

Abstract
The bed nucleus of the stria terminalis (BNST) is thought to generate anxiety-like states via its projections to autonomic and neuroendocrine regulatory structures of the brain. However, because most BNST cells are GABAergic, they are expected to inhibit target neurons. In contrast with this, infusion of calcitonin gene-related peptide (CGRP) into BNST was reported to potentiate anxiety while activating BNST targets. The present study aimed to shed light on this paradox. The CGRP innervation of BNST originates in the pontine parabrachial nucleus and targets its anterolateral sector (BNST-AL). Thus, we investigated the effects of CGRP on BNST-AL neurons using patch recordings in vitro in male rats. CGRP did not alter the passive properties of BNST-AL cells but increased the amplitude of IPSPs evoked by stimulation of the stria terminalis (ST). However, IPSP paired-pulse ratios were unchanged by CGRP, and there was no correlation between IPSP potentiation and variance, suggesting that CGRP acts postsynaptically. Consistent with this, CGRP hyperpolarized the GABA-A reversal of BNST-AL cells. These results indicate that CGRP increases ST-evoked GABA-A IPSPs and hyperpolarizes their reversal potential through a postsynaptic change in Cl− homeostasis. Overall, our findings suggest that CGRP potentiates anxiety-like behaviors and increases neural activity in BNST targets, by inhibiting BNST-AL cells, supporting the conclusion that BNST-AL exerts anxiolytic effects.
Introduction
The bed nucleus of the stria terminalis (BNST) is a poorly understood brain structure thought to play a critical role in fear and anxiety. Indeed, electrolytic (Gewirtz et al., 1998; Sullivan et al., 2004) or neurotoxic (LeDoux et al., 1988; Hammack et al., 2004; Duvarci et al., 2009) BNST lesions as well as reversible inactivation of BNST (Walker and Davis, 1997) impair the expression of classically conditioned fear responses to contexts. In keeping with this, BNST projects to hypothalamic (Prewitt and Herman, 1998; Dong et al., 2001; Dong and Swanson, 2006) and brainstem (Sofroniew, 1983; Holstege et al., 1985; Moga et al., 1989; Sun and Cassell, 1993) structures that generate the neuroendocrine, behavioral, and cardiovascular correlates of fear and anxiety.
Currently, it is unclear whether activation or inhibition of BNST is required to produce anxiety-like behaviors. On the one hand, the fact that lesion or inactivation of BNST produces anxiolytic effects suggests that increased BNST activity generates negative emotional states. On the other hand, BNST is mainly comprised of GABAergic neurons (Esclapez et al., 1993; Hur and Zaborszky, 2005; Poulin et al., 2009) and therefore presumably exerts inhibitory effects on its targets. In contrast with this, however, Sink et al. (2011) reported that intra-BNST injections of calcitonin gene-related peptide (CGRP) augment acoustic startle while increasing activity in targets of BNST. The present study was undertaken to address this apparent contradiction.
CGRP is a 37 amino acid peptide involved in autonomic functions and pain processing (for review, see van Rossum et al., 1997). The sole CGRP input to BNST originates in the pontine parabrachial nucleus (Shimada et al., 1985), which projects heavily to the anterolateral portion of BNST (BNST-AL) (Gustafson and Greengard, 1990; Alden et al., 1994; Dobolyi et al., 2005). Thus, to shed light on how BNST regulates anxiety, we studied the effect of CGRP on BNST-AL neurons recorded with the patch method in brain slices in vitro.
Materials and Methods
Slice preparation.
Procedures were approved by the Institutional Animal Care and Use Committee of Rutgers University, in compliance with the Guide for the Care and Use of Laboratory Animals (Department of Health and Human Services). Male Lewis rats (4–7 weeks old) were anesthetized with avertin (300 mg/kg, i.p.), followed by isoflurane. After abolition of reflexes, they were perfused with an ice-cold solution containing (in mm) as follows: 126 choline chloride, 2.5 KCl, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 10 glucose. The brains were sliced with a vibrating microtome (300 μm) while submerged in the same solution. The slices were then kept in an oxygenated chamber containing artificial CSF (aCSF; as above except for the substitution of 126 mm NaCl for choline chloride, pH 7.2, 300 mOsm). The temperature of the chamber was kept at 34°C for 20 min and then returned to room temperature. One hour later, slices were then transferred to a recording chamber perfused with oxygenated aCSF at 32°C (6 ml/min).
Electrophysiological recordings.
Whole-cell recordings of BNST-AL neurons were obtained under visual guidance using 5–8 mΩ pipettes pulled from borosilicate glass capillaries. The intracellular solution contained (in mm): 130 K-gluconate, 10 HEPES, 10 KCl, 2 MgCl2, 2 ATP-Mg, and 0.2 GTP-tris(hydroxymethyl)aminomethane, pH 7.2, 280 mOsm. The liquid junction potential was 10 mV with this solution. However, membrane potential (Vm) values mentioned below were not corrected for the junction potential. We used an Axoclamp-2B amplifier (Molecular Devices) and digitized the data at 10 kHz with a Digidata-1200 interface controlled by pClamp-8.1 (Molecular Devices).
