ACP was highly specific. The same results were observed when the experiments were performed on HEK293 cells. In that case, the ACP tag was appended to the N-terminus of a recombinant human G protein-coupled receptor, neurokinin-1, one of the most important classes of therapeutic targets.
The size of the PCP or ACP is relatively large (75-80 amino acids; compared to small peptide recognition tags summarized in table 1), thus Walsh and coworkers tried to identify smaller peptide substrates for Sfp-catalyzed reactions. By probing a genomic library from B. subtilis using phage display, they found that truncated forms of the predicted ybbR protein are substrates for Sfp phosphopantetheinyltransferase.68 Sfp-catalyzed biotin labeling of phage-displayed ybbR truncates was confirmed by ELISA. The site of the modification on JY529, the shortest ybbR truncated protein selected by phage display, was mapped by Fourier-transform MS to Ser-274 (following the numbering in the full length ybbR). JY529 showed no obvious sequence homology with ACPs or PCPs, the known substrates of Sfp. Therefore, short peptides corresponding to the flanking sequence of Ser-274 in JY529 were synthesized and evaluated as substrates for Sfp. Eventually, Walsh and coworkers identified a series of peptides, the smallest being an 11-residue molecule with the sequence DSLEFIASKLA, as efficient substrates for Sfp phosphopantetheinyl transferase using small molecule–CoA conjugates. The peptides were named “ybbR tags” because part of their sequence is derived from the ybbR ORF in the B. subtilis genome. Importantly, they also demonstrated that the ybbR tags can be fused to the N- or C-termini of target proteins or inserted into flexible loop regions in the middle of a target protein for site-specific protein labeling by Sfp, highlighting the versatility of this approach.
Table 1.
Kinetic parameters of different enzymes used for protein labeling.
| Enzyme | Encoded tag peptide | Kcat min−1 | KM μM | Kcat/KM min−1 μM−1 | Site of the peptide |
|---|---|---|---|---|---|
| Formylglycine generating enzyme |
CXPXR or 13-mer LCTPSRGSLFTGR |
- | - | - | C or N terminus |
| Phosphopantetheinyl transferase | ACP, PCP or ybbR tags (11 mer: DSLEFIASKLA; 13 mer: VLDSLEFIASKLA; 17 mer: GSQDVLDSLEFIASKLA) | (for 500 μM ybbR 13 mer and biotin-CoA) | C or N terminus or flexible loops | ||
| 14.7 | 60.8 | 0.242 | |||
| Sortase | LPXTG | 16.2 | 5,500 | 0.003 | Any section |
| Transglutaminase | XXQXX | 45 | 7 | 6 | Any section |
| Farnesyl transferase | CaaX | (for yPFTase and 2.4 μM CVIA) for FPP: | C terminus | ||
| 31.2 | 1.71 | 18.2 | |||
| for FPP-aldehyde: | |||||
| 7.8 | 1.87 | 4.17 | |||
| Biotin ligase | GLNDIFEAQKIEWHE | (for Escherichia coli biotin ligase and biotin) | C or N terminus | ||
| 9.6 | 4.2 | 2.3 | |||
| Lipoic acid ligase | GFEIDKVWYDLDA | 2.88 | - | - | C or N terminus or flexible loops |
Overall, enzymatic labeling via Sfp by various small molecule probes is highly specific and efficient. Sfp has broad substrate specificity with respect to the small molecule probes conjugated to CoA, including fluorescent substrates such as AlexaFluor dyes and fluorescein, affinity substrates such as glutathione and biotin, redox substrates such as porphyrin and also sugar derivatives of CoA. The PPTase strategy can also be used for direct site-selective covalent protein immobilization onto solid supports derivatized with CoA.69
O-GlcNAc post-translational modification
The glycosylation of nuclear and cytoplasmic proteins at serine and threonine residues by the addition of a single GlcNAc residue (O-GlcNAc) is a widespread post-translational modification (Figure 4). This modification has been found on numerous proteins such as nuclear pore proteins, enzymes and transcription factors. The enzyme catalyzing the O-GlcNAc modification of cellular proteins, UDP-Glc -NAc: polypeptidyltransferase (OGTase), is essential for cell viability in mammals and is conserved in a range of organisms from Arabidopsis thaliana to humans. Glycosylation levels are typically monitored by immunoblotting with a general O-GlcNAc antibody, which detects only a limited number of O-GlcNAc-modified proteins and makes it difficult to identify proteins undergoing changes in glycosylation. Bertozzi and coworkers described a chemical strategy directed toward identifying O-GlcNAc-modified proteins from living cells or proteins modified in vitro.70 They demonstrated, in vitro, that each enzyme in the hexosamine salvage pathway, and the enzymes that affect this dynamic modification (UDP-Glc-NAc: polypeptidyltransferase and O-GlcNAcase), tolerate structural modifications in which the N-acyl side chain of the natural substrate is functionalized with a bioorthogonal azide moiety. Cells treated with N-azidoacetylglucosamine resulted in the metabolic incorporation of the azidosugar into nuclear and cytoplasmic proteins. These O-azidoacetylglucosamine-modified proteins can then, theoretically, be covalently derivatized with various biochemical probes at the site of protein glycosylation using the Staudinger ligation reaction.71
Figure 4.
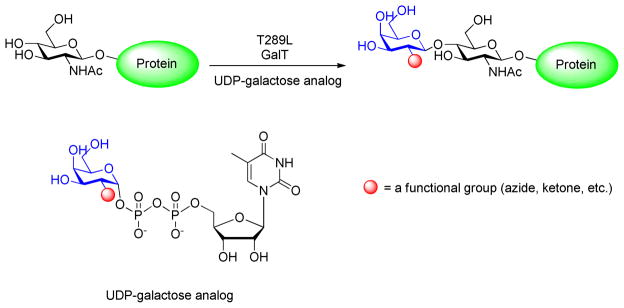
Chemoenzymatic labeling of O-GlcNAc-containg proteins using a T289L mutant -galactosyl transferase and UDP-galactose analogue. The functionalized protein can then be used for chemoselective reactions, such as oxime or hydrazone ligations or azide-alkyne [3+2] cycloaddition.
The incorporation of N-azidoacetylglucosamine (GlcNAz) onto proteins within cultured human cells was also investigated. Jurkat cells were cultured in the presence of a derivative of GlcNAz for 3 days. Nuclear protein extracts were prepared and reacted with a FLAG-containing phosphine-based reagent. Subsequently, FLAG-modified glycoproteins were identified by Western blot using a mouse anti-FLAG-mAb conjugated to HRP. As a control, cells that were not treated with the azide-modified sugar were analyzed and no signal was detected for this sample. To further validate this approach, they selected a specific protein, the nuclear pore protein p62, which is known to be post-translationally modified with O-GlcNAc, and analyzed it from the GlcNAz-treated cells. Using this strategy, protein p62 was successfully labeled with the GlcNAz.
Overall, the promising result from this approach was that they could analyze O-GlcNAc-modified proteins in the proteome. This strategy will prove useful for both the identification of O-GlcNAc-modified proteins and the elucidation of the specific residues that bear this saccharide. Additionally, this approach could be used to derivatize O-GlcNAc-modified proteins with a variety of other probe molecules.
Enzymatic methods using an engineered mutant of β-1,4-galactosyltransferase have also been used to detect O-GlcNAc post-translational modifications. Hsieh-Wilson and coworkers reported an enzymatic strategy for a rapid and sensitive detection of O-GlcNAc post-translational modifications.72–75 Their approach exploited the ability of an engineered mutant of β-1,4-galactosyltransferase to selectively transfer a ketone or an azide onto O-GlcNAc glycosylated proteins (Figure 4) using a suitably functionalized carbohydrate precursor. Next, using a hydrazone or oxime ligation or an alkyne-azide cycloaddition reaction employing the transferred ketone or azide, they could site-specifically attach a biotin or a fluorophore to these proteins.
In other work, using this same approach, Hsieh-Wilson and coworkers were able to rapidly visualize proteins that were present at the limits of detection of traditional methods.73 They examined whether O-GlcNAc-modified proteins could be chemoenzymatically tagged and then imaged in cells. They fixed HeLa cells and cultured cortical neurons, permeabilized them, and then labeled them with GlcNAc-azide using the Y289L GalT-engineered enzyme. Next, they reacted the cells with biotin-alkyne or TAMRA-alkyne, creating a covalent bond via azide-alkyne cycloaddition. The biotin-alkyne-treated cells were further incubated with a streptavidin-AlexaFluor488 conjugate. Results showed that O-GlcNAc-glycosylated proteins were found in both the nucleus and cytoplasm in both types of cells. Moreover, they observed robust staining of proteins along neuronal processes. They labeled nuclear and cytosolic protein fractions from rat forebrain with azide-functionalized O-GlcNAc and subsequently modified the material with a TAMRA-alkyne derivative. After enrichment of O-GlcNAc proteins by immunoprecipitation using an anti-TAMRA antibody, the precipitates were resolved by 1D or 2D gel electrophoresis, visualized by in-gel fluorescence imaging and then proteolytically digested and subjected to nano-LC-MS/MS analysis. These experiments showed minimal nonspecific labeling with the TAMRA-alkyne dye and also resulted in the identification of 213 proteins, among which 67 were previously known and 146 were novel, putative O-GlcNAc-modified proteins. Most of the proteins identified participate in neuronal signaling and synaptic function, which suggests important functional roles for O-GlcNAc in neuronal communication. In summary, this exciting method enables the direct fluorescence detection of O-GlcNAc proteins in gels, which in turn facilitates proteomic studies and extends the reach of existing technologies. This new tool for imaging O-GlcNAc-glycosylated proteins also enables the monitoring of expression and dynamics of the modification in cells or tissues.
Sortagging
Recently, the use of the enzyme sortase for labeling proteins has become an area of great interest. Sortase catalyzes a transpeptidase reaction between a specific internal sequence of a protein and an amine group present on the N-terminus of glycine. This method of labeling proteins has been denoted as “Sortagging”. One of the most common sortases is Sortase A (SrtA) derived from S. aureus76 which recognizes the pentapeptide sequence LPXTG and cleaves the bond between the threonine and glycine, creating a thioester bond between a SrtA-derived cysteine residue and the substrate. This covalent intermediate then undergoes a subsequent aminolysis reaction with the N-terminus of an oligoglycine, thus creating a new covalent bond between the two substrates (Figure 5).77 Additional work has shown that sortagging can be carried out ex vivo and cell surface proteins expressing the LPXTG motif can be fluorescently labeled using a small fluorophore.78 Popp et al. showed that they could take HEK293T cells expressing CD40L fused with GFP and an LPETG tag, and incubate them with sortase and a TAMRA-labeled oligoglycine. They found that such proteins could be labeled ex vivo. This type of ex vivo labeling can be performed using a wide variety of cell types as well.
Figure 5.
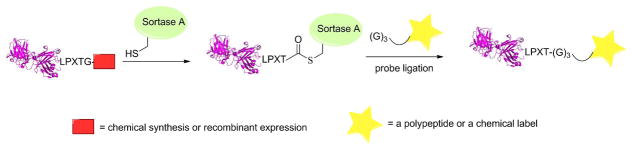
Schematic representation of sortase-mediated ligation. First, sortase A recognizes a LPXTG sequence within a polypeptide and forms a covalent acyl-enzyme intermediate. Next, the thioester intermediate is attacked by the N-terminal amino group of a (G)3-containing probe and forms a native peptide bond between the two.
