

Abstract
Cataracts is a misfolding protein disease in which one of its major components is the γD-crystallin protein. The conformational structure of the aggregated γD-crystallin and the interactions that cause aggregation are largely unknown. A recent experimental two-dimensional infrared (2DIR) spectroscopy study determined that the C-terminal domain has a high propensity to form β-sheets whereas the N-terminal domain forms a disordered structure in the fiber state. We present a combined computational molecular dynamics (MD) and infrared spectroscopy study of the local dynamics of these domains. The computed 2DIR signals agree remarkably well with experiment. We show that both domains having a Greek key structural fold experience different electrostatic environments, which may be related to the fact that the C-terminal domain is more structurally stable than the N-terminal domain. We correlate the vibrational couplings to known energy dissipation mechanisms and reveal their origin.
Keywords: Cataracts, Aggregation, Greek Key Motifs, Misfolding, Isotope Labelling, Molecular Dynamics
Introduction
Many NMR, FTIR and X-ray Crystallography studies have been used to characterize the structures of the Crystallin proteins aggregates (1–5). Cataracts are misfolded protein diseases that cause blindness and affect elderly population over the age of 65 (National Eye Institute). Its treatment is invasive, expensive and performed only when the disease has severely damaged the eye lens. Pathology of human cataractous lenses reveals that cataracts are composed of protein aggregates called Crystallin proteins. These proteins are degenerative during adulthood, and are damaged by radiation exposure and other chemical modifications that obstruct the passage of light through the lens which cause blurred vision that worsens with time (6–9). Even though the causes of cataracts are known, the aggregation mechanism and the three-dimensional structure of these aggregates remain unknown.
There are three classes of Crystallins: α, β and γ-Crystallins. Their solubilities and stabilities in their native states are critical to keep the transparency of the lens for a lifetime since Crystallin are not regenerated (7, 9, 10). α-Crystallins are oligomers that exhibit chaperone activity, interact with unfolded proteins and prevent aggregation (11–15). β and γ-Crystallins proteins are believed to be structural proteins involved in aggregation and share similar structures composed of antiparallel β-sheets. Their secondary structures are separated into two domains, labeled as N-terminal domain (N-td) and C-terminal domain (C-td) and further clustered into four subdomains called Greek Key Motifs that contain antiparallel β-strand (See Fig. 1A). A key biological difference between β and γ Crystallin proteins is that β-Crystallin forms domain-swapped dimers while the γ-Crystallins are monomeric in solution (1–3, 16).
Figure 1.

A: Structure of the γD-Crystallin protein showing its four Greek Motifs divided into the two domains Nt (blue and red) and Ct (green and yellow) connected by a loop (purple) and their respective sequence as shown in the crystal structure (PDB ID: 1KHO) with the mutation S84C indicated (red). Residues located in the interdomains (in licorice) are responsible in the stability of the entire structure. B: Linear absorption (LA) (top) in the infrared regime of the mutant S84C-HγD-Crystallin for the full sequence (black), N-td labeled (red) and C-td labeled (blue).
Wild-type γ-Crystallins are stable eye lens proteins in vitro with higher free-energies of unfolding than the other Crystallin proteins. The two domains of a γ-Crystallin protein interact intermolecularly across a hydrophobic domain interface that helps stabilize the protein (17, 18). Human γD-Crystallin (HγD-Crys) is the third most abundant γ-Crystallin found in young human eyes. HγDCrys is a 173 amino-acid protein with a high structural homology with other γ-Crystallins (10, 19). Its secondary structure is composed of antiparallel β-sheets and loops that are grouped in two domains connected by a loop linker. Mutagenesis studies revealed that single amino acid substitutions in the HγD-Crys sequence are linked to aggregation and juvenile-onset cataracts (20–26). The aggregation mechanism in the aging lens differs from that of the juvenile-onset cataracts because they present a large number of modifications such as Methionine and Cysteine residues oxidation, deamidation, and glycation (27–30)
Fluorescence studies in triple tryptophan mutants of HγD-Crys by Kosinski-Collins et al. (31) found that the C-td is more structurally stable than the N-td. Studies of unfolding/folding experiments under physiological conditions demonstrated that the protein refolds through sequential structuring of its domains, where the C-td folds first and domain interface residues subsequently mediate the refolding of the N-td. Unfolding/folding kinetics studies by Flaugh et al. (17, 18) revealed the formation of partially folded intermediate structure containing the C-td in its native state and the N-td in its random state. They further suggested that hydrophobic residues in the intercore domain are critical in the unfolding/refolding process where the refolded C-td plays the role as a nucleating center for the N-td.
Two-dimensional ultrafast spectroscopy has been used to characterize geometrically ordered structures such as hairpins, amyloid fibrils and helical dimers for many proteins (32–38). One of the advantages of this techinique is its high temporal resolution; detailed information about the kinetics of the proteins can be obtained on timescale regimes inaccessible by others techniques. It can also be applied continously to study amyloid and protein folding kinetics on timescales of months to hours. The QM/MM computation of 2D spectroscopy signals has become a valuable tool in the study of the internal dynamics occurring in biological complexes such as amyloid fibrils (37, 39–43). Often, computations are necessary to fully interpret experimental 2D spectra. A recent study of γD-Crystallin using isotope labeling of domains and 2DIR spectroscopy by Moran et al. (44), determined that the fibril nucleation of the Crystallin occurs exclusively in the C-td. The authors further proposed a structural model of the fibrils composed of a segment of β-sheets formed by the C-td and another segment of random structure formed by the N-td.
