

Abstract
Microtubule-poisoning drugs, such as Paclitaxel (or Taxol, PTX), are powerful and commonly used anti-neoplastic agents for the treatment of several malignancies. PTX triggers cell death, mainly through a mitotic arrest following the activation of the spindle assembly checkpoint (SAC). Cells treated with PTX slowly slip from this mitotic block and die by mitotic catastrophe. However, cancer cells can acquire or are intrinsically resistant to this drug, posing one of the main obstacles for PTX clinical effectiveness. In order to override PTX resistance and increase its efficacy, we investigated both the enhancement of mitotic slippage and the block of mitotic exit.
To test these opposing strategies, we used physiological hyperthermia (HT) to force exit from PTX-induced mitotic block and the anaphase-promoting complex/cyclosome (APC/C) inhibitor, proTAME, to block mitotic exit. We observed that application of HT on PTX-treated cells forced mitotic slippage, as shown by the rapid decline of cyclin B levels and by microscopy analysis. Similarly, HT induced mitotic exit in cells blocked in mitosis by other antimitotic drugs, such as Nocodazole and the Aurora A inhibitor MLN8054, indicating a common effect of HT on mitotic cells. On the other hand, proTAME prevented mitotic exit of PTX and MLN8054 arrested cells, prolonged mitosis, and induced apoptosis. In addition, we showed that proTAME prevented HT-mediated mitotic exit, indicating that stress-induced APC/C activation is necessary for HT-induced mitotic slippage.
Finally, HT significantly increased PTX cytotoxicity, regardless of cancer cells’ sensitivity to PTX, and this activity was superior to the combination of PTX with pro-TAME. Our data suggested that forced mitotic exit of cells arrested in mitosis by anti-mitotic drugs, such as PTX, can be a more successful anticancer strategy than blocking mitotic exit by inactivation of the APC/C.
Keywords: mitotic block, mitotic catastrophe, hyperthermia, APC/C, proTAME, taxanes, aurora A inhibitors, antimitotic drug resistance
Introduction
Paclitaxel (PTX), a taxane that was originally isolated from the Pacific yew, belongs to a family of drugs known as mitotic poisons.1 PTX is one of the most commonly used drugs for the treatment of several human malignancies, including breast, ovarian, lung, Kaposi sarcoma, and head and neck cancers.2,3 PTX cytotoxic activity relies on the molecule ability to bind reversibly tubulin, with a net effect of microtubule hyperstabilization.2 The following inhibition of microtubules dynamics leads to the activation of the spindle assembly checkpoint (SAC), which prompts a persistent mitotic arrest. Cells sensitive to the action of PTX will not be able to satisfy the SAC and eventually will slip through mitosis by a mechanism known as mitotic catastrophe, an event biochemically characterized by slow and steady degradation of one of the anaphase-promoting complex/cyclosome (APC/C) substrates, cyclin B.4,5 The mechanism of this degradation in the presence of an active SAC is not yet understood; however, it is well established that as soon as levels of cyclin B (among other APC/C substrates) drop below a threshold, cells exit mitosis in an aberrant G1 stage as micronucleated. These cells often fail the next round of cell division by undergoing apoptosis, necrosis, or senescence.6-8 The mitotic slippage and subsequent catastrophe seems to be in competition with the apoptotic machinery that instead prompts cell death directly from mitosis.9-11 Thus, a cell treated with PTX will eventually succumb to mitotic slippage or apoptosis.
Mutations in these pathways, which are necessary for mitotic cell death, as well as defects in the SAC and cell cycle regulation, are among the most common causes of PTX resistance.12-14 Indeed, up to 50% of cancer patients are resistant or become resistant to PTX during drug administration.15-17 Therefore, it is compelling to overcome taxane resistance or to find alternative therapeutic strategies that would kill tumor cells more efficiently than taxanes. Over the past decades great translational effort has been directed toward the development of drugs that target the mitotic spindle assembly or function (such as Eg5, Aurora, Plk1, or cyclin-dependent kinases). Such drugs have shown scarce efficacy in clinical application in spite of outstanding results in preclinical studies.18-23 Thus, despite of the toxicity and high resistance rates, taxanes are a mainstay in the clinical landscape.24
In order to overcome resistance mechanisms and more efficiently kill tumor cells, it was recently proposed to target mitotic exit.25-28 Therefore, we decided to compare two opposing strategies to synergize with PTX and potentially circumvent PTX resistance, one that would cause mitotic exit and another that would enhance the mitotic block.
As first strategy, we forced mitotic exit using physiological hyperthermia (HT, also referred as heat shock) to promote mitotic slippage from PTX-induced mitotic block. We observed that mild hyperthermia (1–2 h at 42 °C) induced an exit from mitosis, in agreement with reported effects of hyperthermia on the disruption of microtubules network and microtubule organizing centers.29 These results prompted us to further investigate the efficacy of HT to efficiently induce mitotic exit from the drug-mediated mitotic arrest.
Physiological hyperthermia (39.5–45 °C) has been used as enhancer of radiotherapy and of many chemotherapeutic agents,30 including PTX,31-33 with limited toxicity to normal tissues.34 Hyperthermia is mainly applied locally, regionally, interstitially, or to the whole body according to the tumor type, stage, depth, localization, and presence of spread metastatic disease.35 The interest for the use of hyperthermia in cancer treatment arose more than 30 years ago, but its application in clinics has been limited, until recent years, mainly due to the lack of appropriate equipment.36 Another reason for the hesitancy toward the use of hyperthermia, alone or as PTX sensitizer, can be attributed to conflicting outcomes of preclinical studies.31,37-39 However, these differences can be explained by the substantial variations in the HT protocols applied (e.g., temperature, length of HT treatment, concentration, and length of PTX exposure).