Tanaka et al. added a sortase recognition tag, LPETGG, to osteoclast differentiation factor (ODF), which is expressed as a cell membrane protein, and transfected it into HEK293T cells.79 They showed that upon incubating the cells with sortase and biotin connected to a three-glycine oligomer, they were able to biotinylate on the surface of the cell. They expanded on this initial observation by demonstrating that this approach can work in multiple cell types, including CHO and HeLa cells, and that large molecules, such as GFP containing a five glycine N-terminus, can be conjugated to the protein on the cell surface. Strijbis et al. showed that labeling can be done in both S. cerevisiae and in the lumen of the ER or cytosol of the HEK293T cells.80
Recently, significant progress has been made in creating protein conjugates using SrtA to facilitate covalent attachment. Harrenga et al. were able to covalently link antibody Fab fragments to a small molecule fluorophore using SrtA at micromolar concentrations. In this case, they linked the light chain of L19 Fab fragment, which is specific for fibronectinED-B and is a promising target for cancer therapies, to a small peptide containing the fluorophore DY-647.81 Additionally, their work showed that ε-amino groups on lysines can also react causing undesirable protein aggregates. They found that these side reactions were dependent on both the position of the residue within the tertiary structure and on the pH of the reaction. Other examples of creating protein-molecule conjugates using sortagging include site-specifically PEGylating proteins,82 creating site-specific protein-lipid conjugates,83 and constructing peptides and glycosylphosphatidylinositol chimeras.84 Sortase has also been used in peptide synthesis to cyclize peptides to create macrocyclic peptides and glycopeptides.85
In addition to small molecule-protein conjugates, other types of protein conjugates have also been designed. Buti et al. were able to immobilize a library of proteins from an influenza virus onto glass plates using SrtA.86 In particular, they showed that the LPETG motif could be appended onto the C-terminal ends of proteins, which could in turn be site-specifically and covalently attached to the surfaces of glass slides that had been derivatized with a triglycine peptide. Once the proteins were immobilized on the glass plate, they were probed with H1N1 antiserum. Bound H1N1 antibodies were then visualized with a secondary antibody coupled to AlexaFluor-647, whose fluorescence intensity could be quantified. By examining the difference in fluorescence intensities, they were able to discern which proteins from the influenza virus were antigens for the H1N1 antiserum. This not only demonstrates that proteins can be efficiently conjugated to surfaces using sortase but also that these protein arrays can then be used for probing specific biological interactions.
Another application of sortagging is for the creation of protein-protein conjugates. Witte et al. showed that N-to-N and C-to-C protein conjugates could be made using a variant of sortagging.87 By expressing a protein with triglycine at the N-terminus and then incubating that protein with SrtA and a small LPETGG peptide incorporating either an azide or alkyne, a bioorthogonal handle was added to the N-terminus of proteins. To create N-to-N protein-protein conjugates they reacted two complementary handles to prepare the desired dimer. To create C-to-C protein conjugates, the target protein was expressed to contain a C-terminal LPETGG sequence that was then reacted with an azide- or alkyne-functionalized triglycine peptide. Using this method, Witte et al. prepared homodimeric N-to-N fusions of ubiquitin vinylmethylester (UbVME). They showed that UbVME was still able to bind its target protein, in this case ubiquitin carboxyl-terminal hydrolase isozyme L3 (UCHL3). Because the dimer UbVME was able to bind two UCHL3 proteins, this confirmed that the UbVME retained its biological activity. Similarly, they also constructed C-to-C fusions using a heavy chain antibody, VHH, which binds to GFP. They created this protein conjugate as described above and were able to confirm that it retained biological activity based on the ability of the dimer to bind two GFP proteins. This method demonstrates how sortagging can be used to create protein dimers without compromising the biological activity of the proteins being conjugated.
One of the problems with using sortase as a labeling catalyst is that the reaction is reversible; the glycine residue that is released in the first step can act as a nucleophile to reform the original species. This means that an excess of nucleophile is needed to drive the reaction towards product formation. This is especially problematic if the nucleophile is costly or labor intensive to prepare. To address this problem, Williamson et al. developed a depsipeptide, where the amide bond between the threonine and the glycine of the LPETG sequence was replaced with an ester.88 Their hypothesis was that the peptide would still be a substrate for the enzyme but that the alcohol product could not efficiently undergo the reverse reaction. They used this peptide first to label a small peptide with the sequence GGSEFG and showed that they were able to obtain almost 100% conversion to product using a 1:1 ratio of the two peptides. Next, they used this method to fluorescently label both human mannose binding protein and mouse pumilio-2 Puf RNA-binding domain at the N-terminus, which contained a glycine. Significantly, it was possible to achieve quantitative labeling of the protein using only 1.5 equivalents of the corresponding dansylated-LPETG peptide. This strategy overcomes one of the limitations of using sortagging in that proteins can be labeled at their N-termini without having to use excess amounts of the protein or peptide to obtain sufficient levels of labeling.
More recently (2013), the Ploegh lab has used sortase to create a ubiquitin-based probe to study deubiquitylating enzymes (DUBs).89 Using the sortase strategy they were able to selectively modify a ubiquitin that contained three glycines at the N-terminus with a R1-LPRTGG peptide in which R1 is a biotin derivative with a cleavable linker. They, went on to use these probes to retrieve and identify various DUBs and their interacting partners. Their results revealed that DUB2, a ubiquitin-specific protease, is expressed during the C. trachomatis infection. The presence and activity of this protein had not been previously detected in infected cells. In this strategy, sortase was used as a synthetic reagent to probe molecules used in subsequent biological applications.
In summary, sortagging offers a versatile and powerful tool for the site-specific labeling and modification of proteins. Sortagging can be accomplished by directly conjugating proteins to surfaces or other biologically relevant molecules or it can be used to attach bioorthogonal functional groups which can then be used to perform a secondary reaction. As research continues to enhance and refine sortagging, its utility will undoubtedly increase.
Transglutaminase
Transglutaminases (TGases) are a family of enzymes that catalyze acyl transfer reactions between the carboxamide groups of glutamine residues (when present in flexible loops in proteins) and a wide variety of unbranched primary amines, commonly the ε-amino group of lysine. Due to their promiscuity with regard to the primary amine substrate, transglutaminases are attractive catalysts for generating protein conjugates (Figure 6).5
Figure 6.
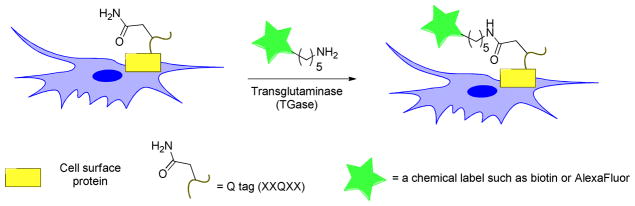
Schematic representation of site-specific labeling of proteins using transglutaminase (TGase) catalyzed ligation of cadaverine-functionalized probes (green star) to cell surface proteins recombinantly fused to a Q-tag (yellow rectangle). TGase site-specifically incorporates a probe into the protein by ligating the glutamine of the Q-tag to the amine probe and forms a new amide bond between the two while releasing ammonia into solution.
One of the ways transglutaminases can be used to create therapeutic proteins is through the preparation of antibody-small molecule conjugates. Jeger et al. used a bacterial transglutaminase to covalently link antibodies generated against tumor antigens with small molecule chelators that can deliver metals for use as imaging agents.90 Specifically they conjugated anti-L1-CAM mAb chCE7 antibody and a commercially available anti-CD20 antibody, rituximab. Initial experiments to label the antibodies using transglutaminase failed even though both antibodies had glutamine residues which should have reacted with the enzyme. It was not until the authors employed a mutant form of the antibodies that conjugation was observed. When this was investigated, it was concluded that since the mutation changes the ability of the antibody to be glycosylated and since antibody glycosylation caused the loop containing the targeted glutamine to be in a less accessible conformation, the efficiency of modification is highly dependent on the accessibility of the glutamine which is modified by the enzyme. They were able to follow-up on these findings by treating the antibody with a deglycosylase enzyme, N-glycosidase F, and showed that once the antibodies were deglycosylated, they would then react with transglutaminase. In this way they were able to generate site-specific conjugates of the antibodies.
In addition to creating small molecule-protein conjugates, another area of active research has been in using transglutaminases to create protein polyethylene glycol (PEG) conjugates. One of the drawbacks to using transglutaminases to PEGylate proteins is that the promiscuity of the enzyme towards glutamine residues makes it difficult to predict the site(s) of modification. This requires using MS methods to determine the site of conjugation, which can be laborious although progress is being made in this field.91 Additionally, efforts to lipidate proteins using transglutaminase have been reported. Recently (2011), Abe et al. reported a new substrate for a S. mobaraensis transglutaminase which can be used to attach lipids to proteins.92 This substrate consists of an eight residue sequence (GGGSLLQG) that is functionalized with an alkyl chain. This small peptide-alkyl chain molecule can then act as the amine-accepting substrate for the enzyme and become attached to a lysine residue via enzymatic reaction resulting in the introduction of a lipid modification onto various proteins. This new substrate was used to create lipid-conjugated GFPs. The GFP contained the enzyme recognition sequence MRHKGS, where the lysine acted as the amine donator, and the lipid substrates ranged between 14 and 18 carbons long. They were then able to show that these conjugates could anchor the modified proteins into the cellular membrane and that the degree of membrane anchoring increased with the length of the alkyl chain. In this way they were able to effectively create protein lipid conjugates using transglutaminase and employ these conjugates to study cellular systems.
Beyond the creation of protein-small molecule conjugates, work has also been performed to create site-specific protein conjugates. Tanaka et al. used a transglutaminase from S. mobaraensis to create GFP-dihydrofolate reductase (DHFR) protein conjugates.93 This was accomplished by including a pentaglycine linker at the N-terminus of GFP and adding a myc tag, which contained the glutaminase recognition sequence, to DHFR. They were then able to show that they could create a covalent linkage between the glycine residue of the N-terminus of GFP and a glutamine residue near the N-terminus of DHFR. This works illustrates how transglutaminase can be used to create heterodimeric protein conjugates.
Transglutaminase can also be used to covalently attach proteins to a solid support. Tominaga et al. were able to conjugate E. coli alkaline phosphatase (AP) to casein-grafted agarose beads.94 They were able to do this in a site-specific manner by adding the TGase recognition tag to the N terminus of AP. Importantly they found that immobilized AP had a very similar specific activity when compared to the soluble enzyme.
Finally, transglutaminases can be used for ex vivo labeling as well. Lin et al. used this enzyme to label proteins within living cells.95 Of particular significance, they used a guinea pig transglutaminase (gpTGase) because it exhibits a high specificity for its glutamine containing protein substrate but also a high promiscuity for the amine substrate. In this way they could specifically label proteins with what was termed as a Q-tag. Three different amino acid tags were examined including PNPQLPF, PKPQQFM, and GQQQLG, and all were found to be efficiently labeled by gpTGase with a cadaverine-biotin- or AlexaFluor-568-containing analogue. They showed that this labeling could also be accomplished ex vivo by labeling cyan fluorescent protein (CFP) containing a cell surface targeting sequence using their transglutaminase method. Finally they examined how this method could be used to create an assay for difficult-to-assay targets. They chose to assay for the presence of the NF-κB homodimer, which can form in the presence of DNA damage but is difficult to quantify. Their transglutaminase method was employed to label NF-κB with a benzophenone spermine analogue, which acts as a photoaffinity reagent, creating a covalent bond between the protein subunits upon photolysis. The presence of the homodimer was easily detected using via SDS-PAGE. This work highlights the utility of transglutaminase for protein labeling since it can be used in a site-specific manner to function in both in vitro and ex vivo environments. Overall, transglutaminase manifests high potential for use as a site-specific labeling agent. While these enzymes can be somewhat non-selective, as was noted above, ways to overcome this limitation have been developed.
