

Abstract
Pulsed field gel electrophoresis (PFGE) offers a high-resolution approach to quantify chromosomal fragmentation in bacteria, measured as percent of chromosomal DNA entering the gel. The degree of separation in PFG depends upon the size of DNA, as well as various conditions of electrophoresis, such as electric field strength (FS), time of electrophoresis, switch time and buffer composition. Here we describe a new parameter, the structural integrity of the sample DNA itself, that influences its migration through PFGs. We show that sub-chromosomal fragments containing both spontaneous and DNA damage-induced nicks are prone to breakage during PFGE. Such breakage at single strand interruptions results in artefactual decrease in molecular weight of linear DNA making accurate determination of the number of double strand breaks difficult. While breakage of nicked sub-chromosomal fragments is FS-independent, some high molecular weight sub-chromosomal fragments are also trapped within wells under the standard PFGE conditions. This trapping can be minimized by lowering the field strength and increasing the time of electrophoresis. We discuss how breakage of nicked DNA may be mechanistically linked to trapping. Our results suggest how to optimize conditions for PFGE when quantifying chromosomal fragmentation induced by DNA damage.
Keywords: field strength, time of electrophoresis, DNA trapping, single-strand breaks, double-strand breaks
Introduction
Chromosomal lesions lead to cell cycle arrest in eukaryotes and block progression of replication forks in prokaryotes, posing serious problems for the accurate and timely duplication of the genome and its physical integrity [1; 2; 3]. While all DNA lesions, including single stranded nicks and gaps (SSNs and SSGs) are harmful, unrepaired double-strand breaks (DSBs) are the most deleterious ones and can cause genome loss and cell death [4; 5; 6; 7]. Studies on the mechanisms of chromosomal fragmentation are of critical importance for two reasons. First, all known DNA damaging agents, both naturally occurring and man-made, induce chromosomal fragmentation in replicating cells [8; 9; 10]. Second, cells that cannot repair fragmented chromosome, die, while those that can, experience genome instability.
Quantification of chromosomal fragmentation relies on physical methods with a long linear range of detection, and pulsed field gel electrophoresis has proven to be the best technique for this purposes. Conceived and developed in 1980’s, PFGE fractionates chromosome-size DNA molecules ([11; 12], for reviews see [13; 14; 15]), and has been used extensively in the areas of genome characterization ([16; 17; 18], reviewed in [19; 20]), and epidemiological studies ([21; 22], reviewed in [23; 24]). In PFGE, the DNA is subjected to alternating electric fields with a constant reorientation angle over a long period of time, and the migration of DNA is controlled by a variety of factors including field strength (FS), pulse time, gel concentration and buffer composition, and temperature and time of electrophoresis [25; 26]. A peculiar characteristic of the PFGE is that even 30 kbp relaxed circular DNA molecules cannot enter the gel [27; 28; 29; 30], whereas linear fragments of up to 10 Mbp do [31; 32; 33]. This property makes PFGE an ideal technique for chromosomal fragmentation studies in bacteria, where the linearized chromosomes and chromosomal fragments are efficiently separated from the (unbroken) circular chromosome because the former enter the gel, whereas the latter stays within the wells [5; 6; 34; 35; 36; 37].
The use of pulsed field gel electrophoresis to quantify chromosomal fragmentation in bacteria was initiated by Michel et al in 1997 [37] who based her approach on an earlier demonstration in yeast that 1) a circular 305 kbp chromosome does not enter the gel; 2) it does enter the gel when it is linearized [33]. The technique has since been successfully used in a number of chromosomal fragmentation studies [5; 6; 34; 35; 38; 39; 40]. However, with the exception of the high energy ionizing radiation (gamma- and X-rays), DNA damaging conditions rarely generate direct DSBs. At the same time, no one has ever asked if single-strand interruptions in duplex DNA, generated in vivo either in the course of repair of DNA damage or due to DNA metabolism defects, could influence its electrophoretic behavior during PFGE.
Recently, while studying the mechanisms of UV-induced chromosomal fragmentation in polA and ligA mutants of E. coli that are defective in joining of Okazaki fragments in the newly-replicated DNA, we suspected that long DNA molecules were broken at the nicks due to high electric field strength (FS) used during standard PFGE conditions. While testing this idea, we found that nicked linear DNA indeed was further fragmented during PFGE. However, contrary to our expectation, reducing FS did not reduce the conversion of nicks into breaks, demonstrating that this phenomenon was FS-independent. While we detected FS-independent breakage of high-molecular weight (HMW) DNA at nicks in a variety of experiments, including assays with defined sub-chromosomal fragments, we found that one particular batch of agarose caused especially prominent nick-dependent trapping of large chromosomal fragments within wells. These nicked HMW species could be forced into the gel by lowering the FS and increasing the time of electrophoresis, but once these nicked species entered the gel, they were further broken, likely at pre-existing nicks, mechanistically linking breakage at nicks with trapping inside the gel. Our findings demonstrate that care should be taken when interpreting molecular weight distribution of the chromosomal fragmentation results if the DNA is expected to contain nicks.
Materials and methods
Bacterial Strains, Growth Conditions and Chemical Reagents
All E. coli strains used in this study are K-12 derivatives and are described in Table 1. All strains were grown in LB (10 g of tryptone, 5 g of yeast extract, 5 g of NaCl/liter of broth, pH to 7.4 with 250 μl of 4 M NaOH; LB agar contained 15 g of agar/liter of LB broth) at 28 °C unless stated otherwise. When required, antibiotics were added to the following final concentrations in μg/ml: ampicillin, 100; kanamycin, 50; tetracycline, 10. DNA-damaging agents were used at the following final concentrations: AZT, 500 ng/ml; phleomycin, 2.5 or 10 g/ml; nalidixic acid, 25 g/ml; fluorouracil, 100 g/ml; hydroxyurea, 100 mM. Agarose used in this study was obtained from Denville Scientific, Fisher and Gibco BRL, whereas enzymes used in various assays were purchased from New England Biolabs.
Table 1.
Strains used in this study.
| Strain | Relevant genotype | Source or reference | Properties |
|---|---|---|---|
|
| |||
| AB1157 | wild-type strain | [58] | Repair proficient |
| SK129 | recB270(Ts) recC271(Ts) | [59] | Defective in DSB repair and DNA degradation |
| AK107 | SK129 dut-1 zic4901::Tn10 | [38] | Defective in DSB repair and also incorporates uracil in DNA |
| AK25 | AB1157 polA12(Ts)::Tn10 | Lab Collection | Defective in excision DNA repair |
| GR501 | GR523 ligA251(Ts) | [60] | Defective in sealing DNA nicks |
| LA20 | GR501 ypeB::kan | [61] | Defective in sealing DNA nicks |
| SRK312 | SK129 polA12(Ts)::Tn10 | SK129 × P1 AK25 | Defective in DSB repair and excision repair |
| SRK322 | SK129 ligA251(Ts) | SK129 × P1 LA20 | Defective in DSB repair and sealing DNA nicks |
| SRK301 | SK129 ΔuvrA277::Tn10 | [5] | Defective in DSB- and nucleotide excision repair |
Chromosomal Fragmentation
Labeling of the chromosomes of growing cultures with 32P, as well as UV treatments, were done exactly as described in Khan and Kuzminov [5]. Briefly, cells were grown aerobically at 28 °C in the presence of 2–4 μCi/ml [32P] orthophosphoric acid (MP Biomedicals) until they reached A600 ~ 0.3. At this point, DNA damaging agents were added directly to the cultures, which were then transferred to 37°C shaker for 2 hours. For UV assays, the cultures were harvested, and the cell pellets were suspended in volumes of sterile 1% NaCl containing 0.01% Triton X-100 (Sigma) to yield an absorbance of approximately 0.6. The UV-irradiation was performed at 28°C in a dark room lit by yellow lamps (F15T8-GO, General Electric) to avoid photoreactivation. Following irradiation, the cultures were diluted 1:1 with a sterile no-salt 2xLB solution and transferred to 37°C shaker for either 10 min or 2 hours. For spontaneous chromosomal fragmentation studies, the strains were labeled, harvested and concentrated in Triton X-100 + NaCl as described above for the UV assay. After normalizing the suspensions to absorbance of ~0.6, the cultures were diluted 1:1 with 2xLB (without NaCl) and transferred to 37°C shaker for 2 hours.
Agarose plugs and PFGE
After completion of the required incubations, cultures from a volume of 0.5 ml were harvested for each plug. The cell pellets were washed once in 1 ml of sterile TE buffer (10 mm Tris-HCl, 1 mm EDTA, pH 8.0) and resuspended in 60 μl of TE. To make agarose plugs, 5 μl of 5 mg/ml proteinase K solution (Roche Applied Science; final concentration in plugs 200 μg/ml) and 65 μl of molten agarose in lysis buffer (1.2% agarose in 1% lauroylsarcosine, 50 mm Tris-HCl, 25 mm EDTA, pH 8.0) were mixed with the cells, and the suspensions were transferred to plug molds (Bio-Rad). The solidified agarose plugs were submerged in 1 ml of the lysis buffer (1% lauroylsarcosine, 50 mm Tris-HCl, 25 mm EDTA, pH 8.0) and incubated overnight at 60°C. After completion of digestion, the lysis buffer was replaced with TE, and the plugs were stored at 4°C until used. Unless specifically mentioned, ½ plugs were used for electrophoresis.
