

Abstract
Shiverer-immunodeficient (Shi-id) mice demonstrate defective myelination in the central nervous system (CNS) and significant ataxia by 2 to 3 weeks of life. Expanded, banked human neural stem cells (HuCNS-SCs) were transplanted into three sites in the brains of neonatal or juvenile Shi-id mice, which were asymptomatic or showed advanced hypomyelination, respectively. In both groups of mice, HuCNS-SCs engrafted and underwent preferential differentiation into oligodendrocytes. These oligodendrocytes generated compact myelin with normalized nodal organization, ultrastructure, and axon conduction velocities. Myelination was equivalent in neonatal and juvenile mice by quantitative histopathology and high-field ex vivo magnetic resonance imaging, which, through fractional anisotropy, revealed CNS myelination 5 to 7 weeks after HuCNS-SC transplantation. Transplanted HuCNS-SCs generated functional myelin in the CNS, even in animals with severe symptomatic hypomyelination, suggesting that this strategy may be useful for treating dysmyelinating diseases.
INTRODUCTION
Disorders of central nervous system (CNS) dysmyelination are a diverse group of pediatric and adult disorders that result in severe neurological deterioration because of the vital roles that myelin plays in rapid conduction of nerve impulses and maintenance of axonal integrity (1). Currently, no approved drug or biologic can produce new myelin; hence, there is growing interest in neural stem cell–based therapies for the treatment of dysmyelinating disorders.
Homozygous shiverer (Shi) mice are widely used as a model of dysmyelinating disorders (2). Shi mice have a spontaneous deletion of multiple exons in the gene encoding myelin basic protein (MBP), which results in pronounced ataxia by 2 to 3 weeks of age and the onset of fatal seizures by ~8 to 14 weeks (3–6). One variant of this model is the immunodeficient Shi × RAG2−/− mouse, which displays a milder CNS phenotype and a longer life span of ~18 to 21 weeks. Delivery of freshly isolated human oligodendrocyte progenitor cells from the brain tissue of fetuses at 9 to 22 weeks of gestation into multiple CNS sites in asymptomatic newborn Shi × RAG2−/− mice resulted in diffuse CNS myelination and markedly prolonged survival in some of the animals (7, 8). This study demonstrated the potential of human fetal oligodendrocyte progenitor cells to produce myelin, but clinical application would require a continuing source of these cells, which would be difficult.
One alternative is to use banked human CNS neural stem cells (HuCNS-SCs) grown as neurospheres that are purified, expanded, and then banked and cryopreserved under conditions suitable for clinical application. These nongenetically modified HuCNS-SCs were prospectively isolated from human brain tissue of fetuses at 16 to 20 weeks of gestation using antibody-based fluorescence-activated cell sorting, which yielded a highly purified stem cell population of CD133+ CD24−/lo cells (9). Using a process compliant with Good Manufacturing Practice (GMP), HuCNS-SCs purified from tissue from a single fetal brain can be expanded to create cryopreserved cell banks of sufficient size with which to perform all of the required nonclinical safety studies as well as clinical trials. Our ongoing studies show that the cells are viable and stable for greater than 10 years.
In previous works (10–17), these banked HuCNS-SCs were evaluated for their preclinical safety and efficacy before testing for clinical application in several CNS disorders. When transplanted into rodents, HuCNS-SCs migrated within the brain or spinal cord and displayed region-appropriate multilineage differentiation. Moreover, they were capable of protecting host neurons from cell death under conditions of progressive CNS neurodegeneration or injury without evidence of tumor formation or microglial cell activation (10–15). In a rodent model of acquired spinal cord contusion injury, HuCNS-SCs restored a measure of locomotor function, which required the continued presence of human cells (11, 14, 17). Cell fate analysis revealed that a high proportion of transplanted HuCNS-SCs preferentially differentiated into oligodendrocytes or neurons without any evidence of glial scar formation (11, 14, 17), suggesting the potential of these cells to treat inherited or acquired myelinating disorders.
Here, we demonstrate the potential of HuCNS-SCs to preferentially generate oligodendrocytes and functional myelin after transplant into juvenile shiverer-immunodeficient (Shi-id) mice that display symptomatic dysmyelination 3 weeks after birth.
RESULTS
Transplant of HuCNS-SCs into neonatal and juvenile Shi-id mice generates oligodendrocytes
HuCNS-SCs were prospectively isolated and expanded under serum-free conditions. Selective expansion of neural stem cells was promoted, resulting in the formation of neurospheres. Cells in the neurospheres expressed neural stem markers, including CD133, Sox2, and nestin (18), but did not express O4, the oligodendrocyte progenitor marker, before transplantation (fig. S1, A to C). Engraftment and migration of HuCNS-SCs were first evaluated after bilateral injection into three distinct CNS sites—the corpus callosum, the fimbria of the fornix, and the cerebellar white matter—of asymptomatic neonatal Shi-id mice. At 8 to 12 weeks after transplant, widespread dense immunoreactivity for the human-specific marker SC121 demonstrated that human cells had migrated diffusely from the three transplant sites (Fig. 1A, arrows) and into the adjacent cerebral cortex, corpus callosum, anterior commissure, and striatum (Fig. 1A, inset).
Fig. 1.

Migration and differentiation of HuCNS-SCs after transplantation into neonatal Shi-id mouse. (A) Cells were transplanted bilaterally at three coronal levels (arrows) that targeted the forebrain areas (striatum, rostral corpus callosum, and anterior cortex), the midbrain areas (hippocampus, body of the corpus callosum, and posterior cortex), and the hindbrain areas (cerebellum and brainstem). SC121 staining (brown) at 8 weeks after transplant demonstrated extensive migration of HuCNS-SCs within white matter tracts that included the corpus callosum, fimbria of the fornix, and those in the cerebellum. (Inset) Detail of SC121 staining from an adjacent section from the same animal revealed robust engraftment at the injection core and migration of the HuCNS-SC progeny into the cortex (CTX), corpus callosum (CC), and striatum (ST) 8 weeks after transplant. Regional boundaries are delineated with methyl green counter-stain. (B to D) Quantification of the percentage of human cells committed to the oligodendrocyte lineage at successive time points after transplantation. Human oligodendrocytes were identified by coexpression of the human nuclear marker SC101+ and the nuclear transcription factor Olig2 in the cerebellum (B), corpus callosum (C), and fimbria (D). (E to G) Immunofluorescent staining for Olig2 (green), human-specific cytoplasmic marker SC121 (red), and Hoechst 33324 (nuclear stain, blue) in cerebellum (E), corpus callosum (F), and olfactory bulb (G).
The time course of HuCNS-SC commitment to the oligodendrocyte lineage was followed from 2 to 7 weeks after transplant by immunohistochemical colocalization of the human-specific nuclear marker SC101 and the nuclear transcription factor and pan-oligodendrocyte marker Olig2. At 2 to 3 weeks after transplant (Fig. 1, B to D), ~50% of human engrafted cells had differentiated into Olig2+ cells in the cerebellum, corpus callosum, and fimbria of the fornix. In adult white matter, at 6 to 7 weeks, ~70% of human cells were committed to the oligodendrocyte lineage. With the human-specific cytoplasmic marker SC121, the morphology of the human Olig2+ cells was defined by confocal microscopy. The SC121+ cells also colabeled with Olig2 in the cerebellum (Fig. 1E and fig. S1D) and corpus callosum (Fig. 1F) at 12 weeks after transplant. In contrast, no colabeled cells were observed in the olfactory bulb (Fig. 1G), suggesting that commitment to the oligodendrocyte lineage occurred in a site-appropriate manner. The long-term maintenance of oligodendrocytes and oligodendrocyte progenitors was demonstrated by the presence of SC121+Olig2+ cells in heterozygous Shi−/+-id mice 25 weeks after transplant (fig. S1, E and F).
Triple staining for Olig2, SC121, and MBP confirmed that HuCNS-SCs differentiated into mature oligodendrocytes that initiated myelination of multiple axons, which was examined 12 weeks after transplant (Fig. 2A). Double staining for SC121 and the mature oligodendrocyte marker CC1 confirmed the terminal differentiation of the HuCNS-SCs into myelinating oligodendrocytes (Fig. 2B). Analysis of HuCNS-SCs derived from 10 different donors confirmed that HuCNS-SCs from different donor cell banks generated mature human oligodendrocytes that produced myelin (fig. S2).