PFTase post-translational modification
In nature, numerous proteins are post-translationally modified on the sulfur atom present in a cysteine residue near the C-termini of proteins by protein farnesyltransferase (PFTase). PFTase employs farnesyl pyrophosphate (FPP) as its natural substrate to transfer a farnesyl group to the cysteine of a “CaaX-box” tetrapeptide sequence (C is cysteine, a is an aliphatic amino acid, X is one of a variety of amino acids). The reaction can also be performed in vitro. PFTase is promiscuous in the nature of its substrates, in that it can tolerate a variety of CaaX sequences as well as some modifications to the FPP structure. Recently, a number of groups have used this feature to site-specifically modify proteins using analogues of FPP. Various isoprenoid surrogates containing azide,96, 97 alkyne,98 aldehyde,42, 99 biotin,100 or aryl groups101 have been synthesized and shown to be successfully processed by PFTase. The PFTase-catalyzed transfer of synthetic alkyne- or azide-functionalized farnesyl analogues in combination with the Huisgen [3+2] cycloaddition or Staudinger ligation has been used for site-specific fluorophore labeling and oriented immobilization of proteins.96 Importantly, CaaX-box sequences such as CVIA, or CVIM can be genetically encoded into the C-termini of many proteins making them efficient substrates for PFTase.
In early work, Poulter and coworkers used this strategy to immobilize protein on solid surfaces.7 They synthesized alkyne-functionalized FPP and incorporated the alkyne moiety into the proteins (GFP and GST) at their C-termini using PFTase. A glass-slide was functionalized with an azide group and covalent immobilization of the proteins to the slide was subsequently achieved using copper (I)-catalyzed [3+2] cycloaddition. In subsequent work they synthesized a collection of FPP analogues containing either alkyne or azide moieties and analyzed their kinetic parameters (KM and kcat) as substrates for PFTase.102 Out of the eleven synthetic analogues, two of them gave steady-state kinetic parameters (KM and kcat) very similar to those of the natural substrate FPP.
Waldmann and coworkers used the PFTase strategy to modify proteins including H-, N-, and K-Ras GTPases, which are important in cellular signaling and are among the human oncogene products, at their C-termini with a farnesyl group. Subsequently they used thiol-functionalized surfaces to react photochemically with the alkenes present in the farnesyl groups introduced into proteins as noted above. In this way, proteins were covalently immobilized in a site-specific manner under mild conditions in about 10 minutes.103
In other work, they developed several azide-, biotin- and diene-functionalized analogues of FPP. After kinetic analysis of the enzymatic reactions with PFTase, they concluded that all of these new substrates, except the biotin-containing analogue, can be transferred onto proteins that possess a CaaX box.104 Subsequently, proteins functionalized with azide or diene groups were successfully modified by Staudinger ligation or Diels–Alder cycloaddition. In related work, Alexandrov and coworkers described a structure-guided design of an engineered farnesyl transferase and geranylgeranyl transferase-I that could both efficiently use biotin-geranyl pyrophosphate as a substrate.100 Using these mutant enzymes and biotin-geranyl pyrophosphate as a substrate, they could detect femtomolar levels of prenylatable proteins in cells and organs and identified their cognate protein prenyltransferases.
Distefano and coworkers have exploited the PFTase-catalyzed reaction for a number of applications. In early work, they synthesized an analogue of geranyl diphosphate containing an azide group at C-8 and showed that PFTase could transfer that shorter isoprenoid onto a peptide containing a CaaX box.105 The resulting peptide was further modified by selective Staudinger ligation between the peptidyl azide and a suitable triphenylphosphine derivative; they also found that a longer azide-containing analogue based on FPP could be similarly transferred and reacted.106
One problem with these azide-containing analogues is that they undergo thermal rearrangements to produce a mixture of isomers due to the allylic azide moiety. Thus, proteins modified with these reagents can potentially produce similar mixtures abrogating one of the advantages of site-selective modification. To avoid this problem, Xu et al. synthesized a dihydroazide analogue based on geraniol that replaces the allylic azide with an aliphatic one and demonstrated that the resulting molecule is an alternative substrate for PFTase.107 That work was extended by synthesizing a similar analogue based on FPP. In this latter case, the analogue was used to tag GFP appended with a CaaX box with an azide that was subsequently used to immobilize the protein onto alkyne-functionalized agarose beads. Importantly, this two-step enzymatic modification/immobilization sequence was accomplished with both purified protein as well as target present in crude E. coli extract at levels as low as 1% of total protein.96 The versatility of the proteins functionalized with azides incorporated via PFTase was further demonstrated in a subsequent publication where protein-DNA conjugates were prepared.108 In that case, a simple GFP-DNA species was first generated by Cu-catalyzed click reaction between GFP-azide and an oligonucleotide equipped with an alkyne. Upon hybridization with a second complementary oligonucleotide containing a Texas Red fluorophore, FRET was observed confirming the production of the desired product. That was extended to the preparation of a more complex protein-DNA tetrahedron composed of four oligonucleotides and four GFP molecules assembled in an analogous manner which was characterized by a combination of electrophoretic and single molecule techniques. The ability to create nanoscale sized protein-based objects with defined architecture highlights the utility of this approach. Recently (2012) this strategy for preparing protein-DNA assemblies has been extended by using the related strain-promoted azide-alkyne cyclization reaction. By incorporating a dibenzocyclooctyne moiety into the oligonucleotide, a number of proteins were conjugated to DNA including GFP, mCherry, GIP and HIV NC.109 GFP-mCherry heterodimers were also prepared using this strategy by conjugating complementary alkyne-modified DNA oligonucleotides to the two respective protein-azides followed by hybridization.
In performing the Cu-catalyzed click reaction, it has been noted that lower levels of background reaction are often obtained when the azide component is the one used in excess.110 Accordingly, Distefano and coworkers prepared an alkyne-containing analogue using a GPP scaffold and showed that it was a good alternative substrate for PFTase.98 They then showed that more nonspecific immobilization occurred when unfunctionalized GFP was reacted under typical click reaction conditions with alkyne-functionalized agarose beads compared to a similar experiment with azide-containing beads. Based on those results, GFP-alkyne, produced via PFTase-catalyzed incorporation of the alkyne-containing analogue noted above, was labeled with excess Texas Red azide in high (81%) yield. FRET experiments allowed a distance of 37 Å between the GFP and Texas Red fluorophores to be measured which was in good agreement with the distance determined from computer modeling. These experiments highlight the utility of this approach for selective labeling for biophysical experiments. Additional work with a longer alkyne-containing isoprenoid diphosphate based on FPP showed that this new compound was an efficient substrate for both PFTase and the related enzyme protein geranylgeranyltransferase (GGTase) opening up the possibility for transferring larger probes.111 Conversely, one problem concerning the addition of isoprenoids to proteins is that this modification increases the hydrophobicity of the protein and thereby decreases its solubility. Accordingly, Wollack et al. prepared a more polar PFTase substrate analogue consisting of a single isoprenoid unit linked to an alkyne.112 While this analogue was incorporated significantly less efficiently (relative to the natural substrate, FPP), it was still possible to achieve essentially quantitative modification of mCherry and Hcp using higher amounts of enzyme. Interestingly, modification of Hcp with the C15-alkyne analogue resulted in a protein that was insoluble; in contrast, the C5-alkyne modified protein was completely soluble highlighting the utility of this less lipophilic modification. In that same report, the authors also demonstrated that the three C-terminal residues following the cysteine residue of the CaaX box could be proteolytically removed by treatment with carboxypeptidase Y after a peptide or protein was modified with the C5 analogue. This means that while PFTase-catalyzed protein modification is not traceless, the size of the tag remaining in the final protein product can be limited to a single prenylated cysteine residue. This has important implications in the area of protein therapeutics where the antigenicity of any added element may prove deleterious.
Recently (2010), FPP analogues that incorporate an aldehyde functionality have also been developed. Rashidian et al. showed that a simple aldehyde-containing analogue of FPP could be used to incorporate an aldehyde into GFP. The resulting aldehyde was used to either immobilize the protein or to label it with a fluorophore via oxime formation.99 A subsequent study demonstrated that such aldehyde-modified proteins could be prepared and captured from crude extract of E. coli by incubation with a hydrazide resin. The hydrazone linkage, though stable, is not irreversible and can be converted to a more stable oxime linkage. The authors used this to release, and simultaneously label, the captured protein into the solution by elution with an aryl amine catalyst and an alkoxyamine.42 Using this strategy, the fluorescently labeled or PEGylated GFP was obtained without prior purification of the starting protein. A PEGylated form of glucose-dependent insulinotropic polypeptide (GIP), a protein with potential as a therapeutic agent for diabetes, was also prepared in an analogous fashion. A detailed protocol for performing this capture and release strategy has recently (2013) been published.113 In the aforementioned strategy, the release of the protein, via a hydrazone-oxime reaction, is a particularly slow process even in presence of aniline40, 114 as a catalyst. Recently (2013), Rashidian et al. improved this strategy using a new, more efficient catalyst, m-phenylenediamine (mPDA), which can significantly increase the rate of the hydrazone-oxime exchange as the protein-release step. While mPDA is only modestly more effective than aniline when used in equal concentrations (~ 2-fold), its much greater aqueous solubility relative to aniline allows it to be used at higher concentrations, resulting in significantly more efficient catalysis.115 Related catalysts have been recently reported for oxime ligation reactions involving nucleic acids.116 In general, this approach for protein modification could be particularly useful for large-scale production of protein conjugates for therapeutic or industrial applications.
Biotin ligase
Biotin ligases catalyze the attachment of biotin groups onto proteins and other biomolecules (Figure 8). These enzymes possess several useful attributes for protein labeling. They can be used to site-specifically modify proteins by genetic fusion of a ligase recognition domain onto the target protein of interest. Enzymatic labeling of the protein with biotin allows for the subsequent use of avidin (and related proteins) resulting in the formation of very strong non-covalent conjugates because of the low dissociation constant between biotin and avidin (~10−15 M).117
Figure 8.
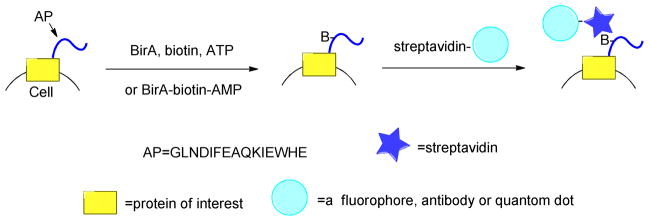
Schematic representation of targeting different species including quantum dots, fluorophores or antibodies to surface proteins in living cells using biotin ligase. The acceptor peptide (AP) tag is genetically encoded either at the C- or N- terminus of a protein. Biotinylation of AP is performed by adding recombinantly expressed biotin ligase (BirA), ATP and biotin (B) to the cell medium. After removing excess biotin by washing, fluorescently labeled streptavidin is added to visualize the biotinylated surface proteins.
A number of different groups have used biotin ligase to site-specifically modify proteins. One common method is to use the E. coli biotin ligase, BirA, to attach biotin to proteins. BirA is convenient because it links biotin onto the lysine of a small, 15-residue, acceptor peptide (AP). This sequence, GLNDIFEAQKIEWHE, was specifically developed as the minimalist structural element that can be efficiently recognized by BirA.