In this article, we present a simulation study of 2DIR spectroscopy of natively folded γD-Crystallin and compared with the experimental results of Moran et al. (44). This comparison of experiment and simulation provides new insight into differences in how the native domains are predisposed to misfolding and aggregation.
To the best of our knowledge, this is the first successful comparison of 2DIR simulation with experiment on a protein of this size. We find excellent agreement between the two. Details of the experimental conditions may be found in ref. (44).
Materials and Methods
Protein Model
The HγD-Crys crystal structure at 1.25 Å resolution was solved and is available in the Protein Data Bank (PDB ID: 1HKO) (19). Its sequence consists of 173 amino acids which are grouped in the N-td and C-td domains which each has 2 sub-groups called Greek Key Motif with the labels: N-td1(2–40), N-td2(41–83), C-td3(88–128), and C-td4 (129–171) as shown in 1A. In order to mirror the experimental studies, the mutant S84C HγD-Crys(S84C-HγD-Crys) was created and the MET residue added in their N-Terminus using the MODELLER (45) package.
Simulation Protocol
Molecular dynamics (MD) trajectories of the S84C-HγD-Crys were obtained by using the NAMD Package 2.6 (46) with the CHARMM27 force field (47). Simulations were carried out in aqueous solutions (TIP3P water model) (48) with cubic periodic boundary conditions. The particle-mesh Ewald protocol was used to calculate the long-range electrostatic interactions and a 12 cutoff distance was used for nonbonded interactions. 20 ns MD simulations were performed after 2 ns equilibration with a time step of 1 fs at T=300K. 100 MD snapshots with a time interval of 2 ps from the last 2 ns were harvested to compute the signals. The simulated linear absorption (LA) and the 2DIR signals were computed using the SPECTRON package (49, 50).
A 2DIR experiment uses four input pulses k1, k2, k3 with ks satisfying ks = ±k1 ±k2 ±k3 that interact with the protein sample with delays t1, t2 and t3. The signal is defined as the transmission change of the ks beam is recorded. The signals were computed using the Fluctuating Vibrational-Exciton Hamiltonian (FVE) (51) which is based on the Frenkel Exciton Model. The FVE Hamiltonian, H●FVE, has the form:
| (1) |
where H●S is the hamiltonian of the system:
| (2) |
and H●F is the interaction with the optical field, E(t):
| (3) |
and B●m are the bosonic annihilation and creation operators for the m-th amide I mode, εm is the frequency, Δm is the anharmonicity, Jmn are the harmonic intermode couplings between modes m and n, and μm is the transition dipole moment. Jmn remains the same for all isotopomers, as known from experiments on HγD-Crys.
H●S and H●F were evaluated for each snapshot using the fluctuating parameters for each amide I unit using the vibrational map of Hayashi et al. (52). Vibrational maps connect the frequencies and transition dipoles of molecular vibrations with the fluctuating local electrostatic environment. Δm = 16 cm−1 were used fro the anharmonicities (53). The intermode coupling, Jmn, is calculated using the transition dipole coupling scheme (54). Once the Hamiltonian is constructed and diagonalized, the signal is computed using equations 115, 119 and 121 in ref. (50):
where Ie(Ω) and
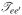 are the one and two-exciton Green’s functions, respectively. In this study, we set the broadening factor γe = 5.5 cm−1. Γe4e3e2e1 (Ω) is the exciton scattering matrix with two incoming (e1 and e2) and two outcoming excitons (e3 and e4) as defined in ref. (50), equation 325.
are the one and two-exciton Green’s functions, respectively. In this study, we set the broadening factor γe = 5.5 cm−1. Γe4e3e2e1 (Ω) is the exciton scattering matrix with two incoming (e1 and e2) and two outcoming excitons (e3 and e4) as defined in ref. (50), equation 325.
The signals are plotted by using two-dimensional Fourier transform of the times t1 → Ω1 and t3→Ω3. The signals can be either non-chiral or chiral depending on the polarization configuration of the laser pulses. In the dipole approximation, there are three independent signals to leading zeroth order in k: xyxy, xyyx, and xxyy (where x and y are the polarization of the laser pulses). The absorptive all-parallel signal, xxxx, is given by the linear combination xxxx = xyxy + xyyx + xxyy. Some combinations of non-chiral induced signals can be used to enhance desired features. The xyxy-xyyx combination supresses the population pathways and reveals the coherent quantum dynamics through crosspeaks (50). The combination xxyy-xyxy highlights the population dynamics and eliminates the coherent pathways in stimulated emission. For t2 = 0, we have xyxy=xxyy, and the combination xyxy-xyyx would only reveal excited state absorption.
We have simulated the absorptive 2DIR signals SkI + SkII. Here kI = −k1 + k2 + k3 and kII = k1−k2 + k3. We focused on the amide I band, which corresponds to the backbone carbonyl vibrational motions. The xxxx, the xxyy signals are compared with experiment. We further predict the combination xyxy-xyyx signal. The 2D non-chiral spectra used to compare with the experimental signals are plotted using a linear scale.