In some assays, in which plugs were to be restriction-digested, a modified method of plug lysis was used. In this procedure, cell pellets were washed and suspended in 65 μl of TE, mixed with 65 μl of molten plain agarose (1.2% agarose in water) and transferred into plug molds. Once solidified, these plugs were shifted to glass tubes containing 1 ml of 1 mg/ml lysozyme in TE and incubated at 37°C for 1 hour. Following this, the plugs were removed from TE and transferred to fresh tubes containing 1 ml of 1 mg/ml ProteinaseK in lysis buffer (1% lauroylsarcosine, 50 mm Tris-HCl, 25 mm EDTA, pH 8.0). The tubes were shifted to 60°C and plug lysis was continued for 15–18 H.
All electrophoresis runs were performed on 1% agarose gels in 0.5X Tris-borate-EDTA buffer at 12°C, in a Bio-Rad CHEF-DRII pulsed field gel electrophoresis system operating with initial and final switch times of 60 and 120 s, respectively. Field strengths between 1 V/cm and 6 V/cm and time of electrophoresis between 10 hours and 150 hours are specified in description of individual experiments. In most of the gels, yeast chromosome PFG marker or lambda ladder PFG marker (both from New England Biolabs) were also included.
Following the electrophoresis, the ladder lanes were cut off, stained with ethidium bromide, and photographed. The rest of the gel was dried under vacuum, exposed to a phosphorimager screen and scanned with an FLA-3000 fluorescent image analyzer (FujiFilm). The data were processed using Image Gauge version 3.41 software (FujiFilm).
Calculations
The percentage of chromosomal fragmentation was calculated as the signal in the lane below the well divided by the combined signal of the lane plus well and multiplied by 100. The percentage of signal within the wells was calculated as the signal in the wells divided by the combined signal of the lane plus the well and multiplied by 100. In order to dissect the fragmented chromosomes into size zones under various conditions of electrophoresis, scanned gels were aligned with the stained ladder. The percentage of signal within a particular zone was calculated as the signal in this zone divided by the combined signal of the lane plus the well and multiplied by 100.
Restriction digestions of agarose plugs
Lysed plugs were washed and processed as described [35]. Briefly, overnight-digested plugs were transferred to fresh tubes and washed for 2–3 hours with 6–8 changes of 1 ml of TE buffer. After completion of washing, the plugs were submerged in 100–150 μl of 1X NEBuffer 3 (NEB) containing BSA and 50–150 units of NotI (New England Biolabs) and incubated at 37°C for 15–18 H. The reactions were stopped by adding 1 ml of ice-cold TE, and digested plugs were stored at 4°C until ready to load.
Results
Strains with nicked DNA yield fragmented chromosomes
While PFGE offers excellent separation of chromosome-size linear fragments, it is not known how introduction of ss-nicks and -gaps in these fragments will influence their integrity during electrophoresis. Since in our chromosomal fragmentation studies we specifically work with strains that are expected to have single-strand interruptions in their chromosomal DNA, either as a result of ongoing repair of massive DNA damage, or because of defects in sealing DNA nicks, we were concerned that these nicks and gaps could be converted into DSBs during our standard PFGE (6 V/cm for 24 H, also see Materials and methods). Suspecting that 6 V/cm field strength might be too harsh for nicked DNA, we expected that reducing the field strength would decrease chromosomal fragmentation in strains containing nicks in their DNA.
To verify this reasoning, we measured spontaneous chromosomal fragmentation in a set of strains with varying number of expected single-strand interruptions in their DNA: wild type, recBC (Ts), recBC(Ts) polA12 (Ts), recBC(Ts) ligA251(Ts) and recBC(Ts) dut-1. Since in recBC(Ts) background, there is no repair of DSBs and no degradation of linear DNA at the non-permissive temperature [41], the full extent of chromosomal fragmentation is preserved and can be quantified. No nicks were expected in DNA of wild type and recBC(Ts) control strains, and chromosome fragmentation is at the background level in these strains [5]. Based on the strength of other mutant alleles, recBC(Ts) ligA251(Ts) was expected to have the highest density of nicks in the chromosomal DNA, whereas the recBC(Ts) dut-1 and recBC(Ts) polA12(Ts) strains were expected to have intermediate number of nicks.
When we compared the chromosomal fragmentation among these strains after running at 6 V/cm for 24 H, we found that recBC(Ts) ligA251(Ts) indeed showed the highest spontaneous chromosomal fragmentation over the wild type and recBC(Ts) controls, with both controls showing similar, low levels of fragmentation (Fig. 1A, the white bars). The recBC(Ts) polA12(Ts) and recBC(Ts) dut-1 strains showed intermediate fragmentation, (Fig. 1A, the white bars). This data indicated a direct relationship between the expected densities of nicks and gaps in the chromosomal DNA of the mutant and its chromosomal fragmentation. Such a direct relationship between the number of nicks and chromosomal fragmentation has been generally interpreted to represent replication fork collapse at preexisting nicks in template DNA [42; 43]. However, it was also possible that DSBs were generated at the nicks as artifacts of PFGE, because the field strength was too high (6 V/cm). If true, reducing the field strength would eliminate or reduce chromosomal fragmentation in strains containing higher density of nicks and gaps in their DNA.
Figure 1.
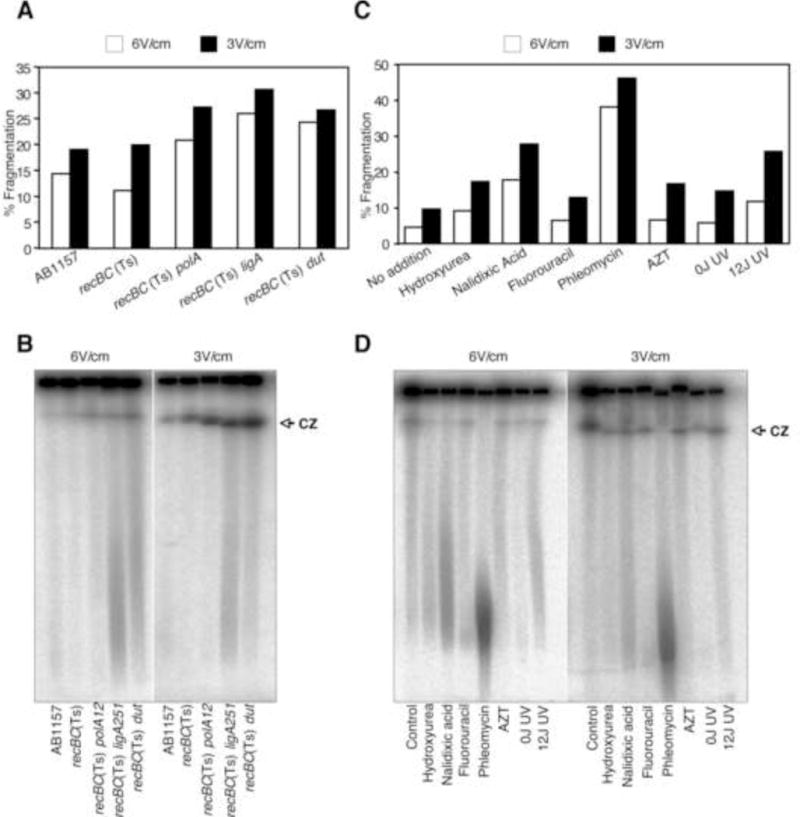
Effect of electrophoretic conditions on the extent of spontaneous and DNA damage-induced chromosomal fragmentation. Samples were run on a 1% agarose gel at 12°C in 0.5X TBE with 60–120 seconds switch time, either at 6 V/cm for 24 H, or at 3 V/cm for 72 H.
(A) Quantification of spontaneous fragmentation in AB1157, recBC(Ts), recBC(Ts) polA12, recBC(Ts) ligA251 and recBC(Ts) dut-1 after growth of the strains at 37°C for 2 hours.
(B) The corresponding gel from which the quantification in “A” was derived. CZ, compression zone.
(C) Quantification of chromosomal fragmentation in recBC(Ts) cells upon exposure to a variety of DNA-damaging agents. Cells were treated with clastogens as described in materials and methods and transferred to 37°C for 2 hours before the preparation of plugs.
(D) The corresponding gel from which the quantification in “C” was derived. CZ, compression zone.
To test the idea, we ran the remaining, identical plugs from the same experiment at reduced field strength (3 V/cm) for 70 H (“long run at low field strength” or LLFS conditions), so that the fastest chromosomal species migrated to about same distance as with 6 V/cm for 24 H (“short run at high field strength” or SHFS conditions) (Fig. 1B). Contrary to our expectation, we observed an overall increase in chromosomal fragmentation in LLFS conditions compared to SHFS conditions in all strains. The relative fragmentation stayed the same, with recBC(Ts) ligA251(Ts) still exhibiting the highest fragmentation (Fig. 1A, the black bars). Since the pulling forces were lower at 3 V/cm, the increase in fragmentation made it unlikely that fragmentation seen at 6 V/cm electrophoresis was due to high field strength-dependent breakage of chromosomal DNA at the nicks. A set of experiments done using a different batch of agarose from the same source (“DBA experiments”), showed an even higher increase in strains with nicked chromosomes in LLFS conditions (Fig. S1A).
Irrespective of agarose, compression zone showed the biggest difference between the two running conditions (Fig. 1B, and S1B). First, there was more DNA in the compression zone after LLFS electrophoresis, and this compression zone enhancement seemed nick-dependent, as AB1157 showed the least absolute increase. And second, while the compression zone of all strains migrated equally when the electrophoresis was done at 6 V/cm, the compression zones of the strains showing high fragmentation, recBC(Ts) ligA251(Ts) and recBC(Ts) dut-1, migrated faster than the rest when ran at 3 V/cm. This indicated that the chromosomal fragments running in the compression zones were of different sizes and were generally shorter when the expected density of nicks in the chromosomal DNA was higher.
