

Abstract
E-DNA sensors are a reagentless, electrochemical oligonucleotide sensing platform based on a redox-tag modified, electrode-bound probe DNA. Because E-DNA signaling is linked to hybridization-linked changes in the dynamics of this probe, sensor performance is likely dependent on the nature of the self-assembled monolayer coating the electrode. We have investigated this question by characterizing the gain, specificity, response time and shelf-life of E-DNA sensors fabricated using a range of co-adsorbates, including both charged and neutral alkane thiols. We find that, among the thiols tested, the positively charged cysteamine gives rise to the largest and most rapid response to target and leads to significantly improved storage stability. The best mismatch specificity, however, is achieved with mercaptoethanesulfonic and mercaptoundecanol, presumably due to the destabilizing effects of, respectively, the negative charge and steric bulk of these co-adsorbates. These results demonstrate that a careful choice of co-adsorbate chemistry can lead to significant improvements in the performance of this broad class of electrochemical DNA sensors.
Keywords: DNA, Sensor, Electrochemical, Thiol, Self-assembled monolayers
1. Introduction
E-DNA sensors, the electrochemical analog of optical molecular beacons [e.g., 1–4], are based on the hybridization-induced folding of an electrode-bound, redox-tagged DNA probe. In their original implementation, the concentration of a target oligonucleotide is recorded when it hybridizes to a stem-loop DNA probe, leading to the formation of a rigid, double stranded duplex that sequesters the redox tag from the interrogating electrode [1]. Follow-on E-DNA architectures have dispensed with the stem-loop probe in favor of linear probes, leading to improved binding thermodynamics and, thus, improved gain [5], as well as strand-invasion, hairpin and pseudoknot probes producing signal-on sensors [6–8]. Because E-DNA sensors are reagentless, electronic (electrochemical) and highly selective (they perform well even when challenged directly in complex, multi-component samples such as blood serum or soil) [e.g., 9], E-DNA sensors appear to be a promising and appealing approach for the sequence-specific detection of DNA and RNA [see, e.g., 10,11].
The signaling mechanism of E-DNA sensors is linked to a binding-specific change in the flexibility of the redox-tagged probe; upon hybridization, the relatively rigid target/probe duplex hampers the collision of the electrochemical tag thus decreasing the observable amperometric signal [5,12]. This, in turn, suggests that E-DNA signaling may be sensitive to changes in surface chemistry which, due to surface charge and steric bulk effects, would likely alter the dynamics of a negatively charged DNA probe. However, despite rapid growth in the E-DNA literature [reviewed in 13] the extent to which surface chemistry affects E-DNA signaling has not been established; all previous E-DNA sensors were fabricated using hydroxyl-terminated alkane thiol self-assembled monolayers (SAMs) [e.g., 1,3,5,7,9]. Here we address this question and describe a study of E-DNA sensors fabricated using co-adsorbates of various lengths and charges in an effort to further optimize E-DNA performance.
2. Materials and methods
2.1. Probe DNA and sensor fabrication
A 27-base, 3′ thiol-, 5′ methylene blue (MB)-modified probe DNAwas obtained from Biosearch Technologies (Novato, CA) and employed as the probe DNA. The 17 internal bases of this sequence are complementary to the Salmonella gyrB gene. The MB redox moiety was conjugated to the 3′ end of the oligonucleotide via succinimide ester coupling to a 3′ amino modification (MB-NHS, EMP Biotech, Berlin) producing the probe sequence: 5′-HS-(CH2)6-CGTCAATCTTCTATTTCTCCACACTGC-(CH2)7-NH2-MB-3′. Reagent grade chemicals, including 6-mercapto-1-hexanol (C6-OH), β-mercaptoethanol (C2-OH), 11-mercapto-1-undecanol (C11-OH), cysteamine (C2-amine), mercaptoethanesulfonic acid (C2-sulfonate), sulfuric acid, potassium phosphate monobasic, dibasic, and sodium chloride (all from Sigma-Aldrich, St. Louis, MO), were used without further purification.