Results
Linear Absorption Spectra of the γD-Crys Protein
The linear absorption (LA) signals in the IR regime for the γD-Crys protein with its respective labeled domains are displayed in 1B. The labeling protocol was implemented in this study as described by Moran et al. (44): One domain is 13C isotopically labeled and the other domain remains 12C intact. In the LA signal, the unlabelled protein displays a peak centered at 1638 cm−1. When one of the domains is isotope labeled, the signal acquires a bimodal shape with two peaks around at 1595 cm−1 and 1638 cm−1 (1B). The LA signal for the labeled Ct-d shows that the peak at 1638 cm−1 moves slightly to the higher frequency making the bimodal shape more pronounced than that of the labeled Nt-d.
2DIR Signals
The 2DIR signals for the S84C-HγD-Crys and their respective labeled domain samples were computed at t2 = 0. The experimental and simulated all parallel xxxx spectra of the mutant protein and its respective labeled domains are compared in 2. For the unlabeled and labeled full protein (2, 12C Nt/Ct and 13C Nt/Ct, respectively), a pair of negative and positive peaks are observed in each spectrum: At Ω3 = 1638 cm−1 for the unlabeled protein and at Ω3 = 1595 cm−1 for the labeled protein. These frequencies correspond to a structure with antiparallel β-sheets (55). The negative peak results from ground state bleach and excited state emission pathways while the positive peak results from excited state absorption. The broadening of these peaks is due to contributions of other secondary structures formed in the protein. These peaks are reproduced in the simulations (2, right panels). These pairs are shifted around by 40 cm−1 which is consistent with the effective mass replacement of the carbon. The node slopes (44) were calculated for both of these states to compare the contributions of inhomogeneous versus homogeneous broadening (Table S1). A node slope of one represents a perfectly inhomogeneously broadened peak while a value of infinity represents a perfectly homogeneously broadened peak. The node slope of the unlabeled peak pair is in good agreement with experiment, while the node slope for the labeled full protein shows a 15% deviation from experiment. This deviation may be because while the isotope labeling was accounted for in the Hamiltonian (equation 2), the MD simulations used for conformational sampling did not use a heavy carbon, which may have a slight influence on the dynamics.
For the proteins with labeled domain, two pairs of positive and negative peaks appear in the spectra. The experimental spectra of the labeled domains (2, Nt-d and Ct-d) show two pairs of peaks near Ω3 = 1640 cm−1 and Ω3 = 1605 cm−1 in each spectrum. Differences between the experimental signals can be seen. The peak pairs in the labeled Nt-domain signal are less intense and more separated than those of the labeled Ct-domains (see Table S1), where the high frequency pair in the labeled Ct-domain are elongated and broader than those of in the labeled Nt-domain signal. For the low frequency pair, the labeled Nt-domain signal shows difused peaks, especially the negative peak, while the positive peak is well defined and centered at Ω3 = 1605 cm−1. In the labeled Ct-domain signal, the low frequency peak pair is intense and are centered at Ω3 = 1605 cm−1.
Some of these features are reproduced in the simulated labeled domain signals (2, right panels third and fourth rows). Both show a similar profile with two pairs of peaks separated by 40 cm−1. In the labeled Nt-domain signal, the negative peaks are less broad than those of in the labeled Ct-domain signal. The peak pairs in the labeled N-td signal are more separated than those of the labeled C-td signal, as observed in experiment and the simulated linear absorption spectra. Comparing the simulated and experimental node slopes, we see that the node slopes attributed to the unlabeled domains are in good agreement with experiment while those attributed to the labeled domains show a larger deviation from experiment. Again, we believe this larger deviation from experiment may be because all MD simulations used for conformational sampling used 12C in the amide backbone.
We next turn to the xxyy signal (3, left panels). All spectra share similar profile as in the xxxx spectra but with less intense peaks at Ω3 = 1640 cm−1 and Ω3 = 1605 cm−1. This pattern is observed in the simulated data as well. However, the peak pairs in the labeled full protein are stronger and broader than in the unlabeled full protein which indicates that the N-td follows different dynamics where its eigenstates exhibit a wider frequency range despite the high homology in the backbone geometry. We note that in the simulated spectra, we are able to better resolve the low intensity cross peaks better than in experiment because of the lack of experimental (white) noise in the simulations.
The simulated combination xyxy-xyyx spectrum (4) for labeled domains share the same pairs of peaks as observed in the xxxx combination. However, the N-td shows more elongated positive peaks along the diagonal than the positive peaks in the C-td. The spectra also show cross-peaks at Ω1 = 1610, 1660 cm−1 that are more diffuse in the Ct-Domain than the N-td.
Both experiment and simulation show clear differences in the 2DIR spectra when the N-t or C-t domain are labeled, despite their high structural homology. The experimental and simulated spectra show some weak cross peaks which suggest couplings between the backbone amide states. These couplings depend strongly on the geometry of the protein. The domains are connected by a flexible linker loop and one domain is essentially the reflection of the other domain. The secondary structure propensity for the protein displayed in 5 showed that reflection among domains especially where the propensity to form β-strand is high except in the residues 40–41 and 60–61 from the N-td2 motif, and their corresponding reflections in the C-td2 motif (residues 129–130 and 147–148). Other differences are observed in the turn propensities where the loop between N-td1 and N-td2 motifs are higher than that of among C-td1 and C-td2 motifs. These differences in secondary structure indicate that the interactions among the sub-domains may affect the overall stability of each domain.