To characterize the electroresponsive properties of the cells, we applied series of current pulses (±10 pA increments; 500 ms; 0.2 Hz) from −55 and −70 mV; this revealed that all of the neurons recorded in this study correspond to the previously described Type 1 or Type 2 neurons (Hammack et al., 2007; Rodríguez-Sierra et al., 2013), with no difference in CGRP responsiveness between them. A pair of tungsten-stimulating electrodes (intertip spacing, 200 μm) was placed in the stria terminalis (ST; see Fig. 1A) and used to deliver brief current pulses (0.1 ms; 0.03 Hz). Cells were kept at −55 mV unless stated otherwise. When testing the effects of CGRP on ST-evoked EPSP amplitudes, stimulation intensity (0.1–0.8 mA) was adjusted to obtain the highest subthreshold response amplitudes. When testing CGRP effects on IPSPs, the stimulation intensity was adjusted to elicit IPSPs of ∼5 mV amplitude so that the IPSP peak would not approach the GABA-A reversal potential. Input resistance (Rin) was calculated as the average voltage response to −10 pA current injections during the stimulation protocol. A 10 min baseline recording was obtained before CGRP application. CGRP was then applied for 10 min. Before versus after peptide comparisons were made using responses obtained 5–10 min before versus 15–20 min after onset of CGRP application (separate averages of 10 ST-evoked responses). Antagonists were added to the perfusate solution 15 min before CGRP application and were present throughout the recordings.
Figure 1.

CGRP potentiates ST-evoked IPSPs. A, Scheme showing stimulating (Stim.) and recording (circles) sites. B, Time course of CGRP effect on IPSPs (solid squares represent CGRP 0.5 μm; empty squares represent control cells). CGRP steadily increases ST-evoked IPSP amplitudes, and this effect outlasts the period of CGRP application. Insets on left, Examples of ST-evoked responses in a control cell (top) and one exposed to CGRP (bottom). Inset on top right, Examples of EPSPs isolated with picrotoxin (100 μm) before versus after a 10 min application of CGRP (1 μm). C, Top, Dose dependency of CGRP effect on IPSPs. After CGRP application (15 min), IPSP amplitudes increased to 260 ± 49% for 2 μm (n = 9, p = 0.002), 174 ± 22% for 1 μm (n = 4, p = 0.01), and 170 ± 15% for 0.5 μm (n = 5, p = 0.0005) of preapplication values. At a concentration of 250 nm CGRP had no significant effect (n = 8, p = 0.41). Eight cells were tested with no CGRP application. C, Bottom, Two CGRP receptor Type 1 antagonists reduced the effect of CGRP, compared with when CGRP (500 nm, n = 5) was applied alone (SB268262, 50 μm, n = 12, p < 0.001; 500 μm, n = 7, p < 0.01; CGRP8–37, 1 μm, n = 4, p < 0.001; 2 μm, n = 5, p = 0.01). AC, Anterior commissure; IC, internal capsule.
Drugs.
CGRP (rat), SB-268262 (N-methyl-N-(2-methylphenyl)-3-nitro-4-(2-thiazolylsulfinyl)-benzamide), picrotoxin, CNQX disodium salt, and (±)-3-(2-carboxypiperazin-4-yl)propyl-1-phosphonic acid (CPP) were obtained from Sigma. CGRP8–37 was obtained from Tocris Bioscience.
Data analysis.
Data were analyzed offline using Clampfit version 9.2. All data are reported as mean ± SEM. SEM calculations were modified for repeated designs as described by Cousineau (2005). For statistical analyses, we conducted one-way ANOVA with Tukey's Honestly significant difference paired post hoc tests as well as paired t tests.
Results
Effect of CGRP on the electroresponsive properties and synaptic responses of BNST-AL cells
We obtained patch recordings of 184 BNST-AL cells (Fig. 1A) that had stable resting potentials and generated overshooting action potentials upon depolarization. We first tested whether CGRP (0.5–2 μm) alters the passive properties, firing pattern, or spike characteristics of BNST-AL cells but found no effect (Table 1). Because it was reported that CGRP increases EPSP amplitudes in central amygdala neurons at their resting potential (Han et al., 2005, 2010), we next examined the impact of CGRP on ST-evoked EPSPs in the presence of picrotoxin (100 μm). Picrotoxin completely abolished ST-evoked IPSPs in 90% of tested neurons (36 of 40), allowing us to examine the influence of CGRP (1 μm) on isolated EPSPs. However, EPSP amplitudes were unaffected by CGRP (difference of −0.7 ± 0.6 mV, n = 12, p = 0.25; Fig. 1B, top right inset).