Fig. 2.

HuCNS-SCs preferentially differentiate into oligodendrocytes. (A and B) Confocal images demonstrate the myelination of axons by human cells 12 weeks after transplant in neonatal mice. (A) In cerebellum, a SC121+ human cell (red) coexpressing Olig2+ (green) appeared contiguous with several MBP+ myelin sheaths (light blue). Dark blue, Hoechst nuclear counterstain. (B) The mature oligodendrocyte marker CC1 (green) is coexpressed with SC121 (red) in the corpus callosum and demonstrates the typical morphology of a human myelinating cell. Note the diffuse colocalization of CC1 and SC121 in the cytoplasm (yellow-orange). (C to F) Cell fate analysis of human cells 10 weeks after neonatal Shi-id transplantation. Representative immunohistochemical staining in the cerebellum of neonatal Shi-id mice is shown for the human marker SC121 (C), MBP (D), human nestin (hNestin) (E), and human GFAP (hGFAP) (F). Arrowheads indicate immunoperoxidase staining of human markers. Regional boundaries are delineated with methyl green counterstain.
We have previously shown the multipotentiality of banked human neural stem cells by demonstrating their in vivo differentiation into neurons, astrocytes, and oligodendrocytes (9, 11, 13, 14, 19). To address the differentiation potential of HuCNS-SCs into neurons and glial cells after transplantation into hypomyelinated white matter areas of Shi-id mice, we examined the fate of the engrafted human cells in the mouse cerebellum at 9 weeks after transplant. In the transplanted cerebellum, SC121+ cells (Fig. 2C) differentiated into oligodendrocytes expressing MBP (Fig. 2D). Rare cells stained for human nestin, which was consistent with the maintenance of an immature pool of cells (Fig. 2E, arrowhead). Little human-specific staining for the astrocyte marker glial fibrillary acid protein (GFAP) was observed (Fig. 2F, arrowhead), prompting an analysis using the glial progenitor cell marker S100β, which stains cells of both the oligodendroglial and the astrocytic lineages (20, 21). A quantitative human cell fate analysis was performed by costaining with the anti-human nuclear antigen SC101, S100β, and other lineage markers. The S100β antibody labeled about 15.7 ± 3.5% of the human cells (n = 3; Fig. 3, A and H). These cells typically were immature-appearing (Fig. 3, A to E, arrows) with a simple multipolar or pseudo-unipolar morphology, consistent with glial progenitors. Furthermore, three-color confocal analysis revealed an additional discrete subset of human GFAP+ astrocytes in the cerebellum (Fig. 3, B and C, arrowhead) and corpus callosum (Fig. 3E, arrowheads) that were distinct from the S100β-labeled cells (Fig. 3, B to E, arrows). In the cerebellum, cells expressing human GFAP had little to no S100β expression (Fig. 3, B and C), whereas those cells in the corpus callosum did express S100β (Fig. 3E, arrowhead). Only 4.1 ± 2.5% of human cells in white matter expressed the neuronal marker NeuN (Fig. 3, F and H).
Fig. 3.

Analyses of cell fate and proliferation status. Confocal images of engrafted human cells 9 weeks after transplant into neonatal Shi-id mice. (A to C, F, and G) Cerebellum. (D and E) Corpus callosum. All confocal images (A to G) were counterstained with Hoechst 33324 nuclear stain (dark blue). (A and D) Two-color staining for SC101 (green) and S100β (red) in the cerebellum (A) and corpus callosum (D). These cells typically had an immature morphology with few processes. (B, C, and E) Triple staining for SC101 (green), S100β (red), and hGFAP (SC123, light blue) in the cerebellum (B and C) and corpus callosum (E). Arrows indicate cells with immature pathology that were SC101+ (green), S100β+ (red), and hGFAP−. hGFAP+ cells in the cerebellum expressed low levels of S100β (B and C). More hGFAP+ cells were present in the corpus callosum (E), and they expressed higher S100β compared to the cerebellum [arrowheads in (E)]. Arrows in (A) to (E) indicate the immature morphology of the SC101+ (green), S100β+ (red), and hGFAP− cells that were also seen in the corpus callosum. (F) Confocal images of human cells in cerebellum stained with SC101 (green) and the neuronal marker NeuN (red). (G) Two-color staining for SC101 (green) and the proliferation marker Ki67 (red). Arrows indicate occasional proliferating human cells. (H) Quantitative analysis of HuCNS-SC cell fate after transplant of three neonatal mice. Quantitation was in cerebellar white matter (S100β, NeuN, and Ki67) or in corpus callosum (CC) (Ki67). (I) In vivo differentiation of HuCNS-SCs into oligodendrocytes is time-dependent. Percentage of total animals studied (n = 69) that displayed human cell engraftment and were immunoreactive (IR) for MBP in the cerebellum and brainstem at successive times after transplantation.
We also assessed the proliferation status of the transplanted HuCNS-SCs both to test for the persistence of residual stem/progenitor cells and to visualize concentrated foci of proliferating cells that might be indicative of tumorigenic potential or abnormal cell growth. Quantification of human cell proliferation with Ki67 and SC101 revealed that 5.0 ± 0.79% and 7.3 ± 2.4% of the cells expressed Ki67 in the cerebellum and corpus callosum, respectively (n = 3; Fig. 3H). Thus, consistent with our previous findings, HuCNS-SCs proliferate slowly to maintain a pool of stem/progenitor cells (13).
HuCNS-SCs generate myelin in the CNS of transplanted neonatal mice
To estimate the time course of CNS myelin production, we scored MBP immunostaining in cerebellar white matter at successive time points after neonatal transplantation (Fig. 3I). Myelination was rarely observed before 3 weeks when only rare cells expressing human MBP (1 to 10 cells per brain; n = 16) were detected. Between 4 and 6 weeks after transplant, 4 of 15 Shi-id mouse brains had scattered myelinated axons. By week 6, robust human MBP expression was detected in all animals (n = 14), which persisted at later time points (n = 24). Hence, in asymptomatic neonates, progressive myelination that became widespread after a delay of about 6 to 7 weeks was observed.
In major white matter tracts, the diffuse engraftment of human SC121+ cells (Fig. 4, A, C, and E) coincided with extensive donor-derived myelination (Fig. 4, B, D, and F). For example, HuCNS-SC transplants in the anterior cortex and corpus callosum migrated diffusely within the cortex and corpus callosum (Fig. 4E), but MBP staining was restricted to the corpus callosum (Fig. 4F). Thus, site-specific differentiation of human oligodendrocytes occurred in the white matter tracts of Shi-id mice, where human oligodendrocytes derived from HuCNS-SCs competed with endogenous mutant mouse oligodendrocytes to myelinate host axons.
Fig. 4.

Production of myelin after HuCNS-SC transplant into Shi-id mice. Representative images from neonatal Shi-id mice after HuCNS-SC transplantation. (A) SC121 staining (brown) revealed diffuse human stem cell engraftment in cerebellar white matter 9 weeks after transplant. (B) MBP staining (brown) in an adjacent section of the cerebellum revealed extensive myelination. (C and E) SC121 staining (brown) revealed diffuse engraftment of HuCNS-SCs in the fimbria of the fornix (C) and in the cortex (CTX) and corpus callosum (E) at 12 weeks after transplant. (D and F) Corresponding MBP staining in the fimbria of the fornix and in the cortex and corpus callosum. Regional boundaries are delineated with methyl green counterstain.
The onset of myelination in all CNS white matter tracts occurred progressively after an initial delay associated with migration and engraftment of HuCNS-SCs. In the cerebellum, human cells engrafted robustly at 2 weeks after transplant (fig. S3A), but no MBP+ oligodendrocytes were detected (fig. S3B) and few were present at 4 weeks (fig. S3, C and D). By 8 weeks, human cells were dispersed widely in the white matter, granule cell layer, and molecular cell layer (fig. S3E), but diffuse human MBP expression was restricted to the white matter (fig. S3F).