The Ting group has adapted this method for preparing a wide range of biotinylated proteins which can be used for a variety of applications. In one study the AP recognition site was fused to a cyan fluorescent protein (CFP) that contains a transmembrane domain which should traffic the protein to the cell surface of HeLa cells.118 They found that they could biotinylate the protein using BirA and then conjugate it to either a streptavidin-AlexaFluor-568 dye or a streptavidin-quantum dot and determined that the quantum dot (CdSe) gave a better signal then the AlexaFluor-568 dye did. In additional work, they noted that they could label epidermal growth factor receptor using this same method and image it in a similar fashion. The applicability of this work to primary cells was also studied. Hippocampal neurons were transfected with an AP-tagged GluR2 protein which is a subunit of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptor. They were able to analyze AMPA receptor trafficking by monitoring fluorescent GLuR2. More recent work has focused on refining this method so that the quantum dots are smaller and only contain monovalent streptavidin moieties, which can then better label site-specifically biotinylated proteins in cells.119
Ting and coworkers also explored whether different biotin ligases could catalyze the attachment of biotin analogues that contain alternative functionality suitable for bioorthogonal reactions.120 A panel of eight different biotin ligases was screened for activity using an array of biotin analogues leading to the discovery that a biotin ligase from P. horikoshii could act on analogues that contained either an azide or alkyne group. They were then able to label a domain of a biotin acceptor protein with an azide derivative of biotin and conjugate that protein to a FLAG peptide via the Staudinger ligation. In this way they were able to show that biotin ligases could be used to attach bioorthogonal handles to proteins.
Biotin ligase has also been used to prepare modified proteins suitable for studies of protein-protein interactions. In one example, an AP peptide with a high KM for BirA was engineered and incorporated into FKBP.121 The interaction between that protein and a BirA-FRB fusion was then examined. Consistent with the fact that FRB and FKBP interact only in the presence of rapamycin, they observed that labeling of the AP domain by the BirA-FRB fusion occurred only in the presence of both biotin and rapamycin but not in the absence of the rapamycin. These experiments were performed both in vitro and ex vivo emphasizing the utility of their approach for monitoring protein-protein interactions. To expand the utility of this method they also examined the interaction of Cdc25C with 14-3-3ε phosphoserine/threonine binding protein. Cdc25C was produced as a AP fusion and 14-3-3ε was fused to BirA; both were expressed in HEK cells. Importantly, biotinylation was observed when a wild type-like Cdc25C was used. However, when a non-phosphorylatable Cdc25C mutant was employed, no biotinylation was seen, suggesting that the interaction between Cdc25C and 14-3-3ε is phosphorylation dependent.
Sueda et al. used a biotin ligase from S. tokodaii to modify proteins for imaging applications.122 This enzyme biotinylates the lysine residue of a biotin carboxyl carrier protein (BCCP), and interestingly, upon biotinylation, forms a tight non-covalent complex between the ligase and BCCP. Sueda et al. used this unique property to modify proteins by creating fusion proteins between the target of interest and a truncated version of the BCCP protein. They found that the biotin ligase would accept a truncated 69-residue polypeptide derived from BCCP. A fusion was then created between this tag and the bradykinin B2 receptor (B2R), which is a transmembrane protein. They then expressed this protein in HEK293 cells, and incubated these cells in the presence of S. tokodaii biotin ligase that had been labeled previously with four fluorescein molecules. Fluorescent labeling was observed around the cell membrane, confirming that the N-terminus is localized on the outer side of the plasma membrane. In a second experiment, a C-terminus B2R fusion protein with the BCCP tag was expressed in HEK293 cells together with a GFP-biotin ligase fusion protein and were again able to detect fluorescence at the cell membrane. In this case, fluorescent labeling occurred because the C terminus of the B2R protein positioned on the inner side of the membrane was accessible to the GFP-biotin ligase fusion produced intracellularly.
More recently (2012), Roux et al. used a mutant version of BirA that allows for the promiscuous biotinylation of primary amines that are in close proximity to the enzyme.123 By fusing this BirA mutant to a protein of interest, they could detect other proteins that are in close proximity to the protein of interest. Using this proximity-dependent labeling method, they identified proteins that interact with or are in close proximity to the lamin-A protein in living cells.
As a final remark, it should be noted that while most of the research to date with biotin ligases has focused on ex vivo imaging, its site and substrate specificity makes it an attractive tool for creating site-specific protein conjugates for other applications as well. Examples of this will undoubtedly be reported in the future.
Lipoic acid ligase
Lipoic acid ligase catalyzes the acylation of lysine residues of proteins with lipoic acid (Figure 9). Significant effort has gone into the development of this enzyme for use in site-specific labeling. Fernández-Suárez et al. designed and synthesized ten different lipoic acid analogues functionalized with either an azide or alkyne and tested them as alternative substrates for E. coli lipoic acid ligase (LplA).124 While all analogues exhibited some degree of activity, the authors focused on an azide-containing analogue due to its superior activity. Additionally they sought to develop a smaller peptide substrate to minimize the size of the fusion that would have to be used to decrease the possibility of interference with the function of protein under study. By analyzing the structure of E2p, a naturally lipoylated protein, they were able to design a series of small peptides, which mimicked the β-turn where the lipoylated lysine residue is located on the protein. After further refinement the tag was reduced to just 22 amino acids (termed LAP). They were then able to show that a CFP-LAP fusion could be labeled with a small fluorophore in HEK cell lysates. Additionally they added a transmembrane domain to the CFP-LAP protein and were able to show that they could label the protein in living HEK cells and obtain conjugation using a cyclooctyne fluorophore, either Cy3 or AlexaFluor568. A LAP tag was also added to the low-density lipoprotein receptor (LDLR) and visualized using the same lipoic acid labeling methodology. Finally they tested the cross compatibility of using both lipoic acid labeling and biotin ligase. One population of cells was transfected with a LAP-LDLR producing construct and a different population was treated with an AP-fused epidermal growth factor receptor (AP-EGFR) producing vector. Once transfected, the different cell populations were mixed together and subjected to both lipoic acid ligase and biotin ligase conjugation conditions. The authors observed that only cells containing AP-EGFR showed streptavidin conjugation and cells expressing LAP-LDLR showed only Cy3 labeling. Finally cells were transfected with both constructs and it was shown that the resulting proteins could be conjugated simultaneously using the two different bioconjugation techniques.
Figure 9.

Schematic representation of protein labeling using lipoic acid ligase (LplA). (A) Natural ligation reaction catalyzed by LplA. (B) Site-specific labeling of a cell-surface protein performed by fusion of a 22 residue LAP domain on the protein of interest followed by enzymatic transfer of an azide-containing lipoic acid analogue. The azide can be used for further modification via a [3+2] azide-alkyne cycloaddition reaction or Staudinger ligation. (C) Engineered coumarin ligase produced by mutagenesis of LplA can be used to site-specifically modify a protein with a coumarin probe. This can be used to directly incorporate fluorophores to mammalian cells.
The use of other lipoic acid analogues for selective protein labeling has also been investigated. Through analysis of the LplA crystal structure, a tryptophan and a glutamate residue in the lipoate binding pocket of the enzyme were identified that when mutated to smaller residues allow the mutant enzymes to accept a wider range of lipoic acid analogues. Using that strategy, it was possible to create a mutant of LplA that accepts lipoate analogues that incorporate fluorescent moieties including Pacific Blue or 7-hydroxycoumarin. Thus, fluorescently labeled proteins could be produced ex vivo without having to perform a secondary conjugation reaction.125, 126 These mutant enzymes were also used to incorporate azide analogues containing longer spacers, which were then conjugated ex vivo to more red-shifted fluorophores, which have a higher signal to background ratio when compared to other fluorescent moieties excited at lower wavelengths. Overall, these developments improved the ability to image the localization and distribution of proteins within living cells.
More recently (2011), Ting and coworkers have applied this lipoic ligase tagging technique to examine protein-protein interactions in experiments similar to those described above with biotin ligase.127 A lipoic acid peptide tag was created with a high KM value such that the peptide would only bind when the enzyme was held in close proximity via non-covalent forces. This method was implemented by fusing lipoic acid ligase to one target protein and then fusing the peptide tag to a putative binding partner. This technique was employed to study the rapamycin-dependent binding of FKBP and FRB as well as to probe the interaction between the transcription factors Jun and Fos. They also used this approach to study the interaction between neuroligin-1 and PSD-95, which is involved in excitatory synapse formation and maturation. These examples illustrate the versatility of the lipoic acid ligase method for labeling proteins for the analysis of many different types of protein-protein interactions ex vivo.
The lipoic acid ligase labeling method has also been adapted to work with lipoic acid tags that contain aryl aldehydes and aryl hydrazines.128 Ting and coworkers synthesized lipoic acid derivatives containing an aryl aldehyde and an aryl hydrazine and screened these two analogues against a panel of lipoic acid ligase mutants each containing a mutation of W37. They found that a tryptophan to isoleucine mutation significantly increased the extent of conversion when using these alternative substrates compared to that obtained with the wild type enzyme. This mutant enzyme was then used with the aforementioned analogues to label neurexin-1β and then react it with complementary aldehyde- and hydrazine-containing reagents, which contain either a fluorophore or a chromophore; these reactions were performed ex vivo using HEK cells. In a final set of experiments, they used this method to image single low density lipoprotein receptors on the surface of COS7 cells.
More recent work in the Ting lab has examined the use of lipoic acid ligase mutants to attach a wide variety of chemical “handles” onto proteins, which can then be used to attach other small molecules. These include trans-cyclooctenes derivatives, which can then be used with tetrazine-conjugated fluorophores to rapidly label proteins.37 They have also introduced copper-chelating azides onto proteins and found that these azides can undergo the cycloaddtion reaction with alkynes using much lower copper concentrations, which are, in turn, less toxic to cells.129 Another strategy developed by Ting and coworkers was to label proteins with terminal azides using lipoic acid ligase and perform a subsequent conjugation reaction using ring-strained cyclooctynes, eliminating the need for copper as a catalyst.130
Overall lipoic acid ligase is a powerful tool for creating site-specific protein conjugates. As was noted above, in reference to biotin ligase, work using LplA has also centered around the creation of protein-fluorophore conjugates for monitoring cellular processes ex vivo and for probing protein-protein interactions. However, this enzyme has significant potential for use in creating other types of protein conjugates including protein-DNA, protein-drug, and protein-protein chimeras. It is likely that such applications will be reported soon.
N-myristoylation
Protein N-myristoylation involves the acyl transfer of myristate from myristoyl-CoA to the amino group of an N-terminal glycine residue of a protein to form an amide bond.131 The enzyme that performs this modification, N-myristoyl transferase (NMT), recognizes the sequence GXXXS/T where X can be a variety of amino acids and is found in eukaryotes.131, 132 Approximately, 0.5% of proteins in eukaryotes are myristoylated.133, 134 Protein N-myristoylation is thought to occur during protein translation although recent evidence indicates that it can also occur post-translationally. Some important examples of myristoylated proteins include the Src family of tyrosine kinases, HIV-1 matrix protein, HIV-1 Gag and the ADP-ribosylating factors (ARFs). Because of the involvement of these proteins in various diseases,135 NMT has considerable potential as a therapeutic target.
Tate and coworkers, used the substrate promiscuity of NMT to site-specifically label recombinant proteins with azide and alkyne derivatives of myristoyl-CoA both ex vivo and in vitro.136, 137 They used a well-characterized and widely-used NMT cloned from C. albicans to transfer alkyne- and azide-containing myristate analogues that incorporated the bioorthogonal groups at the distal end of the lipid. First they evaluated the substrate efficiency using a model peptide substrate GLYVSRLFNRLFQKK-NH2 containing a canonical N-terminal myristoylation motif. Incubation of the peptide with either of the two acyl-CoA analogues in the presence of a catalytic quantity of NMT for 2 h resulted in complete transfer of the acyl group to the target peptide.