We employed 3 mm diameter gold screen-printed electrodes as the basis for our sensors, fabricated as previously described [14,15]. Jenkins has previously demonstrated excellent E-DNA performance on such electrodes [16]. Prior to use the electrodes were cleaned using a series of oxidation and reduction cycles in 0.5 M H2SO4, 0.01 M KCl/0.1 M H2SO4, and 0.05 M H2SO4 [4]. The thiol-containing oligonucleotide we have employed is supplied as a mixed disulfide of 6-mercapto-1-hexanol in order to minimize the risk of oxidation. Thus the first step in sensor fabrication is the reduction of the probe DNA (100 μM) for 1 h in a solution of 0.4 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) in 100 mM NaCl/10 mM potassium phosphate pH 7 followed by the use of a spin column to eliminate any 6-mercapto-1-hexanol residue. The effectiveness of this purification procedure has been tested using a spectrophotometric test (Ellman’s test [17]), which confirmed the absence of free thiol in the resulting solution (results not shown). The freshly cleaned electrode surface was then covered with a 50 μl drop of the purified probe solution, incubated for 1 h and rinsed with distilled, deionized water. The electrode was then passivated by incubating in 1 mM of the relevant thiol (in 1 M NaCl/10 mM potassium phosphate buffer, pH 7) for 2 h followed by a final rinse with deionized water.
2.2. Target DNA sequences
We employed the following target DNA sequences, all of which were obtained via commercial synthesis (Sigma Genosys, St. Luis, MO):
T-17 (normal target, 17 bases, 5′-GTG GAG AAA TAG AAG AT-3′)
M17 (three T–T mismatched target, 17 bases, 5′-GTG GAG TTT TAG AAG AT-3′)
T-27 (long target, 27 bases, 5′-GCAGT GTG GAG AAA TAG AAG AT TGACG-3′)
2.3. Electrochemical measurements
The sensor response was measured by incubating the electrodes in 100 nM of the appropriate target DNA. The sensors were interrogated at different intervals in the same target solution until a stable current peak was obtained (typically after 20 to 45 min). The ratio between the stabilized current peak (A) in the presence of target DNA (Itarget) and the current peak (A) in absence of target DNA (Ibuffer) gives the measure of the signal suppression caused by the target. Before being used to detect the next target, the electrodes were rinsed with deionized water and interrogated in target free buffer. This also provides a measure of the extent to which each sensor can be regenerated. All measurements were performed at room temperature using an Autolab (EcoChemie, Utrecht, The Netherlands). Square wave voltammetry (SWV) was recorded from −0.45 V to −0.05 V versus the internal reference electrode. All experiments were conducted in 1 M NaCl/10 mM potassium phosphate buffer, pH 7.
3. Results
As a test-bed for our studies we have employed linear probe E-DNA sensors [5] fabricated on screen-printed electrodes (Fig. 1). Similar screen-printed E-DNA electrodes have been described previously and exhibit excellent signaling properties and stability [16]. Consistent with this, we observe that sensors fabricated on these electrodes using the commonly employed six-carbon, hydroxyl terminated co-adsorbate (6-mercapto-1-hexanol; C6-OH) give rise to a sharp, well-defined peak at ~260 mV (vs. Ag/AgCl) consistent with the formal potential of the methylene blue redox moiety (Fig. 1). This current is significantly reduced in the presence of the fully complementary, 17-base target, and is completely recovered via a 30 s wash with room temperature, distilled water. We note that the use of screen-printed electrodes offers a number of potential advantages over their more traditionally fabricated counterparts, including ease of mass production, low cost and minimal working volumes.
Fig. 1.

E-DNA sensors and coadsorbates used: Here we have employed linear probe E-DNA sensors (top left) as a test bed with which to characterize the effects of surface chemistry on the properties of an electrochemical E-DNA sensor. Because hybridization reduces the rate with which the terminal redox tag collides with the electrode surface and transfers electrons the Faradaic current arising from such linear probes is significantly reduced in the presence of a complementary target sequence (top, right). It is thus likely that this suppression and the motion of the unbound and bound probe will be linked to the nature, steric hindrance and charge of the co-adsorbate used for sensor fabrication. We have tested the effects of a range of thiol co-adsorbates (bottom) differing in their length and/or terminal functional groups and chosen to cover a range of charges and sterics (bottom).