PFGE similarly affects DNA damage-induced chromosomal fragmentation
The uniform increase in fragmented DNA upon LLFS PFGE encouraged us to test the effect of conditions of electrophoresis on the DNA damage-induced chromosomal fragmentation. Unlike spontaneous fragmentation in the mutant strains, in DNA damage-induced fragmentation assays we could control the extent of fragmentation by varying the clastogen doses. For this assay, we exposed recBC(Ts) strain to a variety of DNA damaging agents (Fig. 1C and D, and S1C and D). These agents range from nalidixic acid, which generates pure DSBs [44; 45], to AZT, which is proposed to produce SSGs in the newly-replicated DNA [46], to UV lesions, excision of which could produce SSNs, while encounter with replication forks could generate SSGs and DSBs [5; 47]. As shown in Fig 1C, various DNA damaging treatments resulted in varying levels of chromosomal fragmentation, with phleomycin being the most potent agent in SHFS electrophoresis conditions. Hydroxyurea, fluorouracil and AZT yielded low levels of chromosomal fragmentation under these conditions. Similar to the profile obtained in spontaneous fragmentation assay, reducing the field strength to 3 V/cm resulted in a uniform increase in fragmentation in all samples irrespective of the expected density of nicks. In an identical DBA experiment, the increase in fragmentation remained low in strains without DNA damage, or with treatments that failed to yield fragmentation at 6 V/cm, but showed significant increase with nalidixic acid- and phleomycin-treated cells (Fig. S1C). Overall, and similar to the results observed with spontaneous fragmentation (Fig. 1B), there was a general enhancement in the compression zone signal in LLFS PFGE (Fig. 1D and S1D). Moreover, treatment with DNA-damaging agents again produced faster-migrating compression zone under LLFS PFGE.
At face value, the data in figure 1 indicate that reducing the field strength while increasing the time of electrophoresis results in higher detectable fragmentation irrespective of the presence of additional damage. Comparison of data in figures 1 and S1 also implies that additional factors, including agarose, influence the level of detectable chromosomal fragmentation. The results in figure 1 were reproducible with agarose from three different sources (see Methods).
Enhancement in fragmentation is accompanied by signal loss from the wells
The general increase in fragmentation in LLFS PFGE conditions prompted us to look for the source of additional linear DNA. We noticed some decrease in the absolute signal within the wells with corresponding increase in chromosomal fragmentation (Fig. 1B and D, compare 6 V/cm with 3 V/cm). Quantification of signal within wells indeed showed some signal loss after LLFS under both spontaneous and DNA-damaging conditions (Fig. 2AB). The reduction in signal intensity within wells was significantly higher in DBA experiment and was correlated with the presence of nicks (Fig S2AB). Specifically, recBC(Ts) ligA251(Ts), which had shown the highest increase in chromosomal fragmentation, exhibited the highest decrease in signal within the wells (Fig. S2AB). Taken together, the data in Figs. 2 and S2 show that increase in the chromosomal fragmentation during LLFS PFGE is due to release of chromosomal DNA from wells.
Figure 2.
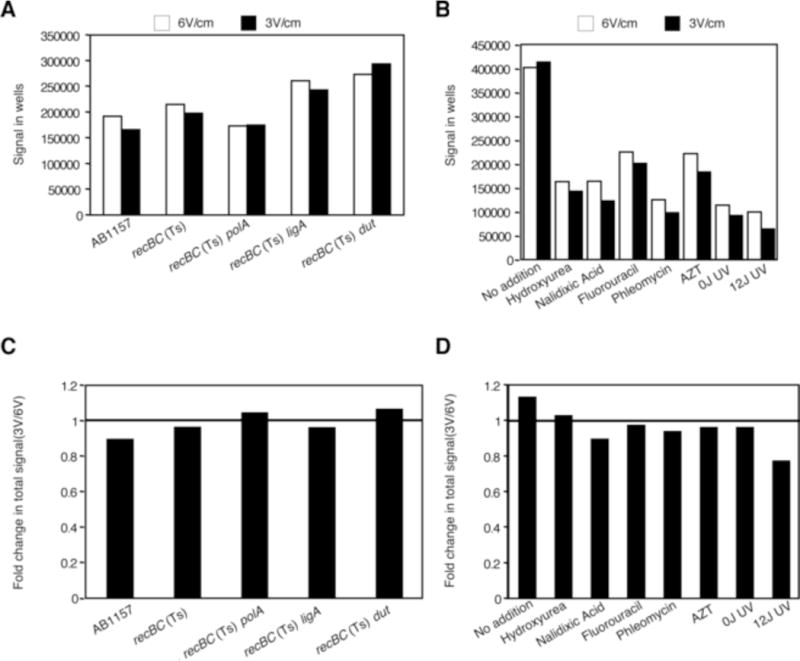
Effect of electrophoretic conditions on the amount of signal staying within the wells (A and B); and total signal (well+lane) (C and D). Values are derived from the gels in Fig 1.
(A) Differences in the absolute amount of signal (arbitrary units) within wells in strains undergoing spontaneous fragmentation.
(B) Differences in the absolute amount of signal (arbitrary units) within wells in recBC(Ts) cells exposed to a variety of DNA damaging agents.
(C) Decrease in the total signal in strains undergoing spontaneous fragmentation, presented as ratios of the total signal in the LLFS conditions divided by the total signal from the same plug in the SHFS conditions.
(D) Decrease in the total signal in strains exposed to various clastogens (calculated as in “C”).
There is a total loss of DNA with LLFS PFGE
The decrease of signal in the wells in the LLFS conditions, while explaining the corresponding signal appearance in the lanes, meant that some linear species were trapped in the wells during the 6 V/cm electrophoresis, but entered the gel during the 3 V/cm electrophoresis. While trapping of HMW DNA during PFGE was shown before [26], whether DNA damage also causes trapping of low-molecular weight (LMW) species has not been determined. If LMW species were indeed retained within wells during 6 V/cm PFGE, we expected them to be released and the smallest fragments to run out of the gel during LLFS electrophoresis. In order to ascertain if there is an overall signal loss coupled with the release of signal from wells, we compared ratios of the total signals obtained at 3 V/cm to those at 6 V/cm. We found no systematic signal loss upon 3 V/cm electrophoresis under spontaneous fragmentation conditions (Fig. 2C). However, the clastogen-treated samples, specifically UV, AZT, nalidixic acid and phleomycin, showed minor systematic decrease in total signal upon LLFS compared to SHFS conditions (Fig. 2D). Also, there was a significant and increasing loss of the total signal under conditions of increasing DNA nicking in the DBA experiments (Fig. S2CD). These results, combined with the fragmentation and loss of signal from the wells, suggested generation of LMW species from the nicked DNA, that run out of the gels during slower-longer electrophoresis. We conclude that some of the species released from the wells are either low molecular weight to begin with or are converted into low molecular weight species during slower-longer electrophoresis.
Resolution of PFG depends upon field strength
The facts that DNA-damaging conditions resulted in faster migrating compression zone, but only upon LLFS PFGE (Fig. 1B, D and S1B, D), and that the compression zone completely disappeared in polA and ligA mutants upon treatment with high UV doses at both field strengths (see below), prompted us to analyze the composition of the compression zone. Although in both electrophoresis conditions, 6 V/cm for 20–24 H and 3 V/cm for 70–72 H, the overall pattern of the smear of fragmented chromosomal DNA appeared the same, inclusion of molecular weight standards revealed the difference in resolution. Specifically, while the faster migrating bands indeed traveled the same distance under the two conditions, there was a significant loss of resolution of high MW species upon LLFS PFGE (Fig. S3). Molecular weight markers showed that LLFS PFGE resolved DNA fragments in the size range of 48.5–680 kbp, with DNA fragments of size 0.7 MB–2MB migrating as one thick band, forming the compression zone. This result explained the thicker compression zone upon LLFS PFGE as merely reflecting a broader compressed region than the one seen with SHFS PFGE.
UV dose-dependent fragmentation is affected by nicks and PFGE conditions
After initially testing a variety of clastogens for chromosomal fragmentation, we chose UV irradiation as our standard DNA-damaging treatment, because the mechanism of nucleotide-excision repair of UV lesions, as well as the mechanism of UV-induced chromosomal fragmentation, are both known [5 and references therein; 47]. To test if PFGE conditions affected the size distribution and quantity of fragmented chromosomes in a manner that depended upon the expected number of repair nicks, we exposed cultures of recBC(Ts), recBC(Ts) polA12(Ts) and recBC(Ts) ligA251(Ts) mutants to various doses of UV and incubated the UV-exposed cells in growth conditions at 37 C for 10 min, concentrating on the so-called “early fragmentation”. In the polA and ligA mutant cells, where excision repair cannot be completed, the final density of accumulating nicks may be reaching the density of UV-lesions (which is ~30 pyrimidine dimers per genome equivalent per 1 J/m2[1]). The plugs were then electrophoressed at 6 V/cm for 20 H or at 3 V/cm for 70 H. As shown in figure 3A, with recBC(Ts), a UV dose-dependent faster migrating compression zone was observed upon 3 V/cm PFGE. Quantification of the gel showed an increase in fragmentation at all UV doses (Fig. 3B). With recBC(Ts) polA12(Ts) (Fig. 3C) and recBC(Ts) ligA251(Ts) (Fig. 3E), however, the compression zone disappeared with UV doses higher than 8 J/m2 at both field strengths, indicating significant additional fragmentation of subchromosomal fragments in response to the increased density of excision repair events. The compression zone observed with 0 J/m2 and 4 J/m2, on the other hand, showed increased signal with LLFS PFGE. The disappearance of compression zone was not accompanied by decrease in chromosomal fragmentation, as both strains showed dose-dependent increase in fragmentation upon 6 V/cm PFGE, which was further enhanced upon 3 V/cm PFGE (Fig. 3D and F). While overall UV-induced fragmentation was higher in recBC(Ts) polA and recBC(Ts) ligA mutants, as compared to recBC(Ts) parent strain (compare Fig. 3 B, D and F), the enhancement in fragmentation at 3 V/cm did not show nick-dependence.