HuCNS-SCs induce myelination in symptomatic juvenile Shi-id mice
HuCNS-SC transplants into asymptomatic neonatal mice resulted in robust commitment of these cells to the oligodendrocyte lineage. Next, we investigated whether the white matter of juvenile Shi-id mice (post-natal day 21), which had advanced dysmyelination and progressive ataxia, would be similarly permissive for differentiation of HuCNS-SCs into mature human myelinating oligodendrocytes. HuCNS-SC–derived myelination was analyzed in the corpus callosum, fimbria of the fornix, and cerebellar white matter of transplanted juvenile mice. The engraftment and migration patterns of HuCNS-SC transplants at each injection site in juvenile mice were very similar to those of transplanted neonatal mice. All transplanted animals with engraftment confirmed by SC121 staining exhibited robust staining for human MBP in the cerebellum and corpus callosum by 6 and 10 weeks, respectively, after transplantation (fig. S4, A to D).
To obtain an unbiased measure of staining for human MBP in the transplanted neonatal and juvenile animals at successive time periods after transplant, we generated high-resolution two-dimensional (2D) montages of MBP staining (Fig. 5A) to quantify the fraction of the cerebellar white matter that expressed human MBP (Fig. 5B). Whereas low concentrations of MBP were detected at 2 to 3 weeks after transplant in both neonatal and juvenile animals, there was a progressive increase in MBP expression at 5 to 6 weeks that persisted at 7 to 9 weeks. There was no significant difference in human-derived MBP expression between neonatal and juvenile mice at any time point [general linear model (GLM); P > 0.38].
Fig. 5.
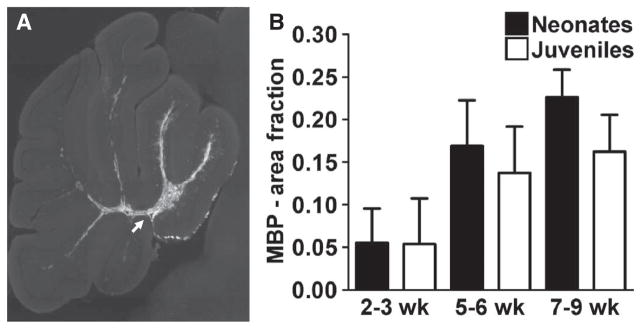
Oligodendrocyte differentiation and myelination after HuCNS-SC transplant. (A) A representative section showing MBP staining of cerebellar white matter tracts 6 weeks after transplant in a neonatal animal (arrow). (B) Quantitation of MBP staining of cerebellar white matter in transplanted neonatal and juvenile mice over time. There were no significant differences in the magnitude of MBP staining between neonates and juveniles at any time point. Neonate: n = 2 (2 to 3 weeks), n = 6 (5 to 6 weeks), n = 3 (7 to 9 weeks); juvenile: n = 3 (2 to 3 weeks), n = 5 (5 to 6 weeks), n = 5 (7 to 9 weeks).
We next assessed whether dysmyelination within the brains of juvenile Shi-id mice induced microglial activation, which could be further exacerbated by HuCNS-SC transplants. We visualized expression of the microglial marker Iba1, a calcium-binding protein that is commonly used to monitor the status of microglial activation (fig. S5). Transplantation of HuCNS-SCs into the brains of either neonatal (fig. S5, A to D) or juvenile (fig. S5, E to H) Shi-id mice resulted in similar microglial activation compared to nontransplanted Shi-id animals (fig. S5, I and J), consistent with results obtained in other animal models (12, 13, 15). Nontransplanted brains of Shi-id mice stained more intensely for Iba1 expression than did nontransplanted brains of wild-type id mice that did not carry the Shi mutation (fig. S5, K and L).
HuCNS-SC–derived myelin normalizes protein distribution at the nodes of Ranvier
We next analyzed whether there was reorganization of protein distribution at the nodes of Ranvier of axons because of HuCNS-SC–derived myelination. We visualized expression of the paranodal contactin-associated protein (Caspr), a component of axonal-glial junctions situated adjacent to the nodes of Ranvier, and of the potassium channel Kv1.2, situated in the juxtaparanodal region flanking Caspr distribution. In both transplanted neonatal and juvenile Shi-id mice, Caspr was sparsely distributed within cerebellar white matter (Fig. 6A) and showed a restricted distribution adjacent to the nodes of Ranvier (Fig. 6, B and C). In contrast, Kv1.2 was often diffusely distributed along the internodes (Fig. 6B), consistent with previous observations in the Shi brain (22) and spinal cord (23). In response to HuCNS-SC–derived myelination, there was a pronounced increase in the amount and distribution of Caspr in the cerebellar white matter (Fig. 6D) in both groups. Caspr was more extensively expressed along the paranodal region (Fig. 6, E and F), whereas Kv1.2 was more focally distributed. HuCNS-SC–derived myelination was associated with normalization of the nodes of Ranvier in both neonatal and juvenile animals.
Fig. 6.

HuCNS-SC–derived myelination in transplanted Shi-id mice. (A to F) HuCNS-SC–derived myelination resulted in a pronounced increase in the amount and distribution of the node of Ranvier-associated proteins, Caspr and Kv1.2, in cerebellar white matter. Data are from 8-week-old Shi-id mice [nontransplanted control (A to C); transplanted juvenile (D to F)]. The transition from the internal granule cell layer to the white matter is delineated with Hoechst fluorescent nuclear counterstain (blue) in (A) and (D). High-power image showing (C) restricted distribution of Caspr in a nontransplanted mouse and (F) a more diffuse distribution of Caspr in the paranodal region of axons in myelinated white matter of a transplanted mouse. (D and E) In transplanted animals, there was more Caspr (red) and Kv1.2 (green) staining in myelinated white matter compared to nontransplanted control animals (A and B). (G) Electron microscopic analysis of the cerebellar white matter of a nontransplanted Shi-id mice showing typical hypomyelination with a few loose myelin sheaths around axons. (H) Electron microscopic analysis of the cerebellar white matter of a Shi-id mouse brain 8 weeks after a HuCNS-SC transplant, showing host axons ensheathed with compact myelin derived from human oligodendrocytes. (I) Inset from (H) showing compact myelin with ~16 major dense lines. Magnifications, ×20,000 (G and H) and ×200,000 (I).
HuCNS-SC–derived oligodendrocytes generate compact myelin
To confirm that the myelination detected by MBP staining corresponded to axons ensheathed with compact myelin, we visualized by transmission electron microscopy the ultrastructural features of the myelin sheaths produced by human oligodendrocytes in the white matter tracts of the cerebellum of transplanted Shi-id mice. The normal myelin sheath consists of 20 to 30 lamellae of myelin, with major dense lines where oligodendroglial cytoplasmic membranes are in apposition (24). Formation of the major dense line requires MBP expression and is, therefore, lacking in shi mice (25). Nontransplanted Shi-id mice displayed severe hypomyelination, with zero to nine loose myelin wrappings with frequent disruption of the intraperiod lines (Fig. 6G). Shi-id mice engrafted with HuCNS-SCs displayed ultrastructurally normal myelin sheaths that had ~16 layers with no disruptions of the major dense line (Fig. 6, H and I). Thus, transplanted HuCNS-SCs were able to engraft and differentiate into oligodendrocytes in a site-specific manner, which then formed normal-appearing myelin sheaths with major dense lines confirming that host myelin was donor-derived.
Transplanted HuCNS-SCs generate myelin that enhances CNS conduction velocity
Normalization of the expression and distribution of nodal proteins suggested that transplanted HuCNS-SCs generate functional myelin. To assess the functionality of newly formed myelin, we performed compound action potential (CAP) recordings with in vitro tissue slices containing the corpus callosum of the Shi-id mouse brain. Fluorescent dextran beads were coinjected with HuCNS-SCs into the rostral corpus callosum of neonatal and juvenile Shi-id mice to guide the placement of stimulating and recording electrodes (Fig. 7A). CAP recordings in the corpus callosum of wild-type mice typically produce robust signals that exhibit a double peak waveform: one short latency peak corresponding to fast conducting myelinated axons and a longer latency peak corresponding to slowly conducting unmyelinated axons (26). The CAP recordings from the vehicle control (Fig. 7C) or from control human liver–derived mesenchymal stromal cells (hlMSCs) injected into Shi-id mouse brains were small, and responses were not readily detected if the electrodes were placed 1.0 mm or farther apart. At 1.0-mm separation, the response often required signal averaging to resolve it, suggesting a high degree of conduction failure. At a separation distance of 0.5 mm, small single peaked CAP responses were typically detected (for example, Fig. 7B). Large fast CAP responses were always observed in corpus callosum sections that contained transplanted HuCNS-SCs (Fig. 7, B and C). Although the CAP responses were smaller at 1.0-mm electrode separations, they were above baseline and did not require signal averaging to resolve them. With few exceptions, the CAP responses in tissues with transplanted HuCNS-SCs had a single peak.