As a second and alternative strategy of mitosis manipulation, we decided to block mitotic exit. This approach was chosen based on recent findings indicating that block of mitotic exit downstream of the SAC, by Cdc20 silencing, efficiently killed mitotic cells.27 Cdc20 silencing caused SAC-independent cell death by apoptosis from mitosis, which would seem optimal to circumvent PTX resistance by defective SAC.27 For these reasons, to block mitotic exit, we opted for the pharmacological inhibition of the APC/C with a previously characterized inhibitor, proTAME.40
Since manipulation of mitotic block seems to be a promising approach to kill cancerous cells, in this paper we attempted to answer the questions: What is the best strategy to induce mitotic death, by extending the mitotic block or by enhancing mitotic exit? Which one would synergize best with antimitotic drugs? Our results, produced on several cell lines, including PTX-resistant breast cancer cells, suggest that forced mitotic exit could be the preferred strategy to elevate cytotoxic effect of PTX treatment.
Results
Hyperthermia (HT) forces mitotic catastrophe in PTX arrested cells
Recent studies have suggested that targeting the mitotic block in cells treated with antimitotic drugs can elevate chemotherapy efficacy.27 We decided to test this model by identifying conditions to induce mitotic exit. We found that HEp2 cells treated with PTX, were rapidly exiting mitosis after HT application. Microscopy analysis showed that, after undergoing HT treatment, cells were forced out of mitosis as micronucleated rather than apoptotic (Fig. 1, right panel), while cells that were exposed to PTX for 18 hours (h) at 37 °C were blocked in mitosis (Fig. 1, central panel). Next, we examined several conditions (Fig. 1B) in which we tested PTX washout (W) and several time points of post-HT recovery at 37 °C (R). We observed that the formation of micronuclei (MN) did not occur immediately after HT application; it started at 2 h and was completed by 4 h of HT recovery (Fig. 1B and C). The maximal HT effect, measured as MN percentile, was observed after exposing cells to PTX for 12 h, stressed by HT for 2 h, and released for 4 h at 37 °C. Almost no MN and/or apoptosis were detected in samples that were treated for the same time with PTX only. At 18 h of PTX treatment, cells were still blocked in mitosis as shown by DNA staining (Fig. 1A), analysis of morphology (Fig. 1B), and immunofluorescence staining (Fig. 1C). Our results indicated that HT forced mitotic exit of PTX-treated cells toward mitotic slippage/catastrophe, resulting in formation of MN. Similar results were obtained whether PTX was washed or remained in culture media during HT and recovery (Fig. 1B), suggesting that the presence of PTX in the media is not necessary for HT-induced mitotic slippage. We also observed that post-stress recovery at 37 °C is necessary to complete the slippage process, as (1) minimal MN formation was observed immediately after HT application, and (2) extended time of HT treatment did not result in MN formation (not shown), indicating that HT recovery is a prerequisite to the exit process.
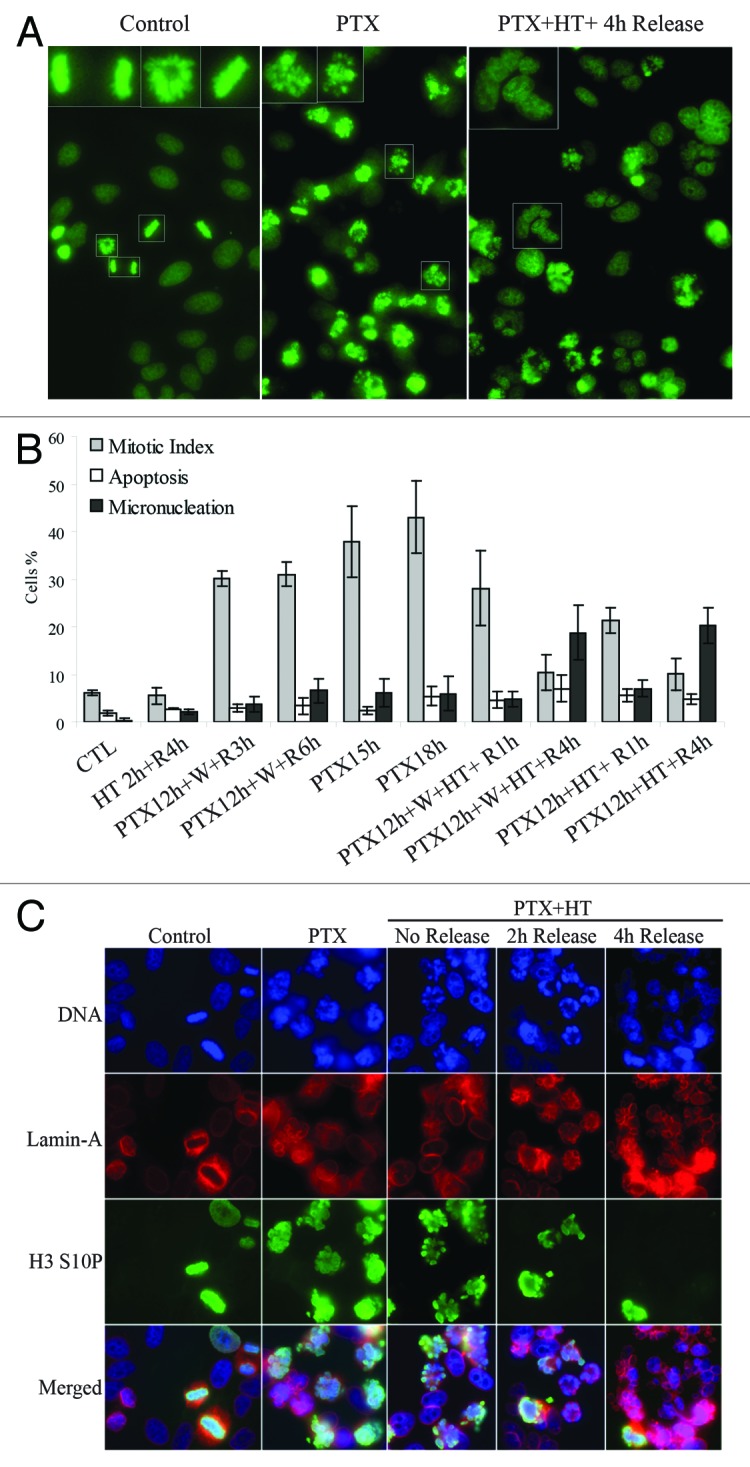
Figure 1. HT forces mitotic catastrophe in PTX-arrested cells. (A) DNA staining of HEp2 cells (control, left) treated with 10 nM PTX for 18 hours (h) at 37 °C (PTX) or treated with PTX at 37 °C for 12 h followed by a 42 °C HT for 2 h and returned at 37 °C for 4 h (PTX + HT + 4 h release). (B) Cellular morphology was analyzed to study the effects of PTX and HT. PTX treatment: 15 or 18 h, HT: 2 h, 42 °C; R, release at 37 °C for 1–6 h; W, PTX washout. At least 500 cells per sample were counted, (SD ± 3). HT forces mitotic exit and induces micronucleation in PTX-arrested cells. (C) Detailed analysis of HT-induced mitotic catastrophe. Immunoflourescence staining of HEp2 cells treated as in (A); Lamin A, marker of nuclear envelope; H3 S10P, mitotic marker H3 phospho-serine 10.