The coupling matrix (shown in 6) may help reveal how the various residues are relevant in the folding and stability of the domains because the couplings are sensitive to distance and orientation. Couplings are observed from intramolecular interactions between residues that form β-sheets structures (anti-parallel to the diagonal). Residues in the N-td (residue indexes 1 to 83) have weaker couplings when compared with the corresponding section in the C-td (residue indexes 88 to 168). In the N-td, R36 is strongly coupled to T4 and K2. These surface residues are solvent-exposed and it is known that mutant R36S is linked to congenital juvenile onset-cataracts (56). Interestingly, Q54 and R58 show a strong coupling with those residues in the segment where R36 is located. The image residue for Q54 in the C-td is Q143. Q54 and R58 are solvent-exposed residues that form one of the β-strands in their respective domains and belong to the set of residues in the interface core. Deamidation of Q54 and Q143 introduce a negative charge at physiological pH into the domain interface and destabilizes HγD-Crys, lower the unfolding kinetic barrier and leads to the formation of partially unfolded intermediate with structured C-td and a unstructured N-td (29). The R58H mutation, which results in aculeiform cataract (20), and the R36S mutant have lower solubilities and crystallize more readily than the wild-type (19).
A cluster of couplings seen in the map corresponds to the hydrophobic interdomain region located between Nt-d2 (residues M43, Q54, F56, R79, I81) and Ct-d4 (V132, Q143, L145, and M147). These residues play a role in the stabilization of the domains and a decrease of the refolding rate of the N-td as indicated in alanine screening experiments (17).
HγD-Crys protein has, on the other hand, a large number of aromatic residues (14 tyrosine, 6 phenylalanine, and 4 tryptophan), few of them are lone residues and the rest are distributed in pairs or set of three residues that maintain the protein structure (57). In the coupling map, we indicate two pairs of tryptophan of interest: W42 and W68 in the N-td and their images residues, W131 and W157, in the C-td. W42 and its image W131 are buried at the bottom of their respective domains. The map indicates that W42 couples with Y28, and W131 couples with D114 but the pair (W42,Y28) displays a weaker coupling than that of the pair (W131,D114). W42 and its image W131 have coupling with W68 and W157, respectively. (W42,W68) has a weaker coupling intensity than (W131,W157) where the latter is surrounded by strong couplings generated by other residues. The other two tryptophan, W68 and its image W157, located at the top of the structure, exhibit some couplings. For W68, we have the pairs (W68,Y6), (W68,Y28) and (W68,Y50) and for W157, we have (W157,H88), (W157,F117), and (W157,W132). It is well established that UV radiation may contribute to the origin of cataract due to the absorption by aromatic residues (57). These tryptophan residues, W42, W68, W131 and W157, have been studied using fluorescence quenching (31) and determined that the local environment around W68 and W157 originate the native state quenching of the protein fluorescence emission. During the refolding/unfolding process, W42 and W68 unfolded at lower concentrations of GdnHCl than their images residues, W131 and W157, respectively. Futher analysis of unfolding and refolding of the tryptophan mutants determined an intermediate state along the wt-HγD-Cryst folding pathway with a structured C-td and unstructured N-td which could be a suitable conformational model for cataract formation.
Even though the two domains are geometrically homologous, they make different contributions to the 2DIR signals. The large separation between densities of states for the pair Q54 and Q143 could be due to their exposure to the solvent and their location in the interface domain which could change the strengths of the interactions with other residues. The tryptophan residues considered here are buried in their respective domains. Although both domains share basically identical backbone geometry, the frequency shifts upon domain labelling suggest that the kinetics of these domains are different where the folding in the N-td depends on the transitions occurring in the C-td, which agrees well with experimental reports (17, 30, 59)
Discussion and Conclusions
Moran et al. used a combined approach of isotope labeling and 2DIR spectroscopy (44) to study the γD-Crystallin protein. They demonstrated that the use of this combined tool is valuable for the characterization of the kinetics and structure in proteins that help to obtain additional information not available from standard methods. In their studies, they expressed labeled and unlabeled domains of the protein separately, and the full length of γD-Crystallin protein was constituted by protein ligation using a covalent linker residue located in the connecting loop. They determined tha C-td of the γD-Crystallin protein is the leading domain to form amyloid fibrils and the use of this covalent linkage between domains plays a role in determining the fibril structure which is a striking result considering that each individual domain can form fibrils on its own (58).
We note that our simulations are limited to the computation of 2DIR spectra of the monomeric form of HγD-Crys, and do not offer a direct comparison with the 2DIR kinetics experiments. We used a computational approach to compliment these results at the level of the native state of the mutant S84C-γD-Crystallin monomers. The computed spectra are in excellent agreement with experiment. Although the N-t and C-t domains are geometrically similar with highly conserved residues in their sequences, the average coupling map analysis shows an asymmetric contribution of some relevant residues and their corresponding image residue to the signals when one of the domains is isotopically labeled. Experiments made by Mills et al. (59) concluded that the isolated N-td was less stable than the full-length proteins and less stable than the C-td. A computational study by Das et al. (60) confirmed that the stability and the folding mechanism of the N-td are regulated by the interdomain interactions.