Fig. 7.

CAP recordings. CAP recordings were made in in vitro slice cultures of white matter axons within the corpus callosum taken from transplanted Shi-id mice. hlMSCs were injected into the brains of Shi-id mice to serve as a non-CNS stem cell control. (A) Typical placement of the stimulation (s) and recording (r) electrodes in the corpus callosum. Fluorescein-dextran beads (green) were coinjected with the cells in all studies to mark the injection sites for subsequent recording. (B) A sample recording from the corpus callosum taken from juvenile Shi-id mice transplanted with HuCNS-SCs or hlMSCs. (C) (Left panel) Quantification of CAP amplitudes in the corpus callosum from neonatal and juvenile Shi-id mice transplanted with HuCNS-SCs or vehicle control. (Right panel) Quantification of CAP amplitudes in the corpus callosum from juvenile Shi-id mice transplanted with HuCNS-SCs or hlMSCs as a control (*P < 0.05, Student’s t test).
After CAP recordings, tissue sections were processed for immunohistochemistry to visualize mature human oligodendrocytes expressing human MBP. Neither vehicle control–injected animals nor hlMSC control–injected animals demonstrated any MBP immunoreactivity. However, there was extensive MBP labeling in sections containing transplanted HuCNS-SCs that demonstrated increased conduction velocities (fig. S6, A and B). These data confirm that mice receiving HuCNS-SC transplants showed increased conduction velocities that were linked to human-derived myelination.
HuCNS-SC myelination results in fractional anisotropy changes detected by magnetic resonance imaging
Increases in water diffusion anisotropy [quantified using fractional anisotropy (FA)] have been used as a marker to determine the magnitude and extent of myelination in the developing human CNS (27, 28). Magnetic resonance imaging (MRI) studies have been problematic in adult Shi-id mice because they are susceptible to fatal seizures triggered by excessive stimulation. To circumvent this limitation, we used ex vivo high-field (11.7 T) MRI to analyze changes in tissue microstructure associated with HuCNS-SC–derived myelination at 5 to 7 weeks after transplant in neonatal and juvenile Shi-id mice.
To determine whether early myelin-associated changes in water diffusion could be detected by diffusion-weighted MRI, we acquired unbiased measurements of FA derived from cerebellar white matter without knowledge of the extent of myelination in animals transplanted as neonates or juveniles. Subsequent to the FA measurements, we estimated the magnitude of myelination by quantification of MBP. Human myelination resulted in microstructural changes in cerebellar white matter tracts that were detected as an increase in FA. Figure 8A shows a representative parasagittal FA map at one level of the entire brain from a neonatal animal at 6 weeks after transplantation. White matter segmentation for the FA map was generated based on the corresponding T2-weighted image (fig. S7) and was used to identify regions of white matter that corresponded to human myelination (red) or to an absence of myelin (green) (Fig. 8B), as defined by the corresponding high-resolution montage of cerebellar MBP staining (Fig. 8C, arrow).
Fig. 8.

High-field MRI analysis of HuCNS-SC–derived myelination. Human myelination after HuCNS-SC transplant into Shi-id mice resulted in microstructural changes in cerebellar white matter tracts identified as an increase in FA. (A) Representative low-power parasagittal FA map at one level of the entire brain from a neonatal animal at 6 weeks after transplant. Arrow indicates the cerebellar region shown in (B). (B) Detailed view of the FA map of the cerebellum. A white matter segmentation for the FA map was generated based on the corresponding T2-weighted image and was used to identify the regions of cerebellar white matter that corresponded to human myelination (red) or to an absence of myelin (green). (C) A high-resolution image of cerebellar MBP staining (arrow) at the level of the FA maps in (A) and (B). (D) FA data from regions of white matter segmentation corresponding to myelin (MBP+) or absence of myelin (MBP−). Data are derived from MRI analyses of neonatal (n = 4) and juvenile (n = 4) transplanted Shi-id mice. (E) There was an increase in FA in regions of human myelination (MBP+; red) relative to regions without myelin (MBP−; green) in both neonatal and juvenile transplanted mice (*P < 0.0001; F = 56; mixed-effects model; **P < 0.047, ***P < 0.001).
When FA data were analyzed across an entire cohort of eight transplanted neonatal and juvenile animals (Fig. 8D), there was a significant increase in FA for the regions corresponding to myelination (MBP+) relative to the un-myelinated regions (MBP−) (*P < 0.0001; F = 56; mixed-effects model). In Fig. 8E, similar significant increases in FA in regions of human myelination (MBP+; red) relative to regions without myelin (MBP−; green) were observed for both the neonatal (**P < 0.047) and the juvenile (***P < 0.001) mice. Hence, early white matter microstructural changes associated with HuCNS-SC–derived myelin can be detected by MRI-based FA measurements at 5 to 7 weeks after transplantation in both neonatal and juvenile Shi-id mice.
DISCUSSION
We show that purified, expanded, and banked fetal human neural stem cells generate mature compact myelin in hypomyelinated Shi-id mice with severe CNS dysmyelination. Expanded, cryopreserved, banked HuCNS-SCs, prepared using methods similar to those used for clinical applications, remained competent for engraftment. They showed migration along major white matter tracts of the mouse forebrain and hindbrain and preferential differentiation into oligodendrocytes in a site-appropriate manner, resulting in production of mature compact myelin. HuCNS-SC–derived oligodendrocytes showed a similar competence for generating functional myelin both in asymptomatic neonatal mice and in juveniles that displayed severe dysmyelination and early-onset progressive ataxia.
These findings suggest that it may be possible to extend the potential window for human CNS stem cell therapy to patients with myelin disorders beyond infancy. Particularly in young children, dysmyelinating disorders such as Pelizaeus-Merzbacher disease (PMD) or Krabbe disease often trigger a progressive irreversible clinical deterioration after a relatively occult period of dysmyelination (29). We tested HuCNS-SC transplants in the Shi-id mouse model of severe dysmyelination, where the occult period in neonates was compared with a period of rapid progressive decline in juveniles. Symptomatic dysmyelination in Shi mice is accompanied by disrupted axonal function, despite a lack of overt axonal loss (1, 30, 31). Shi mice display disrupted maturation of the axonal cytoskeleton (30) and increased numbers of axonal mitochondria (32), which may predispose to abnormal axon trafficking, metabolic stress, and increased susceptibility to excitotoxicity (33). The normalization of the nodes of Ranvier and the increase in axonal conduction velocities in the brains of transplanted juvenile Shi-id mice suggest that there is a potential window for axonal rescue during the initial phase of progressive dysmyelination. Future studies are needed to further define the developmental window during which therapeutic benefit can be achieved.
Dysmyelination also is associated with a compensatory increase in oligodendrocyte progenitor cell proliferation. Increased rates of oligodendrocyte progenitor cell proliferation occur in the Shi mouse relative to wild-type animals (34). Our results suggest that after transplant into mouse brains, human oligodendrocyte progenitors derived from HuCNS-SCs may respond similarly to host oligodendrocyte progenitors and undergo proliferation in dysmyelinated white matter. A low level of cell division was observed in the transplanted human cells, suggesting that HuCNS-SCs may slowly replenish the stem cell reservoir and provide a renewable source of oligodendrocytes for myelination. Notably, myelination by human oligodendrocytes in the Shi-id mouse model (and in a spinal cord injury model) requires that the functionally competent human wild-type oligodendrocytes outcompete the non-myelinating shiverer oligodendrocytes to myelinate and repair the affected tissue. It will be important to determine the cellular and molecular mechanisms underlying successful xenogeneic oligodendrocyte myelination of mouse axons and how these mechanisms are disrupted in shiverer oligodendrocytes.
