

Abstract
The development of a general and practical zinc-catalyzed enantioselective alkyne addition methodology is reported. Commercially available ProPhenol ligand (1) has facilitated the addition of a wide range of zinc alkynylides to aryl, aliphatic and α,β-unsaturated aldehydes in high yield and enantioselectivity. New insights into the mechanism of this reaction have resulted in a significant reduction in reagent stoichiometry, enabling the use of precious alkynes and avoiding the use of excess dimethylzinc. The enantioenriched propargylic alcohols from this reaction serve as versatile synthetic intermediates and have enabled efficient syntheses of several complex natural products.
Keywords: asymmetric catalysis, nucleophilic addition, alkynes, alkynylide, total synthesis
Introduction
The design and development of the ProPhenol ligand, 1,[1] as an enantioselective catalyst for base-mediated nucleophilic addition has led to the discovery of a number of highly efficient transformations.[2] The combination of the ProPhenol ligand and a dialkylzinc reagent has been shown to catalyze asymmetric Mannich and Henry reactions,[3],[4] the desymmetrization of meso 1,3-diols,[5] and the direct aldol reaction.[6] The success of this catalyst system with stabilized nucleophiles, such as enolates and ntironates, prompted the investigation of zinc alkynylides and their addition to aldehydes (Scheme 1). Herein, we provide a full account of the development of a practical and general methodology for zinc-catalyzed enantioselective alkynylation of aldehydes using the commercially available ProPhenol ligand, 1.[7]
Scheme 1.
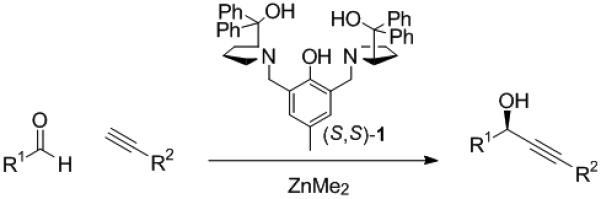
ProPhenol-Catalyzed Alkyne Addition.
Propargyl alcohols serve as robust and versatile intermediates in the synthesis of fine chemicals, natural products and therapeutic agents (Figure 1).[8] The broad synthetic utility of this motif lies in the bifunctional reactivity of alkynes. The terminal alkyne can act as a nucleophile via deprotonation and subsequent alkylation or metal-catalyzed cross coupling. Conversely, the latent electrophilicity of alkynes can be chemoselectively activated by complexation with a transition metal. Further, the reactivity of propargyl alcohols toward SN2 displacement extends the activation to the propargylic position as well. This synthetic versatility makes the catalytic enantioselective preparation of propargylic alcohols especially valuable. Three general approaches have been utilized for the synthesis of secondary propargyl alcohols: enantioselective ynone reduction (A),[9] asymmetric alkyne addition to aldehydes (B),[10] and ynal alkylation (C).[11] Although a number of catalysts have been developed to facilitate both ynone reduction and ynal alkylation, the application of these methods is limited by the propensity of these alkynes to decompose, isomerize, and act as Michael acceptors. The addition of a terminal acetylene to an aldehyde avoids these problems and provides a convergent approach to the desired propargyl alcohol.
Figure 1.
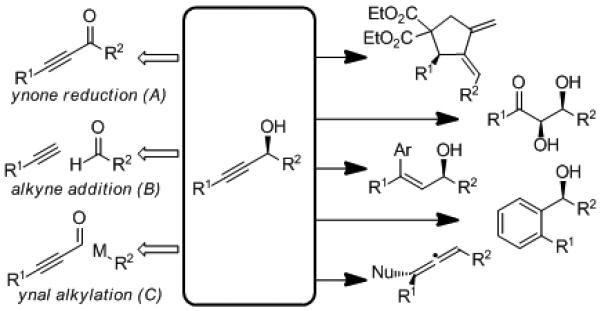
Formation and Reactivity of Propargylic Alcohols.[12]
The mild reactivity of organozinc reagents has enabled the enantioselective addition of alkyl, vinyl, and alkynyl groups to a variety of carbonyl compounds with excellent functional group tolerance.[13] The asymmetric addition of alkynylzinc nucleophiles to aldehydes has recently generated a large amount of interest in the chemical community.[14] Early reports by Carreira demonstrated that stoichiometric (+)-N-methyl ephedrine, Zn(OTf)2, and triethylamine could be used to achieve alkyne metalation and addition to aliphatic aldehydes under particularly mild conditions.[15] High enantioselectivity and yield were obtained with a variety of alkynes, although aryl and α,β-unsaturated aldehydes typically gave lower yields. The initial conditions requiring stoichiometric zinc and ephedrine were ultimately rendered catalytic by increasing the reaction temperature to 60 °C.[15f] Pu and Chan independently reported the use of (S)-BINOL, in conjunction with Ti(OiPr)4 and either Et2Zn or Me2Zn to facilitate nucleophilic addition of alkynes to aldehydes.[16],[17] These conditions require an excess of alkyne and dialkylzinc but ultimately provide good yield and enantioselectivity with a range of substrates. A number of other chiral zinc catalysts have also been reported to enable the enantioselective addition of alkynes to aldehydes.[18] Efficient asymmetric alkyne addition often requires the use of relatively high catalyst loadings and an excess of alkyne and dialkylzinc reagents.[19] Our aim was to develop an efficient chiral catalyst system capable of adding functionalized alkynes to a wide range of aldehydes while minimizing the use of excess reagents and stoichiometric additives, improving the atom economy of this transformation.[20] This would ultimately enable facile access to chiral propargyl alcohols and entry into alkyne-based strategies in the synthesis of natural products.
Results and Discussion
Initial optimization
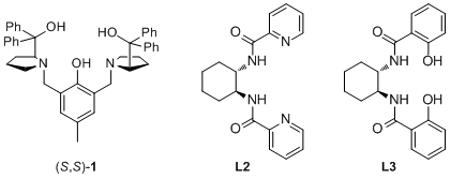
Optimization of the enantioselective addition of phenyl- and TMS-acetylene to p-anisaldehyde (2) commenced with the screening of several C2 symmetric ligands (S,S)-1, L2 and L3 designed in our group (Table 1). Stoichiometric zinc alkynylide was required for adequate enantioselectivity, and all optimization was initially carried out using nearly three equivalents of dialkylzinc and alkyne. Further experiments to improve the atom economy of this alkynylation methodology will be discussed below. Ligand screening revealed that the ProPhenol ligand, (S,S)-1, provided the best results in terms of both yield and enantioselectivity, with the desired propargyl alcohol being isolated in 78% yield and 80% ee (entry 1). Ligands L2 and L3, resembling a Salen ligand and the backbone of the Trost phosphine ligands for Pd-catalyzed asymmetric allylic alkylation, also provided the desired product, albeit with a lower enantioselectivity of 35% and −66% ee, respectively (entries 2–3). These results are in contrast to Cozzi’s asymmetric alkyne addition to ketones, which utilizes a similar Salen ligand to obtain excellent results.[21] Enantiomeric induction from (S,S)-1 was found to be a robust process and provided excellent selectivity for the (R)-propargylic alcohol 3a, across a range of temperatures and catalyst loadings (entries 4–7).[22] Consequently, the majority of optimization experiments were focused primarily on improving reactivity and catalyst turnover. Reducing the catalyst loading to 10 mol % and increasing the reaction time to 48 h produced the desired product in 77% yield and 83% ee (entry 5). These results are similar to those obtained in entry 1 with 20 mol % (S,S)-1. In an attempt to obtain even better enantioselectivity the alkyne addition was performed at −20 °C with both 5 and 10 mol % (S,S)-1 (entries 6–7). These reactions provided similar levels of enantioselectivity but resulted in a substantial decrease in reactivity. TMS-acetylene was found to be significantly less reactive in ProPhenol-catalyzed alkyne additions relative to phenyl acetylene. However, by increasing the reaction concentration, improved reactivity could be obtained with TMS-acetylene to ultimately provide the desired product in 74% yield and 85% ee (entry 10). The optimal alkyne concentration was found to be 0.38 M and a further increase in concentration, to 0.69 M, resulted in decreased enantioselectivity (entry 11). At higher reaction concentrations (ca. 2 M) the ligand-free background reaction proceeds readily, and is presumably the cause of the slight decrease in enantioselectivity.[23] Unfortunately, decreasing the catalyst loading further (< 10 mol %) provided lower yield and significantly lower ee (entries 13–14). The optimization of reaction temperature, time, concentration and catalyst loading has enabled the addition of either TMS-acetylene or phenylacetylene to p-anisaldehyde in good yield (>70%) and ee (>70%) with just 10 mol % catalyst loading. Interestingly, similar results were generally observed for dimethyl- and diethylzinc – however, others have noted that alkyl transfer from dimethylzinc is significantly slower than from diethylzinc, and therefore dimethylzinc was chosen to avoid potential alkyl addition side products.[24] Utilizing the optimized conditions outlined in entries 10 and 12, we set out to investigate the scope of this reaction.
Table 1.
