

1. Introduction
The mitomycins (Figure 1) are a unique family of natural products with a rich history. Hata and coworkers at Kwoya Hakko Kogyo Company in Japan first isolated mitomycins A and B from Streptomyces caespitosus found in soil samples in 1956.[1-3] Although the mitomycins were isolated by researchers at Kwoya Hakko Kogyo, the absolute structure of the mitomycins was determined by Webb and Coworkers at American Cyanamid Company.[4,5] X-ray crystal structures of the mitomycins by Tulinsky also proved critical in determination of the structures of the mitomycins.[6,7] Since that time a number of other members of this family of natural products have been isolated, including FR-900482 and FR-66979 from Streptomyces sandaensis in 1988 (Figure 1).[8, 9]
Figure 1.
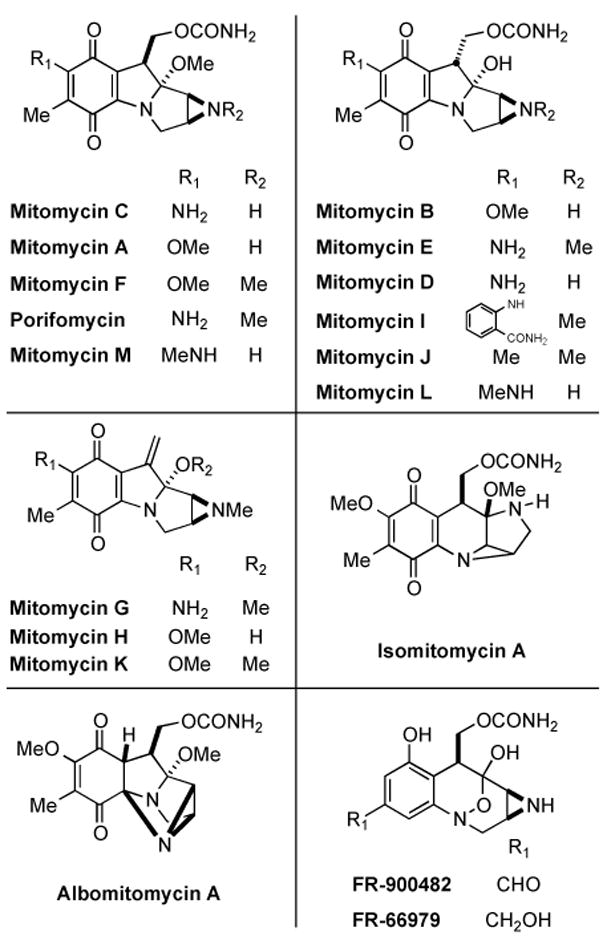
Members of the mitomycin family of natural products.
The biological activity attributed to this family of natural products is a manifestation of their ability to form both inter- and intra-strand DNA cross-links in the minor groove with a specificity favoring 5′-CG-3′ steps.[10,11] At the time of this discovery, no previous examples of natural products acting as DNA cross-linking agents were known. It was after this initial finding that several other natural products were found to also form cross-links with DNA.[12-14] Recent studies have shown that in addition to the formation of DNA cross-links, this family of compounds is also capable of forming cross-links with minor groove-binding nuclear proteins, such as high mobility group I/Y (HMG I/Y now named HMG A1) proteins.[15,16] Additionally, it was shown that monoalkylation and not cross-linking was the major adduct found when an FR-900482 derivative was incubated with nucleosomes. This result could suggest alternate modes of action of this family of natural products in the context of cellular chromatin.
2. Mode of Action
2.1. Mode of Action of the Mitomycins
The reductive pathway by which Mitomycin C (MMC) is activated to cross-link DNA has now been well established.[17,18] Researchers focused on the nature of the electron-transfer step to the quinone of MMC, and the kinetics[19] and mechanism[19-27] of ensuing chemical transformations leading to the ultimate DNA-reactive electrophile, and on enzymes involved in the activation of MMC in tumor cells has been thoroughly investigated and reported. As shown in Scheme 1, initial direct or stepwise one-electron reduction[28] of the quinone moiety of 1 (MMC) by either enzymatic or chemical means may lead to the formation of hydroquinone[29] 4 via a variety of possible intermediates 2, 3, and 5. Expulsion of the methoxy group gives iminium ion 6, which quenches itself via deprotonation/tautomerization to afford the net elimination of methanol from 4 and yielding intermediate leuco-aziridinomitosene 7. Electron donation from the hydroquinone indole core opens the aziridine ring to the intermediate quinone methide 8.
Scheme 1.
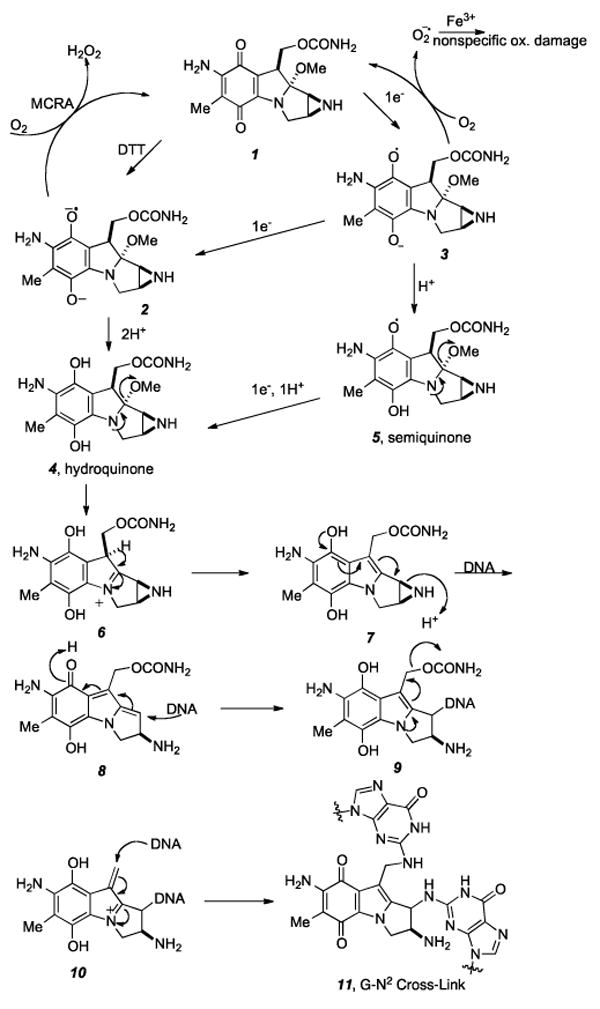
Proposed mechanism of DNA cross-linking by reductive activation of mitomycin C.
Nucleophilic attack by the exocyclic nitrogen of guanine (N2) at the C1 position leads to the first alkylation of DNA and regenerates the hydroquinone, forming 9. In a manner similar to expulsion of the C9a methoxy group, loss of the carbamate to produce the iminium species 10 unveils the second alkylation site and subsequent conjugate addition by the mono-alkylated DNA adduct affords the bis-N2-guanosine cross-link 11.
A number of intracellular flavin reductases utilizing NADH or NADPH as electron donors have been implicated in the enzymatic activation of MMC, which may proceed by either one- or two-electron reduction mechanisms. A multitude of other enzymes have also been shown to catalyze MMC activation, leading to DNA cross-linking and cytotoxicity. These include cytosolic (NQO1, GRP58[30] and other unknown cytosolic proteins) and microsomal enzymes (P-450 reductase, NADPH-cytochrome c reductase, NADH-cytochrome b5 reductase, xanthine oxidase, oxidoreductase, and dehydrogenase), in addition to DT-diaphorase [NAD(P)H:quinone-acceptor oxidoreductase].[23,30-38] DT-diaphorase (DTD) presents an exception, however, in that it catalyzes a two-electron transfer to quinones.[39, 40]MMC activation by DTD is not inhibited in air since the initially formed MMC hydroquinone is not susceptible to the fast reoxidation observed with the semiquinone.[41]
It has been shown that MMA[42] and more recently, MMC[43] are capable of being reductively activated not only by simple thiols but also dithiols, the latter with an efficiency two fold greater than the former. There exist reported modes of involvement of biological dithiols in the modulation of MMC cytotoxicity. Thioredoxin overexpression in Franconi anemia fibroblasts prevents the cytotoxic and DNA damaging effects of mitomycin C[44-46] thus, dithiols could detoxify MMC by activating the drug in the cytosol, where it would be hydrolyzed to inactive mitosenes. Recent studies have revealed that a glucose regulatory protein (GRP58) requires thioredoxin-like domains to catalyze metabolic reduction of MMC to 2,7- diaminomitosene, which then cross-links DNA.[30,47] Therefore, in contrast to performing a detoxification role, dithiols could generate cytotoxic metabolites by reducing MMC in close proximity to the nucleus.[43] The proposed mechanism of reduction of MMC by dithiols should help elucidate the specific interactions of analogous biomolecules with MMC. Scheme 2 depicts MMC (1) being attacked by a thiolate, giving rise to the intermediate 12. Intramolecular cyclization of the tethered thiol with the proximate α-thioether of 12 furnishes a cyclic disulfide and hydroquinone 4, which may proceed through the alkylation and cross-linking cascade shown in Scheme 1. Interestingly, the higher cellular toxicity of MMA compared to MMC can be attributed to more efficient activation of MMA by intracellular thiols due to its higher redox potential.[42, 48]
Scheme 2.
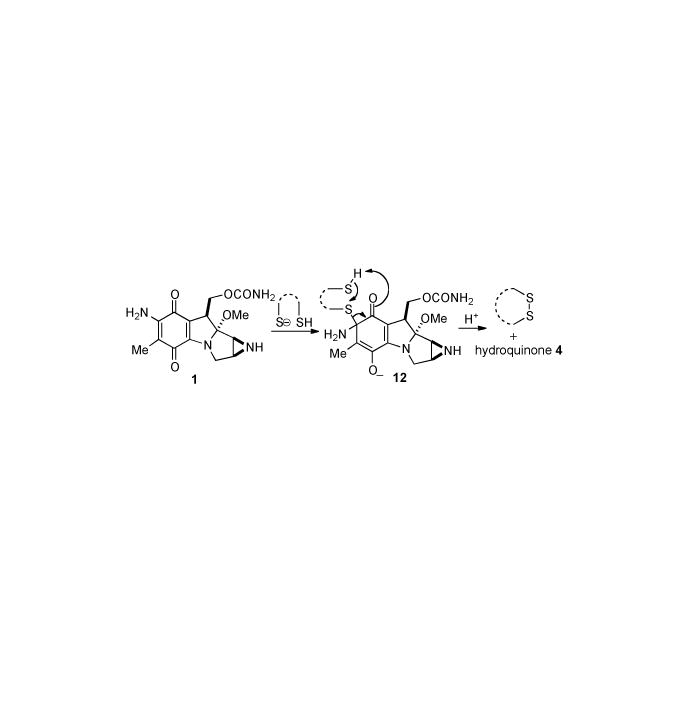
Proposed mechanism of reductive activation of MMC by dithiols.
Alternatively, a series of mitomycinoids substituted at N7 with ethyl-tethered disulfides are able to be nonenzymatically activated via thiols such as glutathione (Scheme 3). A mechanism involving intramolecular electron transfer from the disulfide to the quinone was proposed in which initial attack of glutathione on the disulfide 13 liberates thiolate intermediate 14. An internal redox reaction consisting of cyclization to form 15 followed by homolytic S-C bond cleavage gives the semiquinone radical anion 16. Finally, a second equivalent of glutathione effects reduction to the hydroquinone 17 which can continue along the activation cascade to the quinone methide and eventually alkylate DNA.[49]
Scheme 3.
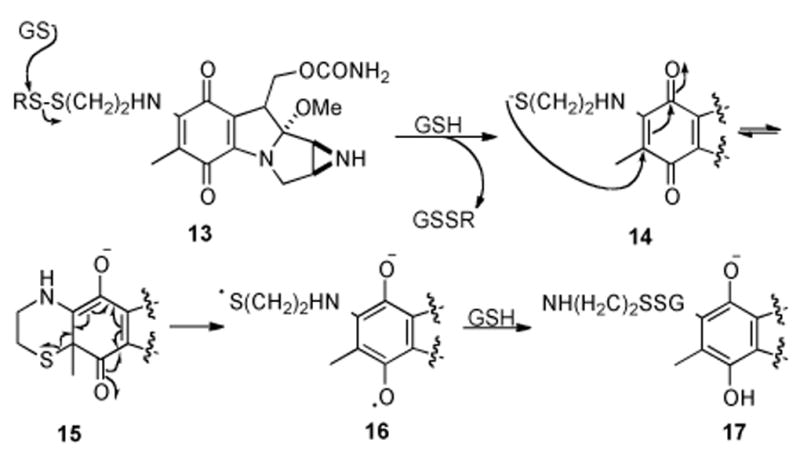
Proposed mechanism of reductive activation of dithiol mitomycin derivatives by GSH.
2.2 FR900482 Mode of Action
Studies by Rajski and Williams[50-53] and by Hopkins and coworkers[54-58] have determined that FR900482 and FR66979 cross-link duplex DNA in the minor groove with the same 5′-CG-3′ sequence specificity as MMC. Both research groups demonstrated that the cross-link occurred on the N2 exo-cyclic amino groups of the opposing guanidines. In addition, both families of compounds show the same preference for flanking base sequences in the order 5′-ACGT-3′ ≫ 5′-TCGA-3′ ∼ 5′-CCGG-3′.[51,54] MMC has additionally demonstrated a preference to cross-link 5′-mCG-3′ sites with the cytosine methylated at C5 over the unmodified sequence.[59,60] Methylated cytosine 5′-CG-3′ sequences represent a statistically rare but important sequence in gene expression, and whether this predilection for the methylated 5′-mCG-3′ sequences translates to the FR compounds remains to be determined.
Based on their analogous dependence on an exogenous reducing agent and their comparable cross-linking ability, a mechanism of action for the FR series of compounds that is similar to that proposed for MMC was first suggested by Fukuyama and Goto in 1989 (Scheme 4).[61] Initial two-electron reduction of the hydroxylamine hemiketal of FR900482 (18) results in ring opening to the eight-membered benzazocinone 19, which is in equilibrium with 20 via transannular opening and closure. Elimination of water from 20 brings into being mitosene intermediate 21. In a manner similar to MMC, a cascade of events ensues, beginning with the opening of the aziridine ring giving 22, which is then monoalkylated by DNA. The monoalkylated compound 23 is subject to expulsion of the carbamate group forming the final electrophilic iminium species 24. This allows for nucleophilic substitution by DNA at the N2 of guanosine to give the final compound, lethal cross-link 25.
Scheme 4.
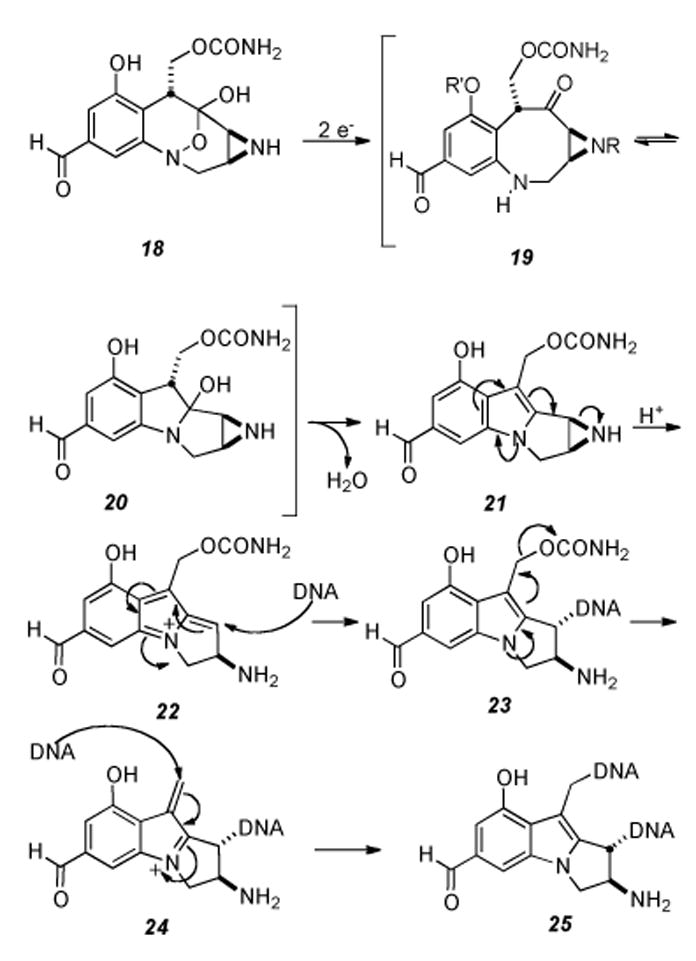
Proposed mechanism of DNA cross-linking ensuing from reductive activation of FR900482.
While the enzyme responsible for the in vivo reduction of MMC remains elusive, in studies with FK317 the reductase responsible for activation of the FR compounds has been shown to be DT diaphorase.[62] Additionally, it should be noted that the semi-synthetic derivatives FK317 (26) and FK973 (27) require deacetylation of the hydroxylamine hemiketal before any reduction event can occur.[62] A detailed study of the differential effects of FR900482 and FK317 on apoptosis, IL-2 gene expression, and induction of vascular leak syndrome was carried out collaboratively by Williams and Reeves.[63]
The proposed reductive pathway represents the generally accepted mechanism and was supported by work of Hopkins and coworkers.[55,58] These in vitro experiments included characterization of various intermediates and derivatives in the reaction cascade,[58] along with isolation and structural elucidation of the covalently cross-linked lesion by both FR900482 and FR66979 after reductive activation.[55] The latter experiment followed precepts introduced by Tomasz et al. in the structure elucidation of MMC cross-linked DNA.[64] These protocols included enzymatic digestion of cross-linked DNA, HPLC purification of the guanosine lesion, acylation, and finally structure determination by means of spectroscopic analysis. By utilizing a palindromic sequence containing a single 5′-CG-3′ site, Hopkins et al. were able to isolate the covalent cross-link, establishing not only the site of alkylation, but also the existence and structure of the activated mitosene species.
Recent studies with the clinical candidate FK317 (26) have revealed an additional structural motif allowing for selective mitosene formation under hypoxic conditions.[65] The oxidation state of the C12 position may play a key role in the reduction of the hydroxylamine hemi ketal functionality. Examination of the metabolites of FK317 in vivo revealed that the C12 aldehyde was reduced to the corresponding alcohol 27, which is converted into free aziridine 28 (Scheme 5). The alcohol oxidation state at C12 enhances the effectiveness of the cross-linking event as demonstrated by the higher relative activity of FR66979 over that of FR900482 in in vitro studies. Moreover, in healthy cells, the majority of the drug was metabolized to the carboxylic acids 29 and 30, derivatives which have been shown to be chemotherapeutically inactive.[65]
Scheme 5.
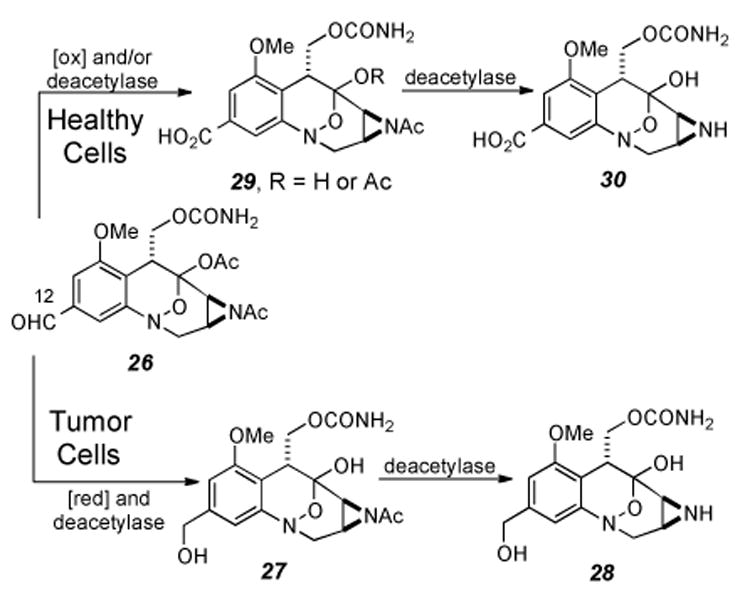
Metabolism of FK317 in both neoplastic and healthy cells.
The origin of the 5′-CpG-3′ sequence specificity for both the MMC and FR series of molecules relies predominately on the ideal geometric fit of the resulting activated mitosene intermediate in the minor groove of DNA.[64, 66]The basis for this ideal positioning is the distance between the exo-cyclic amine group (N2) of the guanosines in the 5′-CpG-3′ sequence in B-DNA (3.1 Å) compared to the distance of the two sites of alkylation on the mitosene species (3.4 Å) (Figure 2).
Figure 2.
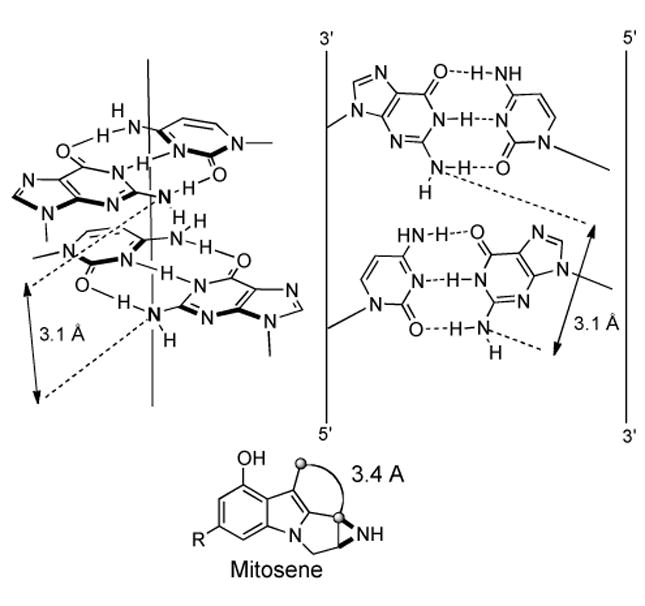
Ideal geometric fit of the mitosene in the 5′-CpG-3′ sequence of B-DNA in the minor groove.
In contrast to the various monoalkylation events seen with MMC, the interstrand cross-link is specific to only 5′-CG-3′ steps in the minor grove.[67-70] The consequence of geometric fit dictating both sequence selectivity and site of alkylation prevails over alternative factors, including greater nucleophilicity and general accessibility, which in the case of CG base pairs resides at the N7 of guanosine in the major groove.[53, 66]
Although the initial evaluation of the natural products MMC and FR900482 demonstrated their capacity to induce protein-DNA cross-links in addition to DNA-DNA cross-links, the majority of biochemical studies for both compounds have solely focused on the latter event. In an effort to evaluate the FR compounds’ ability to form covalent DNA-protein cross-links, Rajski and Williams studied the minor groove binding protein HMGA1 (formerly named HMG I/Y). High mobility group (HMG) proteins have been implicated in the regulation of genes associated with the immune system and cell growth, as well as other processes. They are therefore preferentially expressed in rapidly proliferating cells such as those found in tumor masses and represent a relevant drug target.[71] Rajski and Williams successfully used FR66979 to cross-link a synthetic peptide sequence of the binding domain of this protein to the corresponding synthetic oligonucleotide duplex containing the known HMG1A AT binding sequence.[15,16] Following this initial biochemical study, Tepe and Williams, in collaboration with Reeves and coworkers, isolated both FR900482- and FK317-induced cross-links of HMGA1, HMGB1, and HMGB2, all minor groove-binding proteins, with DNA from human Jurkat cells in vivo.[71] This study demonstrated the viability of such an event. The clinical implications and significance of the DNA-protein cross-link, compared to DNA-DNA cross-linking, are currently being investigated.
Although much discussion has detailed the similarities between the MMC and the FR series of compounds, a profound and clinically significant difference between these two families of compounds resides within the two separate structural motifs masking the active mitosene core. The redox activity of the quinone of MMC has two significant consequences, namely, superoxide production leading to nonspecific oxidative DNA damage and arbitrary arrest of the reductive pathway leading to monoalkylation.[68,72] Following production of superoxide from both the initial one-electron reduction and other quinone redox reactions in the cascade, Haber-Weiss/Fenton cycling produces hydroxyl radicals and related reactive oxidants capable of mediating DNA single strand breaks and other indiscriminate cellular damage.[68,72] Additionally, reversion of the hydroquinone intermediates to the quinone species along the reaction cascade depicted in Scheme 6 allows for alternative reaction pathways to the potentially less therapeutically significant monoalkylations.[68]
Scheme 6.
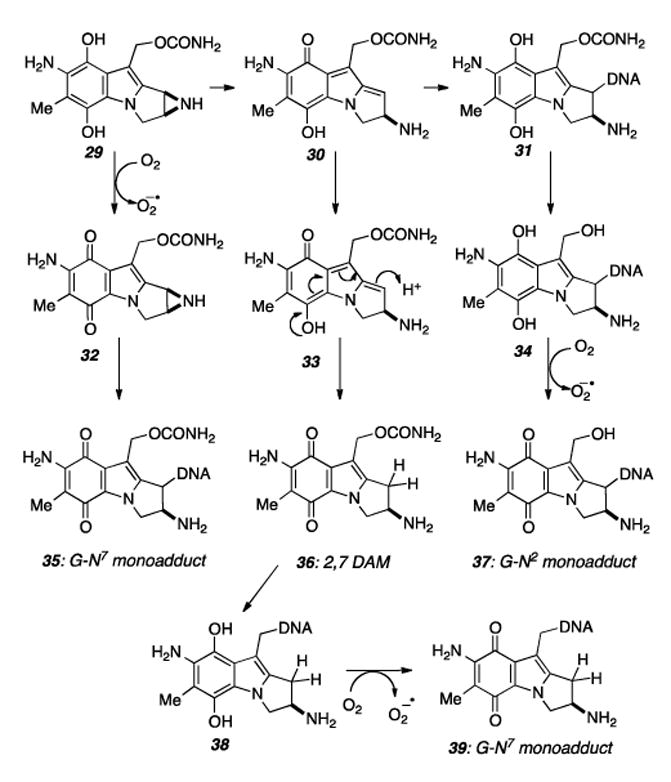
Mechanism of various monoalkylation events by MMC.
Indeed, the majority of alkylation by MMC in vivo results in monoadducts and not the more lethal cross-links.[68, 73] Under aerobic conditions, these include both N2 adducts resulting from the exocyclic amine of guanosine. In tumors and other hypoxic areas of therapeutic interest, the majority of alkylation occurs via the innocuous monoalkylated species 39, resulting from SN2 displacement of the C10 carbamate of 2,7 diaminomitomycin (2,7 DAM) by the N7 of guanine.[68]
In contrast to MMC, the reductive activation pathway of FR900482 (and by analogy FR66979, FK973, and FK317) circumvents the production of superoxide, therefore obviating the detrimental non-specific oxidative damage pathways, while maintaining a more efficient ability to cross-link DNA. These attributes distinguish the FR series of compounds as viable candidates for chemotherapeutic applications.
3. Biochemistry
3.1. Elucidation of MMC Biochemistry
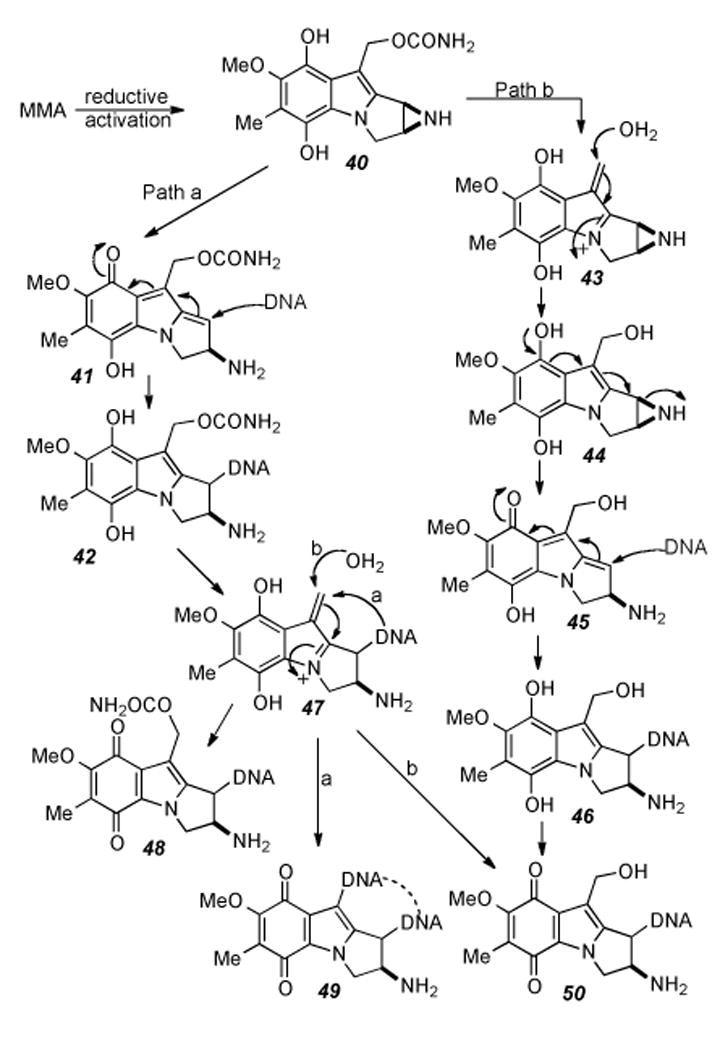
Iyer and Szybalski′s original 1964 proposal concerning the bioreductive activation of the mitomycins stated that they became bifunctional alkylating agents upon chemical or enzymatic reduction, and that a high guanine (G) and cytosine content favors the cross-linking reaction, which is the basis for the lethal effects of these antibiotics. The degree of the cross-linking of DNA was determined from the proportion of spontaneously renaturable DNA molecules under conditions of irreversible denaturation, by the method of equilibrium density-gradient centrifugation in CsCl or Cs2SO4, or by biological assay of residual transforming activity.[11] In 1970, through tritium release assays, it was shown that MMC does not alkylate DNA at the N7 of guanine.[74] At the time, alkylation of DNA at the O6 position of guanine was known[75,76] and indeed this mode of covalent bonding with mitomycins was implicated in 1974.[77] The high specificity of G-MM interactions was shown in 1978 for the first time by Tomasz.[78] Using Remer′s synthetic analogs, the aziridine was shown to be a covalent bonding site essential for cross-linking. The following year saw a report detailing the first characterization of alkylation products of MMC with biological model nucleophiles; nucleotide mono, di, and triphosphates. All reactions occurred at low pH and resulted in aziridine cleavage by the phosphate at C1. The structure of these products was proven by quantitative phosphate analysis, ultraviolet spectra, and enzymatic degradation into known products.[79] Although acidic conditions and phosphate adducts hardly represent the paradigm of the mitomycin mode of action, the Tomasz group developed techniques which ultimately aided in the characterization of minute quantities of mitomycin-DNA adducts.
Experiments using rat liver microsomes confirmed the reductive metabolism of MMC in that C1-phosphates similar to those isolated from the acidic reactions, along with 2,7-DAM were characterized and represented first known bioreductive alkylation products.[20] Thus, as the only natural product known which existed as a prodrug, then reduced in vivo to its active form, MMC was assigned the distinctive title of the “prototype bioreductive alkylating agent”.[20,80] The first direct support that DNA bonds the mitomycins at N2 of guanine was garnered from reactions run between the antibiotic and poly(G) analogs in which N7 or O6 positions were blocked by methylation, or the 2-amino group was lacking. Only in the case of hypoxanthine substitution for guanine was the bonding ratio significantly diminished.[81]
A combination of NMR and second derivative FTIR characterization of the products of the reaction between reductively activated MMC (H2/PtO2) and d(GpC) or 2′-deoxyguanosine provided unambiguous evidence for the covalent bond formed as that between C1 of MMC and N2 of guanine.[82] Reaction of calf thymus DNA with reductively activated MMC and digestion of the subsequently formed complex with DNase I, snake venom phosphodiesterase, or alkaline phosphatase yielded a single mitomycin deoxyguanosine adduct as the major DNA alkylation product.[83] The major products isolated from both types of experiments were assigned the identical structures N2-(2″, β, 7″-diaminomitosen-l″a-yl) 2′-deoxyguanosine.[82,83] Interestingly, it was found that MMC activated by acidic conditions preferentially alkylates the guanine N7 position.[84] This differential reactivity was explained by invoking the argument that the product of acidic activation has considerable localized cationic character due to the electron-withdrawing effect of the quinone.[19,85] Thus, as a hard alkylating agent, reactivity should be greatest with the site of highest electron density (most nucleophilic) in guanine, which is N7.[86] In contrast, the quinone methide intermediate generated upon reductive activation is a highly delocalized species and thus reacts with DNA in a soft fashion similar to that of other highly delocalized electrophiles known for their preferred attack at N2 of guanine.[20, 87-89]
The first isolation of the lethal cross-link adduct was accomplished in 1987 and characterization thereof entailed implementation of the aforementioned methods of 1H NMR, differential FTIR, and a CD method amenable to determination of the stereochemistry of the adduct at the C1 position of MMC.[64,85] Additionally, computer generated models lended support for the bisadduct having a snug fit in the minor groove with minimal distortion of the DNA structure. Previous modeling studies which conveyed the possibility of guanine O6 and N7 bonding modes of the MMC bisadduct were subsequently rejected and revised computer models to fit the data of the Tomasz lab were published.[90] These data also corroborate the conclusions of an earlier report that MMC is bound in a groove, and does not intercalate between base pairs, as previously suggested.[91,92] One difficulty in MMC/DNA adduct characterization is the lack of diagnostic protons on the guanine residue. To address this issue, authentic guanosine O6-MC derivatives were synthesized and comparisons showed that the N2-adducts and not O6-adducts are the major products from reactions of reduced MMC with DNA.[93]
As a result of these seminal reports on the isolation and characterization of MMC/DNA adducts (only three adducts resulting from reductive activation were known at the time)[94], a working model for the mechanism of monofunctional and bifunctional alkylation was established, upholding and expanding upon Iyer and Szybalski′s original proposal.[10,11,95] Reacting MMC with the short slice of duplex DNA d(Tl-A2-C3-G4-T5-A6)·d(T7-A8-C9-GlO-T11-A12), and characterization by NOE data confirmed the structural assignment of the cross-link as that between the exocyclic amines (N7) of adjacent guanine residues on opposing strands.[96] Various oligodeoxyribonucleotides cross-linked by reductively activated MMC were prepared, purified, and the cross-linked products were structurally characterized by nucleoside and MCnucleoside adduct analysis. The cross-links are stable to heat at neutral pH but are removed by treatment in hot piperidine or by the reducing agents Na2S2O4 and dithiothreitol.[97]
The explosion of simultaneous publications aimed at elucidating the sequence specificity of the DNA-MMC crosslink in the late 80′s mostly supported the notion of selectivity for 5′CpG sequences. Determining the site of MM bonding by locating the stop sites induced by the processive enzyme λ-exonuclease led to the conclusion that monoalkylation of DNA by MMC occurred preferentially at guanine residues within 5′CG and 5′GG sequences.[98] A multienzyme complex from E. Coli, UVRABC endonuclease, was shown to incise the DNA around N-methyl MMA adducts by Rupp.[99] Improving on the λ-exonuclease assay, Kohn then performed an analysis of MMC-induced incision sites from five different DNA restriction fragments after UVRABC treatment revealed that all drug bonding was localized at guanine residues and that bonding proceeded in a highly sequence-selective manner. The densitometric data indicated that both the 5′ and 3′ nearest neighbor bases surrounding the guanine site affect it’s susceptibility to drug modification and that MM lesions occurred predominantly at 5′CG sequences.[100] Phillips, White, and Cullinane implemented an in vitro transcription assay exhibiting blockage of transcription by E. coli RNA polymerase at specific DNA sites when mitomycin C was reduced by xanthine oxidase/NADH, that showed selectivity for XpC sequences of the non-coding strand, corresponding to G selectivity of the coding strand.[101] Through experiments in the Hopkins lab involving sequence-random cleavage of MMC cross-linked DNA, the sequence specificity of the cross links was determined at single nucleotide resolution to be at the G of 5′- d(CG) in strong preference to 5′-d(GC).[102] Crothers simultaneously utilized a gel electrophoresis assay yielding identical results.[69]
AT-rich duplex oligonucleotides, containing a single central CG·CG, gave high yields of cross-links between the two guanines while those having GC·GC, instead, gave none. In another series, the central sequences CGC·GCG and CGC·ICG both yielded 50% cross-link while CGC·GCI was completely resistant. Oligonucleotides substituted μονοϕυνχτιοναλλψ by MMC at guanine at either a CG or GC sequence were annealed with their complementary strands followed by reductive reactivation of the bound MMC to form a cross-link. The CG oligomers were cross-linked quantitatively while the GC ones were again resistant. These results show unambiguously that the MMC cross-link is absolutely specific to the CG·CG duplex sequence.[67] It was later ascertained that the ratio of the orientation isomers of a MMC interstrand cross-link in non-self-complimentary DNA was approximately 1:1. In this study five different DNA duplexes of 8-bp length were cross-linked by MMC via a procedure of first C1-monoalkylation of each strand followed by conversion to a cross-linked duplex, by annealing the monoalkylated strand to its complement in the presence of a reducing agent.[103]
Reaction of MMC with a series of oligonucleotides (5′-NGT′) under reductive conditions which restricted MMC to monofunctional alkylating activity gave the following yields: 2% at 5′-AGT′, 4.1% at 5′-TGT′, 14.7% at 5′-GGT′, and 36% at 5′-CGT′. MMC activated under acidic pH also displayed 5′-CG alkylation specificity and DMC activated by Na2S2O4showed the same 5′-CG specificity as MMC. Replacement of deoxyguanosine by deoxyinosine in the opposite strand at a 5′-CG or 5′-GG site abolished the enhancement of alkylation. Based on these results, a precovalent binding model was proposed such that at 5′-CG a H-bond is formed between the 2-amino group of guanine in the opposite strand and the 10-O atom of activated MMC, facilitating alkylation at such a sequence.[104] Thus the monoalkylation specificity guides the drug preferentially to the sites of DNA where the most lethal action of the drug may be realized (cross-linking).
Studies supporting the idea of H-bond directed specificity commenced with the report by Millard and Beachy that MMC displays a preference for CpG sequences in which the cytosine is replaced by 5-methylcytosine (m5CpG), a common epigenetic modification formed by the action of DNA methyltrasferases which is present in the CpG sequences of mammalian DNA to an extent of 60-90%.[105,106] Although the preference is modest (∼1.5-2 fold) it was suggested that the difference could arise from local charge effects; a focus of ensuing publications.[59] The formal mechanistic proposal explaining increased reactivity as due to an electronic effect of the 5-methyl group of cytosine (a small pKa increase) transmitted via G·C H-bonding to N2 of guanine (increasing nucleophilicity) in the first, monoalkylation step, was put forth two years later.[107] Support for this mode of binding was furnished by results from DNA alkylation reactions of the unnatural enantiomer of MMC (ent-MMC). These reactions of ent-MMC exhibited selectivity for the CpG sequence in analogy to that of natural MMC. This reactivity was attributed to common precovalent orientation of the two MM active forms relative to the target guanine in the minor groove, reinforcing the earlier proposal that, prior to covalent attachment, the 10′-carbamate group recognizes the CpG sequence by formation of a specific H-bond.[108] Further evidence for electronic effects transmitted through the base pairs was observed in that substituting fluorine for hydrogen at the 5-position of cytosine in CpG steps showed that the extent of cross-linking increased in the order 5-fluoro-C<C<5-methyl-C.[109]
The role of the C-10 substituent in the initial MMC-DNA monoalkylation event was further investigated by comparing the bonding specificity of MMC with C-10 chloro (8) and C-10 bromo (9) deoxycarbamoylporfiromycins under reductive conditions and N-methyl-7-methoxyaziridinomitosene (10) with 7-methoxy-10-noraziridinomitosene (11) under nonreductive conditions.[110] The C-10 halogenated MM derivatives modified DNA at guanines but with significantly diminished 5′CG* sequence selectivity. In addition, mitosene 10 selectively modified 5′CG* sites in DNA, while 11 did not, supporting the aforementioned H-bonding hypothesis depicted in Figure 3. Supplementary support was provided by the solution structure of the monoalkylated MMC–DNA complex, in which such H-bonding interactions were directly observable.[111] An interesting study addressing MMC-DNA recognition probed the interaction of these two reactants in a novel way by substituting 2,6-diaminopurine (DAP, D) as an analog of guanine. DAP, like adenine, forms a specific base pair with thymine, and the minor groove is widened at D·T basepairs, as compared to A·T basepairs. In this respect D·T mimics the G·C basepair and may be utilized as a positive test for the role of the 2-amino group of guanine in drug binding as was invented by Waring and Bailly.[112-114] The results of these elegant studies were completely analogous to those obtained with Gcontaining DNA and support the H-bonding mode of sequence recognition of DNA by MMC. Another conclusion that was drawn is that the sole determinant for alkylation site recognition is the 2-amino group of a purine in the minor groove.[115]
Figure 3.
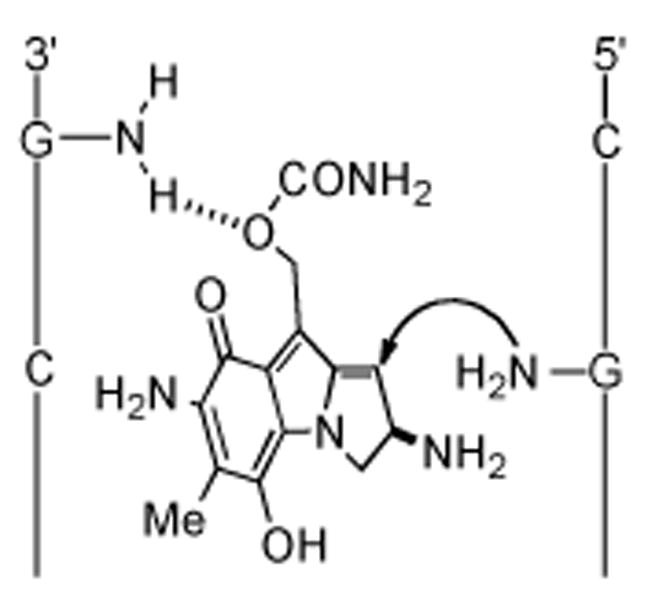
Precovalent H-bond complexation model explaining the origin of CpG sequence selectivity.
A beautiful computational study (DFT) of the protonation of R5G·C (R=F, H, CH3) base pairs resulted in a model in which the carbonyl of cytosine, the hydrogen bond acceptor to the exocyclic amine of guanine, covalently accepts the H-bonding proton (Scheme 7). The resulting positive charge is delocalized through the ring of the cytosine and thus cytosine of the G·C base pair replaces the guanine in the role of the base by loaning it′s basicity to the guanine. The mode of reaction was termed hydrogen-bond acid/base catalysis (HBA/BC) and defined as a reaction in which a proton that is involved in an H-bond is (a) first transferred from the H-bond donor to the H-bond acceptor within the H-bond and (b) then transferred back to its original position in a later stage of the reaction.[116]
Scheme 7.
Hydrogen bond acid/base catalysis model for the initial alkylation of MMC by guanine.
3.2. Cytotoxicity
One factor affecting the cytotoxicity of a mitomycinoid is the reduction potential of the aromatic ring. The less negative the reduction potential, the more facile is the enzymatic reduction. The site of substitution differences on the aromatic portion of the natural mitomycins is C-7. It has been shown that compounds with electron-withdrawing substituents at this position are more easily reduced by virtue of exerting a stabilizing effect on the semiquinone radical anion formed, and helping disperse electron density into the ring.[117] Correlations between the reductive potential and lipophilicity of mitomycinoids and their antitumor activity have been drawn, with the most easily reduced and the most lipophilic compounds being the most potent.[118]
3.3. Miscellaneous Biochemical Studies
Some minor, atypical MMC-DNA adducts have been isolated, for example an intrastrand cross link at d(GpG) sequences derived from synthetic oligonucleotides, Micrococcus luteus, and calf thymus DNAs.[119] At the time only Pt drugs were known to link two bases in the same strand. The bonding sites on MMC and guanine are the same as in the interstrand cross link, however the exocyclic amines of the two linked guanines are forced much closer together than in the unmodified strands (3.4 Å vs. 4.3 Å). This pinching of adjacent guanines causes a 14.6 ± 2.0° DNA bend per lesion, possibly stimulating recognition by DNA binding proteins.[120]
The first non-guanine adduct of MMC and DNA was isolated in 1997 and assigned as bound between adenine N6/MMC C1, but no cross-linking was observed under any conditions.[121] At the carbamate carbon (C10) 2,7-DAM (36) monoalkylates DNA in the major groove at guanine N7 of (G)n tracts.[122,123] The selectivity correlates with the sequence specificity of the negative molecular electrostatic potential of the major groove, suggesting that the alkylation selectivity of 2,7-DAM is determined by sequence-specific variation of the reactivity of the DNA.[123] This may not be surprising considering that N7 is the most nucleophilic position of guanine[86,124] and that the less reactive carbamate alkylating function has been shown to follow patterns of reactivity characteristic of SN2 displacement.[125-127] This adduct is non-toxic and since reduction of MC in vivo[73] or in vitro converts most of the drug to the nontoxic quinone 2,7-DAM, this process may be regarded as an additional detoxification mechanism.[20, 128]
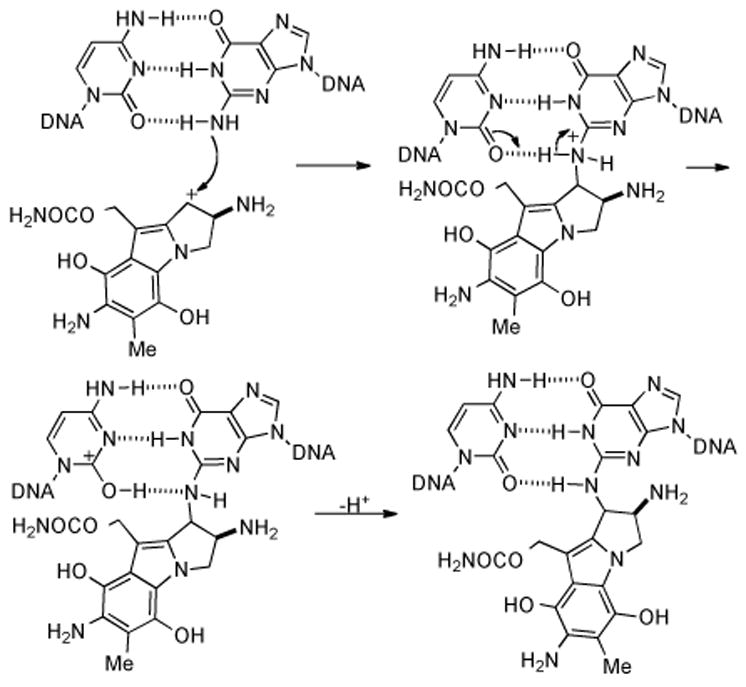
The MMC-activating enzyme DTD is inactivated by MMC at pH 7.8 but not at pH 5.8, and studies using 3H-MMC confirmed that an alkylation of the protein by the drug was responsible for the inactivation. A product consistent with the molecular weight of a DTD dimer cross-linked by MMC was detected by SDS-PAGE. A mechanistic proposal explaining the pH-dependent inhibition of DTD by MMC as a function of the ionization state of groups at the active site of the enzyme was proposed and is depicted in Scheme 8. Nucleophilic groups, such as sulfhydryl or perhaps more likely imidazole substituents, may be protonated at pH 5.8 while at pH 7.8, a greater percent may be in their ionized forms which would favor alkylation by the quinone methide.[129] Since at low pH bioactivation of MMC and porfiromycin is favored, while at higher pH enzyme alkylation and inactivation predominate, the efficacy of exploiting the elevated DTD content of certain human tumors for improved chemotherapeutic response will depend on intracellular pH.[130]
Scheme 8.