There are limitations to using the Shi-id mouse model, the principal one being that neurobehavioral outcomes after HuCNS-SC transplant could not be assessed. We encountered a relatively narrow window between the onset of early HuCNS-SC–derived myelination at 5 to 6 weeks after transplantation and the death of the animals. Previous studies have shown that diffuse myelin synthesis in the white matter of Shi mice requires the backcross of homozygous Shi mice into a Rag2−/− background that prolongs survival to twice the survival time of the Shi-id mice used in this study (8). With the milder phenotype of the Shi-Rag2−/− mice, progressive myelination by freshly isolated fetal human oligodendrocyte progenitor cells continued to occur for a period of several months (8). Freshly isolated human oligodendrocyte progenitors did not alleviate seizures in the Shi-Rag2−/− mice at 18 to 21 weeks of age; full rescue from seizures was not achieved until 1 year of age. Thus, both banked HuCNS-SCs and freshly isolated fetal human oligodendrocyte progenitor cells displayed a similar delay after transplantation before the initiation of myelination.
It is unclear whether this 5- to 6-week interval between stem cell engraftment and myelination is related to factors intrinsic to the murine CNS or to the human-derived oligodendrocytes. In rodents, there is normally a relatively short period of ~2 weeks between birth and the onset of myelination (35). The factors that regulate the tempo of oligodendrocyte lineage progression in humans are unknown, but premyelinating oligodendrocytes are detected in human cerebral white matter ~4 to 8 weeks before the onset of myelination in parietal white matter (36). In contrast to rodents, human oligodendrocytes may be programmed for a more prolonged period between oligodendrocyte commitment and the onset of myelination. By contrast, migration and oligodendrocyte lineage commitment of HuCNS-SCs transplanted into mouse brain appears to be site-directed rather than autonomous. In the mouse olfactory bulb, for example, engrafted human cells did not differentiate into oligodendrocytes, consistent with the notion that the local environment (that is, white matter versus non–white matter) influenced HuCNS-SC cell fate decisions. Dysmyelinated white matter represents an abnormal and degenerative microanatomy; transplantation of HuCNS-SCs into these regions could lead to microglial activation after transplantation. However, transplantation of HuCNS-SCs did not induce additional inflammation or microglial activation in this hypomyelination model. This is consistent with our previous studies in mice with ischemic stroke (12, 15) or neuronal ceroid lipofuscinosis with massive neuronal loss (13), where transplanted HuCNS-SCs likely provided both a regenerative and an immunomodulatory response that coincided with decreased microglial reactivity.
Consistent with previous MRI studies with rodent-derived neural stem cells (37), we found that transplant-derived myelin was not associated with a reduction in the T2 signal despite an increase in FA. Although previous MRI studies have detected the migration of rodent neural stem cells labeled with contrast agents or nanoparticles (12, 38, 39), our study shows de novo generation of human neural stem cell–derived myelin in both neonatal and juvenile mice detected by MRI without cell labeling. Currently, human neural stem cell engraftment and differentiation in clinical trials can only be confirmed at autopsy. Our findings raise the possibility that FA measurements in serial MRIs could serve as a surrogate outcome measure to follow the progression of CNS myelination in living patients.
The myelination of mouse axons by HuCNS-SC–derived oligodendrocytes indicates that the transplanted human neural stem cells were viable and able to compete successfully with the numerous endogenous mutant oligodendrocytes present in the brains of the Shi-id mice (3). A pronounced increase in human-derived myelin resulted in diffuse normalization of the architecture of the nodes of Ranvier and adjacent paranodes. Moreover, the axonal sheaths that were generated displayed the normal ultrastructure of compact myelin and had enhanced axonal conduction velocities. In a related study in this issue, Gupta et al. (40) transplanted HuCNS-SCs (that is, the clinical product manufactured under GMP) into patients with PMD, a severe progressive dysmyelinating disorder. Their MRI studies demonstrated consistent durable and progressive myelin production after transplant. Collectively, our data suggest the potential of human neural stem cell transplants for treating patients with PMD or other progressive dysmyelinating disorders. There are a number of nonprogressive myelin-associated disorders that also may be appropriate candidates for donor cell–derived myelination. These include acquired disorders of white matter injury such as cerebral palsy in preterm survivors, where disturbances in oligodendrocyte progenitor maturation occur (41), and demyelination after spinal cord injury.
MATERIALS AND METHODS
Transplantation of HuCNS-SCs
HuCNS-SCs were generated as described (9, 11, 19) under non-GMP conditions. Cells were cultured at a density of 105/ml in X-VIVO 15 medium (Lonza) supplemented with N2, heparin, N-acetylcysteine, fibroblast growth factor 2, epidermal growth factor (20 ng/ml), and leukemia inhibitory factor (10 ng/ml; Millipore). When neurosphere size reached 200 to 250 μm, cultures were passaged by collagenase treatment and replated in the same medium.
For transplantation, a suspension of HuCNS-SCs (105 cells in 1 μl per site) was prepared and transplanted bilaterally into the corpus callosum, fimbria of the fornix, and cerebellar white matter of neonatal and juvenile Shi-id mice (13). Detailed procedures are provided in Supplementary Materials and Methods.
Immunohistochemical analysis of transplanted mouse brains
Transplanted mice were anesthetized and perfused, and brain sections were processed as described in the Supplementary Materials and Methods. Brain sections of transplanted mice were stained with various antibodies to reveal the distribution and morphology of transplanted cells (table S1). Protocols for ultrastructural studies are described in the Supplementary Materials and Methods.
The percentage of HuCNS-SCs committed to the oligodendrocyte lineage was quantified by double staining for SC101 and Olig2. Oligodendrocytes derived from HuCNS-SCs were identified by immunofluorescent double staining overnight at 2 to 4°C for the proprietary mouse monoclonal antibody SC101, a human-specific nuclear marker (1:250), and a rabbit antiserum to the nuclear transcription factor Olig2 (1:20,000; gift of J. Alberta, Dana-Farber Cancer Institute, Boston, MA). No antigen retrieval or detergents were required to optimize staining. Cell counts were done from a minimum of 10 randomly acquired images that were acquired with a 20× objective on a Leica DMRA upright fluorescence microscope. Each image was acquired as a z stack that captured the full depth of immunohistochemical labeling for both markers. Counts of Olig2+ cells that were single- or double-labeled for SC101 in each transplant site were obtained from a minimum of three adjacent sections. For each site, a target count of 100 to 300 cells was obtained per animal, and the percentage of SC101-labeled cells that also labeled for Olig2 was calculated.
Cell fate and proliferation status measurements were performed by immunofluorescent double staining using the human nuclear marker SC101, with antibodies against NeuN, S100β, or Ki67. Ki67 is a commonly used marker expressed in actively proliferating cells in G1, S, G2, or M phases, but not in G0 (42, 43). Confocal immunofluorescence microscopy was performed on a Leica SP2 AOBS microscope (Leica Microsystems). For characterization of cell fate and proliferation status, the resulting image stacks were analyzed with Volocity software (Improvision). To confirm the colocalization of the labeled antigens within a cell, we inspected the fluorescent staining in the z dimension using the orthogonal view tool.
Analysis of node of Ranvier formation was performed with a rabbit polyclonal antiserum against Caspr (a gift from E. Peles, Weizmann Institute of Science, Rehovot, Israel) and a mouse monoclonal antibody against the potassium channel Kv1.2 (NeuroMab) (see Supplementary Materials and Methods).
CAP recordings
CAP recordings were obtained with methods similar to those described previously (26, 44). Mice were anesthetized with isoflurane (Novaplus) and decapitated. Brains were rapidly removed and submerged in ice-cold artificial cerebrospinal fluid (aCSF; 124 mM NaCl, 5 mM KCl, 1.25 mM NaH2PO4, 26 mM NaHCO3, 1.3 mM MgSO4, 2 mM CaCl2, 10 mM glucose, pH 7.4) saturated with 95% O2/5% CO2. Coronal slices, 400 μm thick, were cut on a vibratome (Leica) and maintained in aerated aCSF at room temperature for 1 to 3 hours before recording.