Table 1.
Effect of CGRP (1 μm) on the electroresponsive properties of BNST-AL neuronsa
| Control | CGRP | p | n | |
|---|---|---|---|---|
| Resting potential (mV) | −61.5 ± 1.5 | −60.9 ± 1.4 | 0.27 | 26 |
| Input resistance (mΩ) | 715.7 ± 74.1 | 731.2 ± 74.6 | 0.38 | 26 |
| Time constant (ms) | 46.9 ± 4.6 | 50.6 ± 4.0 | 0.25 | 26 |
| Rheobase (pA) | 40.8 ± 6.1 | 42.5 ± 5.7 | 0.44 | 12 |
| Spike threshold (mV) | −48.1 ± 2.2 | −49.4 ± 2.3 | 0.31 | 12 |
| Spike latency (ms) | 98.2 ± 13.3 | 104.3 ± 15.9 | 0.7 | 12 |
| Spike amplitude (mV) | 87.8 ± 3 | 83.6 ± 3.63 | 0.16 | 12 |
| Spike duration at half amplitude (ms) | 0.48 ± 0.03 | 0.49 ± 0.01 | 0.8 | 12 |
| Firing rate at rheobase (Hz) | 0.75 ± 0.21 | 0.75 ± 0.14 | 1 | 12 |
aValues are mean ± SEM. We restricted our analysis of passive properties and spike characteristics to cells tested in the same conditions (before and after CGRP), with no other drugs present. Also, 8 additional cells tested with 250 nm CGRP were not included because this dose had no effect. Finally, for the spike characteristics, in 14 of the 26 cells meeting our selection criteria, no responses to positive current pulses were recorded.
In contrast, in the absence of picrotoxin, CGRP significantly increased the amplitude of ST-evoked IPSPs (Fig. 1B, solid squares and bottom inset) in a dose-dependent fashion (n = 26; ANOVA, F(4,29) = 5.48, p = 0.02; Fig. 1C, top). The potentiation of ST-evoked IPSPs outlasted the CGRP application period by >20 min (Fig. 1B). Because the increase in IPSP could have resulted from the gradual equilibration of the intracellular Cl− concentration with that of the pipette solution, we examined whether the IPSP amplitudes increased spontaneously over time (n = 8), with no peptide application. However, no significant time-dependent changes were observed (Fig. 1B, empty squares, inset; n = 8, p = 0.35). Furthermore, two different CGRP Type 1 receptor antagonists, SB268262 (n = 19) and CGRP8–37 (n = 9), reduced or completely abolished the IPSP potentiation produced by CGRP (Fig. 1C, bottom).
Mechanisms underlying the potentiation of ST-evoked IPSPs by CGRP
To investigate whether the increase in ST-evoked IPSPs produced by CGRP is dependent on presynaptic or postsynaptic mechanisms, we conducted three analyses. First, we compared the paired-pulse ratio (PPR) of ST-evoked IPSCs before versus after CGRP application (Fig. 2A). In such analyses, two stimuli of equal intensity are applied in brief succession (50 ms), leading to an enhancement or reduction of the response elicited by the second stimulus. Changes in PPR are commonly thought to reflect alterations in transmitter release probability (Creager et al., 1980; Manabe et al., 1993). PPR tests were performed in the presence of the glutamate receptor antagonists CNQX (10 μm) and CPP (10 μm). In control conditions, a small average paired-pulse facilitation was observed, but it did not reach significance (p = 0.32). Addition of CGRP (1 μm) did not alter the PPR of ST-evoked IPSPs (Fig. 2A; PPR difference of 0.1 ± 0.1; n = 6; p = 0.43), suggesting that CGRP acts postsynaptically to potentiate the IPSPs.
Figure 2.

CGRP potentiates ST-evoked IPSPs through a postsynaptic mechanism. A1, PPR before and 15 min after CGRP application (n = 6). A2, A3, Average traces from the same cell before (2) and after (3) CGRP application. Despite strong ST-evoked IPSC potentiation, there is no difference in the PPR. B, CV2 analysis. Ratio of CV2 during CGRP to control (y-axis) versus ratio of CGRP to control IPSP amplitudes (x-axis, n = 18). C, CGRP causes a negative shift of the GABA-A reversal potential. CNQX (10 μm) and CPP (10 μm) present throughout. Linear fits of IPSP amplitude (y-axis) as a function of Vm (x-axis) before (empty squares) and after (solid squares) CGRP (mean ± SEM, 14 cells). Insets, Average IPSP amplitude of a representative cell before (top right) and after (bottom left) CGRP. D, Linear fits of Vm (y-axis) versus current (x-axis) before the ST stimulus (solid squares) and at the IPSP peak (×), before (blue) and after (red) CGRP. E, CGRP-induced changes in IPSP amplitudes (y-axis) and GABA-A reversal potentials (x-axis) are correlated.