Our findings are consistent with the conclusions that Moran et al. stated about the role of the covalent linkage between domains in determinining the fibril structure and the prevalent dynamics that the C-td has over the N-td in the fibril formation. It will be interesting to use 2DIR spectroscopy to analyze the coupling between domains at different environmental conditions and mutations.
As stated in the introduction, we can consider HγD-Crys in terms of two coupled domains. In a very simple approximation, it is possible to think of this as a coupled two-mode exciton system:
where εi is the mean site-energy of domain i and Jij is the coupling between modes i and j. The eigenvalues are:
When neither domain is isotope labeled or both domains are labeled, εi ≈ε j, and we have (roughly) a linearly coupled homodimer. When either the N-td or the C-td is isotope labeled, we then have a linearly coupled heterodimer. In this approximation, we expect to see two peaks at ε+ and ε− in the linear absorption spectrum. These peaks are separated by . If the system is symmetric, then (εi − ε j)2 = (ε j − εi)2 and the N-td and C-td labeled isotopomers will produce equivalent linear absorption spectra (61). However, as seen from the calculated linear absorption spectra, this is not the case as the peaks in the N-td labeled isotopomer are less separated compared to the peaks in the C-td labeled isotopomer. Since Jij is the same for the two isotopomers, this indicates that (εi − ε j)2 is greater for the C-td labeled isotopomer. This is likely related to the fact that the C-td has a better defined structure, which leads to a lower approximate site energy
While the diagonal elements of the Hamiltonian are decreased by isotopic substitution, the off-diagonal elements remain the same because the transition dipoles of the individual amide I modes, which determine the coupling, do not change upon isotopic substitution. This was verified experimentally by comparing the diagonal cross-sections of the 2DIR spectra of the unlabeled and uniformly labeled 13C proteins.
However, as noted by Fang et. al (62), the cross peaks are a direct measure of the off-diagonal an-harmonic coupling. Since we assume zero mechanical off-diagonal anharmonicity, all off-diagonal anharmonicity must result from electrostatic coupling. In the weak-coupling limit, which is appropriate here, this off-diagonal anharmonicity can be approximated as:
Where Δij is the off-diagonal anharmonicity and Δii is the diagonal anharmonicity (16 cm−1). Therefore, systems with a smaller (εi − ε j)2 will have stronger cross peaks. In our case, the N-td labeled isotopomer has the smaller value and therefore has stronger cross peaks.
Using this very simple model, we conclude that the asymmetry in the linear absorption and 2DIR spectra is caused by the differences in the site-energies in the two domains. The differences in these site-energies are caused by the differing local electrostatic environments of residues within these two domains and is likely related to the secondary structure propensity in each domain.
Supplementary Material
Figure 2.

All-parallel signal xxxx SkI + SkII experiment (left) and simulation (right). From top to bottom: full mutant protein (12C Nt/Ct), labeled full mutant protein (13C Nt/Ct), labeled N-td and the labeled C-td.
Figure 3.

Same as in 2 but for polarization configuration xxyy.
Figure 4.

Simulated xyxy-xyyx signals of the isotopically labeled domains.
Figure 5.

Secondary structure propensity of the domains of γD-Crystallin indicates the segments of the sequence that have the probability to form β-strands or turns.
Figure 6.

Coupling matrix of the γD-Crystallin protein. Inter-domains interactions are represented by a red-blue color scale indicating their strength.
Acknowledgments
We gratefully acknowledge the support of the National Institutes of Health (Grant GM059230 and GM091364), and the National Science Foundation (Grant CHE-1058791). NKP is supported by a National Science Graduate Research Fellowship. MTZ acknowledges NIH DK79895.