Next, E. coli engineered to co-express NMT and a substrate protein, PfARF1, was used for in vivo analysis. The bacterial cells were transfected with a pair of plasmids encoding the substrate protein and the NMT together with orthogonal antibiotic resistance marker genes. The cells were incubated with one of the aforementioned analogues and induced by addition of IPTG to express both proteins. Importantly, this strategy capitalized on endogenous E. coli enzymatic activity to convert the myristate analogues to the corresponding CoA derivatives. After metabolic incorporation, the cells were lysed and reacted with bioorthogonal biotin reagents to detect the tagged protein including azide-, alkyne- and triarylphosphine-containing compounds. Analysis by SDS-PAGE and subsequent detection with streptavidin-HRP showed that this method could specifically tag PfARF1 when labeling was performed under these conditions within the cell. This strategy has been explained in detail in a recently (2012) reported protocol.138 Overall, this method provides a convenient and potentially general method for N-terminal recombinant protein labeling.
Perspectives and Conclusions
In summary, a number of enzymes have been studied and employed for site-specific labeling of proteins. Each of these enzymes manifests differences in substrate specificity, size of the tag needed for protein targeting, position of modification and kinetics of incorporation. These differences must be considered when deciding which enzyme to use for protein modification applications. Table 1 summarizes a number of different parameters for the many of the labeling enzymes described in this review including the size/length of the tag, the position where the tag can be incorporated within a target protein and the catalytic parameters of the labeling enzymes. Inspection of Table 1 shows that a range of options are available using the repertoire of enzymes currently available. One of the main factors to consider is the structural sensitivity of the protein that is to be modified because the addition of a large genetic tag may result in a perturbation of the protein’s activity, and hence methods that use a large recognition tag would not be appropriate. These include formylglycine generating enzyme (13-mer, LCTPSRGSLFTGR) lipoic acid ligase (13 mer, GFEIDKVWYDLDA), biotin ligase (15 mer, GLNDIFEAQKIEWHE) and phosphopantetheinyltransferase (in the case of using ACP or PCP, recognition tags are 75-80 amino acids in length). Conversely, farnesyltransferase (4 mer, CaaX), transglutaminase (5 mer, XXQXX) or sortase (5 mer, LPXTG) would be better options to use for protein modification due to their inherently small recognition sequences.
Another key consideration is the location of the site of modification in the protein. Many of the methods mentioned in this review can be used only for C- or N-terminal modification such as farnesylation, biotin ligation, etc. While C- and N-terminal modifications are commonly used for protein modification, there are instances when the site of modification needs to be located within interior sequences such as between protein domains or when the C- or N-terminus is important for protein activity. In these situations, using enzymes such as transglutaminase or sortase would be particularly useful because there are no limitations for placement of the site of modification within their cognate targets.
The size of the modification to be added must also be considered when selecting an enzymatic modification strategy. If, for example, the protein of interest is intended to be modified with a large peptide, polymer or a protein, then sortase may be particularly useful. This is because in sortagging, there is no limit in the size of the group that can be added to the protein. Phosphopantetheinyltransferase is also relatively flexible in terms of accommodating large modifications. Other methods, including farnesyltransferase, N-myristoyltransferase, lipoic acid ligase, biotin ligase, and transglutaminase are limited by the fact that alternative substrates must be similar in size and shape to their natural substrates; significantly larger substrates cannot be accommodated into their active sites. These methods typically use a substrate analogue which contains a bioorthogonal handle that can be further functionalized in a subsequent reaction making the process more laborious
The kinetic features of specific protein modifying enzymes are also important factors in choosing a particular enzyme. If the protein of interest degrades quickly or loses activity over time, then minimizing the reaction time required for the modification can be very important. In this case, farnesyltransferase is particularly attractive since it manifests a rapid enzymatic reaction rate with a kcat/KM of 18 min−1 μM−1 for the enzyme’s natural substrate (farnesyl diphosphate) and 4 min−1 μM−1 for an aldehyde-analog (Figure 7A). Conversely, sortase has relatively slow enzyme kinetics with a kcat/KM of 0.003 min−1 μM−1.
Figure 7.
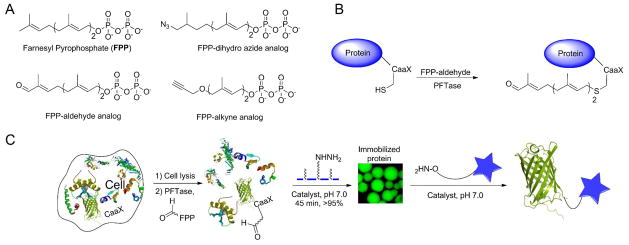
(A) Structures of farnesyl diphosphate (FPP), farnesyl dihydro azide diphosphate (FPP-dihydro azide) farnesyl aldehyde diphosphate (FPP-aldehyde) and farnesyl-alkyne diphosphate (FPP-alkyne) as examples of FPP analogues. (B) Schematic representation of prenylation of a protein containing a CaaX-box positioned at its C-terminus with aldehyde-containing analogue (FPP-aldehyde) to yield the prenylated product. (C) Chemoenzymatic site-specific tagging of proteins by FPP-aldehyde using PFTase followed by capture of the aldehyde-functionalized protein in the crude cell lysate via hydrazide-functionalized beads. The immobilized protein is then released into solution using aminooxy-containing reagents to yield fluorescently labeled or PEGylated protein.
The main disadvantage of many of these protein modification methods is the fact that their respective enzymatic modifications result in addition of organic molecules to the protein of interest, which can perturb the activity and/or solubility of the protein. For example, the addition of a farnesyl (a C15), a myristoyl (a C14) or a lipoic acid (a C8) group can result in decreased solubility and precipitation of the modified protein. Thus, if the protein is not initially highly water soluble to, these modifications can result in precipitation and loss of the modified protein. Thus, the solubility of target protein should be considered when selecting an enzymatic labeling approach to use.
Current methods of site-specific enzymatic protein modification offer a diverse array of labeling approaches that can be performed to modify proteins: everything from modification with small molecules all the way up to linkages with other large proteins. While there has been much progress in site-specific labeling strategies, there still remains much work to be done so that these processes can be more widely utilized. Many of these methods described here have been used primarily in studies of in vitro and ex vivo processes but have yet to be employed in biotechnological applications. The types of methods can be easily adapted to create protein-drug, protein-PEG, or protein conjugates that overcome some of the disadvantages of traditional protein labeling methods. It is expected that in near future we will see multiple pharmaceutical companies start to take advantage of the aforementioned enzymatic site-specific modification approaches to synthesize modified proteins of biological and medical importance.
As the future of protein modification progresses, it will be up to researchers to develop new types of site-specific enzymatic modifications that overcome some of the present challenges and create methods that result in minimum change in functionality of the protein of interest, increased protein pharmacokinetics, increased solubility, increased stability, decreased immunogenicity, and minimal structural perturbance. In addition to finding new types of enzymes to create alternative protein modifications, new types of bioorthogonal reactions would also be of great advantage since many of the current methods for protein modification rely on the synergy created by combining enzymatic labeling with bioorthogonal chemistry. Given the progress made in the last
Figure 10.
Schematic representation of in vivo protein labeling via N-myristoyl transferase (NMT) using an E. coli co-expression system. Cells are incubated with a myristic acid analogue and plasmids encoding NMT and a recombinant target protein. Within the cells, the analogue is converted to its corresponding CoA derivative and incorporated into the desired target protein using NMT. Tagged proteins may then be captured using Staudinger ligation or [3+2] cycloaddition to introduce a secondary label for subsequent applications.
Acknowledgments
This work was supported by the National Institutes of Health (GM058842 and GM084152) and the University of Minnesota.
References
- 1.Hackenberger CPR, Schwarzer D. Chemoselective Ligation and Modification Strategies for Peptides and Proteins. Angew Chem Int Ed. 2008;47:10030–10074. doi: 10.1002/anie.200801313. [DOI] [PubMed] [Google Scholar]
- 2.Rabuka D. Chemoenzymatic methods for site-specific protein modification. Curr Opin Chem Biol. 2010;14:790–796. doi: 10.1016/j.cbpa.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carrico IS. Chemoselective modification of proteins: hitting the target. Chem Soc Rev. 2008;37:1423. doi: 10.1039/b703364h. [DOI] [PubMed] [Google Scholar]
- 4.Wong SS, Jameson DM, Wong SS. Chemistry of protein and nucleic acid cross-linking and conjugation. Taylor & Francis/CRC Press; Boca Raton: 2012. [Google Scholar]
- 5.Fontana A, Spolaore B, Mero A, Veronese FM. Site-specific modification and PEGylation of pharmaceutical proteins mediated by transglutaminase. Adv Drug Deliv Rev. 2008;60:13–28. doi: 10.1016/j.addr.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 6.Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, Lu Y, Meng YG, Ng C, Yang J, Lee CC, Duenas E, Gorrell J, Katta V, Kim A, McDorman K, Flagella K, Venook R, Ross S, Spencer SD, Lee Wong W, Lowman HB, Vandlen R, Sliwkowski MX, Scheller RH, Polakis P, Mallet W. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26:925–932. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 7.Gauchet C, Labadie GR, Poulter CD. Regio- and Chemoselective Covalent Immobilization of Proteins through Unnatural Amino Acids. J Am Chem Soc. 2006;128:9274–9275. doi: 10.1021/ja061131o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kochendoerfer GG. Site-specific polymer modification of therapeutic proteins. Curr Opin Chem Biol. 2005;9:555–560. doi: 10.1016/j.cbpa.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Chen I, Howarth M, Lin W, Ting AY. Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat Meth. 2005;2:99–104. doi: 10.1038/nmeth735. [DOI] [PubMed] [Google Scholar]