Initial Optimization.[a]
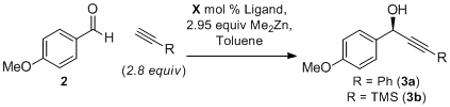
| R | conc.[b] | temp. | time | X | yield | ee[c] | |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Ligand Screening | |||||||
|
| |||||||
| 1 | Ph | 0.18 | rt | 22 h | 20 (1) | 78% | 80% |
| 2 | Ph | 0.18 | 0 °C | 16 h | 20 (L2) | 26% | 35% |
| 3 | Ph | 0.18 | 0 °C | 16 h | 20 (L3) | 36% | −66% |
|
| |||||||
| Time, Temperature and Catalyst Loading, X = (S,S)-1 | |||||||
|
| |||||||
| 4 | Ph | 0.18 | rt | 72 h | 20 | 95% | 79% |
| 5 | Ph | 0.18 | rt | 48 h | 10 | 77% | 83% |
| 6 | Ph | 0.18 | −20 °C | 45 h | 10 | 60% | 77% |
| 7 | Ph | 0.18 | −20 °C | 45 h | 5 | 32% | 72% |
|
| |||||||
| Reaction Concentration, X = (S,S)-1 | |||||||
|
| |||||||
| 8 | TMS | 0.18 | 3 °C | 21 h | 10 | 35% | 85% |
| 9 | TMS | 0.26 | 3 °C | 21 h | 10 | 50% | 85% |
| 10 | TMS | 0.38 | 3 °C | 24 h | 10 | 74% | 85% |
| 11 | TMS | 0.69 | 3 °C | 21 h | 10 | 87% | 75% |
|
| |||||||
| Catalyst Loading at Higher Concentration, X = (S,S)-1 | |||||||
|
| |||||||
| 12 | Ph | 0.38 | 3 °C | 24 h | 10 | 86% | 74% |
| 13 | Ph | 0.38 | 3 °C | 24 h | 5 | 73% | 58% |
| 14 | Ph | 0.38 | 3 °C | 21 h | 2.5 | 68% | 46% |
Reactions run on a 0.325 mmol scale with 2.7 or 2.8 equiv alkyne and 2.6 or 2.95 equivalents of dimethylzinc respectively. Isolated yields are reported.
Reaction concentration (in molarity) is reported with respect to the alkyne and includes the toluene added as part of the dimethylzinc solution.
Enantiomeric excess determined by chiral HPLC analysis.
Alkynylation of Aryl Aldehydes
A variety of aryl aldehydes underwent efficient alkyne addition using the previously optimized conditions (Table 2). High yields and enantioselectivies were obtained in additions to benzaldehydes containing both electron-donating and electron-withdrawing substituents. Substitution on each aromatic carbon was tolerated and particularly good results were obtained with ortho-substituted benzaldehydes. However, the steric encumbrance of 2,6-dimethylbenzaldehyde resulted in decreased yield and ee (entry 20) although 2,6-dimethoxybenzaldehyde performed very well (entry 16). Good functional group tolerance was observed, with only N-(4-formylphenyl)-acetamide providing poor results (entry 7). Good results were obtained with a variety of alkynes and only small variations in yield and enantioselectivity were observed.
Table 2.
Alkynylation of Aryl Aldehydes.

| entry | R1 | R2 | yield[a] | ee[b] |
|---|---|---|---|---|
|
| ||||
| 1 | H | Ph | 95% | 81% |
| 2 | 2-NO2 | Ph | 84% | 92% |
| 3 | 3-NO2 | Ph | 91% | 68% |
| 4 | 4-NO2 | Ph | 78% | 83% |
| 5[c][d] | 4-F | Ph | 77% | 85% |
| 6[c][d] | 4-Cl | Ph | 98% | 83% |
| 7 | 4-NHAc | Ph | trace | - |
| 8 | 2-furyl | TMS | 81% | 84% |
| 9[c] | 2-furyl | Ph | 90% | 85% |
| 10 | C4H4 (2-naphth) | Ph | 89% | 75% |
| 11[c][d] | 2-MeO | Ph | 100% | 86% |
| 12 | 2-MeO | −CH2OMe | 86% | 84% |
| 13[c][d] | 3-MeO | Ph | 86% | 76% |
| 14 | 4-MeO | TMS | 74% | 85% |
| 15[c] | 4-MeO | Ph | 86% | 74% |
| 16 | 2,6-(MeO)2 | Ph | 87% | 99% |
| 17 | 2,6-(MeO)2 | TMS | 79% | 97% |
| 18 | 3,5-(MeO)2 | −CH2CH(OEt)2 | 90% | 82% |
| 19 | 3,5-(MeO)2 | −CH2CH2OTBS | 82% | 87% |
| 20[c] | 2,6-(Me)2 | Ph | 27% | 35% |
| 21 | 2,4-(MeO2-3-Me | Ph | 87% | 92% |
Isolated yield. Reactions performed on a 0.325 mmol scale.
Enantiomeric excess determined by chiral HPLC analysis.
Reaction performed with 2.5 equiv alkyne and 2.5 equiv Me2Zn.
Reaction run at a concentration of 0.2 M with respect to alkyne.
Alkynylation of α,β-unsaturated aldehydes
Alkyne addition to α,β-unsaturated aldehydes provides a particularly valuable extension of substrate scope. The resulting alkenyl alkynyl carbinols contain three orthogonal functional groups primed for further synthetic manipulation. ProPhenol-catalyzed addition of TMS-acetylene to a range of α,β-unsaturated aldehydes proceeded in excellent yield and enantioselectivity (Table 3). The substitution of the α,β-unsaturated aldehyde has a significant effect on the enantioselectivity of alkyne addition. (E)-Cinnamaldehyde provides the desired propargyl alcohol in 91% ee (entry 1). However, replacement of the phenyl substituent with a methyl or hexyl group produces the desired product in less than 40% ee (entries 2–3, but also vide infra Table 6, entry 2) which was restored to over 90% by addition of an α-bromo group (vide infra). The difference between these β-substituents seems to result primarily from steric interactions. Following this notion, the incorporation of an isopropyl group at the β-position restored the excellent enantioselectivity (entry 4). Interestingly, the absence of substitution at the β-position also led to high enantioselectivity (entry 5, 8). A number of functionalized substrates gave good results, including: α-bromoenals (entries 10–11), β-formyl propionates (entry 12), propiolaldehyde diethyl acetal (entries 14–15) and aliphatic alkynes (entry 16).
Table 3.
Alkynylation of α,β-Unsaturated Aldehydes.

| entry | R1 | R2 | R3 | R4 | yield[a] | ee[b] |
|---|---|---|---|---|---|---|
| 1 | H | Ph | H | TMS | 89% | 91% |
| 2[c] | H | Me | H | TMS | 89% | 39% |
| 3 | H | −C6H13 | H | TMS | 90% | 36% |
| 4 | H | iPr | H | TMS | 100% | 94% |
| 5 | Me | H | H | TMS | 74% | 91% |
| 6 | Me | Et | H | TMS | 67% | 87% |
| 7 | −(CH2)4− | H | TMS | 81% | 90% | |
| 8 | H | H | H | −(CH2)2OTBS | 55% | 90% |
| 9 | H | Ph | Ph | TMS | 80% | 76% |
| 10 | Br | −C6H13 | H | TMS | 66%[d] | 91% |
| 11 | Br | Ph | H | TMS | 68% | 95% |
| 12 | Me | −CO2Et | H | TMS | 75% | 86% |
| 13 | H | Ph | H | BDMS | 100% | 73% |
| 14 | H | Ph | H | −CH(OEt)2 | 85% | 82% |
| 15 | H | iPr | H | −CH(OEt)2 | 85% | 87% |
| 16 | H | Ph | H | −C6H13 | 100% | 77% |
Isolated yield. Reactions performed on a 0.325 mmol scale.
Enantiomeric excess determined by chiral HPLC analysis.
Reaction performed with 2.7 equiv alkyne and 2.6 equiv Me2Zn.
14% of the methyl addition side product was also isolated.
Table 6.
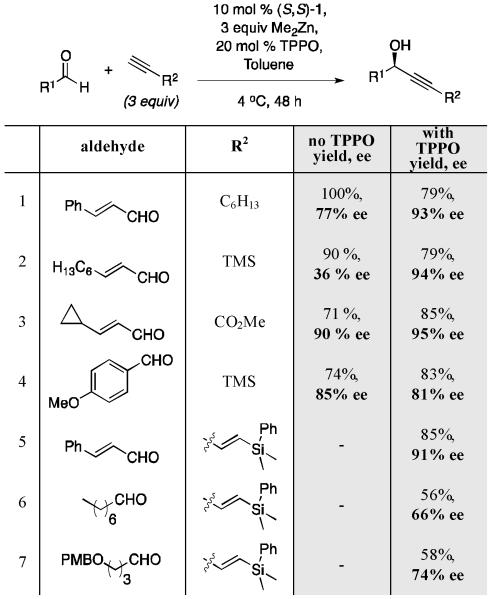
|
Isolated yield.
Enantiomeric excess determined by chiral HPLC analysis.
Methyl propiolate represents a particularly attractive class of nucleophiles for alkyne addition.[25] The resulting γ-hydroxy- α,β-acetylenic esters are extremely versatile synthetic intermediates and have been used in a number of total syntheses.[26] Propiolate donors have traditionally been difficult substrates for asymmetric alkynylation due to their propensity to decompose in the presence of Lewis acids and nucleophiles.[27] Methyl propiolate ultimately proved to be one of the most effective alkynes under Zn-ProPhenol alkynylation conditions (Table 4). Excellent results were obtained with a range of α,β-unsaturated aldehydes, including (E)-non-2-enal, which provided 97% ee with methyl propiolate (entry 2), a major improvement from the 36% ee obtained with TMS-acetylene. The superior results are presumed to be a consequence of the stabilization of the alkynylide of this alkyne along with potential coordination of the propiolate ester with the bimetallic catalyst.