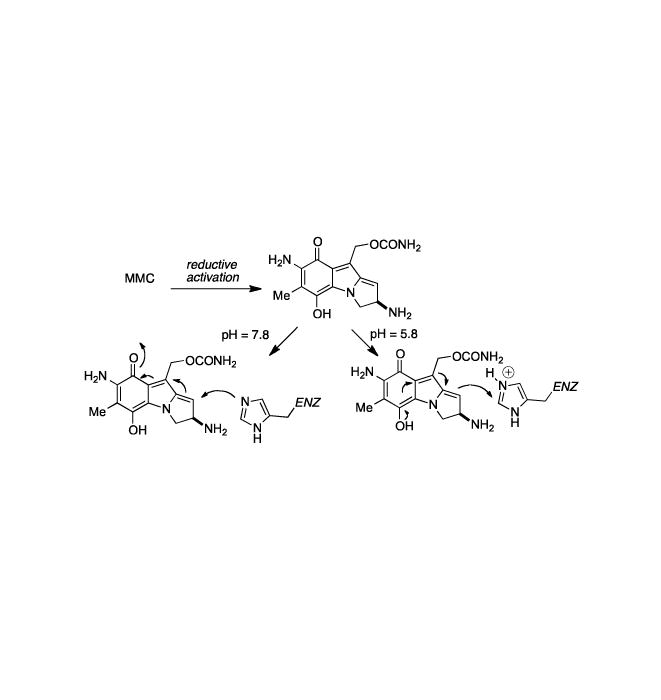
Proposed explanation of pH-dependent inhibition of DTD by MMC.
Slightly simpler mitomycinoid analogs, mitosenes with good leaving groups at the C-1 and C-10 positions have been shown to be able to be reductively activated[131] and are capable of cross-linking DNA under hypoxic conditions.[132] Compounds of the type depicted in Figure 4 were shown to display antitumor activity which correlated primarily with lipophilicity.[133] The bis-acetoxy compound named WV15 (R1= Me, R2=R3=OCOCH3, n=1) presents a different pattern of reactivity from that of MMC as inferred from the structures of two dimeric WV15-DNA monoadducts in which the bond formed is between the mitosene C-10 and deoxyguanosine N-2 atoms.[134]
Figure 4.
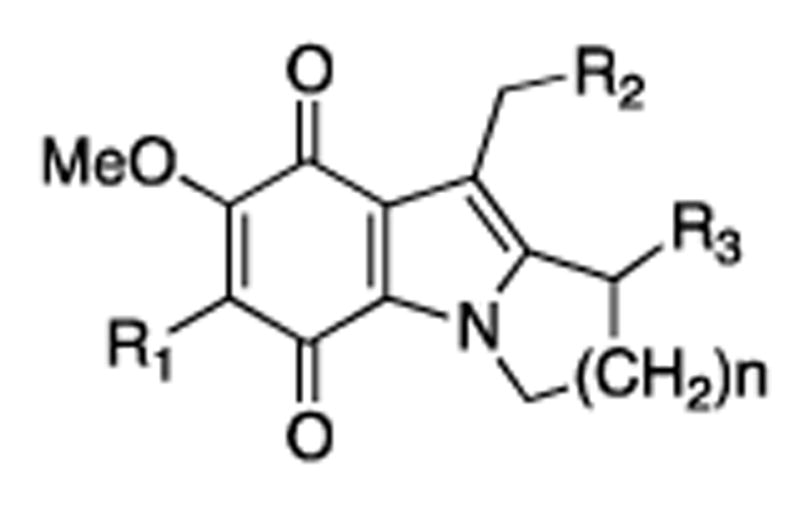
Mitosene core nucleus.
One factor affecting the relative reactivity is that the acetoxy group at C-1 is less reactive than an aziridine. Additionally, the C-7 substituent has been shown to affect the relative reactivity of the crosslinking sites of mitomycins[135] and mitosenes.[125,131] This affect as it relates to the comparison between MMC and MMA has been proposed to be a direct consequence of their different redox potentials, with MMA showing an initial indiscriminate activation of either electrophilic site (Scheme 9).[136]
Scheme 9.
Differential biochemical pathways of MMA upon reductive activation.
Thought previously to be exclusively a monofunctional DNA alkylating agent, dicarbamoyl mitomycin C (DMC) was shown to also be capable of forming interstrand cross-links. This work helps explain DMC′s slightly higher toxicity to hypoxic EMT6 mouse mammary tumor cells, as well as Chinese hamster ovary (CHO) cells, as the total frequency of alkylation by DMC is much greater than that of MMC under identical conditions. Most of the alkylation events induced by DMC result in monofunctionalization of DNA. However, DMC does bifunctionally alkylate at a frequency slightly greater than that of MMC.[137]
Comparison of the ability of MMC, DMC to activate p53 reflected the relatively benign nature of 2,7-DAM monoadducts in that 2,7-DAM was the only substance which did not activate p53 and induce apoptosis. DMC and MMC were shown to activate differential cell death pathways; DMC and MMC both induce apoptosis in cell lines with a functional p53 pathway.[138] In cell lines lacking wild-type p53 DMC is more cytotoxic than MMC, but not as a consequence of increased cross- linking. Instead, the cause of toxicity was found to be associated with early poly(ADP-ribose) polymerase (PARP) activation of Chk1 kinase depletion.[139]
An interesting study probed the efficacy of mitomycin dimers of the structures shown in Figure 5 as polyfunctional cross-linkers of DNA. The dimeric compounds did indeed cross link DNA with the same sequence specificity as MMC, and were shown to form interstrand cross links more efficiently than MMC. One explanation proposed for this enhanced cross-link formation is that in addition to the well known mode of cross-link formation (vide infra), an additional mode of cross-linking exists in which a monofunctional alkylation event occurs at each unit of the dimer on opposite strands of DNA. Furthermore, evidence for triand tetrafunctional alkylation of DNA by meso-compound 51 was gathered.[140]
Figure 5.
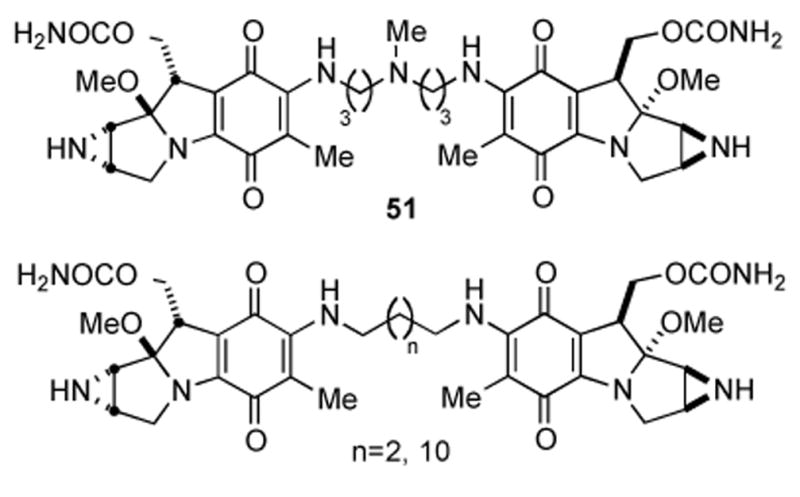
MMC tethered via C-7 amine.
Recently, Rink, et al. reported sequence-specific DNA interstrand cross-linking of a C6-C7 unsubstituted aziridinomitosene for which exogenous reductants are not required to form interstrand cross-linked DNA, specifically at 5′-d(CG) sites. This suggests that structurally related aziridinomitosene intermediates generated from reductive activation of mitomycin C may function as bifunctional DNA alkylating species.[141]
3.4. What is the Primary Target of Mitomycin C in vitro?
MMC which is bioactivated near the nucleus of a cell results in a large increase in cell kill efficacy. This was demonstrated in Chinese hamster ovary (CHO) cells in which the mitochondrial enzyme NADH:cytochrome b5 reductase (FpD) was directed into the nucleus of cells by the fusion of the SV40 large T antigen nuclear localization signal sequence to the amino terminus of an FpD gene that lacked the membrane anchor domain. Treatment of such cells with MMC showed marked increase in drug sensitivity, proposed to be a result of bioactivation occurring in close proximity to nuclear DNA.[142] CHO cells transfected with NAD(P)H:quinone oxidoreductase 1 (NQO1) cDNA that overexpress NQO1 in either the nucleus or the cytosol were both more sensitive to MMC than the parental cells. Furthermore, cells with nuclear localization of transfected NQO1 activity showed the greatest sensitivity, not because of increased metabolic activation of MMC, but again, because of its activation in close proximity to nuclear DNA. Thus, the subcellular localization of the bioactivating reducing enzymes, as well as the levels of these enzymes, is important in determining the cytotoxicity of MMC.[143] In contrast to demonstrating toxicity via cross-linking of nuclear DNA, it was recently shown that MMC inhibits ribosomal RNA, resulting in the assertion of a novel cytotoxic mechanism for bioreductive drugs. Pristos has suggested that nuclear DNA may be an unlikely target for bioreductive drugs in vivo due to the highly speculative nature of translocation and that other nucleic acids may be more important physiologically.[144]
3.5 Endogenous Resistance to Mitomycins in Streptomyces lavendulae
In 1994 David Sherman and colleagues isolated and cloned a genetic locus in the producing organism, S. lavendulae that conferred high-level resistance when cloned into S. lividans.[145] Two genes, mcrA and mcrB were identified as required for cellular self-protection, and comparison of the sequence similarity of the deduced protein product of mcrA (MCRA) with other proteins showed significant similarity with certain oxygen oxidoreductases. In particular, MCRA includes the known flavin adenine dinucleotide (FAD)-binding site.[146] This discovery led to the proposal that MCRA includes a covalently bound FAD molecule that mediates the oxidation of reduced MMC in vivo. Additionally, it was shown that expression of the mcr locus is induced specifically by mitomycins and their metabolites; the strongest inducers have an aziridine NH (as opposed to N-CH3). Protein levels increase concomitantly with increasing amounts of endogenously produced MMC, thus suggesting that induction of mcr occurs in the producing organism as a response to increasing levels of MMC.[147] An oxygen-dependent redox relay mechanism for the regeneration of MMC from the reduced hydroquinone was proposed as the mode of cellular resistance conferred by MCRA, the first reported example of a redox-mediated resistance mechanism (Scheme 10).[148] MCRA expression in mammalian cells (Chinese hamster ovary) greatly reduced the toxicity of the mitomycins under aerobic conditions but had little effect on toxicity under hypoxia, lending further support to the oxygen dependent nature of MCRA as a redox cycling protein.[149] Since the reoxidation of the semiquinone anion radical to MMC with molecular oxygen is extremely fast,[150] approaching diffusion-controlled rates, it would be prevented from reacting with other nucleophiles; whereas the hydroquinone intermediate is comparatively oxygen insensitive, the latter species must be largely responsible for the cytotoxicity of the mitomycins under aerobic conditions.[149] Horseradish peroxidase (HRP), myeloperoxidase (MP), and lactoperoxidase (LP) also oxidize mitomycin C hydroquinone in the presence of a source of hydrogen peroxide, and prevent to various degrees, the cross linking of T7 DNA.
Scheme 10.
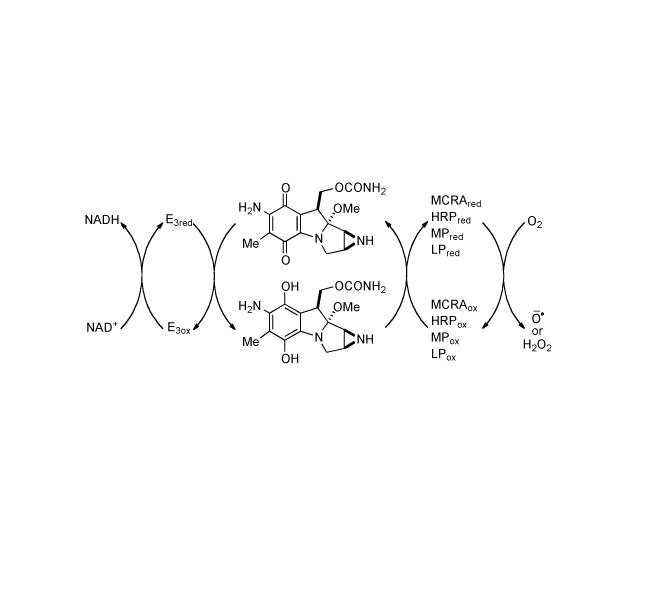
MCRA-catalyzed redox relay reaction. E3: CDP-6-deoxy-L-threo-D-glycero-4- hexulose-3-dehydrase reductase.
The cloning and expression of another gene, mrd, from S. lavendulae resulted in the synthesis of a soluble protein MRD, a drug-binding protein that prevents reductive activation of MMC through specific binding of the pro-drug to give a complex.[151] S. Lavendulae mutants lacking mrd exhibit levels of MMC sensitivity approximately ten-fold higher. Sequence analysis of the DNA adjacent to the mitomycin resistance locus mrd lead to the characterization of a third MMC resistance determinant which encodes for a membrane-associated protein Mct involved in excretion of MMC from S. Lavendulae.[152]
Cellular protection afforded by Mct is suggested to be a function of drug transport from the cytoplasm. Coexpression of mrd and mct in E. coli resulted in dramatically increased resistance (150 fold) to exogenously added MMC as well as reduced intracellular accumulation thereof. The binding protein MRD may be considered an accessory component, a specific adaptation required for optimal drug resistance. Together, MRD and Mct act as components of a novel drug transport system which sequesters the intact pro-drug for efficient excretion to the environment. This represents a unique cellular strategy for self-preservation by the MMC-producing organism.[152] Characterization of MRD revealed that it slowly reductively transforms MMC into 1,2-cis-1-hydroxy-2,7-diaminomitosene 52 (Scheme 11), a compound that is produced via the reductive activation cascade. Seemingly paradoxical is the situation in which a protein involved in cellular resistance is also a drug activator. The unique quinone reductase activity of MRD perhaps represents evolutionary switching from a potential drug-activating enzyme to a drug-binding component of the MMC export system. Indeed, functional switching may be achieved primarily through the replacements of two amino acid residues at the active site of MRD. MRD appears to be a unique reductase that independently catalyses the direct transfer of hydride from NADH to the quinone moiety of MMC regardless of aerobic or anaerobic conditions, thus providing evidence that the reaction catalyzed entails an O2-independent two-electron reduction. Slow reduction by MRD actually results in prolonged association of intact MMC with the protein. If a drug transport protein such as MCT is coupled with MRD it is possible that the rate of export could exceed the rate of reductive activation resulting in rapid removal of MMC from the cell instead of transformation to a cytotoxic species. Support for this idea is garnered by the observation that MMC, and not mitosenes, are the major product generated, secreted, and isolated from S. Lavendulae.[153] Subsequently, the crystal structure of MRD was reported, including MRD with bound 1,2-cis-1-hydroxy-2,7-diaminomitosene. A striking structural similarity of the active site with that of bleomycin resistance protein (BRP) was revealed, as well as a very similar mode of drug binding, despite the low amino acid sequence similarity between the proteins and the chemically dissimilar binding substrates.[154] Thus, the remarkable ability of S. lavendulae to mediate the potential DNA cross-linking activity of the mitomycins may be traced to a combination of at least three genetic determinants which encode for proteins performing multiple diverse functions: reoxidation of hydroquinones to the corresponding less toxic pro-drug mitomycin forms (MCRA), drug binding and transport (MRD), and finally expulsion from the cell (MCT).
Scheme 11.
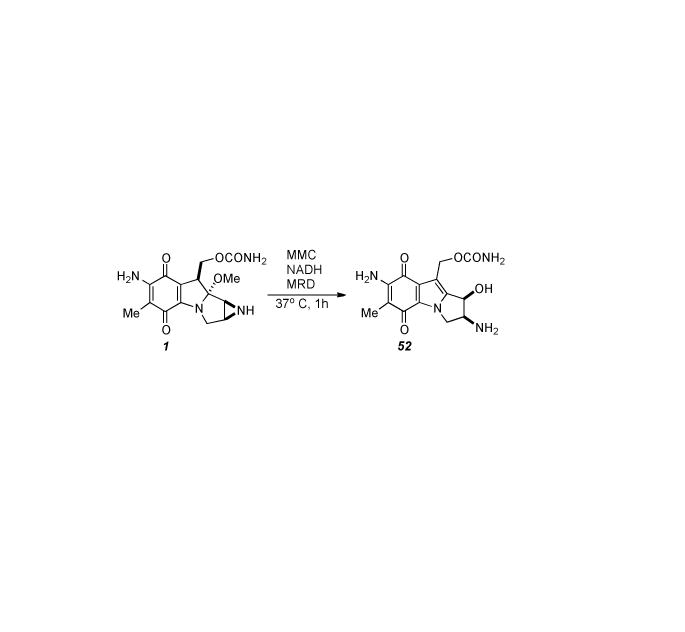
Transformation of MMC into 1,2-cis-1-hydroxy-2,7-diaminomitosene.
4. Biosynthesis
Initial experiments to elucidate the biosynthesis of the mitomycins were carried out in the laboratory of Kirsch. Methionine-S-methyl-14C was fed to Streptomyces verticillatus and 1.1% radioactivity was incorporated into MMA. Essentially all radioactivity incorporated was distributed evenly between the C7 and C9a methoxy functionalities. Acidic hydrolytic removal of C9a effects a nearly 50% decrease in specific radioactivity of the apo-MMA chromophore and alkaline treatment of apo-MMA (54) selectively removes the C7-methyl group which abrogates the remaining radioactivity. Thus the C6-methyl group is not derived from methion ine (Scheme 12).[155]
Scheme 12.
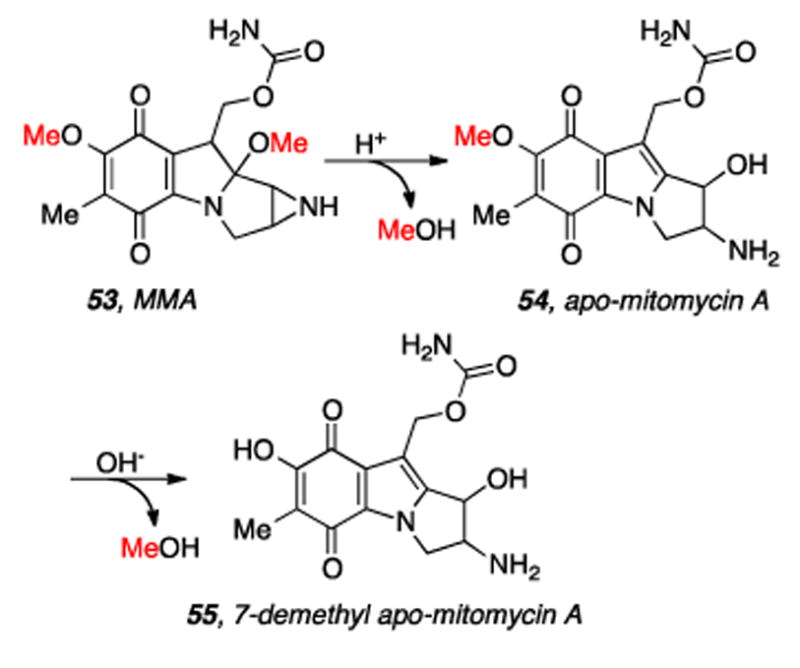
Sequential loss of radioactive methyl groups derived from Methionine-S-methyl-14C.
More extensive feeding studies were executed in Hornemann′s laboratories. All potential aromatic precursors and many smaller molecules were fed to S. verticillatus as radiolabeled substrates; all showed incorporation below 0.1 percent. Insightful experiments included eliminating peripheral groups on TLC plates and examining the loss of radioactivity with various labeled substrates. In addition to supporting the conclusions of Kirsch and Korshalla,[155] several other important inferences were made.
MMA labeled from[guanido-14C]-L-arginine loses relatively little radioactivity upon treatment with ammonia but nearly all after exposure to concentrated HCl. Thus, radioactivity from[guanido-14C]-Larginine was shown to be incorporated into the carbamoyl group. [1-14C, 6-3H]-D-glucosamine and [1-14C, 1-3H]-D-glucosamine were incorporated at rates of 1.9% and 2.7% respectively, with tritium retention (91% and 78% respectively). Kuhn-Roth oxidation (H2SO4/CrO3) gave non-radioactive acetic acid. However, the C7 unit was labeled by glucose and ribose: Kuhn-Roth oxidation of MMA obtained from [U-14C]-D-ribose yielded acetic acid with 13.3% of the specific radioactivity of the antibiotic. A rough biosynthetic picture now emerged in which the C7 unit of the mitomycins can be formed from glucose and/or ribose, possibly via a heptose intermediate, and condensation with glucosamine would provide the pyrroloindoline scaffold.[156] Concurrently, Bezanson and Vining performed feeding studies with radioactive supplements, the results of which primarily reinforced the dominant biosynthetic role of glucose as a precursor for heptulose and aminohexose building blocks. Furthermore, their experiments supported conclusions that the carbon skeleton is not assembled from an aromatic precursor derived from either the shikimate or polyketide pathways, and is not a disguised sesquiterpene.[157]
Radiolabeled MMB (56) isolated from S. verticillatus which was fed D-[1-14C,15N]-glucosamine was subjected to mass spectral analysis in order to track the biosynthetic fate of these atoms (Scheme 13). The ion peak clusters corresponding to the fragments 57, 58, and 59 were analyzed. The data on the specific incorporation of 14C and of 15N in the experiments with D-[1-14C,15N]glucosamine show that the 14C incorporation into MMB and the 15N incorporation into the aziridine ring are similar and nearly parallel the amount of the precursor added.
Scheme 13.
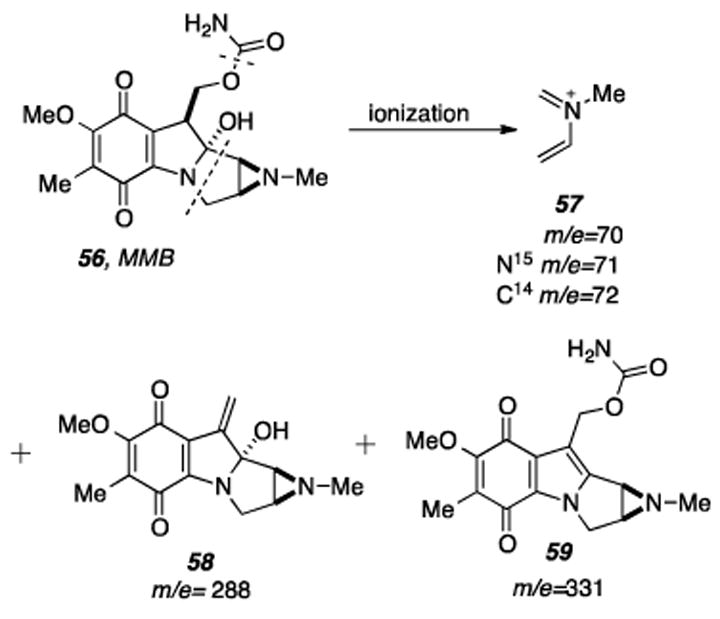
Mass spectral fragments from MMB that may be analyzed to expose the location of the radiolabels originating from D-[1-14C,15N] glucosamine fed to the MMB producing organism.
This suggests that both isotopes are predominantly incorporated without separation and it was concluded that D-glucosamine can provide the nitrogen atom of the aziridine ring. However, some breakdown of D-glucosamine seems to take place, since a fraction of the 15N is incorporated also into the carbamoyl group and into the ’indolic’ nitrogen of MMB.[158] The latter conclusion should not be surprising in light of the fact that bacteria readily interconvert sugars. The C4H8N (57) ion cluster was also examined for MMB isolated upon feeding D-[1-13C, 15N]glucosamine to S. verticillatus. The doubly labeled fragment C313CH815N (m/e=73) comprised 31% of the relative abundance of ions belonging to the cluster with singly labeled fragments C313CH8N and C4H815N comprised only 4.9% and 8.4% of the relative abundance, respectively, lending further creedence to the notion that glucosamine is utilized intact as a biosynthetic precursor.
Indeed, Hornemann has shown that the three dextrorotatory sugars glucosamine, galactosamine, and mannosamine are all incorporated into mitomycins when fed as 1-14C labeled substrates to S. verticillatus. The radiolabel from D-[6-14C]glucosamine 60 was shown to be exclusively incorporated into the mitomycin C10 position (Scheme 14). As such, substrate feeding and isolation of MMC (61) was followed by some degradative steps to furnish triol 62, whereupon periodate oxidation gave radiolabeled formaldehyde 63. Crystallization of the radioactive formaldehyde as the dimedone derivative 64 allowed the determination to be made that 72% of the specific radioactivity had been retained.
Scheme 14.
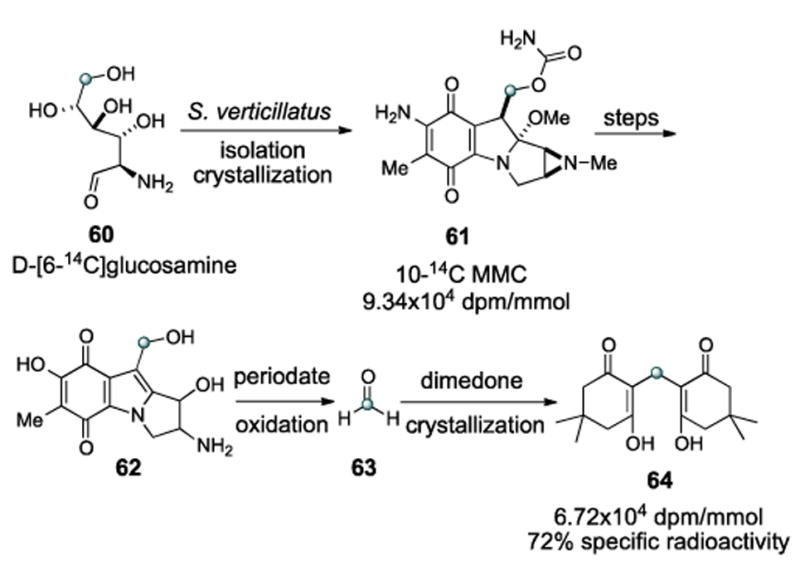
Tracking the origin of the carbamate carbon.
Competition feeding experiments between L-[guanidino-14C]arginine 65 and L-[ureido-14C]citrulline 66 suggested L-citrulline to be the more proximate precursor to the carbamate of the mitomycins (Scheme 15).[159]
Scheme 15.
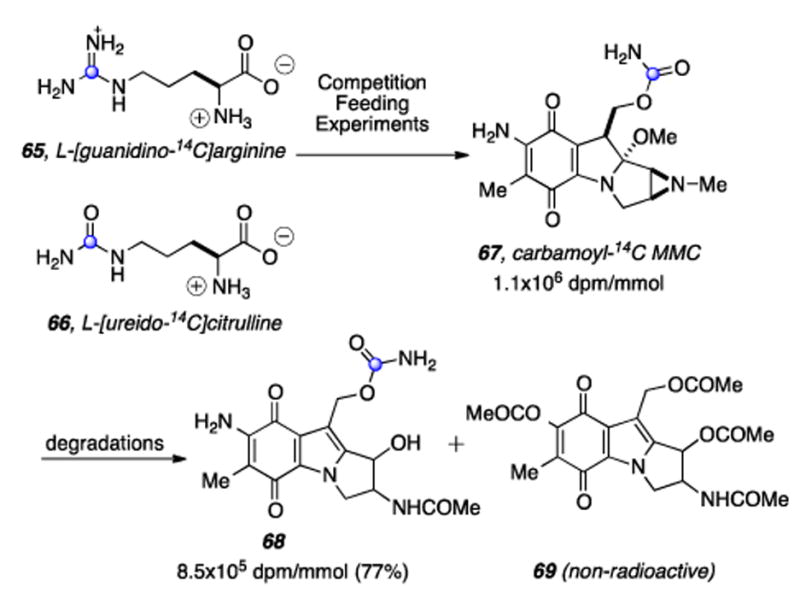
The C6 position of glucosamine labels C10 of the mitomycinoids.
Labeled MMC 67 was then subjected to degradative chemistry to provide a mixture of compound 68, retaining a majority (77%) of the radioactivity of 67, and the non-radioactive tetra-acetylated derivative 69. Feeding experiments with doubly-labeled L-[NH2CO-13C,15N]citrulline (not shown) ruled out the possibility that the carbamoyl group of L-citrulline was undergoing extensive metabolic conversion leading to a separaion of its carbon and nitrogen atoms prior to their individual incorporation into the carbamoyl group of the mitomycins.[160] The mystery of the biosynthetic origin of the exocyclic methyl C6a was investigated by Hornemann and coworkers and it was found that D-[3,4-14C]glucose (70) and [1-14C]pyruvic acid (71) specifically label this atom (Scheme 16).[161] Hypothesizing that the condensation of pyruvic acid with eyrythrose-4-phosphate (E4P) may give rise to an intermediary “C7N unit” 75 via 4-amino-4-deoxy-D-arabinohepulosonic acid 7-phosphate (74), it was subsequently shown that D-[4-14C]erythrose (73) and D-[3-14C]pyruvate (72) are incorporated into the mC7N unit of MMC (76) with the label from erythrose residing at C-7 and that from pyruvate at C-5. (Scheme 16).[162] At this point a major curiosity was the assembly of the meta-C7N moiety. This structural element is present in a number of diverse natural products, for example, geldanamycin, pactamycin, rifamycin, and validamycin A.[163-165]
Scheme 16.
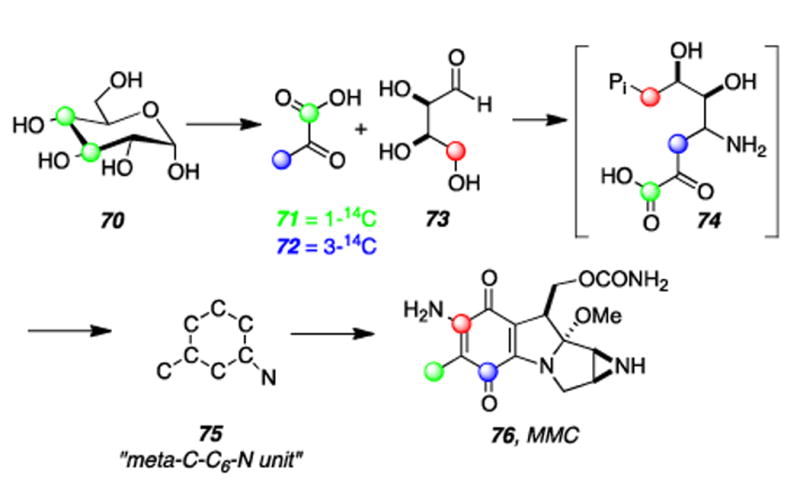
Results of two of Hornemann′s radiolabeling studies show that the mC7N unit of MMC is derived from erythrose and pyruvate.
Although initially the possibility was promulgated that mC7N units may arise through a shikimic acid type route, neither of the radiolabeled shikimic acid biosynthetic precursors [7-14C]-3-dehydroquinic acid or [1,6-14C2]-shikimic acid methyl ester, nor [U-14C]-shikimic acid were incorporated when fed to S. verticillatus, although their biosynthetic precursors (pyruvate and erythrose) were.[157,162]
Shortly after the identification of 3-amino-5-hydroxybenzoic acid (AHBA) as the key biogenetic precursor which initiates polyketide formation in the macrocyclic lactam antibiotics ansamycins and maytansinoids, Rickards demonstrated the intermediacy of AHBA in mitomycin biosynthesis by detecting an 8% enrichment at C6a of porfiromycin (78) when [carboxy-13C]-3-amino-5-hydroxybenzoic acid (77) was used as a feeding substrate (Scheme 17).[166,167] Shortly thereafter AHBA was determined to be a naturally occurring amino acid via an isotope dilution assay performed on a fermentation broth which was fed [carboxy-13C]-AHBA·HCl.[168] Deuterated substrate 79 was not incorporated by S. Verticillatus, confirming that AHBA is reduced after condensation with glucosamine.[168]
Scheme 17.
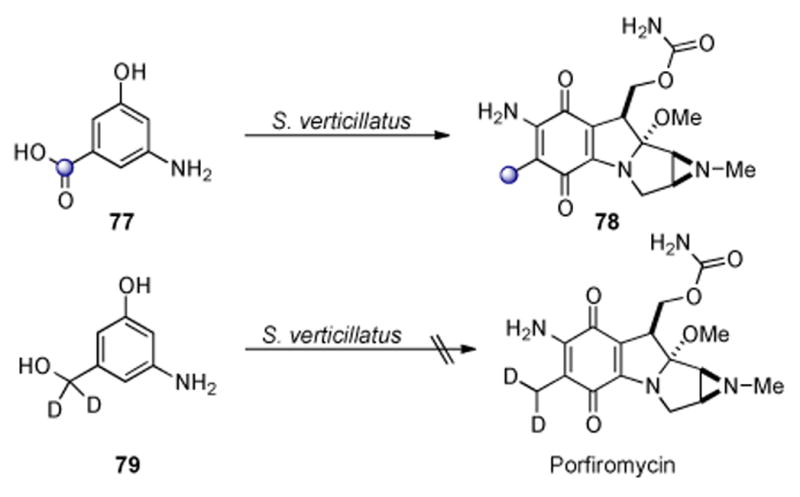
AHBA is reduced after condensation with glucosamine.
In 1989 Floss developed a “speculative” scheme describing the biosynthesis of AHBA which accounted for the published data (vide infra). Thus, transamination of E4P with glutamine to gives imine 86 followed by condensation with PEP (87) would give rise to amino-3-deoxy-D-arabino-heptulosonic acid 7-phosphate (ADAHP, 88, Scheme 18). Cyclization and loss of inorganic phosphate (Pi) give rise to aminodehydro-quinic acid (ADHQ, 89), followed by dehydration to form aminodehydro-shikimic acid (ADHS, 90). Finally a second dehydration and aromatization would give AHBA (91).[169] In time, it would be shown that this scheme is correct save for the formation of imino-E4P (86), which derives from kanosamine biosynthesis (vide infra).
Scheme 18.
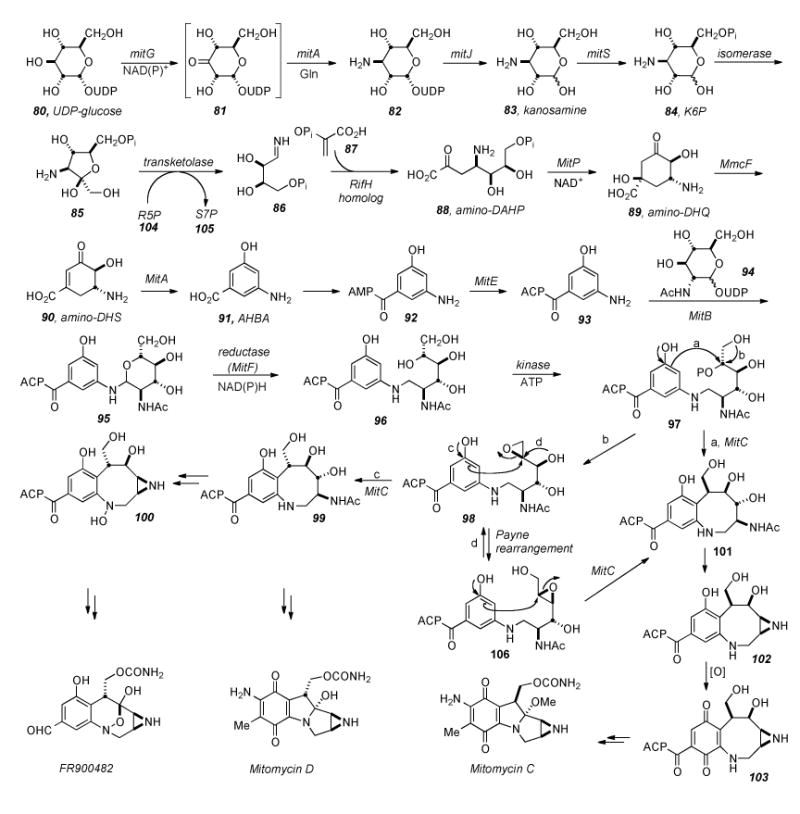
Putative biosynthesis of the Mitomycinoids and FR series of compounds from UDP-glucose. Functionality of many enzymes in the pathway has been ascertained, yet several elude assignment of their roles, or the specific order in which functions are excecuted. For example, the authors have proposed that MitC be involved in deacetylation of an early adduct of AHBA and glucosamine, but it is not known at which point in the biosynthesis that the enzyme acts.
ADAHP (88), synthesized in ten steps from glucose, was fed to the rifamycin B-producing organism Nocardia mediterranei, and demonstrated by isotope dilution assay to be converted into AHBA to an extent of 45%. Furthermore, ADHS (90) was synthesized from SA in five steps and demonstrated 95% conversion to AHBA (91) in 10 hours. In contrast, amino-shikimic acid (ASA) (not shown) was essentially not incorporated.
Given the demonstration of ASA as an acceptable substrate for shikimate dehydrogenase in the synthesis of ADHS, this enzyme was apparently lacking in N. mediterranei and thus an independent pathway was assumed to be operative.[170] Further experiments indicated that formation of AHBA must diverge from the normal shikimate pathway in the very first reaction.[171] Isolation, purification, and X-ray crystal structure analysis of AHBA synthase showed it to be a dimeric PLP-dependent enzyme which catalyzes the aromatization of ADHS to AHBA (90 to 91).[172, 173]
One ambiguity in mitomycin biosynthesis which remained was the nitrogen source of AHBA. Frost demonstrated the biosynthesis of 1-deoxy-1-imino-D-erythrose 4-phosphate (AE4P, 86) from aminofructose-6-phosphate (AF6P, 85) through a transketolase catalyzed ketol transfer to D-ribose-5-phosphate (R5P, 104). Radiolabeled 85 (3-[15N]-amino-3-deoxy-d-6,6-[2H2]-fructose 6- phosphate) incubated in Amycolatopsis mediterranei crude cell lysate with R5P (104) and PEP (87) produced ADAHP, (88) retaining the radiolabels and indicating that AF6P (85) is serving as a sequestered form of AE4P (86).[174] Kanosamine 6-phosphate (84) incubated in A. mediterranei cell-free lysate along with D-ribose 5-phosphate (not shown) and PEP (87) formed ADAHP (88) and AHBA (91). Subsequent incubation of glutamine and NAD with UDP-glucose resulted in the formation of kanosamine (83), suggesting that kanosamine biosynthesis is the source of the aminoshikimate pathway′s nitrogen atom.[175] The discovery that the RifN protein specifically converts kanosamine (83) to kanosamine-6 phosphate (84) solidified the notion that these compounds are intermediates in AHBA biosynthesis.[176]
The Sherman group has identified the genes mitA and mitB, required for mitomycin biosynthesis in S. lavendulae, which yield proteins related to AHBA synthases (MitA) and a group of glycosyltransferases (MitB).[177] Nucleotide sequence analysis of the mitomycin biosynthetic gene cluster revealed 47 genes spanning 55 kilobases of S. lavendulae DNA which govern MMC biosynthesis.[178] Included are homologs of the seven genes which are necessary and sufficient for AHBA formation in rifamycin producers A. mediterranei and S. Coelicolor (rifG, -H, -J, -K, -L, -M, -N). All are required for AHBA production when expressed under the control of an external promoter/regulator system, suggesting none have regulatory function, but they all encode proteins with catalytic functions in the biosynthetic pathway.[179]
Taking together what is known about AHBA biosynthesis[180], and Sherman′s assignment of the functions of mitomycin biosynthetic genes (based on homology with rifamycin biosynthetic genes), an emerging representation of mitomycin biosynthesis is depicted in Scheme 18.
Beginning with UDP-glucose (80), the kanosamine biosynthesis pathway ensues via NAD(P)+ dependent 3-dehydrogenation catalyzed by MitG to give intermediate ketone 81. Ketone 81 undergoes mitA mediated transamination with glutamine as the amino donor to UDP-kanosamine (82), and hydrolysis via MitJ to kanosamine (83).[181] MitS is responsible for phosphorylation of 83 to 84, and then 84 is isomerized to amino-fructose-6-phosphate (85) by an unidentified isomerase. The conversion from 85 to imino-eyrythrose-4-phosphate (86) likely involves a primordial transketolase. Ensuing coupling with phosphoenolpyruvate (87) to give amino-DAHP (88) would proceed via a RifH homolog. Cyclization of 88 by aminoDHQ synthase (MitP) gives 89, water is then eliminated by dehydratase MmcF to give 90, and finally, AHBA synthase (MitA) catalyzes aromatization to 91. Interestingly, AHBA synthase has been shown to be closely related to glutamine-dependent aminotransferases and is ascribed to have the dual functionality of recruiting nitrogen to the oxidized UDP-glucose to give UDP-kanosamine. After conversion of 91 into the AMP ligated species 92 and linking to the acyl carrier protien by MitE, 93 is condensed with N-acetyl-UDP-glucose (94) in the presence of glycosyltransferase MitB giving putative intermediate 95. NAD(P)H dependent reductase (MitF) catalyzed ring-opening to give 96, followed by kinase mediated phosphoylation with ATP would give 97, a prime candidate as a precursor to cyclization events which would lead to either the core of the mitomycins or the FR series of compounds. Several reactive modes ensuing from compound 97 may be envisioned: direct SN2 displacent of the phosphate via nucleophilic aromatic substitution would give benzazocine 101. Aziridine formation to give 102 followed by oxidation to the quinone 103 and finally tetracycle formation and tailoring steps would give compounds in the mitomycin series with the absolute configuration of MMC. Alternatively, if 97 underwent epoxide formation by displacement of the phosphate with the alcohol beta to it, 98 would be formed. As with 97, multiple options as to the reactivity of 98 exist.
Williams, et al. have suggested that the formation of compound 98 would explain the differential stereochemistry observed in the natural products at C9. Opening of the epoxide 98 by nucleophilic attack of the aromatic ring (path c) would give 99, bearing the alpha stereochemistry at C9. As described for 101, compound 99 would undergo aziridine formation, oxidation, and tailoring steps to arrive at the C9-alpha series of mitomycins. Additionally, the aniline amine of 99 may be oxidized to arrive at the FR-type compounds via the intermediacy of aziridino hydroxylamine 100. The FR-series of natural products also bear the alpha-configuration at C7, corresponding to C9 of the mitiomycinoids. Terminal epoxide 98 might also undergo Payne rearrangement to internal epoxide 104 (path d), effecting stereochemical inversion at C9 (MM numbering). Subsequent opening of the epoxide in the manner shown would effect a second inversion about the C9 atom to arrive at compound 101, with net retention of stereochemistry as compared to 98. Williams’ proposal accounts for the two different sterochemistries observed in the natural products as arising from a single biogenetic precursor. Currently, synthetic probes are being investigated to elucidate this part of the biosynthesis. Another intriguing question which remains is that of assembly of the aziridine ring, a topic yet to be investigated.
Aside from the biosynthetic studies on pre-tetracyclic substrates, Sherman has studied some of the enzymes involved in the late stage modifications of the aziridinomitosene core, i.e. MitM, MitN, and MmcR. MitM is an aziridine N-methyltransferase which seems to accept mitomycins with a C7-methoxy group such as 109 and MMA, but not those with a C7-amino group such as MMC (Scheme 19).[182] MmcR, obtained by cloning and overexpression of the corresponding mmcR gene, has been shown to be a 7-O methyltransferase which will catalyze the 7-O-methylation of both C9 β- and C9 α-configured 7-hydroxymitomycins in vitro (Scheme 20). An mmcR gene deletion mutant strain of S. lavendulae allowed for the identification two new biosynthetic intermediates, 7-demethylmitomycin A (111) and 7- demethylmitomycin B, (112).[183] Furthermore, a mutant mitN deletion strain of S. lavendulae was unable to produce MMB and accumulated a new mitomycin analogue, 9-epi-mitomycin C (116), however, the production of MMC and MMA was unaffected. In the presence of MitN and SAM, 9-epi-MMC was converted to a new product, mitomycin E (Scheme 21).
Scheme 19.
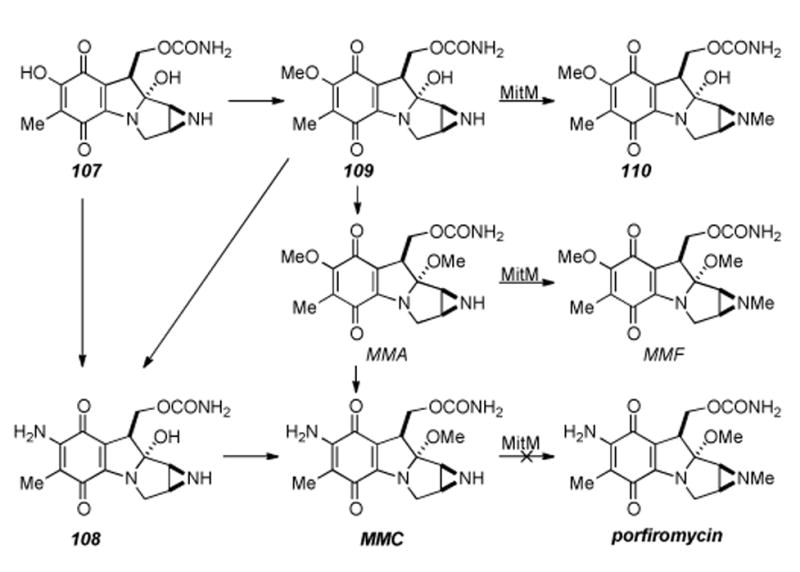
Functional analysis of the MitM aziridine N-methyltransferase.