- 10.Köhn M. Immobilization strategies for small molecule, peptide and protein microarrays. J Pept Sci. 2009;15:393–397. doi: 10.1002/psc.1130. [DOI] [PubMed] [Google Scholar]
- 11.Wong LS, Khan F, Micklefield J. Selective Covalent Protein Immobilization: Strategies and Applications. Chem Rev. 2009;109:4025–4053. doi: 10.1021/cr8004668. [DOI] [PubMed] [Google Scholar]
- 12.Tiefenbrunn TK, Dawson PE. Chemoselective ligation techniques: Modern applications of time-honored chemistry. Biopolymers. 2010;94:95–106. doi: 10.1002/bip.21337. [DOI] [PubMed] [Google Scholar]
- 13.Wu BY, Hou SH, Huang L, Yin F, Zhao ZX, Anzai JI, Chen Q. Oriented immobilization of immunoglobulin G onto the cuvette surface of the resonant mirror biosensor through layer-by-layer assembly of multilayer films. Mater Sci Eng, C. 2008;28:1065–1069. [Google Scholar]
- 14.Hermanson GT. Bioconjugate techniques. Academic Press; Amsterdam: 2008. [Google Scholar]
- 15.Dixon HB. Transamination of peptides. Biochem J. 1964;92:661–666. doi: 10.1042/bj0920661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu P, Brand L. N-terminal modification of proteins for fluorescence measurements. Meth Enzymol. 1997;278:321–330. doi: 10.1016/s0076-6879(97)78017-0. [DOI] [PubMed] [Google Scholar]
- 17.Scheck RA, Dedeo MT, Iavarone AT, Francis MB. Optimization of a biomimetic transamination reaction. J Am Chem Soc. 2008;130:11762–11770. doi: 10.1021/ja802495w. [DOI] [PubMed] [Google Scholar]
- 18.Geoghegan KF, Stroh JG. Site-directed conjugation of nonpeptide groups to peptides and proteins via periodate oxidation of a 2-amino alcohol. Application to modification at N-terminal serine. Bioconjugate Chem. 1992;3:138–146. doi: 10.1021/bc00014a008. [DOI] [PubMed] [Google Scholar]
- 19.Akgul C, Moulding DA, White MR, Edwards SW. In vivo localisation and stability of human Mcl-1 using green fluorescent protein (GFP) fusion proteins. FEBS Lett. 2000;478:72–76. doi: 10.1016/s0014-5793(00)01809-3. [DOI] [PubMed] [Google Scholar]
- 20.Dundr M, McNally JG, Cohen J, Misteli T. Quantitation of GFP-fusion proteins in single living cells. J Struc Biol. 2002;140:92–99. doi: 10.1016/s1047-8477(02)00521-x. [DOI] [PubMed] [Google Scholar]
- 21.Kanda T, Sullivan KF, Wahl GM. Histone–GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol. 1998;8:377–385. doi: 10.1016/s0960-9822(98)70156-3. [DOI] [PubMed] [Google Scholar]
- 22.Marguet D, Spiliotis ET, Pentcheva T, Lebowitz M, Schneck J, Edidin M. Lateral diffusion of GFP-tagged H2Ld molecules and of GFP-TAP1 reports on the assembly and retention of these molecules in the endoplasmic reticulum. Immunity. 1999;11:231–240. doi: 10.1016/s1074-7613(00)80098-9. [DOI] [PubMed] [Google Scholar]
- 23.Lisenbee CS, Karnik SK, Trelease RN. Overexpression and mislocalization of a tail-anchored GFP redefines the identity of peroxisomal ER. Traffic. 2003;4:491–501. doi: 10.1034/j.1600-0854.2003.00107.x. [DOI] [PubMed] [Google Scholar]
- 24.De Graaf AJ, Kooijman M, Hennink WE, Mastrobattista E. Nonnatural Amino Acids for Site-Specific Protein Conjugation. Bioconjugate Chem. 2009;20:1281–1295. doi: 10.1021/bc800294a. [DOI] [PubMed] [Google Scholar]
- 25.Wang L, Xie J, Schultz PG. Expanding the genetic code. Annu Rev Biophys Biomol Struct. 2006;35:225–249. doi: 10.1146/annurev.biophys.35.101105.121507. [DOI] [PubMed] [Google Scholar]
- 26.Chen PR, Groff D, Guo J, Ou W, Cellitti S, Geierstanger BH, Schultz PG. A Facile System for Encoding Unnatural Amino Acids in Mammalian Cells. Angew Chem Int Ed. 2009;48:4052–4055. doi: 10.1002/anie.200900683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo J, Melancon CE, Lee HS, Groff D, Schultz PG. Evolution of Amber Suppressor tRNAs for Efficient Bacterial Production of Proteins Containing Nonnatural Amino Acids. Angew Chem Int Ed. 2009;48:9148–9151. doi: 10.1002/anie.200904035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chatterjee A, Xiao H, Schultz PG. Evolution of multiple, mutually orthogonal prolyl-tRNA synthetase/tRNA pairs for unnatural amino acid mutagenesis in Escherichia coli. Proc Natl Acad Sci. 2012;109:14841–14846. doi: 10.1073/pnas.1212454109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huisgen R, Szeimies G, Mobius L. Chemische Berichte-Recueil. Chemische Berichte. 1967;100:2494. [Google Scholar]
- 30.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 31.Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 32.Köhn M, Breinbauer R. The Staudinger Ligation—A Gift to Chemical Biology. Angew Chem Int Ed. 2004;43:3106–3116. doi: 10.1002/anie.200401744. [DOI] [PubMed] [Google Scholar]
- 33.Song W, Wang Y, Qu J, Madden MM, Lin Q. A Photoinducible 1,3-Dipolar Cycloaddition Reaction for Rapid, Selective Modification of Tetrazole-Containing Proteins. Angew Chem Int Ed. 2008;47:2832–2835. doi: 10.1002/anie.200705805. [DOI] [PubMed] [Google Scholar]
- 34.Song W, Wang Y, Qu J, Lin Q. Selective Functionalization of a Genetically Encoded Alkene-Containing Protein via “Photoclick Chemistry” in Bacterial Cells. J Am Chem Soc. 2008;130:9654–9655. doi: 10.1021/ja803598e. [DOI] [PubMed] [Google Scholar]
- 35.De Araújo AD, Palomo JM, Cramer J, Seitz O, Alexandrov K, Waldmann H. Diels–Alder Ligation of Peptides and Proteins. Chem Eur J. 2006;12:6095–6109. doi: 10.1002/chem.200600148. [DOI] [PubMed] [Google Scholar]
- 36.De Araújo AD, Palomo JM, Cramer J, Köhn M, Schröder H, Wacker R, Niemeyer C, Alexandrov K, Waldmann H. Diels-Alder Ligation and Surface Immobilization of Proteins. Angew Chem Int Ed. 2006;45:296–301. doi: 10.1002/anie.200502266. [DOI] [PubMed] [Google Scholar]
- 37.Liu DS, Tangpeerachaikul A, Selvaraj R, Taylor MT, Fox JM, Ting AY. Diels–Alder Cycloaddition for Fluorophore Targeting to Specific Proteins inside Living Cells. J Am Chem Soc. 2012;134:792–795. doi: 10.1021/ja209325n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blackman ML, Royzen M, Fox JM. Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels-Alder Reactivity. J Am Chem Soc. 2008;130:13518–13519. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schoch J, Wiessler M, Jäschke A. Post-Synthetic Modification of DNA by Inverse-Electron-Demand Diels-Alder Reaction. J Am Chem Soc. 2010;132:8846–8847. doi: 10.1021/ja102871p. [DOI] [PubMed] [Google Scholar]
- 40.Cordes EH, Jencks WP. Nucleophilic Catalysis of Semicarbazone Formation by Anilines. J Am Chem Soc. 1962;84:826–831. [Google Scholar]
- 41.Dirksen A, Yegneswaran S, Dawson PE. Bisaryl Hydrazones as Exchangeable Biocompatible Linkers. Angew Chem Int Ed. 2010:2023–2027. doi: 10.1002/anie.200906756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rashidian M, Song JM, Pricer RE, Distefano MD. Chemoenzymatic Reversible Immobilization and Labeling of Proteins without Prior Purification. J Am Chem Soc. 2012;134:8455–8467. doi: 10.1021/ja211308s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller LW, Cornish VW. Selective chemical labeling of proteins in living cells. Curr Opin Chem Biol. 2005;9:56–61. doi: 10.1016/j.cbpa.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 44.Sunbul M, Yin J. Site specific protein labeling by enzymatic posttranslational modification. Org Biomol Chem. 2009;7:3361. doi: 10.1039/b908687k. [DOI] [PubMed] [Google Scholar]
- 45.Roeser D, Preusser-Kunze A, Schmidt B, Gasow K, Wittmann JG, Dierks T, von Figura K, Rudolph MG. A general binding mechanism for all human sulfatases by the formylglycine-generating enzyme. Proc Natl Acad Sci USA. 2006;103:81–86. doi: 10.1073/pnas.0507592102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rush JS, Bertozzi CR. New Aldehyde Tag Sequences Identified by Screening Formylglycine Generating Enzymes in Vitro and in Vivo. J Am Chem Soc. 2008;130:12240–12241. doi: 10.1021/ja804530w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu P, Shui W, Carlson BL, Hu N, Rabuka D, Lee J, Bertozzi CR. Site-specific chemical modification of recombinant proteins produced in mammalian cells by using the genetically encoded aldehyde tag. Proc Natl Acad Sci. 2009;106:3000–3005. doi: 10.1073/pnas.0807820106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rabuka D, Rush JS, deHart GW, Wu P, Bertozzi CR. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat Protoc. 2012;7:1052–1067. doi: 10.1038/nprot.2012.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bertozzi CR, Kiessling LL. Chemical glycobiology. Science. 2001;291:2357–2364. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]
- 50.Angata T, Varki A. Chemical diversity in the sialic acids and related alpha-keto acids: an evolutionary perspective. Chem Rev. 2002;102:439–469. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]
- 51.Bork K, Horstkorte R, Weidemann W. Increasing the sialylation of therapeutic glycoproteins: the potential of the sialic acid biosynthetic pathway. J Pharm Sci. 2009;98:3499–3508. doi: 10.1002/jps.21684. [DOI] [PubMed] [Google Scholar]
- 52.Anthony RM, Ravetch JV. A novel role for the IgG Fc glycan: the anti-inflammatory activity of sialylated IgG Fcs. J Clin Immunol. 2010;30(Suppl 1):S9–14. doi: 10.1007/s10875-010-9405-6. [DOI] [PubMed] [Google Scholar]
- 53.Yu H, Chokhawala H, Karpel R, Yu H, Wu B, Zhang J, Zhang Y, Jia Q, Chen X. A Multifunctional Pasteurella multocida Sialyltransferase: A Powerful Tool for the Synthesis of Sialoside Libraries. J Am Chem Soc. 2005;127:17618–17619. doi: 10.1021/ja0561690. [DOI] [PubMed] [Google Scholar]
- 54.Yu H, Huang S, Chokhawala H, Sun M, Zheng H, Chen X. Highly Efficient Chemoenzymatic Synthesis of Naturally Occurring and Non-Natural α-2,6-Linked Sialosides: AP. damsela α-2,6-Sialyltransferase with Extremely Flexible Donor–Substrate Specificity. Angew Chem Int Ed. 2006;45:3938–3944. doi: 10.1002/anie.200600572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khedri Z, Muthana MM, Li Y, Muthana SM, Yu H, Cao H, Chen X. Probe sialidase substrate specificity using chemoenzymatically synthesized sialosides containing C9-modified sialic acid. Chem Commun. 2012;48:3357–3359. doi: 10.1039/c2cc17393j. [DOI] [PubMed] [Google Scholar]