HT forces mitotic exit of cells treated with anti-mitotics by activating mitotic catastrophe
To test whether HT-induced mitotic exit is unique in cells treated with the microtubule hyperpolymerizer PTX or occurs in presence of other anti-mitotic drugs, we decided to test the Aurora A kinase inhibitor MLN805441 and the microtubule-depolymerizing agent Nocodazole (Noc). Cells were first treated with these drugs for 15 h and then were exposed to HT for 2 h at 42 °C and returned at 37 °C for additional 4 h. We chose to monitor cyclin B levels, as an APC/C substrate, to follow mitotic dynamics. Our analysis revealed that HT triggered degradation of this protein in cells arrested in mitosis by all three antimitotic drugs (Fig. 2A). In addition, we did not observe substantial apoptosis elevation in post-HT cells, as marked by the absence of cleaved Poly [ADP-ribose] polymerase 1 (PARP, Fig. 2A). These findings were confirmed by microscopy analysis of mitotic index (MI), apoptosis, and MN. Application of HT forced cells arrested in mitosis by any of these drugs (see 15 h time points, Fig. 2B) to exit mitosis as micronucleated rather than apoptotic (see 15 h + HT + R 4 h time points, Fig. 2B). The mitotic index in these samples strikingly dropped compared with the pre-HT samples or to cells that were kept at 37 °C for the same treatment times, where very little apoptosis and MN were observed (see 21 h time points, Fig. 2B). These results indicate that HT significantly shortens the time of mitotic block regardless of the anti-mitotic drug used.

Figure 2. In cells treated with anti-mitotic drugs, HT forces exit toward mitotic catastrophe. (A) Western blot analysis of PARP cleavage and cyclin B (CycB) in HEp2 cells treated for a total time of 21 h with paclitaxel (PTX), Nocodazole (Noc), or MLN8054, a selective Aurora A kinase inhibitor. After 15 h of treatment some samples were exposed to 2 h heat shock at 42 °C (HT) and then returned to 37 °C for 4 h (R4h). Treatment with Doxorubicin and Etoposide induce apoptosis and were used as positive controls of PARP cleavage. (B) Average values for microscopy analysis of mitotic index, apoptosis, and mitotic catastrophe (as micronucleation). Microscopy analysis based on DNA morphology of HEp2 cells that were treated as in (A). At least 500 cells were counted for each sample in 3 independent experiments.
proTAME delays mitotic exit of cells treated with anti-mitotics and prompts apoptosis
To test the effects of a prolonged mitotic arrest, we decided to employ proTAME, a small-molecule inhibitor of the APC/C,40 which we expected to prevent mitotic slippage in cells treated with anti-mitotic drugs. HEp2 cells were treated with PTX or MLN8054 for 18 and 8 h, respectively, which were the times of highest mitotic accumulation according to cell cycle and mitotic index analyses (Fig. S1). proTAME was then added for 6, 12 and 18 h, timings which caused maximal mitotic arrest in proTAME-treated cells (Fig. S2). Addition of proTAME to cells pre-treated with PTX and MLN8054 caused a further accumulation of these cells in mitosis, as shown by slowed cyclin B degradation (Fig. 3A; Fig. S3A and B) and increase of the mitotic index (Fig. 3B). As expected, proTAME prevented mitotic exit by mitotic slippage as shown by the decrease of MN in PTX-treated cells. Next, we analyzed mitotic timing of cells treated with PTX and MLN8054 and in combination with proTAME. Average mitotic times of cells exposed to PTX and MLN8054 alone were 378 and 215 min, respectively, compared with 56 min of untreated cells (Table S1). APC/C inhibition with proTAME combined with PTX and MLN8054 prolonged mitotic timings to 543 and 270 min, respectively (Fig. 3C; Table S1).

Figure 3. proTAME delays mitotic exit of cells treated with anti-mitotic drugs. (A) Cyclin B stability evaluated in HEp2 cells treated with PTX (left panel) or MLN8054 (right panel) with or without the addition during the last 6, 12, or 18 h of treatment of the APC inhibitor proTAME at 12 µM by western blot analysis (see Fig. S3 for proTAME treatment dynamics). Graphs represent cyclin B values normalized over Actin and the control sample. (B) Mitotic index was evaluated by DNA staining to study the effects of 12 µM proTAME alone for 6 h, 10 nM PTX alone for 24 h, 4 µM MLN8054 for 14 h and 24 h PTX or 14 h MLN8054, where proTAME was added for the last 6 h of treatment. Figure represents the average counts of 5 fields. (C) Left: scatter plots representing mitotic timing monitored by time-lapse microscopy of GFP-H2B HEp2 cells pre-treated with PTX for 12 h in presence or absence of proTAME (P value < 0.001, ± SEM). Right: scatter plots representing mitotic timing of GFP-H2B HEp2 cells pre-treated with MLN8054 for 8 h in presence or absence of proTAME (P value < 0.05, ± SEM).
