

Abstract
Motivation: The increasing availability of metabolomics data enables to better understand the metabolic processes involved in the immediate response of an organism to environmental changes and stress. The data usually come in the form of a list of metabolites whose concentrations significantly changed under some conditions, and are thus not easy to interpret without being able to precisely visualize how such metabolites are interconnected.
Results: We present a method that enables to organize the data from any metabolomics experiment into metabolic stories. Each story corresponds to a possible scenario explaining the flow of matter between the metabolites of interest. These scenarios may then be ranked in different ways depending on which interpretation one wishes to emphasize for the causal link between two affected metabolites: enzyme activation, enzyme inhibition or domino effect on the concentration changes of substrates and products. Equally probable stories under any selected ranking scheme can be further grouped into a single anthology that summarizes, in a unique subnetwork, all equivalently plausible alternative stories. An anthology is simply a union of such stories. We detail an application of the method to the response of yeast to cadmium exposure. We use this system as a proof of concept for our method, and we show that we are able to find a story that reproduces very well the current knowledge about the yeast response to cadmium. We further show that this response is mostly based on enzyme activation. We also provide a framework for exploring the alternative pathways or side effects this local response is expected to have in the rest of the network. We discuss several interpretations for the changes we see, and we suggest hypotheses that could in principle be experimentally tested. Noticeably, our method requires simple input data and could be used in a wide variety of applications.
Availability and implementation: The code for the method presented in this article is available at http://gobbolino.gforge.inria.fr.
Contact: pvmilreu@gmail.com; vincent.lacroix@univ-lyon1.fr; marie-france.sagot@inria.fr
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
One of the main goals of metabolic studies is to understand the metabolic processes involved in the adaptation to an environmental change. Recently, metabolomic techniques gained the spotlight by providing a way to monitor metabolism by measuring the concentration of metabolites in different conditions or at different time points. A typical result from such an experiment is a list of metabolites whose concentrations significantly changed when the cell or organism was exposed to some stress. How to interpret this list became then a new research topic, consisting in identifying the metabolic processes that link the metabolites of interest, possibly explaining the observed variations in their concentrations. This topic goes in the literature by the name of ‘metabolite set enrichment analysis’, and is an extension to metabolism of work that was initiated in the context of transcriptomics and then proteomics under the name of ‘gene set enrichment analysis’ [see (Subramanian et al., 2005) for what is possibly the first work on this and (Khatri et al., 2012) for a recent survey]. The simplest idea one may think of is to highlight the set of metabolites identified in the experiment that have significantly changed their concentration, let us call them discriminating compounds, and then to visually analyze their interconnections. This is what is done notably in Xia et al. (2012). However, like a number of other approaches on metabolite set enrichment analysis, the projection of enriched metabolites is done on pathways instead of the whole network, thereby missing (alternative) pathways not annotated in current databases, or more generally paths traversing several pathways.
For genome-scale networks, the metabolism of a whole organism is considered, which may be large (Thiele and Palsson, 2010), whereas a metabolic perturbation caused by some stress condition may impact only a small portion of this complex network. Even if it is sometimes possible to visually identify the pathways that explain some of the variations in the monitored metabolites, getting an overall explanation for all the observed variations usually cannot be performed by visual inspection.
Recently, automatic methods have been proposed to deal with this kind of data (Antonov et al., 2009; Dittrich et al., 2008; Faust et al., 2010; Leader et al., 2011). A natural idea is to try to link all discriminating compounds through chains of reactions. One possible model for this is by means of a Steiner tree, which is a minimum cost tree that connects all nodes belonging to a predefined subset called terminals, which in the case of metabolism would be the discriminating compounds (Dittrich et al., 2008; Scott et al., 2005). However, any pair of metabolites may be connected through several alternative paths within a network, and each of these paths may validly explain the observed changes of concentration. In this context, the extraction of subgraphs appears to be more relevant than the extraction of subtrees. The number of alternative paths between two metabolites may, however, be large and restricting the search to all the shortest or lightest (the weight is given by the sum of the out-degrees of the vertices in the path) paths between pairs of metabolites seems to be a realistic compromise.
This is the approach followed by (Croes et al., 2006; Faust et al., 2010; van Helden et al., 2002) where the authors concentrate on a pair of discriminating compounds and search for subgraphs corresponding to source-to-sink paths between them. In Antonov et al. (2009), this approach is pushed one step further as the authors consider all pairs of metabolites and unify all the shortest paths, this time with a maximum length k. In practice, this may lead to large networks (if k is too big) or to disconnected ones (if k is too small).
The aforementioned methods are based only on the topology of the network, but one could consider different approaches based on flux distributions over the set of reactions, such as elementary modes (Schuster and Hilgetag, 1994; Schuster et al., 1999) that are minimal subnetworks working at steady state. One difficulty in this case is that flux-based models need stoichiometric values as well as a definition of the boundaries of the system under analysis, which are not always simple to identify, particularly in the case of a metabolomics experiment in which the list of discriminating compounds does not directly define such boundaries. Moreover, flux approaches are focused on reactions and are not designed to take into account endogenous metabolite concentrations. The very same metabolites may play different roles in different metabolic processes, being source in one, intermediate in a second and target in a third one. The inability and the unwillingness to tell, a priori, the role of the discriminating compounds in each scenario to be proposed is a key factor of our approach: we are interested not only in connecting the discriminating compounds but also in establishing their individual role for each scenario.
Our approach is a subgraph extraction technique in which we want to find maximal directed acyclic subgraphs (DAGs) whose set of sources and targets are discriminating compounds. We call such subgraphs metabolic stories, or for short, simply stories. In practice, for a given set of discriminating compounds, the number of stories may be large. Because we do not have a clear criterion for choosing which of these stories is the most relevant, we first aim at enumerating them all. In a second step, we discuss ways to rank them based on how the concentration of the discriminating compounds is observed to vary in the experiment. This procedure allows a good filter of the solutions, selecting stories that best fit the experimental data.
2 MODELS
2.1 Modeling metabolic stories
In this section, we introduce the notion of story and give a rationale for its definition. Briefly, stories are subgraphs that summarize the flow of matter from a set of source metabolites to a set of target metabolites. The candidates to be the endpoints (sources or targets) of a story should belong to the set of discriminating compounds. To guarantee that stories will have at least one source and one target, we introduce the acyclicity constraint. These two combined constraints lead us to search for DAGs with sources and targets contained in the given set of discriminating compounds. Then, because there can be several paths connecting two discriminating compounds and we want the story to contain all these alternative paths, we impose a constraint of maximality, that is, we search for maximal DAGs, in the sense that alternative pathways between all the nodes should be included, if their addition does not create cycles. In other words, a DAG is maximal if by adding any arc makes it not a DAG anymore, meaning that it contains at least one cycle.