- 56.Cane DE, Walsh CT, Khosla C. Harnessing the biosynthetic code: combinations, permutations, and mutations. Science. 1998;282:63–68. doi: 10.1126/science.282.5386.63. [DOI] [PubMed] [Google Scholar]
- 57.Marahiel MA, Stachelhaus T, Mootz HD. Modular Peptide Synthetases Involved in Nonribosomal Peptide Synthesis. Chem Rev. 1997;97:2651–2674. doi: 10.1021/cr960029e. [DOI] [PubMed] [Google Scholar]
- 58.Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 59.Wakil SJ, Stoops JK, Joshi VC. Fatty acid synthesis and its regulation. Annu Rev Biochem. 1983;52:537–579. doi: 10.1146/annurev.bi.52.070183.002541. [DOI] [PubMed] [Google Scholar]
- 60.Walsh CT, Gehring AM, Weinreb PH, Quadri LE, Flugel RS. Post-translational modification of polyketide and nonribosomal peptide synthases. Curr Opin Chem Biol. 1997;1:309–315. doi: 10.1016/s1367-5931(97)80067-1. [DOI] [PubMed] [Google Scholar]
- 61.Yin J, Liu F, Li X, Walsh CT. Labeling Proteins with Small Molecules by Site-Specific Posttranslational Modification. J Am Chem Soc. 2004;126:7754–7755. doi: 10.1021/ja047749k. [DOI] [PubMed] [Google Scholar]
- 62.Zhou Z, Cironi P, Lin AJ, Xu Y, Hrvatin S, Golan DE, Silver PA, Walsh CT, Yin J. Genetically Encoded Short Peptide Tags for Orthogonal Protein Labeling by Sfp and AcpS Phosphopantetheinyl Transferases. ACS Chem Biol. 2007;2:337–346. doi: 10.1021/cb700054k. [DOI] [PubMed] [Google Scholar]
- 63.Clarke KM, Mercer AC, La Clair JJ, Burkart MD. In Vivo Reporter Labeling of Proteins via Metabolic Delivery of Coenzyme A Analogues. J Am Chem Soc. 2005;127:11234–11235. doi: 10.1021/ja052911k. [DOI] [PubMed] [Google Scholar]
- 64.Worthington AS, Burkart MD. One-pot chemo-enzymatic synthesis of reporter-modified proteins. Org Biomol Chem. 2006;4:44. doi: 10.1039/b512735a. [DOI] [PubMed] [Google Scholar]
- 65.Kosa NM, Haushalter RW, Smith AR, Burkart MD. Reversible labeling of native and fusion-protein motifs. Nat Methods. 2012;9:981–984. doi: 10.1038/nmeth.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.George N, Pick H, Vogel H, Johnsson N, Johnsson K. Specific Labeling of Cell Surface Proteins with Chemically Diverse Compounds. J Am Chem Soc. 2004;126:8896–8897. doi: 10.1021/ja048396s. [DOI] [PubMed] [Google Scholar]
- 67.Cappellaro C, Baldermann C, Rachel R, Tanner W. Mating type-specific cell-cell recognition of Saccharomyces cerevisiae: cell wall attachment and active sites of a- and alpha-agglutinin. EMBO J. 1994;13:4737–4744. doi: 10.1002/j.1460-2075.1994.tb06799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yin J, Straight PD, McLoughlin SM, Zhou Z, Lin AJ, Golan DE, Kelleher NL, Kolter R, Walsh CT. Genetically encoded short peptide tag for versatile protein labeling by Sfp phosphopantetheinyl transferase. Proc Natl Acad Sci. 2005;102:15815–15820. doi: 10.1073/pnas.0507705102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wong LS, Thirlway J, Micklefield J. Direct Site-Selective Covalent Protein Immobilization Catalyzed by a Phosphopantetheinyl Transferase. J Am Chem Soc. 2008;130:12456–12464. doi: 10.1021/ja8030278. [DOI] [PubMed] [Google Scholar]
- 70.Vocadlo DJ, Hang HC, Kim E, Hanover JA, Bertozzi CR. A chemical approach for identifying O-GlcNAc-modified proteins in cells. Proc Natl Acad Sci. 2003;100:9116–9121. doi: 10.1073/pnas.1632821100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saxon E, Bertozzi CR. Cell surface engineering by a modified Staudinger reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 72.Khidekel N, Arndt S, Lamarre-Vincent N, Lippert A, Poulin-Kerstien KG, Ramakrishnan B, Qasba PK, Hsieh-Wilson LC. A Chemoenzymatic Approach toward the Rapid and Sensitive Detection of O-GlcNAc Posttranslational Modifications. J Am Chem Soc. 2003;125:16162–16163. doi: 10.1021/ja038545r. [DOI] [PubMed] [Google Scholar]
- 73.Clark PM, Dweck JF, Mason DE, Hart CR, Buck SB, Peters EC, Agnew BJ, Hsieh-Wilson LC. Direct In-Gel Fluorescence Detection and Cellular Imaging of O-GlcNAc-Modified Proteins. J Am Chem Soc. 2008;130:11576–11577. doi: 10.1021/ja8030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC. Exploring the O-GlcNAc proteome: Direct identification of O-GlcNAc-modified proteins from the brain. Proc Natl Acad Sci. 2004;101:13132–13137. doi: 10.1073/pnas.0403471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khidekel N, Ficarro SB, Clark PM, Bryan MC, Swaney DL, Rexach JE, Sun YE, Coon JJ, Peters EC, Hsieh-Wilson LC. Probing the dynamics of O-GlcNAc glycosylation in the brain using quantitative proteomics. Nat Chem Biol. 2007;3:339–348. doi: 10.1038/nchembio881. [DOI] [PubMed] [Google Scholar]
- 76.Mazmanian SK, Liu G, Ton-That H, Schneewind O. Staphylococcus aureus Sortase, an Enzyme that Anchors Surface Proteins to the Cell Wall. Science. 1999;285:760–763. doi: 10.1126/science.285.5428.760. [DOI] [PubMed] [Google Scholar]
- 77.Tsukiji S, Nagamune T. Sortase-Mediated Ligation: A Gift from Gram-Positive Bacteria to Protein Engineering. ChemBioChem. 2009;10:787–798. doi: 10.1002/cbic.200800724. [DOI] [PubMed] [Google Scholar]
- 78.Popp MW, Antos JM, Grotenbreg GM, Spooner E, Ploegh HL. Sortagging: a versatile method for protein labeling. Nat Chem Biol. 2007;3:707–708. doi: 10.1038/nchembio.2007.31. [DOI] [PubMed] [Google Scholar]
- 79.Tanaka T, Yamamoto T, Tsukiji S, Nagamune T. Site-Specific Protein Modification on Living Cells Catalyzed by Sortase. ChemBioChem. 2008;9:802–807. doi: 10.1002/cbic.200700614. [DOI] [PubMed] [Google Scholar]
- 80.Strijbis K, Spooner E, Ploegh HL. Protein Ligation in Living Cells Using Sortase. Traffic. 2012;13:780–789. doi: 10.1111/j.1600-0854.2012.01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Möhlmann S, Mahlert C, Greven S, Scholz P, Harrenga A. In vitro Sortagging of an Antibody Fab Fragment: Overcoming Unproductive Reactions of Sortase with Water and Lysine Side Chains. ChemBioChem. 2011;12:1774–1780. doi: 10.1002/cbic.201100002. [DOI] [PubMed] [Google Scholar]
- 82.Popp MW, Dougan SK, Chuang TY, Spooner E, Ploegh HL. Sortase-catalyzed transformations that improve the properties of cytokines. Proc Natl Acad Sci. 2011;108:3169–3174. doi: 10.1073/pnas.1016863108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Antos JM, Miller GM, Grotenbreg GM, Ploegh HL. Lipid Modification of Proteins through Sortase-Catalyzed Transpeptidation. J Am Chem Soc. 2008;130:16338–16343. doi: 10.1021/ja806779e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guo X, Wang Q, Swarts BM, Guo Z. Sortase-Catalyzed Peptide-Glycosylphosphatidylinositol Analogue Ligation. J Am Chem Soc. 2009;131:9878–9879. doi: 10.1021/ja903231v. [DOI] [PubMed] [Google Scholar]
- 85.Wu Z, Guo X, Guo Z. Sortase A-catalyzed peptide cyclization for the synthesis of macrocyclic peptides and glycopeptides. Chem Commun. 2011;47:9218. doi: 10.1039/c1cc13322e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sinisi A, Popp MW-L, Antos JM, Pansegrau W, Savino S, Nissum M, Rappuoli R, Ploegh HL, Buti L. Development of an Influenza virus Protein Array Using Sortagging Technology. Bioconjugate Chem. 2012;23:1119–1126. doi: 10.1021/bc200577u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Witte MD, Cragnolini JJ, Dougan SK, Yoder NC, Popp MW, Ploegh HL. Preparation of unnatural N-to-N and C-to-C protein fusions. Proc Natl Acad Sci. 2012;109:11993–11998. doi: 10.1073/pnas.1205427109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Williamson DJ, Fascione MA, Webb ME, Turnbull WB. Efficient N-Terminal Labeling of Proteins by Use of Sortase. Angew Chem Int Ed. 2012;124:9511–9514. doi: 10.1002/anie.201204538. [DOI] [PubMed] [Google Scholar]
- 89.Claessen JHL, Witte MD, Yoder NC, Zhu AY, Spooner E, Ploegh HL. Catch-and-Release Probes Applied to Semi-Intact Cells Reveal Ubiquitin-Specific Protease Expression in Chlamydia trachomatis Infection. ChemBioChem. 2013;14:343–352. doi: 10.1002/cbic.201200701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jeger S, Zimmermann K, Blanc A, Grünberg J, Honer M, Hunziker P, Struthers H, Schibli R. Site-Specific and Stoichiometric Modification of Antibodies by Bacterial Transglutaminase. Angew Chem Int Ed. 2010;49:9995–9997. doi: 10.1002/anie.201004243. [DOI] [PubMed] [Google Scholar]
- 91.Mero A, Spolaore B, Veronese FM, Fontana A. Transglutaminase-Mediated PEGylation of Proteins: Direct Identification of the Sites of Protein Modification by Mass Spectrometry using a Novel Monodisperse PEG. Bioconjugate Chem. 2009;20:384–389. doi: 10.1021/bc800427n. [DOI] [PubMed] [Google Scholar]
- 92.Abe H, Goto M, Kamiya N. Protein Lipidation Catalyzed by Microbial Transglutaminase. Chem Eur J. 2011;17:14004–14008. doi: 10.1002/chem.201102121. [DOI] [PubMed] [Google Scholar]
- 93.Tanaka T, Kamiya N, Nagamune T. N-terminal glycine-specific protein conjugation catalyzed by microbial transglutaminase. FEBS Lett. 2005;579:2092–2096. doi: 10.1016/j.febslet.2005.02.064. [DOI] [PubMed] [Google Scholar]
- 94.Tominaga J, Kamiya N, Doi S, Ichinose H, Maruyama T, Goto M. Design of a Specific Peptide Tag that Affords Covalent and Site-Specific Enzyme Immobilization Catalyzed by Microbial Transglutaminase. Biomacromolecules. 2005;6:2299–2304. doi: 10.1021/bm050193o. [DOI] [PubMed] [Google Scholar]
- 95.Lin CW, Ting AY. Transglutaminase-Catalyzed Site-Specific Conjugation of Small-Molecule Probes to Proteins in Vitro and on the Surface of Living Cells. J Am Chem Soc. 2006;128:4542–4543. doi: 10.1021/ja0604111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Duckworth BP, Xu J, Taton TA, Guo A, Distefano MD. Site-Specific, Covalent Attachment of Proteins to a Solid Surface. Bioconjugate Chem. 2006;17:967–974. doi: 10.1021/bc060125e. [DOI] [PubMed] [Google Scholar]
- 97.Kho Y. A tagging-via-substrate technology for detection and proteomics of farnesylated proteins. Proc Natl Acad Sci USA. 2004;101:12479–12484. doi: 10.1073/pnas.0403413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Duckworth BP, Zhang Z, Hosokawa A, Distefano MD. Selective Labeling of Proteins by Using Protein Farnesyltransferase. ChemBioChem. 2007;8:98–105. doi: 10.1002/cbic.200600340. [DOI] [PubMed] [Google Scholar]