References
- 1.Wistow G, Turnell B, Summers L, Slingsby C, Moss D, Miller L, Lindley P, Blundell T. X-ray Analysis of the Eye Lens Protein γ-II Crystallin at 1.9 Å Resolution. J Mol Biol. 1983;170(1):175–202. doi: 10.1016/s0022-2836(83)80232-0. [DOI] [PubMed] [Google Scholar]
- 2.Cooper PG, Carver JA, Aquilina JA, Ralston GB, Truscott RJA. 1H NMR Spectroscopic Comparison of γS-and γB-Crystallins. Exp Eye Res. 1994;59(2):211–20. doi: 10.1006/exer.1994.1099. [DOI] [PubMed] [Google Scholar]
- 3.Slingsby C, Norledge B, Simpson A, Bateman OA, Wright G, Driessen HPC, Lindley PF, Moss DS, Bax B. X-ray Diffraction and Structure of Crystallins. Prog Ret Eye Res. 1997;16(1):3–29. [Google Scholar]
- 4.Carver JA. Probing the Structure and Interactions of Crystallin Proteins by NMR Spectroscopy. Prog Retin Eye Res. 1999;18(4):431–462. doi: 10.1016/s1350-9462(98)00027-5. [DOI] [PubMed] [Google Scholar]
- 5.Purkiss AG, Bateman OA, Goodfellow JM, Lubsen NH, Slingsby C. The X-ray Crystal Structure of Human γS-Crystallin C-terminal Domain. 2002;277(6):4199–4205. doi: 10.1074/jbc.M110083200. [DOI] [PubMed] [Google Scholar]
- 6.Hoenders HJ, Bloemendal H. Lens Proteins and Aging. J Gerontol. 1983;38(3):278–286. doi: 10.1093/geronj/38.3.278. [DOI] [PubMed] [Google Scholar]
- 7.Slingsby C, Clout NJ. Structure of the Crystallins. Eye. 1999;13(Pt3b):395–402. doi: 10.1038/eye.1999.113. [DOI] [PubMed] [Google Scholar]
- 8.Bloemendal H, de Jong W, Jaenicke R, Lubsen NH, Slingsby C, Tardieu A. Ageing and Vision: Structure, Stability and Function of Lens Crystallins. Prog in Biophys and Mol Biol. 2004;86(3):407–485. doi: 10.1016/j.pbiomolbio.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 9.Graw J. Genetics of Crystallins: Cataract and Beyond. Exp Eye Res. 2009;88(2):173–189. doi: 10.1016/j.exer.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 10.Jaenicke R, Slingsby C. Lens Crystallins and Their Microbial Homologs: Structure, Stability, and Function. Crit Rev Biochem Mol Biol. 2001;36(5):435–499. doi: 10.1080/20014091074237. [DOI] [PubMed] [Google Scholar]
- 11.Horwitz J. α-Crystallin Can Function As A Molecular Chaperone. Proc Natl Acad Sci USA. 1992;89(21):10449–10453. doi: 10.1073/pnas.89.21.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groenen PJ, Merck KB, de Jong WW, Bloemendal H. Structure and Modifications of the Junior Chaperone α-Crystallin. From Lens Transparency to Molecular Pathology. Eur J Biochem. 1994;225(1):1–19. doi: 10.1111/j.1432-1033.1994.00001.x. [DOI] [PubMed] [Google Scholar]
- 13.Clark JI, Muchowski PJ. Small Heat-Shock Proteins and Their Potential Role in Human Disease. Curr Opin Struc Biol. 2000;10(1):52–59. doi: 10.1016/s0959-440x(99)00048-2. [DOI] [PubMed] [Google Scholar]
- 14.Andley UP. Effects of α-Crystallin on Lens Cell Function and Cataract Pathology. Curr Mol Med. 2009;9(7):887–892. doi: 10.2174/156652409789105598. [DOI] [PubMed] [Google Scholar]
- 15.Kumar PA, Reddy GB. Modulation of α-Crystallin Chaperone Activity: A Target to Prevent or Delay Cataract? IUBMB Life. 2009;65(5):485–495. doi: 10.1002/iub.176. [DOI] [PubMed] [Google Scholar]
- 16.Cooper PG, Carver JA, Truscott RJ. 1H-NMR Spectroscopy of Bovine Lens β-Crystallin. The Role of the β B2-Crystallin C-Terminal Extension in Aggregation. Eur J Biochem. 1993;213(1):321–328. doi: 10.1111/j.1432-1033.1993.tb17765.x. [DOI] [PubMed] [Google Scholar]
- 17.Flaugh SL, Kosinski-Collins MS, King J. Interdomain Side-Chain Interactions in Human γD Crystallin Influencing Folding and Stability. Protein Sci. 2005;14(8):2030–2043. doi: 10.1110/ps.051460505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flaugh SL, Kosinski-Collins MS, King J. Contributions of Hydrophobic Domain Interface Interactions to the Folding and Stability of Human γD-crystallin. Protein Sci. 2005;14(3):569–581. doi: 10.1110/ps.041111405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Basak A, Bateman O, Slingsby C, Pande A, Asherie N, Ogun O, Benedek GB, Pande J. High-Resolution X-Ray Crystal Structures of Human γD Crystallin (1.25 Å) and the R58H Mutant (1.15 Å) Associated with Aculeiform Cataract. J Mol Biol. 2003;328(5):1137–1147. doi: 10.1016/s0022-2836(03)00375-9. [DOI] [PubMed] [Google Scholar]