For recording, slices were transferred to a recording chamber mounted on a Zeiss upright microscope equipped with epifluorescence to confirm the location of fluorescent dextran beads used to identify the injection sites of HuCNS-SCs. Recording and stimulating electrodes were positioned in the corpus callosum on either side of the cell injection sites. The bipolar stimulating electrodes were fashioned from Teflon-insulated tungsten wires (Word Precision Instruments), with the tips positioned ~0.3 mm apart. The stimulating electrodes were connected to a Grass stimulation isolation unit and a Grass S88 stimulator. Stimulus intensity was adjusted to obtain a maximal CAP response. Recording electrodes were fashioned from glass tubing (Word Precision Instruments) with a Flaming/Brown P-97 microelectrode puller. When filled with aCSF, they had a resistance of 3–5 megohm. Recordings were obtained from three sites across the thickness of the corpus callosum at 0.5 and 1.0 mm from the stimulating electrode. Ten responses were averaged at each recording site. Recordings were amplified with a Multiclamp 700B (Axon Instruments) and filtered at 10 kHz. The magnitude of the CAP waveform was determined by measuring the maximum negative deflection with respect to a tangent drawn between the adjacent positive deflections (44). The amplitudes of the CAP recordings were compared between Shi-id mice injected with HuCNS-SCs, vehicle control, or hlMSC control. hlMSCs were derived from liver tissue of gestational 16 weeks of age. hlMSCs were CD105+, CD90+, CD73+, CD166+, CD34−, and CD45−. Recordings were performed in a blinded fashion.
Ex vivo high-field MRI
Brains were collected from four neonatal animals that survived until 6 (n = 2) or 7 weeks (n = 2) and from four juvenile animals that survived until 5 (n = 1), 6 (n = 2), or 7 weeks (n = 1). Brains were hemisected in the sagittal plane. Tissue was embedded alongside a corresponding control tissue block in 0.5% agarose and immersed in phosphate-buffered saline within a 4-cm-diameter plexiglass tube. A custom single-turn solenoidal coil (diameter, 5 cm; length, 5 cm) was used for radio-frequency transmission and reception. Experiments were performed with an 11.7-T magnet interfaced with a 9–cm–inner diameter magnetic field gradient coil (Bruker). Detailed scanning and image segmentation procedures are provided in the Supplementary Materials and Methods, and generally followed the previously published strategy that used diffusion tensor imaging to characterize postmortem brain tissue from other species (45, 46).
Generation of histopathology images
2D montages (5×) were generated from entire tissue sections stained for MBP with a Leica DMIRE2 inverted fluorescence microscope (Leica Microsystems) and Stereo Investigator stereology system (MBF Bioscience) and saved as an 8-bit tiff image.
Calculation of MBP area fraction
MBP quantification was calculated as an area fraction (MBP+ pixels/ white matter pixels) (41). Montages were cropped to analyze only the cerebellum. A cerebellar white matter region of interest (ROI) was traced with ImageJ. The MBP area was determined in ImageJ with image thresholding that created a binary mask of MBP+ versus MBP− pixels. Slices not showing MBP staining were excluded unless they stained positively for SC121, which confirmed successful stem cell engraftment.
Definition of ROIs
Each montage was visually matched to a corresponding sagittal MRI slice. A white matter ROI was drawn on each MRI slice with the amplitude map and ITK Snap. Histological MBP+ segmentations were superimposed over the MRI white matter segmentation with the histopathalogical montage as a reference. Any white matter ROI not marked as MBP+ was designated MBP−.
Quantification of MBP in brains imaged by MRI
MBP was visualized with SMI-99P (1:1000; Covance) (41). SMI-99P does not recognize epitopes encoded by deleted exons in the MBP gene. It does not cross-react with the abnormal myelin present in the shi/shi CNS but detected MBP derived from human donor cells. The proprietary monoclonal antibody SC121 (STEM121 SC Proven; Stem Cell Sciences AB-121-U-050) visualized engraftment and migration of human cells.
After MRI, tissue blocks were serially sectioned at 50 μm with a VTS1000 vibrating microtome (Leica) and stained for MBP, which was analyzed in an unbiased fashion as adapted from previously described methods (41, 53). Using a motorized x, y stage, we captured montages of the entire MBP-stained section at ×5 magnification (DMIRE2 inverted fluorescence microscope, Leica) using an Orca ER–cooled charge-coupled device camera (Hamamatsu) and Stereo Investigator (MicroBrightField Inc.). MBP staining was analyzed within the boundaries of the entire white matter tract defined by Hoechst 33342 fluorescent counterstain. MBP area fraction was determined from a pixel intensity histogram, generated for each ROI with ImageJ (National Institutes of Health; http://rsbweb.nih.gov/ij/index.html), as described (41).
Statistical analysis
Data analysis was performed with SAS software 9.2 (SAS Institute Inc.). Box and whisker plots represent the 25th and 75th percentiles, median (central line), and mean (central box). The necessary assumptions (for example, normality and equal variance) were tested before the following analyses were performed. Analysis of MBP area fraction for neonatal and juvenile animals was performed with a GLM, where the main-effect term was group designation (neonate versus juvenile) and the interaction term was the survival time after transplant. Pairwise comparisons of CAP amplitude were performed with unpaired two-tailed t tests. For analysis of the MRI-MBP association studies, a mixed-effects model was used to analyze the entire cohort of neonatal and juvenile MRI studies. Group analyses were tested for independent effects of age, survival time, transplant group, and number of MRI slices analyzed, and no confounding effects were found. Analysis of the separate cohorts of neonatal versus juvenile animals for differences in MBP+ versus MBP− ROIs was analyzed by pairwise comparisons. All statistical tests were considered significant at a P = 0.05 level of error.
Supplementary Material
Acknowledgments
We thank D. Anderson, A. Capela, F. Gage, T. Gorba, and S. Huhn for expert advice and E. Apilado, E. Pali, J. Tang, R. Ibeid, and M. Piechowiak for technical support. Funding: Supported by NIH grants P51RR000163 (to C.D.K.) and NCRR P51 RR000113 (to L.S.S.), National Institute of Neurological Disorders and Stroke grants 1RO1NS054044 and R37NS045737-06S1/06S2 (to S.A.B.) and 1F30NS066704 (to A.R.), a Bugher Award from the American Heart Association (to S.A.B.), March of Dimes Birth Defects Foundation (to S.A.B.), and Friends of Doernbecher Children’s Hospital Foundation (to S.A.B.). High-field MRI instrumentation used in this work was purchased with support from the W. M. Keck Foundation.
Footnotes
Author contributions: N.U., B.J.C., C.D.K., A.J.A., S.J.T., A.T., I.L.W., L.S.S., and S.A.B. conceived and designed the experiments and coordinated the work presented. S.J.T. generated Shi-id mice and performed transplantation. M.D. and X.G. were responsible for histological studies and related fluorescent immunohistochemical double-labeling studies. M.D. performed immunohistochemistry, confocal microscopy, cell fate, and proliferation analyses. K.D.H. and J.D. analyzed nodes of Ranvier. D.J.B. and Y.W. performed cell counts to analyze commitment to the oligodendrocyte lineage. T.N. performed biostatistical analyses related to the MRI studies and quantification of myelin. B.J.C. and A.J.A. directed the ultrastructural studies. Electrophysiolgy studies were performed and analyzed by S.G.M. and L.S.S. MRI studies were designed by C.D.K., and data were collected and analyzed by K.C., A.R., and J.R.B. A.J.A, A.T., and I.L.W. commented on and edited the manuscript. N.U., L.S.S., C.D.K., and S.A.B. wrote the manuscript.
Competing interests: N.U., M.D., and A.T. are employees of StemCells Inc.; S.A.B. was previously a paid consultant to StemCells Inc.; I.L.W. is Chair of the Scientific Advisory Board, co-founder, and Director of StemCells Inc. StemCells Inc. holds patents related to this work regarding human CNS neural stem cell lines and neurospheres. The other authors declare that they have no competing interests.
www.sciencetranslationalmedicine.org/cgi/content/full/4/155/155ra136/DC1
Materials and Methods
Table S1. Antibodies used for histological and immunohistochemical studies.
Fig. S1. HuCNS-SC characteristics before and after transplantation.
Fig. S2. Production of myelin from 10 different HuCNS-SC lots.
Fig. S3 Time course of HuCNS-SC engraftment and oligodendrocyte differentiation in vivo.
Fig. S4. Juvenile transplantation of HuCNS-SCs.