- 99.Rashidian M, Dozier JK, Lenevich S, Distefano MD. Selective labeling of polypeptides using protein farnesyltransferase via rapid oxime ligation. Chem Commun. 2010;46:8998–9000. doi: 10.1039/c0cc03305g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nguyen UTT, Guo Z, Delon C, Wu Y, Deraeve C, Fränzel B, Bon RS, Blankenfeldt W, Goody RS, Waldmann H, Wolters D, Alexandrov K. Analysis of the eukaryotic prenylome by isoprenoid affinity tagging. Nat Chem Biol. 2009;5:227–235. doi: 10.1038/nchembio.149. [DOI] [PubMed] [Google Scholar]
- 101.Subramanian T, Pais JE, Liu S, Troutman JM, Suzuki Y, Leela Subramanian K, Fierke CA, Andres DA, Spielmann HP. Farnesyl diphosphate analogues with aryl moieties are efficient alternate substrates for protein farnesyltransferase. Biochemistry. 2012;51:8307–8319. doi: 10.1021/bi3011362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Labadie GR, Viswanathan R, Poulter CD. Farnesyl Diphosphate Analogues with ω-Bioorthogonal Azide and Alkyne Functional Groups for Protein Farnesyl Transferase-Catalyzed Ligation Reactions. J Org Chem. 2007;72:9291–9297. doi: 10.1021/jo7017747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Weinrich D, Lin PC, Jonkheijm P, Nguyen UTT, Schröder H, Niemeyer CM, Alexandrov K, Goody R, Waldmann H. Oriented Immobilization of Farnesylated Proteins by the Thiol-Ene Reaction. Angew Chem Int Ed. 2010;49:1252–1257. doi: 10.1002/anie.200906190. [DOI] [PubMed] [Google Scholar]
- 104.Nguyen UTT, Cramer J, Gomis J, Reents R, Gutierrez-Rodriguez M, Goody RS, Alexandrov K, Waldmann H. Exploiting the Substrate Tolerance of Farnesyltransferase for Site-Selective Protein Derivatization. ChemBioChem. 2007;8:408–423. doi: 10.1002/cbic.200600440. [DOI] [PubMed] [Google Scholar]
- 105.Rose MW, Xu J, Kale TA, O’Doherty G, Barany G, Distefano MD. Enzymatic incorporation of orthogonally reactive prenylazide groups into peptides using geranylazide diphosphate via protein farnesyltransferase: implications for selective protein labeling. Biopolymers. 2005;80:164–171. doi: 10.1002/bip.20239. [DOI] [PubMed] [Google Scholar]
- 106.Rose MW, Rose ND, Boggs J, Lenevich S, Xu J, Barany G, Distefano MD. Evaluation of geranylazide and farnesylazide diphosphate for incorporation of prenylazides into a CAAX box-containing peptide using protein farnesyltransferase. J Pept Res. 2005;65:529–537. doi: 10.1111/j.1399-3011.2005.00261.x. [DOI] [PubMed] [Google Scholar]
- 107.Xu J, DeGraw AJ, Duckworth BP, Lenevich S, Tann CM, Jenson EC, Gruber SJ, Barany G, Distefano MD. Synthesis and Reactivity of 6,7-dihydrogeranylazides: Reagents for Primary Azide Incorporation into Peptides and Subsequent Staudinger Ligation. Chem Biol Drug Des. 2006;68:85–96. doi: 10.1111/j.1747-0285.2006.00420.x. [DOI] [PubMed] [Google Scholar]
- 108.Duckworth BP, Chen Y, Wollack JW, Sham Y, Mueller JD, Taton TA, Distefano MD. A Universal Method for the Preparation of Covalent Protein–DNA Conjugates for Use in Creating Protein Nanostructures. Angew Chem. 2007;119:8975–8978. doi: 10.1002/anie.200701942. [DOI] [PubMed] [Google Scholar]
- 109.Khatwani SL, Kang JS, Mullen DG, Hast MA, Beese LS, Distefano MD, Taton TA. Covalent protein–oligonucleotide conjugates by copper-free click reaction. Bioorg Med Chem. 2012;20:4532–4539. doi: 10.1016/j.bmc.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Speers AE, Cravatt BF. Profiling Enzyme Activities In Vivo Using Click Chemistry Methods. Chem Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 111.Hosokawa A, Wollack JW, Zhang Z, Chen L, Barany G, Distefano MD. Evaluation of an Alkyne-containing Analogue of Farnesyl Diphosphate as a Dual Substrate for Protein-prenyltransferases. Int J Pept Res Ther. 2007;13:345–354. [Google Scholar]
- 112.Wollack JW, Silverman JM, Petzold CJ, Mougous JD, Distefano MD. A Minimalist Substrate for Enzymatic Peptide and Protein Conjugation. ChemBioChem. 2009;10:2934–2943. doi: 10.1002/cbic.200900566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mahmoodi MM, Rashidian M, Dozier JK, Distefano MD. Chemoenzymatic reversible immobilization and labeling of proteins from crude cellular extract. Curr Protoc Chem Biol. 2013:89–109. doi: 10.1002/9780470559277.ch120247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dirksen A, Hackeng TM, Dawson PE. Nucleophilic Catalysis of Oxime Ligation. Angew Chem Int Ed. 2006;45:7581–7584. doi: 10.1002/anie.200602877. [DOI] [PubMed] [Google Scholar]
- 115.Rashidian M, Mahmoodi MM, Shah R, Dozier JK, Wagner CR, Distefano MD. A highly efficient catalyst for oxime ligation and hydrazone-oxime exchange suitable for bioconjugation. Bioconjugate Chem. 2013;24:333–342. doi: 10.1021/bc3004167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Crisalli P, Kool ET. Water-Soluble Organocatalysts for Hydrazone and Oxime Formation. J Org Chem. 2013;78:1184–1189. doi: 10.1021/jo302746p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Algar WR, Prasuhn DE, Stewart MH, Jennings TL, Blanco-Canosa JB, Dawson PE, Medintz IL. The Controlled Display of Biomolecules on Nanoparticles: A Challenge Suited to Bioorthogonal Chemistry. Bioconjugate Chem. 2011;22:825–858. doi: 10.1021/bc200065z. [DOI] [PubMed] [Google Scholar]
- 118.Howarth M, Takao K, hayashi Y, Ting AY. Targeting quantum dots to surface proteins in living cells with biotin ligase. Proc Natl Acad Sci. 2005;102:7583–7588. doi: 10.1073/pnas.0503125102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Howarth M, Liu W, Puthenveetil S, Zheng Y, Marshall LF, Schmidt MM, Wittrup KD, Bawendi MG, Ting AY. Monovalent, reduced-size quantum dots for imaging receptors on living cells. Nat Meth. 2008;5:397–399. doi: 10.1038/nmeth.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Slavoff SA, Chen I, Choi YA, Ting AY. Expanding the Substrate Tolerance of Biotin Ligase through Exploration of Enzymes from Diverse Species. J Am Chem Soc. 2008;130:1160–1162. doi: 10.1021/ja076655i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fernández-Suárez M, Chen TS, Ting AY. Protein-Protein Interaction Detection in Vitro and in Cells by Proximity Biotinylation. J Am Chem Soc. 2008;130:9251–9253. doi: 10.1021/ja801445p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sueda S, Yoneda S, Hayashi H. Site-Specific Labeling of Proteins by Using Biotin Protein Ligase Conjugated with Fluorophores. ChemBioChem. 2011;12:1367–1375. doi: 10.1002/cbic.201000738. [DOI] [PubMed] [Google Scholar]
- 123.Roux KJ, Kim DI, Raida M, Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol. 2012;196:801–810. doi: 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fernández-Suárez M, Baruah H, Martínez-Hernández L, Xie KT, Baskin JM, Bertozzi CR, Ting AY. Redirecting lipoic acid ligase for cell surface protein labeling with small-molecule probes. Nat Biotechnol. 2007;25:1483–1487. doi: 10.1038/nbt1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cohen JD, Thompson S, Ting AY. Structure-Guided Engineering of a Pacific Blue Fluorophore Ligase for Specific Protein Imaging in Living Cells. Biochemistry. 2011;50:8221–8225. doi: 10.1021/bi201037r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Uttamapinant C, White KA, Baruah H, Thompson S, Fernandez-Suarez M, Puthenveetil S, Ting AY. From the Cover: A fluorophore ligase for site-specific protein labeling inside living cells. Proc Natl Acad Sci. 2010;107:10914–10919. doi: 10.1073/pnas.0914067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Slavoff SA, Liu DS, Cohen JD, Ting AY. Imaging Protein–Protein Interactions inside Living Cells via Interaction-Dependent Fluorophore Ligation. J Am Chem Soc. 2011;133:19769–19776. doi: 10.1021/ja206435e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cohen JD, Zou P, Ting AY. Site-Specific Protein Modification Using Lipoic Acid Ligase and Bis-Aryl Hydrazone Formation. ChemBioChem. 2012;13:888–894. doi: 10.1002/cbic.201100764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Uttamapinant C, Tangpeerachaikul A, Grecian S, Clarke S, Singh U, Slade P, Gee KR, Ting AY. Fast, cell-compatible click chemistry with copper-chelating azides for biomolecular labeling. Angew Chem Int Ed. 2012;51:5852–5856. doi: 10.1002/anie.201108181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yao JZ, Uttamapinant C, Poloukhtine A, Baskin JM, Codelli JA, Sletten EM, Bertozzi CR, Popik VV, Ting AY. Fluorophore targeting to cellular proteins via enzyme-mediated azide ligation and strain-promoted cycloaddition. J Am Chem Soc. 2012;134:3720–3728. doi: 10.1021/ja208090p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Farazi TA, Waksman G, Gordon JI. The biology and enzymology of protein N-myristoylation. J Biol Chem. 2001;276:39501–39504. doi: 10.1074/jbc.R100042200. [DOI] [PubMed] [Google Scholar]
- 132.Towler DA, Gordon JI, Adams SP, Glaser L. The biology and enzymology of eukaryotic protein acylation. Annu Rev Biochem. 1988;57:69–99. doi: 10.1146/annurev.bi.57.070188.000441. [DOI] [PubMed] [Google Scholar]
- 133.Price HP, Menon MR, Panethymitaki C, Goulding D, McKean PG, Smith DF. Myristoyl-CoA:protein N-myristoyltransferase, an essential enzyme and potential drug target in kinetoplastid parasites. J Biol Chem. 2003;278:7206–7214. doi: 10.1074/jbc.M211391200. [DOI] [PubMed] [Google Scholar]
- 134.Martinez A, Traverso JA, Valot B, Ferro M, Espagne C, Ephritikhine G, Zivy M, Giglione C, Meinnel T. Extent of N-terminal modifications in cytosolic proteins from eukaryotes. Proteomics. 2008;8:2809–2831. doi: 10.1002/pmic.200701191. [DOI] [PubMed] [Google Scholar]
- 135.Wright MH, Heal WP, Mann DJ, Tate EW. Protein myristoylation in health and disease. J Chem Biol. 2010;3:19–35. doi: 10.1007/s12154-009-0032-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Heal WP, Wickramasinghe SR, Bowyer PW, Holder AA, Smith DF, Leatherbarrow RJ, Tate EW. Site-specific N-terminal labelling of proteins in vitro and in vivo using N-myristoyl transferase and bioorthogonal ligation chemistry. Chem Comm. 2008:480. doi: 10.1039/b716115h. [DOI] [PubMed] [Google Scholar]
- 137.Heal WP, Wickramasinghe SR, Leatherbarrow RJ, Tate EW. N-Myristoyl transferase-mediated protein labelling in vivo. Org Biomol Chem. 2008;6:2308. doi: 10.1039/b803258k. [DOI] [PubMed] [Google Scholar]
- 138.Heal WP, Wright MH, Thinon E, Tate EW. Multifunctional protein labeling via enzymatic N-terminal tagging and elaboration by click chemistry. Nat Protoc. 2012;7:105–117. doi: 10.1038/nprot.2011.425. [DOI] [PubMed] [Google Scholar]