Our goal is to have an algorithm that enumerates all stories, i.e. to provide all possible scenarios that explain the observed transformations. Because our focus is on the relation between discriminating compounds, we use a representation of metabolic networks focused on metabolites, the so-called compound network (Lacroix et al., 2008), that is a directed graph in which vertices are compounds and there is an arc from a compound to another compound if there is a reaction that consumes the first to produce the second.
More formally, we introduce a constrained version of the problem of enumerating all maximal DAGs of a graph G (Schwikowski and Speckenmeyer, 2002). Let 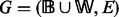 be a directed graph such that
be a directed graph such that 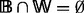 . We write
. We write 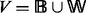 . Nodes in
. Nodes in 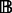 are said to be black nodes and correspond to the discriminating compounds, whereas those in
are said to be black nodes and correspond to the discriminating compounds, whereas those in 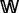 are said to be white nodes. Let
are said to be white nodes. Let 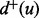 and
and 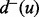 denote, respectively, the in-degree and the out-degree of a node u. Node u is called a source if
denote, respectively, the in-degree and the out-degree of a node u. Node u is called a source if 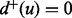 and
and 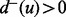 and a target if
and a target if 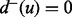 and
and 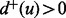 .
.
A metabolic story of G is a maximal acyclic subgraph 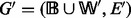 of G with
of G with 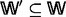 and
and 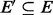 and such that, for each node
and such that, for each node 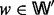 , w is neither a source nor a target red in
, w is neither a source nor a target red in 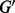 . Maximality means that it is not possible to add other arcs or nodes without creating cycles, or white sources or targets. We denote by
. Maximality means that it is not possible to add other arcs or nodes without creating cycles, or white sources or targets. We denote by 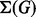 the set of stories of G.
the set of stories of G.
2.2 Enumerating metabolic stories
A first step of our algorithm to enumerate 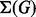 is to apply compression operations on the input graph obtaining a more compact representation, which is equivalent in terms of story sets. The operations are (i) white source and target removal that consists in removing iteratively white nodes that are either sources or targets, as such nodes cannot appear in any story; (ii) self-loop removal that consists in removing all arcs of the form
is to apply compression operations on the input graph obtaining a more compact representation, which is equivalent in terms of story sets. The operations are (i) white source and target removal that consists in removing iteratively white nodes that are either sources or targets, as such nodes cannot appear in any story; (ii) self-loop removal that consists in removing all arcs of the form 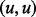 : because stories are acyclic, such arcs do not appear in any story; (iii) forward and backward bottleneck removal, that consists in removing a white node v whose out-degree (respectively, in-degree) is equal to 1, and directly connecting any predecessor (respectively, successor) of v to the unique successor (respectively predecessor) of v (without creating multiarcs). Our preprocessing algorithm consists in applying operations (i), (ii) and (iii) successively until no more white sources and targets, self-loops and bottlenecks are present in the graph. We call the resulting graph a compressed network.
: because stories are acyclic, such arcs do not appear in any story; (iii) forward and backward bottleneck removal, that consists in removing a white node v whose out-degree (respectively, in-degree) is equal to 1, and directly connecting any predecessor (respectively, successor) of v to the unique successor (respectively predecessor) of v (without creating multiarcs). Our preprocessing algorithm consists in applying operations (i), (ii) and (iii) successively until no more white sources and targets, self-loops and bottlenecks are present in the graph. We call the resulting graph a compressed network.
In (Acuna et al., 2012), we proposed a first method to enumerate stories based on a polynomial-time algorithm to compute one story. This is briefly recalled in Supplementary Material S1. More recently we developed a much faster enumerator for stories based on a linear-time enumeration algorithm for non-maximal stories (Borassi et al., 2013) that allows us to explore the whole set of solutions even for genome-scale metabolic networks. This is the enumerator algorithm we use here.
2.3 Scoring function
From a formal point of view, there is no qualitative difference between any two stories. In this sense, whether a given discriminating compound is a source, an intermediate node or a target in a story is indifferent for the enumeration process, as all possible scenarios satisfying the three properties given by the definition, namely, maximality of paths, acyclicity and source/target constraint, have to be computed.
However, in practice, the number of stories can be large and being able to rank them greatly facilitates their analysis. To do this, we propose the following score function:
where the score s(S) of a story S is the sum, for each black transformation 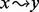 , of the product of the node weights
, of the product of the node weights
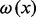 and
and 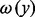 of the nodes x and y, times the path weight
of the nodes x and y, times the path weight
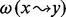 . A black transformation is defined as an arc or a simple white path between two black nodes. A simple white path is a simple path (i.e. containing no cycles) composed of only white nodes between two black ones. The values assigned to the node and path weights will depend on the data available and are thus perfectly suited for the integration of various omics data. For our analysis, we used the topology of the stories and additional data from the metabolomics experiments as described in more detail in the Section 3 (see Table 2).
. A black transformation is defined as an arc or a simple white path between two black nodes. A simple white path is a simple path (i.e. containing no cycles) composed of only white nodes between two black ones. The values assigned to the node and path weights will depend on the data available and are thus perfectly suited for the integration of various omics data. For our analysis, we used the topology of the stories and additional data from the metabolomics experiments as described in more detail in the Section 3 (see Table 2).
Table 2.
Weights for different score functions of a story
| (a) Enzyme activation first |
(b) Concentration Change first |
(c) Enzyme inhibition first |
||||||
|---|---|---|---|---|---|---|---|---|
| Outgoing arcs | Outgoing arcs | Outgoing arcs | ||||||
| Arc | To red | To green | Arc | To red | To green | Arc | To red | To green |
| From red | 0 | 1 | From red | 1 | −1 | From red | 0 | −1 |
| From green | −1 | 0 | From green | −1 | 1 | From green | 1 | 0 |
Note: Table exhibiting the arc weights for interactions between green and red nodes used for computing the score of a story in the context of a metabolomics experiment: (a) weights used to privilege enzyme activation, (b) weights used to privilege concentration change and (c) weights used to privilege enzyme inhibition.
2.4 Yeast metabolic network
For the analysis of the metabolics experiment (Madalinski et al., 2008), we used the metabolic reconstruction of Saccharomyces cerevisiae s288c available in MetExplore (Cottret et al., 2010) (the metabolic model was built based on the YeastCyc database). The procedure followed is briefly described in Supplementary Material S2.