We have tested the effects of a range of thiol co-adsorbates, differing in length and/or terminal functional groups and chosen to cover a range of charges and sterics (see Fig. 1), on the signal gain, specificity, stability and selectivity of the E-DNA sensor. We have also investigated the effects of these coadsorbates on the electrochemistry of the methylene blue tag using Square Wave Voltammetry (SWV), which provides a convenient means of evaluating electron transfer rates and reversibility for species confined to the electrode surface [18,19]. As background to this discussion, we note that co-adsorbates are necessary to achieve proper E-DNA function: the absence of co-adsorbates significantly degrades sensor reproducibility, producing sensors with wildly varying, poorly equilibrating signal suppression (ranging from 10 to 50% in the presence of saturating target-data not shown). The poor performance observed in the absence of co-adsorbates presumably arises due to poor ordering of the DNA probe and/or non-specific interactions between the probe DNA and the electrode surface.
Sensors fabricated using all the different co-adsorbates tested in this study produce reversible behavior of the methylene blue label (Fig. 2) and give rise to well-defined, reproducible peak currents (RSD % values range from 5 to 15%). SWV peaks with wave-width at half-height are in fact indistinguishable from the 90 mV expected for ideal reversible electron transfer to a surface confined species [20], and the peak potentials of both forward and reverse scans and the net response (the difference between reverse and forwards scan) are, as expected for perfectly reversible redox couples, independent of frequency [19]. Moreover, a plot of the SWV frequency measurement (f) versus the peak current (Ip) always shows the hallmark behavior of a surface confined redox species. Likewise peak currents vary linearly (R=0.997) with the logarithm of the SWV frequency over the entire frequency range we have investigated, a behavior that is typical for an electrochemical label adsorbed on the electrode surface [19,21,22].
Fig. 2.
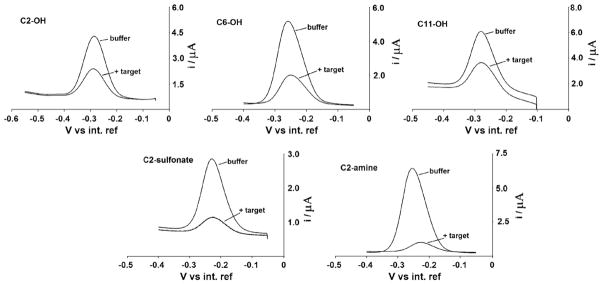
Coadsorbate effect on E-DNA signaling: Because E-DNA signaling is linked to a binding-specific change in the collision efficiency of the probe-bound redox tag with the electrode surface, the nature (i.e., length and charge) of the co-adsorbate used for sensor production is a determining factor in the performance of E-DNA sensors. Shown are SW voltammograms of sensors fabricated with each of five coadsorbates before and after the addition of the relevant 17-base target (at 100 M). We find that, among the thiols tested, the positively charged C2-amine (cysteamine) gives rise to the largest and most rapid response to target.