Table 4.
Addition of Methyl Propiolate to α,β-Unsaturated Aldehydes.
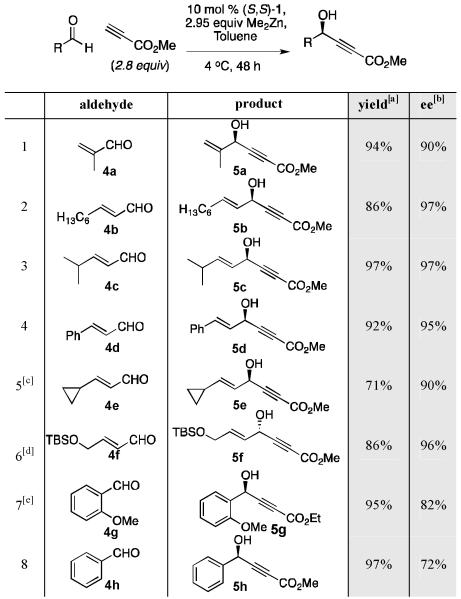
|
Isolated yield.
Enantiomeric excess determined by chiral HPLC.
Reaction performed using 3 equiv alkyne and 3 equiv Me2Zn.
Reaction performed using (R,R)-1.
Reaction performed using ethyl propiolate.
Aliphatic aldehydes
While the initial communication disclosed only the use of unsaturated aldehydes, a variety of aliphatic aldehydes are suitable electrophiles for asymmetric alkynylation with the ProPhenol ligand, (S,S)-1 (Table 5). Addition of methyl propiolate to α- and β-substituted aliphatic aldehydes proceeded in good yield and enantioselectivity. The addition of methyl propiolate to cyclopropanecarboxaldehyde gave the desired propargylic alcohol in 88% yield and 94% ee. n-Aliphatic aldehydes are particularly challenging substrates for alkynylation due to their propensity to enolize and undergo cross-aldol side reactions.[28]
Table 5.
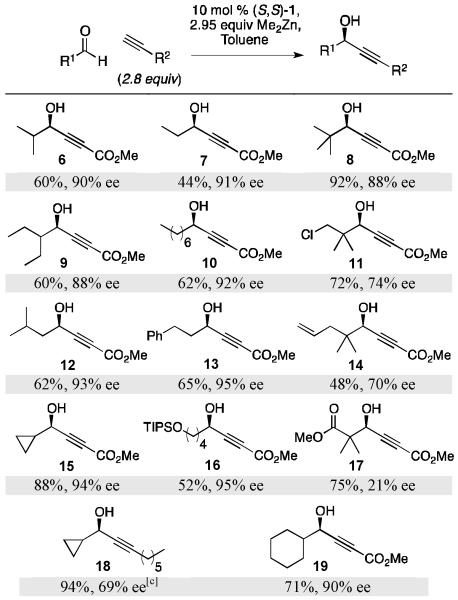
|
Isolated yield.
Enantiomeric excess determined by chiral HPLC analysis.
Enantiomeric excess determined by chiral HPLC analysis of the corresponding benzoate.
Modest yield and excellent enantioselectivity were ultimately achieved when utilizing the more acidic alkyne, methyl propiolate (see Table 5). Aliphatic aldehydes with an increased propensity to enolize, such as cyclopentane carboxaldehyde and 3-methylbut-3-enal, did not produce the desired products. On the other hand, the less easily enolized aldehyde, cyclohexanecarboxaldehyde, performed exemplarily. The increased steric hindrance of 2,2-dimethyl-substituted aldehydes often results in decreased enantioselectivity although addition of methyl propiolate to pivaldehyde provided the desired product in 88% ee. However, the analogous 3-chloro- and 3-oxopropanoate substrates provided significantly lower enantioselectivity. Extension of this methodology to aliphatic aldehydes further demonstrates the generality of the ProPhenol-catalyzed alkynylation with respect to both nucleophile and electrophile.
Proposed mechanism
The proposed mechanism for ProPhenol-catalyzed alkyne addition involves the formation of the dinuclear zinc species A (Scheme 2). This complex contains both Brønsted basic and Lewis acidic sites, and can therefore act as a bifunctional catalyst, activating two reagents simultaneously. The relative acidity of a terminal alkyne (pKa (DMSO) PhC≡CH = 28.7)[29] enables the formation of an alkynylzinc nucleophile, which undergoes nucleophilic addition to the si face of an aldehyde. Product dissociation via metal exchange liberates a propargylic zinc alkoxide and regenerates the active catalyst. Turnover of the catalyst in this way necessitates the use of a stoichiometric amount of organozinc reagent. In contrast, the ProPhenol-catalyzed direct aldol reaction requires only a catalytic amount of dialkylzinc reagent and dissociation of the product alkoxide is postulated to occur via proton exchange.[6]
Scheme 2.
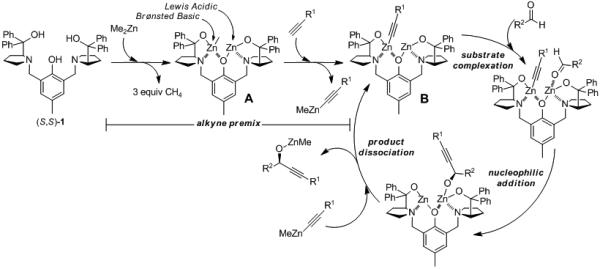
Proposed Mechanism for ProPhenol-Catalyzed Alkynylation of Aldehydes.
Additive effects and TPPO
During research into the ProPhenol-catalyzed addition of diynes, it was discovered that the presence of acetate groups in the substrate, far from the reacting alkyne terminus, resulted in significantly higher enantioselectivity.[30] It was hypothesized that this Lewis basic group interacts with the dinuclear zinc catalyst and reinforces the chiral pocket created by the ProPhenol ligand. The proposed coordination is supported by the X-ray crystal structure of the ProPhenol dinuclear zinc complex reported by Ding and coworkers (Figure 2).[31] The crystal structure contains a molecule of THF (dashed circle) coordinated to each of the zinc atoms, analogous to the postulated interaction with a Lewis base.
Figure 2.
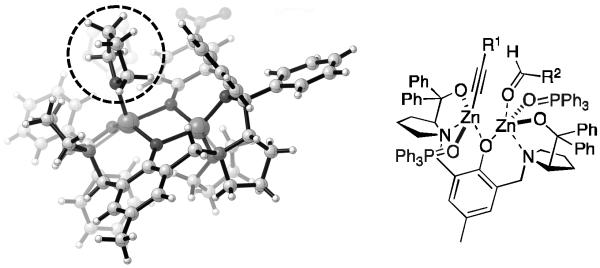
Zn-ProPhenol Crystal Structure and Proposed Interaction with Lewis Basic Additives.
Screening a variety of substoichiometric Lewis basic additives revealed that the addition of 20 mol % triphenylphosphine oxide (TPPO) provided optimal results. The largest improvements were observed in cases where the substrates lacked potential Lewis basic sites (Table 6). A large improvement in enantioselectivity was observed for the addition of TMS-acetylene to (E)-nonenal (entry 2).
Methyl propiolate, an alkyne containing a Lewis basic ester, often shows little or no improvement with the addition of TPPO (Entry 4). The addition of TPPO to the reaction has enabled the successful addition of a silylated enyne to both unsaturated (Entry 5) and saturated aldehydes (Entries 6-7).
Reagent stoichiometry
To maximize the practicality and synthetic efficiency of this methodology we sought to reduce reagent stoichiometry to a minimum. Ideally, the alkyne addition could be catalytic in metal with the resultant zinc alkoxide serving as a base for subsequent alkyne deprotonations. Carreira and coworkers were able to achieve alkynylation catalytic in zinc triflate in some cases by increasing the reaction temperature to 60 °C.[15f] Attempts to use a catalytic amount of dimethylzinc and elevated reaction temperatures in the ProPhenol-catalyzed alkynylation resulted in only recovered starting material (Table 7, entry 4). Previous attempts to use just a slight excess of alkyne and dimethylzinc have resulted in a significant drop in enantioselectivity. Zinc alkoxides are known to form aggregates,[32] and it was hypothesized that the aggregation state of the reactive zinc species plays an important role in enantioselectivity. Since the aggregation state is likely to be concentration dependent, a number of experiments were performed with the concentration of alkyne and dimethylzinc held constant, while reducing the stoichiometry of each. Consequently, the ProPhenol-catalyzed addition of methyl propiolate was found to give the same high enantioselectivity with less than half the amount of alkyne (entry 3). 1-Octyne addition using 1.5 equivalents of alkyne also provided good enantioselectivity, although a drop in yield was observed. The lower yield is presumably a consequence of decreased reactivity and to counter this, the reaction concentration was increased to 0.5 M (entry 6). When this adjustment was used in conjunction with 20 mol % TPPO, the desired product was obtained in 80% yield and 93% ee using just 1.2 equivalents of alkyne. TMS-acetylene also required higher reaction concentration and 20 mol % TPPO to provide high yield and enantioselectivity. The concentration dependence of this reaction led us to investigate the possibility of a dimeric active catalyst.[33] A number of dimeric zinc amino alcohol catalysts have been reported previously and display characteristic non-linear asymmetric induction in alkyl addition to aldehydes.[34] To evaluate potential non-linear effects in ProPhenol-catalyzed alkyne additions, the addition of methyl propiolate to (E)-nonenal was performed under the original superstoichiometric conditions with (S,S)-1 of varying optical purity (Figure 3, see SI for full experimental details). The enantiomeric excess of the product displayed a linear correlation (R2 = 0.99) with the enantiopurity of the ProPhenol ligand used to catalyze the reaction. When the ligand is not enantiomerically pure a dimeric active catalyst could potentially form three different association complexes: Zn-[(S,S)-1+(S,S)-1], Zn-[(S,S)-1+(R,R)-1], and Zn-[(R,R)-1+(R,R)-1]. It is likely that the homo- and heterochiral complexes would display significantly different catalytic activities and, therefore, display non-linear asymmetric induction. The results shown in Figure 3 suggest that a single ProPhenol ligand is present in the enantiodetermining step.