Our study revealed also that proTAME reduced mitotic slippage in PTX- and MLN8054-treated cells and activated apoptosis. The addition of proTAME increased the percentage of apoptotic events from 6.7 to 63.3% for PTX and from 0 to 22% in MLN8054-treated cells (Table S1). Interestingly, apoptosis occurred mainly in cells with the longest mitotic times (Fig. S3C).
proTAME blocks HT induced mitotic exit
Mitotic slippage occurs trough degradation of APC/C substrates, such as cyclin B.4,5,42 We showed that HT accelerated mitotic slippage in PTX-arrested cells, and that post-stress recovery at 37 °C is necessary to complete the slippage process, including cyclin B degradation. We next asked whether APC/C controls HT-mediated mitotic slippage. To answer this question, proTAME was added to HEp2 cells pre-treated with PTX for 12 h in absence of HT (Fig. 4, PTX + proTAME 5 or 6 h) or during 1 or 2 h of HT (Fig. 4, PTX + HT2h + proTAME 5 h or proTAME 6 h). Microscopy analysis of MN and mitotic index (MI) (Fig. 4A) showed that proTAME prevented HT-induced micronucleation in PTX-treated cells (Figs. 1C and 2B). These results were confirmed by the study of cyclin B levels (Fig. 4B), where proTAME addition stopped the HT-mediated cyclin B degradation that we observed after HT exposure of cells arrested in mitosis by PTX (Fig. 2A). Our results indicated that APC/C-dependent proteolysis is required to achieve mitotic slippage/micronucleation in cells that underwent hyperthermia.

Figure 4. proTAME blocks HT induced mitotic exit. (A) Microscopy analysis based on DNA morphology of mitotic index (MI) and mitotic catastrophe (MN). HEp2 cells were treated with 12 µM proTAME for 5 and 6 h or 10 nM PTX for a total time of 18 h. After 12 or 13 h of PTX exposure proTAME was added and cells were heat shocked for 1 or 2 h at 42 °C and then returned to 37 °C for additional 4 h. A minimum of 1000 cells were counted for each sample. (B) Western blot analysis of cyclin B stability in cells treated as in (A). Addition of proTAME inhibits HT induced mitotic exit by micronucleation.
HT increases PTX cytotoxicity
We observed that HT forced mitotic slippage of PTX-treated cells, suggesting that HT increases drug cytotoxicity. Therefore we next sought to understand whether HT combined with PTX could trigger cell death in HEp2 cells. We compared cell viability among cells that were treated with PTX and those that were exposed to HT at the end of PTX treatment. Cell exposure to HT after PTX pre-treatment significantly increased cell death in colony formation assay (Fig. 5A) compared with PTX or HT alone. HT cytotoxic effect was much higher at the end of PTX treatment, when cells were arrested in mitosis, compared with HT application in the first 2 h of PTX treatment, when mitotic index is low. Our results suggest that treatment with HT is much more efficient when cells are accumulated in mitosis by anti-mitotic drugs (Fig. 1B), as this cell cycle stage is extremely sensitive to HT (Fig. 5A). In addition, we observed that application of HT significantly decreased viability of cells resistant to PTX, such as the breast cancer cell line T47D43 (Fig. S4A).

Figure 5. HT increases significantly PTX cytoxicity. (A) Colony formation assay of HEp2 cells treated with 10 nM PTX for a total time of 18 h with or without heat shock for 1 or 2 h at the end of PTX treatment or with 2 h HT at the beginning of PTX treatment (SD ± 3). (B) Survival assay of MCF7 cells, wild type (WT) and a taxane-resistant (TR) derivative,43 heat shocked as in (A) and treated with 12 µM proTAME alone or in combination with PTX (SD ± 3).
Recent studies have proposed that targeting mitotic exit should be a more promising strategy to eradicate tumor cells.25-28 Therefore we next sought to investigate which one of the two approaches would kill cells more efficiently: (1) forcing mitotic exit with HT or (2) prolonging the mitotic block with inhibition of APC/C. To this end, we employed the MCF-7 breast cancer cell line and an MCF-7-derivative line, which acquired PTX resistance.44 PTX alone was very efficient in killing more than 80% of the MCF-7 parental cells but had only a marginal effect (11% death) in the resistant line (Fig. 5B). However, proTAME treatment alone resulted in minor cytotoxicity on both parental and resistant cell lines. The combination of PTX and 2 h HT had a significant effect on the resistant cell line viability, enhancing cell death from a mere 11 to 55% (Fig. 5B). On the opposite, proTAME did not significantly enhance PTX-induced cells death of either parental or resistant cell lines (Fig. 5B). Similar results were produced when the effects of the 2 strategies on the survival of HEp2 cells were compared (Fig. S4B). These data suggest that forcing mitotic exit represents an efficient strategy to increase PTX cytotoxicity in cells that are either sensitive or resistant to PTX.
Discussion
Therapeutic strategies that interfere with mitotic progression have been successfully used for cancer treatment. Microtubules poisons, such as taxanes, are cytotoxic drugs that have proven their efficacy over the past 30 years in a number of malignancies. However, the clinical success of these agents is limited by toxicity and high resistance rates.
To address these issues, extensive work has been done to develop novel and targeted anti-mitotic agents. Drugs inhibiting mitotic kinases (Aurora, Plk1, and cycling dependent kinases) or kinesins yielded very promising results in cell culture.45 Yet, the clinical outcomes of these drugs are very disappointing due to severe side effects and poor anticancer activity when compared with the taxane Paclitaxel (PTX).18-23
Besides the efforts toward novel drugs development, several groups focused on understanding how we can predict the response to PTX and thus circumvent the resistance mechanisms. Studies in cell lines demonstrated that the cell fate after exposure to PTX is extremely variable.9 The authors proposed that cells arrested in mitosis by PTX will eventually fall in one of two competing pathways: apoptosis or mitotic slippage.9,10 If we can understand how to push cells in one or the other direction from the PTX-induced mitotic block, we should be able to kill cancer cells more efficiently.
To this end, we investigated and compared 2 approaches: the first, forced exit from mitosis, and the second, preventing it.