3.1. Effects of co-adsorbate length on E-DNA signaling
Hydroxyl-terminated alkane thiols of lengths 2, 6 or 11 carbons (C2-OH, C6-OH and C11-OH respectively) were used to evaluate the effect on the sensor’s performance of co-adsorbates of different length and were found to generate very different signaling properties. For example, we find that C6-OH (6-mercapto-1-hexanol) SAMs gives rise to greater signal suppression (Itarget/Ibuffer) than either longer or shorter hydroxyl-terminated coadsorbates (Fig. 2 and Table 1) (Here Table 1). The use of C11-OH (11-mercapto-1-undecanol) as a co-adsorbate produces a similar current in the absence of target but significantly degrades signaling: whereas we observe 47% signal suppression for sensors fabricated using C11-OH SAMs (100 nM of the 17-base target), under the same conditions, sensors fabricated with C6-OH achieve a 70% signal change (Table 1). We should note, however, that our DNA probe is linked to the electrode via a six-carbon linker, and thus C11-OH co-adsorbate likely buries the terminus of the probe within the monolayer. The steric effects of this could account for the poorer signal suppression. The signal suppression observed when C2-OH (β-mercaptoethanol) is employed is also slightly poorer than that observed using C6-OH, presumably because, with this very thin SAM, both the probe DNA is flexible enough to transfer electrons to the electrode irrespective of whether or not it is hybridized. Alternatively, it has been reported that short thiols produce defective monolayers, allowing electrochemistry to occur directly on the underlying gold electrode [23] and, perhaps, enhancing electron transfer from both the hybridized and unhybridized probe. This hypothesis seems to be confirmed by the study of the electron transfer rate. The rate of electron transfer through the SAM is in fact, as expected [24–26], inversely related to the thickness of the SAM: as the co-adsorbate lengthens from C2-OH to C11-OH the transfer rate constant decreases from 94.4 s−1 to 17.7 s−1 (Table 2).
Table 1.
Effect of co-adsorbate on E-DNA signaling, specificity and reusability.
| Thiol | Chemical formula | Initial signal/μAa | (Itarget/Ibuffer)/% T-17b | (Itarget/Ibuffer)/% T-27b | (Itarget/Ibuffer)/% M-17b | Specificity ratio Rsc | Regeneration (Ireg/Ibuffer)/%d | |
|---|---|---|---|---|---|---|---|---|
| 2-mercaptoethanol |
|
3.9±0.3 | 62±3 | 70±4 | 50±3 | 1.2 | 85±5 | |
| 6-mercaptohexanol |
|
4.7±0.5 | 71±4 | 80±3 | 55±3 | 1.3 | 98±6 | |
| 11-mercaptoundecanol |
|
4.5±0.4 | 47±3 | 60±4 | 25±2 | 1.9 | 96±8 | |
| Cysteamine (2-mercaptoethylamine) |
|
5.9±0.7 | 90±6 d | 90±4 | 77±5 | 1.1 | 89±6 | |
| Mercaptoethane sulfonic acid |
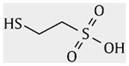
|
2.6±0.4 | 80±5 | 52±3 | 36±6 | 2.2 | 91±7 |
Signal in the presence of buffer.
% values of signal suppression (ratio of current signal obtained in target–Itarget–and current signal obtained in buffer -Ibuffer) as a result of target hybridization are the average and standard deviation of measurements performed with four independent sensors.
Rs is the ratio of the signal suppression (Itarget/Ibuffer) obtained with the fully complementary target (T-17) to that observed with the mismatched target (M-17).
% values of regeneration (ratio of current signal obtained in buffer after regeneration–Ireg–and current signal obtained in buffer–Ibuffer) are the average and standard deviation of measurements performed with four independent sensors.
Table 2.
Effects of co-adsorbate on electron transfer kinetics and reversibility of the methylene blue tag.
| Thiol | ks/s−1 | ΔEp/2/mV |
|---|---|---|
| β-Mercaptoethanol | 94.4±5 | 96±1 |
| 6-mercapto-1-hexanol | 23.6±2 | 89±1 |
| 11-mercapto-1-undecanol | 17.7±2 | 95±1 |
| Cysteamine (2-mercaptoethylamine) | 17.7±2 | 99 |
| Mercaptoethane sulfonic acid | 9.5±1 | 107 |
3.2. Effects of co-adsorbate charge on E-DNA signaling
We have also investigated the effects of the positively and negatively charged co-adsorbates C2-amine (cysteamine) and C2-sulfonate (mercaptoethanesulfonic acid) respectively on E-DNA signaling (Fig. 2 and Table 1). In the case of C2-amine we find that, while the signal in absence of target is of the same order of magnitude of that observed for its neutral, C2-OH counterpart, the response to target differs significantly: the signal suppression observed with these sensors is, at 90%, much greater than that observed for any of the sensors fabricated using uncharged monolayers (Fig. 2, Table 1). The signal suppression observed with C2-sulfonate, however, is, at 80%, also higher than that observed for simple, hydroxyl-terminated SAMs. The origins of these effects are unclear but it is likely that both the positively charged methylene blue and the highly negatively charged DNA strand could be affected by the presence of a charged SAM on the electrode surface. Perhaps consistent with this, both C2-sulfonate and C2-amine exhibit the slowest electron transfer rates of our five SAMs (9.5 and 17 s−1 respectively), presumably due to repulsions between the negatively charged DNA probe and C2-sulfonate or the positively charged methylene blue with C2-amine. Perhaps also consistent with this, only the negatively charged C2-sulfonate SAM produces less signal gain with a longer, 27-base target than with a shorter, 17 target (Table 1).