- 20.Heon E, Priston M, Schorderet DF, Billingsley GD, Girard PO, Lubsen N, Munier FL. The γ-Drystallins and Human Cataracts: A Puzzle Made Clearer. Am J Hum Genet. 1999;65(5):1261–1267. doi: 10.1086/302619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nandrot E, Slingsby C, Basak A, Cherif-Chefchaouni M, Benazzouz B, Hajaji Y, Boutayeb S, Gribouval O, Arbogast L, et al. Gamma-D Crystallin Gene (CRYGD) Mutation Causes Autosomal Dominant Congenital Cerulean Cataracts. J Med Genet. 2003;40:262. doi: 10.1136/jmg.40.4.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santhiya ST, Manohar MS, Rawlley D, Vijayalakshmi P, Namperumalsamy P, Gopinath PM, Löster J, Graw J. Novel Mutations in the γ-Crystallin Genes Cause Autosomal Dominant Congenital Cataracts. J Med Genet. 2002;9(5):352–358. doi: 10.1136/jmg.39.5.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sandilands A, Hutcheson AM, Long HA, Prescott AR, Vrensen G, Löster J, Klopp N, Lutz RB, Graw J, Masaki S, et al. Altered Aggregation Properties of Mutant γ-Crystallins Cause Inherited Cataract. The EMBO J. 2002;21(22):6005–6014. doi: 10.1093/emboj/cdf609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans P, Wyatt K, Wistow GJ, Bateman OA, Wallace BA, Slingsby C. The P23T Cataract Mutation Causes Loss of Solubility of Folded γD-Crystallin. J Mol Biol. 2004;343(2):435–444. doi: 10.1016/j.jmb.2004.08.050. [DOI] [PubMed] [Google Scholar]
- 25.Kumar M, Agarwal T, Khokhar S, Kumar M, Kaur P, Roy TS, Dada R. Mutation Screening and Genotype Phenotype Correlation of α-Crystallin, γ-Crystallin and GJA8 Gene in Congenital Cataract. Mol Vis. 2011;17:693–707. [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W, Cai HC, Li FF, Xi YB, Ma X, Yan YB. The Congenital Cataract-linked G61C Mutation Destabilizes γD-Crystallin and Promotes Non-Native Aggregation. PLoS One. 2011;6(5):e20564. doi: 10.1371/journal.pone.0020564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stevens A. The Contribution of Glycation to Cataract Formation in Diabetes. J Am Optom Assoc. 1998;69(8):519–530. [PubMed] [Google Scholar]
- 28.Hanson SR, Hasan A, Smith DL, Smith JB. The Major In Vivo Modifications of the Human Water-Insoluble Lens Crystallins are Disulfide Bonds, Deamidation, Methionine Oxidation and Backbone Cleavage. Exp Eye Res. 2000;71(2):195–207. doi: 10.1006/exer.2000.0868. [DOI] [PubMed] [Google Scholar]
- 29.Flaugh SL, Mills IA, King J. Glutamine Deamidation Destabilizes Human γD-Crystallin and Lowers the Kinetic Barrier to Unfolding. 2006;281(41):30782–30792. doi: 10.1074/jbc.M603882200. [DOI] [PubMed] [Google Scholar]
- 30.Goulet DR, Knee KM, King JA. Inhibition of Unfolding and Aggregation of Lens Protein Human γD Crystallin by Sodium Citrate. Exp Eye Res. 2011;93(4):371–381. doi: 10.1016/j.exer.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kosinski-Collins MS, Flaugh SL, King J. Probing Folding and Fluorescence Quenching in Human γD Crystallin Greek Key Domains using Triple Tryptophan Mutant Proteins. Protein Sci. 2004;13(8):2223–2235. doi: 10.1110/ps.04627004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith AW, Tokmakoff A. Amide I Two-Dimensional Infrared Spectroscopy of β-Hairpin Peptides. J Chem Phys. 2007;126(4):045109-1–10. doi: 10.1063/1.2428300. [DOI] [PubMed] [Google Scholar]
- 33.Kim YS, Liu L, Axelsen PH, Hochstrasser RM. Two-dimensional Infrared Spectra of Isotopically Diluted Amyloid Fibrils from Aβ40. Proc Natl Acad Sci USA. 2008;105(22):7720–7725. doi: 10.1073/pnas.0802993105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strasfeld DB, Ling YL, Shim S-H, Zanni MT. Tracking Fiber Formation in Human Islet Amyloid Polypeptide with Automated 2D-IR Spectroscopy. J Am Chem Soc. 2008;130(21):6698–6699. doi: 10.1021/ja801483n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strasfeld DB, Ling YL, Gupta R, Raleigh DP, Zanni MT. Strategies for Extracting Structural Information from 2DIR Spectroscopy of Amyloid: Application to Islet Amyloid Polypeptide. J Phys Chem B. 2009;113(47):15679–15691. doi: 10.1021/jp9072203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shim S-H, Gupta R, Ling YL, Strasfeld DB, Raleigh DP, Zanni MT. Two-Dimensional IR Spectroscopy and Isotope Labeling Defines the Pathway of Amyloid Formation with Residue-Specific Resolution. Proc Natl Acad Sci USA. 2009;106(16):6614–6619. doi: 10.1073/pnas.0805957106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reddy AS, Wang L, Lin YS, Ling Y, Chopra M, Zanni MT, Skinner JL, Pablo JJD. Solution Structures of Rat Amylin Peptide: Simulation, Theory, and Experiment. Biophys J. 2010;98(3):443–451. doi: 10.1016/j.bpj.2009.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Remorino A, Korendovych IV, Wu YB, DeGrado WF, Hochstrasser RM. Residue-Specific Vibrational Echoes Yield 3D Structures of a Transmembrane Helix Dimer. Science. 2011;332(6034):1206–1209. doi: 10.1126/science.1202997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhuang WDA, Voronine DV, Mukamel S. Simulation of Two-Dimensional Infrared Spectroscopy of Amyloid Fibrils. Proc Natl Acad Sci USA. 2007;104(36):14233–14236. doi: 10.1073/pnas.0700392104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang J, Abramavicius D, Falvo C, Bulheller BM, Hirst JD, Mukamel S. Simulation of Two Dimensional Ultraviolet (2DUV) Spectroscopy of Amyloid Fibrils. J Phys Chem B. 2010;114(37):12150–12156. doi: 10.1021/jp1046968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhuang W, Sgourakis NK, Zhenyu L, Garcia A, Mukamel S. Discriminating Early Stage Aβ42 Monomer Structure Using Chirality-Induced 2DIR Spectroscopy in a Simulation Study. Proc Natl Acad Sci USA. 2010;107(36):15687–15692. doi: 10.1073/pnas.1002131107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lam A, Jiang J, Mukamel S. Distinguishing Amyloid Fibril Structures in Alzheimer’s Disease (AD) by Two-Dimensional Ultraviolet (2DUV) Spectroscopy. Biochemistry. 