Scheme 20.
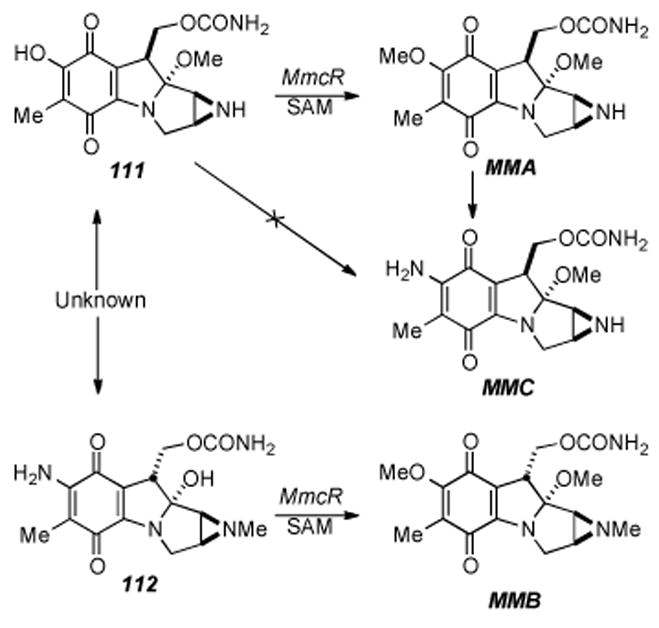
Hydroxyquinone O-methylation in mitomycin biosynthesis.
Scheme 21.
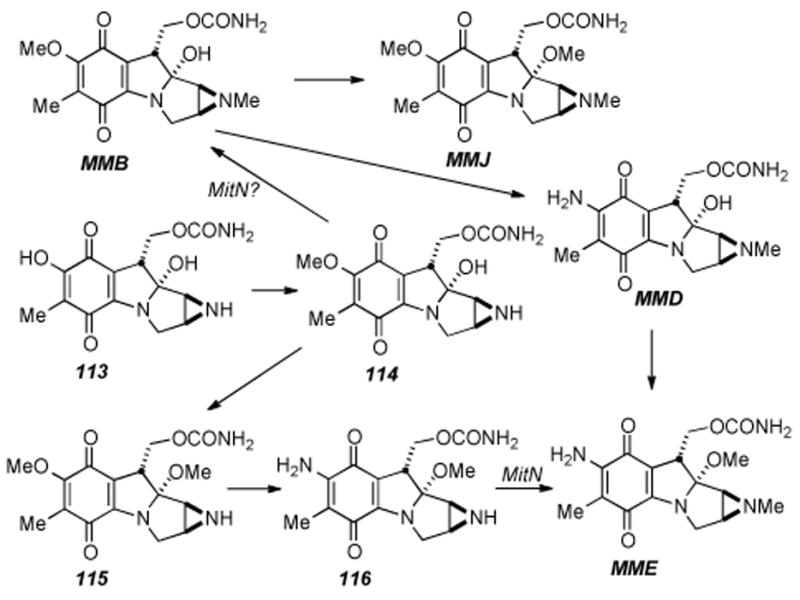
Proposed late stage modifications of 9 αbranch of mitomycins.
Thus, Sherman has concluded that MitN is an aziridine N- methyltransferase that is involved in tailoring of the 9 α-series of mitomycins and that separate aziridine N-methyl transferases catalyze the stereochemically defined pathways; C9 α (mitN) and C9 β (mitM).[184]
5. The Mitomycins: Successful Total Syntheses
5.1. Synthetic Challenges of the Mitomycins
One of the challenges associated with the synthesis of these compounds is the hemiaminal at the C9a position. Under various conditions mitomycins will suffer rapid elimination of methanol to form the corresponding mitosene (Scheme 22). This transformation appears to be even more facile in certain leucomitomycin systems. With the double bond of mitosene in place, the tetracycle is more prone to opening of the aziridine.
Scheme 22.
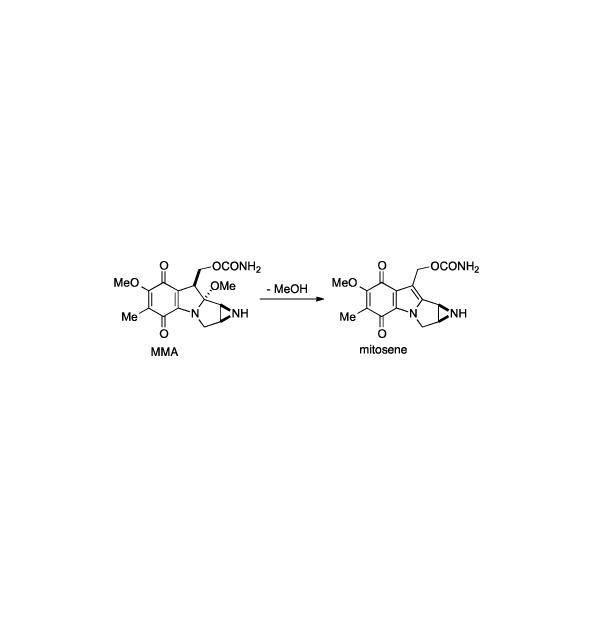
Mitosene formation shown for mitomycin A.
Another challenge with the mitomycins is the installation and preservation of the aziridine ring. Given the mode of action of these compounds, it is not surprising that the aziridine is prone to opening in certain cases. One might think of it as going through a synthetic sequence with a spring-loaded compound, ready to unleash itself to its active and more stable form at any moment. The fact that no enantioselective or enantiospecific total synthesis of the mitomycins has been accomplished is largely due to the inability to introduce the aziridine at an early stage and in an enantiospecific fashion.
The quinone moiety in the mitomycins also presents a problem. Once the quinone is in place, certain transformations (like removal of a protecting group from the aziridine) are difficult. Additionally as a synthesis unfolds, it is easy to get differences in oxidation states throughout the system that make for a challenge.
Arguably the most difficult challenge in the total synthesis is the compact, densely functionalized nature of these compounds. Danishefsky eloquently put the predicament this way:
“The complexity of the problem arises from the need to accommodate highly interactive functionality in a rather compact matrix and to orchestrate the chemical progression such as to expose and maintain vulnerable structural elements as the synthesis unfolds. The synthesis of a mitomycin is the chemical equivalent of walking on egg shells.”[17]
Each of these challenges associated with the synthesis of this family of natural products will manifest themselves as the total syntheses and approaches to these molecules are presented.
5.2. Kishi′s Synthesis of Mitomycins A, B, C and Porfiromycin
In 1977 Kishi and co-workers reported a landmark and first total synthesis of mitomycins A, B, C and porfiromycin.[185-188] This synthesis is very impressive even by today′s standards and represented a quantum leap in the field of natural product synthesis. The synthesis started with commercially available 2,6-dimethoxy toluene (117) (Scheme 23), and was elaborated in thirteen steps to arrive at ketone 118. From ketone 118, ten more steps were required to from the ketal, install the diol, and reduce the terminal cyanide to the corresponding acetate to give diol 119. Many of these steps were various protecting group manipulations in order to get the right functionality in place for subsequent transformations. With diol 119 in hand, the next task was installation of the aziridine ring and conversion to a suitable cyclization precursor. Thirteen steps were needed to accomplish these tasks and obtain ketal 120. The majority of these thirteen steps were employed in the synthesis of the protected aziridine found in 120. Due to the nature of the precursors, 120 was not prepared enantioselectively. Ketal 120 was ready to be subjected to the key steps in the synthesis. Treatment of 120 under hydrogenation conditions simultaneously removed the benzyl groups on the aromatic, the hydroxyl group adjacent to C10 and the terminal amine. Treatment of the intermediate hydroquinone/primary amine compound under oxygen in methanol promoted quinone formation, Michael addition of the primary amine into the quinone, and re-oxidation to provide the protected benzazocane 121 in moderate yield (42% for two steps).
Scheme 23.
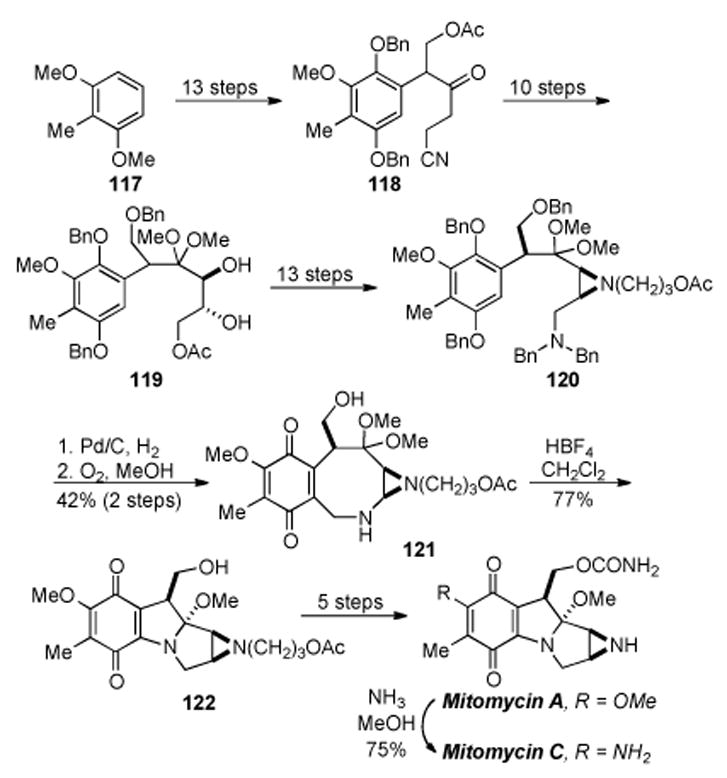
Kishi′s synthesis of mitomycins A and C.
The final key step of the synthesis entailed conversion of protected benzazocane 121 to tetracycle 122 (Scheme 23). This transformation was accomplished by treatment of 121 with HBF4 in dichloromethane to provide tetracycle 122 in good yield. With tetracycle 122 in hand, all that remained was conversion of the alcohol to the carbamate and removal of the protecting group on the aziridine. The carbamate was made in two steps and the free aziridine was obtained through a three-step procedure involving removal of the acetate, oxidation of the alcohol to the aldehyde, and treatment with a Lewis Acid to effect a retro-Michael addition and furnish mitomycin A in a forty-four step linear sequence with an overall yield of 0.19%. Conversion of mitomycin A to mitomycin C was accomplished according to the procedure of Webb and co-workers.[189, 190]
This method was also applied to the total synthesis of porfiromycin (Scheme 24) starting from intermediate 123.[187] Methylation of the free aziridine followed by quinone formation and Michael addition as before provided quinone 124. Lewis acid promoted transannular cyclization under the previously developed conditions provided tetracycle 125, which was subsequently converted to porfiomycin in three additional steps involving carbamate and amine formation. The overall yield for porfiomycin is about the same as for mitomycin A.
Scheme 24.
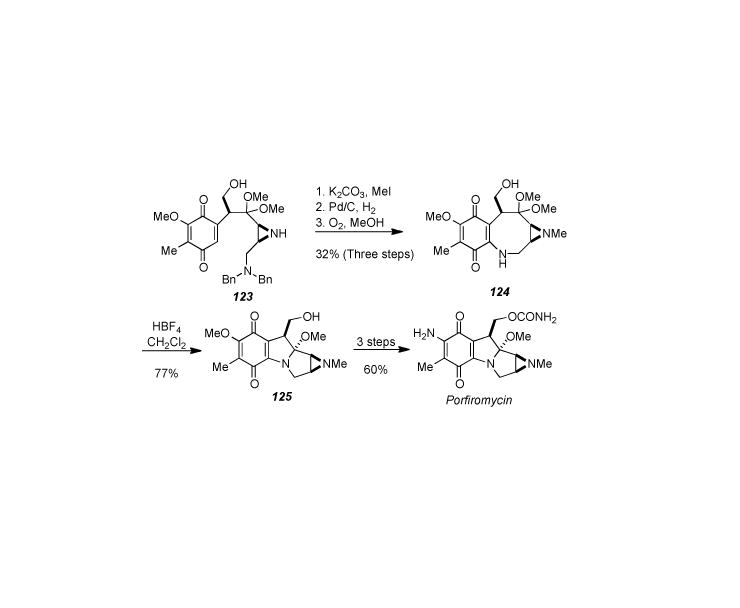
Kishi′s synthesis of porfiomycin.
The Kishi laboratory also accomplished the total synthesis of mitomycin B in a similar manner as mitomycins A, C and porfiomycin (Scheme 25). At the outset of the synthesis, it was not known which diastereomer of C10, resulting from a hydroxymethylation reaction, corresponded to mitomycins A and B respectively. Having established the correct diastereomer for the total synthesis of mitomycins A and C, the other diastereomer was brought through the same sequence of steps to arrive at quinone 126. Reduction of 126 by hydrogenation followed by protection of the benzazocane nitrogen with benzyl chloroformate gave hydroquinone 127. Subjecting 127 to hydrogenation conditions and then oxygen effected removal of the Cbz carbamate, oxidation to the quinone and subsequent cyclization to give tetracycle 128 in a poor overall yield (5%) from quinone 126. Conversion of tetracycle 128 to mitomycin B proved difficult, but it was eventually found that addition of potassium carbonate in methanol in the second step did provide mitomycin B in 55% yield for the two steps. Kishi′s total synthesis of the mitomycins represented a landmark achievement at the time and remains as the standard against which all future synthetic work in this area is measured. The synthetic route, though lengthy and linear, allowed for the synthesis of multiple members of the mitomycins from common intermediates.
Scheme 25.
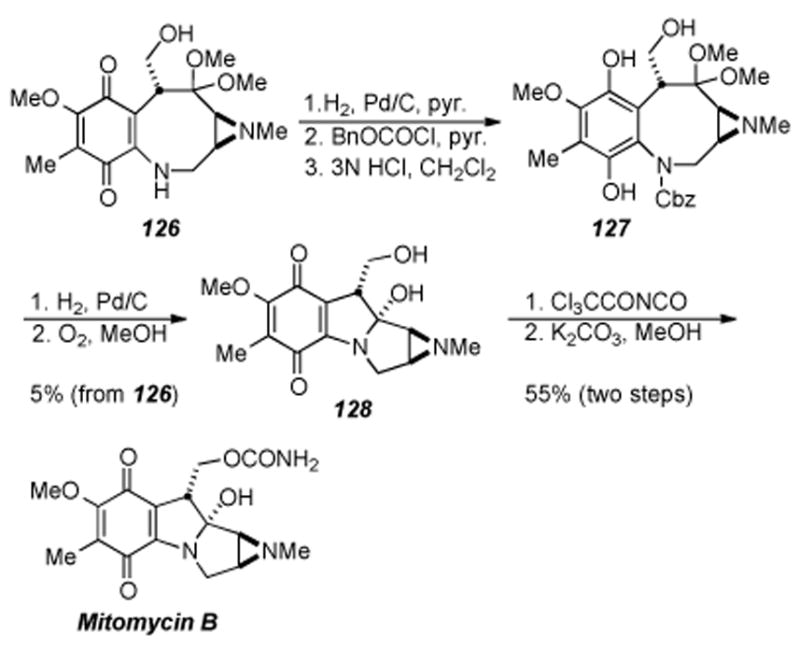
Kishi′s synthesis of mitomycin B.
5.3. Fukuyama′s Synthesis of Mitomycins A and C
In 1987 and later in 1989 Fukuyama and Yang published a racemic total synthesis of isomitomycin A, mitomycin A, and mitomycin C.[191-193] Their approach took advantage of the mitomycin rearrangement reaction discovered earlier by Kono and co-workers (Scheme 26).[194] This novel approach could be advantageous with regard to being able to avoid the unstable tetracyclic skeleton of the mitomycins until the last step, at which the unstable natural product would be formed. As shown is Scheme 27, the synthesis commenced by use of 2,6-dimethoxy toluene (117), the same starting material used by Kishi, and elaborated in thirteen steps to chalcone 129. Chalcone 129 was then reacted with siloxy furan 130 (prepared in one step) with tin tetrachloride at -78 °C to cleanly give (after the addition of pyridine) azide 131 in excellent yield.
Scheme 26.
Mitomycin rearrangement as discovered by Kono et. al.
Scheme 27.
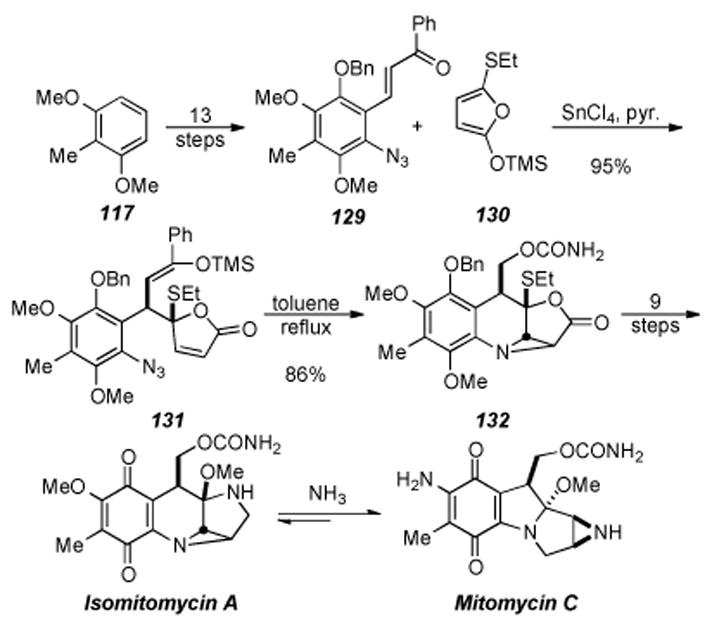
Fukuyama′s synthesis of Mitomycin C.
Azide 131 was then heated in toluene at reflux to induce an intramolecular azide-olefin cycloaddition that smoothly gave the tetracyclic aziridine 132 in good yield. Ten steps were used to transform tetracyclic aziridine 132 to isomitomycin A. It was found that subjection of isomitomycin A to ammonia in methanol cleanly provided mitomycin C (presumably through isomitomycin C as an intermediate), concluding the synthesis of twenty-six steps and 10% overall yield.
Mitomycin A was also obtained by treatment of isomitomycin A to the mitomycin rearrangement conditions (Al(OiPr)3, MeOH, room temperature, 2 days) in 91% yield. This elegant synthesis has set the benchmark for total synthesis of the mitomycins that has yet to be matched. The authors also reported that each step in the synthesis is scalable, allowing for gram amounts of the natural products to be made.
5.4. Danishefsky′s Synthesis of Mitomycin K
Danishefsky and co-workers have contributed significantly to both the synthesis of the mitomycins and their analogues as well as to an understanding of their mode of action.[17,195,196] As part of this work, Danishefsky developed a synthesis of mitomycin K using novel transformations never before applied to the mitomycins. The synthesis starts from nitro aldehyde 133 (prepared in six steps from commercial material) which was reacted with the known 1-methoxylithiobutadiene (134) to form alcohol 135 (Scheme 28). Reaction of 135 under intramolecular Diels-Alder (IMDA) conditions provided tricycle 137 in modest yield. Presumably the nitrosodieneone is the active IMDA substrate and forms the fused intermediate 136 (Scheme 28), which upon rearrangement gives ketone 137. Ketone 137 was smoothly converted to triazoline 138 in five more steps.
Scheme 28.
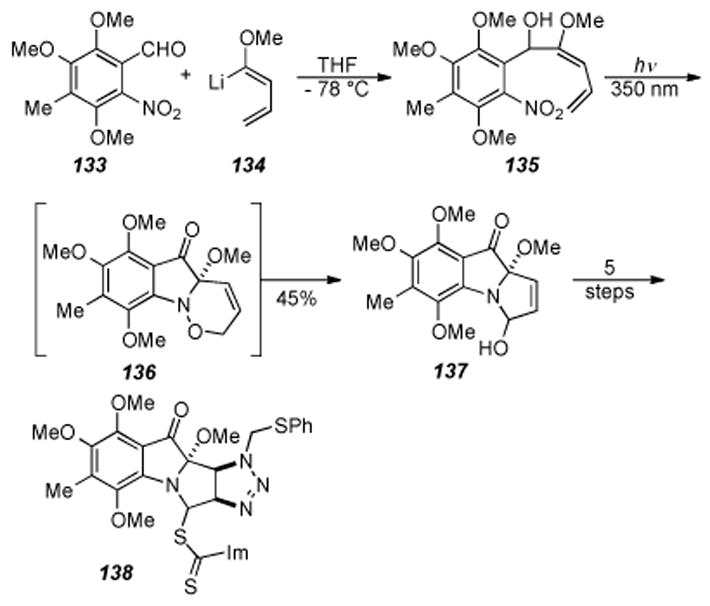
Danishefsky and coworkers′ route to mitomycin K.
The tetracyclic core of the mitomycins was formed first by elimination of the thiocarbonyl moiety from 138 under radical conditions, followed by formation of the aziridine via photolysis to form ketone 139 (Scheme 29). De-sulfurization of 139 gave the N-methyl aziridine, reaction of the ketone with trimethylsilylmethyl lithium, and quinone formation provided quinone 140. It should be noted that oxidation to the quinone with the para-methoxy groups was accomplished, albeit in poor yield (8-16%). Treatment of 140 under PPTS conditions effected a Peterson elimination and provided mitomycin K (141), in twenty-one steps and 0.1% overall yield.
Scheme 29.
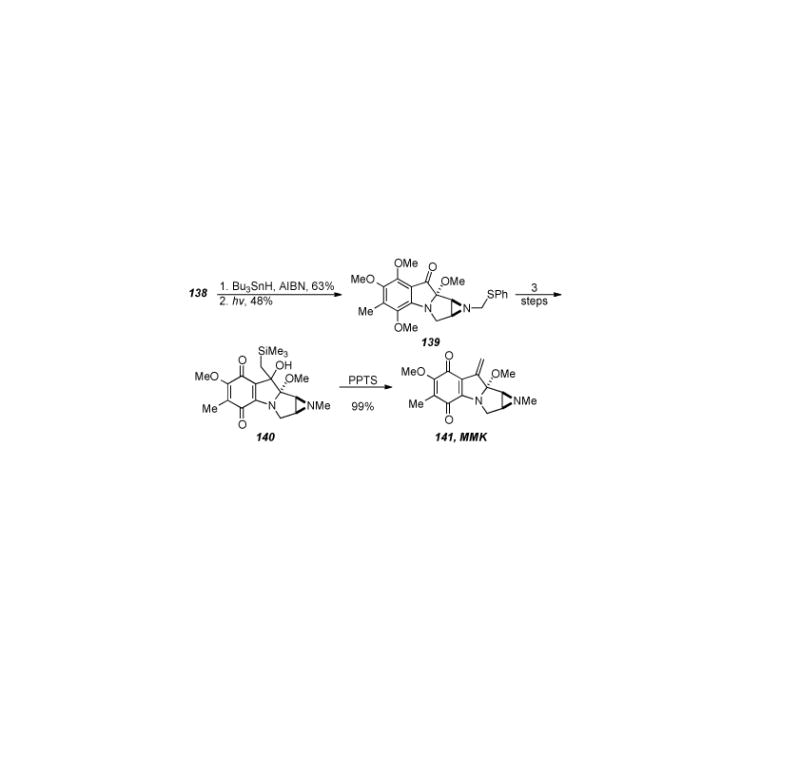
Completion of Danishefsky′s synthesis of mitomycin K.
Despite the low yield in the quinone-forming reaction, the synthesis of mitomycin K by Danishefsky is an elegant piece of work that employs several novel transformations. Key steps in the synthesis include formation of the tricyclic core by an IMDA reaction, aziridine formation by photolysis of the intermediate triazoline derived from treatment of 138 with radical forming conditions (compound not shown), and Peterson elimination to form the exocyclic olefin found in mitomycin K.
5.5. Jimenez′s Synthesis of Mitomycin K
The most recently completed total synthesis of a natural mitomycin is the synthesis of mitomycin K by Jimenez and Wang (Scheme 30).[197,198] The synthesis starts from dinitro arene 142 (prepared in one step from 2,5-dimethylanisole) and, after alkylation and reduction, provided indole 143. Reaction of indole 143 with Fremy′s salt gave the corresponding quinone, which was subsequently reduced to the hydroquinone, protected as the bis-TBS arene ether, followed by reduction of the methyl ester to give aldehyde 144. Reaction of aldehyde 144 with dimethylvinylsulfonium iodide in the presence of sodium hydride, followed by ring-opening of the tetracyclic epoxide with sodium azide furnished tricyclic azido alcohol. Mesylation of the tricyclic azido alcohol provided pyrroloindole 145, which was ready for the last key step of the synthesis.
Scheme 30.
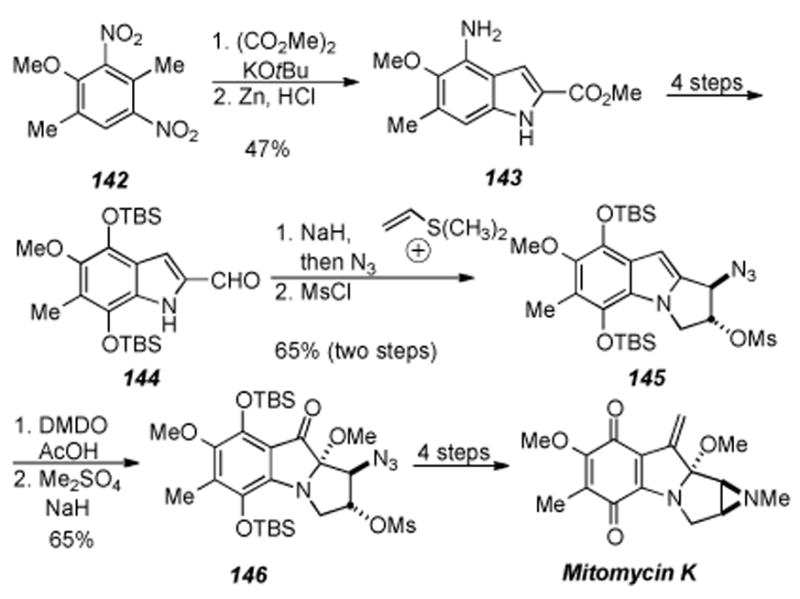
Synthesis of mitomycin K by Jimenez and Wang.
The last key step of the synthesis entailed oxidation of the indole double bond of 145. It was found that treatment of indole 145 with dimethyldioxirane and acetic acid followed by methylation of the resulting alcohol gave ketone 146 in good yield for the two steps as a single diastereomer. Staudinger reduction of 146 to form the free aziridine and methylation thereof gave a tetracyclic ketone (not shown). Conditions used by Danishefsky then effected formation of both the quinone and the exocyclic olefin to provide mitomycin K in fifteen steps and 9% overall yield.
Jimenez and Wang accomplished the total synthesis of mitomycin K in a straightforward and efficient manner without the excess use of protecting groups. The key step in the synthesis is the oxidation of the C2-C3 indole double bond in 145 to form ketone 146 with concomitant installation of the requisite methylaminal. This novel transformation gave access to substrates where chemistry developed earlier by Danishefsky and coworkers could be used to complete the synthesis.
5.6. Conclusion
As may be inferred from the relatively few total syntheses of the mitomycins reported in the literature over the 55 years since the first report of their isolation, the compact tetracyclic famework of these facinating alkaloids bears a deceptive guise of simplicity. Scores of competent and innocent synthetic organic chemists have been taken advantage of by the allure of the prospect of building these molecules in the laboratory from inexpensive petrochemical feedstocks. In the following chaper the reader may find that there are multitudinous successful methods for constructing pyrroloindole tricycles and also aziridinomitosene/sane frameworks. However, the modus operandi of organic synthesis has apparantly not evolved to the point at which all the peripheral functional groups of the natural products may be installed upon core frameworks without painstaking forethought and fortuitous insght.
6. The Mitomycins: Synthetic Studies
6.1 General Remarks
Surveyed in this section are the approaches towards mitomycinoids which have not culminated in the production of a known natural product. Excluded are semisyntheses and reports in which an advanced compound was but menially elaborated. The primary focus is on reports in which polycyclic scaffolds are created from acyclic or monocyclic precursors. Also surveyed is the work of groups which have combined and built upon the methodology of others to bear out significant synthetic advances.
6.2. Benzazocine/Mitosane Synthesis by Williams
Our laboratory recently developed a novel reductive aminocyclization approach towards the mitomycins whereby late-stage intermediates were obtained in the context of developing an asymmetric synthesis of the mitomycins. This approach, shown in Scheme 31, relies on an aldol coupling of nitro arene 147 with aziridine aldehyde 148 followed by oxidation of the secondary alcohols (obtained as a mixture of diastereomers from the coupling reaction) to afford ketone 149.
Scheme 31.
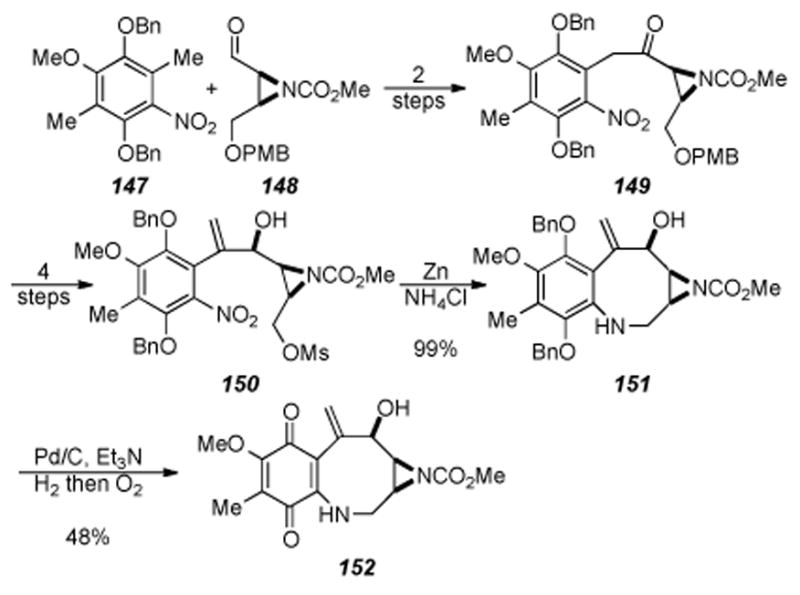
Benzazocine formation by reductive aminocyclization.
Four straightforward steps of methylenation, PMB ether removal, mesylation, and ketone reduction provided allylic alcohol 150. Treatment of allylic alcohol 150 under zinc ammonium chloride conditions did provide the aniline as well as another product that was identified as benzazocine 151. It was found that prolonged treatment (3 hours as opposed to 30 minutes) of allylic alcohol 150 under zinc ammonium conditions gave essentially a quantitative yield of benzazocine 151. Quinone 152 was obtained by treatment of benzazocine 151 to hydrogenation conditions followed by air oxidation. This reductive aminocyclization reaction (i.e. 150→ 151) is the first example in the mitomycin literature where cyclization and benzazocine formation occurs without the need for prior activation of the aniline nitrogen.
The reductive aminocyclization strategy used above for formation of benzazocine 151 was also employed in constructing the tetracyclic core of the mitomycins in one step from an acyclic precursor (Scheme 32). Treatment of bis-mesylate 154 (obtained from mesylation of allylic alcohol 150) under the reductive aminocyclization conditions used previously gave a 55% yield of mitosane 155. Quinone formation under conditions used previously gave quinone 156. Allylic oxidation of quinone 156 followed by N- methyl aziridine formation, if feasible, has potential for the first asymmetric total synthesis of mitomycin K and also would be the first asymmetric total synthesis of any member of the mitomycins. Efforts to convert benzazocine 151 and mitosane 156 into mitomycin K are now in progress.
Scheme 32.
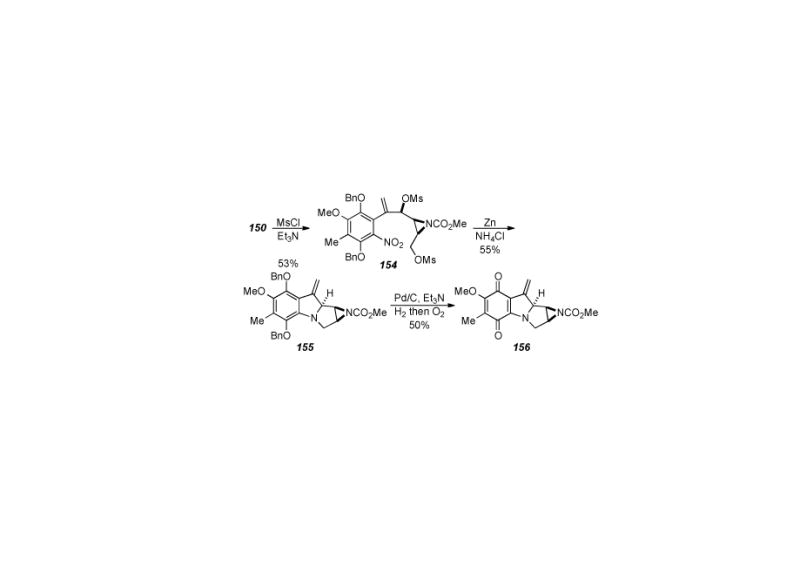
Mitosane formation via reductive aminocyclization.
6.3. Aziridinomitosene Synthesis by Vedejs
Vedejs and coworkers have developed an innovative dipolar cycloaddition approach to access the tetracyclic core of the mitomycins.[199-201] This work grew out of the feasibility of pyrrole synthesis by intramolecular cycloaddition of azomethine ylides starting from oxazole precursors.[202-206] The key step of the synthesis, shown in Scheme 33, entailed treatment of alkyne 158 (prepared in four steps from oxazole 157) with silver triflate to give tricycle 159. Tricycle 159 was not isolated and treated in situ with the organic soluble cyanide source BnMe3N+CN− to provide aziridinomitosene 160 in 33% yield. It is presumed that formation of aziridinomitosene 160 proceeds through intermediate 161, upon which a dipolar cycloaddition provides the observed product. This method for the formation of aziridinomitosene 160 employs several novel transformations heretofore unseen in the construction of the mitocenes. Several late-stage intermediates as well as mitosene analogues of mitomycins A and C have been constructed utilizing this approach. Inherent in this approach, is that mitosenes are directly accessed and the conversion of a mitosene into a natural mitomycin or FR900482 system has proven extremely difficult. In addition, Vedejs found that removal of the N-trityl protecting group could not be achieved without aziridine cleavage,[201] but this problem has been subsequently addressed utilizing bulky silicon-based protecting groups.[207]
Scheme 33.
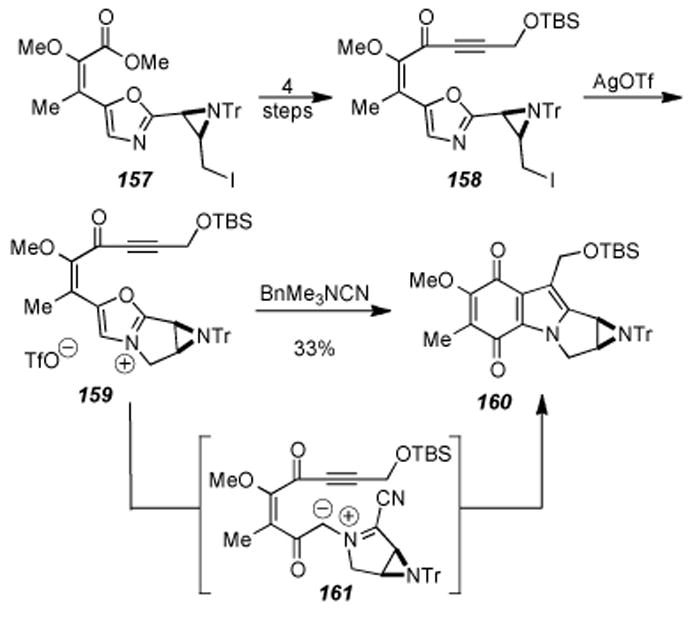
Synthesis of aziridinomitosene 131by Vedejs.
6.4. Johnston′s Synthesis of an Advanced Intermediate
Johnston and coworkers recently developed an efficient approach to an advanced intermediate 165 by regioselective coupling of enamine 163 with quinone 164 (Scheme 34).[208, 209] Enamine 163 is formed in situ from alkynylamine 162, which is constructed by a base-promoted aza-Darzens reaction between the diphenylmethyl amine Shiff base of ethyl glyoxylate and tert-butyl chloroacetate.
Scheme 34.
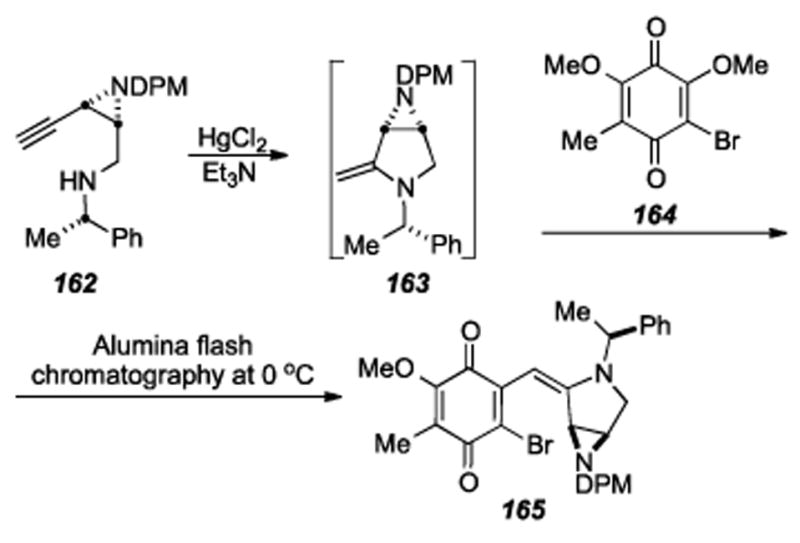
Johnston′s synthesis of advanced intermediate 165.
Despite the low yield of 165 (26% over two steps), the synthetic sequence requires only nine steps from the commercially available ethyl glyoxylate. If successful this synthesis would be a huge step forward in the quest to find a concise and synthetically viable route for the efficient construction of the mitomycins and analogues.
One drawback to Johnston′s synthesis is the instability of intermediate 165 (t1/2 = 1.5 days at -15 °C). This will undoubtedly make the next steps in the synthesis quite challenging. Hurdles yet to overcome in the synthesis include removal of the protecting groups on both the aziridine and pyrrolidine nitrogens, as well as installation of the C10 hydroxymethyl moiety.
6.5. Michael′s Formal Synthesis of 7-Methoxyaziridinomitosene
After reporting on a tetracyclic model system,[210] in 2006 J. P. Michael and coworkers disclosed a formal synthesis of 7-methoxy-aziridinomitosene (Scheme 35).[211] The synthesis was actually a formal total synthesis relying on the work of Jimenez and Dong[212] (and detailed elsewhere in this article) for late stage installation of the aziridine and completion of the target. The synthesis started from 2-methylresorcinol (166) which was converted to lactam 167 in nine steps. The first key step of the sequence was lactam formation accomplished by mesylation of a primary alcohol and subsequent nucleophilic attack and ring closure by the amide nitrogen. Two more steps were required to make enamine 168. The next key step entailed treatment of enamine 168 to intramolecular Heck cyclization conditions, which afforded tetracycle 169 in 82% yield. Ten more steps involving aziridine, quinone, and carbamate formation were required to access 7-methoxyaziridinomitosene A 170. In addition to intercepting a key intermediate reported by Jimenez and Dong, this synthesis builds off of earlier precedent in the mitomycin literature where enamines were used in tetracycle formation.[213]
Scheme 35.

Michael′s formal synthesis of aziridinomitosene 170.
6.6. Benzazocine Construction by Ciufolini
Ciufolini recently disclosed an approach to the mitomycins[214] that takes advantage of the homo-Brook rearrangement methodology for eight-membered ring formation previously used in their total synthesis of FR66979.[215] The synthesis starts with aldehyde 171 being converted to aziridine 174 in three steps through the intermediacy of cycloaddition precursor 172 and triazoline 173, now primed for the key steps of the synthesis (Scheme 36). Photoinduced ring contraction of triazoline 173 effected a net loss of nitrogen and delivered aziridine 174. Homo-Brook rearrangement of aziridine 174 proceeds smoothly by treatment with tetrabutylammonium hydroxide and, as in the case of FR66979, provides benzazocine 175 in reasonable yield. Reaction of benzazocine 175 with benzyl azide smoothly produced triazoline 176, whereupon photolysis of 176 should yield the N-benzyl protected aziridine. From this point, a number of straightforward transformations should give the mitomycins. Potential pitfalls include quinone oxidation and transannular cyclization to access the tetracyclic core of the mitomycins. This approach demonstrates the effective application of the homo-Brook rearrangement methodology to access the core structure of the mitomycins and highlights the utility of this approach in the formation of benzazocines.
Scheme 36.
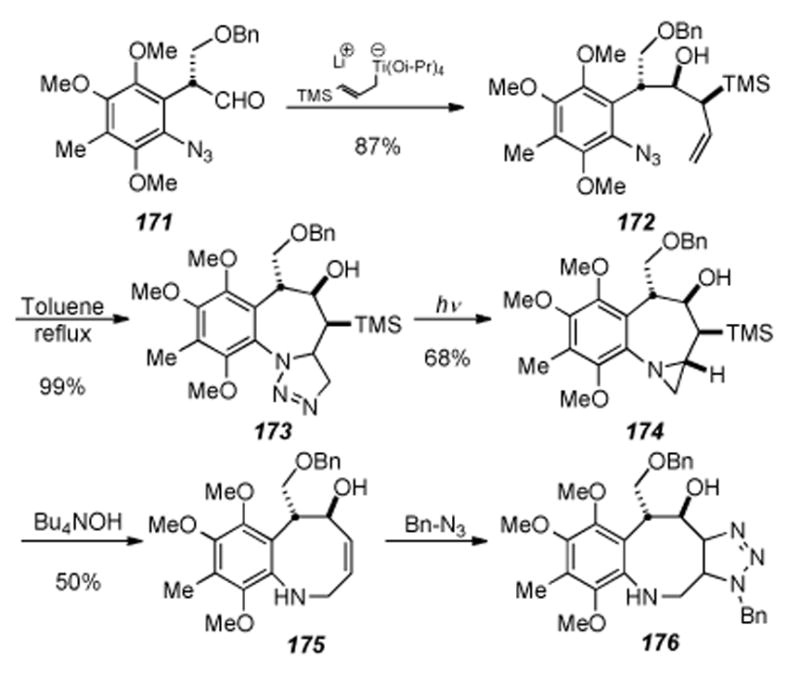
Ciufolini′s approach towards the mitomycins.
6.7. Coleman′s Synthesis of (+)-9a-Desmethoxymitomycin A
In 2004 Coleman and coworkers reported the enantiospecific synthesis (+)-9a-desmethoxymitomycin A (181), containing all the required functionality for mitomycin A except the C9a methoxy moiety (Scheme 37).[216] The key step of the sequence is the addition of allylstannane 179 to iminium ion 178 (formed in situ from pyrrolidine 177) to form alcohol 180. Aziridine formation by use of a Mitsunobu reaction, carbamate formation, Boc removal, quinone formation, and cyclization provided (+)-9-adesmethoxymitomycin A 181 in good overall yield.
Scheme 37.
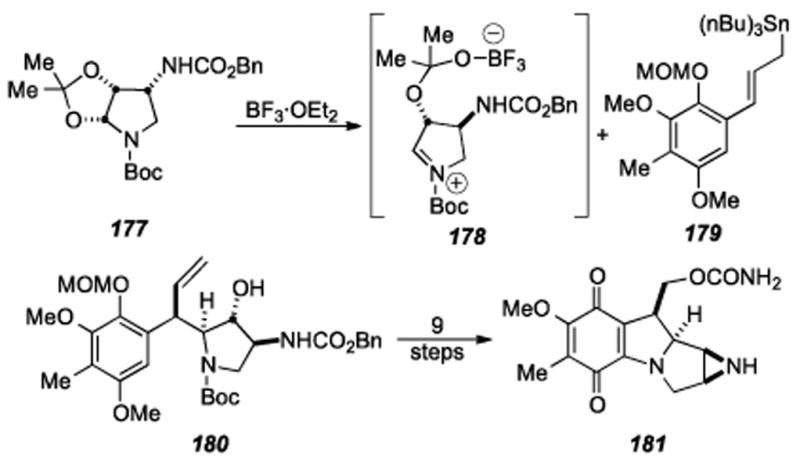
Coleman′s enantiospecific synthesis of (+)-9a-desmethoxymitomycin A (181).
The authors report that attempts to install the required C9a oxygen for completion of the synthesis is now in progress. If late-stage introduction of oxygen fails, significant re-tooling of the synthetic route would be required so as to account for earlier installation of the required oxygen.
6.8. Miller′s Enantiospecific Synthesis of the Tetracyclic Core
Miller and co-workers have recently developed a novel route to the tetracyclic core of the mitomycins starting from isatin (182) as shown in Scheme 38.[217] Ring-opening of isatin (182) and further elaboration led to allyl ester 183. Conversion of allyl ester 183 to vinyl ketone 184 proceeded smoothly in two steps and provided material for the key step of ring-closing metathesis (RCM) to form enone 185. It was found that the Grubbs II catalyst effected this transformation in 83% yield. Although Miller and Grubbs have shown the feasibility of constructing eight-membered rings using RCM,[218] the use of RCM in the construction of the benzazocane core of this family of natural products was first demonstrated by Martin and coworkers (see section 7.4).[219,220] Conversion of enone 185 to the enantiomerically pure epoxide 186 required four steps. The key step of the sequence involved kinetic resolution of the secondary alcohol (resulting from reduction of the ketone) using a peptide-based asymmetric acyl transfer catalyst developed previously by Miller.[221-224] With successful procurement of epoxide 186, the next major task was the transannular cyclization of 186 and tetracycle formation. It was found that treatment of 186 under dilute acid with heating effected deprotection of the ketal to the ketone, Boc removal, subsequent cyclization to the tetracycle, and installation of the methyl aminal to afford 187 in a single step in good yield (81%). Formation of the methyl aminal is proposed to arise from methanol addition to the corresponding iminium ion formed in situ under the reaction conditions. Treatment of tetracycle 187 to four more synthetic transformations furnished aziridine 188, the furthest intermediate reported by these authors to date. Published attempts by Miller, et al. to functionalize the aromatic ring in order to complete the total synthesis of the mitomycins have been unsuccessful to date. Nevertheless, the above approach is elegant, concise, and contains several new transformations to access the tetracyclic core of the mitomycins. The authors report that further studies are underway in order to access the natural products and have since developed a mild, efficient azide conjugate addition to an eight-membered ring enone substrate allowing for rapid entry into mitomycin-like ring systems.[225]
Scheme 38.