Second, we studied IPSP variability using the data obtained in the dose–response experiments of Figure 1C. This variability is known to reflect the probabilistic process underlying transmitter release and can be estimated by computing the coefficient of variation (CV, SD/mean). By plotting the ratio of experimental to control CV2 against the ratio of experimental to control response amplitudes, the dependence of presynaptic versus postsynaptic function can be determined (Bekkers and Stevens, 1990; Manabe et al., 1993). In these prior studies, a positive correlation between the two was shown to reflect a presynaptic mechanism, whereas a horizontal regression is indicative of a purely postsynaptic action. We found no significant relationship between CV2 and IPSP amplitudes (Fig. 2B; r = −0.06, p = 0.77, n = 14), again pointing to a postsynaptic locus of CGRP action.
Third, we tested whether CGRP alters the reversal potential (EGABA-A) of ST-evoked IPSPs in the presence of the glutamate receptor antagonists CNQX (10 μm) and CPP (10 μm). CGRP (1 μm) caused a significant negative shift of EGABA-A (Fig. 2C; difference 4.31 ± 1.25 mV; control, −72.66 ± 1.9 mV; CGRP, −76.97 ± 1.99 mV; p = 0.004, n = 14). Importantly, this effect was not associated with a change in the Rin drop caused by the IPSPs (Fig. 2D; −11 ± 11 mΩ; p = 0.33), suggesting that the CGRP-induced augmentation in IPSP amplitude is largely dependent on an increased Cl− driving force. Indeed, there was a significant correlation between the CGRP-induced changes in IPSP amplitudes and EGABA-A (Fig. 2E; n = 14, r = −0.59, p = 0.03). In contrast, in a different sample of control cells without CGRP application, no time-dependent shift in GABA-A reversal potential was observed (−0.1 ± 0.9 mV, n = 14, p = 0.89).
If CGRP acts postsynaptically to enhance IPSPs by increasing the Cl− driving force, one would expect manipulations that interfere with the cells' Cl− homeostasis to disrupt CGRP's effects. A major regulator of the intracellular Cl− concentration in neurons (for review, see Kaila, 1994) is the potassium-chloride cotransporter (KCC), which mediates Cl− extrusion (Misgeld et al., 1986; Thompson et al., 1988). Consistent with this, VU-0240551 (40 μm), a selective KCC2 blocker, depolarized EGABA-A by 4.1 ± 1.1 mV (n = 9, p = 0.007) and prevented the effects of CGRP on IPSP amplitudes (0.5 ± 0.5 mV; n = 7, p = 0.31) and EGABA-A (−0.3 ± 0.7 mV; p = 0.72; Fig. 3).
Figure 3.
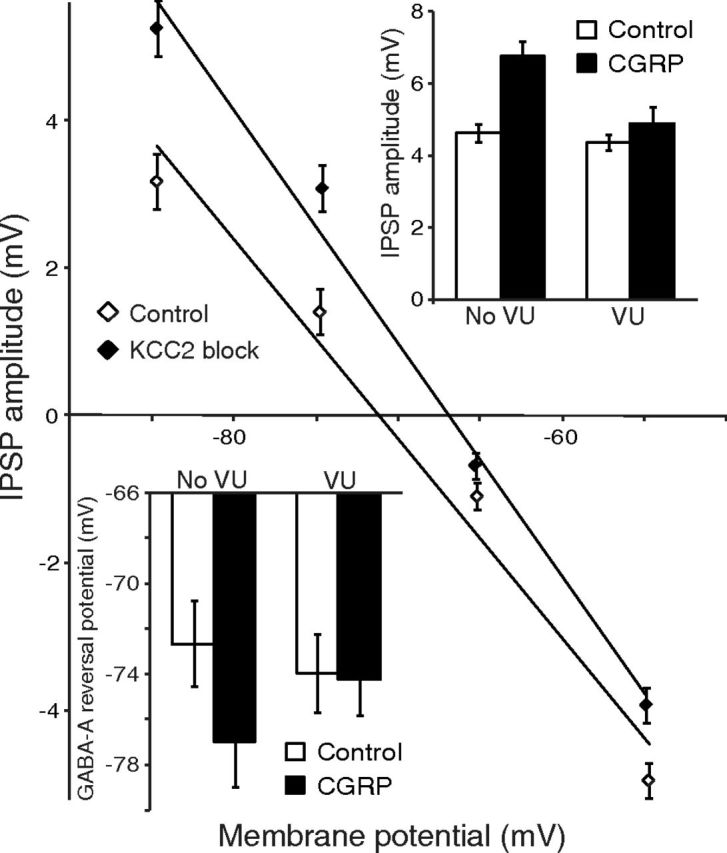
KCC2 antagonist VU-0240551 (VU) prevents the effects of CGRP on IPSP amplitudes and reversal potentials. IPSP amplitude (y-axis) as a function of membrane potential (x-axis) in cells recorded in the absence (empty diamonds) versus the presence (filled diamonds) of the KCC2 antagonist VU-0240551 (40 μm). Top right inset, Amplitude of IPSPs before (empty bars) versus after CGRP (filled bars) in the absence (left) versus presence (right) of VU-0240551. Bottom left inset, GABA-A reversal potential before (empty bars) versus after CGRP (filled bars) in the absence (left) versus presence (right) of VU-0240551.