3.3. Effects of co-adsorbate on E-DNA specificity and equilibration time
The nature of co-adsorbate also strongly affects the specificity of the E-DNA sensor. To demonstrate this we have tested a mismatch target (M-17) containing three T–T mismatches dispersed throughout the 17-base recognition element. The ratios (Rs) of the suppression obtained with the fully complementary target of the same length (T-17) to that observed with the mismatched target (both at 100 nM) then provide a measure of the sensor specificity. We find that, while these ratios are similar with C2-amine and C2-OH (Rs =1.1 and 1.2 respectively), they are dramatically higher (Rs =2.2) for the negatively charged C2-sulfonate (Table 1). This presumably occurs when repulsion arising from the negatively charged surface destabilizes the probe-target duplex, leading to improved discrimination. Similar specificity is observed for C11-OH-based sensors (Rs =1.9) (Table 1), apparently because steric hindrance arising from the co-adsorbate similarly destabilizes the duplex (Fig. 1).
Alterations of the surface chemistry also affect the speed with which our sensors equilibrate. Sensors fabricated with C6-OH SAMs exhibit an equilibration half-life (time for half of the signal change to occur) of 25 min (Fig. 3) (Here Fig. 3). In contrast, the positive charge of the C2-amine accelerates hybridization: with a 17 base, fully complementary target we achieve half-life of ~5 min. C2-OH and C2-sulfonate produce slower kinetics (22.5 and 27.5 min half-lives respectively), and C11-OH produces kinetics similar to those observed for C2-amine (17.5 min). The origins of this latter acceleration are unclear, especially given the presumably increased steric clashes associated with this SAM. We note, however, that thicker SAMs lead to reduced electric double layers [27,28], which could accelerate diffusion of the negatively charged target to the electrode surface.
Fig. 3.

Coadsorbate effect on E-DNA response time: Nature and charge of co-adsorbate affect the response times (the time needed for the stabilization of the current signal in the presence of target). While all five probes equilibrate similarly rapidly, the positive charge of C2-amine SAMs leads to the most rapid hybridization: with a 17 base, fully complementary target (T-17) we observe an equilibration half-life (time for half of the signal to occur) of ~15 min. Conditions are as for Fig. 2. Shown are the signal suppression values (% ratio of current signal—A—obtained in target—Itarget—and current signal obtained in buffer—Ibuffer) vs. time after target (T-17) injection.
3.4. Effects of co-adsorbate on E-DNA stability
E-DNA shelf life, an important parameter to consider vis a vis real-world sensor applications, can be strongly affected by the nature of the co-adsorbate [29,30]. To test this we studied the dry storage stability of our sensors by periodically taking them up in buffer and evaluating their initial (no target) peak currents (Fig. 4). With regard to co-adsorbate length we find that C6-OH leads to slightly improved stability (about 50% of the initial signal is retained after 3 weeks of storage) relative to that of the equivalent sensor fabricated using C2-OH, where a complete loss of signal was observed after the same period. Given that, as previously demonstrated [23], the C2 leads poorly-ordered SAMs this is perhaps not surprising. We do not, however, observe any further improvement in stability when C11-OH is employed as the co-adsorbate, presumably because our probes are anchored via a six-carbon thiol and thus C11-OH co-adsorbate leads to partial burial of the DNA probe. As is true for co-adsorbate length, the nature of the terminating group on the alkane thiol also affects E-DNA stability. For example, cystamine-based sensors lose only 55% of their initial signal, and retain their target sensitivity, after five weeks dry storage. In contrast sensors fabricated using C2-sulfonate complete loss of signal after only 2 weeks of storage.