Figure 3.
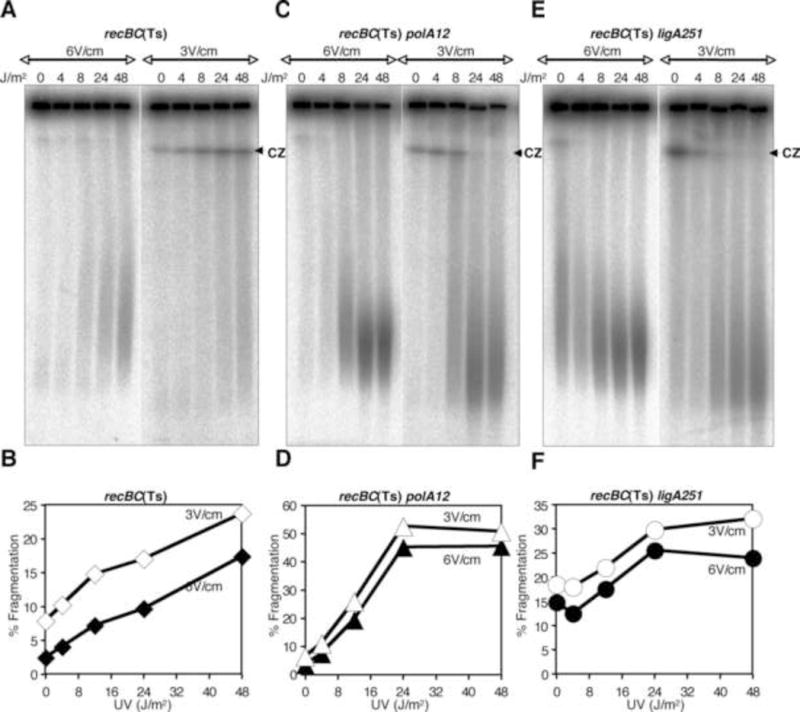
UV dose-dependence of chromosomal fragmentation in recBC(Ts) or its derivatives harboring additional mutations in polA or ligA genes. Agarose plugs were made after incubating the UV-exposed cultures at 37°C for 10 minutes. Plugs were run either at 6 V/cm for 24 H or at 3 V/cm for 72 H as described in Fig. 1. Panels A, C and E are radiograms of the gels. Panels B, D and F show quantification of fragmentation from the corresponding gels. CZ, compression zone.
In an identical experiment done with DBA, in which similar qualitative changes in the compression zones were observed (Fig. S4A, C, E), the increase in fragmentation seen at 3 V/cm was significantly higher in recBC(Ts) polA and recBC(Ts) ligA mutants, as compared to control (Fig. S4B, D, F). As expected, higher signal in the lanes was due to release of signals from the wells (Fig. S5A). Also, there was decrease in the total signal when samples were electrophoressed at 3 V/cm compared to 6 V/cm, but only in polA and ligA mutants (Fig. S5B). The general interpretation of both sets of results is that the defect in repair of single-strand DNA interruptions, when combined with the increasing amount of DNA damage, increases chromosomal fragmentation (Figs. 3BDF and S4BDF). In terms of PFGE, we conclude that some of the generated linear DNA is trapped in the wells in standard field strength of 6 V/cm, but is released at reduced field strength of 3 V/cm, which is especially evident in the DBA conditions. Interestingly, while trapping of nicked DNA could be influenced by FS, dramatic reduction in the size of the fragments is not (Fig. 3ACE and S4ACE). We suggest that trapping at high FS could be conditional, but the presence of nicks always causes the additional fragmentation of linear DNA.
Zonal dissection of chromosomal fragmentation shows the importance of nicks and gaps
Since compression zone was absent in polA and ligA mutants exposed to high doses of UV under both conditions of electrophoresis, and there was an enhancement in total fragmentation upon LLFS PFGE (Fig. S4), qualitative distribution of fragmented DNA must have changed between the LLFS and SHFS PFGE conditions. To quantify this change in distribution, we subdivided total fragmentation shown in figure S4 into two regions: the region covering fragmented DNA ≤0.7 Mbp in size and the complementary region covering everything that was ≥ 0.7 Mbp up to the wells. The recBC(Ts) strain, which shows an overall modest dose-dependent increase in fragmentation (Fig. S4B), showed a UV dose-dependent enhancement in both regions of fragmentation upon SHFS-and LLFS-conditions (Fig. S6A). In contrast, both recBC(Ts) polA and recBC(Ts) ligA mutants showed a slight UV dose-dependent reduction in signal from the ≥ 0.7MB region when ran under the LLFS conditions (Fig. S6B and C) and no dose-dependent change when the electrophoresis was performed in the SHFS conditions. Signal in the region containing DNA of size range ≤0.7 Mbp, however, showed significant UV dose-dependent increase upon SHFS PFGE, and the signal in this fragmentation zone was further enhanced when field strength was reduced (Fig. S6B and C). We conclude that the main increase in LMW fragmented species at LLFS PFGE is due to the release of the nicked HMW chromosomal fragments from the wells. However, once these species were released from the wells in LLFS PFGE conditions, they were further broken, presumably at the nicks, leading to accumulation of faster migrating fragments. While our interpretation is supported by the observation that, upon LLFS PFGE, the amount of high molecular weight DNA is reduced only in polA and ligA mutants that accumulate nicks in their DNA (Fig. S6ABC), release of well-trapped LMW species could also contribute to the accumulation of faster migrating species.
Time of electrophoresis is important
So far we have been changing two electrophoretic parameters at the same time, field strength and time of electrophoresis, and observing their combined effect on chromosomal fragmentation. While reducing the field strength from 6 V/cm to 3 V/cm and increasing time of electrophoresis from 20–24 H to 70–72 H resulted in significant enhancement in chromosomal fragmentation in strains that contain nicks and ss-gaps in their chromosomal DNA, it remained unknown which of the two parameters, time of electrophoresis or field strength, played the major role. Since the trapping and fragmentation enhancement upon LLFS was most significant with DBA experiments, we used these conditions to further characterize the phenomenon.
To test the effect of field strength, we exposed recBC(Ts), recBC(Ts) polA12(Ts) and recBC(Ts) ligA251(Ts) to 0 and 48 J/m2 of UV and ran the samples on DBA at 6 V/cm, 5 V/cm, 4 V/cm and 3 V/cm for the fixed time of 20 H. We detected no significant change in the amount of fragmented DNA when the electrophoresis was performed for 20 H irrespective of the field strength or the expected density of nicks in the chromosomal DNA (Fig. 4A). Corresponding gels (Fig. 4B) feature significant compression zones only upon electrophoresis at field strengths of 3 V/cm and 4 V/cm (compare 0 J/m2 lanes). This data indicate that field strength does not influence fragmentation when electrophoresis is performed for the same, short time.
Figure 4.
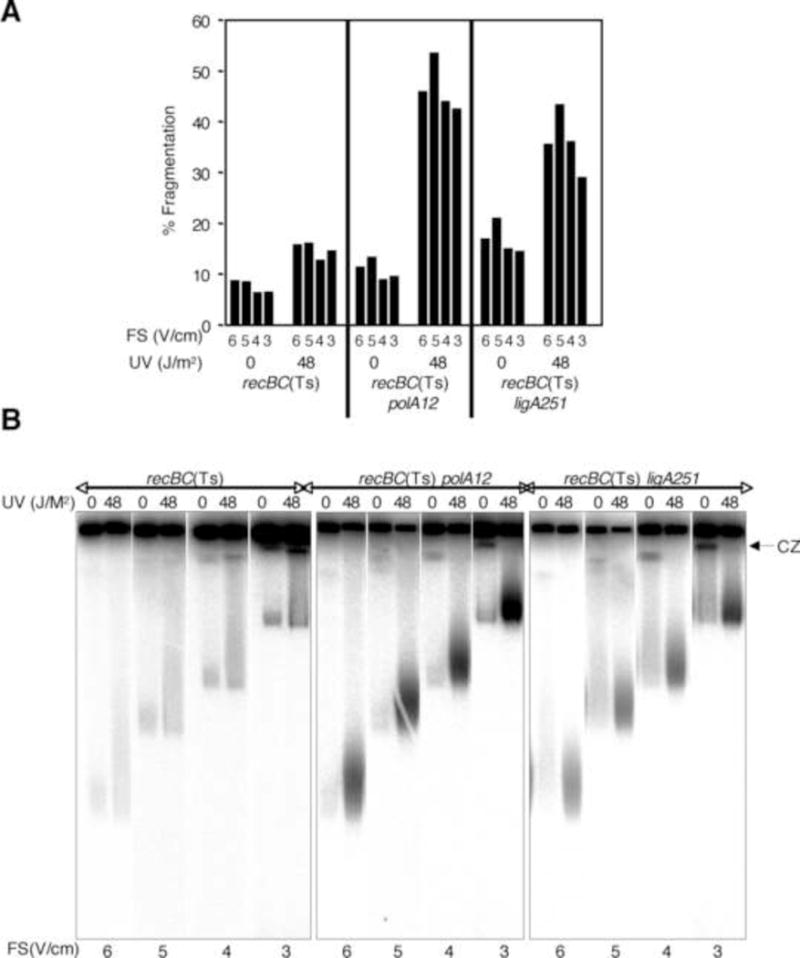
The effect of field strength (at constant run time) on chromosomal fragmentation.