An alternative interpretation for CGRP effects, namely, that it results from an enhancement of GABA-B IPSPs, appears unlikely for the following reasons. First, as mentioned just above, inhibiting KCC2 blocked the CGRP effect. Second, our IPSP measurements were performed at the peak of the GABA-A IPSPs, ∼30 ms from response onset, well before the development of GABA-B responses in other cell types. Third, most BNST-AL cells lacked overt GABA-B responses, with picrotoxin abolishing ST-evoked IPSPs in 36 of 40 cells. Last, in the rare BNST-AL cells with picrotoxin-resistant IPSP components, addition of CGRP failed to alter the residual IPSP amplitudes (reduction of 0.4 ± 0.04 mV, n = 2).
Discussion
This study aimed to characterize the influence of CGRP on BNST neurons. Pontine parabrachial neurons constitute the sole source of CGRP to BNST, and they project to its anterolateral sector (Alden et al., 1994) where there are no glutamatergic, only GABAergic/peptidergic, cells (Poulin et al., 2009). In light of these data, the finding that intra-BNST infusions of CGRP enhance startle and neuronal activation in BNST-AL targets (Sink et al., 2011) suggested that CGRP inhibits BNST-AL neurons. However, this inference is in apparent contradiction with the generally accepted view that BNST activity exerts an anxiogenic influence (Davis et al., 2010). Here, we observed that CGRP inhibits BNST-AL neurons. Below, we consider the mechanisms and significance of CGRP's inhibitory influence on BNST-AL for the regulation of fear and anxiety.
CGRP potentiates GABA-A inhibition through a postsynaptic regulation of Cl− homeostasis
To the best of our knowledge, there are no prior reports of CGRP's influence on BNST neurons. However, in other parts of the nervous system, a variety of cell type-specific effects were reported. For example, CGRP inhibits high-threshold voltage-gated Ca2+ currents in neurons of nucleus tractus solitarius (Hosokawa et al., 2010) but enhances them in DRG cells (Ryu et al., 1988). It causes a membrane hyperpolarization in some cell types (Kajekar and Myers, 2008) and the opposite (Gokin et al., 1996) or no change in others (Meng et al., 2009). In CA1 pyramidal cells, CGRP inhibits the slow Ca2+-dependent K+ current (Haug and Storm, 2000). Consistent with this, in central amygdala neurons, CGRP reduces spike frequency adaptation. In the same cell type, CGRP also causes a postsynaptically mediated increase in glutamatergic EPSCs at rest (Han et al., 2005, 2010).
Given the functional kinship and anatomical similarities between BNST and the central amygdala, one might expect CGRP to exert similar effects at the two sites. Yet, this is not what we observed. In BNST-AL cells, firing rate/pattern, passive properties, spike characteristics, and EPSP amplitudes were unaffected by CGRP. Instead, CGRP produced a robust potentiation of ST-evoked IPSPs, and this effect was reduced or blocked by CGRP Type 1 receptor antagonists.
Several observations indicate that the IPSP potentiation produced by CGRP is dependent on a postsynaptic mechanism. First, CGRP did not alter the PPR, and there was no correlation between the ratios of experimental to control IPSP variance versus amplitude. Second, the Rin drop associated with the IPSP was not altered by CGRP. Third, CGRP shifted EGABA-A negatively, and the amplitude of this shift correlated with the magnitude of IPSP potentiation. Last, CGRP's effect on IPSP amplitudes and reversal potentials was blocked by prior application of a KCC inhibitor. Although the mechanisms of CGRP action on chloride homeostasis are currently unclear, given the results obtained in other cells types (for review, see Kahle et al., 2010), an upregulation of potassium chloride transporters by phosphoregulation is likely involved.