Fig. 4.

Coadsorbate effect on E-DNA storage stability: Nature and charge of co-adsorbate strongly affect the storage stability of E-DNA sensors. Indicated are the % ratio of the signal obtained in buffer solution (no target) after different intervals of dry room temperature storage (Itime) and immediately after fabrication (Iinitial) vs the storage time (weeks).
4. Conclusion
The properties of the E-DNA sensor are strongly dependent on the surface chemistry of the interrogating electrode. We find, for example, that the positive charged C2-amine (cysteamine) co-adsorbate supports optimal signal suppression, the most rapid response time and the best storage stability, while best mismatch specificity is achieved with negatively charged C2-sulfonate (mercaptoethansulfonic) and longer-chain C11-OH (mercaptoundecanol) SAMs, presumably because these SAMs destabilize the target-probe duplex.
The results we have observed here are consistent with our earlier claims that the signaling mechanism of E-DNA sensors is, at least in part, due to the change in the efficiency with which the attached redox tag strikes the electrode. From this perspective, the effect of coadsorbate length on signaling is significant. Optimal signal suppression is obtained at intermediate coadsorbate length (C6-OH), presumably because the flexibility of the probe with a shorter coadsorbate (C2-OH) leads to enhanced collision dynamics for the probe-target duplex. Conversely, the longer C11-OH coadsorbate reduces collision efficiency for the single-stranded probe, also leading to lower signal suppression upon target hybridization. The interpretation of the effects of coadsorbate charge is more subtle due to the possible concomitant effect of the positively charged methylene blue tag and of the highly negatively charged DNA strands. As a result both the positively charged C2-amine and the negatively charged C2-sulfonate produce significant signal suppression upon target hybridization. Of note, however, the storage stability of the C2-amine based E-DNA sensors is greatly enhanced and it appears likely that electrostatic interactions among the positively charged amine group and the negatively charged probe DNA support stabilization in storage conditions thus preventing the leakage of the probe from the electrode surface. When compared with previously reported results obtained with similar sensing platforms employing alkanethiols as coadsorbates [4,5,29], the C2-amine based sensors exhibit improved signal gain and storage stability, but they also exhibit slightly poorer specificity. Despite this, detection limit (10 nM) and working range (10–200 nM) of the C2-amine sensor is comparable to other alkanethiols based E-DNA sensors [4,5]. Of note, reproducibility of the E-DNA signaling upon target hybridization is not affected by coadsorbate’s nature with RSD% values ranging from 4 to 6%.
Many electrochemical, electronic, mechanical and optical sensors have been reported that rely on the hybridization properties of SAM-bound DNA probes [reviewed in 23,31–34]. Exhaustive efforts to survey the literature, however, suggest that the effects of altering the chemistry of the SAM layer seen only very limited explorations [e.g., 35]. The results we have presented here indicate, in turn, that such explorations may prove of value in the optimization of many of these technologies.
Contributor Information
Francesco Ricci, Email: francesco.ricci@uniroma2.it.
Felice Caprio, Email: felice.caprio@uniroma2.it.
Aziz Amine, Email: azizamine@yahoo.fr.
Danila Moscone, Email: moscone@uniroma2.it.
Giuseppe Palleschi, Email: palleschi@uniroma2.it.
Kevin W. Plaxco, Email: kwp@chem.ucsb.edu.