(A) Quantification of fragmentation in recBC(Ts) strain, or its polA or ligA derivatives, exposed to 0- or 48 J/m2 of UV and ran at different field strengths, but all for 20 hours. Data are averages of two independent measurements.
(B) Composite radiograms of the gels from which the data in (A) are derived. FS, field strength; CZ, compression zone.
We then determined the effect of duration of electrophoresis on the enhancement of chromosomal fragmentation. For this assay we again tested both the UV-induced chromosomal fragmentation (Fig. 5A and B) and spontaneous fragmentation (Fig. 5C and D). Increasing the time of electrophoresis from 24 H to 72 H at constant field strength (3 V/cm) resulted in gradual increase in chromosomal fragmentation (Fig. 5A). This increase was observed in all strains, but the overall fragmentation, as expected, was significantly higher in polA and ligA mutants. Corresponding gels presented in figure 5B show significant reduction in the amount of signal within wells with increase in the time of electrophoresis. As described previously, there was less release of the signal from the wells in UV-exposed recBC(Ts) mutant that harbored minimal nicks and gaps, while the released DNA represented all molecular weights. On the other hand, the release of DNA from the wells contributed exclusively to the faster migrating species in the UV-treated recBC(Ts) polA12(Ts) and recBC(Ts) ligA251(Ts) double mutants, that showed no compression zone (Fig. 5B). Such enhancement in chromosomal fragmentation with electrophoresis time was not specific to DNA damage, as similar enhancement in spontaneous fragmentation was also detected upon longer electrophoresis (Fig. 5C and D). Based on the data of figure 5, we conclude that, under the conditions when high FS cause nick-dependent trapping within wells, time of electrophoresis at 3 V/cm determines the release of DNA from the wells. Further breakage of the fragments (presumably at nicks) within the lane does not seem to be dependent upon field strength or duration of the run, though. We propose that, once released from the wells, the DNA can be further fragmented, if it contained nicks, irrespective of the field strength applied. Unfortunately, in this set of experiments, we could not generate the kinetics of chromosomal fragmentation upon electrophoresis at 6 V/cm past 24 hours, as chromosomal fragments started running out of the gel at that time.
Figure 5.
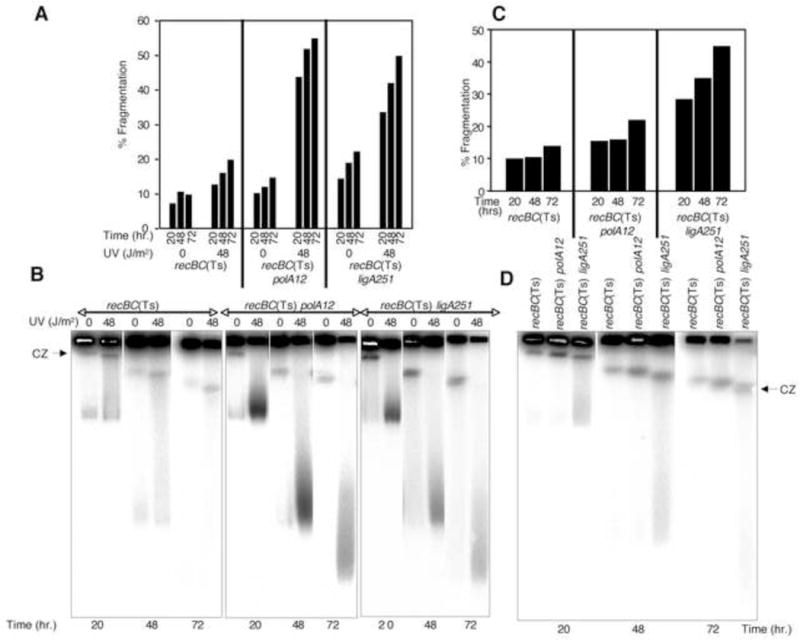
The effect of time of electrophoresis on chromosomal fragmentation.
(A) Quantification of fragmentation in recBC(Ts) strain, or its polA or ligA derivatives, exposed to 0- or 48 J/m2 of UV and ran at 3 V/cm for 20-, 48- or 72 H. Data are averages of two independent measurements.
(B) Composite radiograms of the gels from which the data in (A) are derived. CZ, compression zone.
(C) Quantification of spontaneous fragmentation in recBC(Ts), recBC(Ts) polA12, and recBC(Ts) ligA251 mutants. Corresponding cultures were initially grown at 28°C and then transferred to the non-permissive temperature of 37°C for 2 hours before removal of samples for plugs. After the lysis, the agarose plugs were electrophoressed at a constant field strength of 3 V/cm for a period of 20- 48- or 72 H.
(D) Composite radiograms of the gels from which the data in (C) are derived. CZ, compression zone.
Field strength of electrophoresis is important
The results so far indicated that, by increasing the time of electrophoresis from 24 H to 72 H, at 3 V/cm, we could detect more chromosomal fragmentation under conditions of both spontaneous or induced DNA damage. But was this increase in fragmented DNA only due to increase in time of electrophoresis, or the reduced field strength also played a role? Since we could not directly compare the level of fragmentation between 3 V/cm and 6 V/cm samples ran for 72 H (in the latter, most DNA would run out), we decided to compare the signal remaining in the wells under the two conditions, based on the above finding that increase in fragmentation reflects the decrease in the signal in wells (Fig. S2A and B).
For this particular experiment, we used recBC(Ts), recBC(Ts) polA12(Ts) and recBC(Ts) ligA251(Ts) strains exposed to 0 and 48 J/m2 and subsequently incubated under growth conditions for 10 minutes (the “early” UV-induced fragmentation). We also measured spontaneous fragmentation in these mutants (no UV exposure) after incubation at 37°C for 2 hours. We found that running the samples at 6 V/cm for 20 H or 72 H did not result in any loss of signal from the wells in either UV-irradiated samples (Fig. S7A) or spontaneous fragmentation samples (Fig. S7B), indicating no DNA release from wells during high field strength PFGE. In contrast, when identical plugs were run at 3 V/cm for 20 H and 72 H, there was a significant loss of signal from the wells in UV-exposed samples, especially in UV-exposed recBC(Ts) ligA251(Ts) double mutant (Fig. S7A). Spontaneous fragmentation samples in the recBC(Ts) polA and recBC(Ts) ligA double mutants also showed significant loss of signal from the wells when ran at 3 V/cm for 72 H (Fig. S7B). These results confirmed that both the longer time of electrophoresis and the lower field strength are required to increase the amount of linear nicked DNA entering the gels.
PFGE break the nicked chromosomes
So far our experiments showed that, as compared to 6 V/cm for 20H (SHFS) electrophoresis, 3 V/cm for 70H (LLFS) PFGE resulted in an overall increase in fragmentation, the magnitude of which depended upon a variety of factors including the integrity of the genome and the extent of linear DNA trapping in the wells. However, irrespective of the electrophoresis conditions, comparison of the size of fragmented DNA showed somewhat unexpected results. Upon electrophoresis at both field strengths, the UV-treated cultures of recBC(Ts) polA and recBC(Ts) ligA mutants exhibited same quality (LMW) of fragmented DNA, but the quantity was significantly increased upon LLFS electrophoresis when DNA was maximally trapped (DBA data, Fig. S4 and S5). A similar, though less striking effect was observed when DNase treatment was used to introduce nicks (not shown). The fact that electrophoresis at high field strength also yielded LMW sub-chromosomal fragments suggested that the breakage of nicked DNA is FS-independent. We decided to directly test if breaking the nicked linear DNA is a characteristic of PFGE and also if this phenomenon is influenced by the fragment size.
We addressed these questions by exposing stationary phase cultures (all UV treatments so far have been done with growing cells) of recBC(Ts) and its uvrA mutant derivative to ultraviolet light. Stationary phase cells do not have any active replication forks, so the UV-induced, replication-dependent chromosomal fragmentation should stay low. On the other hand, excision repair of thymine dimers in Uvr+ cells will introduce nicks in the chromosomes in a UV-dose-dependent manner. Thus, stationary cells should have allowed us to test whether nicked circular DNA is broken during PFGE and enters gels as sub-chromosomal fragments. In the second part of the experiment, we digested one set of plugs with NotI to create precise sub-chromosomal fragments and electrophoressed the samples at 3 V/cm and 6 V/cm to determine if specific nicked fragments lose intensity in a size- and FS-dependent manner. Use of stationary cells allowed us to quantitatively detect the 1 Mbp NotI fragment, which is underrepresented in growing cells, most likely due to replication forks going through it [35].