3 RESULTS
3.1 Metabolic stories to analyze metabolomics data
To illustrate how to use our method, we concentrate on the study of the exposition of S.cerevisiae to the toxic cadmium (Cd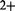 ) reported in Madalinski et al. (2008). A widely studied metabolic pathway in S.cerevisiae is the one responsible for glutathione biosynthesis, as it is related to the detoxification process of the cell when exposed to high concentrations of cadmium (Fauchon et al., 2002; Lafaye et al., 2005; Madalinski et al., 2008). Previous studies demonstrated that the presence of such a metal in the environment has a huge impact in terms of gene expression and metabolism, showing that there is a strong response both at the metabolomic and proteomic levels. Basically, glutathione needs to be produced because it is a thiol metabolite linked to the detoxification of cadmium through a process called chelation (Li et al., 1997). Plants are the natural biotope of S.cerevisiae and it is known that they are able to tolerate cadmium and other metals up to 1% of their dry weight, which is believed to provide defense against herbivores and pathogenic microorganisms (Fauchon et al., 2002). This exposition to cadmium in natural conditions provides a reason for yeast to keep a detoxification pathway. However, the biosynthesis of glutathione requires high quantities of sulfur. To save sulfur, there is a replacement of abundant sulfur-rich proteins related to other metabolic processes by sulfur-depleted isozymes (i.e. other enzymes that have the same function). Such is the case for the enzymes pyruvate decarboxylase (Pdc1p), enolase (Eno2p) and aldehyde dehydrogenase (Ald6p) that are replaced by isozymes containing less sulfur amino acids (i.e. methionine and cysteine) that are mobilized in the glutathione pathway and are less available for protein synthesis (Fauchon et al., 2002). This response affects a large portion of the metabolic network and represents the mechanism used by the cell to survive under this specific stress condition. Sulfur limitation conditions slow down the growth rate but do not induce this same sulfur-sparing response (Fauchon et al., 2002). A schema of the known glutathione biosynthesis metabolic pathway is presented in Figure 1.
) reported in Madalinski et al. (2008). A widely studied metabolic pathway in S.cerevisiae is the one responsible for glutathione biosynthesis, as it is related to the detoxification process of the cell when exposed to high concentrations of cadmium (Fauchon et al., 2002; Lafaye et al., 2005; Madalinski et al., 2008). Previous studies demonstrated that the presence of such a metal in the environment has a huge impact in terms of gene expression and metabolism, showing that there is a strong response both at the metabolomic and proteomic levels. Basically, glutathione needs to be produced because it is a thiol metabolite linked to the detoxification of cadmium through a process called chelation (Li et al., 1997). Plants are the natural biotope of S.cerevisiae and it is known that they are able to tolerate cadmium and other metals up to 1% of their dry weight, which is believed to provide defense against herbivores and pathogenic microorganisms (Fauchon et al., 2002). This exposition to cadmium in natural conditions provides a reason for yeast to keep a detoxification pathway. However, the biosynthesis of glutathione requires high quantities of sulfur. To save sulfur, there is a replacement of abundant sulfur-rich proteins related to other metabolic processes by sulfur-depleted isozymes (i.e. other enzymes that have the same function). Such is the case for the enzymes pyruvate decarboxylase (Pdc1p), enolase (Eno2p) and aldehyde dehydrogenase (Ald6p) that are replaced by isozymes containing less sulfur amino acids (i.e. methionine and cysteine) that are mobilized in the glutathione pathway and are less available for protein synthesis (Fauchon et al., 2002). This response affects a large portion of the metabolic network and represents the mechanism used by the cell to survive under this specific stress condition. Sulfur limitation conditions slow down the growth rate but do not induce this same sulfur-sparing response (Fauchon et al., 2002). A schema of the known glutathione biosynthesis metabolic pathway is presented in Figure 1.
Fig. 1.

Glutathione biosynthetic pathway. Compounds in bold are discriminating in Madalinski et al. (2008) and are involved in the synthesis of glutathione. Source: adapted from Figure 1 in Lafaye et al. (2005)
The metabolic network used for this analysis (see the Section 2 for a description) contains 600 metabolites and 949 arcs. Madalinski et al. (2008) identified a list of 24 metabolites whose concentration significantly changed after cadmium exposure, shown in the table given in Supplementary Material S3.
It is important to notice here that identification of the metabolites that have changed their concentration is based on a minimum of two orthogonal criteria relative to an authentic compound analyzed under identical experimental conditions: retention time and mass spectrum or retention time and 1H nuclear magnetic resonance (NMR) spectra, accurate mass and tandem mass spectra or accurate mass and related isotopic clusters or 1H and/or 13C NMR with 2D NMR spectrum (Sumner et al., 2007). However, many metabolites are not commercially available and many of them may require tedious and expensive chemical synthesis, which often hampers their definitive metabolite identification. Thus, such compounds remain putatively annotated or characterized.
We decided to perform two analyses to explore the effect of cadmium exposure on S.cerevisiae cells. We first enumerated metabolic stories using a set of black nodes restricted to the measured metabolites that are known to participate to the biosynthesis of glutathione. The idea is to check whether our method is able to recover one or more stories that correspond to the known metabolic pathway. In a second step, we enumerated metabolic stories using the entire list of 24 discriminating compounds identified in the metabolomics experiments. In this case, the goal is to analyze both the response of glutathione biosynthesis, but also the potential response of other pathways and the side effects of these responses in the rest of the network.
3.2 First analysis: local response to cadmium exposure, biosynthetic pathway of glutathione
We first consider the aforementioned metabolic pathway directly involved in cadmium detoxification, namely, the glutathione biosynthetic pathway, to enumerate stories and check whether we are able to recover one that fits our current knowledge of the biological process. We thus selected as black nodes for this first analysis only the metabolites that were measured in the experiment (Madalinski et al., 2008) and that are also known to participate in the glutathione biosynthetic pathway (Fauchon et al., 2002). These eight compounds are presented in the table given in Supplementary Material S3 with the third column marked as ‘yes’: glutathione, O-acetylhomoserine, methionine, glutamate, glutamylcysteine, serine, glycine and cystathionine.
3.2.1. Compressed network
A first practical result that follows directly from the properties of our definition of stories is the compressed representation of the subnetwork in which all interactions between the discriminating compounds are captured. The compression is obtained in two steps. In the first step, we extract all biologically relevant routes between the black nodes. In our case, we computed lightest paths between black nodes using the out-degree of a node as its weight, which has been defended as being more biologically sound than a simple shortest-path approach (Blum and Kohlbacher, 2008). The second step is to apply the four compression rules that were previously described briefly (see Section 2) and that are fully detailed in Acuna et al. (2012).