Significance for the role of BNST in fear and anxiety
Although its name implies otherwise, BNST is in fact comprised of a collection of nuclei that form contrasting connections with fear effector neurons (Bota et al., 2012). As reviewed above, early lesion and inactivation studies targeting BNST as a whole supported the view that BNST promotes the genesis of long-lasting anxiety-like states (Walker and Davis, 1997). However, more recent reports using techniques that allow selective manipulations of different BNST regions (Kim et al., 2013) or of different cell types within these regions (Jennings et al., 2013) revealed that BNST is functionally heterogeneous. Consistent with this, neurons in BNST-AL and the anteromedial part of BNST (BNST-AM) show opposite behaviors in relation to classically conditioned fear (Haufler et al., 2013): BNST-AL neurons fire at lower rates during high than low fear states, whereas BNST-AM cells do the opposite. The reduced activity of BNST-AL cells during fear is consistent with our observations that CGRP inhibits BNST-AL neurons and the fact that intra-BNST CGRP infusions potentiate acoustic startle while increasing neuronal activity in BNST-AL targets (Sink et al., 2011). Together, these findings suggest that BNST-AM and BNST-AL exert opposite influences on fear output networks, with BNST-AM neurons promoting and BNST-AL cells inhibiting fear and anxiety. Moreover, because BNST-AL contributes inhibitory projections to BNST-AM (Turesson et al., 2013), it is likely that antagonistic interactions take place between the two regions.
In contrast to the above, a different interpretation was recently offered for CGRP's anxiogenic effects. Indeed, Sink et al. (2013) reported that virally mediated knockdown of CRF expression as well as systemic or intra-BNST infusions of CRF1 antagonists interfered with the startle potentiation produced by intra-BNST CGRP infusions. Based on these results, the authors suggested that CGRP acts by increasing the activity of CRF-positive BNST neurons. Given that these cells constitute a small subset of BNST-AL cells and that the CGRP effects described here were seen in the vast majority of tested cells, it remains unclear what the relative importance of the CRF and GABA effects is in mediating CRGP's anxiogenic influence.
Footnotes
This work was supported by National Institutes of Mental Health Grant R01 MH-098738 to D.P.
The authors declare no competing financial interests.
References
- Alden M, Besson JM, Bernard JF. Organization of the efferent projections from the pontine parabrachial area to the bed nucleus of the stria terminalis and neighboring regions: a PHA-L study in the rat. J Comp Neurol. 1994;341:289–314. doi: 10.1002/cne.903410302. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Presynaptic mechanism for long-term potentiation in the hippocampus. Nature. 1990;346:724–729. doi: 10.1038/346724a0. [DOI] [PubMed] [Google Scholar]
- Bota M, Sporns O, Swanson LW. Neuroinformatics analysis of molecular expression patterns and neuron populations in gray matter regions: the rat BST as a rich exemplar. Brain Res. 2012;1450:174–193. doi: 10.1016/j.brainres.2012.02.034. [DOI] [PubMed] [Google Scholar]
- Cousineau D. Confidence intervals in within subject designs: a simpler solution to Loftus and Masson's method. Tutorials Quant Methods Psychol. 2005;1:42–45. [Google Scholar]
- Creager R, Dunwiddie T, Lynch G. Paired-pulse and frequency facilitation in the CA1 region of the in vitro rat hippocampus. J Physiol. 1980;299:409–424. doi: 10.1113/jphysiol.1980.sp013133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Walker DL, Miles L, Grillon C. Phasic vs sustained fear in rats and humans: role of the extended amygdala in fear vs anxiety. Neuropsychopharmacology. 2010;35:105–135. doi: 10.1038/npp.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobolyi A, Irwin S, Makara G, Usdin TB, Palkovits M. Calcitonin gene related peptide containing pathways in the rat forebrain. J Comp Neurol. 2005;489:92–119. doi: 10.1002/cne.20618. [DOI] [PubMed] [Google Scholar]
- Dong HW, Swanson LW. Projections from bed nuclei of the stria terminalis, anteromedial area: cerebral hemisphere integration of neuroendocrine, autonomic, and behavioral aspects of energy balance. J Comp Neurol. 2006;494:142–178. doi: 10.1002/cne.20788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong HW, Petrovich GD, Watts AG, Swanson LW. Basic organization of projections from the oval and fusiform nuclei of the bed nuclei of the stria terminalis in adult rat brain. J Comp Neurol. 2001;436:430–455. doi: 10.1002/cne.1079. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Bauer EP, Paré D. The bed nucleus of the stria terminalis mediates inter-individual variations in anxiety and fear. J Neurosci. 2009;29:10357–10361. doi: 10.1523/JNEUROSCI.2119-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esclapez M, Tillakaratne NJ, Tobin AJ, Houser CR. Comparative localization of two forms of glutamic acid decarboxylase with nonradioactive in situ hybridization methods. J Comp Neurol. 1993;331:339–362. doi: 10.1002/cne.903310305. [DOI] [PubMed] [Google Scholar]