References
- 1.Fan C, Plaxco KW, Heeger AA. Electrochemical interrogation of conformational changes as a reagentless method for the sequence-specific detection of DNA. J Proc Natl Acad Sci USA. 2003;100:9134–9137. doi: 10.1073/pnas.1633515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mao T, Luo C, Ouyang Q. Studies of temperature-dependent electronic transduction on DNA hairpin loop sensor. Nucleic Acids Res. 2003;1:108–172. doi: 10.1093/nar/gng108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Immoos CE, Lee SJ, Grinstaff MW. DNA-PEG-DNA triblock macromolecules for reagentless DNA detection. J Am Chem Soc. 2004;126:10814–10815. doi: 10.1021/ja046634d. [DOI] [PubMed] [Google Scholar]
- 4.Xiao Y, Lai RY, Plaxco KW. Preparation of electrode-immobilized, redox-modified oligonucleotides for electrochemical DNA and aptamer-based sensing. Nature Protoc. 2007;2:2875–2880. doi: 10.1038/nprot.2007.413. [DOI] [PubMed] [Google Scholar]
- 5.Ricci F, Lai RY, Plaxco KW. Linear, redox modified DNA probes as electrochemical DNA sensors. Chem Commun. 2007;2007:3768–3770. doi: 10.1039/b708882e. [DOI] [PubMed] [Google Scholar]
- 6.Xiao Y, Lubin AA, Baker BR, Plaxco KW, Heeger AJ. Single-step electronic detection of femtomolar DNA by target-induced strand displacement in an electrode-bound duplex. Proc Natl Acad Sci USA. 2006;103:16677–16680. doi: 10.1073/pnas.0607693103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Immoos CE, Lee SJ, Grinstaff MW. Conformationally gated electrochemical gene detection. Chembiochem. 2004;5:1100–1104. doi: 10.1002/cbic.200400045. [DOI] [PubMed] [Google Scholar]
- 8.Xiao Y, Qu X, Plaxco KW, Heeger AJ. Label-free electrochemical detection of DNA in blood serum via target-induced resolution of an electrode-bound DNA pseudoknot. J Am Chem Soc. 2007;129(39):11896–11897. doi: 10.1021/ja074218y. [DOI] [PubMed] [Google Scholar]
- 9.Lubin AA, Lai RY, Baker BR, Heeger AJ, Plaxco KW. Sequence-specific, electronic detection of oligonucleotides in blood, soil, and foodstuffs with the reagentless, reusable E-DNA Sensor. Anal Chem. 2006;78(16):5671–5677. doi: 10.1021/ac0601819. [DOI] [PubMed] [Google Scholar]
- 10.Palecek E. Surface-attached molecular beacons light the way for DNA sequencing. Trends Biotechnol. 2004;22:55–58. doi: 10.1016/j.tibtech.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Holden Thorp H. Reagentless detection of DNA sequences on chemically modified electrodes. Trends Biotechnol. 2003;21(12):522–524. doi: 10.1016/j.tibtech.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 12.Ricci F, Lai RY, Heeger AJ, Plaxco KW, Sumner JJ. Effect of molecular crowding on the response of an electrochemical DNA sensor. Langmuir. 2007;23:6827–6834. doi: 10.1021/la700328r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ricci F, Plaxco KW. E-DNA sensors for convenient, label-free electrochemical detection of hybridization. Microchim Acta. 2008;163(3–4):149–155. [Google Scholar]
- 14.Carpini G, Lucarelli F, Marrazza G, Mascini M. Oligonucleotide-modified screen-printed gold electrodes for enzyme-amplified sensing of nucleic acids. Biosens Bioelectron. 2004;20(2):167–175. doi: 10.1016/j.bios.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 15.Ricci F, Amine A, Moscone D, Palleschi G. A probe for NADH and H2O2 amperometric detection at low applied potential for oxidase and dehydrogenase based biosensor applications. Biosens Bioelectron. 2007;22(6):854–862. doi: 10.1016/j.bios.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Jenkins DM, Chami B, Kreuzer M, Presting G, Alvarez AM, Liaw BY. Hybridization probe for femtomolar quantification of selected nucleic acid sequences on a disposable electrode. Anal Chem. 2006;78(7):2314–2318. doi: 10.1021/ac051619s. [DOI] [PubMed] [Google Scholar]