To force mitotic exit, we used a short physiological hyperthermia (HT) as a trigger of mitotic slippage/catastrophe. It is well established that, in the presence of a mitotic stress or a mitotic poisonous drug (including PTX), the SAC temporarily blocks cells in mitosis; however, the mitotic exit cannot be prevented indefinitely. Thus, cells escape mitosis in presence of an active SAC by triggering apoptosis11,46,47 or by a slow and steady degradation of APC/C substrates, including cyclin B, which leads to a form of mitotic exit known as mitotic slippage.48 We observed that HT induced slippage in drug-arrested mitotic cells, while cells exposed to antimitotic drugs alone were arrested in mitosis (Fig. 2). As well, HT marginally affected cells not treated with drugs. Interestingly, we found that HT promptly activated mitotic slippage in cells treated with 3 anti-mitotic drugs with different mode of action: the microtubule hyperpolymerizer PTX, the microtubule depolymerizer Nocodazole (Noc), and the Aurora A inhibitor MLN8054 (Fig. 2). These results indicated that HT triggers mitotic exit independently from the modality of mitotic blockage and also emphasized mitosis as a HT-sensitive stage of cell cycle.
The accelerated slippage observed after HT can be explained by the disruption of the microtubules network and microtubule-organizing centers (MTOC), as it was previously described29; indeed, we observed similar HT-induced structural changes in our experimental conditions (not shown). However, given the multitude of intra-cellular targets of HT, microtubules collapse and MTOC/centrosome abnormalities may only partially explain the HT-induced mitotic slippage. Indeed, similar dynamics of mitotic exit were observed in both heat shocked Noc-treated cells, where microtubules are depolymerized, and in heat shocked MLN8054-treated cells (Fig. 2), where centrosomes function is compromised,49 suggesting that HT activates mitotic catastrophe by additional mechanisms.
We also observed that the recovery at 37 °C post-HT was necessary to induce mitotic slippage: at 4 h of recovery most of mitotic cells slipped into micronucleation (Fig. 1A and B), while extension of HT time did not induce mitotic catastrophe (not shown). This suggests that the mitotic exit process is most likely activated during HT, but must be executed during the post-stress recovery at 37 °C.
It is of great interest to understand the molecular mechanism leading to mitotic slippage. Our results indicate that cyclin B becomes promptly degraded during HT recovery, hinting to the APC/C participation. To understand whether HT-mediated slippage is APC/C dependent, we employed the APC/C inhibitor proTAME.40 We found that proTAME inhibited HT-induced mitotic slippage (Figs. 2 and 4), suggesting that APC/C activation by HT occurs without satisfaction of the SAC. The detailed mechanism of this activation can be the subject for future studies.
As second strategy, we inhibited the APC/C to investigate the block of mitotic exit. proTAME extended the average mitosis times of cells treated with both PTX and the Aurora A inhibitor (Fig. 3). Moreover, time-lapse microscopy experiments demonstrated that proTAME activated apoptosis in combination with PTX or MLN8054 (Table S1). Noteworthy, this form of cell death was preferred in cells with prolonged mitotic arrest (Fig. S3C and Table S1).
Combination of these results allowed us to propose a model for drug-induced mitotic block manipulation, where application of hyperthermia/heat shock almost exclusively forces mitotic catastrophe, potentially via APC/C activation, while block of mitotic exit by APC/C inhibition can also activate apoptosis (Fig. 6).
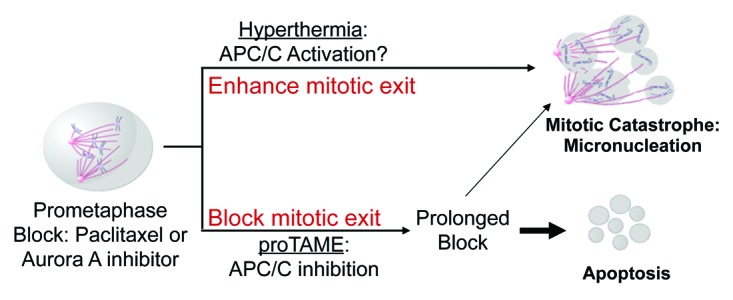
Figure 6. Model. Antimitotic drugs such as taxanes and Aurora A inhibitors have been developed and used to target and kill cancerous cells. However, chemotherapy resistance, both intrinsic and acquired, limits the efficacy of these drugs. We found that promoting mitotic exit by applying hyperthermia after exposing cancerous cells to antimitotic drugs, dramatically enhances cell death by mitotic catastrophe of either PTX sensitive or resistant cells. Our results indicate that block of mitotic exit can trigger cell death by apoptosis. Thus, studies of mitotic manipulation will help to rationally design novel combinatorial regimens for cancer treatment.
Regarding the efficacy of the 2 opposing strategies, our results indicate that, at least in the cell lines studied, the activation of mitotic exit is favorable over the prolongation of the mitotic arrest to increase PTX cytotoxicity (Fig. 5; Fig. S4B). In addition, we showed that HT triggers mitotic slippage and re-sensitizes drug-resistant cell to PTX (Fig. 5; Fig. S4A).
Since HT and PTX both target the microtubule network, this combination has been investigated to treat cancerous cells.31,37,39,50,51 Both in vitro and in vivo studies proved that this combination induced a “thermal enhancement” for PTX cytotoxicity.31,50,51 In current clinical trials, PTX is administered with hyperthermic perfusion for the treatment of ovarian and lung cancers and gastrointestinal neoplasms (http://www.clinicaltrial.gov).
Our results suggest a novel combination of PTX/HT, where PTX (and potentially other antimitotic drugs) can be used in a “first phase” to arrest cancer cells in mitosis, and then HT can be used in a “second phase” of treatment to force mitotic exit and cell death. In the future, in vivo studies are necessary to support this concept, especially in light of a recent report that showed decidedly lower mitotic index in response to PTX administration in xenograft settings compared with that achieved in cell culture.52 Although lower mitotic rates are observed in human specimens as well,53 the analysis of fine-needle aspiration biopsies of breast cancer patients demonstrated that the tumor mitotic index was increased up to 10-fold after PTX treatment and lasted up to 2–3 days after PTX administration.54 These results suggest that the combinatorial treatment regimen that we propose is feasible. Further studies of mitotic manipulation and pathways of mitotic cell death will help to rationally design novel combinatorial regimens for cancer treatment.