Table 7.
Alkynylation with Reduced Stoichiometry.
| Alkyne R1 | R2 | X/Y | comments[a] | yield[b] | ee[c] | |
|---|---|---|---|---|---|---|
|
| ||||||
| 1 | CO2Me | iPr | 3.0/3.0 | - | 97% | 97% |
| 2 | CO2Me | iPr | 1.5/1.5 | - | 79% | 97% |
| 3 | CO2Me | Ph | 1.2/1.5 | 0.48 M | 81%d | 94% |
| 4 | CO2Me | Ph | 1.2/0.2 | 60 °C, 24 h | - | - |
| 5 | (CH2)5CH3 | Ph | 3.0/3.0 | - | 83% | 81% |
| 6 | (CH2)5CH3 | Ph | 1.5/1.5 | - | 60% | 75% |
| 7 | (CH2)5CH3 | Ph | 1.2/1.5 | 20 mol % TPPO, 0.48 M |
80% | 93% |
| 8 | TMS | iPr | 3.0/3.0 | - | 100% | 94% |
| 9 | TMS | iPr | 1.2/1.2 | - | 71% | 50% |
| 10 | TMS | Ph | 3.0/3.0 | - | 75% | 90% |
| 11 | TMS | Ph | 1.2/1.2 | - | 52%[e] | 50% |
| 12 | TMS | Ph | 1.2/1.5 | 20 mol % TPPO, 0.48 M |
83% | 88% |
Reaction concentration is reported with respect to alkyne.
Isolated yield.
Enantiomeric excess determined by chiral HPLC analysis.
Isolated along with 16% yield of the methyl addition side product.
17% recovered starting material.
Figure 3.
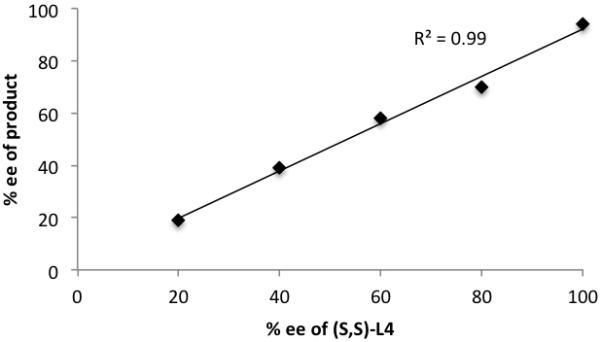
The Absence of Non-Linear Effects in the Addition of Methyl Propiolate to (E)-Nonenal with (S,S)-1.
Alkynylzinc formation and analysis of the alkyne premix
Reducing the equivalents of alkyne used in additions to enolizable aldehydes represents a much more challenging prospect. These reactions produce undesired cross aldol side products and reducing the equivalents of alkyne exacerbates the problem (Table 8, Entries 1-3). It was hypothesized that this side reaction occurs as a result of incomplete alkynylzinc formation and the presence of unreacted dimethyl zinc in the reaction mixture. Formation of the alkynylzinc species is driven, in part, by the entropically favored release of methane gas. However, a number of additional factors also contribute to the overall composition of the alkyne premix. Non-polar solvents have been shown to disfavor formation of the alkynylzinc species. In the case of phenylacetylene in heptane, no formation of the methylalkynylzinc species is observed.[35] A number of other Zn-alkyne addition methodologies rely on the use of additives to aid in the formation of the alkynylzinc nucleophile.[36] A stepwise 1H-NMR analysis of the methyl propiolate/Me2Zn/(S,S)-1 premix in [D8]toluene was used to evaluate alkyne deprotonation. Using a 1.2/1.5/0.1 ratio of methyl propiolate/Me2Zn/(S,S)-1, integration of the peaks at 0.17 (s, 3H, CH3ZnC≡C) and −0.69 (s, 6H, (CH3)2Zn) ppm revealed that approximately 30% of the desired alkynylzinc species was being formed during the standard 1 hour premix at room temperature (Figure 4, see SI for full experimental details).[37] Deprotonation of the terminal alkyne was not observed in the absence of the ProPhenol ligand. Analysis of the alkyne premix by volumetric gas titration of methane revealed that the initial rate of deprotonation is rapid but tapers off after about 15 minutes. The volume of methane obtained over the standard premix time was in good agreement with the 1H-NMR results.
Table 8.
Optimization of Alkyne Addition with an Enolizable Substrate.
| CHO | X/Y | conditions | yield[a] | ee[b] | |
|---|---|---|---|---|---|
|
| |||||
| 1 | 23a | 2.8/2.95 | - | 35%[c] | 45% |
| 2 | 23a | 2.8/2.95 | 20 mol % TPPO | 39% | 62% |
| 3 | 23a | 1.2/1.5 | 20 mol % TPPO | 22% | 54% |
| 4 | 23a | 1.2/1.3 | 30 mol % NMI | 11% | 72% |
| 5 | 23a | 1.2/1.4 | 4 equiv DMSO | 4% | 14% |
| 6 | 23a | 1.2/1.4 | 4 equiv DMF | 10% | 17% |
| 7 | 23b | 2.8/2.95 | 20 mol % TPPO, 24 hour alkyne premix |
58% | 50%[d] |
| 8 | 23b | 2.8/2.95 | 20 mol % (S,S)-1, 40 mol % TPPO, 24 hour alkyne premix |
69% | 67%[d] |
Isolated yield. All reactions were run on a 0.1625 mmol scale.
Enantiomeric excess determined by chiral HPLC analysis.
Isolated along with 19% yield of the cross aldol side product 25
Enantiomeric excess determined by 1H-NMR analysis of the corresponding (S)-methyl mandelate.
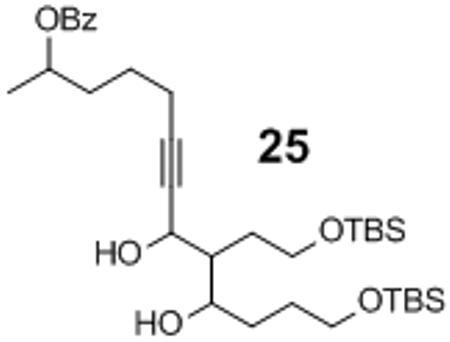
Figure 4.
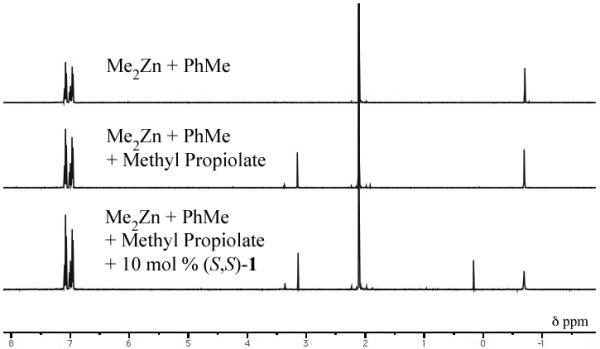
1H-NMR Analysis of the Alkyne Premix.
In contrast to enolizable aldehydes, the presence of significant amounts of dimethylzinc has little effect on the outcome of alkyne additions to aryl and α,β-unsaturated aldehydes.[38] Minor amounts of methyl addition side products, such as compound 21, have been isolated in only a small number of cases (Scheme 3). ProPhenol-catalyzed methyl addition is a relatively slow process and required 3 days to react with approximately half of the starting aryl aldehyde (Scheme 4).
Scheme 3.
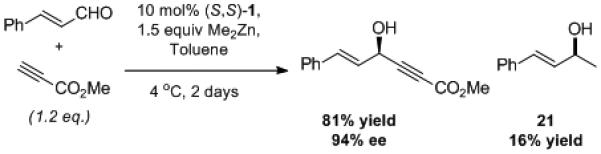
ProPhenol-Catalyzed Methyl Addition.
Scheme 4.
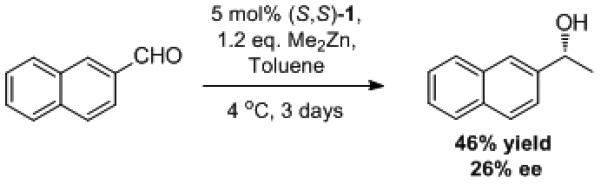
Competing Methyl Addition.
These observations prompted the investigation of methods to facilitate the formation of the alkynylzinc nucleophile and, therefore, reduce the amount of dimethylzinc present (Table 8). The addition of aliphatic alkyne 22 to the enolizable aldehyde 23a provided the desired propargylic alcohol 24a in low yield under the original super stoichiometric conditions (Entry 1). The low yield is a consequence of competing aldol side reactions, which provide a complex mixture of oligomers including compound 25, which was isolated in 19% as a mixture of diastereomers from entry 1. The use of Lewis basic additives such as N-methyl imidazole (NMI), DMSO and DMF to aid the formation of the desired alkynylzinc nucleophile provided low yield of the desired product 24a (Entries 4–6).[36] However, extending the alkyne premix time from 1 hour to 24 hours provided a 19% increase in yield (Entry 7). Combining this discovery with a higher catalyst loading (20 mol %) led to an optimized result of 69% yield and 67% ee for this challenging alkyne-aldehyde pairing (Entry 8). Applying our mechanistic understanding led to a modest improvement in the addition of a non-stabilized alkyne nucleophile (22) to enolizable aliphatic aldehyde (23), the most challenging combination for this methodology.
