

Abstract
Neutrophils act as the first line of defence in the human immune system by migrating to the site of abnormal events and performing their designated roles. One major signalling pathway that drives neutrophil action in vivo is the p38 mitogen-activated protein kinase (MAPK)-dependent pathway. Herein, a microfluidic platform is employed to explore the mechanistic role of p38 MAPK in neutrophil chemotaxis. Neutrophils, with and without p38 MAPK inhibition, were exposed to pairwise competing gradients of chemotaxis-inducing molecules. Overall, p38 MAPK inhibitor-treated neutrophils were still capable of moving toward a chemoattractant signal; however, the hierarchy of neutrophil response to various chemoattractants changed and more deviation from direct movement toward a chemoattractant signal occurred in p38 MAPK-blocked cells. In a parallel fluorescence imaging study, neutrophil expression of surface receptors (CXCR1, FPR2, BLTR, CD11b and CD66b) changed when comparing untreated and p38 MAPK-blocked cells. All results demonstrate that the p38 MAPK-dependent pathway plays a critical role in neutrophil chemotaxis and this role is, in part, through the regulation of surface receptor expression. These data regarding how receptor expression and chemotaxis are influenced by the p38 MAPK pathways lend insight into neutrophil behaviour in physiological environments and the potential manipulation of p38 MAPK for therapeutic purposes.
Introduction
Neutrophils are in the polymorphonuclear cell family with basophils and eosinophils. They make up roughly 70% of the white blood cell population in humans, and circulate in the blood scanning for signalling cues (foreign invaders, dead/dying host cells, or even small abnormalities on the endothelium). Once neutrophils sense a signal, they migrate to the site of abnormal events by following an increasing concentration of chemical messengers known as chemoattractants. This directed migration of neutrophils, called chemotaxis, is a crucial component in the human immune system, and abnormalities in neutrophil populations or the levels of neutrophil chemoattractants have been measured in several inflammation,1, 2 infection,3 and disease models including cancer4–6 and asthma.7–9 Unfortunately, however, the molecular mechanisms governing neutrophil migration are not well understood and thus detailed characterization of the controlling factors in chemotaxis will enable deeper understanding of neutrophil immune response in both healthy and diseased systems.
With no doubt, neutrophils are surrounded by a complex mixture of signalling molecules during immune response. Upon activation qby surrounding signals, neutrophils coordinate a variety of signalling cascades to interpret the input signals and regulate their chemotaxis toward a particular signal. Stimulation of chemotaxis requires phosphorylation of protein kinase B (PKB), and many previous studies have demonstrated that phosphatidylinositol 3-kinase (PI3K)- and p38 mitogen-activated protein kinase (MAPK)-involved signalling cascades are critical to achieve this phosphorylation.10, 11 p38 MAPK is a protein kinase that governs a wide array of cell functions such as survival, differentiation and proliferation.12–21 Diverse cytokines, including chemoattractants, have been shown to phosphorylate p38 MAPK in neutrophils.14, 16 In general, it is clear that inhibition of p38 MAPK impairs neutrophil chemotaxis, but the mechanism of this impairment has not been clearly established. Literature precedent suggests the potential involvement of p38 MAPK in providing directional guidance to the cells.14–18 For example, Heit et al showed the role of phosphatase and tensin homolog (PTEN) in prioritizing a certain chemical signal16 soon after Shen et al. demonstrated the role of p38 MAPK in the regulation of PTEN.22 It is critical for neutrophils to navigate through complex signals in pursuit of bacteria or to the site of injurious events; thus, deeper understanding of the role played by p38 MAPK in chemotaxis will facilitate both fundamental understanding of chemotaxis and the development of potential therapeutic treatments for the diseases mentioned above.