Materials and Methods
Cell culture and drugs treatment
HEp2 human larynx carcinoma and T47D human ductal breast epithelial tumor cell lines were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM glutamine and 100 U/mL penicillin, and 100 µg/mL streptomycin (Gibco BRL) and grown in a humidified 5% CO2 incubator. MCF7 parental and PTX-resistant were a kind gift from Dr. Parissenti and were cultured as previously described.44
Paclitaxel (PTX, Sigma) was added to a final concentration of 10 nM . Aurora A selective inhibitor MLN8054 (Selleck) was used at 4 µM. proTAME (Boston Biochem) was used at 12 µM. Nocodazole (EMD Millipore) was used at 10 µM. Doxorubicin and Etoposide (Sigma) were used at 5 µM and 100 µM, respectively.
Cell cycle analysis
HEp2 cells were incubated with drugs or DMSO for 8, 16, 24, and 32 h. Cells were trypsinized, washed, and gently resuspended in ice-cold 70% ethanol for 15 min on ice. Samples where than carefully washed and incubated with 125 µl of 1 mg/ml RNaseI at 37 °C for 15 min. For labeling, 125 µl of a 50 µg/ml propidium iodide solution in of 1.12% (w/v) of sodium citrate were added to samples for at least 30 min at room temperature before analyzing on the flow cytometer.
Hyperthermia protocol
Cells were grown to 70–75% confluency on either 35 mm plates or on glass coverslips for immunofluorescence experiments. Cells with or without drugs were exposed at 37 °C or 42 °C for 1 or 2 h. Cells were allowed to recover at 37 °C before fixation as indicated in specific experiments and stained for microscopy analysis or collected for protein analysis.
Microscopy analysis
Cells were fixed and stained with cell permeable Hoechst 33342 (Sigma). Images were taken with a Leica DMI4000 B fluorescent microscope. At least 500 cells per condition were analyzed and categorized as mitotic, interphase, apoptotic, and micronucleated cells as described previously.43
Immunofluorescence
The procedure was previously described.55 As primary antibodies H3 Phospho-Ser10 (Cell Signaling) and Lamin-A autoimmune antibodies56 were used in this study. Images were analyzed using a Leica TCS SP5 confocal microscope.
Western blot analysis
Protein samples were separated by 4–20% SDS-PAGE (Biorad), transferred to nitrocellulose membranes (Whatman) and blocked with 3% non-fat milk/PBS, 0.1% Tween (PBST). Primary antibodies cyclin B1 (Santa Cruz Biotechnology), Actin (Sigma), PARP-1 (Santa Cruz Biotechnology), and H3 Phospho-Ser10 were diluted in 3% milk/PBST and incubated overnight at 4 °C. Membranes were then washed 3× with PBST for 1 h at RT with appropriate secondary antibody (Millipore; all 1:2500). Membranes were then washed with PBST and exposed using ECL reagent (Amersham, GE Healthcare). Cyclin B and actin western blots densitometry analysis was performed using the Quantity One software from Bio-Rad.
Survival assays
Cells were seeded for triplicates at 25 000 cells/well, in 6-well plates. Twenty-four hours later cells were exposed to drugs for the indicated times. Hyperthermia was applied for 1 or 2 h at 42 °C during the last hours of treatment unless differently indicated. Cells were then washed twice with PBS to remove drugs; fresh media was added; and cells were replaced at 37 °C. Five days later cells were trypsinized and counted in a hemacytometer by trypan blue exclusion. For the clonogenic assay cells were treated as above and then re-plated in triplicates at 1:1000 dilutions on 6-well plates (Corning). Five to 7 days afterwards, colonies were stained with crystal violet and counted. Each experiment was repeated in triplicate at least 3 times.
Time-lapse microscopy
Time-lapse imaging of cells was performed as described before.57 Briefly, HEp2 cells were stably transfected with GFP-histone H2B (gift from Dr Duane Compton, Dartmouth); cells were treated as described and analyzed by Leica TCS SP5 confocal microscope equipped with environmental chamber; images were taken every 6 min. Mitosis time was determined by chromatin condensation marked as late G2/prophase transition (start) and beginning of chromosome decondensation (end). Thirty to 50 mitotic events were analyzed for each experimental condition. DNA morphology was also used to analyze mitotic outcome as normal division (D), apoptosis (A) or mitotic catastrophe (MC).
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr Parissenti for the kind gift of parental and PTX-resistant MCF-7 cells.
Grant Support
This work was supported by NIH/NCI R01 CA127378-01A1 for SG, DB, VMM, and AMI and Shula Foundation for SG
Glossary
Abbreviations:
- APC/C
anaphase promoting complex/cyclosome
- Cdc20
cell-division cycle protein 20
- HS
heat shock
- HT
hyperthermia
- MI
mitotic index
- MN
micronuclei
- PTX
paclitaxel
- SAC
spindle assembly checkpoint
Supplemental Materials
Supplemental materials may be found here: http://www.landesbioscience.com/journals/cc/article/25591
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25591
References
- 1.Rowinsky EK, Donehower RC. Paclitaxel (taxol) N Engl J Med. 1995;332:1004–14. doi: 10.1056/NEJM199504133321507. [DOI] [PubMed] [Google Scholar]
- 2.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–65. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 3.Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol. 2006;16:1194–200. doi: 10.1016/j.cub.2006.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schnerch D, Follo M, Krohs J, Felthaus J, Engelhardt M, Wäsch R. Monitoring APC/C activity in the presence of chromosomal misalig nM ent in unperturbed cell populations. Cell Cycle. 2012;11:310–21. doi: 10.4161/cc.11.2.18737. [DOI] [PubMed] [Google Scholar]