In contrast to the previous example, enolizable aliphatic aldehydes were shown to be excellent electrophiles in the ProPhenol-catalyzed alkynylation when more stabilized nucleophilic alkynes were employed. For example, the higher acidity of methyl propiolate provides improved results with enolizable aldehydes relative to other alkynes (Table 9). Addition of methyl propiolate to octanal under the initial conditions provides propargylic alcohol 26 in 62% yield. Lowering the reaction stoichiometry from 2.8 to 1.2 equivalents of alkyne results in a significantly decreased yield (Entry 4). Longer alkyne premix time results in visible decomposition of methyl propiolate (Entry 5). Therefore the use of higher catalyst loading was used to aid the consumption of dimethylzinc through alkynylzinc formation (Entries 6 & 7). Ultimately, 57% yield and 90% ee of the desired propargyl alcohol were obtained using 1.2 equivalents of alkyne.
Table 9.
Optimization of Methyl Propiolate Addition to Octanal.
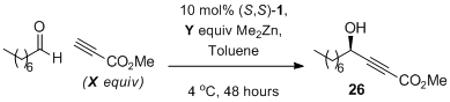
| entry | X/Y | conditions | yield[a] | ee[b] |
|---|---|---|---|---|
|
| ||||
| 1 | 2.8/2.95 | - | 62% | 92% |
| 2 | 1.5/1.5 | - | 37% | 86% |
| 3 | 1.5/1.5 | 20 mol % TPPO |
35% | 90% |
| 4 | 1.2/1.5 | - | 42% | 91% |
| 5 | 2.8/2.95 (Et2Zn) |
4 h alkyne premix |
60% | 70% |
| 6 | 1.2/1.5 | 20 mol % (S,S)-1 |
57% | 90% |
| 7 | 2.8/2.95 | 20 mol % (S,S)-1 |
72% | 87% |
All reactions were run on a 0.325 mmol scale. Isolated yield.
Enantiomeric excess determined by chiral HPLC analysis.
Alkynylation scope with reduced alkynylide stoichiometry
The use of a stoichiometric amount of alkyne would enable the use of precious alkynes to access complex synthetic targets in a highly efficient manner. Thus, we were pleased to observe that the newly developed reaction conditions allowed for the use of only 1.2 equivalents of alkyne (Scheme 5). In line with our previous results, the nature of the alkyne affected the results of the alkynylation. When methyl propiolate was used, good enantioselectivity and yield were observed with each class of aldehydes: aliphatic, α,β-unsaturated and aryl. However, when only 1.2 equivalents of a non-stabilized aliphatic alkyne, such as 22, is added to an α,β-unsaturated aldehyde, TPPO is required as an additive to provide high yield and ee. The addition of 22 to enolizable aldehydes is particularly challenging and gives moderate yield and enantioselectivity when an excess of alkyne is used in conjunction with TPPO, increased catalyst loading and a 24 hour alkyne premix. The synthesis of propargyl alcohol 27 in 88% yield and 90% ee was achieved using a single equivalent of both alkyne and aldehyde.
Scheme 5.
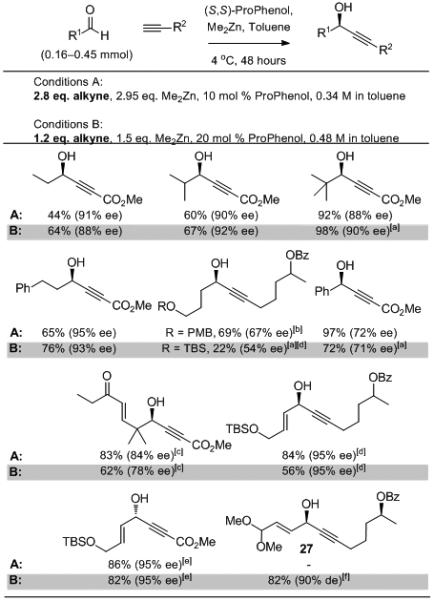
Scope and Evaluation of Alkynylation with Reduced Stoichiometry.
[a] 10 mol % (S,S)-1 used in reaction. [b] 20 mol % (S,S)-1, 40 mol % TPPO used in conjunction with a 24 h premix. [c] 20 mol % (S,S)-1 used in reaction. [d] 20 mol % TPPO used in this reaction. [e] 3.0 Equiv of Me2Zn used in reaction. [f] Reaction performed with (R,R)-1.
Synthetic Applications
The Zn-ProPhenol-catalyzed alkyne addition methodology has enabled the total synthesis of a wide range of complex natural products.[39] The synthetic utility of this transformation is a direct consequence of the mild reaction conditions and broad substrate scope that has become a focal point of this methodology. The propargylic alcohol moiety is present in a number of biologically and structurally interesting polyacetylenic natural products. The ProPhenol ligand enables the direct and convergent installation of this functionality, as shown in the total synthesis of adociacetylene B, 28 (Scheme 6).[40] The key intermediate, bis-enal 30, was prepared from the symmetric propargylic alcohol 29 using a Ru-catalyzed redox-isomerization.[41] This atom-economic transformation avoids the use of protecting groups and multiple redox operations often used with conventional olefination chemistry. Bis-alkynylation of 30 with TMS-acetylene provided the desired product 31 in excellent enantioselectivity and 9:1 mixture of dl and meso diastereomers. Alkynylation with the conditions from the original optimization (3 equiv Me2Zn, 3 equiv alkyne and 10 mol % (S,S)-1) resulted in significant amounts of the mono-alkynylation product and starting material, even when extending the reaction time to 3 days. The superior performance of methyl propiolate as an alkynylzinc nucleophile was once again illustrated by the increased yield and dr obtained relative to TMS-acetylene. The resulting γ-hydroxy-α,β-acetylenic ester 32 was saponified and decarboxylated to provide (−)-adociacetylene B (28) in a highly efficient manner. (R,R)-1 was also used to facilitate this reaction, providing access to both enantiomers of the natural product.
Scheme 6.
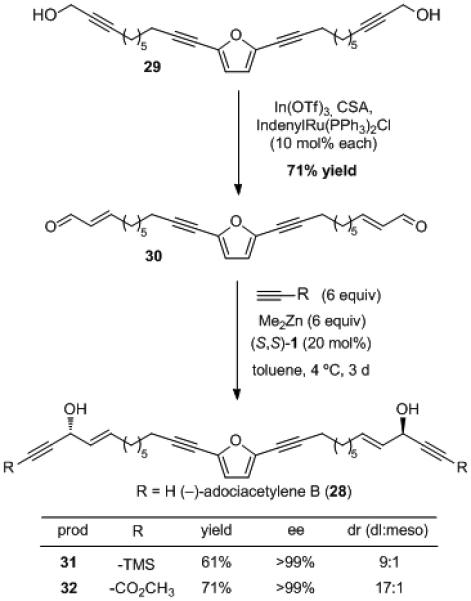
Total Synthesis of Adociacetylene B.
The orthogonal reactivity of alkynes was utilized to great effect in the total synthesis of (+)-spirolaxine methyl ether (Scheme 7).[42] ProPhenol-catalyzed addition of the aliphatic alkyne 33 to 3,5-dimethoxybenzaldehyde provided propargylic alcohol 34 in high ee and yield. Mild and selective alkyne reduction was subsequently achieved using Adam’s catalyst and provided access to the chiral benzyl alcohol 35. An additional asymmetric alkynylation–hydrogenation sequence was used to install a second chiral alcohol in the presence of a relatively sensitive phthalide group. This alkyne strategy ultimately led to the total synthesis of spirolaxine methyl ether in 13 linear steps. This work highlights the utility of asymmetric alkyne addition as a surrogate for a saturated alkyl group addition in the preparation of chiral alcohols.
Scheme 7.
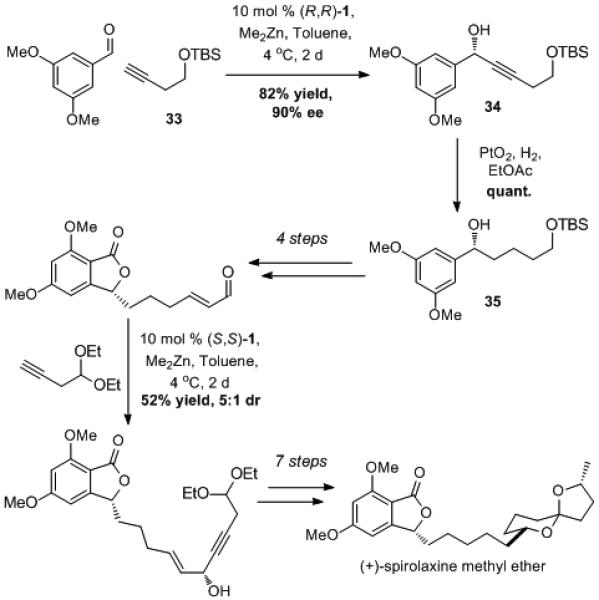
Alkyne Additions in the Total Synthesis of Spirolaxine Methyl Ether.