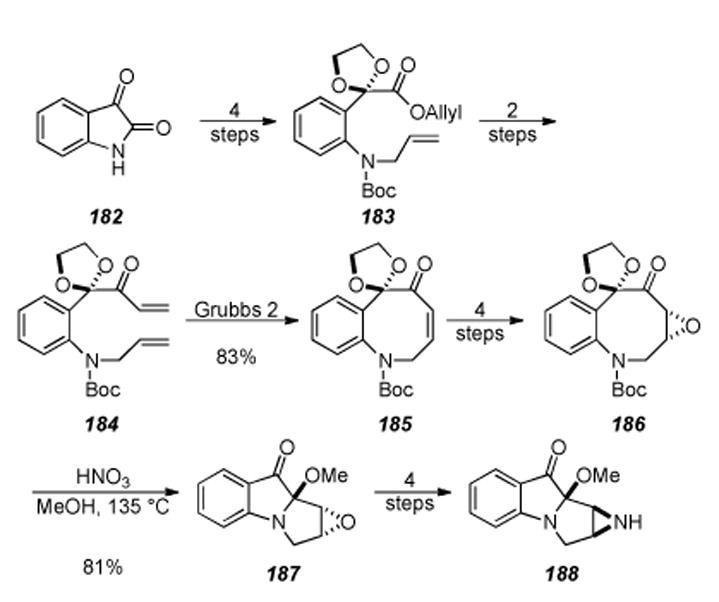
Miller′s enantiospecific synthesis of the tetracyclic core.
6.9. Tandem Radical Cyclization Approach by Parsons et. al.
A tandem radical cyclization approach was developed by Parsons for rapid construction of the tricyclic core of the mitomycins via formation of the C9-C9a bond (Scheme 39).[226] The synthesis commenced with nitro aldehyde 189 and was elaborated four steps to vinyl bromide 190. Treatment of vinyl bromide 190 under standard radical-forming conditions in refluxing toluene provided a 44% yield of tricycle 191. Although lacking key functionality for the natural products, this study demonstrates a rapid method for construction of the mitomycin core, and could be a useful method on an appropriately functionalized substrate.
Scheme 39.
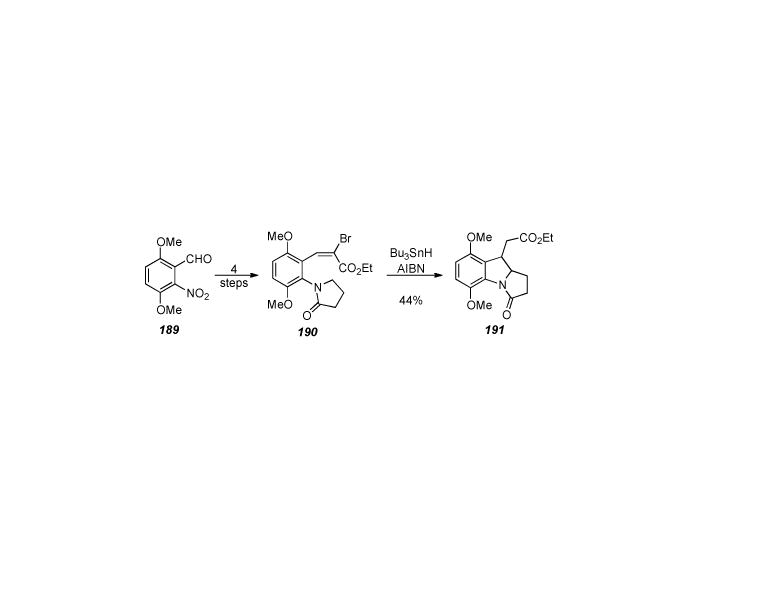
Parson’s formation of tricycle 191by radical cyclization.
6.10. Radical Cyclization Approach by Jones
Jones and coworkers published an approach to the mitomycin core skeleton utilizing a radical cyclization as the key step (Scheme 40).[227] Unlike the work done by Parsons,[226] Jones’ work involves forming the C9a-C1 bond during the radical reaction. Bromination at the C2 position of 5-methoxy indole followed by N-alkylation of the indole nitrogen provided indole 192. Treatment of indole 192 under radical-inducing conditions provided a 91% yield of tricycle 193.
Scheme 40.
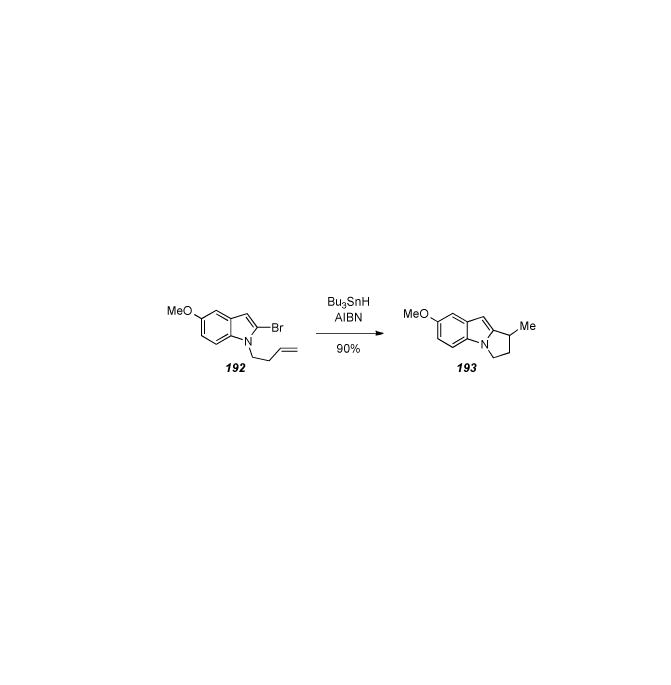
Radical cyclization approach by Jones and coworkers.
6.11. 7-Methoxyaziridinomitosene by Jimenez and Dong
In 1994 and 1996 Jimenez and Wang reported the synthesis of the tetracyclic mitomycin skeleton via a dialkylvinylsulfonium salt,[228] and an aziridinomitosene analog,[229] respectively. In 1999 Jimenez and Dong published the synthesis of 7-methoxyaziridinomitosene A (199), the product obtained by elimination of methanol from mitomycin A.[212] At the time, this synthesis was the first reported in the literature of a fully functionalized mitosene and served as a platform from which the total synthesis of the FR900482 tetracyclic ring system was constructed, as reported by Jimenez and Wang one year later.[230] Treatment of bis-nitro arene 194 to four steps involving alkylation, cyclization and quinone formation furnished quinone 195 (Scheme 41). Hydroquinone formation and protection of the oxygens as the TBS silyl ethers followed by reduction of the ester to the aldehyde provided indolic aldehyde 196. As a key transformation in the synthesis, indole 196 underwent a Corey-Chaykovsky epoxidation reaction upon treatment with diisopropylvinylsulfonium triflate to form the tetracyclic epoxide compound. The epoxide was not isolated but was rather reacted in situ by treatment with sodium azide, prompting ring-opening and formation of the secondary alcohol. Mesylation of the secondary alcohol obtained from the previous step provided tricycle 197 in moderate yield (65%) for the two steps. Elaboration of tricycle 197 over four more steps involving quinone and carbamate formation provided quinone 198 in good yield. To complete the synthesis, quinone 198 was subjected to two more conditions that induced aziridine formation and provided 7-methoxyaziridinomitosene A 199 in a concise and straightforward manner. The late stage transformations in this route were used by Michael and coworkers in their synthesis of 199 as described previously in this article.
Scheme 41.
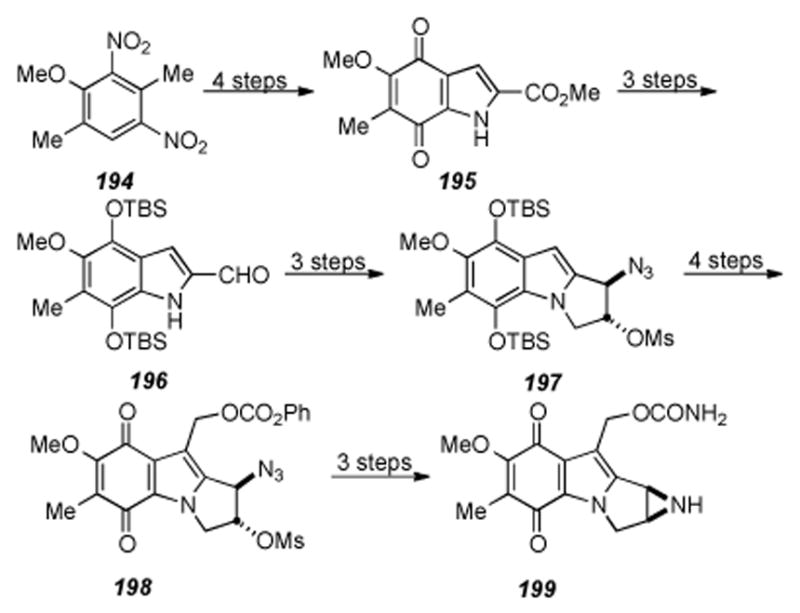
Fully elaborated mitosene by Jimenez and Dong.
6.12. Ziegler′s Synthesis of (+)-9a-Desmethoxymitomycin A
Before the successful synthesis of desmethoxymitomycin A (181) by Coleman and coworkers (Scheme 37),[216] Ziegler and Berlin reported the enantiospecific synthesis of the same molecule by use of a aziridinyl radical cyclization route.[231] This same approach was successfully used in the synthesis of the FR900482 core structure.[232] The synthesis, shown in Scheme 42, commenced with nitro arene 200, which was elaborated in eight steps to indole 201. Indole 201 was alkylated with triflate 203 to afford coupled product 204, which was elaborated four more steps to give bromoaziridine 205. The key step of the synthesis was treatment of bromoaziridine 205 under aziridinyl radical-forming conditions, which cyclized onto the indole 2,3-double bond to give tetracycle 206 in decent yield. It is interesting to note that conversion of 205 to tetracycle 206 is completely diastereoselective. Further manipulation of 206 did provide desmethoxymitomycin A 181 in seven more synthetic transformations. The above work by Ziegler and Berlin highlights the utility of a radical cyclization approach to complete the core of the mitomycins containing all the requisite functionality except the C9a methyl aminal.
Scheme 42.
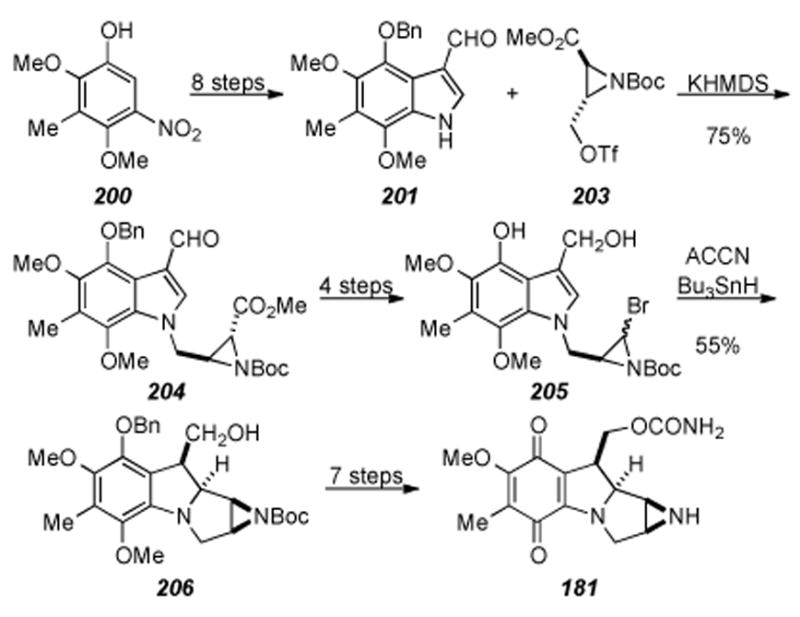
Ziegler′s synthesis of (+)-9a-desmethoxymitomycin A.
6.13. Mitosane Synthesis by Jones and Coworkers
A method to the mitosane core of the mitomycins was reported by Jones and coworkers starting from indole (207) (Scheme 43).[233] Functionalization to tricyclic mitosene 208 was achieved by a three-step procedure. Selective reduction of the C1-C2 double bond followed by complexation with chromium provided the η6 arene chromium carbonyl species 209. As a key step in the synthesis, the authors found that a stereoselective reduction could be performed on complex 209 to arrive at tricycle 210. Lithiation of 210 followed by addition of formaldehyde effected hydroxymethylation at the benzylic position with exclusive anti configuration of the hydroxymethyl moiety relative to the C9a hydrogen installed in the previous step. Deprotection of the chromium arene complex provided alcohol 211 in good yield for the two steps. It was then illustrated that alcohol 211 could be efficiently converted in high yields to either the carbamate present in mitomycins A and C, or to the exocyclic olefin present in mitomycin K (i.e. 212 or 213 respectively).
Scheme 43.
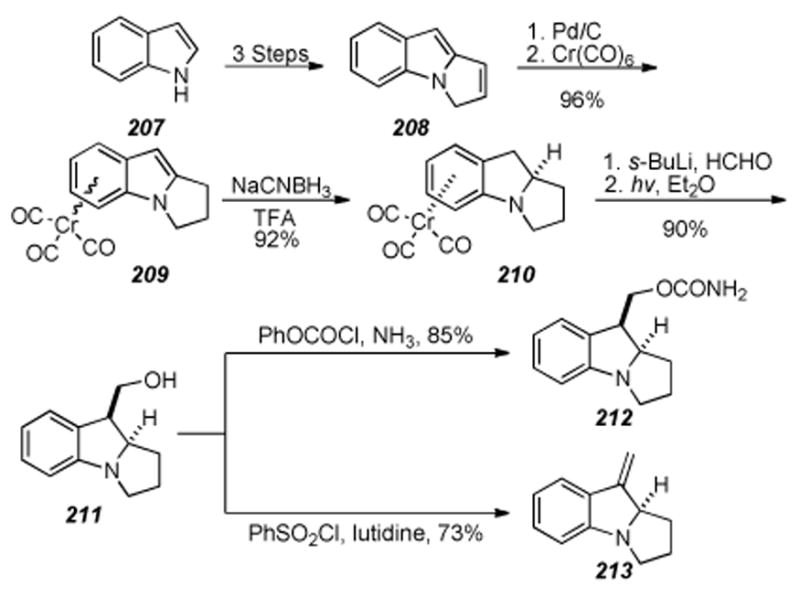
Mitosane synthesis by Jones and coworkers.
6.14. Approach to the Tricyclic Core by Cha and Coworkers
An efficient approach to the tricyclic core of the mitomycins was reported by Cha and coworkers.[234] The key precursor in the synthetic sequence was the methoxy-substituted o-imidostyrene 214. Treatment of styrene 214 to conditions originally developed by Kulinkovich[235] for the cyclopropanation of carboxylic esters provided either tricycle 216 or 217 depending on the workup conditions (Scheme 44). This reaction is believed to proceed through titanium metallocycle 215. This method provides rapid access to the tricyclic core of the mitomycins and application of this methodology to obtain the natural products are underway.
Scheme 44.
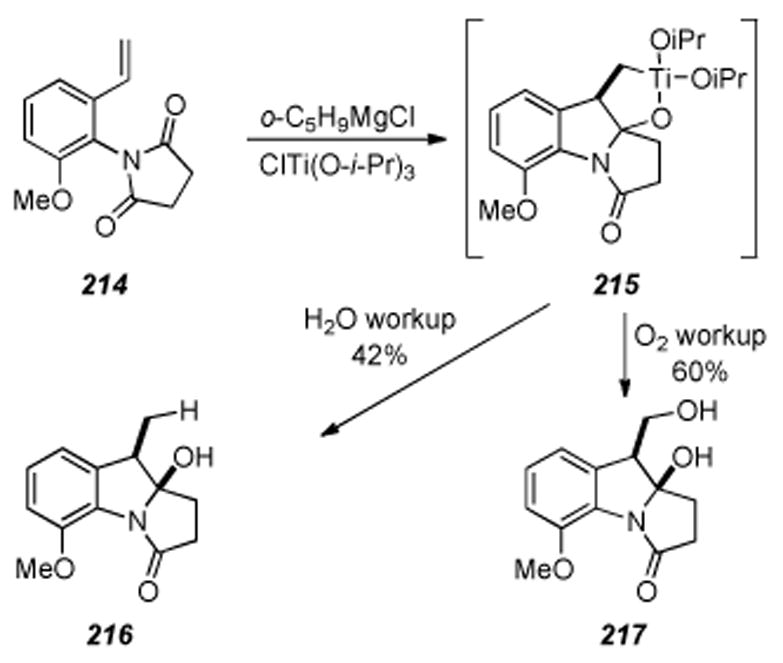
Approach to the mitomycin core by Cha and coworkers.
6.15. Mitosene Synthesis by Sulikowski and Lim
Sulikowski and Lim reported an approach to the tetracyclic core of the mitomycins as illustrated in Scheme 45.[236] Diazo ester 220 was prepared in six steps from the commercially available 2-nitrophenylacetic acid (218) via intermediate 219. The key step of the sequence involved treatment of diazoester 220 with copper (I) bis(oxazoline) complexes to provide mitosanes 221 and 222 in a 5:1 ratio but with poor enantioselectivity (48% ee in best case). Treatment of 221 and 222 with DDQ provided mitosene 223 in good yield. Although lacking the necessary functionality requisite for the natural products, this is a simple and straightforward approach to the mitomycin core structure.
Scheme 45.
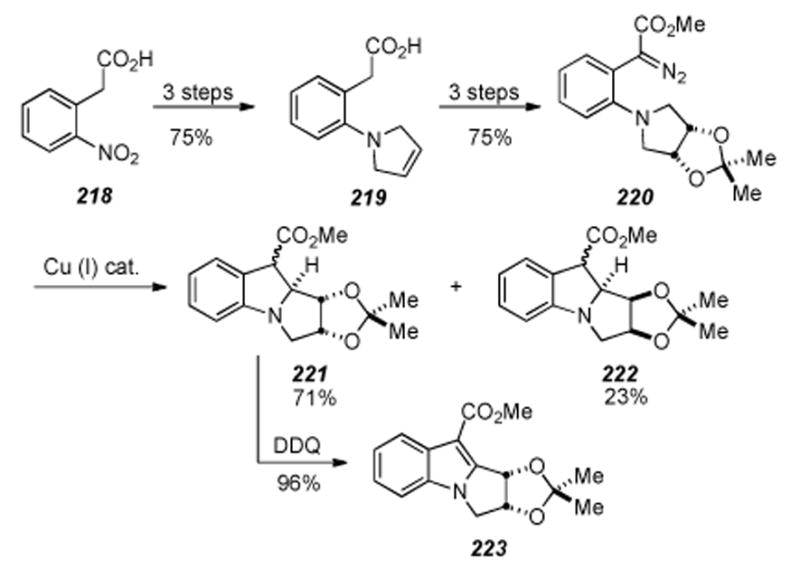
Tetracycle formation by Sulikowski and Lim.
6.16. Ban′s Synthesis of a Decarbamoyloxymitomycin Derivative
The synthesis of a decarbamoyloxymitomycin derivative was reported by Ban and coworkers in 1994 (Scheme 46).[237] Quinone 224 (obtained by a criss-cross annulation reaction[238]) was elaborated to benzazocane 225 in a straightforward manner requiring ten steps. The key step of the synthesis involved treatment of benzazocane 225 with TBSOTf and triethyl amine, which effected a transannular cyclization reaction and provided tetracycle 226 in quantitative yield. This is a curious result in that the tosyl sulfonamide is cleaved under such mild conditions. This transformation was also used by Miller and coworkers where the benzazocine nitrogen was protected as the Boc carbamate and gave similar results (see Scheme 38).[217] Three more steps involving quinone formation and removal of protecting groups gave tetracycle 227 in good overall yield.
Scheme 46.
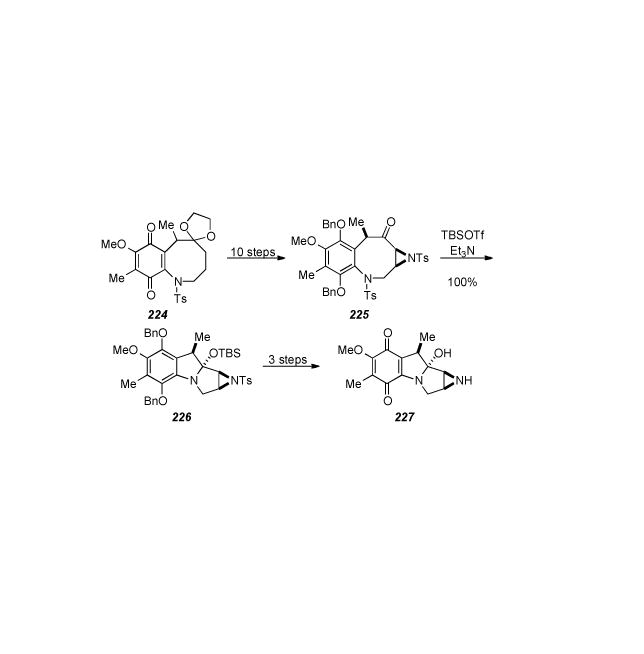
Tetracycle formation via transannular cyclization.
6.17. Kametani: introduction of O at C9a, approach to apo-Mitomycin B, and establishment of the synthetic interconversion of seco-mitosanes with mitosenes
Concurrently with the development of Kishi′s syntheses of mitomycinoids, Kametani and coworkers made several important contributions to the general field of mitomycin synthesis and published two excellent reviews of the state of the art of that time period.[239,240] Kametani′s seminal work in introducing oxygen functionality at the C9a position must be treated with respect, as this is arguably the most difficult transformation of a mitomycin synthesis.[241] A copper mediated amination reaction cyclized 228 in the presence of sodium hydride and cuprous bromide in dimethylformamide at room temperature to provide 229 in 53% yield. Irradiation of 229 in methanol under an oxygen atmosphere in the presence of Rose Bengal as sensitizer with a 200 W tungsten lamp for 72 h gave two products along with starting material which were separated by preparative TLC on silica gel. The structure of the faster running product, obtained as an unstable solid in 21% yield, was assigned as the hydroperoxide 231, while that of the slower running compound, isolated as pale yellowish needles in 21% yield, as C9a-methoxypyrroloindole 232 (Scheme 47). Cyclic peroxide 230 is presumed to be an intermediate in the process. A subsequent publication describes an improved procedure for isolation of 232 as the sole product in 71% yield and introduction of ethyl and isopropyl ethers at the C9a position.[242]
Scheme 47.
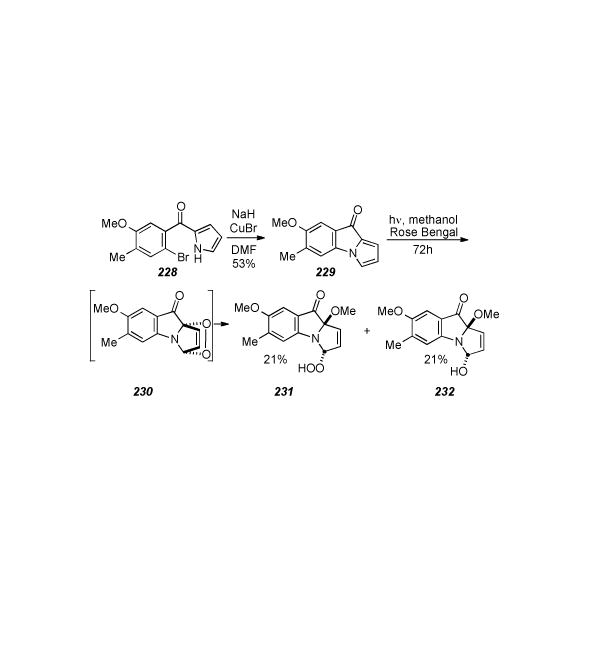
Kametani′s introduction of a methoxy substituent at C9a.
Kametani′s approach to apo-mitomicin B, a biologically relavent mitosene metabolite of mitomicin B which does not posess a C9a functionality, extended the nucleophilic aromatic substitution methodology to more functionalized substrates and represents the author′s ultimate contribution to the field.[243] Benzyl ester 233 was subjected to hydrogenolysis and converted to the thioester via an acid chloride in 99% yield. A second hydrogenolysis using Raney nickel then afforded the required aldehyde 234 in 28.5% yield (Scheme 48). Synthesis of 235 concluded with nitration of the aldehyde 234 with nitric acid, reduction of the intermediate nitorarene with iron powder in aqueous acetic, acid followed by oxidation with Fremy′s salt.
Scheme 48.
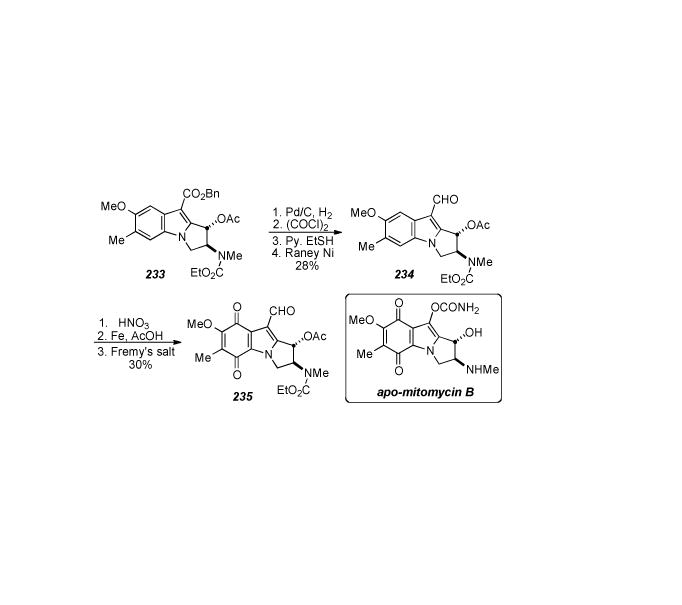
Kametani′s approach towards apo-mitomycin B.
Another of Kametani′s significant contributions regarding mitomycin synthetic studies was establishment of the synthetic pathway of the seco-mitosane, also known as eight membered quinone type compounds, from the mitosene type compounds. Furthermore, Kametani established the synthetic interconversion of these two types of compounds via a Von-Bruaun opening of indolines,[244] and developed several other synthetic procedures for forming benzazocin-5-ones from indoles.[245],[246] These facts are significant because they enabled a connection to be made between the traditional extensive studies on mitomycins and Kishi′s total synthesis procedure according to the biosynthetic pathway.
Thus, protected quinoid 236 was reduced via catalytic hydrogenation to the indoline 237, which was ring opened with TFAA to the benzazocine, and then the ketone 238 was formed via hydrolosis and oxidation (Scheme 49). The one carbon methylene unit of 239 was introduced by treatment of 238 with N,N,N′,N′-tetramethyldiaminomethane (TMDM) and acetic anhydride. Reduction with lithium aluminum hydride then provided recyclization precursor 240, which upon treatment with HCl furnished indole 241.[247] As 241 is the precursor for 236, derived via reduction and acetylation, Kametani′s work exposes the cyclic nature of the transformations between benzazocin-5-ones and the pyrroloindoles characteristic of the mitomycinoid skeleton.
Scheme 49.
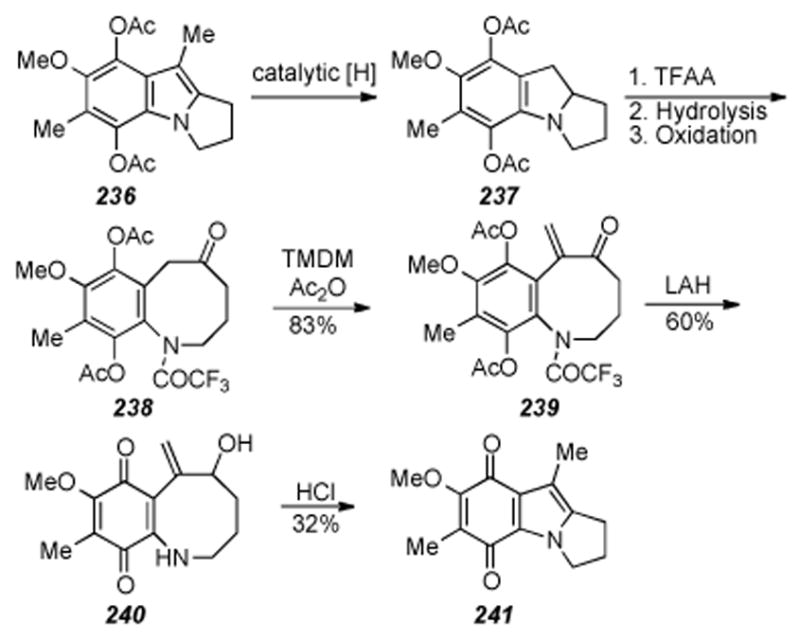
Cyclic nature of the seco-mitosane.
6.18. Danishefsky′s Route to a Mitosane via an Activated Cyclopropane
Four years after Kishi′s first total synthesis of Mitomycin C, Danishefsky and colleagues disclosed their synthesis of mitosane 250 via intramolecular nucleophilic displacement of an activated cyclopropane (Scheme 50).[248] Building on an earlier report[249], the synthesis began with Claisen rearrangement precursor 242, prepared in three steps from commercially available starting materials. Pyrolysis in dimethylaniline effected a very clean Claisen rearrangement, the quality of which is ascribed to the stabilizing effect of the two methoxy groups on the phenoxy radical generated by heterolysis of the allyl group. Methyl ether formation with sodium hydride and iodomethane then gave trimethoxyarene 243. The acetate containing side chain was then elaborated in six steps to furnish the next key intermediate, α-diazo ester 244, which when treated with Cu(acac)2 in chlorobenzene resulted in insertion of the intermediary carbene on the terminal olefin to give the cyclopropane adduct 245, albeit in a modest 35% yield. Ring opening of the lactone under acidic conditions followed by silyl ether protection of the free alcohol and deprotection to the aniline 246 set the stage for formation of dihydroindole 247. This transformation was effected in good yield by an intriguing two-step procedure involving refluxing 246 in dichloromethane and the addition of 2.5 equivalents of monomethylhydrazine over three days. Subsequent chromatography of the resulting residue and heating of the isolate in methanol gave the desired product 247. Tricyclic lactam 248 was prepared in 58% yield by thermolysis of 248 in toluene in the presence of camphorsulfonic acid. The β -dicarbonyl functionality of 248 was exploited to introduce a phenylseleno function α - to the lactam center in three steps, to produce 249. Finally, oxidative elimination with hydrogen peroxide in tetrahydrofuran afforded the final product 250 in 55% yield, isolated exclusively as the N-allylic system as in accordance with a precedent set by Sharpless.[250] This work constitutes an early and innovative approach to the mitosane core, but leaves unaddressed the stereoselective formation of the aziridine.
Scheme 50.
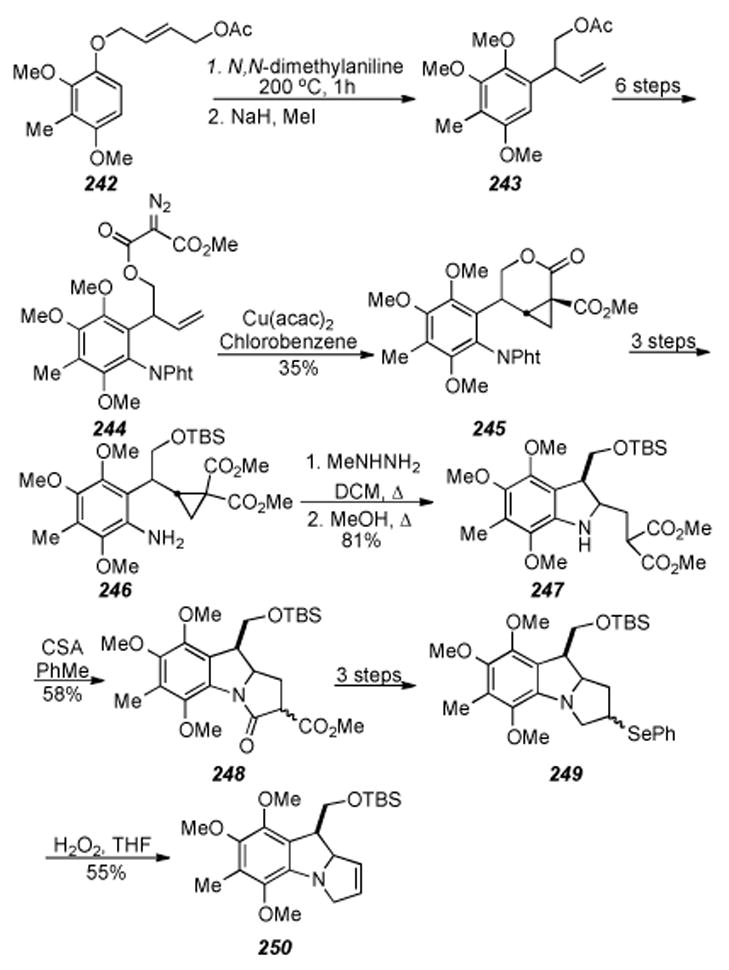
Danishefsky′s Route to a mitosane via an activated cyclopropane.
6.19 Danishefsky′s Stereospecific Route to Aziridinomitosanes
Four years following the publication of their route to mitosane 250, Danishefsky and coworkers had successfully introduced the aziridine onto the mitomycin scaffold, building up the key intermediate in a similar fashion, again utilizing a Claisen rearrangement (Scheme 51).[251] However, here the synthetic pathways diverged markedly, as the subsequent key tandem nucleophilic displacement reaction involving Nicolaou’s[186-188,251,252] reagent N-phenylselenophthalimide (N-PSP) forged the N4-C9a and N4-C3 bonds in a single step to give the tricyclic benzopyrrolizidine core of the mitomycins. The synthesis started from the vinylogous alcohol 251, prepared in six steps from benzaldehyde and (Z)-2-butene-1,4-diol, and the dimethoxyphenol 252, constructed in a slightly modified fashion compared to that previously described.[185]
Scheme 51.
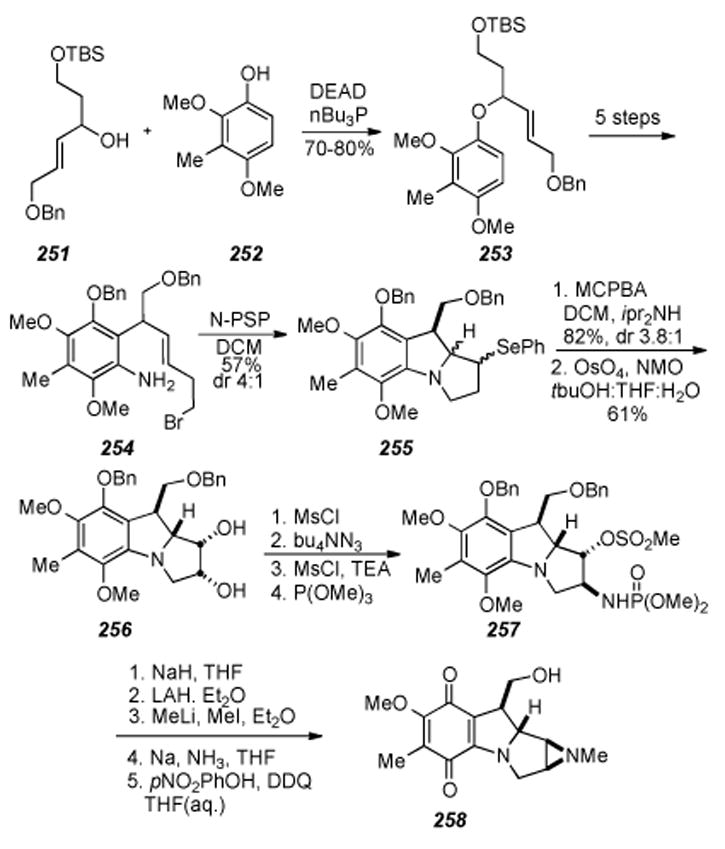
Stereospecific synthesis of aziridinomitosane 258as perfomed in the laboratories of Danishefsky.
Mitsunobu reaction of the two coupling fragments provided the Claisen rearrangement antecedent 253, which following the Claisen rearrangement, was elaborated to biscyclization precursor 254 via a series of protection, nitration, nitro reduction/deprotection, and bromine installation events. At this stage, the authors note: “it was hoped that attack of a suitable electrophile upon the double bond, leading to establishment of the indoline ring, would also set the stage for a second alkylation to generate the complete pyrroloindoline system required.” Furthermore, the trans-double bond in 254 mitigated against intramolecular alkylation of the amino group by the primary carbon bearing the bromide, and reaction with N-PSP in methylene chloride followed by basic extraction afforded a high yield of a diastereomeric mixture of selenides formulated as 255. The minor, undesired stereoisomer was best removed as the corresponding olefin.
After subjection of 255 to oxidative elimination conditions, the olefins were isolated in good yield as a 3.8:1 mixture of diastereomers. Fortuitously, dihydroxylation with osmium tetroxide was only effective on the desired species, giving a mixture of 256 and the unreacted, undesired olefin which was easily removed; apparently osmylation of the minor olefin is less facile. As an alternative procedure, it was found that the bis-cyclization reaction of the C8-aryl acetate analogue of 254 gave exclusively the desired stereoisomer and thus this reaction product was then deacylated with potassium carbonate-methanol and benzylated with benzyl bromide in order to furnish the benzyl ether at the phenolic center, which would provide more suitable protection regarding the installation of the aziridine. Once the stereochemistry across the double bond had been defined via dihydroxylation, a series of four reactions gave the corresponding phosphoramidate ester 257. Intramolecular displacement of the mesylate derived from 256 to form the azide with proper relative stereochemistry for aziridine formation, followed by mesylation of the remaining alcohol, and treatment of the resulting crude azido mesylate with trimethylphosphite gave 257. The free aziridine was then formed by treating 257 with a suspension of sodium hydride in THF to effect displacement of the secondary mesylate, followed by reduction of the phosphoramidate ester with lithium aluminum hydride in anhydrous ether. Finally, treatment of the free aziridine with methyllithium and iodomethane in ether gave the N-methyl derivative in 64% yield. Both benzyl groups were removed under sodium/liquid ammonia conditions, quantitatively yielding a crystalline diol which upon X-ray crystal structure analysis confirmed the relative configuration as that belonging to the target natural products. Quinone formation was effected by the action of DDQ in aqueous THF to give the stereodefined aziridinomitosane 258.
6.20. Coates’ Approaches Towards Pyrroloindole Type Compounds
Coates and colleague developed a novel variation of the Nenitzescu indole synthesis[253] which affords directly 2,3-dihydro-1H-pyrrolo[l,2-a]indole-9-carboxylates with a substitution pattern suitable for elaboration to the pyrroloindole quinone core characteristic of the mitomycins (Scheme 52).[254] Thus, the regioselectivity of the Michael addition of the sodium salt of exocyclic enamino ester 260 was controlled by transforming the quinone of the coupling partner to the quinone monoketal 259. In this manner, the bridged bicyclic adduct 261 was obtained in good yield and subsequently rearranged to the mitosene 262 by treatment with catalytic hydrochloric acid. After a series of six steps the authors intercepted a known[255] intermediate which was elaborated to 7-methoxymitosene 263 in two further steps.
Scheme 52.
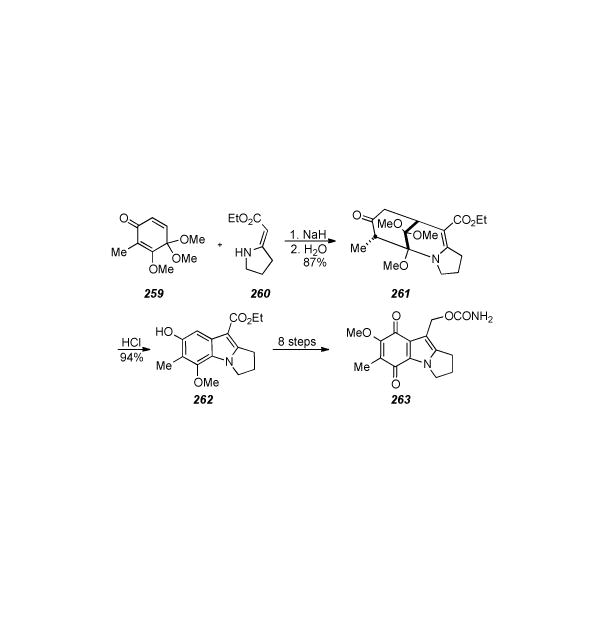
Coates′ synthesis of 7-methoxymitosene 263.
A one-pot synthesis of mitosenes from N-arylhydroxylamines (264) and ethyl 6-oxo-2-hexynoate (265) was also developed in the Coates laboratory (Scheme 53).[256]
Scheme 53.
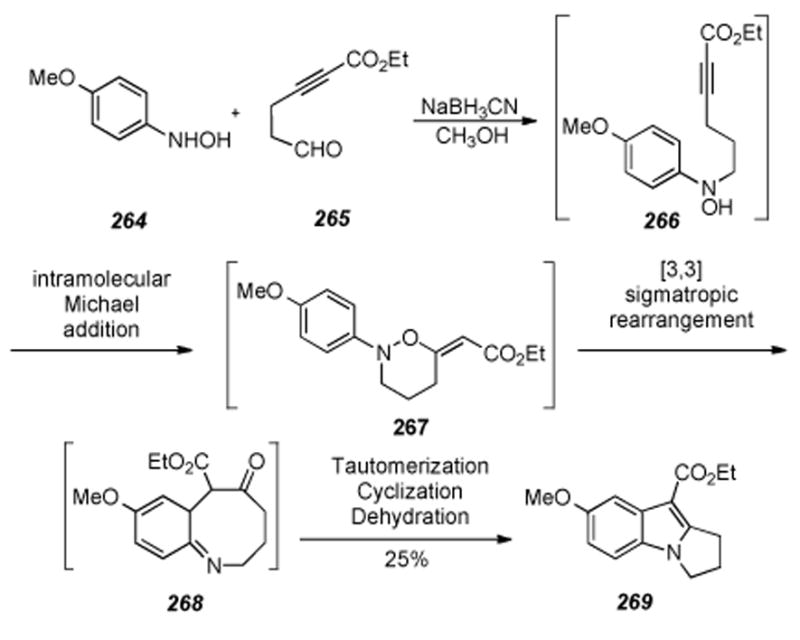
Coates′ one-pot synthesis of mitosenes.
The mechanism involves initial condensation of the hydroxylamine onto the aldehyde to form a nitrone which may be isolated in the absence of the hydride source. Hydride reduction of this nitrone forms alkylated hydroxylamine 266, the oxygen of which performs intramolecular Michael addition into the acytelenic ester to form 267.
Following [3,3] simatropic rearrangement of 267, tautomerization to reform the aromatic ring, transannular cyclization, and dehydration, the mitosene 269 could be isolated in a modest 25% yield. Although the substitution pattern most closley resembled the natural products targeted, all other hydroxylamine substrates screened for this reaction gave higher yields. A Swiss group in Basel used very similar methodology for a one-pot synthesis of the tricyclic mitomycin skeleton.[257] A hetero–Diels-Alder between nitrosobenzene and pentadienal gave an intermediate which proceeded through an analogous hetero-Cope rearrangement to give a hydroxypyrroloindole.
6.21. Remers′ Syntheses of a C1, C9 Functionalized Mitosene and Desammonoapomitomycin A
Remers and co-workers published a series of twenty-three articles describing synthetic approaches to the various functionalities present in the mitomycins. In addition to these early reports, Remers authored several other papers dealing with the synthesis and biological evaluation of mitomycin analogs. Firstly, as part of a comprehensive program for the synthesis of analogs of the mitomycins, Remers developed a useful method for the introduction of the 9-hydroxymethyl substituent into a pyrrolo-[1,2-a]indole having a carbonyl at the 1-position suitable for subsequent elaborations into the aziridine or other functional groups.[258] A direct Vilsmeier-Haack formylation of 270 was realized, albeit in a dismal yield of only 7% (Scheme 54). An alternative, five-step procedure for the transformation of 270 into 271 entailed reduction of the carbonyl to the alcohol and acylation thereof, formylation at C9, and then deacetylation and reoxidation at C1 with an overall yield of 25%. Reacting 271 with the electrophilic reagent diborane afforded 272 in 67% yield. Unfortunately, an aziridine containing compound was not reported from 272.[259]
Scheme 54.
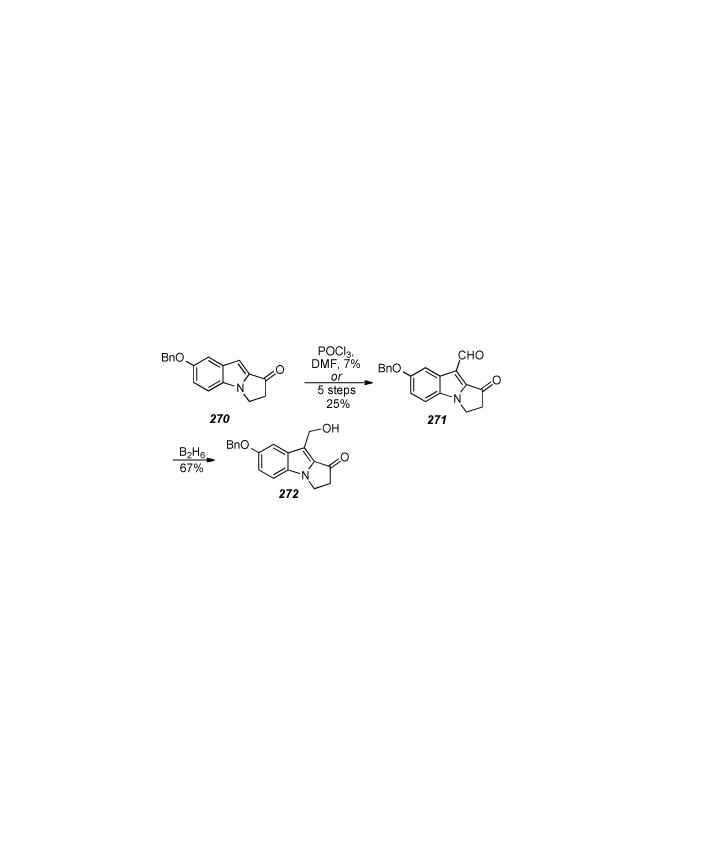
Mitosene synthesis by Remers.