- 6.Erenpreisa JE, Ivanov A, Dekena G, Vitina A, Krampe R, Freivalds T, et al. Arrest in metaphase and anatomy of mitotic catastrophe: mild heat shock in two human osteosarcoma cell lines. Cell Biol Int. 2000;24:61–70. doi: 10.1006/cbir.1999.0466. [DOI] [PubMed] [Google Scholar]
- 7.Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008;15:1153–62. doi: 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]
- 8.Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat. 2001;4:303–13. doi: 10.1054/drup.2001.0213. [DOI] [PubMed] [Google Scholar]
- 9.Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–22. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 10.Gascoigne KE, Taylor SS. How do anti-mitotic drugs kill cancer cells? J Cell Sci. 2009;122:2579–85. doi: 10.1242/jcs.039719. [DOI] [PubMed] [Google Scholar]
- 11.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–4. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 12.McGrogan BT, Gilmartin B, Carney DN, McCann A. Taxanes, microtubules and chemoresistant breast cancer. Biochim Biophys Acta. 2008;1785:96–132. doi: 10.1016/j.bbcan.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 13.Sudo T, Nitta M, Saya H, Ueno NT. Dependence of paclitaxel sensitivity on a functional spindle assembly checkpoint. Cancer Res. 2004;64:2502–8. doi: 10.1158/0008-5472.CAN-03-2013. [DOI] [PubMed] [Google Scholar]
- 14.Wang TH, Wang HS, Soong YK. Paclitaxel-induced cell death: where the cell cycle and apoptosis come together. Cancer. 2000;88:2619–28. doi: 10.1002/1097-0142(20000601)88:11<2619::AID-CNCR26>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 15.Crown J, O’Leary M, Ooi WS. Docetaxel and paclitaxel in the treatment of breast cancer: a review of clinical experience. Oncologist. 2004;9(Suppl 2):24–32. doi: 10.1634/theoncologist.9-suppl_2-24. [DOI] [PubMed] [Google Scholar]
- 16.Bonneterre J, Spielman M, Guastalla JP, Marty M, Viens P, Chollet P, et al. Efficacy and safety of docetaxel (Taxotere) in heavily pretreated advanced breast cancer patients: the French compassionate use programme experience. Eur J Cancer. 1999;35:1431–9. doi: 10.1016/S0959-8049(99)00174-4. [DOI] [PubMed] [Google Scholar]
- 17.Ravdin P, Erban J, Overmoyer B. Phase III comparison of docetaxel and paclitaxel in patients with metastatic breast cancer. Eur J Cancer. 2003;(suppl1):32. [Google Scholar]
- 18.Janssen A, Medema RH. Mitosis as an anti-cancer target. Oncogene. 2011;30:2799–809. doi: 10.1038/onc.2011.30. [DOI] [PubMed] [Google Scholar]
- 19.Schmit TL, Ahmad N. Regulation of mitosis via mitotic kinases: new opportunities for cancer management. Mol Cancer Ther. 2007;6:1920–31. doi: 10.1158/1535-7163.MCT-06-0781. [DOI] [PubMed] [Google Scholar]
- 20.Boss DS, Beijnen JH, Schellens JH. Clinical experience with aurora kinase inhibitors: a review. Oncologist. 2009;14:780–93. doi: 10.1634/theoncologist.2009-0019. [DOI] [PubMed] [Google Scholar]
- 21.Schöffski P, Blay JY, De Greve J, Brain E, Machiels JP, Soria JC, et al. Multicentric parallel phase II trial of the polo-like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network Of Core Institutes (NOCI) Eur J Cancer. 2010;46:2206–15. doi: 10.1016/j.ejca.2010.03.039. [DOI] [PubMed] [Google Scholar]
- 22.Orth JD, Tang Y, Shi J, Loy CT, Amendt C, Wilm C, et al. Quantitative live imaging of cancer and normal cells treated with Kinesin-5 inhibitors indicates significant differences in phenotypic responses and cell fate. Mol Cancer Ther. 2008;7:3480–9. doi: 10.1158/1535-7163.MCT-08-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purcell JW, Davis J, Reddy M, Martin S, Samayoa K, Vo H, et al. Activity of the kinesin spindle protein inhibitor ispinesib (SB-715992) in models of breast cancer. Clin Cancer Res. 2010;16:566–76. doi: 10.1158/1078-0432.CCR-09-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ribeiro JT, Macedo LT, Curigliano G, Fumagalli L, Locatelli M, Dalton M, et al. Cytotoxic drugs for patients with breast cancer in the era of targeted treatment: back to the future? Ann Oncol. 2011 doi: 10.1093/annonc/mdr382. [DOI] [PubMed] [Google Scholar]
- 25.Rieder CL, Medema RH. No way out for tumor cells. Cancer Cell. 2009;16:274–5. doi: 10.1016/j.ccr.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 26.Matson DR, Stukenberg PT. Spindle poisons and cell fate: a tale of two pathways. Mol Interv. 2011;11:141–50. doi: 10.1124/mi.11.2.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang HC, Shi J, Orth JD, Mitchison TJ. Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell. 2009;16:347–58. doi: 10.1016/j.ccr.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nilsson J, Yekezare M, Minshull J, Pines J. The APC/C maintains the spindle assembly checkpoint by targeting Cdc20 for destruction. Nat Cell Biol. 2008;10:1411–20. doi: 10.1038/ncb1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knox JD, Mitchel RE, Brown DL. Effects of hyperthermia on microtubule organization and cytolytic activity of murine cytotoxic T lymphocytes. Exp Cell Res. 1991;194:275–83. doi: 10.1016/0014-4827(91)90365-2. [DOI] [PubMed] [Google Scholar]
- 30.Zaffaroni N, Fiorentini G, De Giorgi U. Hyperthermia and hypoxia: new developments in anticancer chemotherapy. Eur J Surg Oncol. 2001;27:340–2. doi: 10.1053/ejso.2000.1040. [DOI] [PubMed] [Google Scholar]
- 31.Lin Y, Liu Z, Li Y, Liao X, Liao S, Cen S, et al. Short-term hyperthermia promotes the sensitivity of MCF-7 human breast cancer cells to paclitaxel. Biol Pharm Bull. 2013;36:376–83. doi: 10.1248/bpb.b12-00774. [DOI] [PubMed] [Google Scholar]
- 32.Michalakis J, Georgatos SD, de Bree E, Polioudaki H, Romanos J, Georgoulias V, et al. Short-term exposure of cancer cells to micromolar doses of paclitaxel, with or without hyperthermia, induces long-term inhibition of cell proliferation and cell death in vitro. Ann Surg Oncol. 2007;14:1220–8. doi: 10.1245/s10434-006-9305-4. [DOI] [PubMed] [Google Scholar]