The compressed network obtained for the reduced set of black nodes contains 10 nodes and 25 arcs, i.e. represents >98% of compression in terms of nodes and >97% in terms of arcs with respect to the original input size of the S.cerevisiae metabolic network. The resulting compressed network is shown in Figure 2. This compression ratio is spectacular, as it is now much easier to visually inspect the network in which we can highlight the metabolites of interest. This type of visualization is, therefore, already a result in itself, which can readily be used to start proposing causal explanations for the changes of metabolite concentrations. To facilitate this, we further enrich this representation with the information on the direction of the change of concentration (whether the metabolite concentration increased or decreased) and the intensity of this change. Of the 8 metabolites considered, 3 had a significant increase of their concentration (reduced glutathione, cystathionine and glutamylcysteine), whereas the other 5 had a significant decrease of their concentration (methionine, O-acetylhomoserine, glutamate, serine and glycine). From now on, we will denote the first set as green nodes and the second set as red nodes. The other nodes, whose concentration did not change significantly, will remain identified as white nodes. We notice that this distinction between red and green nodes is only possible for applications where two conditions are compared. This is the case we consider in this article. When more than two conditions are compared, our methodology still applies, keeping the terminology of black and white nodes. We can produce the compressed network and enumerate the stories. The ranking scheme described later would, however, need to be adapted. Finally, during the preprocessing of the network, some paths are compressed into a single arc. To distinguish between reactions linking two compounds and these compressed paths, we used solid lines for the former and dashed lines for the latter. Importantly, the compression of the network is lossless as it is easily reversible, for instance if we need to have access to the full path of white nodes that indirectly link two black nodes. Interestingly, in practice, although most white nodes can be compressed, some remain. Their compression would prevent us from being able to enumerate the full set of stories. These compounds, although not detected as discriminating, seem to also play an important role in the studied process as they are at the crossroads between at least two possible routes between discriminating compounds.
Fig. 2.
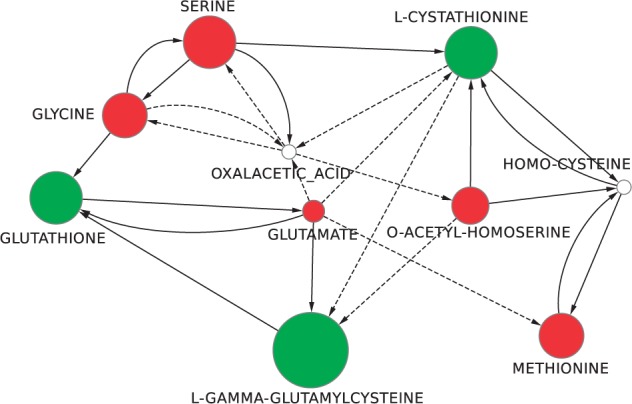
The compressed network computed considering as black nodes the eight compounds of the table in Supplementary Material S3 marked as present in the glutathione biosynthetic pathway. Green nodes are the ones whose concentration significantly increased in the presence of cadmium, whereas the red are the ones whose concentration significantly decreased. The diameter of the nodes is proportional to the concentration change. Solid arcs represent single reactions connecting the two compounds, whereas dashed ones correspond to a chain of at least two reactions
3.2.2. Enumerating and scoring the stories
The compressed network is already a result per se, but its visual inspection remains difficult; the many cycles it contains allow for a reading of the flow of matter in many possible directions, thereby suggesting several possible causal scenarios. Therefore, we go one step further in the analysis and enumerate the metabolic stories. In this analysis, there are a total of 222 stories.
With the aim of classifying the set of computed stories, we have to define how to assign values to the node and arc weights needed by our score function scheme (see Section 2).
There are basically four kinds of interactions that may be observed in a metabolic story (see Table 1). In the following proposal for causal interpretation of each type of arc, we will make the simplifying assumption that each arc is independent from the other ones. In this context, an arc linking a red node to a green node will correspond to the consumption of the red node to the benefit of the green node. If we focus solely on this arc, this can only be explained by an activation of the enzyme catalyzing the reaction linking the two nodes. On the other hand, an arc linking a green node to a red node can be interpreted as the inhibition of the enzyme catalyzing the reaction linking the two nodes. Finally, an arc linking two red nodes can be explained by a domino effect. The simple fact that the substrate concentration decreases causes the product concentration to decrease. This domino effect does not require any enzyme change. It just corresponds to a change in concentration that propagates. The case of green to green arcs can be explained by a similar effect. We additionally need to assume that the enzyme is not present in a limiting amount.
Table 1.
Biological interpretation for arcs in a story
| Arc | To red | To green |
|---|---|---|
| From red | Concentration change | Enzyme activation |
| From green | Enzyme inhibition | Concentration change |
We remind that in this section, our approach is local and focuses on single transformations. We always favor the most parsimonious explanation (the one with fewest enzyme changes), but, in practice, other plausible explanations could be proposed for each arc. Importantly, the notion of enzyme activation or inhibition as used in this article should be understood in a general sense as it captures allosteric regulation of the enzyme or transcriptional regulation of the gene(s) encoding the enzyme. In the application considered here, the time separating the measurements (before and after exposure to cadmium) is large enough to allow to interpret enzyme activations as a change in their concentration through a transcriptional response. Our methodology also applies when the time separating the measurements is shorter. In the following, we propose three ranking schemes for stories. In each of them we favor one type of arc, which means that we look for the stories with a large number of arcs of this type. Even if the individual explanation of each arc is not necessarily correct, the overall optimization of the total number of each arc type makes intuitive sense, and we show that in practice it enables to explore efficiently the space of all stories.
3.2.3 Three scoring schemes
Let us start by defining the arc weights that are restricted to being −1, 0 or +1. The first scoring scheme privileges stories where green nodes are preferentially targets in the story (i.e. are produced) and, on the other hand, red nodes are preferentially sources in the story (i.e. are consumed). Let us call this score function enzyme-activation-first, as it should privilege arcs from red to green nodes and penalize the inverse as shown in Table 2a. Another possibility is to classify first stories in which the concentration change responses are privileged as shown in Table 2b. Let us call this score function concentration-change-first, as it should privilege arcs from red to red nodes or green to green nodes. Finally, we may define a score function in which we privilege arcs going from green nodes to red nodes; in such a case these arcs represent enzyme inhibition, as shown in Table 2c. Let us call this score function enzyme-inhibition-first.
Once an arc weighting scheme has been chosen, we define the node weights. For our experiments, we define the value 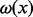 for a given node x as its normalized intensity ratio, which is its intensity ratio divided by the maximum intensity ratio observed in the experiment (if v is a green node) or the minimum intensity ratio observed in the experiment divided by the intensity ratio of the node (if v is a red node). An example is given in the figure in Supplementary Material S4.
for a given node x as its normalized intensity ratio, which is its intensity ratio divided by the maximum intensity ratio observed in the experiment (if v is a green node) or the minimum intensity ratio observed in the experiment divided by the intensity ratio of the node (if v is a red node). An example is given in the figure in Supplementary Material S4.