The first synthesis of desaminoapomitomycin A, a major degradation product of the mitomycins, was accomplished by Remers,[260] and building on this methodology, mitosene derivative 276 was synthesized, bearing functionalization at all the crucial positions of the skeleton relative to mitosenes derived from natural sources (Scheme 55).[261]
Scheme 55.
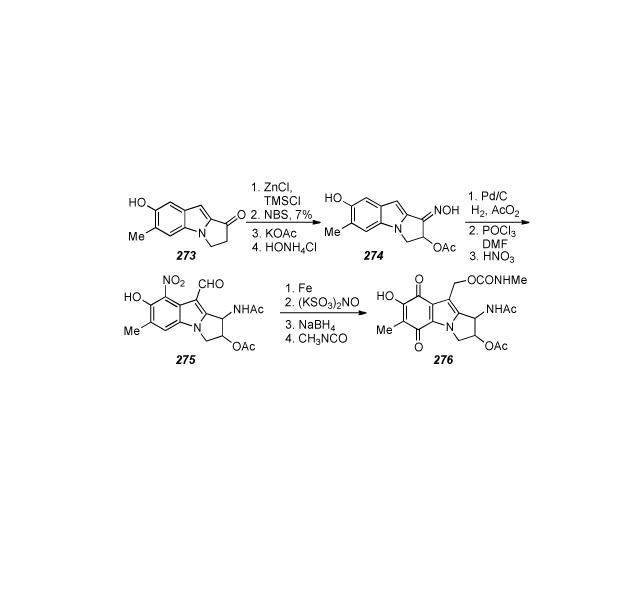
Highly functionalized mitosene synthesis.
Thus treatment of 273 with zinc chloride and chlorotrimethylsilane, followed by N-bromosuccinimide gave the corresponding α-bromo ketone, which was then converted to the oxime acetate 274 by treatment with potassium acetate and hydroxylamine hydrochloride. The oxime was reduced with palladium on charcoal under hydrogen and the resulting amine was then reacted with acetic anhydride. Istallation of the aldehyde commenced with a Vilsmeier-Haack precedure, followed by formation of the quinone in a manner similar to that utilized by Kametani, namely nitration to form 275, reduction to the aniline, and oxidation to the quinone with Fremy′s salt. Finally reduction of the aldehyde with sodium borohydride gave the alcohol, which was then transformed into the methyl carbamate 276 by treatment with methyl isocyanate.
6.22. Iwasawa′s One-Pot Synthesis of Pyrroloindoles
A fairly recent report from the Iwasawa group presents a one-pot synthesis of the tricyclic pyroloindole skeleton of the mitosenes from quite simple monocyclic precursors (Scheme 56).[262] In order to craft a mitosene-specific approach, the authors utilized a starting material that would ultimately give rise to the siloxymethyl derivative 281. The premise of the reaction is that upon electrophilic activation of the internal alkyne with third-row transition-metal complexes, especially PtCl2 and AuBr3, the phenylimino-alkyne precursor 277 undergos a nucleophilic 5-endo cyclization of the imino nitrogen onto the alkyne moiety. The corresponding metal-containing azomethine ylide represented in Scheme 56 as the resonance forms 278 and 279 was generated by this procedure. Successive [3+2] cycloaddition onto vinyl tert-butyl ether forms intermediate 280, and 1,2-alkyl migration allows tricyclic pyrroloindoles such as 281 to be isolated with regeneration of the catalyst. Optimized reaction conditions of 3 mol% of PtCl2, four equivalents of tert-butyl vinyl ether, and 5Å molecular sieves in toluene, gave the desired product bearing the siloxymethyl group in the correct position for elaboration to a more functionalized mitosene in 73% yield as a mixture (3:7) of cis and trans isomers in a single pot. Although the methodology presented builds the mitosane skeleton very efficiently, extensive functionalization of relatively naked mitosenes has been shown to be very difficult and it is questionable whether or not this type of reaction would proceed with more appropriately functionalized, electron-rich substrates.
Scheme 56.
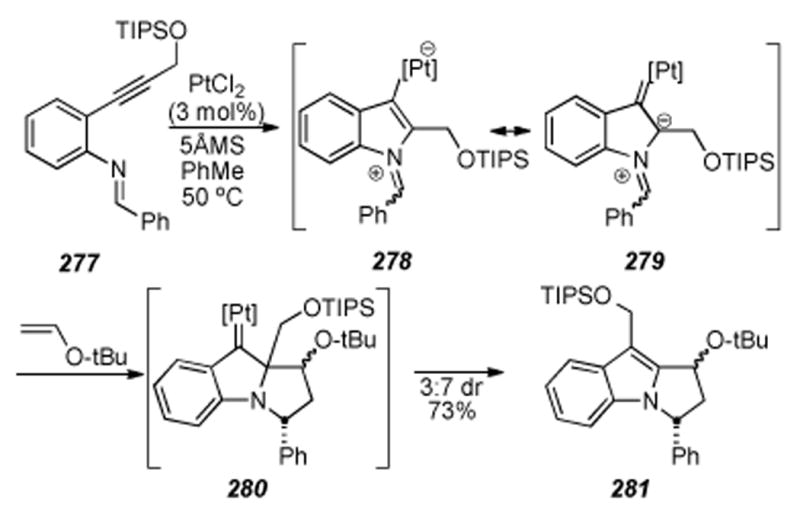
Iwasawa′s one-pot synthesis of mitosenes.
6.23. Takada′s Photocyclization to Form Mitosene Analogs
Early efforts aimed at the total synthesis of the mitomycins occurred in the Takada labs[263-265] and by 1977 methods to build mitosene[266] and mitosane[267] type compounds via thermolysis and photolysis[268] of amino-1,4-quinones possessing an active methylene group at the 2-position to form indoloquinones had been described. In applying this methodology towards mitomycin synthesis,[269] aziridinomitosene derivative 288 was formed by simple irradiation of the bicyclic amine 283 and chloroquinone 282 starting materials in a variety of solvents (Scheme 57).[270],[271] This complex reaction apparrently proceeds through a Michael addition-elimination sequence to give an intermediate aminoquinone 284. Intramolecular γ-hydrogen abstraction by the quinone 284 would produce a diradical which can rearrange via spiroaziridine 285 to an unstable, yet isolable, oxazoline 287.[268] The oxazoline 287 then rearranges to compound 288, bearing the characteristic skeleton of the mitomycins. It was proposed that in polar solvents, zwitterionic intermediate 286 would be favored and that it could undergo intramolecular nucleophilic attack to give ring-closed quinones, arriving at final product 288.
Scheme 57.
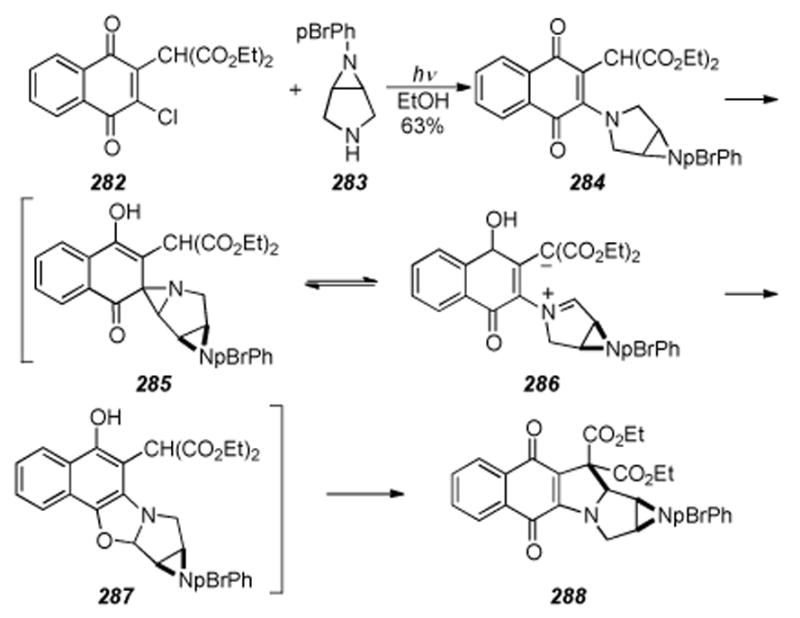
Photoinduced cyclization by Takada.
6.24. Vedejs′s Anionic Michael Approach
In an innovative approach to the tetracyclic skeleton of the aziridinomitosenes, perhaps better suited for approaches to the FR series of compounds[272], Vedejs and coworkers have developed a clever anionic Michael addition for construction of the C1-C9a bond (Scheme 58).[273] Attacking the indole nucleus with C1 of the protected aziridine, this reaction neatly forms the final ring necessary for access to species with possible biological activity. The development of the anionic Michael addition reaction is noteworthy in that the authors overcame the initially very poor yields of this reaction by observing that competitive lithiation at C2 of the indole was preventing cyclization.[199] Based on the concept of kinetic isotope effects, and prior reports[274] where substantial deuterium isotope effects have been encountered in lithiations and were used to address issues of selectivity, lithiation of the indole was effectively suppressed by introducing a deuterium atom at the indole C2. The two coupling partners 289 and 290 were joined in a modest yield of 47% to create the N4-C3 bond via deprotonation of the indole with NaHMDS at -78 °C followed by addition of the mesylate and heating to 60 °C over four days, producing stannane 291. Next, the protective deuterium was installed to form 292 in good yield via deprotonation of 291 with phenyllithium and treatment with deuteroethanol. The anionic Michael cyclization was then realized by affecting a transmetallation of the stannane 292 with methyllithium to give a solution of the intermediate deuterated lithioaziridinomitosane 293. Subsequent treatment with phenylselenium chloride induced elimination to the N-protected aziridinomitosene 294 in 80% yield. Finally, 294 was deprotected to the free aziridine 295 in fairly good yield (65% via a two-step, single pot procedure); first treatment with methanesulfonic acid and triethylsilane formed an amino mesylate in situ(not shown) that secondly, when treated with Hünig’s base caused displacement of the mesylate and formed the naked aziridine 295. The incredibly sensitive nature of free aziridinomitosenes is of course well known, however, some stability in the case of compound 295 is abetted by the conjugated ethyl ester. Additionally, Vedejs’ use of the aforementioned anionic Michael addition methodology with nonaromatic substrates initiated a program designed to prepare other aziridinomitosenes for the eventual study of their role in the activation cascade from FK317.[275]
Scheme 58.
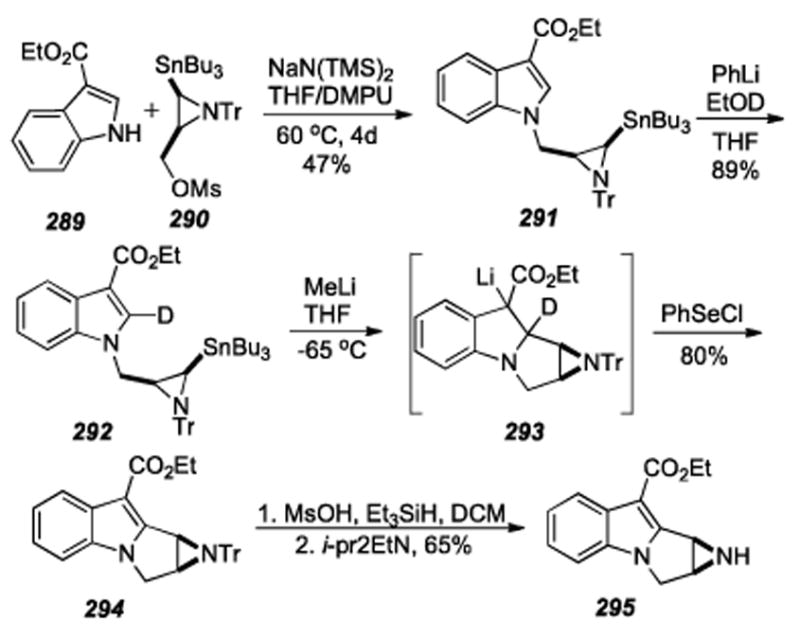
Anionic Michael addition facilitated by protection of the indole C2 from lithiation by deuterium.
6.25. Reinhoudt: Tertiary Amino Effect
One of Reinhoudt′s earlier approaches to the synthesis of less toxic analogues of the mitomycins was based on studies of the “tertiary amino effect”[276] in heterocyclic chemistry: viz., the thermal rearrangement of the 1-(1-pyrrolidinyl)-2-vinylbenzene derivatives to 2,3,9,9a-tetrahydro-1H-pyrrolo-[1,2-a]indoles (the mitosane basic skeleton) and the subsequent introduction of required functional groups.[277]
It was proposed that the conversion of 1-(1-pyrrolidinyl)-1,3-butadienes (297) into mixtures of cisand trans-pyrrolizines (300) can be explained by two consecutive pericyclic reactions (Scheme 59).[278] The first step, a [1,6] hydrogen shift, produces a conjugated 1,5 dipolar species (299) that subsequently undergoes a concerted disrotatory electrocyclization of the 6π- electron system and produces the pyrrolizines. This conjecture was supported by extensive mechanistic analysis including deuterium labeling studies. The studies utilizing this chemistry towards the synthesis of mitomycin C analogs was detailed in a series of two back-to-back publications, the first of which described the introduction of a urethane function at the C10 position, followed by a paper portraying the introduction of a leaving group at the C1 position and oxidation of the aromatic ring (albeit using separate substrates).[279] The urethane was shown to be accessable via the C10 benzylcyanomethyl functionality present in Scheme 59. Compound 297, accessed in eight steps from commercially available 2-hydroxy-4-methylchlorobenzene (296), which was condensed with benzaldehyde under the action of sodium methoxide in methanol afforded a low yield (20%) of cyclization precursor 298. The interesting cyclization reaction took place in refluxing n-butanol and provided 300, via intermediate dipole 299 by the mechanism previously mentioned. Attemps to form a quinone were unsuccessful; the nitro group in 300 was reduced to give 301, but upon treatment with Fremy’s salt, only a trace of the compound 302 was identifiable by mass spectral analysis. Unfortunately, this interesting methodology did not give access to biologically relavent mitomicinoids, as it was not possible to synthesize the corresponding p-quinones with standard methodology. Exploring the possibility of adapting the aforementioned methodology to constructing a compound with potential for aziridine formation, compound 303 was heated in toluene to give the C1-C2 functionalized mitosane 304 (Scheme 60). Thus, a vicinal diol 305 was formed by treating 304 with osmium tetroxide. In the same year it was shown by Danishefsky that a stereodefined aziridinomitosane could be formed via a vicinal diol.[251] To address the issue of quinone formation a synthetic route based on a modified Madelung reaction was developed.[280]
Scheme 59.
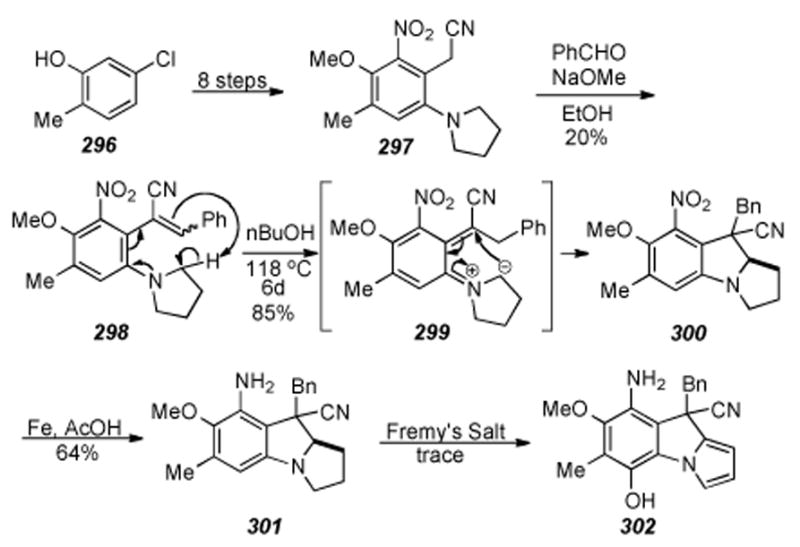
Double pericyclic reaction to form the tricyclic mitomycin skeleton.
Scheme 60.
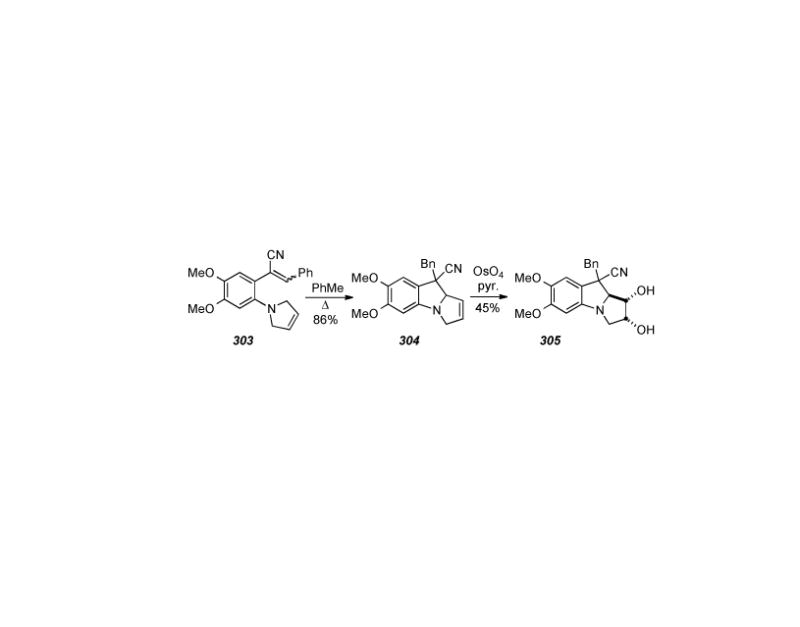
Synthesis of a functionalized hexahydropyrroloindole.
6.26. Kozikowski′s Intramolecular Nitrile Oxide Cycloaddition
Kozikowski developed an intramolecular nitrile oxide cycloaddition (INOC) route to benzazocine 308 in seven steps from 1-aminoacetophenone 306 (Scheme 61).[281] The cyclization was realized in 45% by treatment of a highly diluted solution of oxime 307 with sodium hypochlorite in the presence of a catalytic amount of triethylamine.
Scheme 61.
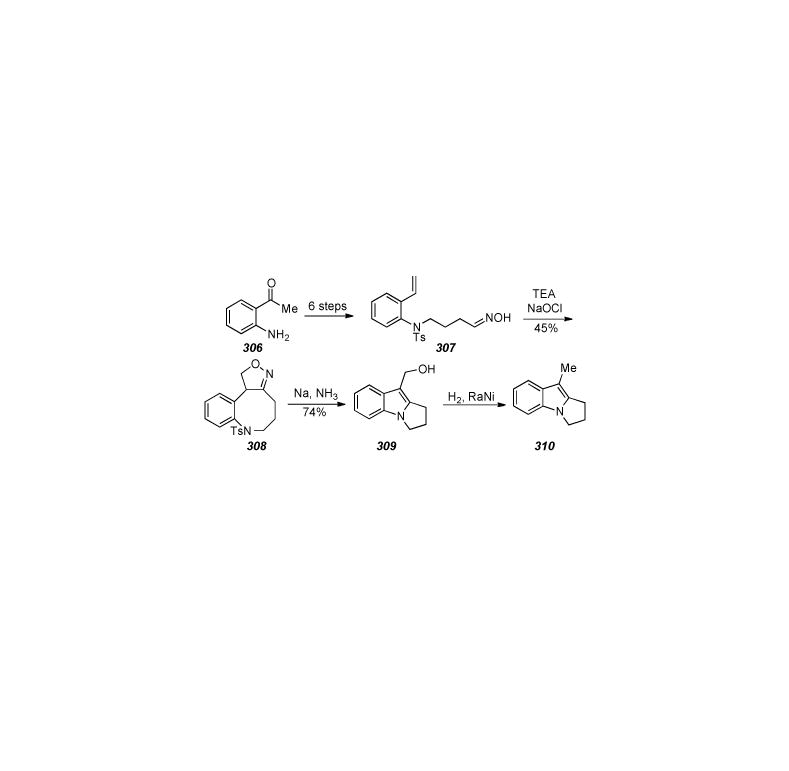
Kozikowski′s INOC to form benzazocine precursors to mitosenes.
Treatment of the cycloadduct 308 with sodium in ammonia gave the product of transannular cyclization, β -hydroxy indole 309. Finally, known[282] mitosene 310 was accessed via deoxygenation with hydrogen and raney nickel.
6.27. Rapoport′s Approaches to Mitosenes and Aziridinomitosenes
In the early 1980s, Rapoport and colleagues published several improved synthetic routes to the model compound 7-methoxymitosene (263) (Scheme 62). Utilizing the iminium salt chemistry applied to the synthesis of naphthoquinone mitosene analogues,[283] the key step in Rapoport′s first synthesis[284] of 263 is the ring closure at C9-C9a, effected by heating of the acyclic dimethyl acetal 311 with phosphoryl chloride to generate the charged intermediate 312. Cyclization with the addition of methanol regenerates the acetal as functionalized pyrrolidino indane 313. The target compound 7-methoxymitosene 263 was then accessed via known chemistry.
Scheme 62.
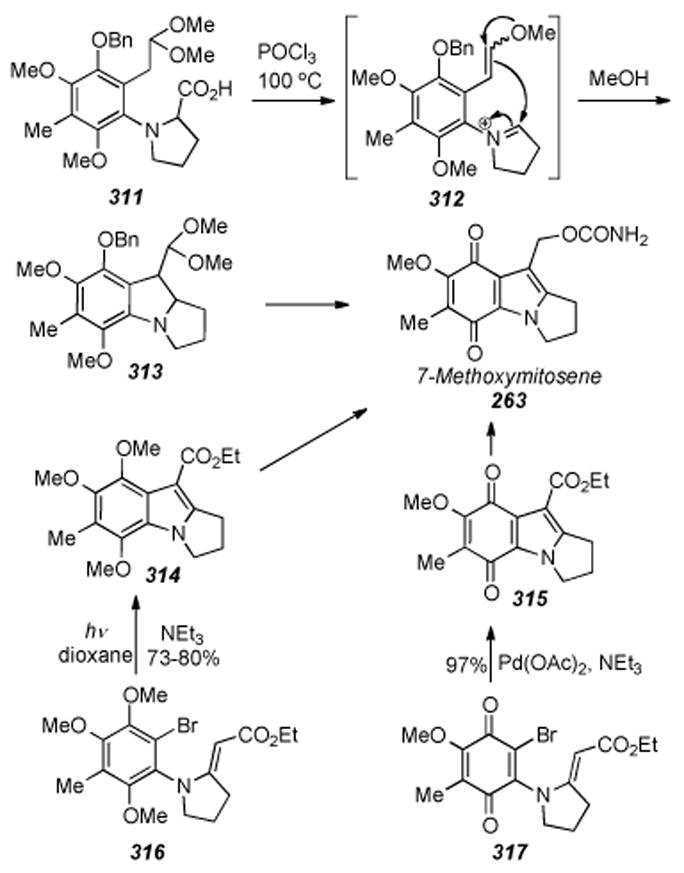
Various approaches towards 7-methoxymitosene developed in Rapoport′s lab.
In another approach[285], the mitosene skeleton was constructed via formation of the C8a-C9 bond, effected by photocyclization of 316 into 314. Finally, the most effiecient synthesis[286] of 263 involved the same bond disconnection as just described, however a palladium-catalyzed ring closure (intramolecular Heck reaction) of the quinone 317 into 315 shortened the synthetic sequence and was able to be realized in 97% yield.
The first enantiospecific synthesis of an aziridinomitosene was accomplished by Rapoport in 1985 wherein the aziridine ring was formed at an early stage (Scheme 63).[287] Thus epoxidation of vinylglycine derivative 318 with m-chloroperoxybenzoic acid gave quantitatively the epoxide 319 as a 1:4 anti:syn ratio of diastereomers. A five-step sequence was then carried out to give cyclization precursor 320 in a combined yield of 31%.
Scheme 63.
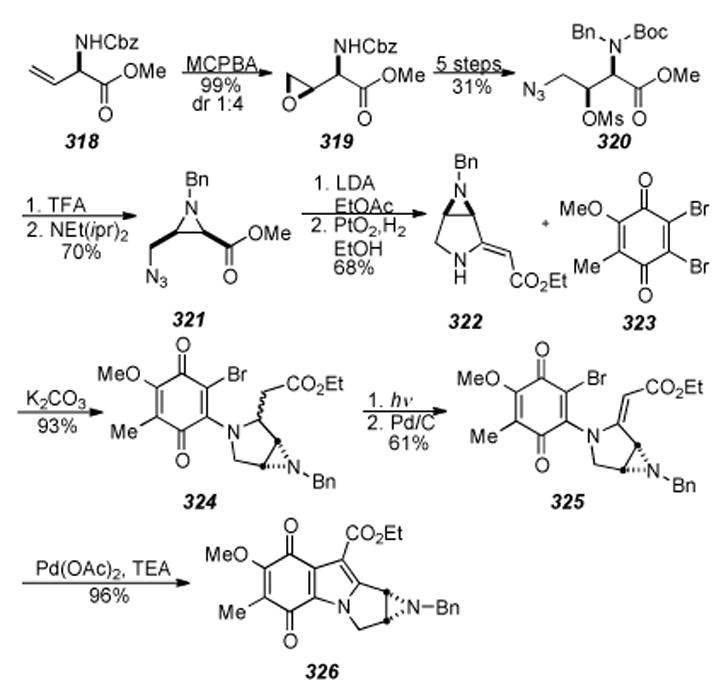
Rapoport′s synthesis of an aziridinomitosene with opposite stereochemistry as that of the natural products.
Aziridine formation commenced with removal of the Boc group via action of trifluoroacetic acid, and then treatment with Hünig’s base to effect aziridination and form 321. Pyrrolidine derivative 322 was then accessed via the intermediate β-keto ester (not shown) derived from treatment of 321 with the lithium enolate of ethyl acetate. The azide was then reduced to the free amine with palladium dioxide under a hydrogen atmosphere. Attack of the in situ-formed amine upon the carbonyl followed by dehydration then gave 322, which was reacted with the dibromoquinone 323 according to precedent[288] in a Michael addition / elimination sequence to give 324 as a single regioisomer in excellent yield. A two-step photochemical oxidation-reduction-reoxidation sequence proceeded in 61% yield giving quinone 325, and palladium-catalyzed ring closure afforded the aziridinomitosene derivative 326. This work was done was at the time that Hirayama solved the absolute stereochemistry of mitomycin C by high precision X-ray crystallographic analysis,[289] and the structures of the mitiomicins were shown to differ stereochemically only at C9 by Hornemann.[290] Thus, Rapoport had in fact synthesized the aziridine with the opposite absolute stereochemistry of the natural substances.
6.28. Flitsch-Dötz Benzannulation, Wittig Olefination, and Reformatsky Approaches to the Tricyclic Core; C9a Oxidized Mitosanes
In the 1980′s Flitsch and colleagues developed multiple approaches towards the mitomycins. Their report of accessing benzopyrrolizines via chromium carbene complexes is noteworthy, as it represents a rare type of approach in which the phenyl ring is actually created synthetically instead of being incorporated as part of the starting materials.[291] With this methodology, known more recently as the Dötz benzannulation reaction,[292] chromium carbene complex 327 was reacted with 1-trimethylsilylpropyne to yield the mitosene 328 in 32% yield (Scheme 64). Attempts to carry out this transformation with more functionalized substrates, such as aziridine containing compounds, have not been reported.
Scheme 64.
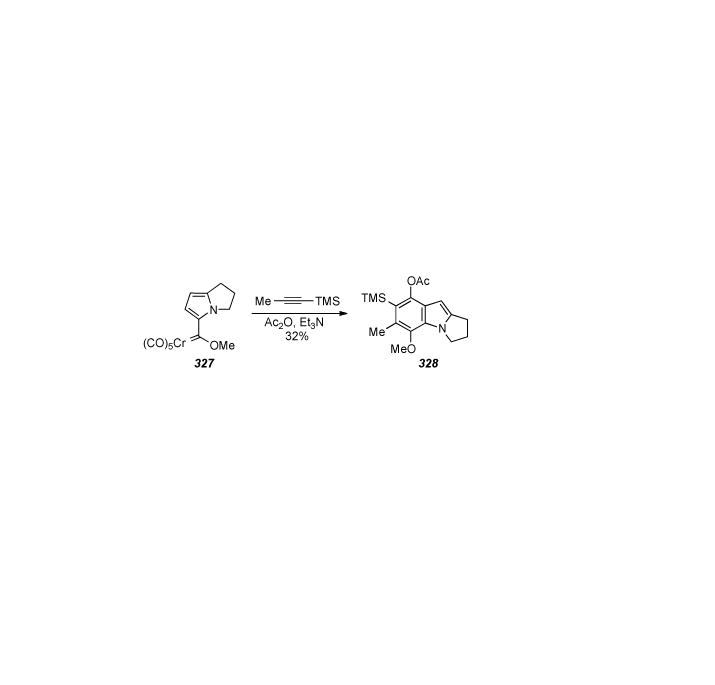
Dötz benzannulation to form a mitosene.
Methodology described in a previous synthesis of a C9, C9a functionalized mitosane[293] was to be used to effect similar functionalization of the protected quinone 330, however the cyclization of N-aryl succinimide 329 via Wittig olefination and subsequent Vilsmeier-Haack fromylation was the most advanced reaction reported (Scheme 65).[294]
Scheme 65.
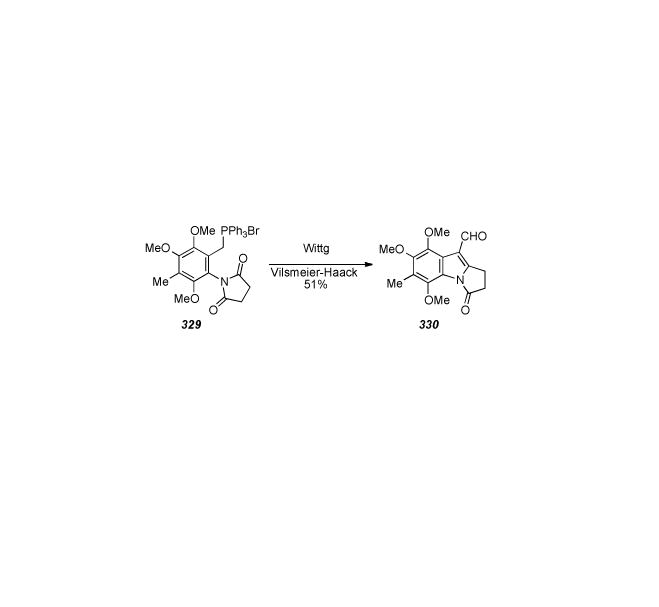
Procedure for forming mitosene lactam 330.
Flitsch also reported two other ways to make C9, C9a oxidized mitosanes.[295] Treatment of the mitosene 331 with N-bromosuccinimide in methanol gave C9a-methoxy mitosane 332, the bromine atom of which was removed with tributyltin hydride giving 333 in 63% overall (Scheme 66). Alternatively, the C9a-hydroxy mitosane 335 was synthesized in 70% yield via a Reformatsky reaction of 334.
Scheme 66.

Strategies for installing C9a functionality.
6.29. Franck′s Syntheses of Aziridinomitosenes
In the early 1970s Franck and coworkers succeeded in constructing the tetracyclic framework of the mitomycins by synthesizing[296] aziridinomitosene 342 (Scheme 67). Previously attempted functionalization of 9H-pyrrolo[1,2-a]indole (338)[297] had remained relatively fruitless.[298] However, singlet oxygen oxidation in the presence of pyridine routinely gave 70% of 338. Presumably the intermediate endoperoxide 337 was formed, and pyridine served to affect ring-opening and elimination to give 338. The C1-C2 double bond (mitomycin numbering) was shown to be amenable to the Scheiner aziridine synthesis.[299],[300]
Scheme 67.
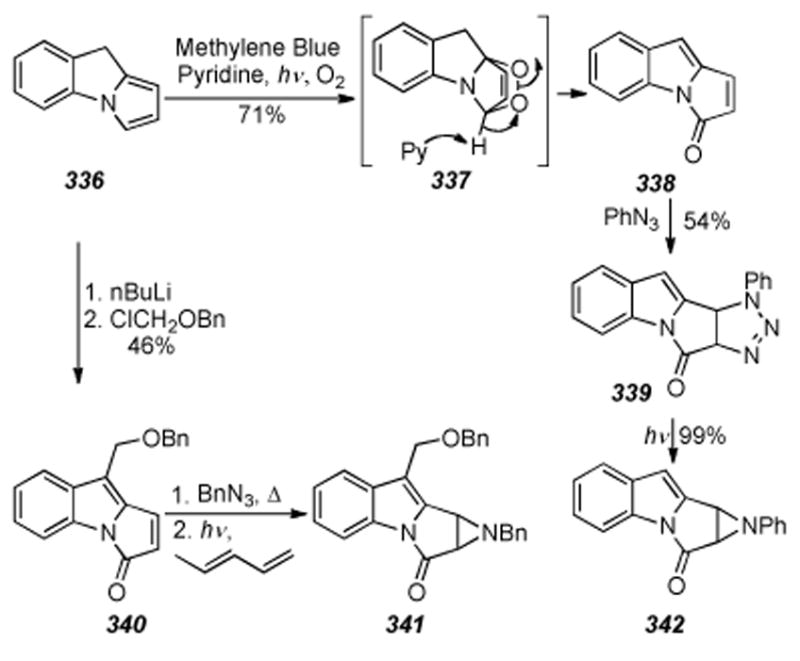
Tetracycles synthesized in the Franck Laboratories.
Thus, dipolar cycloaddition with phenyl azide gave 339, which was then photolytically decomposed to quantitatively access aziridinomitosene 342. A few years later the synthesis of the bis-benzyl protected C9 substituted aziridinomitosene 341 was reported.[301] The C9 substituent was only able to be introduced if the crude alkylation reaction mixture was immediately photooxygenated. Furthermore, Scheiner aziridination of 340 to form 341 was only successful in the presence of piperylene, which acted as a quencher of the exited triplet state of such a reaction as shown by Padwa.[302]
6.30. Hegedus’ Organometallic Tandem Cyclization
In 1985 Hegedus and co-workers developed a palladium-mediated tandem cyclization process for synthesizing pyrroloindolequinones.[303]Bis-N-allyl quinone 343, derived in eight steps from 1,4-benzoquinone, was subjected to the optimized conditions shown to give a combination of the unsaturated tricyclic compounds 345 and 346 in 15% and 20% yields, respectively (Scheme 68).
Scheme 68.
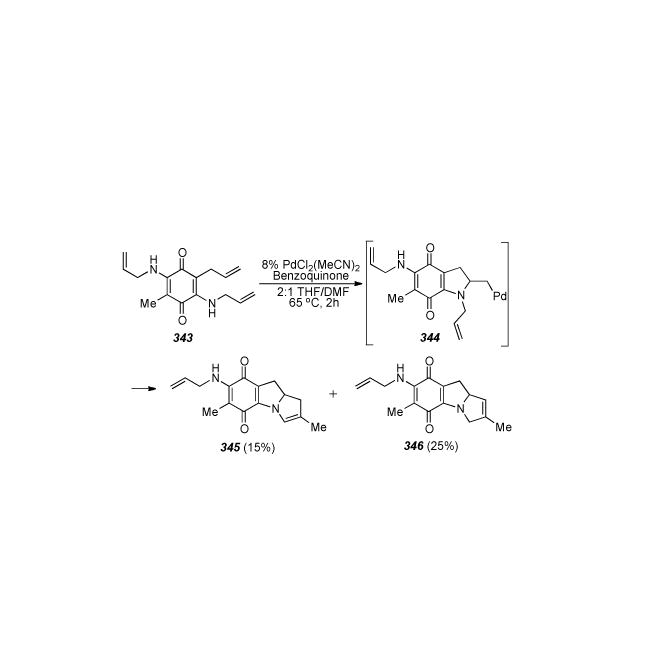
Hegedus′ Palladium mediated bis-cyclization process.
Additionally, in 20% yield, the undesired by-product (not shown) corresponding to β -elimination of palladium from intermediate 344 was isolated.
6.31. Naruta-Maruyama: Double Cyclization of an Azidodienyl-Quinone
In 1988 Naruta and Maruyama disclosed a route to 9a-deoxymitomycin congeners with the key step being a copper-catalyzed double cyclization of an azidopentadienylquinone (349) to form 1,2-unsaturated pyrroloindoloquinone (350, Scheme 69).[304] Thus, beginning with 2-methylresorcinol 347, the fluorinated Claisen rearrangement precursor 348 was accessed in nine steps. The fluorine atom was incorporated as a halogen functionality which would be un-exchaneable with nBuLi during formation of a pentadienyl anion, i.e. in functionalizing what would become C9 of the mitosane skeleton.
Scheme 69.
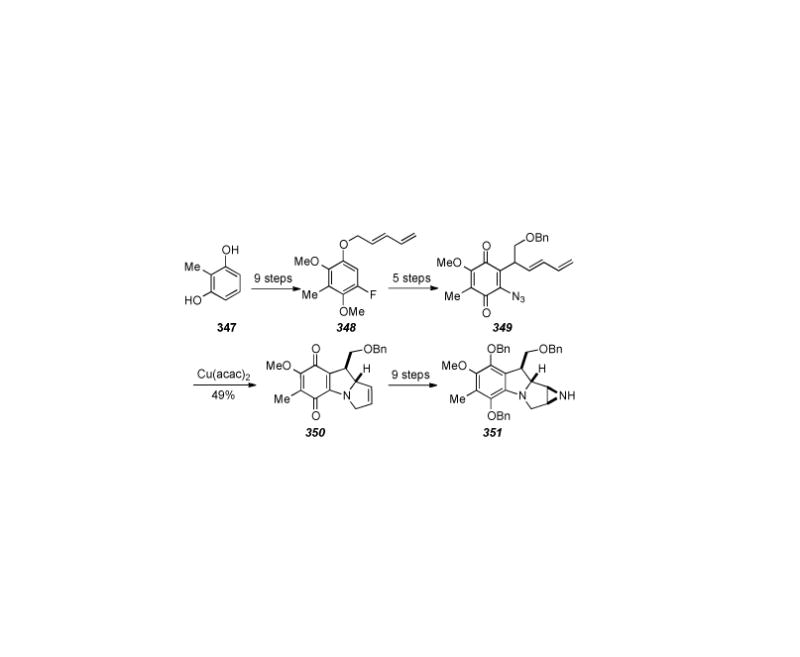
Cu(acac)2 mediated bis-cyclization of an azidodienylquinone.
Claisen-type rearrangement of 348 using AlCl3 as the Lewis acid, protection of the resulting phenol as the MOM ether, and subsequent treatment with nBuLi and benzyloxymethyl chloride gave an intermediate (not shown) containing all the carbon atoms necessary for formation of the target compound. Deprotection of the MOM-group yielded the free phenol, which was oxidized to the quinone with CAN, and finally converted to the cyclization precursor, azidodienylquinone 349, by exchange of the fluorine with azide. Azidodienylquinone 349 was then treated with catalytic Cu(acac)2 to yield 49% of the tricycle 350. A series of nine further synthetic manipulations allowed for installation of the aziridine and ultimately provided the tetracycle 351. Efforts towards oxidation at C9a were noted, yet these have likely gone unrewarded as subsequent reports have not surfaced.
6.32. Nakatsuka′s Synthesis of 7-Methoxymitosene
In 1987 the synthesis of 7-methoxymitosene 263 from 6-methylindole 352 was described by Nakatsuka (Scheme 70).[305] Six steps were required to access cyclization precursor 353 from 352, and thirteen subsequent transformations resulted in the ultimate installation of the 7-methoxy, quinone, and carbamate groups to give 7-methoxymitosene 263 in nineteen total steps.
Scheme 70.
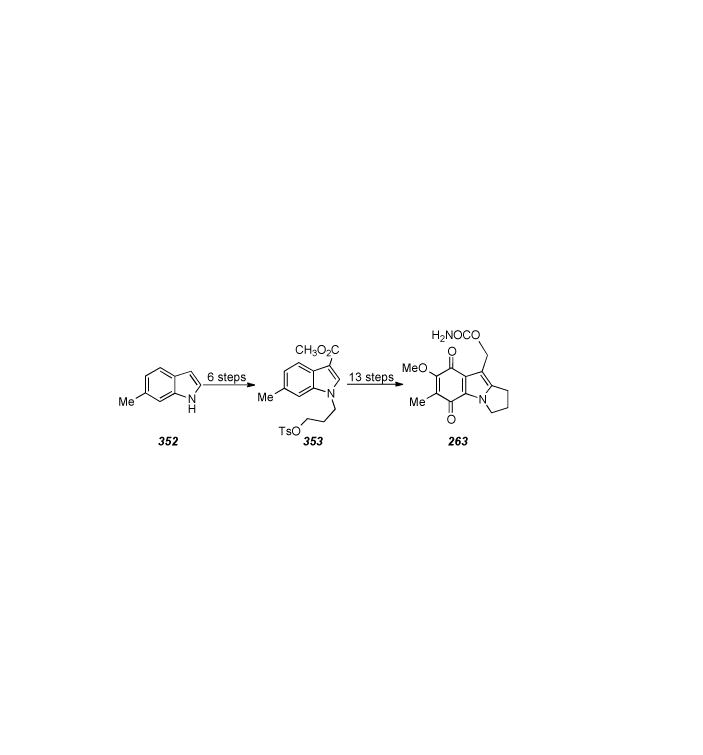
Nakatsuka′s synthesis of 7-methoxymitosene.
6.33. Rebek′s Synthesis of a Mitosene and Corroboration of the Absolute Stereochemistry of the Mitomycins
In 1981 Rebek described the synthesis of 1-methoxy-2,7-DAM (362), the degradation product of acidic methanolysis of MMC, in nineteen steps from L-hydroxyproline (355) in 1% overall yield (Scheme 71).[306] As in their previous description of the synthesis of a mitosene,[307]355 was coupled to glutarate derivative 354 to give the Huisgen [3+2] dipolar cycloaddition precursor 356. Treatment with DMAD then provided pyrrolizidine 357, ripe for Dieckmann cyclization to ketone 358.
Scheme 71.

Rebek′s synthesis of a 1-methoxy-diaminomitosene.
Similar compounds to ketone 358, namely 4-keto-4,5,6,7-tetrahydroindoles, and methodology for their preparation had been previously described by Hershensen.[308] A five-step procedure gave bromohydrin 359, and then a four-step sequence initially involving treatment of 359 with dilute methanolic methoxide to give the 1-methoxy-2-ol derivative via an elusive intermediary epoxide, eventually afforded the mesylate 360. Oxidation to the indole with DDQ and four further synthetic transformations yielded bromoquinone 361. Finally, treatment with sodium azide effects initial displacement of the bromide, forming an orange azidoquinone which turns purple when reduced further to the amine, and then slow displacement of the mesylate occurs. Three more steps complete the formation of the carbamate side chain and reduction to the aliphatic amine 362. Until this point there had been no syntheses of mitosenes obtainable from natural products and the stereochemical assignment of the aziridine ring was in part revised based on the synthesis of compound 362.[309]
6.34. Wender′s Synthesis of a 7-Methoxy-Mitosene via Photolysis of a Triazole
A synthesis of 7-methoxy-mitosene based on triazole photochemistry to form indoles was published in 1987 by Wender and co-workers (Scheme 72).
Scheme 72.
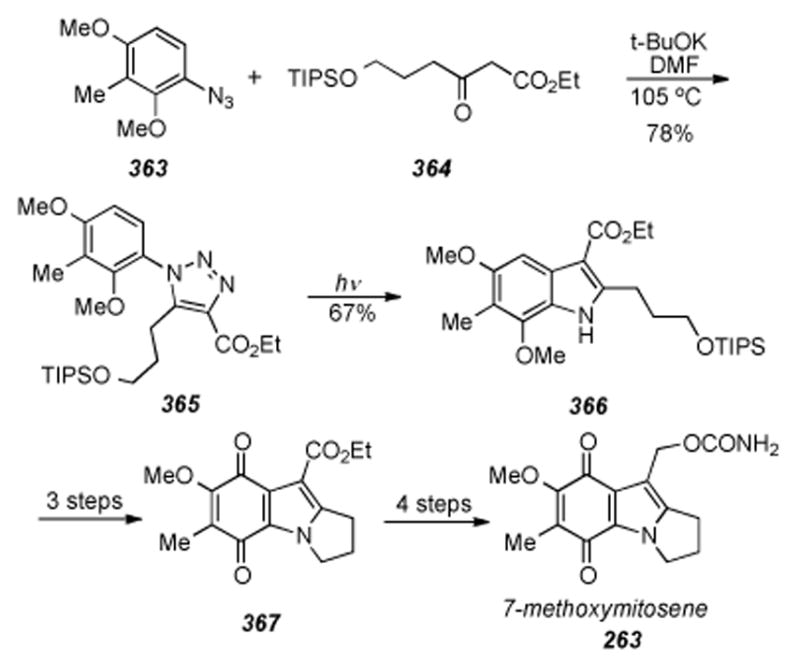
Wender′s synthesis of 7-methoxy-mitosene.
Aryl azide 363, available in three steps from 2,6-dimethoxytoluene, was subjected to Dimroth condensation with β-ketoester 364, to give triazole 365 in 78% yield. Photolysis of 365 then gave the indole 366 via diradical intermediates. The pyrroloindole core was then assembled in a straightforward manner via deprotetion of the alcohol of 366 and conversion to its tosylate followed by intramolecular displacement thereof by the indolic nitrogen to arrive at compound 367. Finally, electrochemical oxidation of 367 to the bis-methoxy protected hydroquinone, oxidation to the quinone, hydrogenolysis of the ester to the alcohol, and carbamoylation with potassium isocyanate gave 7-methoxy-mitosene 263.[310]
6.35. Edstrom′s Synthesis of a Fully Functionalized 7-Aminoaziridinomitosene
Edstrom described the synthesis of a fully functionalized 7-aminoaziridinomitosene in 1997 (Scheme 73).[311] The starting material for the synthesis was constructed via Franck′s two-step procedure for making tricycle 368.[312] To introduce a handle for installation of the C1-C2 double bond this material was subjected to DDQ in the presence of methanol to afford C1 methyl ether 369. Exposure of 369 to methyllithium and then N-phenyltriflamide converted the acetate to the triflate which underwent palladium-catalyzed carbonylative insertion of carbon monoxide in the presence of methanol to give the C10 methyl ester. Finally, exposure to TMSCl and LiBr in acetonitrile at 55 °C effected net elimination of methanol and simultaneous cleavage of the methyl ester to arrive at ketoacid 370. Bromohydrin formation, treatment with sodium azide, simultaneous mesylation of the alcohol and the acid with concomitant displacement of the acid derived mesylate with ethanol to form the ethyl ester, and finally aromatization of the six membered ring with DDQ completes the four-step sequence which transformed 370 into 371. Bromination at C7 gave 372, which was converted to the final compound, 7-aminoaziridinomitosene (373), by displacement of the bromine with azide, reduction thereof to the amine with sodium dithionite, and aziridine formation by reduction of the C1 azide with triphenylphosphine and displacement of the adjacent mesylate.