- 33.Michalakis J, Georgatos SD, Romanos J, Koutala H, Georgoulias V, Tsiftsis D, et al. Micromolar taxol, with or without hyperthermia, induces mitotic catastrophe and cell necrosis in HeLa cells. Cancer Chemother Pharmacol. 2005;56:615–22. doi: 10.1007/s00280-005-1002-7. [DOI] [PubMed] [Google Scholar]
- 34.Fajardo LF. Pathological effects of hyperthermia in normal tissues. Cancer Res. 1984;44(Suppl):4826s–35s. [PubMed] [Google Scholar]
- 35.Wust P, Hildebrandt B, Sreenivasa G, Rau B, Gellermann J, Riess H, et al. Hyperthermia in combined treatment of cancer. Lancet Oncol. 2002;3:487–97. doi: 10.1016/S1470-2045(02)00818-5. [DOI] [PubMed] [Google Scholar]
- 36.van der Zee J. Heating the patient: a promising approach? Ann Oncol. 2002;13:1173–84. doi: 10.1093/annonc/mdf280. [DOI] [PubMed] [Google Scholar]
- 37.Cividalli A, Cruciani G, Livdi E, Pasqualetti P, Tirindelli Danesi D. Hyperthermia enhances the response of paclitaxel and radiation in a mouse adenocarcinoma. Int J Radiat Oncol Biol Phys. 1999;44:407–12. doi: 10.1016/S0360-3016(99)00008-5. [DOI] [PubMed] [Google Scholar]
- 38.Bouquet W, Ceelen W, Fritzinger B, Pattyn P, Peeters M, Remon JP, et al. Paclitaxel/beta-cyclodextrin complexes for hyperthermic peritoneal perfusion - formulation and stability. Eur J Pharm Biopharm. 2007;66:391–7. doi: 10.1016/j.ejpb.2006.11.025. [DOI] [PubMed] [Google Scholar]
- 39.Mohamed F, Marchettini P, Stuart OA, Urano M, Sugarbaker PH. Thermal enhancement of new chemotherapeutic agents at moderate hyperthermia. Ann Surg Oncol. 2003;10:463–8. doi: 10.1245/ASO.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 40.Zeng X, Sigoillot F, Gaur S, Choi S, Pfaff KL, Oh DC, et al. Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell. 2010;18:382–95. doi: 10.1016/j.ccr.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci U S A. 2007;104:4106–11. doi: 10.1073/pnas.0608798104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Toda K, Naito K, Mase S, Ueno M, Uritani M, Yamamoto A, et al. APC/C-Cdh1-dependent anaphase and telophase progression during mitotic slippage. Cell Div. 2012;7:4. doi: 10.1186/1747-1028-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindsay CR, Scholz A, Morozov VM, Ishov AM. Daxx shortens mitotic arrest caused by paclitaxel. Cell Cycle. 2007;6:1200–4. doi: 10.4161/cc.6.10.4244. [DOI] [PubMed] [Google Scholar]
- 44.Hembruff SL, Laberge ML, Villeneuve DJ, Guo B, Veitch Z, Cecchetto M, et al. Role of drug transporters and drug accumulation in the temporal acquisition of drug resistance. BMC Cancer. 2008;8:318. doi: 10.1186/1471-2407-8-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manchado E, Guillamot M, Malumbres M. Killing cells by targeting mitosis. Cell Death Differ. 2012;19:369–77. doi: 10.1038/cdd.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104–9. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Letai AG. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8:121–32. doi: 10.1038/nrc2297. [DOI] [PubMed] [Google Scholar]
- 48.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–20. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoar K, Chakravarty A, Rabino C, Wysong D, Bowman D, Roy N, et al. MLN8054, a small-molecule inhibitor of Aurora A, causes spindle pole and chromosome congression defects leading to aneuploidy. Mol Cell Biol. 2007;27:4513–25. doi: 10.1128/MCB.02364-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Othman T, Goto S, Lee JB, Taimura A, Matsumoto T, Kosaka M. Hyperthermic enhancement of the apoptotic and antiproliferative activities of paclitaxel. Pharmacology. 2001;62:208–12. doi: 10.1159/000056096. [DOI] [PubMed] [Google Scholar]
- 51.Sharma D, Chelvi TP, Kaur J, Ralhan R. Thermosensitive liposomal taxol formulation: heat-mediated targeted drug delivery in murine melanoma. Melanoma Res. 1998;8:240–4. doi: 10.1097/00008390-199806000-00006. [DOI] [PubMed] [Google Scholar]
- 52.Orth JD, Kohler RH, Foijer F, Sorger PK, Weissleder R, Mitchison TJ. Analysis of mitosis and antimitotic drug responses in tumors by in vivo microscopy and single-cell pharmacodynamics. Cancer Res. 2011;71:4608–16. doi: 10.1158/0008-5472.CAN-11-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mitchison TJ. The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell. 2012;23:1–6. doi: 10.1091/mbc.E10-04-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Symmans WF, Volm MD, Shapiro RL, Perkins AB, Kim AY, Demaria S, et al. Paclitaxel-induced apoptosis and mitotic arrest assessed by serial fine-needle aspiration: implications for early prediction of breast cancer response to neoadjuvant treatment. Clin Cancer Res. 2000;6:4610–7. doi: 10.1097/00008390-199806000-00006. [DOI] [PubMed] [Google Scholar]
- 55.Giovinazzi S, Lindsay CR, Morozov VM, Escobar-Cabrera E, Summers MK, Han HS, et al. Regulation of mitosis and taxane response by Daxx and Rassf1. Oncogene. 2012;31:13–26. doi: 10.1038/onc.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nefkens I, Negorev DG, Ishov AM, Michaelson JS, Yeh ET, Tanguay RM, et al. Heat shock and Cd2+ exposure regulate PML and Daxx release from ND10 by independent mechanisms that modify the induction of heat-shock proteins 70 and 25 differently. J Cell Sci. 2003;116:513–24. doi: 10.1242/jcs.00253. [DOI] [PubMed] [Google Scholar]
- 57.Lindsay CR, Giovinazzi S, Ishov AM. Daxx is a predominately nuclear protein that does not translocate to the cytoplasm in response to cell stress. Cell Cycle. 2009;8:1544–51. doi: 10.4161/cc.8.10.8379. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.