- Gewirtz JC, McNish KA, Davis M. Lesions of the bed nucleus of the stria terminalis block sensitization of the acoustic startle reflex produced by repeated stress, but not fear-potentiated startle. Prog Neuropsychopharmacol Biol Psychiatry. 1998;22:625–648. doi: 10.1016/S0278-5846(98)00028-1. [DOI] [PubMed] [Google Scholar]
- Gokin AP, Jennings LJ, Mawe GM. Actions of calcitonin gene-related peptide in guinea pig gallbladder ganglia. Am J Physiol. 1996;271:G876–G883. doi: 10.1152/ajpgi.1996.271.5.G876. [DOI] [PubMed] [Google Scholar]
- Gustafson EL, Greengard P. Localization of DARPP-32 immunoreactive neurons in the bed nucleus of stria terminalis and central nucleus of amygdala: codistribution with axons containing tyrosine hydroxylase, vasoactive intestinal polypeptide, and calcitonin gene related peptide. Exp Brain Res. 1990;1990:447–458. doi: 10.1007/BF00229315. [DOI] [PubMed] [Google Scholar]
- Hammack SE, Richey KJ, Watkins LR, Maier SF. Chemical lesions of the bed nucleus of the stria terminalis blocks the behavioral consequences of uncontrollable stress. Behav Neurosci. 2004;118:443–448. doi: 10.1037/0735-7044.118.2.443. [DOI] [PubMed] [Google Scholar]
- Hammack SE, Mania I, Rainnie DG. Differential expression of intrinsic membrane currents in defined cell types of the anterolateral bed nucleus of the stria terminalis. J Neurophysiol. 2007;98:638–656. doi: 10.1152/jn.00382.2007. [DOI] [PubMed] [Google Scholar]
- Han JS, Li W, Neugebauer V. Critical role of calcitonin gene related peptide 1 receptors in the amygdala in synaptic plasticity and pain behavior. J Neurosci. 2005;25:10717–10728. doi: 10.1523/JNEUROSCI.4112-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Adwanikar H, Li Z, Ji G, Neugebauer V. Facilitation of synaptic transmission and pain responses by CGRP in the amygdala of normal rats. Mol Pain. 2010;6:10. doi: 10.1186/1744-8069-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haufler D, Nagy FZ, Pare D. Neuronal correlates of fear conditioning in the bed nucleus of the stria terminalis. Learn Mem, 2013;20:633–641. doi: 10.1101/lm.031799.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug T, Storm JF. Protein kinase A mediates the modulation of the slow Ca(2+)-dependent K(+) current, I(sAHP), by the neuropeptides CRF, VIP, and CGRP in hippocampal pyramidal neurons. J Neurophysiol. 2000;83:2071–2079. doi: 10.1152/jn.2000.83.4.2071. [DOI] [PubMed] [Google Scholar]
- Holstege G, Meiners L, Tan K. Projections of the bed nucleus of the stria terminalis to the mesencephalon, pons, and medulla oblongata in the cat. Exp Brain Res. 1985;58:379–391. doi: 10.1007/BF00235319. [DOI] [PubMed] [Google Scholar]
- Hosokawa S, Endoh T, Shibukawa Y, Tsumura M, Ichikawa H, Tazaki M, Furusawa M. Calcitonin gene-related peptide- and adrenomedullin-induced facilitation of calcium current by different signal pathways in nucleus tractus solitarius. Brain Res. 2010;1327:47–55. doi: 10.1016/j.brainres.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Hur EE, Zaborszky L. Vglut2 afferents to the medial prefrontal and primary somatosensory cortices: a combined retrograde tracing in situ hybridization study. J Comp Neurol. 2005;483:351–373. doi: 10.1002/cne.20444. [DOI] [PubMed] [Google Scholar]
- Jennings JH, Sparta DR, Stamatakis AM, Ung RL, Pleil KE, Kash TL, Stuber GD. Distinct extended amygdala circuits for divergent motivational states. Nature. 2013;496:224–228. doi: 10.1038/nature12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Rinehart J, Lifton RP. Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochim Biophys Acta. 2010;1802:1150–1158. doi: 10.1016/j.bbadis.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K. Ionic basis of GABA-a receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Kajekar R, Myers AC. Calcitonin gene-related peptide affects synaptic and membrane properties of bronchial parasympathetic neurons. Respir Physiol Neurobiol. 2008;160:28–36. doi: 10.1016/j.resp.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Adhikari A, Lee SY, Marshel JH, Kim CK, Mallory CS, Lo M, Pak S, Mattis J, Lim BK, Malenka RC, Warden MR, Neve R, Tye KM, Deisseroth K. Diverging neural pathways assemble a behavioral state from separable features in anxiety. Nature. 2013;496:219–223. doi: 10.1038/nature12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE, Iwata J, Cicchetti P, Reis DJ. Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci. 1988;8:2517–2519. doi: 10.1523/JNEUROSCI.08-07-02517.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Meng J, Ovsepian SV, Wang J, Pickering M, Sasse A, Aoki KR, Lawrence GW, Dolly JO. Activation of TRPV1 mediates calcitonin gene-related peptide release, which excites trigeminal sensory neurons and is attenuated by a retargeted botulinum toxin with anti-nociceptive potential. J Neurosci. 2009;29:4981–4992. doi: 10.1523/JNEUROSCI.5490-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld U, Deisz RA, Dodt HU, Lux HD. The role chloride transport in postsynaptic inhibition of hippocampal neurons. Science. 1986;232:1413–1415. doi: 10.1126/science.2424084. [DOI] [PubMed] [Google Scholar]