Scheme 73.

Edstrom′s synthesis of aziridinomitosene 373.
7. Successful Total Syntheses of FR900482
7.1. Fukuyama’s Initial Total Synthesis of (±)-FR900482
Fukuyama et al. reported the first total synthesis of (±) FR900482 just five years after its discovery in 1992.[313] In addition to being racemic, the synthesis proved to be quite lengthy, requiring forty-three synthetic steps. Despite this, many of the challenges in the construction of key functionalities, including the aziridine portion and the unique hydroxylamine hemiketal, were addressed. Indeed, much of the synthetic strategy employed is reminiscent of the total syntheses of the related mitomycins, namely Kishi’s historic syntheses of Mitomycin A and C in the late 1970’s and Fukuyama’s improved total synthesis of Mitomycin C that followed a decade later.[188, 314, 315]
The key steps in Fukuyama’s FR900482 synthesis (Scheme 74) involve the formation of the eightmembered ring, a stereospecific aldol reaction to install the C9 hydroxymethyl portion, construction of the hydroxylamine hemiketal, and a late stage installation of the aziridine portion via an epoxide. Lewis acid-mediated addition of 2- (trimethylsilyloxy) furan 376 to the azido arene 375, derived from ethyl benzoate 374, provided the precursor for constructing the eight-membered ring. Thus, following Michael addition of thiophenol, reductive elimination of the secondary alcohol, and zinc-mediated reduction of the azide, substrate 377 was obtained over five synthetic steps.
Scheme 74.
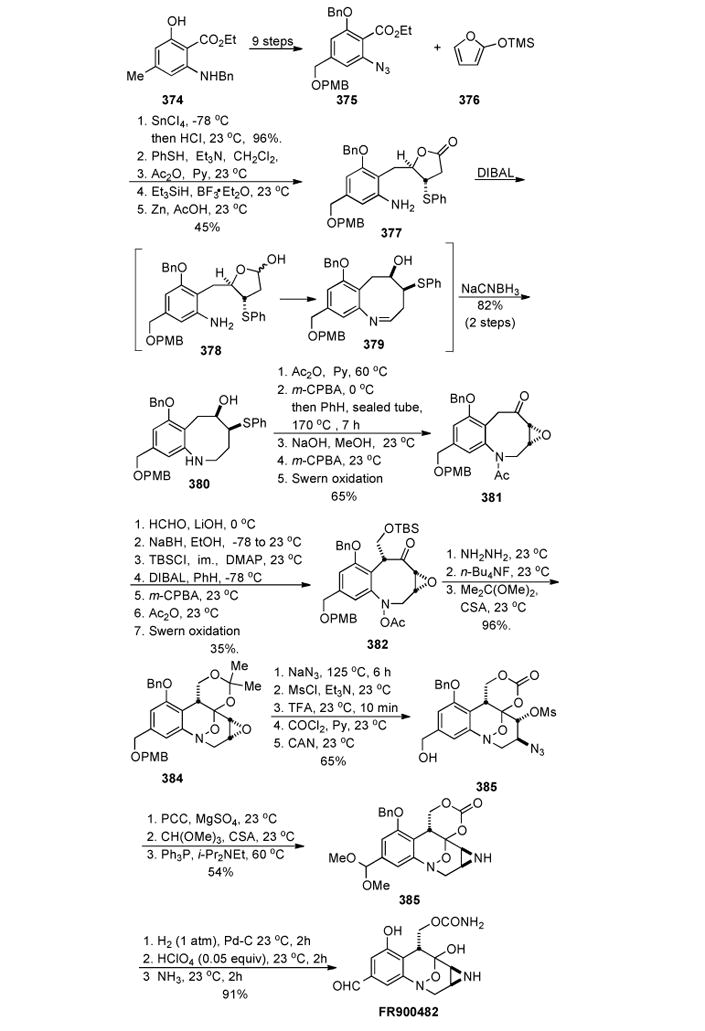
Initial synthesis of FR900482 by Fukuyama.
Reduction with DIBAL afforded the lactol 378, which in turn gave the intermediate imine 379, and subsequent in situ reduction (NaCNBH3/TFA) produced the eight-membered cyclized intermediate 380. Further elaboration gave the epoxy ketone 382 in five steps. Functionalization at C10 was accessed via a stereospecific aldol reaction utilizing LiOH and 38% aqueous formaldehyde, affording the hydroxymethyl analog possessing the desired cis-configuration relative to the epoxide (shown as the TBS ether 382). Presumably, the boat configuration of the eight-membered ring allowed hydrogen bonding between the hydroxyl group and the epoxide, dictating the stereochemical outcome of the reaction. The next challenge proved to be installing the hydroxylamine hemiketal portion of the molecule. The main difficulty encountered at this stage was the need to circumvent unmasking the secondary amine in the presence of the ketone. Such an event would lead to transannular cyclization and formation of the mitosene. Following reduction of the ketone with NaBH4 and selective protection of the primary hydroxyl group, the acetamide was cleaved with DIBAL to afford the free secondary amine. Subsequent oxidation of the secondary amine with mCPBA to the hydroxylamine and protection as the acetate permitted the reinstallation of the ketone, arriving at intermediate 382 by Swern oxidation of the secondary alcohol (Scheme 74).
Finally, cleavage of the acetate group with hydrazine unmasked the hydroxylamine, which spontaneously closed to furnish the bicyclic hydroxylamine hemiketal. Deprotection of the silyl group with TBAF followed by reaction with dimethoxypropane under acidic conditions to effect reprotection, laced up the diol as pentacycle 384 (Scheme 75). The fully protected compound 384 was now poised for manipulation of the epoxide into the desired aziridine. In a manner similar to Kishi’s mitomycin C synthesis, a Staudinger reduction was employed to install the aziridine ring in the final stage of the synthesis.3,4 Due to the lability of the aziridine functionality under acidic conditions, this strategy proved more convenient than having to carry this moiety through the majority of the synthetic route. The epoxide ring of 383 was opened with sodium azide, the resulting secondary alcohol was treated with methanesulfonyl chloride, and the resulting azido mesylate aziridine precursor was carried until the end of the synthesis. Conversion of the acetonide to the cyclic carbonate and deprotection of the PMB group was accomplished in three steps to give 384. Following oxidation of C12 to the acetal, a Staudinger reduction of the azide with triphenyl phosphine resulted in nucleophilic displacement of the mesylate to furnish aziridine 385 in 54% yield over three steps. Removal of the protecting groups and regioselective opening of the carbonate with ammonia furnished (±) FR900482, which shared identical spectroscopic properties and TLC behaviour with an authentic sample of the natural product. Considering that Fukuyama’s work represents the first successful synthesis of the natural product, the difficulties encountered and solutions developed in constructing the various complex functionalities for the first time should not be overshadowed by the lengthy sequence or racemic nature of the synthesis.
Scheme 75.
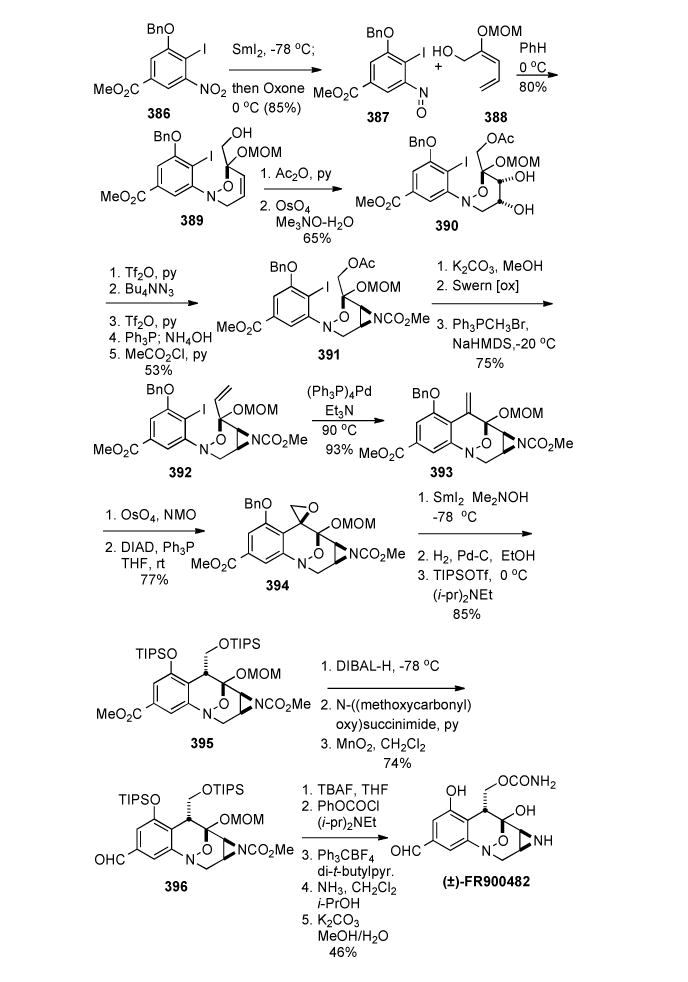
Danishefsky’s Synthesis of (±)-FR900482.
7.2. Danishefsky’s Total Synthesis of (±)-FR900482
Three years after Fukuyama’s initial total synthesis, Danishefsky et al. completed the second total synthesis of D,L-FR900482 in 1995 (Scheme 75).[316] Several key steps in the synthesis allowed for a more concise construction of the molecule and provided the natural product in thirty-four steps, a noteworthy improvement over Fukuyama’s synthesis.
The highlight of the synthesis proved to be a hetero Diels-Alder reaction between the nitroso iodobenzoate species 387 and the requisite diene 388. Compound 387 was prepared from nitro compound 386 by sequential treatment with samarium diiodide and then oxone, and 386 was derived from methyl vanillate in seven steps (32% overall yield). The cycloaddition furnished adduct 389 yielding a MOMprotected hydroxylamine hemiketal functionality in a single operation and thus avoiding the need for the lengthy manipulations employed in Fukuyama’s synthesis. Following acetylation of the hydroxyl group of 389, the aziridine ring was constructed in a manner similar to the initial FR900482 and mitomycin C syntheses.
Oxidation of the olefin with OsO4 produced the diol 390 in racemic fashion, which in turn was selectively converted to the C10 triflate. Displacement of the triflate by reaction with tetrabutylammonium azide, and conversion of the remaining alcohol to a triflate, set the stage for aziridine formation through a Staudinger reduction. Reduction of the azide with triphenyl phosphine furnished the corresponding free aziridine, which was protected as the methyl carbamate 391. Incorporation of the hydroxymethyl portion at C7 proved to be the next challenge of the synthesis. Following ring closure of 391 to 392 by Heck arylation, osmylation of the exocyclic methylene afforded the diol with high selectivity (10:1), presumably due to the approach from the less hindered β-face of the bicyclic ring system. Subsequent epoxide formation under Mitsunobu conditions furnished 394 and reductive opening of the oxirane with samarium iodide in the presence of N,N-dimethylethanolamine secured the hydroxymethyl group with the desired stereochemistry. In addition, the epoxide derived from the minor diastereomer of the osmylation also afforded the identical hydroxymethyl product. This observation led to a proposed sp2-hybridized intermediate (at C7) in the reduction of both epoxides, which in turn undergoes kinetic protonation from the less-hindered β -face.
Hydrogenolysis of the benzyl ether and protection of both hydroxyls as Tips ethers gave compound 395. DIBAL reduction of both carbomethoxy groups, reprotection of aziridine as the methyl carbamate, and oxidation furnished the aldehyde portion of target compound 396. After exchanging the silyl protecting groups for phenyl carbonates the remainder of the synthesis was completed through installation of the urethane segment by treatment of the C14 phenyl carbonate with ammonia, and deprotection of the aziridine and both hydroxyl groups over three steps. The mild conditions employed for the hydrolysis of the methyl carbamate from the aziridine (K2CO3, MeOH/H2O) warrants further comment. Under normal circumstances, liberation of a carbomethoxy-protected nitrogen requires strong reducing/hydrolytic conditions. In the case of an aziridine ring, however, the minimal overlap of the nitrogen lone pair with the carbonyl of a carbamoyl or acyl group allows for easier removal of such groups. In addition to the reduction of synthetic steps and a key hetero Diels-Alder reaction, Danishefsky’s synthesis demonstrates a successful protection strategy for the aziridine functionality, one that will be used in future synthetic studies, including our own.
7.3. Terashima’s Enantiospecific Total Synthesis of FR900482
Terashima et al. reported the first enantiospecific total synthesis of (+)-FR900482 in 1996 and 1997 over a series of seven publications.[317] By starting with L-diethyl tartrate, the template on which the enantiomerically pure aziridine ring could be constructed was present from the beginning of the synthesis. This strategy avoided the problem of constructing the aziridine during the late stages of the synthesis. Although the enantiospecific nature of the synthesis underlies the importance of the work, the sheer number of synthetic steps (fifty-seven steps in total; Scheme 76) detracts from its synthetic appeal.
Scheme 76.
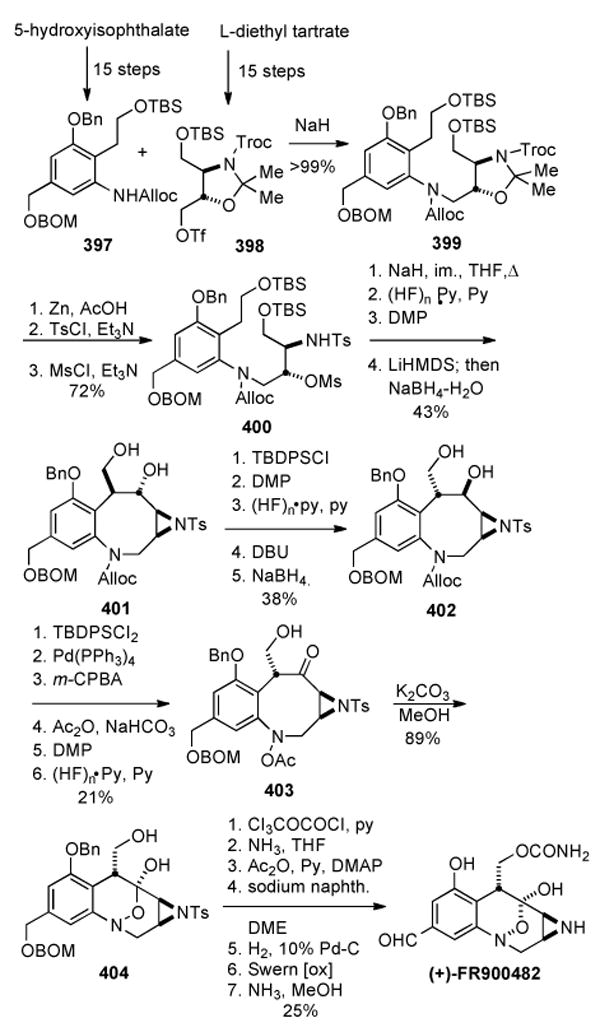
Terashima’s total synthesis of (+)-FR900482.
The synthesis relies on the convergent coupling of the aromatic portion 397 of the target compound with the aliphatic fragment 398 30 steps into the synthesis. The aromatic segment 397 was prepared in fifteen steps from 5-hydroxyisophtalic acid following standard synthetic operations and the aliphatic portion 398 of the molecule was derived from L-diethyl tartrate, which was selectively protected following a known sequence of reactions and subsequently converted to an optically pure epoxide (not shown). Initially, it was realized that the epoxide could be more directly accessed through a Sharpless asymmetric epoxidation of an allylic alcohol in approximately 85% ee, but with an enantiomerically pure synthesis being one of the goals of the work, this alternative strategy was not utilized. Opening of the epoxide with sodium azide, TBDPS protection of the primary alcohol, and Staudinger reduction, gave an intermediate amino alcohol. Not surprisingly, the ring opening gave a poor regioselectivity (3:2) and isolation of the product from the undesired 1,2 diol was accomplished by chemoselective reaction of the mixture with sodium periodate, allowing the resulting azido aldehyde by-product to be readily separated. Further protection of the amino alcohol to the oxazoline and conversion to primary triflate 398 furnished the aliphatic segment. Coupling of the two segments to give 399 was accomplished by SN2 displacement of the triflate 398 with the anion of the Alloc-protected aniline 397. Standard conditions for Troc deprotection also prompted decomposition of the N-O acetal to give the amino alcohol which was first tosylated at nitrogen and then mesylated at oxygen to form 400. Deprotonation of the protected amine prompted displacement of the mesylate and furnished the aziridine ring as the N-protected toluene sulfonamide.
The key step in the synthesis proved to be formation of the eight-membered ring through an intramolecular aldol reaction. Thus, following desilylation and Dess-Martin oxidation, the corresponding dialdehyde underwent the desired intramolecular addition upon treatment with LiHMDS. Following in situ reduction (NaBH4) of the resulting hydroxy aldehyde, diol 401 was isolated in 48% yield along with 33% unreacted substrate (isolated as the reduced uncyclized diol). Unfortunately, this stereoselective reaction resulted in a product possessing exclusively the C7 configuration opposite that of the natural product. Selective silylation of the primary alcohol of 401, oxidation of the remaining secondary alcohol to the ketone, and deprotection of the primary silyl ether with HF-pyridine furnished a hydroxyketone that could be epimerized. Reaction with diazabicyclo[4,5,0]undec-7-ene (DBU) in THF over two hours gave a ∼ 2:1 mixture of diastereomers favouring the product with the desired stereochemistry, and reduction with NaBH4 yielded 402 in 38% from 401. Longer reaction times during the epimerization resulted in formation of the exocyclic methylene derivative resulting from elimination of the hydroxyl component. In nearly the identical manner as in Fukuyama’s synthesis, transannular cyclization precursor 403 was constructed over six steps. Removal of the acetate of 403 with potassium carbonate in methanol gave the intermediate N-oxide anion (not shown), which cyclized to the hydroxylamine hemiketal 404. The completion of the synthesis commenced over seven steps including formation of the carbamate at C13 through the cyclic carbonate and reaction with ammonia, deprotection of the aziridine and hydroxyl functionalities, and oxidation of the C12 hydroxymethyl group to the aldehyde. Notably, oxidation in the presence of the free aziridine was successful only when using Swern conditions; a variety of other oxidative methods explored led to decomposition.
Terashima’s synthesis proved important in providing the natural product in optically pure form, thereby confirming the absolute stereochemistry of the molecule. In addition, epi-FR900482, whose biological activity was previously discussed, was synthesized starting from the antipode of the diethyl tartrate. Finally, the work demonstrates the importance of a well-developed protecting group scheme for the total synthesis of this natural product, as evidenced by the numerous synthetic steps dedicated to protecting group manipulations.
7.4. Martin’s Formal Total Synthesis of FR900482
Martin et al. reported a formal total synthesis of (±)-FR900482 that in principle could be applied to an enantiospecific variant (Scheme 77).[219] The key step utilized a ring-closing metathesis (RCM) reaction to construct an unsaturated eight-membered ring, which in turn could be elaborated to the target compound. The application of the RCM reaction followed preliminary reports on studies with functionally simpler model systems.
Scheme 77.
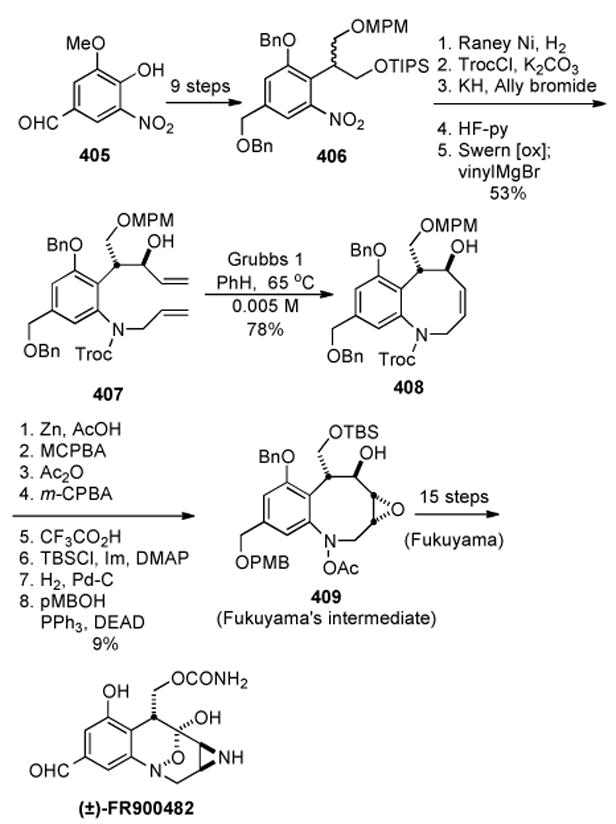
Martin’s formal total synthesis of (±)-FR900482.
Starting from 5-nitrovanillin 405, three synthetic operations gave an intermediate in which the methyl ether had been transformed to the benzyl ether, and the phenol converted to the triflate (compound not shown). This compound was in turn transformed into the malonate following displacement of the triflate with the malonate anion. Reduction with DIBAL afforded the corresponding 1,3-diol in modest yield (38%).
At this stage two separate routes were reported, one in which the ability to derive the protected diol in optically active form was demonstrated, with the alternative scheme producing the protected diol 406 in racemic fashion. Asymmetric desymmetrization of the previously mentioned 1,3-diol with Pseudomonas sp. lipase (PSL) afforded an optically active monoacetate in 68% yield (94% ee, compound not shown). The resulting alcohol was protected as the triisopropylsilyl (TIPS) ether, which, after hydrolysis of the acetate, was converted to the PMB-protected diol corresponding to the optically pure version of 406. Additionally, the 1,3-diol precursor to 406 could be directly protected through similar conditions to give the achiral equivalent. Due to the high cost of the enzymatic desymmetrization, and the low yield in the conversion to the optically pure compound, the racemic version of the protected diol was used for the completion of the formal synthesis. In the event, the nitro group of racemic 406 was elaborated to the N-Troc-protected allyl amine, desilylation, oxidation to the aldehyde, and addition of vinylmagnesium bromide afforded the diene 407 with the resulting secondary alcohol trans to the protected hydroxymethyl group. For the RCM reaction, the ruthenium-based Grubbs’ catalyst was chosen, based on this catalyst’s ability to tolerate free hydroxyl groups. Reaction of diene 407 under dilute conditions afforded the desired unsaturated eight-membered ring in 78% yield. Unable to install the aziridine ring in a stereocontrolled manner, a formal total synthesis was opted for by conversion of 408 into a late stage intermediate of Fukuyama’s synthesis 409 over eight steps. The application of an RCM reaction for the construction of the natural product was successfully demonstrated, providing a slightly improved synthesis of a late stage intermediate in Fukuyama’s racemic synthesis (38 total steps vs. 43 overall).The ability to derive the intermediate in optically active form with the insertion of the enzymatic desymmetrization provides an alternative means of synthesizing (+)-FR900482 in forty steps.
7.5. Fukuyama’s Enantiospecific Total Synthesis of (+)-FR900482
In a second generation synthesis by Fukuyama et al., optically pure acetonide 410 (derived from Ltartaric acid in five steps) provided a scaffold on which to complete an enantiospecific synthesis (Scheme 78).[318] Although much of the latter stages of the synthesis relied on nearly identical strategies as in the original racemic total synthesis, several key novel transformations early in the synthesis allowed for an efficient construction of the core structure. Sonogashira-coupling of the optically pure acetylene 410 with the corresponding aromatic triflate 411 (six steps from methyl vanillate) furnished alkyne 412 in good yield. Taking into account the conjugation of the alkyne functionality with the O-Nitro aromatic portion, selective addition of pyrrolidine followed by aqueous hydrolysis provided an efficient means of providing ketone 413. Following several protecting group manipulations and installation of the epoxide, construction of the eight-membered ring was accomplished by oxidation of the alcohol of 414 and reduction of the nitro group to give N-hydroxybenzazocine 415. Interestingly, this identical strategy for ring-closure had failed during the total synthesis by Judd and Williams on a very similar substrate. The absence of the protected aziridine functionality along with the use of a sterically bulkier silyl protecting group (TIPS in place of DEIPS) on the secondary hydroxyl group may have facilitated ring closure in this case. The hydroxylamine 415 was protected as the methoxy methylethyl ether, TIPS-deprotected to the alcohol and oxidized to α-epoxy ketone 416. Subsequent aldol reaction with LiOH and aqueous formaldehyde as in Fukuyama’s previous total synthesis, and in situ deprotection of the hydroxylamine with aqueous HCl gave the hydroxylamine hemiketal 417 directly, circumventing the need for in situ reduction of the ketone to the diol as in the previous racemic synthesis. Following several synthetic manipulations, hydroxylamine hemiketal 417 was transformed to a similar intermediate 384 (Scheme 75) as in the original synthesis. With intermediate 418 in hand the total synthesis of (+) FR900482 was completed following essentially the same strategy as in the original synthesis.
Scheme 78.
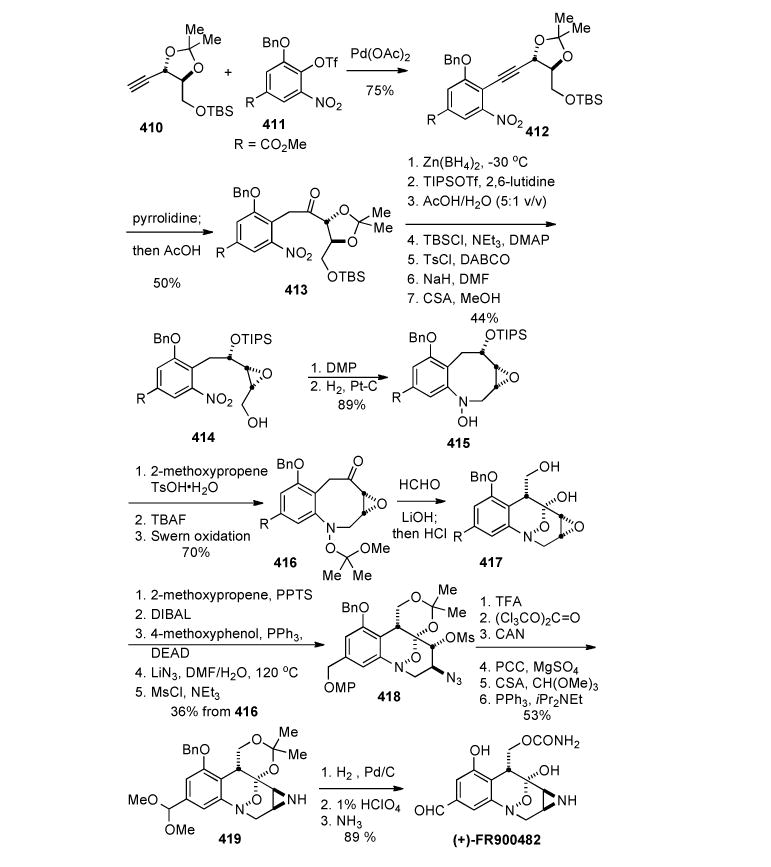
Fukuyama’s enantiospecific synthesis of (+)-FR900482.
In addition to being an enantiospecific synthesis, Fukuyama’s second generation synthesis demonstrated significant improvements over the original synthesis. An efficient coupling strategy through the acetylene substrate 412, followed by direct conversion to the ketone by conjugate addition of pyrolidine, represents a novel approach to accessing the core structure. Additionally, the use of the methoxy methylethyl ether protecting group on the hydroxylamine allowed for in situ production of the hydroxylamine hemi-ketal bond, precluding the need for a multi-step procedure to access this functionality, as in the original synthesis.
7.6. Ciufolini’s Total Synthesis of (±)-FR66979
Following the completion of Fukuyama’s second generation synthesis, Ciufolini and Ducray disclosed their synthesis of (±) FR66979 (Scheme 79).215 Despite being racemic in nature, several key transformations in the middle of the synthetic sequence highlight a unique approach to this family of molecules. Utilizing an initial approach as in Martin’s formal total synthesis, aldehyde 419 was derived from an identical intermediate from Martin’s formal total synthesis (11 steps total from 5-nitrovanillin). Substrate 420 was constructed through allylation of aromatic aldehyde 411via a lithiated allyltrimethylsilane and Ti(O-iPr)4. Heating 420 in toluene induced an intramolecular 1,3-dipolar cycloaddition of the azido allylsilane intermediate affording triazoline 421 as a single diastereomer in good yield. Brief exposure to UV light resulted in direct conversion of the triazoline to the corresponding aziridine 422. In the key synthetic transformation, hydroxide mediated homo-Brook rearrangement of the hydroxyl silyl intermediate 422, accompanied by fragmentation of the aziridine ring, unveiled 423, containing the eight-membered ring core structure of the natural product in a single step. This remarkable transformation, along with the direct accessibility of the precursor through a 3-step sequence from aldehyde 419, demonstrated not only a unique synthetic approach to FR66979 and FR900482, but also to functionalized benzazocines in general.
Scheme 79.
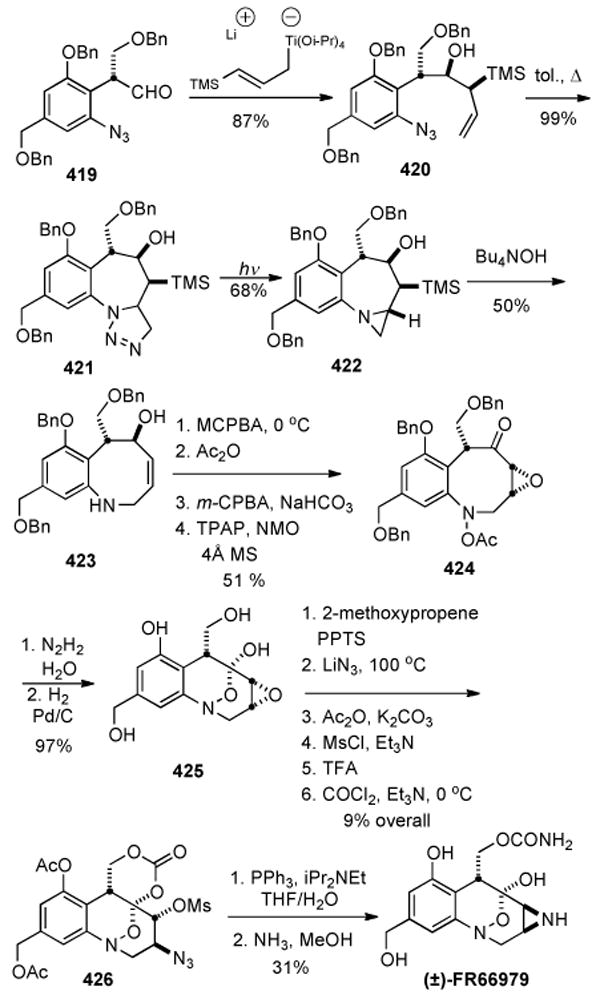
Ciufolini’s racemic synthesis of FR66979.
The completion of the total synthesis of FR66979 from the eight-membered ring 423 utilized nearly the identical overall strategy as in both Fukuyama’s total syntheses including the synthesis of the aziridine and hydroxylamine hemi-ketal functionalities. One interesting feature however, involved using benzyl groups as the sole protecting groups of the various hydroxyl groups in the synthetic sequence, as shown in compound 424. This strategy allowed for a global deprotection near the end of the synthesis to afford the tetraol hydroxylamine hemi-ketal 425. Following selective protection of the primary hydroxyl and hydroxylamine hemi-ketal as the acetonide, the remainder of the synthetic intermediate could be acetylated, allowing for deprotection during ammonolysis of the cyclic carbonate derived from aziridine precursor 426 in the final synthetic step. Despite the moderate yield of this final step (40%), the protecting group strategy thus employed demonstrated the ability to circumvent numerous selective protection-deprotection steps encountered during previous total syntheses.
Finally, although the natural product was synthesized in racemic form, the use of a nitro-benzyl propane diol intermediate as a precursor to aldehyde 419 should allow for an enantioselective version of the synthesis. By example of Martin’s enzymatic resolution of the identical substrate it should be possible to prepare aldehyde 419 in an optically pure form.[219] Overall the total synthesis of (±)-FR66979 by Ciufolini and Ducray details a unique approach to the eight-membered ring core structure of FR66979 and FR900482 and in combination with previous total syntheses represents a relatively quick access to both natural products.
7.7. Williams’ Total Synthesis of (+)-FR66979 and (+)-FR900482[319]
The Williams laboratory reported an enantioselective total synthesis of (+)-FR66979 and (+)- FR900482 utilizing a strategy that constructed the aziridine ring at an early stage – a somewhat daring approach considering the intrinsic reactivity of the aziridine. The aromatic coupling partner 427 was synthesized in four steps from the commercially available 3,5-dinitro-p-toluic acid, and the aziridine aldehyde 428 was accessed in ten steps from (Z)-1,4-butenediol. The two chemical pieces, 427 and 428, were joined by treatment with sodium methoxide to effect deprotonation of the aromatic methyl group of 427 and consequent attack upon the aldehyde of 428 to give compound 429 (Scheme 80). Protection of the alcohol as a silyl ether, subsequent deprotection of the PMB protected alcohol, and oxidation to aldehyde 430 set the stage for benzazocine formation.
Scheme 80.
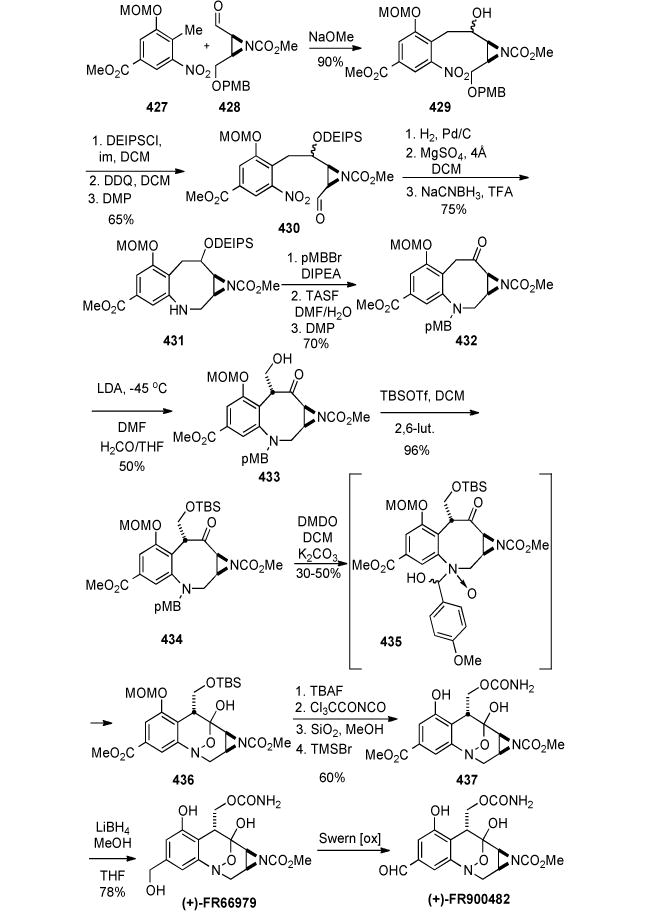
Williams’ Synthesis of (+)-FR66979 and (+)-FR900482.
Thus, treatment of 430 to hydrogenation conditions to reduce the nitro group, imine formation, and finally reduction of the imine with sodium cyanoborohydride gave the tricycle 431. Reaction of 431 with p-methoxybenzyl bromide, followed by removal of the DEIPS group with TASF in DMF/H2O[320] and subsequent oxidation with Dess-Martin periodinane[321] afforded ketone 432. Treatment of 432 with LDA in DMF at -45 °C followed by the addition of an anhydrous formaldehyde solution in THF[322] furnished the aldol adduct 433 as a ∼ 1:1 mixture of diastereomers in 50% yield (45% recovery of unreacted starting material). Separation of the diastereomers by preparative thin-layer chromatography (PTLC) followed by treatment of the undesired adduct with DBU in toluene afforded a 2.5:1 mixture of epimers favoring 433, which has the desired configuration at C7. Treatment of the primary alcohol of 433 with TBSOTf and 2,6-lutidine gave the silyl ether 434 in essentially quantitative yield (Scheme 81). For the construction of 436, a one-step protocol was employed that both cleaved the N-p-methoxybenzyl residue and oxidized the amine to the corresponding hydroxylamine, thus forming the desired hydroxylamine hemiketal. Reaction of 434 with excess dimethyldioxirane (DMDO)[323] in a 1:1 mixture of CH2Cl2 and saturated aqueous K2CO3 furnished 436 as the only isolated product in 30-50% yield, along with recovered starting material (40-50%). Attempts to drive this reaction to completion by varying the stoichiometry of DMDO, time, temperature, etc., proved unsuccessful. The very clean nature of this reaction allowed the practical recycling of recovered 434. The mechanism for the formation of 436 from 434 is presumed to involve initial insertion of the dioxirane into the C-H bond of the N- p-methoxybenzyl methylene residue to form a methanolamine species. The hydroxy group of the methanolamine is invoked to direct the DMDO oxidation of the amine to the corresponding N- oxide species 435.[324] Subsequent collapse of the methanolamine with concomitant loss of p-anisaldehyde and transannular closure of the incipient hydroxylamine on the ketone furnishes 436. Removal of the TBS protecting group followed by reaction of the primary hydroxy group with trichloroacetyl isocyanate (methanol/silica gel workup)[325] installed the urethane moiety at C13.
Scheme 81.
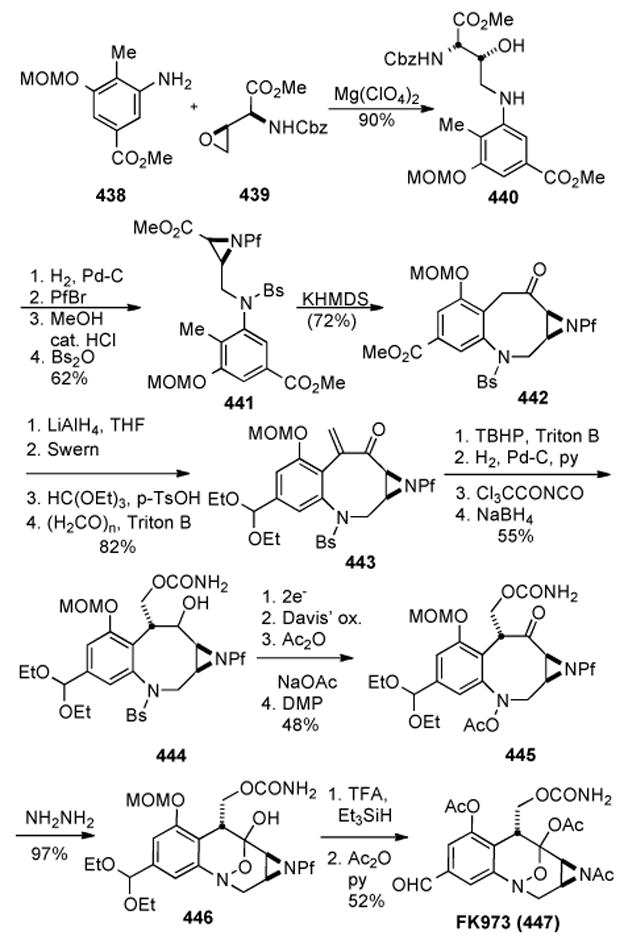
Rapoport’s enantiospecific synthesis of FK973.
TMSBr effected removal of the methoxymethyl ether in the presence of the acid-sensitive aziridine functionality at −45 °C over 3h to afford 437 in 60% yield.[326] Final cleavage of both carbomethoxy groups with LiBH4/MeOH in THF followed by Pd-catalyzed cleavage of the resulting borane amine complex,[327] furnished the natural product FR66979 in 78% yield. Finally, the natural product FR900482 can obtained by Swern oxidation of FR66979 in 33% yield.[328, 329]
7.8. Rapoport’s Formal Enantiospecific Synthesis of (+)-FR900482
Rapoport and co-workers actively pursued the synthetic construction of the FR series of compounds for over twenty years. An initial report detailed the construction of a functionalized benzazocinone, however, the most advanced intermediate reported bore a bare methine at the benzylic position of the benzaocine (C9 of mitomycins).[330] In 2003 this group reported a formal enantiospecific synthesis of (+)-FR900482 (Scheme 81).[331] Amino alcohol 440 was obtained in 90% yield by regioselective ringopening of epoxide 439 with aniline 438 in CH3CN in the presence of Mg(ClO4)2. To preserve the enantiomeric purity of the product through the next steps in the synthesis, a protecting group was needed on the aliphatic nitrogen to inhibit deprotonation α to the carbonyl group; for this purpose 440 was transformed in its N-(9-phenylfluoren-9-yl) derivative via a three step sequence. Subsequent treatment with freshly prepared benzenesulfonic anhydride (Bs2O) in pyridine effected cyclization to the aziridine ring and protection of the aromatic amine in the same step to give 441 in 62% yield over four steps. Deprotonation at the benzylic position with KHMDS in THF and intramolecular condensation of the resulting carbanion with the methyl ester afforded the aziridinobenzazocinone 442 in 72% yield. LAH reduction of 442 in THF gave a diol (97%), which was oxidized to the keto aldehyde under Swern conditions. The aldehyde could be protected selectively as the diethyl acetal by treatment with triethyl orthoformate in THF/ethanol in the presence of p-TsOH. Reaction of the ketone with p-formaldehyde and Triton B afforded enone 443 in 82% yield over four steps. Installation of the carbamate commenced upon treatment of 443 with TBHP and Triton B to give a mixture of epoxides, the major one being isolated in 77% yield.
Catalytic hydrogenation in the presence of pyridine was successfully applied to give an intermediate hydroxy ketone, which when treated with trichloroacetyl isocyanate in THF followed by reduction with NaBH4 in EtOH gave 444 in 55% yield over four steps. The benzenesulfonyl group was then electrochemically reduced, and only with Davis’ oxaziridine was the yield of the unstable hydroxylamine acceptable. Immediately, it was selectively protected with Ac2O/NaOAc and treated with Dess-Martin periodinane, affording keto-carbamate 445 in 48% yield over four steps. Finally, the stage was set for transannular cyclization; the hydrolysis of the N-OAc group was easily accomplished with hydrazine in a 1:1 mixture of CH2Cl2:MeOH to give spontaneously the hemiacetal 446 in almost quantitative yield. Ultimately, 446 was converted into FK973 (447) with TFA:CH2Cl2 in the presence of Et3SiH, followed by acetylation with Ac2O in pyridine. The utility of this synthetic route rests on the relevance of 446 as a precursor to every member of the FR900482 family by careful manipulation of the protected functional groups.
7.9. Trost’s Synthesis of 7-Epi-(+)-FR900482
Trost and co-workers reported the synthesis of 7-epi-(+)-FR900482.[332] While technically not a natural product total synthesis, since 7-epi-FR900482 has not yet been discovered, it is included here as this molecule is functionally active as cytotoxic agent and DNA cross-linking agent and intercepts the same mitosene species produced by the reductive activation of the natural product. Although an earlier report detailed the construction of the benzazocine core of FR900482 via palladium-catalyzed carbonylative lactamization,[333] the latter paper described a different approach. Accesed over a series of seventeen steps in 15% yield from 448 and 449, the stereodefined intramolecular Heck cyclization precursor 450 was converted to exo-methlene compound 451 in excellent yield by treatment with catalytic palladium in the presence of triphenylphosphine and silver carbonate (Scheme 82). Following dihydroxylation of the double bond with osmium tetroxide and formation of the cyclic carbonate as per reaction of triphosgene, the C8 position was oxidized to give the hemiaminal 452 via a Polonovski reaction in a net yield of 47% over the three steps.[334]
Scheme 82.
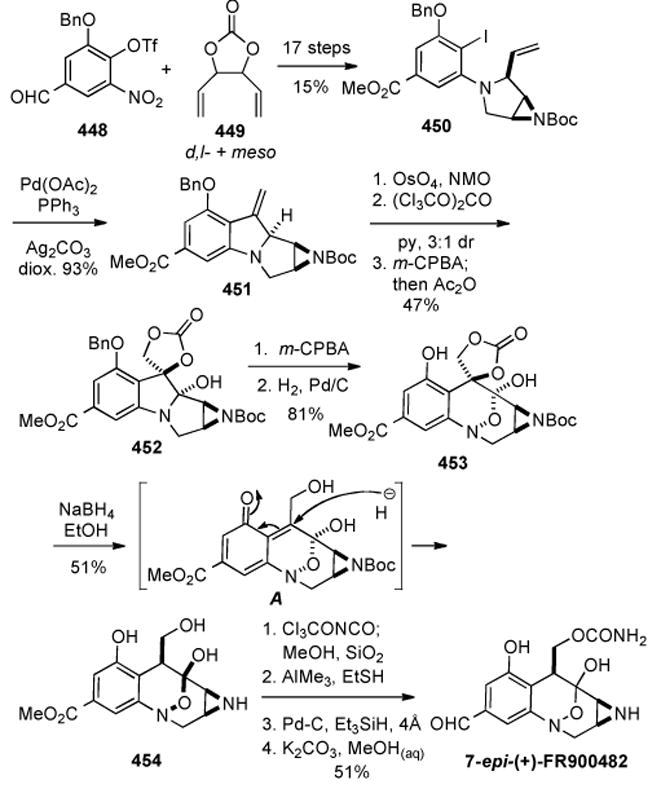
Trost’s synthesis of 7-epi-(+)-FR900482.
Further mCPBA oxidation of the aryl nitrogen of 452 to the zwitterionic N-oxide affected oxidative ring expansion-contraction to give 453 bearing the desired FR900482 core. The epi stereochemistry at C7 may be explained by invoking the possibility that the ensuing reduction of the cyclic carbamate with sodium borohydride occurs through the ortho quinone methide 453 to give the triol 454. Four more steps to elaborate the carbamate and remove protecting groups then gave the final product, 7-epi-(+)-FR900482. It was shown that the compound so prepared displayed essentially identical cytotoxic activity against two human breast cancer cell lines. This fact is not surprising considering that the in vivo reductively activated forms of these drugs, the mitosenes, lack sterochemical information at the C7- stereogenic center.