3.2.4 Application to cadmium stress response in yeast
Using the three presented score functions, we were able to rank the 222 stories previously computed and identify the top scoring stories for each one of the three functions. Figure 3a shows one of the six optimal stories according to the enzyme-activation-first scheme, Figure 3b shows the single optimal story according to the concentration-change-first scheme and Figure 3c shows one of the two optimal stories found according to the enzyme inhibition first scheme. The goal of this first analysis is to try to identify stories that could correspond to the current knowledge on the response of yeast to cadmium exposure, i.e. a story that corresponds to the glutathione biosynthetic pathway previously presented in Figure 1. Among the top scored stories, the one given by the enzyme-activation-first score function (see Figure 3a) agrees well with the current knowledge of yeast response to cadmium. The discussion in Madalinski et al. (2008) presents as a result a flux corresponding to the detoxification of cadmium by glutathione, explaining that the levels of metabolites involved in the glutathione biosynthesis pathway (homocysteine, cystathionine, glutamyl-cysteine and glutathione itself) were increased following cadmium exposure, which is the same flow of matter preserved in the story shown in Figure 3a. The story selected by the concentration-change-first score function, shown in Figure 3b, preferentially preserves arcs between two nodes of a same color. The idea is that an increase (or a decrease) of concentration of a given metabolite could be a side effect of the increase (or decrease) of another one. The goal is to minimize the number of arcs that suggest some enzyme activity change, i.e. arcs that involve red and green nodes. Interestingly, the story that scores best with this ranking scheme does not fit with the current knowledge of the response to cadmium exposure. This means that, in principle, there exists a scenario that uses fewer red to green or green to red arcs than the true response (and therefore fewer enzyme changes), but this scenario is not the one taken in practice. There can be a number of reasons why this optimal scenario is not taken. Although any enzyme can, in principle, be activated or inhibited, in practice, some have more degrees of freedom. In addition, some reactions annotated as reversible in general, happen to have one clearly favored direction in specific conditions. Finally, the story presented in Figure 3c preferentially preserves green to red arcs that could represent an enzyme inhibition. Again, this scenario does not fit with the current knowledge on yeast response to cadmium, which indicates that the response is probably not based mostly on enzyme inhibition.
Fig. 3.
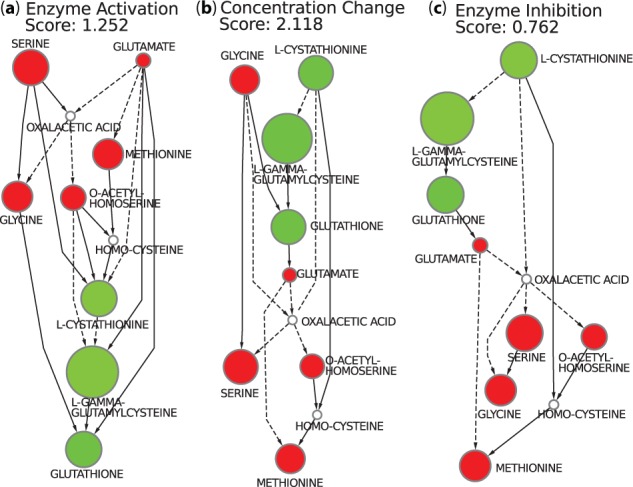
The best stories generated for our analysis taking into account only the metabolites known to be present in the glutathione biosynthesis and whose concentration significantly changed after cadmium exposure. Green nodes are the ones whose concentration significantly increased in the presence of cadmium, whereas the red nodes are the ones whose concentration significantly decreased. The diameter of the nodes is proportional to the concentration change. Solid arcs represent single reactions connecting the two compounds, whereas dashed ones correspond to a chain of at least two reactions
3.2.5 Anthologies
In Figure 3a, a story with score 1.252 for the enzyme-activation-first score function is presented. However, there are other five stories that achieved the same score. These tied optimal stories may be combined into a single graph representation to ease the analysis of their differences as presented in Figure 4. A unique graph representing the union of several different stories is called an anthology. Notice that differently from stories, which are maximal DAGs, an anthology contains at least one cycle. The sources and targets (sinks) of an anthology (if any remains) are, however, black nodes only, as with stories. In this case, the equivalent stories are due to the fact that serine, glycine and oxalacetic acid are all interconnected by reversible paths.
Fig. 4.
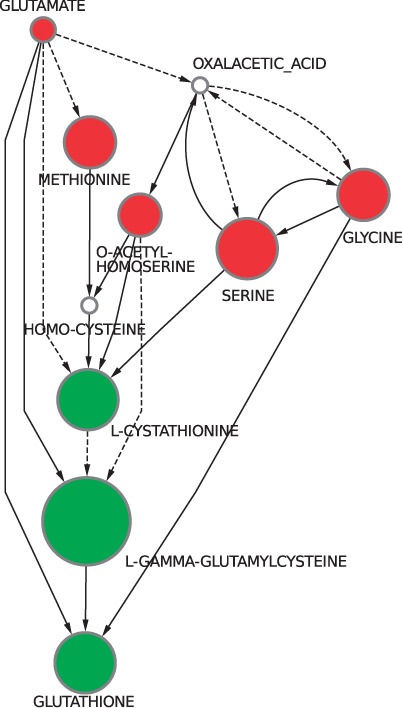
The anthology combining the six maximal stories obtained with the enzyme-activation-first score function. Notice that the anthology preserves the flow of matter observed in the pathway known to be involved in cadmium detoxification by the yeast. Once more, green nodes are the ones whose concentration significantly increased in the presence of cadmium, whereas the red are the ones whose concentration significantly decreased. The diameter of the nodes is proportional to the magnitude of the concentration change as measured by the intensity ratio of the compound. Solid arcs represent single reactions connecting the two compounds, whereas dashed ones correspond to a chain of at least two reactions
3.3 Second analysis: global response to cadmium exposure
For the second analysis, we decided to explore the global response to cadmium exposure and we considered all 24 discriminating compounds. One of them, pyroline-hydroxy-carboxylate, was eliminated when computing the lightest paths between all pairs of black nodes, as it was part of a small disconnected component of the original input graph, most probably due to missing information in the metabolic network reconstruction as the metabolite was present in the metabolome of the strain. The computed compressed network contains 34 nodes and 76 arcs, i.e. a compression of 94% in terms of nodes and 92% in terms of arcs. The resulting compressed network is shown in Figure 5. Again, this compressed network is already a result per se as it enables to visualize jointly all the possible ways of explaining the flow of matter through the network. However, in this case again, and probably even more than before, the readability is complicated, and we, therefore, go one step further and compute all stories.
Fig. 5.