The formal total synthesis of aspergillide B (36) was enabled by an alkyne addition linchpin strategy, whereby (S)-hept-6-yn-2-yl benzoate (22) and methyl propiolate were added to each end of a butane dialdehyde equivalent 37 (Scheme 8).[43] Substrate and reaction optimization enabled alkynylation using just 1 equivalent of the precious alkyne 22 (see Table 8). Subsequent Ru-catalyzed hydrosilylation was used to achieve an E-selective reduction while also differentiating the two alkenes present in the product and enabling selective hydrogenation.[44] The late-stage addition of methyl propiolate to aliphatic aldehyde 38 proceeded in good yield. Chemoselective reduction of 39 was then achieved using our hydrosilylation/protodesilylation protocol for the formation of E alkenes. The basic reactions conditions used for this transformation triggered spontaneous oxy-Michael addition to produce the desired tetrahydrofuran ring and complete the formal total synthesis of aspergillide B. In this case, the asymmetric alkynylation served as a surrogate for a vinylation.
Scheme 8.
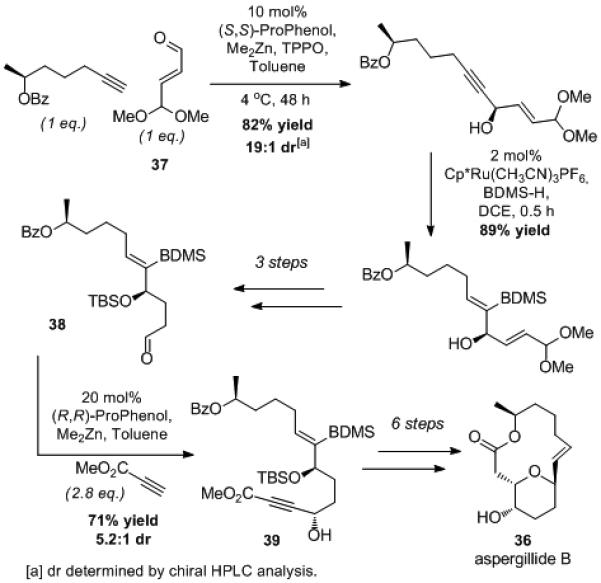
Alkyne Addition in the Synthesis of Aspergillide B.
Conclusion
Continued optimization of Zn-ProPhenol-catalyzed alkyne addition has led to the development of practical and general conditions for the asymmetric alkynylation of aldehydes. This synthetically efficient methodology operates with relatively low catalyst loading and can avoid the use of excess alkyne and dialkylzinc reagents. The chiral propargylic alcohols produced in this reaction are versatile synthetic intermediates and have enabled the synthesis of various natural products. Further research into the asymmetric alkynylation of enolizable aldehydes is currently underway.
Experimental Section
Representative alkynylation procedure with reduced reagent stoichiometry: preparation of 1-phenyl-5-trimethylsilanyl-pent-1-en-4-yn-3-ol
Full experimental details can be found in the Supporting Information.
To a solution of (S,S)-ProPhenol ligand (20.8 mg, 0.0325 mmol, 10 mol%), triphenylphosphine oxide (18 mg, 0.065 mmol, 20 mol %) and TMS-acetylene (56 μL, 0.39 mmol, 1.2 equiv) in anhydrous toluene (0.44 mL) was added dimethyl zinc (406 μL, 1.2 M solution in toluene, 0.488 mmol, 1.5 equiv) at 0 °C (0.813 mL total toluene, 0.48 M alkyne concentration). The reaction was warmed to room temperature and stirred for 60 minutes before addition of the trans-cinammaldehyde (43 mg, 0.325 mmol, 1 equiv) at 0 °C. The reaction was stirred for 48 hours at 4 °C before quenching with saturated, aqueous NH4Cl. The organic phase was extracted three times with Et2O and the combined organics were concentrated in vacuo. The crude product was purified by flash column chromatography. The title compound was isolated as a white solid (67 mg, 83% yield). Melting Point: 57-58 °C. [α]D25 = +2.16° (c = 1.05, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 7.40-7.43 (m, 2H), 7.32-7.36 (m, 2H), 7.25-7.29 (m, 1H), 6.77 (dd, J = 16, 1.2 Hz, 1H), 6.29 (dd, J = 16, 6 Hz, 1H), 5.05 (dt, J = 6, 1.2 Hz, 1H), 1.96 (t, J = 6 Hz, 1H), 0.21 (s, 9H). 13C-NMR (101 MHz, CDCl3): δ 136.0, 132.0, 128.6, 128.1, 127.8, 126.8, 104.1, 91.3, 66.3, −0.2. IR (film): 3300 (br, OH), 2960, 2172, 1654, 1496, 1449, 1407, 1251 cm−1. HRMS–EI (m/z): calculated for C14H18OSi: 230.1127, found: 230.1126, 0.6 ppm. Chiral HPLC: Chiralcel® AD column, heptane/iPrOH = 90/10, 1.0 mL/min, λ = 254 nm: 6.97/8.79 min (88% ee). Characterization data matches literature.[45]
Supplementary Material
Acknowledgements
We thank the National Science Foundation (CHE-0846427) and the National Institutes of Health (GM-33049) for their generous support of our programs. M.J.B. is grateful to Victoria University of Wellington for a Ph.D. scholarship. A.H.W. thanks Glaxo-SmithKline (ACS Division of Organic Chemistry Fellowship) and Bristol–Myers Squibb (for a graduate fellowship). A.J.v.W. thanks the DAAD for a postdoctoral fellowship. We also thank Sigma-Aldrich for donating ProPhenol ligand.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- [1].Trost BM, Ito H. J. Am. Chem. Soc. 2000;122:12003–12004. [Google Scholar]
- [2].For subsequent applications of this ligand, see: Trost BM, Malhotra S, Koschker P, Ellerbrock P. J. Am. Chem. Soc. 2012;134:2075–2084. doi: 10.1021/ja206995s. Trost BM, Hitce J. J. Am. Chem. Soc. 2009;131:4572–4573. doi: 10.1021/ja809723u. and references cited therein.
- [3].Trost BM, Terrell LR. J. Am. Chem. Soc. 2003;125:338–339. doi: 10.1021/ja028782e. [DOI] [PubMed] [Google Scholar]; Trost BM, Jaratjaroonphong J, Reutrakul V. J. Am. Chem. Soc. 2006;128:2778–2779. doi: 10.1021/ja057498v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Trost BM, Yeh VSC. Angew. Chem., Int. Ed. 2002;41:861–863. doi: 10.1002/1521-3773(20020301)41:5<861::aid-anie861>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]; Trost BM, Yeh VSC, Ito H, Bremeyer N. Org. Lett. 2002;4:2621–2623. doi: 10.1021/ol020077n. [DOI] [PubMed] [Google Scholar]; Trost BM, Lupton DW. Org. Lett. 2007;9:2023–2026. doi: 10.1021/ol070618e. [DOI] [PubMed] [Google Scholar]
- [5].Trost BM, Mino T. J. Am. Chem. Soc. 2003;125:2410–2411. doi: 10.1021/ja029708z. [DOI] [PubMed] [Google Scholar]; Trost BM, Malhotra S, Mino T, Rajapaksa NS. Chem. Eur. J. 2008;14:7648–7657. doi: 10.1002/chem.200800623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Trost BM, Ito H, Silcoff ER. J. Am. Chem. Soc. 2001;123:3367–3368. doi: 10.1021/ja003871h. [DOI] [PubMed] [Google Scholar]; Trost BM, Fettes A, Shireman BT. J. Am. Chem. Soc. 2004;126:2660–2661. doi: 10.1021/ja038666r. [DOI] [PubMed] [Google Scholar]; Trost BM, Shin S, Sclafani JA. J. Am. Chem. Soc. 2005;127:8602–8603. doi: 10.1021/ja051526s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].For the initial communication of this methodology, see: Trost BM, Weiss AH, von Wangelin AJ. J. Am. Chem. Soc. 2006;128:8–9. doi: 10.1021/ja054871q.
- [8].Stang PJ, Diederich F, editors. Modern Acetylene Chemistry. VCH; Weinheim: 2008. [Google Scholar]
- [9].For selected examples of catalytic enantioselective ynone reduction, see: Helal CJ, Magriotis PA, Corey EJ. J. Am. Chem. Soc. 1996;118:10938–10939. Matsumura K, Hashiguchi S, Ikariya T, Noyori R. J. Am. Chem. Soc. 1997;119:8738–8739. Parker KA, Ledeboer MW. J. Org. Chem. 1996;61:3214–3217. doi: 10.1021/jo951712o.
- [10].For a recent review on asymmetric alkyne addition to carbonyls, see: Trost BM, Weiss AH. Adv. Synth. Catal. 2009;351:963–983. doi: 10.1002/adsc.200800776.
- [11].For selected examples of asymmetric ynal alkylation, see: Seebach D, Beck AK, Schmidt B, Wang YM. Tetrahedron. 1994;50:4363–4384. Fontes M, Verdaguer X, Sola L, Vidal-Ferran A, Reddy KS, Riera A, Pericas MA. Org. Lett. 2002;4:2381–2383. doi: 10.1021/ol0260268. BouzBouz S, Pradaux F, Cossy J, Ferroud C, Falguieres A. Tetrahedron Lett. 2000:8877–8880. Uraguchi D, Nakamura S, Ooi T. Angew. Chem. Int. Ed. 2010;49:7562–7565. doi: 10.1002/anie.201004072. Belot S, Quintard A, Alexakis A, Krause N. Adv. Synth, Cat. 2010;352:667–695.