- Moga MM, Saper CB, Gray TS. Bed nucleus of the stria terminalis: cytoarchitecture, immunohistochemistry, and projection to the parabrachial nucleus in the rat. J Comp Neurol. 1989;283:315–332. doi: 10.1002/cne.902830302. [DOI] [PubMed] [Google Scholar]
- Poulin JF, Arbour D, Laforest S, Drolet G. Neuroanatomical characterization of endogenous opioids in the bed nucleus of the stria terminalis. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:1356–1365. doi: 10.1016/j.pnpbp.2009.06.021. [DOI] [PubMed] [Google Scholar]
- Prewitt CM, Herman JP. Anatomical interactions between the central amygdaloid nucleus and the hypothalamic paraventricular nucleus of the rat: a dual tract-tracing analysis. J Chem Neuroanat. 1998;15:173–185. doi: 10.1016/S0891-0618(98)00045-3. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Sierra OE, Turesson HK, Pare D. Contrasting distribution of physiological cell types in different regions of the bed nucleus of the stria terminalis. J Neurophysiol. 2013;110:2037–3049. doi: 10.1152/jn.00408.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu PD, Gerber G, Murase K, Randic M. Calcitonin gene-related peptide enhances calcium current of rat dorsal root ganglion neurons and spinal excitatory synaptic transmission. Neurosci Lett. 1988;89:305–312. doi: 10.1016/0304-3940(88)90544-7. [DOI] [PubMed] [Google Scholar]
- Shimada S, Shiosaka S, Emson PC, Hillyard CJ, Girgis S, MacIntyre I, Tohyama M. Calcitonin gene related peptidergic projection from the parabrachial area to the forebrain and diencephalon in the rat: an immunohistochemical analysis. Neuroscience. 1985;16:607–616. doi: 10.1016/0306-4522(85)90195-2. [DOI] [PubMed] [Google Scholar]
- Sink KS, Walker DL, Yang Y, Davis M. Calcitonin gene related peptide in the bed nucleus of the stria terminalis produces an anxiety like pattern of behavior and increases neural activation in anxiety related structures. J Neurosci. 2011;31:1802–1810. doi: 10.1523/JNEUROSCI.5274-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sink KS, Chung A, Ressler KJ, Davis M, Walker DL. Anxiogenic effects of CGRP within the BNST may be mediated by CRF acting at BNST CRFR1 receptors. Behav Brain Res. 2013;243:286–293. doi: 10.1016/j.bbr.2013.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Direct reciprocal connections between the bed nucleus of the stria terminalis and dorsomedial medulla oblongata: evidence from immunohistochemical detection of tracer proteins. J Comp Neurol. 1983;213:399–405. doi: 10.1002/cne.902130404. [DOI] [PubMed] [Google Scholar]
- Sullivan GM, Apergis J, Bush DE, Johnson LR, Hou M, Ledoux JE. Lesions in the bed nucleus of the stria terminalis disrupt corticosterone and freezing responses elicited by a contextual but not by a specific cue-conditioned fear stimulus. Neuroscience. 2004;128:7–14. doi: 10.1016/j.neuroscience.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Sun N, Cassell MD. Intrinsic GABAergic neurons in the rat central amygdala. J Comp Neurol. 1993;330:381–404. doi: 10.1002/cne.903300308. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Deisz RA, Prince DA. Outward chloride/cation co-transport in mammalian cortical neurons. Neurosci Lett. 1988;89:49–54. doi: 10.1016/0304-3940(88)90479-X. [DOI] [PubMed] [Google Scholar]
- Turesson HK, Rodríguez-Sierra OE, Pare D. Intrinsic connections in the anterior part of the bed nucleus of the stria terminalis. J Neurophysiol. 2013;109:2438–2450. doi: 10.1152/jn.00004.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rossum D, Hanisch UK, Quirion R. Neuroanatomical localization, pharmacological characterization and functions of CGRP, related peptides and their receptors. Neurosci Biobehav Rev. 1997;21:649–678. doi: 10.1016/S0149-7634(96)00023-1. [DOI] [PubMed] [Google Scholar]
- Walker DL, Davis M. Double dissociation between the involvement of the bed nucleus of the stria terminalis and the central nucleus of the amygdala in light enhances versus fear-potentiated startle. J Neurosci. 1997;17:9375–9383. doi: 10.1523/JNEUROSCI.17-23-09375.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]