8. Approaches to the FR900482 and FK317 Core Structures
8.1. William’s Synthesis of of a Phototriggered FR900482 Mitosene Progenitor
Our laboratory has focused on the design and synthesis of pro-mitosenes that are activated by alternative chemical signals to the obligate reductive activation pathway necessary for FR900482 and congeners.[335] Optically active aziridine 428, prepared as previously described was coupled with 427 and converted to 431, the same azocine intermediate (431, Scheme 80) used in the total synthesis effort (Scheme 83).[336-337]
Scheme 83.

William’s Synthesis of of a Phototriggered FR900482 Mitosene Progenitor.
Acetylation of 431 with 6-nitroveratryl chloroformate (NVOC-Cl) gave 455 (75%), and removal of the DEIPS group with TBAF and Dess-Martin oxidation, gave ketone 456 in 60% yield over two steps (Scheme 83). Introduction of the crucial hydroxymethyl group was accomplished by reaction of 456 with LDA in dry DMF at -45 °C followed by addition of an anhydrous solution of formaldehyde in THF[338] to give the desired aldol adduct 457 as a 1:1 mixture of diastereomers in 58% yield. Recrystallization from EtOAc gave suitable crystals for X-ray analysis, which revealed that the desired anti-configuration had been secured. The capacity of 457 to cross-link DNA was evaluated and these studies demonstrate the viability of photoactivated pro-mitosenes based on the FR70496 framework to lead to the efficient generation of interstrand DNA cross-link formation. The implication of this study is that compound 457, upon photochemical activation, most likely generates the reactive mitosene intermediate 462 (Scheme 84), which upon successive monoalkylation followed by cross-linking appears to be very similar to the presumed FR70496-derived mitosene 463. In both cases, the respective phenolic alkoxy groups in the aromatic ring are apparently sufficiently electron-rich to activate the acylated aziridine species for DNA adduction.
Scheme 84.
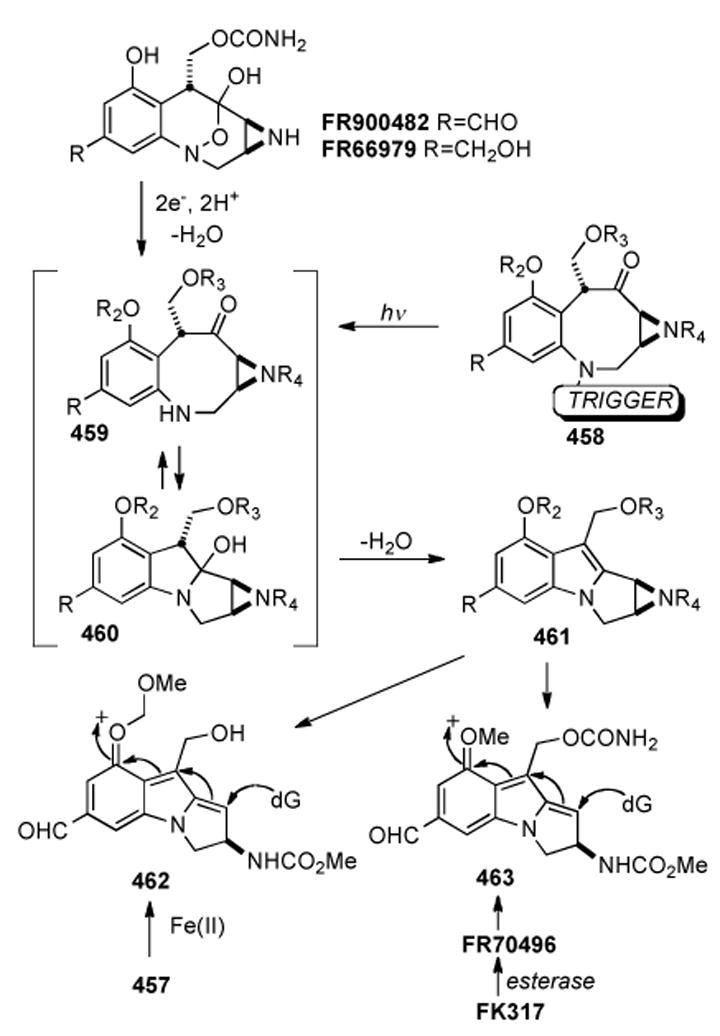
Reactivity of the phototriggered pro-mitosene.
Compound 457 was also used in a study of the interaction of these drugs in nucleosomes by Luger, Williams and co-workers.[339] Significantly, it was found that the organization of DNA into nucleosomes effectively protects the DNA molecule against drug-mediated interstrand crosslinking, while permitting monoalkylation. The authors of this study noted that, histones are transiently removed from DNA during transcription, generating a window of opportunity for efficient crosslinking of nucleosome-free DNA by mitosene-based drugs. Using compound 457 as a probe, since it is very efficiently converted into the reactive mitosene upon photochemical irradiation, it was found using in vitro approaches, that interstrand crosslinking of free DNA by 457 results in a significant decrease in basal and activated transcription. In addition, 457-crosslinked plasmid DNA was found to be inefficiently assembled into chromatin. The bifunctional activity of the mitosene species was concluded to be dependent on the conformation of the DNA template. As the vast majority of biochemical studies on the interaction of MMC- and FR900482-based mitosenes have been conducted on B-form DNA in the absence of histone proteins and nucleosomal structures, much of the published literature on the sequence specificity of these drugs in both mono-alkylation and cross-linking contexts may be less relevant to chromosomal DNA in living cells.[339]
8.2. Dmitrienko’s Oxidative Ring Expansion
Work done in the Dmitrienko lab prior to 1992 first established the feasibility of accessing the hydroxylamine hemiketal ring system of FR900482 via oxidative-ring expansion of an appropriately substituted pyrrolo[1,2a]indole ring system.[340]
Treatment of the starting material 464 (Scheme 85) with a solution of methanolic bromine followed by a basic aqueous workup gave the methoxy hemiaminal 465 in 85% yield (Scheme 85). Tautomerism to the ring opened form 466 allows for the action of Davis’ oxaziridine to convert the secondary amine to the intermediate hydroxylamine 467 which then tautomerizes again to the [3.3.1] hydroxylamine hemiketal ring system of the FR series of compounds togive the final product 468.
Scheme 85.
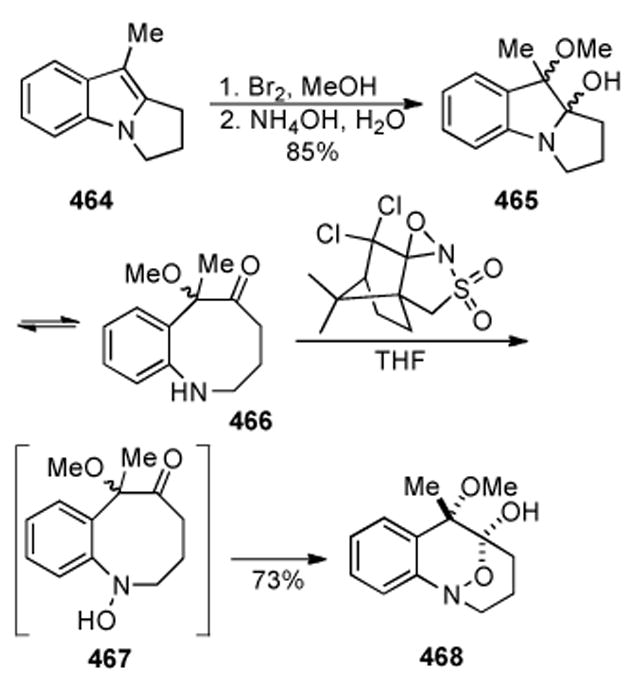
Dmitrienko’s oxidative ring exansion approach.
8.3. Sulikowski’s Copper (I) Mediated Cyclization of Diazoketones
Sulikowski reported the synthesis of a fully funtionalized core structure of FR66979 in 1996, accessed via Dmirienko[340] oxidative expansion of mitosene 472 (Scheme 86).[341] The key transformation in their approach is the copper(I)-mediated cyclization-oxidation of diazoketone 470. Beginning with phenol 469 (available in three steps from commercial material), a series of fourteen steps in 5% overall yield gave access to the diazo compound 470 to be used for the key transformation. In contrast to results described earlier,[236] the cyclization of diazoketone 470 required harsher conditions and unexpectedly provided mitosene 472, in modest yield and poor enantimeric excess. Presumably 472 arose from the oxidation of the intermediate mitosane (not shown). Dihydroxylation of 472 using an excess of osmium tetroxide in pyridine proceeded to provide the corresponding osmate ester, which was not isolated, but directly reduced with hydrogen sulfide to yield a single isomeric diol 473 in 33% yield. Treatment of 473 with an excess of dimethyldioxirane affected oxidative ring expansion to the core structure 474 in 62% yield.
Scheme 86.
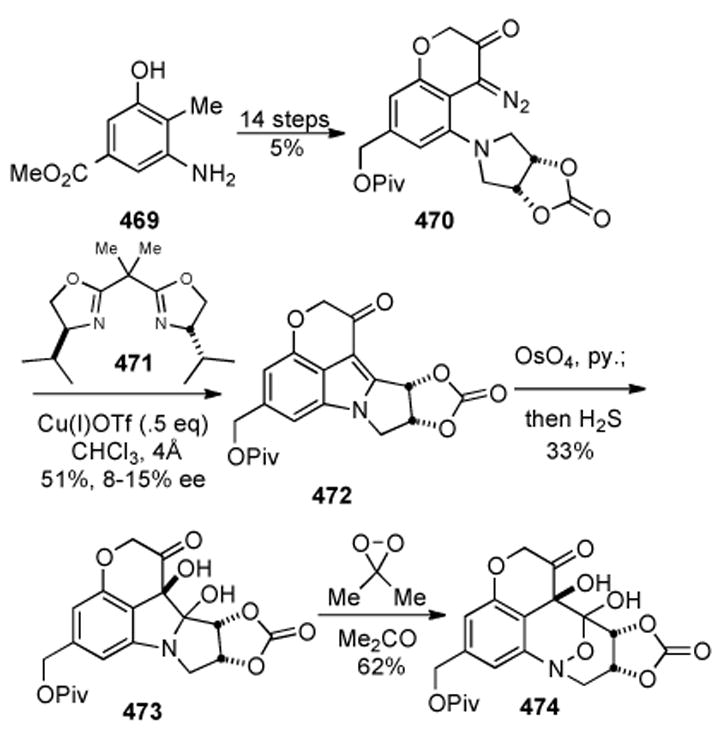
Sulikowski’s cyclization of diazoketone 473 and oxidative ring expansion.
8.4. Ziegler’s Enantiospecificc Route via Chiral Aziridinyl Radicals
Ziegler reported an enantiospecific route to make the fully functionalized core of FR-900482 via chiral aziridinyl radicals in 1997.[232] As with several other syntheses, Ziegler’s strategy entails oxidation of a mitosane 477 via a Polonovski[334] reaction and Hootele[342]-Dmitrienko[340] rearrangement of 478 to 479 (Scheme 87). Drawing on previous studies geared towards synthesis of the mitomycins involving the generation of carbon-centered aziridinyl radicals and their intramolecular addition to an indole nucleus, substrate 475 was synthesized as a cyclization precursor.[343-345] A four-step sequence of reactions gave the cyclized and reduced product 476 in 51% yield. Thus, treatment of 475 with sodium borohydride followed by TBS protection of the resultant alcohol, radical cyclization, and deprotection of the alcohol furnished 476. Oxidation of 476 to the aldehyde followed by a formylation and benzyl carbamateforming sequence provided the penultimate precursor to the well-utilized Polonovski reaction, which underwent m-CPBA oxidation to the N-oxide 477 and Polonovski reaction providing 478 over five steps in 44% yield. Further exposure to m-CPBA induced Hootele[342]-Dmitrienko[340] rearrangement furnishing product 479, which was converted to acetonide 480 via hydrogenolysis of the benzyl groups, acetonide formation, and final decarbonylation with superstoiciometric Wilkinson’s catalyst.
Scheme 87.
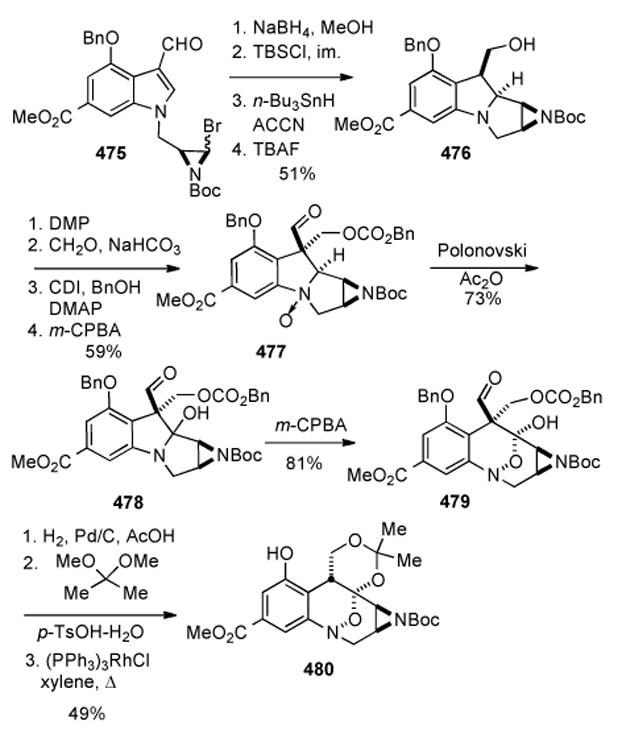
Ziegler’s Polonovski-Hootele-Dmitrienko approach.
8.5. Jimenez: One Pot Oxidation of a Pyrroloindole To a Benzoxazine
Jimenez showed in 2003 that a C9 unfunctionalized mitosene could be oxidized simultaneously at the C9 and C9a positions via the action of dimethyldioxirane in acetone.[198] By analysis of the mechanism of the oxidation taken together with previous results[230] and a report by Dmitrienko[346], it was intuitively reasoned that the DMDO oxidation of a C9 alkylated mitosene performed in the presence of water could lead to the formation of the FR900482 ring system. Thus, C9 methyl mitosene derivative 481, in the presence of DMDO and water in aceone underwent initial oxidation to the intermediates represented by the isomers 482 and 483 (Scheme 88). Interception of the zwitterionic resonance form 483 by water gives hemiaminal 484, subject to further oxidation by DMDO, this time at N4 to give 485. Rearrangement to the FR900482 skeleton then proceeds through intermediate 486 via a ring-opening and recyclization sequence to give the model compound 487 as a single diastereomer in 59% yield. Thus, Jimenez has demonstrated the first example of a 2,3-dihydo-1H-pyrrolo[1,2-a]indole analogue being oxidatively transformed into the FR900482 skeleton in a single step.
Scheme 88.
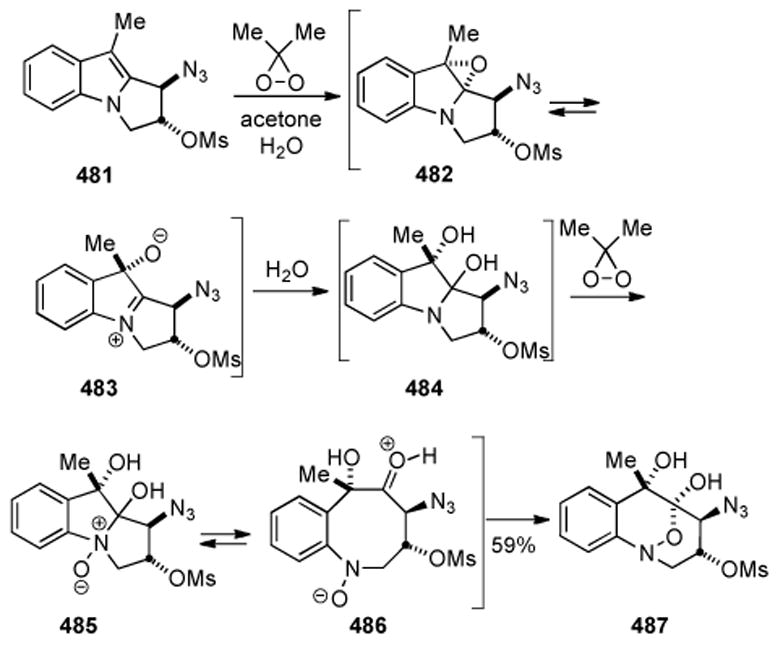
Jimenez’ single-pot procedure.
8.6. Kerr: Heck Reaction to form Tetrahydro-1,2-oxazines
In 2003 the Kerr laboratory developed a very nice method for constructing tetrahydro-1,2-oxazines from the reaction between nitrones and 1,1-cyclopropane diesters and applied it to the synthesis of the FR900482 skeleton (Scheme 89).[347]
Scheme 89.
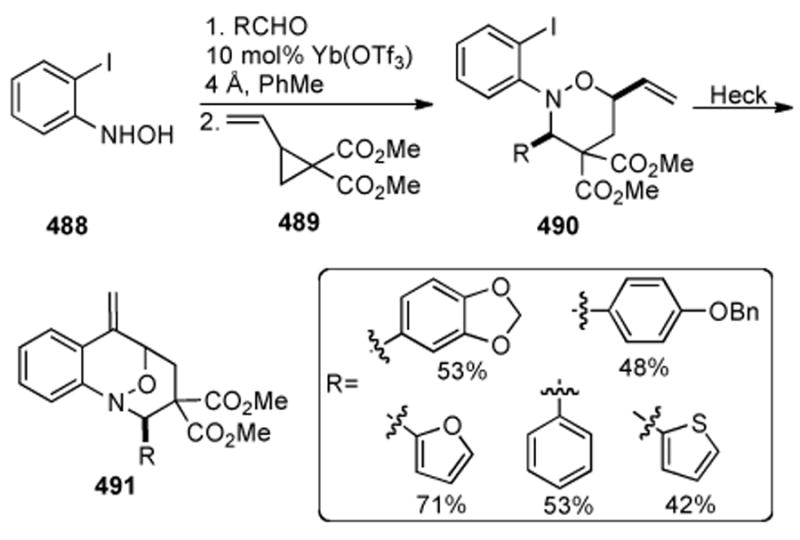
Kerr’s method for constructing tetrahydro-1,2-oxazines.
The following year a report appeared detailing the extension of this methodology to the synthesis of several more oxazines, an additional four congeners of FR900482, and featuring an improved practical procedure; forming the requisite nitrones in situ from the reaction between a hydroxylamine and an aldehyde in the presence of a Lewis acid, thus avoiding the isolation or handling of the nitrone at all.[348] The reaction begins by stirring the hydroxylamine 488 with the desired aldehyde and a catalytic quantity of the lanthanide Lewis acid in toluene under molecular sieves for half an hour. The cyclopropane 489 is then added to yield, after chromatography, the heterocycles 490, where the R groups appear to be chosen as pertinent pharmacophores. The reaction is described as a three component homo [3+2] dipolar cycloaddition as opposed to a [3+3] cycloaddition reaction since the methylene unit of the reacting cyclopropane is not electronically involved. In a fashion ascribed to being akin to Danishefsky’s synthesis of FR900482[316] Heck conditions allowed for cyclization to the bridged oxazines 491.
8.7. Vedejs’ Internal Michael Addition
Vedejs applied the internal Michael addition approach to mitosenes based on lithiated aziridines developed in his lab[199, 273, 275] to the preparation of the N-trityl protected form of the proposed reductively activated metabolite of FK317 responsible for DNA cross linking.[272] A deuterated stannane cyclization precursor 492 was prepared and reacted with methyllithium to give the intermediate lithiodeuterated tricycle 493, followed by phenylselenium chloride mediated elimination to yield aziridinomitosene 494 (Scheme 90; 80%). The ultimate product 495 was accessed in five further transformations and 29% yield. It was noted however that late stage detritylation to afford the free aziridine was not successful.
Scheme 90.
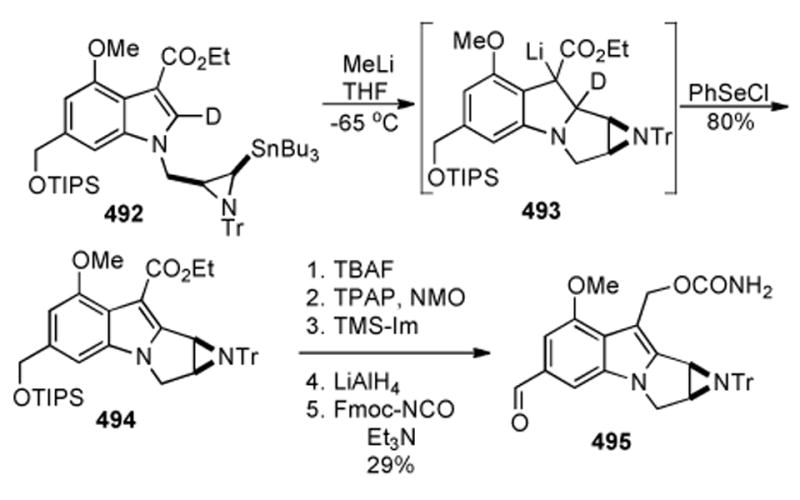
Vedejs’ synthesis of an FK317 mitosene derivative.
8.8. Yamauchi’s Synthesis of the Core
During efforts to develop syntheses of chirally substituted [1,2]-oxazines, a Japanese group succeeded in executing a quick procedure for the construction of the FR900482 skeleton (Scheme 91).[349] A regioselective allylic substitution of the hydroxylamine 496 to dispel the carbonate of 497 in 69% yield was catalyzed by [Ir(cod)Cl]2 with diethyl zinc as the base. Ring-closing methathesis of 498 then secured the oxazine 499 in 76% yield and finally, utilization of the Heck protocol furnished the tricycle 500 nearly quantitatively.
Scheme 91.
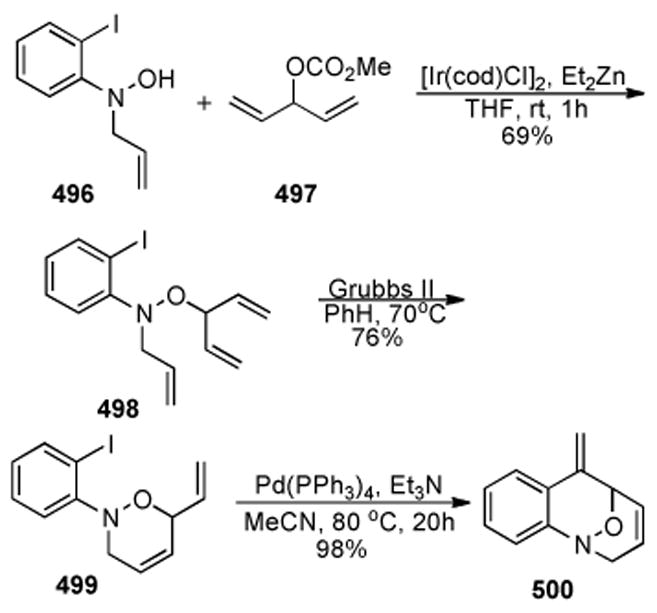
Quick synthesis of the bridged tricyclic oxazine core of the FR series of compounds by Yamauchi.
8.9 Williams’ Synthesis of the Hydroxylamine Hemi-ketal
The first successful construction of the hydroxylamine hemi-ketal functionality present in FR900482 and congeners, was reported by Yasuda and Williams in 1989.[350] As shown in Scheme 92, the nitrotoluene derivative 501 was sequentially homologated to the corresponding nitro keto-aldehyde 504. A new protecting group for aldehydes was devised for this work that involved trapping the cyanohydrin derived from 504 with Alloc-chloride to form the cyanohydrin hemi-allylcarbonate 505. Reduction of the nitro group with zinc dust under carefully controlled conditions, yielded the protected hydroxylamine 506. Removal of the Alloc-carbonate with Pd(0) in the presence of an allyl cation trap, yielded the desired tricyclic species 507, for which a single crystal X-ray analysis confirmed the assigned structure. While not forming the ultimate strategic basis for the enantioselective total synthesis of FR900482 and FR66979 reported by the Williams laboratory years later, this work constituted the first synthetic method to assemble the unique hydroxylamine hemi-ketal ring system of the natural product. It should be further noted that, this functionality is highly unique to the natural product and there were no reports of this functionality present in the literature before the discovery of FR900482. Nature’s impressive creative evolution of the reductively-activatable hydroxylamine hemi-ketal in the FR900482 family of pro-drugs, reveals that this unusual and chemically stable functionality might find use as a pro-drug functional paradigm in other families of drugs.
Scheme 92.

Yasuda and Williams’ synthesis of the hydroxylamine hemi-ketal model system.
8.10. Williams’ Synthesis of the Hydroxylamine Hemi-acetal in a pyrrolizidine alkaloid
One such entrée to using the FR900482 hyroxylamine hemi-ketal as a means of activating other classes of drugs, was published by Tepe and Williams as illustrated in Scheme 93.[351] Commercially available monocrotaline, an oxidatively activated cytotoxic pyrrolizidine alkaloid, was subjected to a von Braun-type of ring-opening and protection to form 508. Removal of the N-Fmoc group followed by oxidation of the resultant secondary amine with m-CPBA and acidic removal of the acetal protecting group provided the chemically stable hydroxylamine hemi-acetal 509. This substance was found to be reductively activated in the presence of Fe(II) to give the highly reactive and cytotoxic DNA cross-linking species dehydromonocrotaline.
Scheme 93.
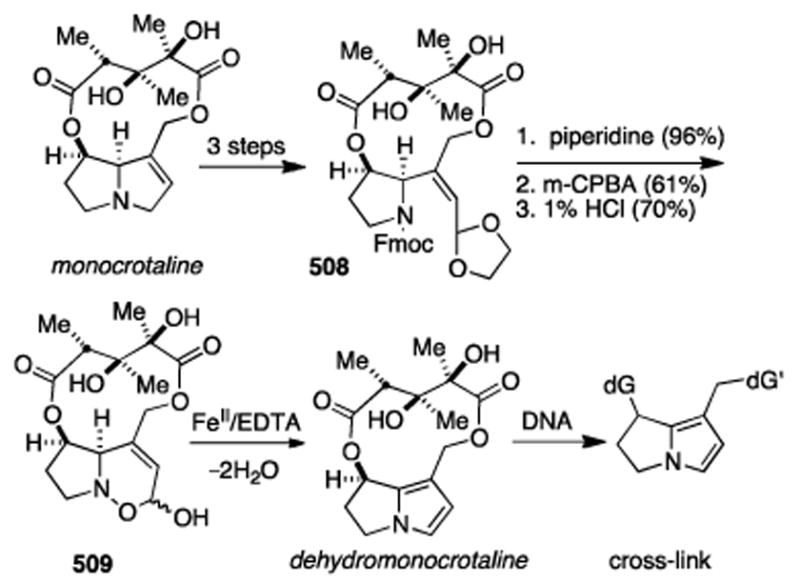
Tepe and Williams’ synthesis of a dehydromonocrotaline progenitor with a hydroxylamine hemi-acetal pro-drug trigger.
This work constitutes the first time that an oxidtively-activated pyrrolizidine alkaloid had been semi-synthetically converted into a reductively-activated congener. The fact that most pyrrolizidine alkaloids are hepatotoxic, as a consequence of being activated by liver cytochrome P450 enzymes, lends further significance to this study.
9. Conclusion
The discovery of the mitomycins over fifty years ago at Kyowa Hakko Kogyo Co. in Japan, has resulted in a vast body of literature covering the biological activity, clinical utility, mechanism of action, synthesis and biosynthesis of these clinically useful anti-tumor drugs. The mitomycins have attracted an unusually high level of interest from the synthetic community, reflecting the myriad of challenges that these small, densely functionalized and unstable molecules pose. Some of the finest minds in chemistry have addressed the synthesis of the mitomycins and FR900482 and it is anticipated that these structures will continue their irresistible allure. It is striking that, despite being the subject of numerous synthetic approaches and total synthesis efforts spanning decades, that there remains no enantiospecific synthesis of any natural mitomycin, and that challenge remains. In light of the absence of a stereodefined total synthesis of a natural mitomycin, the numerous published synthetic approaches attest to the challenge presented by these compounds to the synthetic organic chemist. The closely related FR900482 class of drugs, made their way to Phase II human clinical trials (FK317), but were halted very close to the time that Fujisawa Pharmaceutical Co., the discoverer of these drugs, and Yamanouchi Co. merged to become Astellas. That circumstances of hard business decisions, rather than scientific or clinical deficiencies were responsible for the cessation of the clinical development of FK317, is not publicly accessible information. It is indeed unfortunate that FK317 never realized it’s potential as an anti-cancer chemotherapeutic, despite positive results from the Phase I and II clinical studies, and it’s much improved efficacy and safety relative to the mitomycins.
While still in clinical use today, the world-wide sales of mitomycin C (Mithramycin) and later-developed semi-synthetic analogs, have been steadily declining for the past decade or so and the future clinical utility of these drugs remains uncertain. It is hoped that this review has helped the reader glean an appreciation for the scope of research activities directed at the mitomycinoid antibiotics within the scientific community. Much remains unknown concerning the detailed chemical biology of both classes of drugs and it is this insight, available today through the deployment of a host of powerful tools, that could potentially open new therapeutic windows of opportunity for these natural products as well as their semi-synthetic congeners. Much of the literature concerning the biochemical interaction of these drugs with DNA, may be of diminished importance due to more recent insight into the fascinatingly complex molecular geometry of DNA as it exists in nucleosomes and chromatin.[339] The nucleosomal study by Luger et al.,[339] reveals that we have a very incomplete, and low-resolution understanding of how these drugs interact with chromosomal DNA in living cells and studies of their interactions with other macromolecules, such as RNA have not been explored. It is not unreasonable to anticipate that, such highly reactive electrophilic species such as the mitosenes derived by cellular activation of these drugs, would also alkylate and cross-link other macromolecules such as RNA and protein-nucleic acid complexes; the biological significance of such as yet undiscovered lesions awaits investigation. The authors hope that this review, which has emphasized the synthetic chemistry of these agents, might serve to stimulate future research in to the chemistry and biology of these fascinating antural alkaloids.
Acknowledgments
This paper is dedicated to Professor Yoshito Kishi of Harvard University on the occasion of his 75th birthday. The authors are grateful to the National Institutes of Health (Grant RO1 CA051875) that supported our research in this area between 1992-2009. We are additionally grateful to Fujisawa Pharmaceutical Co., Japan (Astellas) for the generous gifts of natural FR900482, FR66979, FK973 and FK317.
Biographical Sketches
Phillip David Bass was born on March 2nd 1981 in New York, NY, and grew up in Champaign, Illinois. As the progeny of a German mother and an American father, each of whom reside in their respective homeland, the bicultural upbringing of the author has had a significant impact on his perception. Phillip completed his BS in chemistry in 2004 at the University of Illinois at Urbana-Champaign. Following an ensuing year of travel, Phillip engaged in graduate studies at Colorado State University in Fort Collins, Colorado. He is currently pursuing his PhD under the guidance of Professor Robert M. Williams and his research is focused on the asymmetric synthesis of mitomycins.

Daniel A. Gubler was born in 1979 in Provo, Utah and received a B.S. degree in biochemistry from Brigham Young University in 2003. He obtained his Ph.D. degree in 2009 from Colorado State University under the direction of Professor Robert M. Williams for work towards the asymmetric total synthesis of the mitomycins. After completion of his Ph.D., he moved to the California Institute of Technology where he was an American Cancer Society Postdoctoral Fellow in the lab of Professor Peter B. Dervan in the area of bioorganic chemistry. Dr. Gubler is currently an assistant professor at Brigham Young University-Hawaii.

Ted C. Judd was born in 1974 in Croton-on-Hudson, New York and received a B.A. degree in chemistry from SUNY at Buffalo in 1996. He obtained his Ph.D. degree in 2003 from Colorado State University under the direction of Professor Robert M. Williams completing the asymmetric total synthesis of (+)-FR900482 and (+)-FR66979. After completion of his Ph.D., he moved to Harvard University as an NIH postdoctoral fellow working in the laboratory of Professor Yoshito Kishi completing the structural elucidation and total synthesis of Mycolactone C. Dr. Judd is currently working as a medicinal chemist at Amgen in Thousand Oaks, California.

Robert M. Williams was born in New York (1953) and attended Syracuse University obtaining a B.A. in Chemistry in 1975; Professor Ei-ichi Negishi served as his undergraduate research advisor. He obtained the Ph.D. degree in 1979 at MIT under the direction of Professor W.H. Rastetter and was a post-doctoral fellow at Harvard (1979-1980; Professor R.B. Woodward; Professor Yoshito Kishi). He joined Colorado State University in 1980 and was named University Distinguished Professor in 2002. He was a visiting Professor at the University of California, Berkeley in 1990 and a visiting Professor at Harvard University in 1994-95. Co-author of more than 280 publications, his interdisciplinary research program at the chemistry-biology interface concerns the total synthesis, biosynthesis and mechanism of action of biomedically significant natural products, antitumor drug-DNA interactions, histone deacetylase inhibitors, heterocycles, the asymmetric synthesis of amino acids, and peptides. He has been recognized with several honors and awards including the NIH Research Career Development Award (1984); Eli Lilly Young Investigator Award (1986); Alfred P. Sloan Foundation Fellowship (1986); Merck, Academic Development Award (1991); Japanese Society for the Promotion of Science Fellowship (1999), Arthur C. Cope Scholars Award (2002), the ACS Ernest Guenther Award in the Chemistry of Natural Products (2011) and the Japanese Society for the Promotion of Science Long-term Fellowship (2012-2013). Co-founder of five biopharmaceutical companies, he is currently the Director of the Colorado Center for Drug Discovery and President of Cetya Therapeutics.