When we treated these UvrA+ and UvrA− strains with a variety of UV doses, and incubated the exposed cells in spent culture medium (to avoid replication initiation) for 10 min and 2H, the UV-induced fragmentation stayed the same independent of the UV dose (Fig. S8AB) confirming the absence of replication forks, and indicating that circular DNA in the wells does not break at nicks (see discussion). At the same time, the subchromosomal fragments that did enter the gel were additionally broken at excision nicks (Fig. S8B, compare UvrA+ cells irradiated with 0 and 24 J/m2). Then we digested one set of plugs with NotI so that as much as 90% of the DNA entered the gel upon electrophoresis at 6 V/cm for 20H (data not shown). In order to find if nicks, introduced by excision of UV lesions in UvrA+ cells, resulted in the trapping of sub-chromosomal fragments at 6 V/cm electrophoresis, we electrophoressed the NotI digested plugs at 3 V/cm for 70H. As shown in figure S8C, the signal in wells did not show any significant difference under two electrophoresis conditions, indicating that under these conditions the presence of nicks did not significantly trap the largest NotI fragment (1 Mbp) in wells during SHFS. Instead, there was a UV-dependent reduction in the amount of 1 Mbp band upon 6 V/cm electrophoresis in UvrA+ cells (Fig 6A B), suggesting breakage of this long fragment at excision nicks and ss-gaps. This conclusion is further supported by the absence of such decrease in the uvrA mutant strain, which cannot initiate excision repair (Fig. 6B). Unfortunately, due to severe compression of DNA longer than 0.7 MB during LLFS (Fig. S3), the accurate resolution of 1 Mbp fragment was not possible in gels electrophoressed at low field strength. However, since compression zones of NotI-digested samples should primarily consist of 1 Mbp fragment, we compared this region for any UV-dependent signal loss. As shown in figure 6C and D, LLFS also caused breakage of 1 Mbp fragment when the strain was excision-proficient. In order to test if breakage at nicks was restricted to large fragments, we compared smaller NotI fragments for nick-dependent breakage. The drawback of this approach was that evaluation of field strengths was not possible since SHFS didn’t resolve smaller fragments well. Nevertheless, we compared the intensity of the 360 kbp NotI doublet (fragment II in figure 6D) in LLFS and found that this fragment did not show any signal loss between 0 and 24 J/m2 UV doses (Fig S8D). At 24 J/m2, there was some signal loss but the decrease was insignificant and excision-independent, indicating that nicked DNA fragments in the size ranges of 200–300 kb are not readily broken during PFGE. However, when we used UV doses of 48 J/m2, not only the fragments in the size ranges of 200–300 kb were also decreased under LLFS conditions (Fig. S9D and E), but also there was excision-dependent increase in fragmentation of uncut chromosome (Fig. S9AB), suggesting double-strand breaks in circular DNA in the wells. Similar to the profile obtained with lower UV doses, there was excision-dependent loss in the 1 Mbp/CZ signal upon 6 V/cm and 3 V/cm electrophoresis (Fig. S9C). Similar results were obtained with exponentially growing cells (not shown). Taken together, these results show that, given enough nicks, any DNA is prone to breakage when subjected to PFGE. The fact that under both SHFS and LLFS conditions the nicked chromosomal fragments were broken confirms that field strength does not play a role in breaking the nicked sub-chromosomal fragments.
Figure 6.
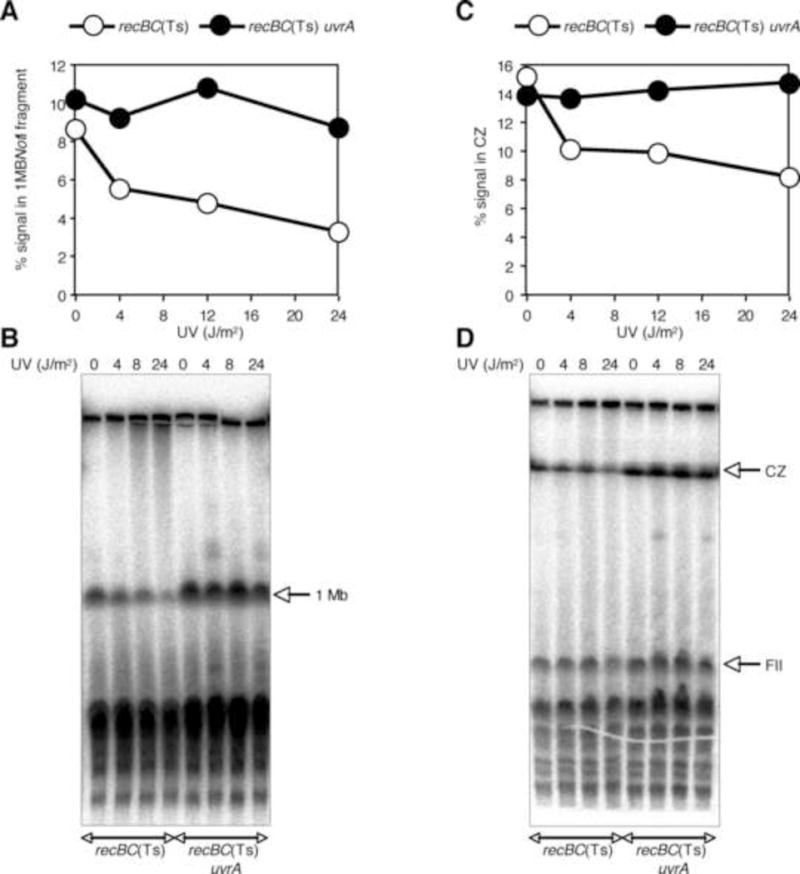
The effect of nicks on the quantity of NotI-digested sub-chromosomal fragments. Stationary cultures of recBC(Ts) and recBC(Ts) uvrA mutants were exposed to a variety of UV doses, and plugs were made after 10 min incubation in spent culture medium. One set of plugs was digested with NotI and electrophoressed, together with non-NotI digested plugs, at 6 V/cm for 20 H and 3 V/cm for 70 H.
(A) Quantification of the amount of signal, presented as % of the total signal, in 1 Mbp NotI fragment after electrophoresis at 6 V/cm for 20 H.
(B) The radiogram of the gel from which the data in (A) are derived.
(C) Qauntification of the amount of signal, presented as % of the total signal, in the compression zone after electrophoresis at 3 V/cm for 70 H.
(D) The radiogram of the gel from which the data in (C) are derived.
Discussion
The present study was undertaken to determine if linear E. coli chromosome, or its sub-chromosomal fragments, further break during their migration through the PFGs because of the presence of nicks. While such breakage could not be visible in ethidium bromide staining of PFGs, or was not of particular concern for those interested in qualitative determination of linear DNA, the occurrence of this breakage would be important for quantification of DNA damage-induced chromosomal fragmentation. Nicked chromosome is an obligatory intermediate during DNA damage repair and is also present in mutants of E. coli defective in either DNA ligase or DNA polymerase I functions that repair these nicks. Specifically, we tested if changing the field strength (the pull force) or the time of electrophoresis (the duration of pulling) caused further breakage of HMW linear DNA containing nicks.
Here we demonstrate that, at the quantitative level, spontaneous or clastogen-induced chromosomal fragmentation depended upon both the field strength and the time of electrophoresis (Table S1, Fig. 4, 5 and S7). Electrophoresis at 3 V/cm, but not at 6 V/cm, showed enhanced fragmentation, but only after 72 H electrophoresis time (Fig. S7), with both field strengths exhibiting comparable fragmentations after 20 H of electrophoresis (Fig. 4). We also observed that LLFS PFGE resulted in a general increase in chromosomal fragmentation, especially if the DNA was expected to have nicks, a result consistent with the trapping of nicked linear DNA subjected to SHFS PFGE [26; 48; 49; 50]. In these experiments, the nick-dependent overall enhancement in fragmented DNA did not influence the general interpretation of fragmentation results, as the relative fragmentation, either spontaneous or DNA damage-induced one, stayed the same (Fig. 1 and S1).
Interestingly, while our original suspicion, that PFGE breaks nicked DNA, was confirmed, our expectation that reduced field strength would preserve the nicked DNA was not validated. We conclude that the release of DNA from the wells is influenced by field strength, but its further breakage during PFGE is determined by the density of nicks. Our conclusion is based on the nick-dependent but FS-independent breakage of chromosomal fragments in three sets of experiments: appearance of LMW DNA upon UV treatments (Fig. 3 and S4), decrease in the intensity of 1 Mbp NotI fragment in excision- and UV dose-dependent manner (Fig. 6, S8 and S9), and enhancement in fragmentation upon treatment with Nb.BbvCI, an enzyme that introduces sequence-specific nicks in the DNA (not shown). Two lines of preliminary evidence indicate that circular chromosomes could also be broken during PFGE if the density of nicks is high: first, at high doses stationary phase cells start showing UV-induced fragmentation (uncoupling of DNA replication and chromosomal fragmentation) (Fig. S9), and second, treatment with nickase Nb.BbvCI, an enzyme that introduces sequence-specific nicks in DNA, results in increased fragmentation (not shown). Further analysis is required to confirm the occurrence- and FS-dependence of the breakage of circular nicked chromosomes.