- [12].1,2-Dialkylidenecyclopentane formation: see reference 6. Syn α,β-dihydroxy ketone formation: Trost BM, Ball ZT, Laemmehold KM. J. Am. Chem. Soc. 2005;127:10028–10038. doi: 10.1021/ja051578h. β-arylation of propargylic alcohols: Trost BM, Machacek MR, Ball ZT. Org. Lett. 2003;5:1895–1898. doi: 10.1021/ol034463w. [2+2+2] cycloaddition: Witulski B, Zimmermann A, Gowans ND. Chem. Commun. 2002:2984–2985. doi: 10.1039/b209573d. Chiral allene formation: Pirkle WH, Boeder CW. J. Org. Chem. 1978;43:1950–1952.
- [13].For reviews on asymmetric addition of organozinc nucleophiles, see: Soai K, Niwa S. Chem. Rev. 1992;92:833–856. Pu L, Yu H-B. Chem. Rev. 2001;101:757–824. doi: 10.1021/cr000411y. Noyori R, Kitamura M. Angew. Chem. Int. Ed. Engl. 1991;30:49–69. Knochel P, Singer RD. Chem. Rev. 1993;93:2117–2188.
- [14].For reviews on zinc-mediated alkyne addition, see: Pu L. Tetrahedron. 2003;59:9873–9886. Cozzi PG, Hilgraf R, Zimmermann N. Eur. J. Org. Chem. 2004:4095–4105. Lu G, Li Y-M, Li X-S, Chan ASC. Coord. Chem. Rev. 2005;249:1736–1744. and reference 10.
- [15].a) Frantz DE, Fässler R, Carreira EM. J. Am. Chem. Soc. 2000;122:1806–1807. [Google Scholar]; b) Boyall D, López F, Sasaki H, Frantz D, Carreira EM. Org. Lett. 2000;2:4233–4236. doi: 10.1021/ol006791r. [DOI] [PubMed] [Google Scholar]; c) Frantz DE, Fässler R, Tomooka CS, Carreira EM. Acc. Chem. Res. 2000;33:373–381. doi: 10.1021/ar990078o. [DOI] [PubMed] [Google Scholar]; d) Sasaki H, Boyall D, Carreira EM. Helv. Chem. Acta. 2001;84:964–971. [Google Scholar]; e) El-Sayed E, Anand NK, Carreira EM. Org. Lett. 2001;3:3017–3020. doi: 10.1021/ol016431j. [DOI] [PubMed] [Google Scholar]; f) Anand NK, Carreira EM. J. Am. Chem. Soc. 2001;123:9687–9688. doi: 10.1021/ja016378u. [DOI] [PubMed] [Google Scholar]; g) Diez RS, Adger B, Carreira EM. Tetrahedron. 2002;58:8341–8344. [Google Scholar]; h) Knöpfel TF, Boyall D, Carreira EM. Org. Lett. 2004;6:2281–2283. doi: 10.1021/ol0491585. [DOI] [PubMed] [Google Scholar]; i) Fässler R, Tomooka CS, Frantz DE, Carreira EM. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5843–5845. doi: 10.1073/pnas.0308326101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gao G, Moore D, Xie R-G, Pu L. Org. Lett. 2002;4:4143–4146. doi: 10.1021/ol026921r. [DOI] [PubMed] [Google Scholar]; Moore D, Pu L. Org. Lett. 2002;4:1855–1857. doi: 10.1021/ol025825n. [DOI] [PubMed] [Google Scholar]; Xu M-H, Pu L. Org. Lett. 2002;4:4555–4557. doi: 10.1021/ol027110q. [DOI] [PubMed] [Google Scholar]; Liu L, Pu L. Tetrahedron. 2004;60:7427–7430. [Google Scholar]; Li Z-B, Pu L. Org. Lett. 2004;6:1065–1068. doi: 10.1021/ol0498139. [DOI] [PubMed] [Google Scholar]; Rajaram AR, Pu L. Org. Lett. 2006;8:2019–2012. doi: 10.1021/ol060377v. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yue Y, Turlington M, Yu X-Q, Pu L. J. Org. Chem. 2009;74:8681–8689. doi: 10.1021/jo9018446. [DOI] [PubMed] [Google Scholar]; Zhou L-H, Yu X-Q, Pu L. J. Org. Chem. 2009;74:2013–2017. doi: 10.1021/jo802592h. [DOI] [PubMed] [Google Scholar]; Turlington M, Yue Y, Yu X-Q, Pu L. J. Org. Chem. 2010;75:6941–6952. doi: 10.1021/jo101545v. [DOI] [PubMed] [Google Scholar]; Turlington M, Du Y, Ostrum SG, Santosh V, Wren K, Lin T, Sabat M, Pu L. J. Am. Chem. Soc. 2011;133:11780–11794. doi: 10.1021/ja204289q. [DOI] [PubMed] [Google Scholar]
- [17].Lu G, Li X, Zhou Z, Chan W, Chan A. Tetrahedron: Asymmetry. 2001;12:2147–2152. [Google Scholar]; Li X, Lu G, Kwok WH, Chan ASC. J. Am. Chem. Soc. 2002;124:12636–12637. doi: 10.1021/ja025541y. [DOI] [PubMed] [Google Scholar]; Lu G, Li X, Chen G, Chan W, Chan A. Tetrahedron: Asymmetry. 2003;14:449–452. [Google Scholar]
- [18].Niwa S, Soai K. J. Chem. Soc., Perkin Trans. 1. 1990:937–943. [Google Scholar]; Tombo GMR, Didier E, Loubinoux B. Synlett. 1990;9:547–548. [Google Scholar]; Ishizaki M, Hoshino O. Tetrahedron Asymmetry. 1994;5:1901–1904. [Google Scholar]; Jiang B, Chen Z, Xiong W. Chem. Commun. 2002:1524–1525. doi: 10.1039/b203984b. [DOI] [PubMed] [Google Scholar]; Lu G, Li X, Chan WL, Chan ASC. Chem. Commun. 2002:172–173. doi: 10.1039/b107817h. [DOI] [PubMed] [Google Scholar]; Chen Z, Xiong WL, Jiang B. Chem. Commun. 2002:2098–2099. doi: 10.1039/b206868k. [DOI] [PubMed] [Google Scholar]; Marshall JA, Bourbeau MP. Org. Lett. 2003;5:3197–3199. doi: 10.1021/ol034918h. [DOI] [PubMed] [Google Scholar]; Xu Z, Wang R, Xu J, Da C-S, Yan W-J, Chen C. Angew. Chem. Int. Ed. 2003;42:5747–5749. doi: 10.1002/anie.200352572. [DOI] [PubMed] [Google Scholar]; Zhou Y-F, Wang R, Xu Z-Q, Yan W-J, Lei L, Gao Y-F, Da C-S. Tetrahedron: Asymmetry. 2004;15:589–591. [Google Scholar]; Kang Y-F, Liu L, Wang R, Yan W-J, Zhou Y-F. Tetrahedron: Asymmetry. 2004;15:3155–3159. [Google Scholar]; Dahmen S. Org. Lett. 2004;6:2113–2116. doi: 10.1021/ol049596b. [DOI] [PubMed] [Google Scholar]; Kang Y, Wang R, Liu L, Da C, Yan W, Xu Z. Tetrahedron Lett. 2005;46:863–865. [Google Scholar]; Ni M, Wang R, Han Z-J, Mao B, Da C-S, Liu L, Chen C. Adv. Synth. Catal. 2005;347:1659–1665. [Google Scholar]; Wolf C, Liu S. J. Am. Chem. Soc. 2006;128:10996–10997. doi: 10.1021/ja062711o. [DOI] [PubMed] [Google Scholar]; Emerson DPG, Hems WP, Davis BG. Org. Lett. 2006;8:207–210. doi: 10.1021/ol052503l. [DOI] [PubMed] [Google Scholar]; Li Z-B, Liu T-D, Pu L. J. Org. Chem. 2007;72:4340–4343. doi: 10.1021/jo070091j. [DOI] [PubMed] [Google Scholar]; Xu J, Mao J, Zhang Y. Org. Biomol. Chem. 2008;6:1288–1292. doi: 10.1039/b719624e. [DOI] [PubMed] [Google Scholar]; Hui XP, Yin C, Chen Z-C, Huang L-N, Xu P-F, Fan GF. Tetrahedron. 2008;64:2553–2558. [Google Scholar]; Wang M-C, Zhang Q-J, Zhao W-X, Wang X-D, Ding X, Jing T-T, Song M-P. J. Org. Chem. 2008;73:168–176. doi: 10.1021/jo701943x. [DOI] [PubMed] [Google Scholar]; Li H, Huang Y, Jin W, Xue F, Wan B. Tetrahedron Lett. 2008;49:1686. [Google Scholar]; Blay G, Cardona L, Fernández I, Macro-Aleixandre A, Muñoz MC, Pedro JR. Org. Biomol. Chem. 2009;7:4301–4308. doi: 10.1039/b911592g. [DOI] [PubMed] [Google Scholar]; Niu J-L, Wang M-C, Lu L-J, Ding G-L, Lu H-J, Chen Q-T, Song M-P. Tetrahedron: Asymmetry. 2009;20:2616–2621. [Google Scholar]; Li Z-Y, Wang M, Bian Q-H, Zheng B, Mao J-Y, Li S-N, Liu S-Z, Wang M-A, Zhong J-C, Guo H-C. Chem. Eur. J. 2011;17:5782–5786. doi: 10.1002/chem.201100535. [DOI] [PubMed] [Google Scholar]; Graham ER, Tykwinski RR. J. Org. Chem. 2011;76:6574–6583. doi: 10.1021/jo2008719. [DOI] [PubMed] [Google Scholar]; Bauer T, Smolinski S, Gawel P, Jurczak J. Tetrahedron Lett. 2011;52:4882–4884. [Google Scholar]; Lu X, Xie G, Li T, Qu X, Mao J. Synthetic Commun. 2012;42:775–783. [Google Scholar]; Boobalan R, Chen C, Lee G-H. Org. Biolmol. Chem. 2012;10:1625–1638. doi: 10.1039/c1ob06683h. [DOI] [PubMed] [Google Scholar]
- [19].For rare examples of zinc-catalyzed asymmetric alkynylation with 1 equivalent of alkyne, see: Kojima N, Nishijima S, Tsuge K, Tanaka T. Org. Biomol. Chem. 2011;9:4425–4428. doi: 10.1039/c1ob05489a. b) reference 15f.