2011;50(45):9809–9816. doi: 10.1021/bi201317c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, Middleton CT, Singh S, Reddy AS, Woys AM, Strasfeld DB, Marek P, Raleigh DP, de JJ, Pablo„Zanni MT, et al. 2DIR Spectroscopy of Human Amylin Fibrils Reflects Stable β-Sheet Structure. J Am Chem Soc. 2011;133(40):16062–16071. doi: 10.1021/ja204035k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moran S, Woys A, Buchanan L, Bixby E, Decatur S, Zanni M. Two-dimensional IR Spectroscopy and Segmental 13C Labeling Reveals the Domain Structure of human γD-Crystallin Amyloid Fibrils. Proc Natl Acad Sci USA. 2012;109(9):3329–3334. doi: 10.1073/pnas.1117704109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fiser A, Sali A. Modeller: Generation and Refinement of Homology-Based Protein Structure Models. Meth Enzymol. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 46.Phillips JC, Braun RWW, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable Molecular Dynamics with NAMD. J Comp Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.MacKerell AD, et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J Comp Chem. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 48.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 49.Zhuang W, Abramavicius D, Mukamel S. Dissecting Coherent Vibrational Spectra of Small Proteins into Secondary Structural Elements by Sensitivity Analysis. Proc Natl Acad Sci USA. 2005;102(21):7443–7448. doi: 10.1073/pnas.0408781102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abramavicius D, Palmieri B, Voronine DV, Sanda F, Mukamel S. Coherent Multidimensional Optical Spectroscopy of Excitons in Molecular Aggregates; Quasiparticle versus Supermolecule Perspectives. Chem Rev. 2009;109(6):2350–2408. doi: 10.1021/cr800268n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhuang W, Abramavicius D, Hayashi T, Mukamel S. Simulation Protocols for Coherent Femtosecond Vibrational Spectra of Peptides. J Phys Chem B. 2006;110(7):3362–3374. doi: 10.1021/jp055813u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayashi T, Zhuang W, Mukamel S. Electrostatic DFT Map for the Complete Vibrational Amide Band of NMA. J Phys Chem A. 2005;109(43):9747–9759. doi: 10.1021/jp052324l. [DOI] [PubMed] [Google Scholar]
- 53.Hamm P, Lim M, Hochstrasser RM. Structure of the Amide I Band of Peptides Measured by Femtosecond Nonlinear-Infrared Spectroscopy. J Phys Chem B. 1998;102:6123–6138. [Google Scholar]
- 54.Torii H, Tasumi M. Model Calculation on the Amide-I Infrared Bands of Globular Proteins. J Chem Phys. 1992;96(5):3379–3387. [Google Scholar]
- 55.Ganim Z, Chung HS, Smith AW, DeFlores LP, Jones KC, Tokmakoff A. Amide I two-dimensional infrared spectroscopy of proteins. Acc Chem Res. 2008;41:432–441. doi: 10.1021/ar700188n. [DOI] [PubMed] [Google Scholar]
- 56.Kmoch S, Brynda J, Asfaw B, Bezouška K, Novák P, Řezáčová P, Ondrová L, Filipec M, Sedláček J, Elleder M. Link Between a Novel Human γD-Crystallin Allele and a Unique Cataract Phenotype Explained by Protein Crystallography. Hum Mol Gen. 2000;9(2):1779–1786. doi: 10.1093/hmg/9.12.1779. [DOI] [PubMed] [Google Scholar]
- 57.Kong F, King J. Contributions of Aromatic Pairs to the Folding and Stability of Long-Lived Human γD-Crystallin. Prot Sci. 2011;20(3):513–528. doi: 10.1002/pro.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Papanikolopoulou K, Mills-Henry I, Thol SL, Wang Y, Gross AAR, Kirschner DA, Decatur SM, King J. Formation of Amyloid Fibrils In Vitro by Human γD-Crystallin and its Isolated Domains. Mol Vis. 2008;14:81–89. [PMC free article] [PubMed] [Google Scholar]
- 59.Mills IA, Flaugh SL, Kosinski-Collins MS, King JA. Folding and Stability of the Isolated Greek Key Domains of the Long-Lived Human Lens Proteins γD-Crystallin and γS-Crystallin. Prot Sci. 2007;16(11):2427–2444. doi: 10.1110/ps.072970207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Das P, King JA, Zhou R. β-Strand Interactions at the Domain Interface Critical for the Stability of Human Lens γD-Crystallin. Prot Sci. 2010;19(1):131–140. doi: 10.1002/pro.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zanni MT, Hamm P. Concepts and Methods of 2D Infrared Spectroscopy. Cambridge University Press; 2011. [Google Scholar]
- 62.Fang C, Senes A, Cristian L, DeGrado WF, Hochstrasser RM. Amide vibrations are delocalized across the hydrophobic interface of a transmembrane helix dimer. Proc Natl Acad Sci USA. 2006;103:16740–16745. doi: 10.1073/pnas.0608243103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.