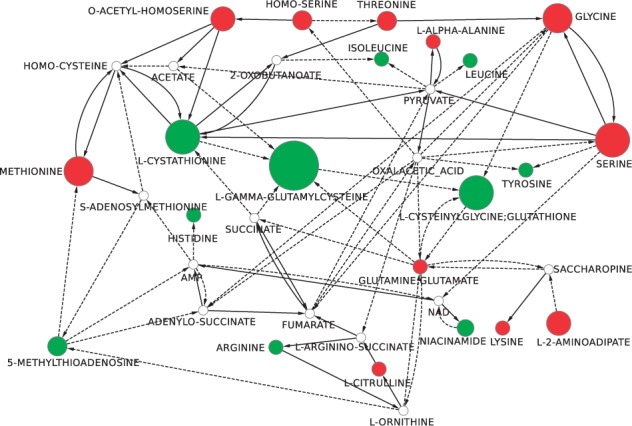
The compressed network computed for the whole list of discriminating compounds of the table in Supplementary Material 3 and the metabolic network of the yeast strain s288c. Green nodes are the ones whose concentration significantly increased in the presence of cadmium, whereas the red are the ones whose concentration significantly decreased. The diameter of the nodes is proportional to the concentration change. Solid arcs represent single reactions connecting the two compounds, whereas dashed ones correspond to a chain of at least two reactions
This time, the number of stories is much larger: there are 3 934 160 in total. In fact, this exact number could only be obtained with the recent improvement we proposed in Borassi et al. (2013). Before that, the computation would not end in reasonable time and we only had an approximate number. In our initial analysis, the score function that selected a story that best fitted the targeted known metabolic pathway of the glutathione biosynthesis was the enzyme-activation-first scoring scheme. For this reason, we used it also to analyze the larger dataset produced in this second analysis, obtaining 20 maximal stories presented as an anthology in Figure 6. Considering all the metabolites that were measured in the experiment as black nodes in our method allows us to have a more global view of the organism’s response to cadmium exposure. This enables to explore whether the other identified paths, apart from the ones involved in the glutathione pathway, are part of this response or simply side effects of the sulfur redirection, as further discussed in the next section.
Fig. 6.
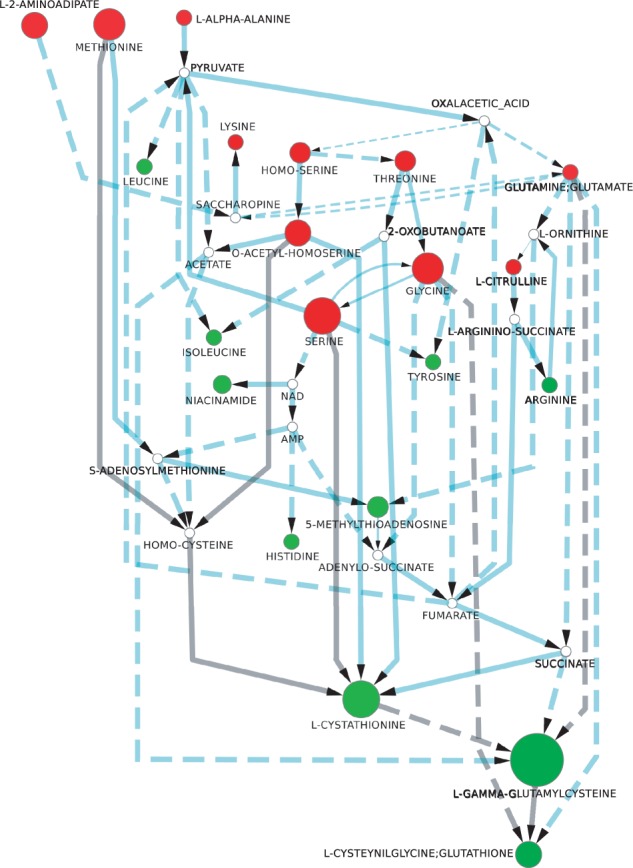
Anthology corresponding to the 20 stories with the maximal score computed for the experiment on yeast s288c exposed to cadmium. Red nodes correspond to metabolites whose concentration decreased and green nodes to those whose concentration increased in the metabolomics experiment. White nodes have their concentration unchanged or it could not be measured. The diameter of the nodes is proportional to the concentration change. Solid arcs represent single reactions connecting the two compounds, whereas dashed ones correspond to a chain of at least two reactions. The arc’s thickness represents the frequency of the arc in the stories making up the anthology, whereas gray arcs correspond to reactions known to be part of the response to cadmium
3.4 Analytic tools
All the compressed networks, stories and anthologies presented in this section were computed using our algorithm called Touché (Borassi et al., 2013). For visualization and analytical purposes, we used Cytoscape (Shannon et al., 2003), which is a software for network visualization, enriched with a plug-in we developed to enable loading, visualizing and inspecting the three aforementioned objects (compressed networks, stories, anthologies) inside Cytoscape. The plug-in applies the given visual properties corresponding to a metabolomics experiment (e.g. colour and diameter of the nodes, the thickness of an arc corresponding to the frequency of the arc in the stories composing the anthology) and allows a zoom-in in the dashed arcs, exhibiting the paths connecting the two nodes. Both Touché and the Cytoscape plug-in are available on demand.
4 DISCUSSION
Focusing specifically on the biological application presented in the previous section, we may see that exploring the topological properties of the stories through the preprocessing of the input network creates a compressed network that captures all the relationships between the discriminating compounds in a much smaller graph than the whole network. This already allows a visual inspection of the observed variations, which is rather difficult, if not impossible, in the entire network. On the other hand, one may easily highlight in a whole metabolic pathway map those metabolites whose concentration were detected as having changed using the YeastCyc database. However, because the pathways are presented as disconnected, it is not possible to follow a path that traverses several pathways (see Figure in Supplementary Material S5). To demonstrate the utility of our approach, we used data from Madalinski et al. (2008) in which the authors monitor changes in metabolite concentration as a response of the yeast S.cerevisiae to cadmium, a toxic chemical. The aim of this study is to analyze the global response of an organism to a stress. Using only the metabolomics experiment data to choose the discriminating compounds and to rank the stories, we are able to obtain stories that correspond well to the current biological understanding of the system under study, as well as to propose new alternatives that could serve as a basis for further experimental validations. Because regulatory information and quantitative information are not needed by the method, this allows it to be used for metabolic network reconstructions even when they are not well refined and where these additional informations may be unavailable or incomplete.
The method herein presented allows visual inspection of a set of discriminating compounds (either local or broader) from metabolomics data in the compressed network, stories and/or anthology with no a priori selected pathways. The metabolic stories may be ranked in different ways depending on which interpretation one wishes to emphasize for the causal link between two affected metabolites: enzyme activation, enzyme inhibition or domino effect on the concentration changes of substrates and products. Equally probable stories under any selected ranking scheme can be further grouped into a single anthology that summarizes in a single subnetwork all equivalently plausible alternative stories.