- [20].Trost BM. Science. 1991;254:1471–1477. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]; Trost BM. Acc. Chem. Res. 2002;35:695–705. doi: 10.1021/ar010068z. [DOI] [PubMed] [Google Scholar]
- [21].Cozzi PG. Angew. Chem. Int. Ed. 2003;42:2895–2898. doi: 10.1002/anie.200351230. [DOI] [PubMed] [Google Scholar]
- [22].Absolute configuration (R) was determined by comparison to the optical rotation reported in the literature.
- [23].Cozzi PG, Rudolph J, Bolm C, Norrby P, Tomasini C. J. Org. Chem. 2005;70:5733–5736. doi: 10.1021/jo050115r. [DOI] [PubMed] [Google Scholar]
- [24].Li Z, Upadhyay V, DeCamp AE, DiMichele L, Reider PJ. Synthesis. 1999:1453–1458. [Google Scholar]
- [25].For successful asymmetric additions of propiolate nucleophiles to aldehydes, see: Gao G, Wang Q, Yu X-Q, Xie R-G, Pu L. Angew. Chem. Int. Ed. 2006;45:122–131. doi: 10.1002/anie.200500469. Rajaram AR, Pu L. Org. Lett. 2006;8:2019–2021. doi: 10.1021/ol060377v. Lin L, Jiang X, Liu W, Qiu L, Xu Z, Xu J, Chan ASC, Wang R. Org. Lett. 2007;9:2329–2332. doi: 10.1021/ol070692x. Turlington M, DeBerardinis AM, Pu L. Org. Lett. 2009;11:2441–2444. doi: 10.1021/ol900667g. Xu T, Liang C, Cai Y, Li J, Li Y-M, Hui X-P. Tetrahedron: Asymmetry. 2009;20:2733–2736. and reference 19a.
- [26].For selected examples of γ-hydroxy-α,β-acetylenic esters as synthetic intermediates, see: Midland MM, Tramontano A, Cable JR. J. Org. Chem. 1980;45:28–29. Midland MM, McDowell DC, Hatch RL, Tramontano A. J. Am. Chem. Soc. 1980;102:867–869. Trost BM, Crawley ML. J. Am. Chem. Soc. 2002;124:9328–9329. doi: 10.1021/ja026438b. Trost BM, Ball ZT. J. Am. Chem. Soc. 2004;126:13942–13944. doi: 10.1021/ja045971j. Molander GA, St. Jean DJ. J. Org. Chem. 2002;67:3861–3865. doi: 10.1021/jo0255936. Meta CT, Koide K. Org. Lett. 2004;6:1785–1787. doi: 10.1021/ol0495366. Albert BJ, Sivaramakrishnan A, Naka T, Koide K. J. Am. Chem. Soc. 2006;128:2792–2793. doi: 10.1021/ja058216u.
- [27].Shahi S, Koide K. Angew. Chem. Int. Ed. 2004;43:2525–2527. doi: 10.1002/anie.200353400. [DOI] [PubMed] [Google Scholar]
- [28].Others have also attributed the modest yield obtained with n-aliphatic aldehydes to competing self-condensation aldol side reactions, see reference 15a.
- [29].Bordwell FG, Drucker GE, Anderen NH, Denniston AD. J. Am. Chem. Soc. 1986;108:7310–7313. [Google Scholar]
- [30].Trost BM, Chan VS, Yamamoto D. J. Am. Chem. Soc. 2010;132:5186–5192. doi: 10.1021/ja910656b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xiao Y, Wang Z, Ding K. Chem. Eur. J. 2005;11:3668–3678. doi: 10.1002/chem.200401159. [DOI] [PubMed] [Google Scholar]
- [32].Coates GE, Ridley D. J. Chem. Soc. 1965:1870–1877. Shearer HMM, Spencer CB. Acta Crystallogr. 1980;B36:2046–2050. For a theoretical discussion on the structure and stability of various zinc alkoxide aggregates, see; Steudel R, Steudel Y. J. Phys. Chem. A. 2006;110:8912–8924. doi: 10.1021/jp060725a.
- [33].For a review on nonlinear effects and polymeric chiral catalysts, see: Girard C, Kagan HB. Angew. Chem. Int. Ed. 1998;37:2922–2959. doi: 10.1002/(SICI)1521-3773(19981116)37:21<2922::AID-ANIE2922>3.0.CO;2-1.
- [34].For selected examples of chiral zinc catalysts that display nonlinear effects, see: Kitamura M, Okada S, Suga S, Noyori R. J. Am. Chem. Soc. 1989;111:4028–4036. Kitamura M, Suga S, Oka H, Noyori R. J. Am. Chem. Soc. 1998;120:9800–9809. Oguni N, Matsuda Y, Kaneko T. J. Am. Chem. Soc. 1988;110:7877–7878.
- [35].Okhlobystin OY, Zakharkin LI. J. Organometal. Chem. 1965;3:257–258. [Google Scholar]
- [36].For selected examples of the use of additives to facilitate the formation of alkynylzinc nucleophiles, see: Gao G, Xie R-G, Pu L. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5417–5420. doi: 10.1073/pnas.0307136101. Yang F, Xi P, Yang L, Lan J, Xie R, You J. J. Org. Chem. 2007;72:5457–5460. doi: 10.1021/jo0707535. Du Y, Turlington M, Zhou X, Pu L. Tetrahedron Lett. 2010;51:5024–5027.
- [37].For related NMR analysis of alkynylzinc nucleophiles, see: Evans DF, Fazakerley GV. J. Chem. Soc. 1971:182–183. De Koning AJ, Van Rijn PE, Boersma J, Van Der Kerk GJM. J. Organometal. Chem. 1979;174:129–140. Fässler R. Ph.D. Thesis. ETH Zürich; Zürich, Switzerland: 2003. The Metalation of Terminal Alkynes by ZnII and Their Addition to Nitrones and Aldehydes. and reference 24.
- [38].In contrast, alkynylation with Me2Zn/Ti(OiPr)4/BINOL suffers from significant amounts of methyl addition if formation of the alkynylzinc nucleophile does not go to completion, see reference 36a.
- [39].Trost BM, Sieber JD, Qian W, Dhawan R, Ball ZT. Angew. Chem., Int. Ed. 2009;48:5478–5481. doi: 10.1002/anie.200901907. [DOI] [PMC free article] [PubMed] [Google Scholar]; Trost BM, O’Boyle BM. J. Am. Chem. Soc. 2008;130:16190–16192. doi: 10.1021/ja807127s. [DOI] [PMC free article] [PubMed] [Google Scholar]; Trost BM, O’Boyle BM, Hund D. J. Am. Chem. Soc. 2009;131:15061–15074. doi: 10.1021/ja906056v. [DOI] [PMC free article] [PubMed] [Google Scholar]; Trost BM, Burns AC, Bartlett MJ, Tautz T, Weiss AH. J. Am. Chem. Soc. 2012;134:1474–1477. doi: 10.1021/ja210986f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Trost BM, Weiss AH. Org. Lett. 2006;8:4461–4464. doi: 10.1021/ol0615836. [DOI] [PubMed] [Google Scholar]
- [41].Trost BM, Livingston RC. J. Am. Chem. Soc. 2008;130:11970–11978. doi: 10.1021/ja804105m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Trost BM, Weiss AH. Angew. Chem. Int. Ed. 2007;46:7664–7666. doi: 10.1002/anie.200702637. [DOI] [PubMed] [Google Scholar]
- [43].Trost BM, Bartlett MJ. Org. Lett. 2012;14:1322–1325. doi: 10.1021/ol300200m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Trost BM, Ball ZT, Jöge T. Angew. Chem. Int. Ed. 2003;115:3537–3540. doi: 10.1002/anie.200351587. [DOI] [PubMed] [Google Scholar]; Trost BM, Ball ZT. J. Am. Chem. Soc. 2005;127:17644–17655. doi: 10.1021/ja0528580. [DOI] [PMC free article] [PubMed] [Google Scholar]; Trost BM, Ball ZT, Jöge T. J. Am. Chem. Soc. 2002;124:7922–7923. doi: 10.1021/ja026457l. [DOI] [PubMed] [Google Scholar]
- [45].Midland MM, Tramontano A, Bazubski A, Graham RS, Tsai DJS, Cardin DB. Tetrahedron. 1984;40:1371–1380. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.