In this paper, we did not specifically address the mechanism of nick-dependent trapping and breakage. However, unusual DNA molecules, including branched recombination intermediates and linear DNA fragments bearing long single stranded tails, have been shown to be electrophoretically retarded when subjected to PFGE [51; 52; 53; 54; 55]. Nicks have been proposed before to affect the PFGE migration of DNA by trapping it within wells, thereby hindering its movement into the gel, or by trapping it within the gel, leading to smearing of DNA bands [48; 56]. We think that the latter type of trapping, while not influencing quantification of the chromosomal fragmentation in our protocol (see methods) [5; 6; 34; 35; 38; 39], leads to the breakage of the moving DNA at the nicks and gaps (Fig. 7). According to the model of Bustamante and colleagues [48; 57], a DNA molecule migrating through the pulsed field agarose gel may bend around a gel fiber in a U shape while sliding past it (Fig. 7A). If the DNA is nicked, its sliding motion around the gel fiber may lead to the local unraveling of the DNA duplex at the nick, locking the DNA at the gel fiber and leading to entrapment of this DNA molecule (Fig. 7B). We would like to extend their model, linking trapping of nicked DNA during agarose gel electrophoresis with breakage at a particular density of nicks. Specifically, we propose that, after the initial encounter, when the electric field-driven unraveling of the DNA duplex reaches the nick in the opposite strand, this causes DNA breakage (Fig. 7B step IV) and subsequent release of the now smaller fragments off the hook by sliding (Fig. 7B, step V). This model not only links together trapping and breakage of nicked DNA, but also explains the nick density-dependence of these processes. Finally, this model predicts that even circular DNA should be eventually broken at high enough density of nicks.
Figure 7.
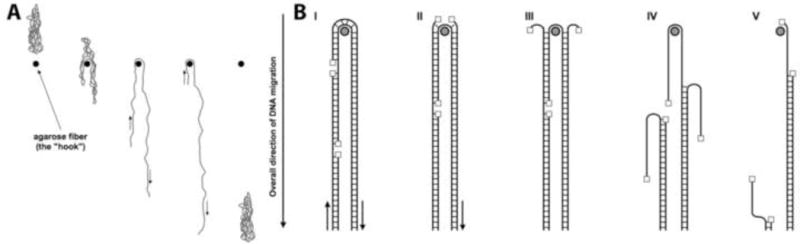
The model.
(A) Migration of DNA through the agarose gel as shown by [48; 57]. In this model, DNA undergoes cyclic motion of alternating elongated and compact conformations, the duration of which depends upon the size of the DNA. The elongated confirmation, in the form of U-shaped structure, is assumed when the DNA molecule is transiently ‘hooked around’ the gel fiber. The small arrows show the direction of DNA sliding off the hook.
(B) Breakage of the transiently trapped DNA at nicks. Nicked DNA stays in the elongated confirmation (I) until a nick reaches the vertex at the hook (II). Subsequent local unraveling of the DNA structure from the nick leads to the formation of SS ends that pull apart, anchoring the DNA at the hook (III). When the electric field-caused unraveling of the DNA duplex reaches another nick in the opposite DNA strand, a double-strand break is formed (IV), and the broken DNA can now slide off the hook (V).
In summary, in this study we have demonstrated that a non-electrophoretic parameter, physical integrity of the DNA itself, affects both the quantity and the quality of linear subchromosomal fragments. The nick-dependent entrapment of linear DNA within wells hinders accurate measurement of fragmented DNA, leading to under-representation of chromosomal fragmentation. We propose that this problem can be counteracted by reducing field strength and increasing the time of electrophoresis. While running the samples for 3 days is not practical for routine use, linear DNA release from the wells provides an option of improving the quantity of fragmented DNA under conditions that yield low levels of fragmentation.
Finally, artificial breakage of DNA at SS interruption is a more serious challenge that seems to be the property of PFGE itself. Unfortunately we could not find a way to minimize it, as both field strengths (6 V/cm and 3 V/cm) exhibited similar levels of breakage. However, we have tested only two parameters, FS and the time of electrophoresis, and more work is needed to find optimal conditions yielding less breakage at the nicks. Till then, care should be taken while analyzing the chromosomal fragmentation data, especially the size of the fragmented DNA that is used to calculate the number of DSBs from strains undergoing extensive DNA damage or harboring endogenous nicks due to mutations.
Supplementary Material
Acknowledgments
This work was supported by grant # GM 073115 from the National Institutes of Health. Authors declare no conflict of interest.
Abbreviations
- PFGE
pulsed-field gel electrophoresis
- LLFS
long run at low field strength
- SHFS
short run at high field strength
- CZ
compression zone
- UV
ultraviolet light
- HMW
high molecular weight
- LMW
low molecular weight
- DBA
different batch of agarose
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Data
Supplementary data file containing table S1, figures S1–S9 and figure legends.
References
- 1.Rupp WD, Howard-Flanders P. Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J Mol Biol. 1968;31:291–304. doi: 10.1016/0022-2836(68)90445-2. [DOI] [PubMed] [Google Scholar]
- 2.Setlow RB, Swenson PA, Carrier WL. Thymine Dimers and Inhibition of DNA Synthesis by Ultraviolet Irradiation of Cells. Science. 1963;142:1464–6. doi: 10.1126/science.142.3598.1464. [DOI] [PubMed] [Google Scholar]
- 3.Siede W, Friedberg AS, Dianova I, Friedberg EC. Characterization of G1 checkpoint control in the yeast Saccharomyces cerevisiae following exposure to DNA-damaging agents. Genetics. 1994;138:271–81. doi: 10.1093/genetics/138.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett CB, Snipe JR, Resnick MA. A persistent double-strand break destabilizes human DNA in yeast and can lead to G2 arrest and lethality. Cancer Res. 1997;57:1970–80. [PubMed] [Google Scholar]
- 5.Khan SR, Kuzminov A. Replication forks stalled at ultraviolet lesions are rescued via RecA and RuvABC protein-catalyzed disintegration in Escherichia coli. J Biol Chem. 2012;287:6250–65. doi: 10.1074/jbc.M111.322990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kouzminova EA, Kuzminov A. Chromosome demise in the wake of ligase-deficient replication. Mol Microbiol. 2012 doi: 10.1111/j.1365-2958.2012.08076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Resnick MA, Skaanild M, Nilsson-Tillgren T. Lack of DNA homology in a pair of divergent chromosomes greatly sensitizes them to loss by DNA damage. Proc Natl Acad Sci U S A. 1989;86:2276–80. doi: 10.1073/pnas.86.7.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chlebowicz E, Jachymczyk WJ. Repair of MMS-induced DNA double-strand breaks in haploid cells of Saccharomyces cerevisiae, which requires the presence of a duplicate genome. Mol Gen Genet. 1979;167:279–286. doi: 10.1007/BF00267420. [DOI] [PubMed] [Google Scholar]
- 9.Dahm-Daphi J, C S, W A. Comparison of biological effects of DNA damage induced by ionizing radiation and hydrogen peroxide in CHO cells. Int J Radiat Biol. 2000;76:67–75. doi: 10.1080/095530000139023. [DOI] [PubMed] [Google Scholar]
- 10.Myung K, Kolodner RD. Induction of genome instability by DNA damage in Saccharomyces cerevisiae. DNA Repair (Amst) 2003;2:243–58. doi: 10.1016/s1568-7864(02)00216-1. [DOI] [PubMed] [Google Scholar]
- 11.Chu G, Vollrath D, Davis RW. Separation of large DNA molecules by contour-clamped homogeneous electric fields. Science. 1986;234:1582–5. doi: 10.1126/science.3538420. [DOI] [PubMed] [Google Scholar]
- 12.Schwartz DC, Cantor CR. Separation of yeast chromosome-sized DNAs by pulsed field gradient gel electrophoresis. Cell. 1984;37:67–75. doi: 10.1016/0092-8674(84)90301-5. [DOI] [PubMed] [Google Scholar]
- 13.Basim E, Basim H. Pulsed-Field Gel Electrophoresis (PFGE) Technique and its use in Molecular Biology. Turk J Biol. 2001;25:405–418. [Google Scholar]
- 14.Chu G. Pulsed-field gel electrophoresis: theory and practice. Methods. 1990;1:129–142. [Google Scholar]
- 15.Maule J. Pulsed-field gel electrophoresis. Mol Biotechnol. 1998;9:107–26. doi: 10.1007/BF02760813. [DOI] [PubMed] [Google Scholar]
- 16.Allardet-Servent A, Michaux-Charachon S, Jumas-Bilak E, Karayan L, Ramuz M. Presence of one linear and one circular chromosome in the Agrobacterium tumefaciens C58 genome. J Bacteriol. 1993;175:7869–74. doi: 10.1128/jb.175.24.7869-7874.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith CL, Econome JG, Schutt A, Klco S, Cantor CR. A physical map of the Escherichia coli K12 genome. Science. 1987;236:1448–53. doi: 10.1126/science.3296194. [DOI] [PubMed] [Google Scholar]
- 18.Van der Ploeg LH, Smits M, Ponnudurai T, Vermeulen A, Meuwissen JH, Langsley G. Chromosome-sized DNA molecules of Plasmodium falciparum. Science. 1985;229:658–61. doi: 10.1126/science.3895435. [DOI] [PubMed] [Google Scholar]
- 19.Lukacsi G, Tako M, Nyilasi I. Pulsed-field gel electrophoresis: a versatile tool for analysis of fungal genomes. A review Acta Microbiol Immunol Hung. 2006;53:95–104. doi: 10.1556/AMicr.53.2006.1.7. [DOI] [PubMed] [Google Scholar]
- 20.Mendez-Alvarez S, Pavon V, Esteve I, Guerrero R, Gaju N. Analysis of bacterial genomes by pulsed field gel electrophoresis. Microbiologia. 1995;11:323–36. [PubMed] [Google Scholar]