References
- 1.Hata T, Sano Y, Sugawara R, Matsumae A, Kanamori K, Shima T, Hoshi T. J Antibiot. 1956;9:141. [PubMed] [Google Scholar]
- 2.Wakaki S, Marumo H, Tomioka K, Shimizu G, Kato E, Kamada H, Kudo S, Fujimoto Y. Antibiot Chemother. 1958;8:228. [PubMed] [Google Scholar]
- 3.Kono M, Saitoh Y, Kasai M, Shirahata K. J Antibiot. 1995;48:179. doi: 10.7164/antibiotics.48.179. [DOI] [PubMed] [Google Scholar]
- 4.Webb JS, Cosulich DB, Mowat JH, Patrick JB, Broschard RW, Meyer WE, Williams RP, Wolf CF, Fulmor W, Pidacks C, Lancaster JE. J Am Chem Soc. 1962;84:3187. [Google Scholar]
- 5.Webb JS, Cosulich DB, Mowat JH, Patrick JB, Broschard RW, Meyer WE, Williams RP, Wolf CF, Fulmor W, Pidacks C, Lancaster JE. J Am Chem Soc. 1962;84:3185. [Google Scholar]
- 6.Tulinsky A. J Am Chem Soc. 1962;84:3188. [Google Scholar]
- 7.Tulinsky A, Van den Hende JH. J Am Chem Soc. 1967;89:2905. doi: 10.1021/ja00988a018. [DOI] [PubMed] [Google Scholar]
- 8.Uchida I, Takase S, Kayakiri H, Kiyoto S, Hashimoto M, Tada T, Koda S, Morimoto Y. J Am Chem Soc. 1987;109:4108. [Google Scholar]
- 9.Masuda K, Nakamura T, Shimomura K, Shibata T, Terano H, Kohsaka M. J Antibiot. 1988;41:1497. doi: 10.7164/antibiotics.41.1497. [DOI] [PubMed] [Google Scholar]
- 10.Iyer VN, Szybalski W. Proc Natl Acad Sci U S A. 1963;50:355. doi: 10.1073/pnas.50.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iyer VN, Szybalski W. Science. 1964;145:55. doi: 10.1126/science.145.3627.55. [DOI] [PubMed] [Google Scholar]
- 12.Wolkenberg SE, Boger DL. Chem Rev. 2002;102:2477. doi: 10.1021/cr010046q. [DOI] [PubMed] [Google Scholar]
- 13.Rajski SR, Williams RM. Chem Rev. 1998;98:2723. doi: 10.1021/cr9800199. [DOI] [PubMed] [Google Scholar]
- 14.Coleman RS. Curr Opin Drug Discov Devel. 2001;4:435. [PubMed] [Google Scholar]
- 15.Rajski SR, Rollins SB, Williams RM. J Am Chem Soc. 1998;120:2192. [Google Scholar]
- 16.Rajski SR, Williams RM. Bioorg Med Chem. 2000;8:1331. doi: 10.1016/s0968-0896(00)00078-x. [DOI] [PubMed] [Google Scholar]
- 17.Danishefsky SJ, Schkeryantz JM. Synlett. 1995;475 [Google Scholar]
- 18.Suresh Kumar G, Lipman R, Cummings J, Tomasz M. Biochemistry. 1997;36:14128. doi: 10.1021/bi971394i. [DOI] [PubMed] [Google Scholar]
- 19.McClelland RA, Lam K. J Am Chem Soc. 1985;107:5182. [Google Scholar]
- 20.Tomasz M, Lipman R. Biochemistry. 1981;20:5056. doi: 10.1021/bi00520a036. [DOI] [PubMed] [Google Scholar]
- 21.Peterson DM, Fisher J. Biochemistry. 1986;25:4077. doi: 10.1021/bi00362a014. [DOI] [PubMed] [Google Scholar]
- 22.Kohn H, Zein N. J Am Chem Soc. 1983;105:4105. [Google Scholar]
- 23.Pan SS, Andrews PA, Glover CJ, Bachur NR. J Biol Chem. 1984;259:959. [PubMed] [Google Scholar]
- 24.Danishefsky S, Ciufolini M. J Am Chem Soc. 1984;106:6424. [Google Scholar]
- 25.Danishefsky SJ, Egbertson M. J Am Chem Soc. 1986;108:4648. [Google Scholar]
- 26.Egbertson M, Danishefsky SJ. J Am Chem Soc. 1987;109:2204. [Google Scholar]
- 27.Schiltz P, Kohn H. J Am Chem Soc. 1993;115:10510. [Google Scholar]
- 28.Galm U, Hager MH, Van Lanen SG, Ju J, Thorson JS, Shen B. Chem Rev. 2005;105:739. doi: 10.1021/cr030117g. [DOI] [PubMed] [Google Scholar]
- 29.Hoey BM, Butler J, Swallow AJ. Biochemistry. 1988;27:2608. doi: 10.1021/bi00407a051. [DOI] [PubMed] [Google Scholar]
- 30.Celli CM, Jaiswal AK. Cancer Res. 2003;63:6016. [PubMed] [Google Scholar]
- 31.Wang X, Doherty GP, Leith MK, Curphey TJ, Begleiter A. Br J Cancer. 1999;80:1223. doi: 10.1038/sj.bjc.6690489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belcourt MF, Hodnick WF, Rockwell S, Sartorelli AC. Proc Natl Acad Sci U S A. 1996;93:456. doi: 10.1073/pnas.93.1.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pritsos CA, Gustafson DL. Oncology Res. 1994;6:477. [PubMed] [Google Scholar]
- 34.Siegel D, Gibson NW, Preusch PC, Ross D. Cancer Res. 1990;50:7293. [PubMed] [Google Scholar]
- 35.Pius J, Yeuhang X, Anil KJ. Int J Cancer. 1996;65:263. [Google Scholar]
- 36.Joseph P, Jaiswal AK. Br J Cancer. 2000;82:1305. doi: 10.1054/bjoc.1999.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cummings J, Spanswick VJ, Smyth JF. Eur J Cancer. 1995;31:1928. doi: 10.1016/0959-8049(95)00364-9. [DOI] [PubMed] [Google Scholar]
- 38.Siegel D, Beall H, Senekowitsch C, Kasai M, Arai H, Gibson NW, Ross D. Biochemistry. 1992;31:7879. doi: 10.1021/bi00149a019. [DOI] [PubMed] [Google Scholar]
- 39.Ernster L, Ronald WE, Maynard EP. Methods Enzymol, Vol. Vol. 10. Academic Press; 1967. p. 309. [Google Scholar]
- 40.Iyanagi T, Yamazaki I. Biochim Biophys Acta, Bioenerg. 1970;216:282. doi: 10.1016/0005-2728(70)90220-3. [DOI] [PubMed] [Google Scholar]
- 41.Siegel D, Gibson NW, Preusch PC, Ross D. Cancer Res. 1990;50:7483. [PubMed] [Google Scholar]
- 42.Paz MM, Tomasz M. Org Lett. 2001;18:2789. doi: 10.1021/ol015517+. [DOI] [PubMed] [Google Scholar]
- 43.Paz MM. Chem Res Toxicol. 2009;22:1663. doi: 10.1021/tx9002758. [DOI] [PubMed] [Google Scholar]
- 44.Ruppitsch W, Meißlitzer C, Hirsch-Kauffmann M, Schweiger M. FEBS Letters. 1998;422:99. doi: 10.1016/s0014-5793(97)01608-6. [DOI] [PubMed] [Google Scholar]
- 45.Kontou M, Adelfalk C, Ramirez MH, Ruppitsch W, Hirsch-Kauffmann M, Schweiger M. Oncogene. 2002;21:2406. doi: 10.1038/sj.onc.1205299. [DOI] [PubMed] [Google Scholar]
- 46.Yokomizo A, Ono M, Nanri H, Makino Y, Ohga T, Wada M, Okamoto T, Yodoi J, Kuwano M, Kohno K. Cancer Res. 1995;55:4293. [PubMed] [Google Scholar]
- 47.Adikesavan AK, Jaiswal AK. Mol Cancer Ther. 2007;6:2719. doi: 10.1158/1535-7163.MCT-07-0160. [DOI] [PubMed] [Google Scholar]
- 48.Paz MM, Das A, Palom Y, He QY, Tomasz M. J Med Chem. 2001;44:2834. doi: 10.1021/jm010072g. [DOI] [PubMed] [Google Scholar]
- 49.He QY, Maruenda H, Tomasz M. J Am Chem Soc. 1994;116:9349. [Google Scholar]
- 50.Williams RM, Rajski SR. Tetrahedron Lett. 1992;33:2929. [Google Scholar]
- 51.Williams RM, Rajski SR. Tetrahedron Lett. 1993;34:7023. [Google Scholar]
- 52.Huang H, Rajski SR, Williams RM, Hopkins PB. Tetrahedron Lett. 199435:9669. [Google Scholar]
- 53.Williams RM, Rajski SR, Rollins SB. Chem Biol. 1997;4:127. doi: 10.1016/s1074-5521(97)90256-8. [DOI] [PubMed] [Google Scholar]
- 54.Woo J, Sigurdsson ST, Hopkins PB. J Am Chem Soc. 1993;115:1199. [Google Scholar]
- 55.Huang H, Pratum TK, Hopkins PB. J Am Chem Soc. 1994;116:2703. [Google Scholar]
- 56.Paz MM, Hopkins PB. Tetrahedron Lett. 1997;38:343. [Google Scholar]
- 57.Paz MM, Hopkins PB. J Am Chem Soc. 1997;119:5999. [Google Scholar]
- 58.Paz MM, Sigurdsson ST, Hopkins PB. Bioorg Med Chem. 2000;8:173. doi: 10.1016/s0968-0896(99)00270-9. [DOI] [PubMed] [Google Scholar]
- 59.Millard JT, Beachy TM. Biochemistry. 1993;32:12850. doi: 10.1021/bi00210a038. [DOI] [PubMed] [Google Scholar]
- 60.Li VS, Tang Ms, Kohn H. Bioorg Med Chem. 2001;9:863. doi: 10.1016/s0968-0896(00)00301-1. [DOI] [PubMed] [Google Scholar]
- 61.Fukuyama T, Goto S. Tetrahedron Lett. 1989;30:6491. [Google Scholar]
- 62.Yoshinori N, Masamichi I, Ikuo K, Fusako N, Susumu T, Sanae M, Toshitaka M, Kyoichi S. Cancer Science. 1998;89:666. [Google Scholar]
- 63.Beckerbauer L, Tepe JJ, Eastman RA, Mixter PF, Williams RM, Reeves R. Chem Biol. 2002;9:427. doi: 10.1016/s1074-5521(02)00122-9. [DOI] [PubMed] [Google Scholar]
- 64.Tomasz M, Lipman R, Chowdary D, Pawlak J, Verdine GL, Nakanishi K. Science. 1987;235:1204. doi: 10.1126/science.3103215. [DOI] [PubMed] [Google Scholar]
- 65.Yoshinori N, Masamichi I, Sanae M, Shoji T, Tomoichi F, Sachiko Y, Ikuo K, Fusako N, Susumu T, Toshitaka M, Kyoichi S. Cancer Science. 1998;89:1306. [Google Scholar]
- 66.Tomasz M, Lipman R. J Am Chem Soc. 1979;101:6063. [Google Scholar]
- 67.Borowy-Borowski H, Lipman R, Tomasz M. Biochemistry. 1990;29:2999. doi: 10.1021/bi00464a016. [DOI] [PubMed] [Google Scholar]
- 68.Tomasz M, Palom Y. Pharmacol Ther. 1997;76:73. doi: 10.1016/s0163-7258(97)00088-0. [DOI] [PubMed] [Google Scholar]
- 69.Teng SP, Woodson SA, Crothers DM. Biochemistry. 1989;28:3901. doi: 10.1021/bi00435a041. [DOI] [PubMed] [Google Scholar]
- 70.Millard JT, Weidner MF, Raucher S, Hopkins PB. J Am Chem Soc. 1990;112:3637. [Google Scholar]
- 71.Beckerbauer L, Tepe JJ, Cullison J, Reeves R, Williams RM. Chem Biol. 2000;7:805. doi: 10.1016/s1074-5521(00)00028-4. [DOI] [PubMed] [Google Scholar]
- 72.(a) Tomasz M. Chem Biol. 1995;2:575. doi: 10.1016/1074-5521(95)90120-5. for another review, see: [DOI] [PubMed] [Google Scholar]; (b) Remers WA. “The Mitomycins”. In: Kingston DGI, Cragg GM, Newman DJ, editors. Anticancer Agents from Natural Products. CRC Press; 2005. Chapter 23. [Google Scholar]
- 73.Chirrey L, Cummings J, Halbert GW, Smyth JF. Cancer Chemother Pharmacol. 1995;35:318. doi: 10.1007/BF00689451. [DOI] [PubMed] [Google Scholar]
- 74.Tomasz M. Biochim Biophys Acta, Nucleic Acids Protein Synth. 1970;213:288. doi: 10.1016/0005-2787(70)90037-7. [DOI] [PubMed] [Google Scholar]
- 75.Lawley PD, Thatcher CJ. Biochem J. 1970;116:693. doi: 10.1042/bj1160693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lawley PD, Shah SA. Biochem J. 1972;128:117. doi: 10.1042/bj1280117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tomasz M, Mercado CM, Olson J, Chatterjie N. Biochemistry. 1974;13:4878. doi: 10.1021/bi00721a002. [DOI] [PubMed] [Google Scholar]
- 78.Lipman R, Weaver J, Tomasz M. Biochim Biophys Acta, Nucleic Acids Protein Synth. 1978;521:779. doi: 10.1016/0005-2787(78)90317-9. [DOI] [PubMed] [Google Scholar]
- 79.Tomasz M, Lipman R. J Am Chem Soc. 1979;101:6063. [Google Scholar]
- 80.Lin Ai J, Cosby Lucille A, Sartorelli Alan C. American Chemical Society. Cancer Chemother. 1976;30:71. [Google Scholar]
- 81.Weaver J, Tomasz M. Biochim Biophys Acta, Gene Struct Expression. 1982697:252. doi: 10.1016/0167-4781(82)90084-7. [DOI] [PubMed] [Google Scholar]
- 82.Tomasz M, Lipman R, Verdine GL, Nakanishi K. Biochemistry. 1986;25:4337. doi: 10.1021/bi00363a024. [DOI] [PubMed] [Google Scholar]
- 83.Tomasz M, Chowdary D, Lipman R, Shimotakahara S, Veiro D, Walker V, Verdine GL. Proc Natl Acad Sci U S A. 1986;83:6702. doi: 10.1073/pnas.83.18.6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tomasz M, Lipman R, Verdine G, Nakanishi K. J Am Chem Soc. 1985;107:6120. [Google Scholar]
- 85.Tomasz M, Jung M, Verdine G, Nakanishi K. J Am Chem Soc. 1984;106:7367. [Google Scholar]
- 86.Pullman A, Pullman B. Q Rev Biophys. 1981;14:289. doi: 10.1017/s0033583500002341. [DOI] [PubMed] [Google Scholar]
- 87.Singer B, Grunberger D. Molecular biology of mutagens and carcinogens. Plenum Press; New York: 1983. [Google Scholar]
- 88.Moore HW. Science. 1977;197:527. doi: 10.1126/science.877572. [DOI] [PubMed] [Google Scholar]
- 89.Tomasz M, Lipman R, Lee MS, Verdine GL, Nakanishi K. Biochemistry. 1987;26:2010. doi: 10.1021/bi00381a034. [DOI] [PubMed] [Google Scholar]
- 90.Remers WA, Rao SN, Wunz TP, Kollman PA. J Med Chem. 1988;31:1612. doi: 10.1021/jm00403a021. [DOI] [PubMed] [Google Scholar]
- 91.Kaplan DJ, Tomasz M. Biochemistry. 1982;21:3006. doi: 10.1021/bi00541a031. [DOI] [PubMed] [Google Scholar]
- 92.Ueda K, Komano T. Nucleic Acids Res. 1984;12:6673. doi: 10.1093/nar/12.17.6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McGuinness BF, Nakanishi K, Lipman R, Tomasz M. Tetrahedron Lett. 1988;29:4673. [Google Scholar]
- 94.Tomasz M, Lipman R, McGuinness BF, Nakanishi K. J Am Chem Soc. 1988;110:5892. [Google Scholar]
- 95.Tomasz M, Chawla AK, Lipman R. Biochemistry. 1988;27:3182. doi: 10.1021/bi00409a009. [DOI] [PubMed] [Google Scholar]
- 96.Norman D, Live D, Sastry M, Lipman R, Hingerty BE, Tomasz M, Broyde S, Patel DJ. Biochemistry. 1990;29:2861. doi: 10.1021/bi00463a032. [DOI] [PubMed] [Google Scholar]
- 97.Borowy-Borowski H, Lipman R, Chowdary D, Tomasz M. Biochemistry. 1990;29:2992. doi: 10.1021/bi00464a015. [DOI] [PubMed] [Google Scholar]
- 98.Li VS, Kohn H. J Am Chem Soc. 1991;113:275. [Google Scholar]
- 99.Pu WT, Kahn R, Munn MM, Rupp WD. J Biol Chem. 1989;264:20697. [PubMed] [Google Scholar]
- 100.Kohn H, Li VS, Tang MS. J Am Chem Soc. 1992;114:5501. [Google Scholar]
- 101.Phillips DR, White RJ, Cullinane C. FEBS Letters. 1989;246:233. doi: 10.1016/0014-5793(89)80289-3. [DOI] [PubMed] [Google Scholar]
- 102.Weidner MF, Millard JT, Hopkins PB. J Am Chem Soc. 1989;111:9270. [Google Scholar]
- 103.Kumar S, Johnson WS, Tomasz M. Biochemistry. 1993;32:1364. doi: 10.1021/bi00056a023. [DOI] [PubMed] [Google Scholar]
- 104.Kumar S, Lipman R, Tomasz M. Biochemistry. 1992;31:1399. doi: 10.1021/bi00120a016. [DOI] [PubMed] [Google Scholar]
- 105.Colot V, Rossignol JL. BioEssays. 1999;21:402. doi: 10.1002/(SICI)1521-1878(199905)21:5<402::AID-BIES7>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 106.Bird AP. Nature. 1986;321:209. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 107.Johnson WS, He QY, Tomasz M. Bioorg Med Chem. 1995;3:851. doi: 10.1016/0968-0896(95)00067-q. [DOI] [PubMed] [Google Scholar]
- 108.Gargiulo D, Musser SS, Yang L, Fukuyama T, Tomasz M. J Am Chem Soc. 1995;117:9388. [Google Scholar]
- 109.Das A, Tang KS, Gopalakrishnan S, Waring MJ, Tomasz M. Chem Biol. 1999;6:461. doi: 10.1016/s1074-5521(99)80064-7. [DOI] [PubMed] [Google Scholar]
- 110.Li VS, Choi D, Wang Z, Jimenez LS, Tang Ms, Kohn H. J Am Chem Soc. 1996;118:2326. [Google Scholar]
- 111.Sastry M, Fiala R, Lipman R, Tomasz M, Patel DJ. J Mol Biol. 1995;247:338. doi: 10.1006/jmbi.1994.0143. [DOI] [PubMed] [Google Scholar]
- 112.Bailly C, Marchand C, Waring MJ. J Am Chem Soc. 1993;115:3784. [Google Scholar]
- 113.Waring MJ, Bailly C. Gene. 1994;149:69. doi: 10.1016/0378-1119(94)90414-6. [DOI] [PubMed] [Google Scholar]
- 114.Bailly C, Payet D, Travers AA, Waring MJ. Proc Natl Acad Sci U S A. 1996;93:13623. doi: 10.1073/pnas.93.24.13623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tomasz M, Das A, Tang KS, Ford MGJ, Minnock A, Musser SM, Waring MJ. J Am Chem Soc. 1998;120:11581. [Google Scholar]
- 116.Dannenberg JJ, Tomasz M. J Am Chem Soc. 2000;122:2062. [Google Scholar]
- 117.Pan SS, Gonzalez H. Mol Pharmacol. 1990;37:966. [PubMed] [Google Scholar]
- 118.Kunz KR, Iyengar BS, Dorr RT, Alberts DS, Remers WA. J Med Chem. 1991;34:2281. doi: 10.1021/jm00111a051. [DOI] [PubMed] [Google Scholar]
- 119.Bizanek R, McGuinness BF, Nakanishi K, Tomasz M. Biochemistry. 1992;31:3084. doi: 10.1021/bi00127a008. [DOI] [PubMed] [Google Scholar]
- 120.Rink SM, Lipman R, Alley SC, Hopkins PB, Tomasz M. Chem Res Toxicol. 1996;9:382. doi: 10.1021/tx950156q. [DOI] [PubMed] [Google Scholar]
- 121.Palom Y, Lipman R, Musser SM, Tomasz M. Chem Res Toxicol. 1998;11:203. doi: 10.1021/tx970205u. [DOI] [PubMed] [Google Scholar]
- 122.Prakash AS, Beall H, Ross D, Gibson NW. Biochemistry. 1993;32:5518. doi: 10.1021/bi00072a005. [DOI] [PubMed] [Google Scholar]
- 123.Subramaniam G, Paz MM, Suresh Kumar G, Das A, Palom Y, Clement CC, Patel DJ, Tomasz M. Biochemistry. 2001;40:10473. doi: 10.1021/bi010965a. [DOI] [PubMed] [Google Scholar]
- 124.Holowachuk EW, Greer MK, Martin DR. Nucleic Acids Res. 1987;15:10551. doi: 10.1093/nar/15.24.10551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Suresh Kumar G, Musser SM, Cummings J, Tomasz M. J Am Chem Soc. 1996;118:9209. [Google Scholar]
- 126.Wurdeman RL, Church KM, Gold B. J Am Chem Soc. 1989;111:6408. [Google Scholar]
- 127.Warpehoski MA, Hurley LH. Chem Res Toxicol. 1988;1:315. doi: 10.1021/tx00006a001. [DOI] [PubMed] [Google Scholar]
- 128.Utzat CD, Clement CC, Ramos LA, Das A, Tomasz M, Basu AK. Chem Res Toxicol. 2005;18:213. doi: 10.1021/tx049813h. [DOI] [PubMed] [Google Scholar]
- 129.Ross D, Siegel D, Beall H, Prakash AS, Mulcahy RT, Gibson NW. Cancer Metastasis Rev. 1993;12:83. doi: 10.1007/BF00689803. [DOI] [PubMed] [Google Scholar]
- 130.Siegel D, Beall H, Kasai M, Arai H, Gibson NW, Ross D. Mol Pharmacol. 1993;44:1128. [PubMed] [Google Scholar]
- 131.Maliepaard M, de Mol NJ, Janssen LHM, Hoogvliet JC, van der Neut W, Verboom W, Reinhoudt DN. J Med Chem. 1993;36:2091. doi: 10.1021/jm00067a006. [DOI] [PubMed] [Google Scholar]
- 132.Maliepaard M, Sitters KAMC, de Mol NJ, Janssen LHM, Stratford IJ, Stephens M, Verboom W, Reinhoudt DN. Biochem Pharmacol. 1994;48:1371. doi: 10.1016/0006-2952(94)90559-2. [DOI] [PubMed] [Google Scholar]
- 133.Maliepaard M, De Mol NJ, Janssen LHM, van der Neut W, Verboom W, Reinhoudt DN. Anticancer Drug Des. 1992;7:415. [PubMed] [Google Scholar]
- 134.Maliepaard M, de Mol NJ, Tomasz M, Gargiulo D, Janssen LHM, van Duynhoven JPM, van Velzen EJJ, Verboom W, Reinhoudt DN. Biochemistry. 1997;36:9211. doi: 10.1021/bi9700680. [DOI] [PubMed] [Google Scholar]
- 135.Subramaniam S, Kohn H. J Am Chem Soc. 1993;115:10519. [Google Scholar]
- 136.McGuinness BF, Lipman R, Goldstein J, Nakanishi K, Tomasz M. Biochemistry. 1991;30:6444. doi: 10.1021/bi00240a015. [DOI] [PubMed] [Google Scholar]
- 137.Palom Y, Suresh Kumar G, Tang LQ, Paz MM, Musser SM, Rockwell S, Tomasz M. Chem Res Toxicol. 2002;15:1398. doi: 10.1021/tx020044g. [DOI] [PubMed] [Google Scholar]
- 138.Abbas T, Olivier M, Lopez J, Houser S, Xiao G, Kumar GS, Tomasz M, Bargonetti J. J Biol Chem. 2002;277:40513. doi: 10.1074/jbc.M205495200. [DOI] [PubMed] [Google Scholar]
- 139.Boamah EK, White DE, Talbott KE, Arva NC, Berman D, Tomasz M, Bargonetti J. ACS Chem Biol. 2007;2:399. doi: 10.1021/cb700060t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Paz MM, Suresh Kumar G, Glover M, Waring MJ, Tomasz M. J Med Chem. 2004;47:3308. doi: 10.1021/jm049863j. [DOI] [PubMed] [Google Scholar]
- 141.Rink SM, Warner DL, Klapars A, Vedejs E. Biochemistry. 2005;44:13981. doi: 10.1021/bi050426w. [DOI] [PubMed] [Google Scholar]
- 142.Holtz KM, Rockwell S, Tomasz M, Sartorelli AC. J Biol Chem. 2003;278:5029. doi: 10.1074/jbc.M209722200. [DOI] [PubMed] [Google Scholar]
- 143.Seow HA, Penketh PG, Belcourt MF, Tomasz M, Rockwell S, Sartorelli AC. J Biol Chem. 2004;279:31606. doi: 10.1074/jbc.M404910200. [DOI] [PubMed] [Google Scholar]
- 144.Snodgrass RG, Collier AC, Coon AE, Pritsos CA. J Biol Chem. 285:19068. doi: 10.1074/jbc.M109.040477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.August PR, Flickinger MC, Sherman DH. J Bacteriol. 1994;176:4448. doi: 10.1128/jb.176.14.4448-4454.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Möhler H, Brühmüller M, Decker K. Eur J Biochem. 1972;29:152. doi: 10.1111/j.1432-1033.1972.tb01969.x. [DOI] [PubMed] [Google Scholar]
- 147.August PR, Rahn JA, Flickinger MC, Sherman DH. Gene. 1996;175:261. doi: 10.1016/0378-1119(96)00172-2. [DOI] [PubMed] [Google Scholar]
- 148.Johnson DA, August PR, Shackleton C, Liu Hw, Sherman DH. J Am Chem Soc. 1997;119:2576. [Google Scholar]
- 149.Belcourt MF, Penketh PG, Hodnick WF, Johnson DA, Sherman DH, Rockwell S, Sartorelli AC. Proc Natl Acad Sci U S A. 1999;96:10489. doi: 10.1073/pnas.96.18.10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kalyanaraman B, Perez-Reyes E, Mason RP. Biochim Biophys Acta, Gen Subj. 1980;630:119. doi: 10.1016/0304-4165(80)90142-7. [DOI] [PubMed] [Google Scholar]
- 151.Sheldon PJ, Johnson DA, August PR, Liu HW, Sherman DH. J Bacteriol. 1997;179:1796. doi: 10.1128/jb.179.5.1796-1804.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sheldon PJ, Mao Y, He M, Sherman DH. J Bacteriol. 1999;181:2507. doi: 10.1128/jb.181.8.2507-2512.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.He M, Sheldon PJ, Sherman DH. Proc Natl Acad Sci U S A. 2001;98:926. doi: 10.1073/pnas.031314998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Martin TW, Dauter Z, Devedjiev Y, Sheffield P, Jelen F, He M, Sherman DH, Otlewski J, Derewenda ZS, Derewenda U. Structure. 2002;10:933. doi: 10.1016/s0969-2126(02)00778-5. [DOI] [PubMed] [Google Scholar]
- 155.Kirsch EJ, Korshalla JD. J Bacteriol. 1964;87:247. doi: 10.1128/jb.87.2.247-255.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hornemann U, Cloyd JC. J Chem Soc D. 1971;301 [Google Scholar]
- 157.Bezanson GS, Vining LC. Can J Biochem. 1971;49:911. doi: 10.1139/o71-131. [DOI] [PubMed] [Google Scholar]
- 158.Hornemann U, Aikman MJ. J Chem Soc, Chem Commun. 1973:88b. [Google Scholar]
- 159.Hornemann U, Kehrer JP, Nunez CS, Ranieri RL. J Am Chem Soc. 1974;96:320. doi: 10.1021/ja00808a087. [DOI] [PubMed] [Google Scholar]
- 160.Hornemann U, Eggert JH. J Antibiot. 1975;28:841. doi: 10.7164/antibiotics.28.841. [DOI] [PubMed] [Google Scholar]
- 161.Hornemann U, Kehrer JP, Eggert JH. J Chem Soc, Chem Commun. 1974:1045. [Google Scholar]
- 162.Hornemann U, Eggert JH, Honor DP. J Chem Soc, Chem Commun. 1980:11. [Google Scholar]
- 163.Weller DD, Rinehart KL. J Am Chem Soc. 1978;100:6757. [Google Scholar]
- 164.Haber A, Johnson RD, Rinehart KL. J Am Chem Soc. 1977;99:3541. doi: 10.1021/ja00452a079. [DOI] [PubMed] [Google Scholar]
- 165.White RJ, Martinelli E. FEBS Letters. 1974;49:233. doi: 10.1016/0014-5793(74)80519-3. [DOI] [PubMed] [Google Scholar]
- 166.Kibby JJ, McDonald IA, Rickards RW. J Chem Soc, Chem Commun. 1980:768. [Google Scholar]
- 167.Anderson MG, Kibby JJ, Rickards RW, Rothschild JM. J Chem Soc, Chem Commun. 1980:1277. [Google Scholar]
- 168.Kibby JJ, Rickards RW. J Antibiot. 1981;34:605. doi: 10.7164/antibiotics.34.605. [DOI] [PubMed] [Google Scholar]
- 169.Floss HG, Beale JM. Angew Chem Int Ed Engl. 1989;28:146. [Google Scholar]
- 170.Kim CG, Kirschning A, Bergon P, Ahn Y, Wang JJ, Shibuya M, Floss HG. J Am Chem Soc. 1992;114:4941. [Google Scholar]
- 171.Kim CG, Kirschning A, Bergon P, Zhou P, Su E, Sauerbrei B, Ning S, Ahn Y, Breuer M, Leistner E, Floss HG. J Am Chem Soc. 1996;118:7486. [Google Scholar]
- 172.Kim CG, Yu TW, Fryhle CB, Handa S, Floss HG. J Biol Chem. 1998;273:6030. doi: 10.1074/jbc.273.11.6030. [DOI] [PubMed] [Google Scholar]
- 173.Eads JC, Beeby M, Scapin G, Yu TW, Floss HG. Biochemistry. 1999;38:9840. doi: 10.1021/bi990018q. [DOI] [PubMed] [Google Scholar]
- 174.Guo J, Frost JW. J Am Chem Soc. 2002;124:528. doi: 10.1021/ja016963v. [DOI] [PubMed] [Google Scholar]
- 175.Guo J, Frost JW. J Am Chem Soc. 2002;124:10642. doi: 10.1021/ja026628m. [DOI] [PubMed] [Google Scholar]
- 176.Arakawa K, Müller M, Mahmud T, Yu TW, Floss HG. J Am Chem Soc. 2002;124:10644. doi: 10.1021/ja0206339. [DOI] [PubMed] [Google Scholar]
- 177.Mao Y, Varoglu M, Sherman DH. J Bacteriol. 1999;181:2199. doi: 10.1128/jb.181.7.2199-2208.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mao Y, Varoglu M, Sherman DH. Chem Biol. 1999;6:251. doi: 10.1016/S1074-5521(99)80040-4. [DOI] [PubMed] [Google Scholar]
- 179.Yu TW, Müller R, Müller M, Zhang X, Draeger G, Kim CG, Leistner E, Floss HG. J Biol Chem. 2001;276:12546. doi: 10.1074/jbc.M009667200. [DOI] [PubMed] [Google Scholar]
- 180.Floss HG, Yu TW, Arakawa K. J Antibiot. 64:35. doi: 10.1038/ja.2010.139. [DOI] [PubMed] [Google Scholar]
- 181.Umezawa S, Shibahara S, Omoto S, Takeuchi T, Umezawa H. J Antibiot. 1968;21:485. doi: 10.7164/antibiotics.21.485. [DOI] [PubMed] [Google Scholar]
- 182.Varoglu M, Mao Y, Sherman DH. J Am Chem Soc. 2001;123:6712. doi: 10.1021/ja015646l. [DOI] [PubMed] [Google Scholar]
- 183.Grüschow S, Chang LC, Mao Y, Sherman DH. J Am Chem Soc. 2007;129:6470. doi: 10.1021/ja0700193. [DOI] [PubMed] [Google Scholar]
- 184.Sitachitta N, Lopanik NB, Mao Y, Sherman DH. J Biol Chem. 2007;282:20941. doi: 10.1074/jbc.M702456200. [DOI] [PubMed] [Google Scholar]
- 185.Kishi Y. J Nat Prod. 1979;42:549. [Google Scholar]
- 186.Nakatsubo F, Cocuzza AJ, Keeley DE, Kishi Y. J Am Chem Soc. 1977;99:4835. doi: 10.1021/ja00456a056. [DOI] [PubMed] [Google Scholar]
- 187.Nakatsubo F, Fukuyama T, Cocuzza AJ, Kishi Y. J Am Chem Soc. 1977;99:8115. doi: 10.1021/ja00456a056. [DOI] [PubMed] [Google Scholar]
- 188.Fukuyama T, Nakatsubo F, Cocuzza AJ, Kishi Y. Tetrahedron Lett. 1977:4295. [Google Scholar]
- 189.Webb JS, Cosulich DB, Mowat JH, Patrick JB, Broschard RW, Meyer WE, Williams RP, Wolf CF, Fulmor W, Pidacks C, Lancaster JE. J Am Chem Soc. 1962;84:3185. [Google Scholar]
- 190.Webb JS, Cosulich DB, Mowat JH, Patrick JB, Lancaster JE, Broschard RW, Meyer WE, Williams RP, Wolf CF, Fulmor W, Pidackas C. J Am Chem Soc. 1962;84:3187. [Google Scholar]
- 191.Fukuyama T, Yang L. Tetrahedron Lett. 1986;27:6299. [Google Scholar]
- 192.Fukuyama T, Yang L. J Am Chem Soc. 1987;109:7881. [Google Scholar]
- 193.Fukuyama T, Yang L. J Am Chem Soc. 1989;111:8303. [Google Scholar]
- 194.Kono M, Saitoh Y, Shirahata K, Arai Y, Ishii S. J Am Chem Soc. 1987;109:7224. [Google Scholar]
- 195.Benbow JW, Schulte GK, Danishefsky SJ. Angew Chem Int Ed Engl. 1992;31:915. [Google Scholar]
- 196.Benbow JW, McClure KF, Danishefsky SJ. J Am Chem Soc. 1993;115:12305. [Google Scholar]
- 197.Wang Z, Jimenez LS. Tetrahedron Lett. 1996;37:6049. [Google Scholar]
- 198.Colandrea VJ, Rajaraman S, Jimenez LS. Org Lett. 2003;5:785. doi: 10.1021/ol026738y. [DOI] [PubMed] [Google Scholar]
- 199.Vedejs E, Little J. J Am Chem Soc. 2002;124:748. doi: 10.1021/ja0120835. [DOI] [PubMed] [Google Scholar]
- 200.Vedejs E, Naidu BN, Klapars A, Warner DL, Li Vs, Na Y, Kohn H. J Am Chem Soc. 2003;125:15796. doi: 10.1021/ja030452m. [DOI] [PubMed] [Google Scholar]
- 201.Bobeck DR, Warner DL, Vedejs E. J Org Chem. 2007;72:8506. doi: 10.1021/jo7013559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Vedejs E, Grissom JW. J Am Chem Soc. 1988;110:3238. [Google Scholar]
- 203.Vedejs E, Dax SL. Tetrahedron Lett. 1989;30:2627. [Google Scholar]
- 204.Vedejs E, Piotrowski DW. J Org Chem. 1993;58:1341. doi: 10.1021/jo0001277. [DOI] [PubMed] [Google Scholar]
- 205.Vedejs E, Monahan SD. J Org Chem. 1997;62:4763. [Google Scholar]
- 206.Vedejs E, Piotrowski DW, Tucci FC. J Org Chem. 200065:5498. doi: 10.1021/jo0001277. [DOI] [PubMed] [Google Scholar]
- 207.Warner DL, Hibberd AM, Kalman M, Klapars A, Vedejs E. J Org Chem. 2007;72:8519. doi: 10.1021/jo7013615. [DOI] [PubMed] [Google Scholar]
- 208.Williams AL, Srinivasan JM, Johnston JN. Org Lett. 2006;8:6047. doi: 10.1021/ol0624676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Cox AL, Johnston JN. Org Lett. 2001;3:3695. doi: 10.1021/ol016625z. [DOI] [PubMed] [Google Scholar]
- 210.Michael JP, de Koning CB, Petersen RL, Stanbury TV. Tetrahedron Lett. 2001;42:7513. [Google Scholar]
- 211.Michael JP, de Koning CB, Mudzunga TT, Petersen RL. Synlett. 2006:3284. [Google Scholar]
- 212.Dong W, Jimenez LS. J Org Chem. 1999;64:2520. [Google Scholar]
- 213.Murphy WS, O'Sullivan PJ. Tetrahedron Lett. 1992;33:531. [Google Scholar]
- 214.Ciufolini MA. Il Farmaco. 2005;60:627. doi: 10.1016/j.farmac.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 215.Ducray R, Ciufolini MA. Angew Chem Int Ed. 2002;41:4688. doi: 10.1002/anie.200290017. [DOI] [PubMed] [Google Scholar]
- 216.Coleman RS, Felpin FX, Chen W. J Org Chem. 2004;69:7309. doi: 10.1021/jo048924i. [DOI] [PubMed] [Google Scholar]
- 217.Papaioannou N, Blank JT, Miller SJ. J Org Chem. 2003;68:2728. doi: 10.1021/jo0269013. [DOI] [PubMed] [Google Scholar]
- 218.Miller SJ, Kim SH, Chen ZR, Grubbs RH. J Am Chem Soc. 1995;117:2108. [Google Scholar]
- 219.Fellows IM, Kaelin DE, Martin SF. J Am Chem Soc. 2000;122:10781. [Google Scholar]
- 220.Martin SF, Wagman AS. Tetrahedron Lett. 1995;36:1169. [Google Scholar]
- 221.Papaioannou N, Evans CA, Blank JT, Miller SJ. Org Lett. 2001;3:2879. doi: 10.1021/ol016372+. [DOI] [PubMed] [Google Scholar]
- 222.Copeland GT, Miller SJ. J Am Chem Soc. 2001;123:6496. doi: 10.1021/ja0108584. [DOI] [PubMed] [Google Scholar]
- 223.Jarvo ER, Copeland GT, Papaioannou N, Bonitatebus PJ, Miller SJ. J Am Chem Soc. 1999;121:11638. [Google Scholar]
- 224.Jarvo ER, Evans CA, Copeland GT, Miller SJ. J Org Chem. 2001;66:5522. doi: 10.1021/jo015803z. [DOI] [PubMed] [Google Scholar]
- 225.Tsuboike K, Guerin DJ, Mennen SM, Miller SJ. Tetrahedron. 2004;60:7367. [Google Scholar]
- 226.Allan GM, Parsons AF, Pons JFo. Synlett. 2002;2002:1431. [Google Scholar]
- 227.Dobbs AP, Jones K, Veal KT. Tetrahedron Lett. 1995;36:4857. [Google Scholar]
- 228.Wang Z, Jimenez LS. J Am Chem Soc. 1994;116:4977. [Google Scholar]
- 229.Wang Z, Jimenez LS. J Org Chem. 1996;61:816. doi: 10.1021/jo9511509. [DOI] [PubMed] [Google Scholar]
- 230.Zhang W, Wang C, Jimenez LS. Synth Commun. 2000;30:351. [Google Scholar]
- 231.Ziegler FE, Berlin MY. Tetrahedron Lett. 1998;39:2455. [Google Scholar]
- 232.Ziegler FE, Belema M. J Org Chem. 1997;62:1083. [Google Scholar]
- 233.Jones GB, Guzel M, Mathews JE. Tetrahedron Lett. 2000;411123 [Google Scholar]
- 234.Lee J, Ha JD, Cha JK. J Am Chem Soc. 1997;119:8127. [Google Scholar]
- 235.O. G. S. (a) Kulinkovich SV, Vasilevskii DA, Pritytskaya TS. Zh Org Khim. 1989;25:2244. [Google Scholar]; (b) Kulinkovich OG, Sviridov SV, Vasilevskii DA, Savchenko AI, Pritytskaya TS. Zh Org Khim. 1991;27:294. [Google Scholar]; (c) Kulinkovich OG, Sviridov SV, Vasilevskii DA. Synthesis. 1991;234 [Google Scholar]; (d) Kulinkovich OG, Savchenko AI, Sviridov SV, Vasilevskii DA. Mendeleev Commun. 1993;192 and references cited therein. [Google Scholar]; (e) de Meijere A, Kozhushkov SI, Spaeth T, Zefirov NS. J Org Chem. 1993;502 [Google Scholar]; (f) Chaplinski V, de Meijere A. Angew Chem Int Ed Engl. 1996;35:413. [Google Scholar]; (g) Corey EJ, Rao SA, Noe MC. J Am Chem Soc. 1994;116:9345. [Google Scholar]; (h) Kasatkin A, Sato F. Tetrahedron Lett. 1995;36:6079. [Google Scholar]; (i) Kasatkin A, Kobayashi K, Okamoto S, Sato F. Tetrahedron Lett. 1996;37:1849. [Google Scholar]
- 236.Lim HJ, Sulikowski GA. J Org Chem. 1995;60:2326. [Google Scholar]
- 237.Ban Y, Nakajima S, Yoshida K, Mori M, Shibazaki M, Date T. Heterocycles. 1994;39:657. [Google Scholar]
- 238.Nakajima S, Yoshida K, Mon M, Ban Y, Shibasaki M. J Chem Soc, Chem Commun. 1990:468. [Google Scholar]
- 239.Takahashi K, Kametani T. Heterocycles. 1979;13:411. [Google Scholar]
- 240.Kametani T, Takahashi K. Heterocycles. 1978;9:293. [Google Scholar]
- 241.Kametani T, Ohsawa T, Takahashi K, Ihara M, Fukumoto K. Heterocycles. 1976;4:1637. [Google Scholar]
- 242.Ihara M, Takahashi K, Kigawa Y, Ohsawa T, Fukumoto K, Kametani T. Heterocycles. 1977;6:1658. [Google Scholar]
- 243.Kametani T, Kigawa Y, Nemoto H, Ihara M, Fukumoto K. Heterocycles. 1980;14:799. [Google Scholar]
- 244.Kametani T, Takahashi K, Ihara M, Fukumoto K. Heterocycles. 1977;6:1371. [Google Scholar]
- 245.Kametani T, Takahashi K, Ihara M, Fukumoto K. Heterocycles. 1978;9:435. [Google Scholar]
- 246.Kametani T, Ohsawa T, Ihara M. Heterocycles. 1979;12:913. [Google Scholar]
- 247.Kametani T, Takahashi K, Ihara M, Fukumoto K. Heterocycles. 1979;12:933. [Google Scholar]
- 248.Danishefsky S, Regan J, Doehner R. J Org Chem. 1981;46:5255. [Google Scholar]
- 249.Danishefsky S, Doehner R. Tetrahedron Lett. 1977;18:3031. [Google Scholar]
- 250.Sharpless KB, Lauer RF. J Org Chem. 1974;39:429. [Google Scholar]
- 251.Danishefsky S, Berman EM, Ciufolini M, Etheredge SJ, Segmuller BE. J Am Chem Soc. 1985;107:3891. [Google Scholar]
- 252.Nicolaou KC. Tetrahedron. 1981;37:4097. [Google Scholar]
- 253.Allen GR. Org React. 1973;20:337. [Google Scholar]
- 254.Coates RM, MacManus PA. J Org Chem. 1982;47:4822. [Google Scholar]
- 255.Allen GR, Poletto JF, Weiss MJ. J Org Chem. 1965;30:2897. doi: 10.1021/jo01020a006. [DOI] [PubMed] [Google Scholar]
- 256.Coates RM, Hutchins CW. J Org Chem. 1979;44:4742. [Google Scholar]
- 257.Defoin A, Fritz H, Geffroy G, Streith J. Tetrahedron Lett. 198627:3135. [Google Scholar]
- 258.Remers WA, Roth RH, Weiss MJ. J Am Chem Soc. 1964;86:4612. [Google Scholar]
- 259.Remers WA, Roth RH, Weiss MJ. J Org Chem. 1965;30:2910. doi: 10.1021/jo01020a008. [DOI] [PubMed] [Google Scholar]
- 260.Leadbetter G, Fost DL, Ekwuribe NN, Remers WA. J Org Chem. 1974;39:3580. doi: 10.1021/jo00938a028. [DOI] [PubMed] [Google Scholar]
- 261.Taylor WG, Leadbetter G, Fost DL, Remers WA. J Med Chem. 1977;20:138. doi: 10.1021/jm00211a029. [DOI] [PubMed] [Google Scholar]
- 262.Kusama H, Miyashita Y, Takaya J, Iwasawa N. Org Lett. 2005;8:289. doi: 10.1021/ol052610f. [DOI] [PubMed] [Google Scholar]
- 263.Takada T, Sadao O. Chem Pharm Bull. 1971;19:977. [Google Scholar]
- 264.Takada T, Sachiko K, Sadao O. Chem Pharm Bull. 1971;19:982. [Google Scholar]
- 265.Takada T, Akiba M. Chem Pharm Bull. 1972;20:1785. [Google Scholar]
- 266.Akiba M, Kosugi Y, Takada T. Heterocycles. 1977;6:1125. [Google Scholar]
- 267.Akiba M, Kosugi Y, Okuyama M, Takada T. Heterocycles. 1977;6:1113. [Google Scholar]
- 268.Akiba M, Kosugi Y, Takada T. J Org Chem. 1978;43:4472. [Google Scholar]
- 269.Akiba M, Kosugi Y, Takada T. Heterocycles. 1978;9:1607. [Google Scholar]
- 270.Akiba M, Kosugi Y, Okuyama M, Takada T. J Org Chem. 1978;43:181. [Google Scholar]
- 271.Akiba M, Takada T. Heterocycles. 1977;6:1861. [Google Scholar]
- 272.Kim M, Vedejs E. J Org Chem. 2004;69:7262. doi: 10.1021/jo040211c. [DOI] [PubMed] [Google Scholar]
- 273.Vedejs E, Little JD. J Org Chem. 2004;69:1794. doi: 10.1021/jo030223i. [DOI] [PubMed] [Google Scholar]
- 274.G. M. (a) Knaus AI. J Org Chem. 1974;39:1192. [Google Scholar]; (b) Jacobs SA, Cortez C, Harvey RG. J Chem Soc, Chem Commun. 1981:1215. [Google Scholar]; (c) Hoppe D, Paetow M, Hintze F. Angew Chem, Int Ed Engl. 1993;32:394. [Google Scholar]; (d) Hoppe D, Kaiser B. Angew Chem, Int Ed Engl. 1995;34:323. [Google Scholar]; (e) Kopach ME, Meyers AI. J Org Chem. 1996;61:6764. doi: 10.1021/jo961272g. [DOI] [PubMed] [Google Scholar]; (f) Ahmed A, Clayden J, Rowley M. Tetrahedron Lett. 1998;39:6103. [Google Scholar]; (g) Clayden J, Pink JH, Westlund N, Wilson FX. Tetrahedron Lett. 1998;39:8377. [Google Scholar]; (h) Anderson DR, Faibish NC, Beak P. J Am Chem Soc. 1999;121:7553. [Google Scholar]; (i) Mathieu J, Gros P, Fort Y. Chem Commun. 2000:951. [Google Scholar]
- 275.Vedejs E, Little JD, Seaney LM. J Org Chem. 2004;69:1788. doi: 10.1021/jo030224a. [DOI] [PubMed] [Google Scholar]
- 276.Meth-Cohn S, H Adv Heterocycl Chem. 1972:14. [Google Scholar]
- 277.Dijksman WC, Verboom W, Egberink RJM, Reinhoudt DN. J Org Chem. 1985;50:3791. [Google Scholar]
- 278.Reinhoudt DN, Visser GW, Verboom W, Benders PH, Pennings MLM. J Am Chem Soc. 1983;105:4775. [Google Scholar]
- 279.Verboom W, Lammerink BHM, Egberink RJM, Reinhoudt DN, Harkema S. J Org Chem. 1985;50:3797. [Google Scholar]
- 280.Verboom W, Orlemans EOH, Berga HJ, Scheltinga HW, Reinhoudt DN. Tetrahedron. 1986;42:5053. [Google Scholar]
- 281.Kozikowski AP, Mugrage BB. J Chem Soc, Chem Commun. 1988:198. [Google Scholar]
- 282.Bailey AS, Scott PW, Vandrevala MH. J Chem Soc, Perkin Trans 1. 1980 [Google Scholar]
- 283.Falling SN, Rapoport H. J Org Chem. 1980;45:1260. [Google Scholar]
- 284.Luly JR, Rapoport H. J Org Chem. 1982;47:2404. [Google Scholar]
- 285.Luly JR, Rapoport H. J Am Chem Soc. 1983;105:2859. [Google Scholar]
- 286.Luly JR, Rapoport H. J Org Chem. 1984;49:1671. [Google Scholar]
- 287.Shaw KJ, Luly JR, Rapoport H. J Org Chem. 1985;50:4515. [Google Scholar]
- 288.Luly JR, Rapoport H. J Org Chem. 1981;46:2745. [Google Scholar]
- 289.Shirahata K, Hirayama N. J Am Chem Soc. 1983;105:7199. [Google Scholar]
- 290.Hornemann U, Heins MJ. J Org Chem. 1985;50:1301. [Google Scholar]
- 291.Flitsch W, Lauterwein J, Micke W. Tetrahedron Lett. 1989;30:1633. [Google Scholar]
- 292.Karl Heinz D. Angew Chem Int Ed Engl. 1984;23:587. [Google Scholar]
- 293.Flitsch W, Rußkamp P, Langer W. Liebigs Ann Chem. 1985:1422. [Google Scholar]
- 294.Flitsch W, Langer W. Liebigs Ann Chem. 1988:391. [Google Scholar]
- 295.Flitsch W, Rußkamp P. Heterocycles. 1984;22:541. [Google Scholar]
- 296.Franck RW, Auerbach J. J Org Chem. 1971:36, 31. [Google Scholar]
- 297.Mazzola VJ, Bernady KF, Franck RW. J Org Chem. 1967;32:486. [Google Scholar]
- 298.Franck RW, Bernady KF. J Org Chem. 1968;33:3050. doi: 10.1021/jo01272a007. [DOI] [PubMed] [Google Scholar]
- 299.Scheiner P. J Org Chem. 1965;30:7. doi: 10.1021/jo01012a002. [DOI] [PubMed] [Google Scholar]
- 300.Scheiner P. Tetrahedron. 1968;24:2757. [Google Scholar]
- 301.Siuta GP, Franck RW, Kempton RP. J Org Chem. 1974;39:3739. doi: 10.1021/jo00939a029. [DOI] [PubMed] [Google Scholar]
- 302.Padwa A, Hamilton L. J Am Chem Soc. 1967;89:102. [Google Scholar]
- 303.Weider PR, Hegedus LS, Asada H. J Org Chem. 1985;50:4276. [Google Scholar]
- 304.Naruta Y, Nagai N, Maruyama K. J Chem Soc, Perkin Trans. 1988;1:1143. [Google Scholar]
- 305.Asano O, Nakatsuka Si, Goto T. Heterocycles. 1987;26:1207. [Google Scholar]
- 306.Rebek JJ, Shaber SH. Heterocycles. 1981;16:1173. [Google Scholar]
- 307.Rebek J, Jr, Gehret JCE. Tetrahedron Lett. 1977;18:3027. [Google Scholar]
- 308.Hershenson FM. J Org Chem. 1975;40:1260. [Google Scholar]
- 309.Rebek J, Shaber SH, Shue YK, Gehret JC, Zimmerman S. J Org Chem. 1984;49:5164. [Google Scholar]
- 310.Wender PA, Cooper CB. Tetrahedron Lett. 1987;28:6125. [Google Scholar]
- 311.Edstrom ED, Tao Y. Tetrahedron. 1997;53:4549. [Google Scholar]
- 312.Friary RJ, Gilligan JM, Szajewski RP, Falci KJ, Franck RW. J Org Chem. 1973;38:3487. [Google Scholar]
- 313.Fukuyama T, Xu L, Goto S. J Am Chem Soc. 1992;114:383. [Google Scholar]
- 314.Kishi Y. J Nat Prod. 1979;42:549. [Google Scholar]
- 315.Fukuyama T, Yang L. J Am Chem Soc. 1989;111:8303. [Google Scholar]
- 316.Schkeryantz JM, Danishefsky SJ. J Am Chem Soc. 1995;117:4722. [Google Scholar]
- 317.T. I. (a) Katoh E, Yoshino T, Terashima S. Tetrahedron Lett. 1996;37:3471. [Google Scholar]; (b) Yoshino T, Nagata Y, Itoh E, Hashimoto M, Katoh T, Terashima S. Tetrahedron Lett. 1996;37:3478. [Google Scholar]; (c) Katoh T, Yoshino T, Nagata Y, Nakatani S, Terashima S. Tetrahedron Lett. 1996;37:3479. [Google Scholar]; (d) Katoh T, Itoh E, Yoshino T, Terashima S. Tetrahedron. 1997;53:10229. [Google Scholar]; (e) Yoshino T, Nagata Y, Itoh E, Hashimoto M, Katoh T, Terashima S. Tetrahedron. 1997;53:10239. [Google Scholar]; (f) Katoh T, Nagata Y, Yoshino T, Nakatani S, Terashima S. Tetrahedron. 1997;53:10253. [Google Scholar]; (g) Katoh T, Terashima S. J Synth Org Chem, Jpn. 1997;55:946. [Google Scholar]
- 318.Suzuki M, Kambe M, Tokuyama H, Fukuyama T. Angew Chem Int Ed. 2002;41:4686. doi: 10.1002/anie.200290016. [DOI] [PubMed] [Google Scholar]
- 319.Judd TC, Williams RM. Angew Chem Int Ed. 2002;41:4683. doi: 10.1002/anie.200290015. [DOI] [PubMed] [Google Scholar]
- 320.H. C. a) Scheidt KA, Follows BC, Chemler SR, Coffey DS. J Org Chem W R Roush. 63:6436. [Google Scholar]; b) Kurosu M, R L, Marcin TJG, Kishi Y. J Am Chem Soc. 1998;120:6627. [Google Scholar]; I. N. c) Noyori R, Sakata J. J Am Chem Soc. 1983;105:1598. [Google Scholar]
- 321.J. C. M. a) Dess DB. J Org Chem. 1983;48:4155. [Google Scholar]; b) Dess DB, Martin JC. J A C S. 113:7277. [Google Scholar]; c) Ireland RE, Liu L. J O C . 58:2899. [Google Scholar]
- 322.Rodriguez L, Lu N, Yang NL. Synlett. 1990;1990:227. [Google Scholar]
- 323.Murray RW, Singh M. Org Synth, Vol Collect Vol IX. Wiley; New York: 1998. p. 228. [Google Scholar]
- 324.Curci R, D'Accolti L, Detomaso A, Fusco C, Takeuchi Ki, Ohga Y, Eaton PE, Chi Yip Y. Tetrahedron Lett. 1993;34:4559. [Google Scholar]
- 325.Kocovský P. Tetrahedron Lett. 1986;27:5521. [Google Scholar]
- 326.Hanessian S, Delorme D, Dufresne Y. Tetrahedron Lett. 1984;25:2515. [Google Scholar]
- 327.Couturier M, Tucker JL, Andresen BM, Dubé P, Negri JT. Org Lett. 2001;3:465. doi: 10.1021/ol006969+. [DOI] [PubMed] [Google Scholar]
- 328.Omura K, Swern D. Tetrahedron. 1978;34:1651. [Google Scholar]
- 329.Ireland RE, Norbeck DW. J Org Chem. 1985;50:2198. [Google Scholar]
- 330.Jones RJ, Rapoport H. J Org Chem. 1990:55, 1144. [Google Scholar]
- 331.Paleo MR, Aurrecoechea N, Jung KY, Rapoport H. J Org Chem. 2002;68:130. doi: 10.1021/jo0206521. [DOI] [PubMed] [Google Scholar]
- 332.Trost BM, O'Boyle BM. Org Lett. 2008;10:1369. doi: 10.1021/ol800127a. [DOI] [PubMed] [Google Scholar]
- 333.Trost BM, Ameriks MK. Org Lett. 2004;6:1745. doi: 10.1021/ol049583y. [DOI] [PubMed] [Google Scholar]
- 334.Grierson D. Org React. 1990;39:85. [Google Scholar]
- 335.Judd TC, Williams RM. Org Lett. 2002;4:3711. doi: 10.1021/ol0266774. [DOI] [PubMed] [Google Scholar]
- 336.Rollins SB, Williams RM. Tetrahedron Lett. 1997;38:4033. [Google Scholar]
- 337.Williams RM, Rollins SB, Judd TC. Tetrahedron. 2000;56:521. [Google Scholar]
- 338.Spada MR, Ubukata M, Isono K. Heterocycles. 1992;34:1147. [Google Scholar]
- 339.Rodriguez L, Lu N, Yang NL. Synlett. 1990;1990:227. [Google Scholar]
- 340.Dmitrienko GI, Denhart D, Mithani S, Prasad GKB, Taylor NJ. Tetrahedron Lett. 1992;33:5705. [Google Scholar]
- 341.Lim HJ, Sulikowski GA. Tetrahedron Lett. 1996;37:5243. [Google Scholar]
- 342.Hootelé C. Tetrahedron Lett. 1969;10:2713. [Google Scholar]
- 343.Ziegler FE, Jeroncic LO. J Org Chem. 1991;56:3479. [Google Scholar]
- 344.Ziegler FE, Harran PG. J Org Chem. 1993;58:2768. [Google Scholar]
- 345.Ziegler FE, Belema M. J Org Chem. 1994;59:7962. [Google Scholar]
- 346.Mithani S, Drew DM, Rydberg EH, Taylor NJ, Mooibroek S, Dmitrienko GI. J Am Chem Soc. 1997;119:1159. [Google Scholar]
- 347.Ian SY, Michael AK. Angew Chem Int Ed. 2003;42:3023. doi: 10.1002/anie.200351573. [DOI] [PubMed] [Google Scholar]
- 348.Young IS, Kerr MA. Org Lett. 2003;6:139. doi: 10.1021/ol0362919. [DOI] [PubMed] [Google Scholar]
- 349.Reddy VK, Miyabe H, Yamauchi M, Takemoto Y. Tetrahedron. 2008;64:1040. [Google Scholar]
- 350.Yasuda N, Williams RM. Tetrahedron Lett. 1989;30:3397. [Google Scholar]
- 351.Tepe JJ, Williams RM. Angew Chem Int Ed. 1999;38:3501. doi: 10.1002/(sici)1521-3773(19991203)38:23<3501::aid-anie3501>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]