Fig. S5. Microglia activation status of chronic transplants in Shi-id mouse brains.
Fig. S6. CAP recordings.
Fig. S7. Representative MRI images and analyses in the Shi-id mouse transplanted with HuCNS-SCs.
REFERENCES AND NOTES
- 1.Nave KA. Myelination and the trophic support of long axons. Nat Rev Neurosci. 2010;11:275–283. doi: 10.1038/nrn2797. [DOI] [PubMed] [Google Scholar]
- 2.Duncan ID, Kondo Y, Zhang SC. The myelin mutants as models to study myelin repair in the leukodystrophies. Neurotherapeutics. 2011;8:607–624. doi: 10.1007/s13311-011-0080-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roach A, Takahashi N, Pravtcheva D, Ruddle F, Hood L. Chromosomal mapping of mouse myelin basic protein gene and structure and transcription of the partially deleted gene in shiverer mutant mice. Cell. 1985;42:149–155. doi: 10.1016/s0092-8674(85)80110-0. [DOI] [PubMed] [Google Scholar]
- 4.Katsuki M, Sato M, Kimura M, Yokoyama M, Kobayashi K, Nomura T. Conversion of normal behavior to shiverer by myelin basic protein antisense cDNA in transgenic mice. Science. 1988;241:593–595. doi: 10.1126/science.2456614. [DOI] [PubMed] [Google Scholar]
- 5.Gansmuller A, Clerin E, Krüger F, Gumpel M, Lachapelle F. Tracing transplanted oligodendrocytes during migration and maturation in the shiverer mouse brain. Glia. 1991;4:580–590. doi: 10.1002/glia.440040605. [DOI] [PubMed] [Google Scholar]
- 6.Mathisen PM, Pease S, Garvey J, Hood L, Readhead C. Identification of an embryonic isoform of myelin basic protein that is expressed widely in the mouse embryo. Proc Natl Acad Sci USA. 1993;90:10125–10129. doi: 10.1073/pnas.90.21.10125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Windrem MS, Nunes MC, Rashbaum WK, Schwartz TH, Goodman RA, McKhann G, II, Roy NS, Goldman SA. Fetal and adult human oligodendrocyte progenitor cell isolates myelinate the congenitally dysmyelinated brain. Nat Med. 2004;10:93–97. doi: 10.1038/nm974. [DOI] [PubMed] [Google Scholar]
- 8.Windrem MS, Schanz SJ, Guo M, Tian GF, Washco V, Stanwood N, Rasband M, Roy NS, Nedergaard M, Havton LA, Wang S, Goldman SA. Neonatal chimerization with human glial progenitor cells can both remyelinate and rescue the otherwise lethally hypomyelinated shiverer mouse. Cell Stem Cell. 2008;2:553–565. doi: 10.1016/j.stem.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, Tsukamoto AS, Gage FH, Weissman IL. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci USA. 2000;97:14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelly S, Bliss TM, Shah AK, Sun GH, Ma M, Foo WC, Masel J, Yenari MA, Weissman IL, Uchida N, Palmer T, Steinberg GK. Transplanted human fetal neural stem cells survive, migrate, and differentiate in ischemic rat cerebral cortex. Proc Natl Acad Sci USA. 2004;101:11839–11844. doi: 10.1073/pnas.0404474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cummings BJ, Uchida N, Tamaki SJ, Salazar DL, Hooshmand M, Summers R, Gage FH, Anderson AJ. Human neural stem cells differentiate and promote locomotor recovery in spinal cord-injured mice. Proc Natl Acad Sci USA. 2005;102:14069–14074. doi: 10.1073/pnas.0507063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guzman R, Uchida N, Bliss TM, He D, Christopherson KK, Stellwagen D, Capela A, Greve J, Malenka RC, Moseley ME, Palmer TD, Steinberg GK. Long-term monitoring of transplanted human neural stem cells in developmental and pathological contexts with MRI. Proc Natl Acad Sci USA. 2007;104:10211–10216. doi: 10.1073/pnas.0608519104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tamaki SJ, Jacobs Y, Dohse M, Capela A, Cooper JD, Reitsma M, He D, Tushinski R, Belichenko PV, Salehi A, Mobley W, Gage FH, Huhn S, Tsukamoto AS, Weissman IL, Uchida N. Neuroprotection of host cells by human central nervous system stem cells in a mouse model of infantile neuronal ceroid lipofuscinosis. Cell Stem Cell. 2009;5:310–319. doi: 10.1016/j.stem.2009.05.022. [DOI] [PubMed] [Google Scholar]
- 14.Salazar DL, Uchida N, Hamers FP, Cummings BJ, Anderson AJ. Human neural stem cells differentiate and promote locomotor recovery in an early chronic spinal cord injury NOD-scid mouse model. PLoS One. 2010;5:e12272. doi: 10.1371/journal.pone.0012272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horie N, Pereira MP, Niizuma K, Sun G, Keren-Gill H, Encarnacion A, Shamloo M, Hamilton SA, Jiang K, Huhn S, Palmer TD, Bliss TM, Steinberg GK. Transplanted stem cell-secreted vascular endothelial growth factor effects poststroke recovery, inflammation, and vascular repair. Stem Cells. 2011;29:274–285. doi: 10.1002/stem.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGill TJ, Cottam B, Lu B, Wang S, Girman S, Tian C, Huhn SL, Lund RD, Capela A. Transplantation of human central nervous system stem cells—Neuroprotection in retinal degeneration. Eur J Neurosci. 2012;35:468–477. doi: 10.1111/j.1460-9568.2011.07970.x. [DOI] [PubMed] [Google Scholar]
- 17.Hooshmand MJ, Sontag CJ, Uchida N, Tamaki S, Anderson AJ, Cummings BJ. Analysis of host-mediated repair mechanisms after human CNS-stem cell transplantation for spinal cord injury: Correlation of engraftment with recovery. PLoS One. 2009;4:e5871. doi: 10.1371/journal.pone.0005871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hook L, Vives J, Fulton N, Leveridge M, Lingard S, Bootman MD, Falk A, Pollard SM, Allsopp TE, Dalma-Weiszhausz D, Tsukamoto A, Uchida N, Gorba T. Non-immortalized human neural stem (NS) cells as a scalable platform for cellular assays. Neurochem Int. 2011;59:432–444. doi: 10.1016/j.neuint.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 19.Tamaki S, Eckert K, He D, Sutton R, Doshe M, Jain G, Tushinski R, Reitsma M, Harris B, Tsukamoto A, Gage F, Weissman I, Uchida N. Engraftment of sorted/expanded human central nervous system stem cells from fetal brain. J Neurosci Res. 2002;69:976–986. doi: 10.1002/jnr.10412. [DOI] [PubMed] [Google Scholar]
- 20.Steiner J, Bernstein HG, Bielau H, Berndt A, Brisch R, Mawrin C, Keilhoff G, Bogerts B. Evidence for a wide extra-astrocytic distribution of S100B in human brain. BMC Neurosci. 2007;8:2. doi: 10.1186/1471-2202-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hachem S, Aguirre A, Vives V, Marks A, Gallo V, Legraverend C. Spatial and temporal expression of S100B in cells of oligodendrocyte lineage. Glia. 2005;51:81–97. doi: 10.1002/glia.20184. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Allen ML, Grigg JJ, Noebels JL, Tempel BL. Hypomyelination alters K+ channel expression in mouse mutants shiverer and Trembler. Neuron. 1995;15:1337–1347. doi: 10.1016/0896-6273(95)90012-8. [DOI] [PubMed] [Google Scholar]
- 23.Sinha K, Karimi-Abdolrezaee S, Velumian AA, Fehlings MG. Functional changes in genetically dysmyelinated spinal cord axons of shiverer mice: Role of juxtaparanodal Kv1 family K+ channels. J Neurophysiol. 2006;95:1683–1695. doi: 10.1152/jn.00899.2005. [DOI] [PubMed] [Google Scholar]
- 24.Trapp B, Pfeiffer S, Anitei M, Kidd J. In: Myelin Biology and Disorders. Lazzarini R, editor. Elsevier Academic Press; San Diego, CA: 2004. pp. 29–55. [Google Scholar]
- 25.Readhead C, Takasashi N, Shine HD, Saavedra R, Sidman R, Hood L. Role of myelin basic protein in the formation of central nervous system myelin. Ann N Y Acad Sci. 1990;605:280–285. doi: 10.1111/j.1749-6632.1990.tb42401.x. [DOI] [PubMed] [Google Scholar]