- 21.Brosch R, Chen J, Luchansky JB. Pulsed-field fingerprinting of listeriae: identification of genomic divisions for Listeria monocytogenes and their correlation with serovar. Appl Environ Microbiol. 1994;60:2584–92. doi: 10.1128/aem.60.7.2584-2592.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Mazurek GH, Cave MD, Eisenach KD, Pang Y, Murphy DT, Wallace RJ., Jr DNA polymorphisms in strains of Mycobacterium tuberculosis analyzed by pulsed-field gel electrophoresis: a tool for epidemiology. J Clin Microbiol. 1992;30:1551–6. doi: 10.1128/jcm.30.6.1551-1556.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goering RV. Pulsed field gel electrophoresis: a review of application and interpretation in the molecular epidemiology of infectious disease. Infect Genet Evol. 2010;10:866–75. doi: 10.1016/j.meegid.2010.07.023. [DOI] [PubMed] [Google Scholar]
- 24.Peters TM. Pulsed-field gel electrophoresis for molecular epidemiology of food pathogens. Methods Mol Biol. 2009;551:59–70. doi: 10.1007/978-1-60327-999-4_6. [DOI] [PubMed] [Google Scholar]
- 25.Noolandi J, Turmel C. Preparation and Separation of Intact Chromosomes of Vertebrates by One-Dimensional Pulsed-Field Gel Electrophoresis (ODPFGE) Methods Mol Biol. 1992;12:135–43. doi: 10.1385/0-89603-229-9:135. [DOI] [PubMed] [Google Scholar]
- 26.Vollrath D, Davis RW. Resolution of DNA molecules greater than 5 megabases by contour-clamped homogeneous electric fields. Nucleic Acids Res. 1987;15:7865–76. doi: 10.1093/nar/15.19.7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mickel S, Arena VJ, Bauer W. Physical properties and gel electrophoresis behavior of R12-derived plasmid DNAs. Nucleic Acids Res. 1977;4:1465–82. doi: 10.1093/nar/4.5.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serwer P, Hayes SJ. A voltage gradient-induced arrest of circular DNA during agarose gel electrophoresis. Electrophoresis. 1987;8:244–6. [Google Scholar]
- 29.Beverley SM. Characterization of the ‘unusual’ mobility of large circular DNAs in pulsed field-gradient electrophoresis. Nucleic Acids Res. 1988;16:925–39. doi: 10.1093/nar/16.3.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levene SD, Zimm BH. Separations of open-circular DNA using pulsed-field electrophoresis. Proc Natl Acad Sci U S A. 1987;84:4054–7. doi: 10.1073/pnas.84.12.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Birren WB, Lai EHC. Pulsed field gel electrophoresis: a practical guide. Academic Press; San Diego: 1993. [Google Scholar]
- 32.Fan JB, Rochet M, Gaillardin C, Smith CL. Detection and characterization of a ring chromosome in the fission yeast Schizosaccharomyces pombe. Nucleic Acids Res. 1992;20:5943–5. doi: 10.1093/nar/20.22.5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Game JC, Sitney KC, Cook VE, Mortimer RK. Use of a ring chromosome and pulsed-field gels to study interhomolog recombination, double-strand DNA breaks and sister-chromatid exchange in yeast. Genetics. 1989;123:695–713. doi: 10.1093/genetics/123.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kouzminova EA, Kuzminov A. Fragmentation of replicating chromosomes triggered by uracil in DNA. J Mol Biol. 2006;355:20–33. doi: 10.1016/j.jmb.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 35.Kouzminova EA, Kuzminov A. Patterns of chromosomal fragmentation due to uracil-DNA incorporation reveal a novel mechanism of replication-dependent double-stranded breaks. Mol Microbiol. 2008;68:202–15. doi: 10.1111/j.1365-2958.2008.06149.x. [DOI] [PubMed] [Google Scholar]
- 36.Kouzminova EA, Rotman E, Macomber L, Zhang J, Kuzminov A. RecA-dependent mutants in Escherichia coli reveal strategies to avoid chromosomal fragmentation. Proc Natl Acad Sci U S A. 2004;101:16262–7. doi: 10.1073/pnas.0405943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michel B, Ehrlich SD, Uzest M. DNA double-strand breaks caused by replication arrest. Embo J. 1997;16:430–8. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kouzminova EA, Kuzminov A. Chromosomal fragmentation in dUTPase-deficient mutants of Escherichia coli and its recombinational repair. Mol Microbiol. 2004;51:1279–95. doi: 10.1111/j.1365-2958.2003.03924.x. [DOI] [PubMed] [Google Scholar]
- 39.Kuong KJ, Kuzminov A. Stalled replication fork repair and misrepair during thymineless death in Escherichia coli. Genes Cells. 2010;15:619–34. doi: 10.1111/j.1365-2443.2010.01405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seigneur M, Bidnenko V, Ehrlich SD, Michel B. RuvAB acts at arrested replication forks. Cell. 1998;95:419–30. doi: 10.1016/s0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- 41.Dillingham MS, Kowalczykowski SC. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol Mol Biol Rev. 2008;72:642–71. doi: 10.1128/MMBR.00020-08. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuzminov A. Collapse and repair of replication forks in Escherichia coli. Mol Microbiol. 1995;16:373–84. doi: 10.1111/j.1365-2958.1995.tb02403.x. [DOI] [PubMed] [Google Scholar]
- 43.Kuzminov A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci U S A. 2001;98:8241–6. doi: 10.1073/pnas.131009198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen CR, Malik M, Snyder M, Drlica K. DNA gyrase and topoisomerase IV on the bacterial chromosome: quinolone-induced DNA cleavage. J Mol Biol. 1996;258:627–37. doi: 10.1006/jmbi.1996.0274. [DOI] [PubMed] [Google Scholar]
- 45.Malik M, Zhao X, Drlica K. Lethal fragmentation of bacterial chromosomes mediated by DNA gyrase and quinolones. Mol Microbiol. 2006;61:810–25. doi: 10.1111/j.1365-2958.2006.05275.x. [DOI] [PubMed] [Google Scholar]
- 46.Cooper DL, Lovett ST. Toxicity and tolerance mechanisms for azidothymidine, a replication gap-promoting agent, in Escherichia coli. DNA Repair (Amst) 10:260–70. doi: 10.1016/j.dnarep.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. ASM Press; Washington, D. C.: 2006. pp. 227–266. [Google Scholar]
- 48.Gurrieri S, Smith SB, Bustamante C. Trapping of megabase-sized DNA molecules during agarose gel electrophoresis. Proc Natl Acad Sci U S A. 1999;96:453–8. doi: 10.1073/pnas.96.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith CL, Cantor CR. Purification, specific fragmentation, and separation of large DNA molecules. Methods Enzymol. 1987;155:449–67. doi: 10.1016/0076-6879(87)55030-3. [DOI] [PubMed] [Google Scholar]
- 50.Turmel C, Brassard E, Slater GW, Noolandi J. Molecular detrapping and band narrowing with high frequency modulation of pulsed field electrophoresis. Nucleic Acids Res. 1990;11:569–75. doi: 10.1093/nar/18.3.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ishioka K, Fukuoh A, Iwasaki H, Nakata A, Shinagawa H. Abortive recombination in Escherichia coli ruv mutants blocks chromosome partitioning. Genes Cells. 1998;3:209–20. doi: 10.1046/j.1365-2443.1998.00185.x. [DOI] [PubMed] [Google Scholar]
- 52.Nakayama K, Kusano K, Irino N, Nakayama H. Thymine starvation-induced structural changes in Escherichia coli DNA. Detection by pulsed field gel electrophoresis and evidence for involvement of homologous recombination. J Mol Biol. 1994;243:611–20. doi: 10.1016/0022-2836(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 53.Rudolph CJ, Upton AL, Lloyd RG. Replication fork collisions cause pathological chromosomal amplification in cells lacking RecG DNA translocase. Mol Microbiol. 2009;74:940–55. doi: 10.1111/j.1365-2958.2009.06909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma W, Westmoreland J, Nakai W, Malkova A, Resnick MA. Characterizing resection at random and unique chromosome double-strand breaks and telomere ends. Methods Mol Biol. 2009;745:15–31. doi: 10.1007/978-1-61779-129-1_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Westmoreland J, Ma W, Yan Y, Van Hulle K, Malkova A, Resnick MA. RAD50 is required for efficient initiation of resection and recombinational repair at random, gamma-induced double-strand break ends. PLoS Genet. 2009;5:e1000656. doi: 10.1371/journal.pgen.1000656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Turmel C, Brassard E, Slater GW, Noolandi J. Molecular detrapping and band narrowing with high frequency modulation of pulsed field electrophoresis. Nucleic Acids Res. 1990;18:569–75. doi: 10.1093/nar/18.3.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gurrieri S, Bustamante C. Purification and staining of intact yeast DNA chromosomes and real-time observation of their migration during gel electrophoresis. Biochem J. 1997;326(Pt 1):131–8. doi: 10.1042/bj3260131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bachmann BJ. Derivations and genotypes of some mutant derivatives of Escherichia coli K-12. in Escherichia coli and Salmonella typhimurium. In: Neidhardt FC, editor. Cellular and Molecular Biology. American Society for Microbiology; Washington, D.C.: 1987. pp. 1190–1219. [Google Scholar]
- 59.Kushner SR. Differential thermolability of exonuclease and endonuclease activities of the recBC nuclease isolated from thermosensitive recB and recC mutants. J Bacteriol. 1974;120:1219–22. doi: 10.1128/jb.120.3.1219-1222.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dermody JJ, Robinson GT, Sternglanz R. Conditional-lethal deoxyribonucleic acid ligase mutant of Escherichia coli. J Bacteriol. 1979;139:701–4. doi: 10.1128/jb.139.2.701-704.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Amado L, Kuzminov A. The replication intermediates in Escherichia coli are not the product of DNA processing or uracil excision. J Biol Chem. 2006;281:22635–46. doi: 10.1074/jbc.M602320200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.