- 17.Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 18.O’Dea JJ, Osteryoung JG. Characterization of quasi-reversible surface processes by square-wave voltammetry. Anal Chem. 1993;65(21):3090–3097. [Google Scholar]
- 19.Lovric M. Square-wave voltammetry. In: Scholz F, editor. Electroanalytical Methods, Guide to Experiments and Applications. Springer-Verlag; Berlin: 2002. pp. 111–133. [Google Scholar]
- 20.Murray RW. In: Electroanalytical Chemistry. Bard AJ, editor. Vol. 13. Marcel Dekker; New York: 1984. pp. 191–368. [Google Scholar]
- 21.Komorsky-Lovric S, Lovric M. Kinetic measurements of a surface confined redox reaction. Anal Chim Acta. 1995;305:248–255. [Google Scholar]
- 22.Lovric M, Komorsky-Lovric S. Square-wave voltammetry of an adsorbed reactant. J Electroanal Chem. 1988;248:239–253. [Google Scholar]
- 23.Lucarelli F, Marrazza G, Turner APF, Mascini M. Carbon and gold electrodes as electrochemical transducers for DNA hybridisation sensors. Biosens Bioelectron. 2004;19:515–530. doi: 10.1016/s0956-5663(03)00256-2. [DOI] [PubMed] [Google Scholar]
- 24.Weber K, Hockett L, Creager S. Long-range electronic coupling between ferrocene and gold in alkanethiolate-based monolayers on electrodes. J Phys Chem B. 1997;101(41):8286–8291. [Google Scholar]
- 25.Creager S, Yu CJ, Bamdad S, O’Connor S, MacLean T, Lam E, Chong Y, Olsen GT, Luo J, Gozin M, Kayyem JF. Electron transfer at electrodes through conjugated “molecular wire” bridges. J Am Chem Soc. 1999;121(5):1059–1064. [Google Scholar]
- 26.Sumner JJ, Creager SE. Redox kinetics in monolayers on electrodes: electron transfer is sluggish for ferrocene groups buried within the monolayer interior. J Phys Chem B. 2001;105(37):8739–8745. [Google Scholar]
- 27.Rentsch S, Siegenthaler H, Papastavrou G. Diffuse layer properties of thiol-modified gold electrodes probed by direct force measurements. Langmuir. 2007;23:9083–9091. doi: 10.1021/la700987u. [DOI] [PubMed] [Google Scholar]
- 28.Hildebrandt P, Murgida DH. Electron transfer dynamics of cytochrome c bound to self-assembled monolayers on silver electrodes. Bioelectrochemistry. 2002;55:139–143. doi: 10.1016/s1567-5394(01)00154-2. [DOI] [PubMed] [Google Scholar]
- 29.Lai RY, Seferos DS, Heeger AJ, Bazan GC, Plaxco KW. Comparison of the signaling and stability of electrochemical DNA sensors fabricated from 6- or 11-carbon self-assembled monolayers. Langmuir. 2006;22(25):10796–10800. doi: 10.1021/la0611817. [DOI] [PubMed] [Google Scholar]
- 30.Phares N, White RJ, Plaxco KW. Improving the stability and sensing of electrochemical biosensors by employing trithiol-anchoring groups in a six-carbon self-assembled monolayer. Anal Chem. 2009;81(3):1095–1100. doi: 10.1021/ac8021983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J. Electrochemical nucleic acid biosensors. Anal Chim Acta. 2002;288:205–214. [Google Scholar]
- 32.Drummond TG, Hill MG, Barton JK. Electrochemical DNA biosensors. Nat Biotechnol. 2003;21:1192–1199. doi: 10.1038/nbt873. [DOI] [PubMed] [Google Scholar]
- 33.Gooding JJ. Electrochemical DNA hybridization biosensors. Electroanalysis. 2002;14:1149–1156. [Google Scholar]
- 34.Wang R, Tombelli S, Minunni M, Spiriti MM, Mascini M. Direct immobilisation of DNA probes for the development of SPR-based sensing. Biosens Bioelectron. 2004;20(5):967–974. doi: 10.1016/j.bios.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 35.Dharuman V, Hahn JH. Effect of short chain alkane diluents on the label free electrochemical DNA hybridization discrimination at the HS-ssDNA/diluent binary mixed monolayer in presence of cationic intercalators. Sens Actuators B. 2007;127:536–544. [Google Scholar]