4.1 First analysis: local response to cadmium exposure
The first analysis performed aimed at locally inspecting the yeast response to cadmium exposure limited to the biosynthetic pathway of glutathione, given in Figure 1. Of the 222 stories found, the ones favoring enzyme activation were clearly closer to our current understanding of this response, where an increased sulfur flux passes through homocysteine, cystathionine, cysteine and glutamyl-cysteine to yield high levels of glutathione (Madalinski et al., 2008). This same flow of matter is captured in the anthology combining the six best stories under this scoring scheme, shown in Figure 4.
Interestingly, we show that there exists one scenario that, in principle, uses fewer enzymes to explain the observed changes in concentration. This is the scenario that favors concentration changes, shown in Figure 5b. However, this scenario does not match the current knowledge of the main pathway of yeast response to cadmium. In fact, it even uses some reactions in the opposite direction. Because these reactions are annotated as reversible, they can be taken in both directions, at least in theory, and this explains that we found these alternative stories. Those are scenarios that are a priori possible. They are not necessarily ‘chosen’ in practice, possibly because the reactions are only reversible under some conditions that are not met in this experiment. Unfortunately, the precise conditions under which a reaction is reversible are in general not well known. The addition of such knowledge would for sure enable to reduce substantially the number of stories we output, as a large part of the combinatorial explosion we observe comes from these ‘cycles’. Conversely, understanding why some possible scenarios are not taken in practice could help to better annotate the reversibility of reactions.
From the list of discriminating compounds identified in Madalinski et al. (2008), the ones that are involved in the glutathione biosynthetic pathway (as shown in bold in Fig. 1) are as follows: O-acetylhomoserine, methionine, serine, cystathionine, glutamate, γ-glutamyl-cysteine, glycine and glutathione. All of them are present in the compressed networks, stories and anthologies herein presented (Figs 2–6). As concerns cysteine and homocysteine, they were either not measured or not discriminating in Madalinski et al. (2008), thus in our analysis they appear as white nodes and may be compressed inside an arc (dashed arcs in Figs 2–6). Cysteine is included in the dashed arc linking cystathionine and L-gamma-glutamyl-cysteine that is its expected place based on the biosynthetic pathway of glutathione. Homocysteine is represented as a white node in all figures. Because it is at a crossroads between three black nodes (cystathione, methionine and O-acetylhomoserine), it could not be compressed.
Interestingly, the compounds involved in the methyl cycle, which is a sulfur salvage pathway (see Fig. 1), were not recovered in the highest score stories found in our first analysis. The reason is that the lightest path found between methionine and cystathione in that analysis passed through the reaction catalyzed by the enzyme homocysteine S-methyltransferase (Mht1), which is assigned as reversible in the YeastCyc database (Caspi et al., 2010) and in our data. This enzyme was described as recycling S-adenosylmethionine (AdoMet) to methionine (Thomas et al., 2000).
4.2 Second analysis: global response to cadmium exposure
This more local view of the behavior of the metabolic network of yeast in this stress condition may be contrasted with the second analysis, where the whole list of discriminating compounds was considered. The anthology combining the 20 best stories under the scoring scheme favoring enzyme activation is presented in Figure 6, where the reactions corresponding to the glutathione biosynthesis are highlighted in gray. This is a strong point of our method, as it allows exploring alternative but close scenarios through the analysis of these (and possibly other) stories altogether, which might provide new insights on the underlying processes that took place under the given conditions.
Among the 35 nodes presented in this anthology, eight have sulfur in their chemical structure: AdoMet, γ-glutamylcysteine, 5-methylthioadenosine (MTA), O-acetyl-L-homoserine, cysteinylglycine, glutathione, cystathionine and L-methionine. Among these sulfured metabolites, the only one that is not involved in the glutathione biosynthesis is MTA, which is instead involved in the MTA cycle, a sulfur salvage pathway (Thomas and Surdin-Kerjan, 1997). This recycles AdoMet to methionine through a chain of reactions, whereas Mht1 (mentioned earlier in the text) can also perform it in one step, which is important for controlling the intracellular ratio between these two metabolites (Thomas et al., 2000) Although there is a redirection of sulfur flux to glutathione biosynthesis after cadmium exposure, the levels of MTA increased as well as those of arginine, which is a precursor for MTA. The metabolites in the methyl cycle are recovered, with the white nodes AdoMet and homocysteine present in the anthology and the metabolite S-adenosyl-homocysteine compressed into the arc between them. We have previously tried to link arginine to sulfur metabolism by emphasizing that it is a precursor of spermidine, a polyamine metabolite that is itself involved in the biosynthesis of MTA, a metabolite associated with the methyl cycle and whose levels are increased after cadmium exposure (Madalinski et al., 2008). However, experimental data lacked to support this assumption. By using the metabolic stories based approach, the increased levels of arginine are linked to decreased concentrations of citrulline, which has not been formally identified in our experimental conditions, and which is itself linked to glutamate. Besides, citrulline was identified as a discriminating compound in Madalinski et al. (2008), but was only indicated as putative, requiring more analysis for final identification. Our results seem to confirm that citrulline was correctly identified. This emphasizes the relevance of using this kind of approach to generate biological hypotheses that have to be further investigated by biologists. Of note, such a link between arginine and sulfur metabolism has been noticed in other organisms (Sekowska et al., 2001) and links between nitric oxide and polyamines have been established with cadmium toxicity in wheat roots (Groppa et al., 2008). Furthermore, this global view of the discriminating compounds links the sulfur metabolism to non-sulfur amino acids and other metabolites through intermediates of the central metabolism. The amino acids that are precursors to the glutathione synthesis have their levels reduced as expected, whereas most of the others increased. This agrees with the fact that global protein synthesis rapidly drops after cadmium exposure (Lafaye et al., 2005), reducing the consumption of amino acids not directly connected to glutathione synthesis.
4.3 Perspectives
We presented a generic method that enables to analyze metabolomics data. This method requires simple input and can be applied to a wide variety of situations. Clearly, the results of the method can be improved with the addition of other types of data. For instance, the use of carbon tracing experiments could help in focusing directly on the stories that are involved in the response to the stress condition, instead of considering the set of all possible stories. Besides, we assumed that the set of discriminating compounds did not need to be questioned. However, these are predicted based on the analysis of peaks in a spectrum. We remark that extracting such information is in itself a bioinformatics challenge. Therefore, a possible extension of the method could be to take into account noisy data, i.e. to deal with a level of confidence for the roles of discriminating compounds and non-discriminating compounds. From the modeling point of view, we enforce that each story corresponds to a flow of matter by the acyclicity constraint. We could relax this constraint by allowing internal cycles, and therefore computing, for each combination of sources and targets, a single story. This will lead to a completely different model and is beyond the scope of this article.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank S. Klamt and J.J. Heijnen for fruitful discussions.