- 26.Crawford DK, Mangiardi M, Tiwari-Woodruff SK. Assaying the functional effects of demyelination and remyelination: Revisiting field potential recordings. J Neurosci Methods. 2009;182:25–33. doi: 10.1016/j.jneumeth.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 27.Mori S, Zhang J. Principles of diffusion tensor imaging and its applications to basic neuroscience research. Neuron. 2006;51:527–539. doi: 10.1016/j.neuron.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 28.Miller SP, Ferriero DM. From selective vulnerability to connectivity: Insights from newborn brain imaging. Trends Neurosci. 2009;32:496–505. doi: 10.1016/j.tins.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Costello DJ, Eichler AF, Eichler FS. Leukodystrophies: Classification, diagnosis, and treatment. Neurologist. 2009;15:319–328. doi: 10.1097/NRL.0b013e3181b287c8. [DOI] [PubMed] [Google Scholar]
- 30.Brady ST, Witt AS, Kirkpatrick LL, de Waegh SM, Readhead C, Tu PH, Lee VM. Formation of compact myelin is required for maturation of the axonal cytoskeleton. J Neurosci. 1999;19:7278–7288. doi: 10.1523/JNEUROSCI.19-17-07278.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenbluth J. Central myelin in the mouse mutant shiverer. J Comp Neurol. 1980;194:639–648. doi: 10.1002/cne.901940310. [DOI] [PubMed] [Google Scholar]
- 32.Andrews H, White K, Thomson C, Edgar J, Bates D, Griffiths I, Turnbull D, Nichols P. Increased axonal mitochondrial activity as an adaptation to myelin deficiency in the Shiverer mouse. J Neurosci Res. 2006;83:1533–1539. doi: 10.1002/jnr.20842. [DOI] [PubMed] [Google Scholar]
- 33.Pitt D, Gonzales E, Cross AH, Goldberg MP. Dysmyelinated axons in shiverer mice are highly vulnerable to α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor-mediated toxicity. Brain Res. 2010;1309:146–154. doi: 10.1016/j.brainres.2009.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bu J, Banki A, Wu Q, Nishiyama A. Increased NG2+ glial cell proliferation and oligodendrocyte generation in the hypomyelinating mutant shiverer. Glia. 2004;48:51–63. doi: 10.1002/glia.20055. [DOI] [PubMed] [Google Scholar]
- 35.Craig A, Ling Luo N, Beardsley DJ, Wingate-Pearse N, Walker DW, Hohimer AR, Back SA. Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Exp Neurol. 2003;181:231–240. doi: 10.1016/s0014-4886(03)00032-3. [DOI] [PubMed] [Google Scholar]
- 36.Back SA, Luo NL, Borenstein NS, Volpe JJ, Kinney HC. Arrested oligodendrocyte lineage progression during human cerebral white matter development: Dissociation between the timing of progenitor differentiation and myelinogenesis. J Neuropathol Exp Neurol. 2002;61:197–211. doi: 10.1093/jnen/61.2.197. [DOI] [PubMed] [Google Scholar]
- 37.Nair G, Tanahashi Y, Low HP, Billings-Gagliardi S, Schwartz WJ, Duong TQ. Myelination and long diffusion times alter diffusion-tensor-imaging contrast in myelin-deficient shiverer mice. Neuroimage. 2005;28:165–174. doi: 10.1016/j.neuroimage.2005.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoehn M, Küstermann E, Blunk J, Wiedermann D, Trapp T, Wecker S, Föcking M, Arnold H, Hescheler J, Fleischmann BK, Schwindt W, Bührle C. Monitoring of implanted stem cell migration in vivo: A highly resolved in vivo magnetic resonance imaging investigation of experimental stroke in rat. Proc Natl Acad Sci USA. 2002;99:16267–16272. doi: 10.1073/pnas.242435499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shapiro EM, Sharer K, Skrtic S, Koretsky AP. In vivo detection of single cells by MRI. Magn Reson Med. 2006;55:242–249. doi: 10.1002/mrm.20718. [DOI] [PubMed] [Google Scholar]
- 40.Gupta N, Henry RG, Strober J, Kang SM, Lim D, Bucci M, Caverzasi E, Gaetano L, Mandelli ML, Ryan T, Perry R, Farrell J, Jeremy RJ, Ulman M, Huhn SL, Barkovich AJ, Rowitch DH. Neural stem cell engraftment and myelination in the human brain. Sci Transl Med. 2012;4:155ra137. doi: 10.1126/scitranslmed.3004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buser JR, Maire J, Riddle A, Gong X, Nguyen T, Nelson K, Luo NL, Ren J, Struve J, Sherman LS, Miller SP, Chau V, Hendson G, Ballabh P, Grafe MR, Back SA. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann Neurol. 2012;71:93–109. doi: 10.1002/ana.22627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133:1710–1715. [PubMed] [Google Scholar]
- 43.Kill IR. Localisation of the Ki-67 antigen within the nucleolus. Evidence for a fibrillarin-deficient region of the dense fibrillar component. J Cell Sci. 1996;109(Pt. 6):1253–1263. doi: 10.1242/jcs.109.6.1253. [DOI] [PubMed] [Google Scholar]
- 44.Reeves TM, Phillips LL, Povlishock JT. Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Exp Neurol. 2005;196:126–137. doi: 10.1016/j.expneurol.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 45.Kroenke CD, Bretthorst GL, Inder TE, Neil JJ. Diffusion MR imaging characteristics of the developing primate brain. Neuroimage. 2005;25:1205–1213. doi: 10.1016/j.neuroimage.2004.12.045. [DOI] [PubMed] [Google Scholar]
- 46.Kroenke CD, Taber EN, Leigland LA, Knutsen AK, Bayly PV. Regional patterns of cerebral cortical differentiation determined by diffusion tensor MRI. Cereb Cortex. 2009;19:2916–2929. doi: 10.1093/cercor/bhp061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun SW, Neil JJ, Liang HF, He YY, Schmidt RE, Hsu CY, Song SK. Formalin fixation alters water diffusion coefficient magnitude but not anisotropy in infarcted brain. Magn Reson Med. 2005;53:1447–1451. doi: 10.1002/mrm.20488. [DOI] [PubMed] [Google Scholar]
- 48.Batchelor PG, Atkinson D, Hill DL, Calamante F, Connelly A. Anisotropic noise propagation in diffusion tensor MRI sampling schemes. Magn Reson Med. 2003;49:1143–1151. doi: 10.1002/mrm.10491. [DOI] [PubMed] [Google Scholar]
- 49.Basser PJ, Pierpaoli C. Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J Magn Reson B. 1996;111:209–219. doi: 10.1006/jmrb.1996.0086. [DOI] [PubMed] [Google Scholar]
- 50.Pieper S, Lorensen B, Schroeder W, Kikinis R. The NA-MIC Kit: ITK, VTK, pipelines, grids and 3D slicer as an open platform for the medical image computing community. Proceedings of the 3rd IEEE International Symposium on Biomedical Imaging: Nano to Macro; Arlington, VA. 6 to 9 April 2006; pp. 698–701. [Google Scholar]
- 51.Gering DT, Nabavi A, Kikinis R, Hata N, O’Donnell LJ, Grimson WE, Jolesz FA, Black PM, Wells WM., III An integrated visualization system for surgical planning and guidance using image fusion and an open MR. J Magn Reson Imaging. 2001;13:967–975. doi: 10.1002/jmri.1139. [DOI] [PubMed] [Google Scholar]
- 52.Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC, Gerig G. User-guided 3D active contour segmentation of anatomical structures: Significantly improved efficiency and reliability. Neuroimage. 2006;31:1116–1128. doi: 10.1016/j.neuroimage.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 53.Back SA, Craig A, Luo NL, Ren J, Akundi RS, Ribeiro I, Rivkees SA. Protective effects of caffeine on chronic hypoxia-induced perinatal white matter injury. Ann Neurol. 2006;60:696–705. doi: 10.1002/ana.21008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.