Funding: European Research Council under the European Community’s Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement no. (247073)10; the French project (ANR MIRI BLAN08-1335497); and the ANR funded LabEx ECOFECT. It was partially supported by the Plateforme Bioinformatique de Toulouse, ANR-BBSRC Systryp, the CIRIC-INRIA Chile line Natural Resources, the NWO-CLS MEMESA project and the ‘DISCO’ PRIN National Research Project.
Conflict of Interest: none declared.
REFERENCES
- Acuna V, et al. Telling stories: enumerating maximal directed acyclic graphs with a constrained set of sources and targets. Theor. Comput. Sci. 2012;457:1–9. [Google Scholar]
- Antonov AV, et al. Ticl – a web tool for network-based interpretation of compound lists inferred by high-throughput metabolomics. FEBS J. 2009;276:2084–2094. doi: 10.1111/j.1742-4658.2009.06943.x. [DOI] [PubMed] [Google Scholar]
- Blum T, Kohlbacher O. Using atom mapping rules for an improved detection of relevant routes in weighted metabolic networks. J. Comput. Biol. 2008;15:565–576. doi: 10.1089/cmb.2008.0044. [DOI] [PubMed] [Google Scholar]
- Borassi M, et al. 12th International Symposium, SEA 2013. Rome, Italy: 2013. Telling stories fast: via linear-time delay pitch enumeration. June 5-7, 2013, Proceedings. [Google Scholar]
- Caspi R, et al. The metacyc database of metabolic pathways and enzymes and the biocyc collection of pathway/genome databases. Nucleic Acids Res. 2010;38:D473–D479. doi: 10.1093/nar/gkp875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottret L, et al. Metexplore: a web server to link metabolomic experiments and genome-scale metabolic networks. Nucleic Acids Res. 2010;38:W132–W137. doi: 10.1093/nar/gkq312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croes D, et al. Inferring meaningful pathways in weighted metabolic networks. J. Mol. Biol. 2006;356:222–236. doi: 10.1016/j.jmb.2005.09.079. [DOI] [PubMed] [Google Scholar]
- Dittrich MT, et al. Identifying functional modules in protein-protein interaction networks: an integrated exact approach. Bioinformatics. 2008;24:i223–i231. doi: 10.1093/bioinformatics/btn161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauchon M, et al. Sulfur sparing in the yeast proteome in response to sulfur demand. Mol. Cell. 2002;9:713–723. doi: 10.1016/s1097-2765(02)00500-2. [DOI] [PubMed] [Google Scholar]
- Faust K, et al. Pathway discovery in metabolic networks by subgraph extraction. Bioinformatics. 2010;26:1211–1218, 2010. doi: 10.1093/bioinformatics/btq105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groppa MD, et al. Benavides. Nitric oxide, polyamines and cd-induced phytotoxicity in wheat roots. Phytochemistry. 2008;69:2609–2615. doi: 10.1016/j.phytochem.2008.07.016. [DOI] [PubMed] [Google Scholar]
- Khatri P, et al. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput. Biol. 2012;8:e1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix V, et al. An introduction to metabolic networks and their structural analysis. IEEE/ACM Trans. Comput. Biol. Bioinform. 2008;5:594–617. doi: 10.1109/TCBB.2008.79. [DOI] [PubMed] [Google Scholar]
- Lafaye A, et al. Combined proteome and metabolite-profiling analyses reveal surprising insights into yeast sulfur metabolism. J. Biol. Chem. 2005;280:24723–24730. doi: 10.1074/jbc.M502285200. [DOI] [PubMed] [Google Scholar]
- Leader DP, et al. Barrett. Pathos: a web facility that uses metabolic maps to display experimental changes in metabolites identified by mass spectrometry. Rapid Commun. Mass Spectrom. 2011;25:3422–3426. doi: 10.1002/rcm.5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z-S, et al. A new pathway for vacuolar cadmium sequestration in Saccharomyces cerevisiae: Ycf1-catalyzed transport of glutathionato cadmium. Proc. Natl Acad. Sci. USA. 1997;94:42–47. doi: 10.1073/pnas.94.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madalinski G, et al. Direct introduction of biological samples into a ltq-orbitrap hybrid mass spectrometer as a tool for fast metabolome analysis. Anal. Chem. 2008;80:3291–3303. doi: 10.1021/ac7024915. [DOI] [PubMed] [Google Scholar]
- Schuster S, et al. Detection of elementary flux modes in biochemical networks: a promising tool for pathway analysis and metabolic engineering. Trends Biotechnol. 1999;17:53–60. doi: 10.1016/s0167-7799(98)01290-6. [DOI] [PubMed] [Google Scholar]
- Schuster S, Hilgetag C. On elementary flux modes in biochemical reaction systems at steady state. J. Biol. Syst. 1994;2:165–182. [Google Scholar]
- Schwikowski B, Speckenmeyer E. On enumerating all minimal solutions of feedback problems. Discrete Appl. Math. 2002;117:253–265. [Google Scholar]
- Scott MS, et al. Identifying regulatory subnetworks for a set of genes. Mol. Cell. Proteomics. 2005;4:683–692. doi: 10.1074/mcp.M400110-MCP200. [DOI] [PubMed] [Google Scholar]
- Sekowska A, et al. Extracting biological information from DNA arrays: an unexpected link between arginine and methionine metabolism in bacillus subtilis. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-6-research0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner LW, et al. Proposed minimum reporting standards for chemical analysis. Metabolomics. 2007;3:211–221. doi: 10.1007/s11306-007-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I, Palsson BO. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010;5:93–121. doi: 10.1038/nprot.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, et al. Reverse methionine biosynthesis from s-adenosylmethionine in eukaryotic cells. J. Biol. Chem. 2000;275:40718–40724. doi: 10.1074/jbc.M005967200. [DOI] [PubMed] [Google Scholar]
- Thomas D, Surdin-Kerjan Y. Metabolism of sulfur amino acids in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1997;61:503–532. doi: 10.1128/mmbr.61.4.503-532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helden J, et al. Bioinformatics and Genome Analysis. In: Mewes H-W, Seidel H, Weiss B, editors. Graph-Based Analysis of Metabolic Networks. Vol. 38. Berlin Heidelberg: Springer; 2002. pp. 245–274. [Google Scholar]
- Xia J, et al. Metaboanalyst 2.0-a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012;40:W127–W133. doi: 10.1093/nar/gks374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.