In this study, the role of p38 MAPK-dependent signalling in neutrophil chemotaxis was investigated in the presence of multiple signals using a microfluidic platform (ESI Fig. S1).23 SB203580 is used in this work as a p38 MAPK inhibitor. SB203580 is a pyridinylimidazole compound that binds selectively to p38 MAPK to inhibit the p38 MAPK signalling cascade.24, 25 Literature precedent has demonstrated that SB203580 is an effective inhibitor for the p38 MAPK pathway-relevant cellular functions by monitoring oxidative burst activity, stress-induced apoptosis, or downstream substrates of p38 MAPK, such as transcription factor 2.26–28 Herein, CXC-motif chemokine 2 and 8 (CXCL2 and CXCL8), leukotriene B4 (LTB4), and a formyl-methionyl-leucyl-phenylalanine (fMLP) are used as neutrophil chemoattractants based on their known roles in neutrophil biology.1, 29–33 In effort to present the neutrophils with a complex environment of these chemoattractants, a microfluidic platform is employed herein to create stable chemoattractant gradients while facilitating single neutrophil trajectory analysis and optical assessment of receptor expression.23, 34, 35 Surface expression of their respective receptors, CXC-motif chemokine receptor 1 (CXCR1), the LTB4-receptor (BLTR), and formyl peptide receptor 2 (FPR2) is fluorescently monitored upon exposure of neutrophils to a chemoattractant gradient. In addition, the surface adhesion molecules CD66b and CD11b are considered based on their purported critical role in neutrophil chemotaxis.36–39 CD66b is a cell surface receptor that regulates neutrophil adhesion to fibronectin in the extracellular matrix or E-selectin on endothelial cells. CD11b is an integrin family receptor that also plays a key role in neutrophil adhesion to endothelial cells by binding to intercellular adhesion molecule-1 (ICAM-1). As such, investigation of these surface markers, in addition to chemotactic behaviour, on p38 MAPK-blocked neutrophils can provide insight into how p38 MAPK regulates those surface markers in neutrophil chemotaxis.
Results and Discussion
The mechanism of neutrophil chemotaxis is generally thought to depend on both PI3K- and p38 MAPK-dependent pathways, whereby exposure of neutrophils to a chemoattractant triggers either or both pathways, localizing relevant intracellular molecules (i.e., phosphatidylinositol phosphates, GTPase protein, and PTEN), polymerizing actin, and directing neutrophils to a designated location.11, 16, 22, 38, 40–44 This study investigates the role of p38 MAPK in chemotaxis and correlates it with relevant surface marker expression while the neutrophils are responding to single or multiple chemoattractant gradients. To note, the error in this manuscript is represented as the standard error of the mean (SEM).
Chemotaxis and p38 MAPK
The p38 MAPK-dependent pathway is known to be essential for neutrophil chemotaxis against fMLP (a bacterially derived chemoattractant); accordingly, neutrophil chemotaxis under a gradient of fMLP concentrations was first investigated by comparing neutrophils with and without p38 MAPK inhibition. The 5.3 μM concentration of SB203580 was chosen based on precedent reports demonstrating the effectiveness of SB203580 at the concentration range of 1 – 10 μM in p38 MAPK inhibition by monitoring secretion of superoxide,21 cytokines such as interleukin 627 or tumor necrosis factor α,26 or downstream proteins of p38 MAPK such as transcription factor 2 or MAPK-activated protein kinase 2.28 To be noted, high concentration SB203580 (above 10 μM) can activate extracellular signal-regulated kinase (ERK), which is also suggested to have a role in neutrophil chemotaxis13, 19, 27, 28, 45–47 but is not a focus of this work. After an hour incubation to facilitate adhesion to the bottom surface of the observation channel, cells were exposed to a gradient of 0 – 10 ng/mL (0 – 22.9 nM) of fMLP (presented versus chemoattractant-free buffer, ESI Fig. S1). As indicators of how active cells were, the motility index (MI, unitless) and average migration rate (pixel/second) were used. The directional migration of neutrophils was assessed with the chemotactic index (CI, unitless), angle distribution, and % population. % population indicates the level of overall deviation from the expected trajectory, and the angle distribution also indicates the level of behaviour deviation, but this parameter is more indicative of the deviation during migration of individual cells. As shown in Figure 1, pre-incubation with SB203580 resulted in no significant difference in MI or CI. On the other hand, the migration rate of neutrophils was hastened slightly in p38 MAPK-blocked neutrophils when compared to untreated neutrophils (0.34 ± 0.013 compared to 0.29 ± 0.012, p = 0.0036). Slightly more p38 MAPK-blocked neutrophils did not set their direction toward fMLP compared to untreated cells (0.2698 ± 0.05088 compared to 0.4198 ± 0.04069, p = 0.0217). The results demonstrate that neutrophils are still capable of recognizing and migrating toward a strong fMLP signal without p38 MAPK activity - during 30 minutes of exposure to the fMLP gradient, a majority population of cells migrated toward the fMLP signal (only 10% of untreated cells and 17% p38 MAPK-blocked cells moved away from the fMLP signal, ESI Fig. S2). In addition, the results indicate that more cells deviate from the dirqection of the gradient when treated with SB203580. Previous studies reported reduced chemotaxis of neutrophils when treated with SB203580,28, 46, 48 and the deviation observed in our results might be the cause of reduced chemotaxis without p38 MAPK activity. The measured migration rate was altered in cells treated with the p38 MAPK inhibitor, which could be explained by the known effect of p38 MAPK in controlling actin polymerization.40, 41 However, despite the single, straightforward chemoattractant signal, the cells did not translate this extra mobility into directed motion.
Fig. 1.
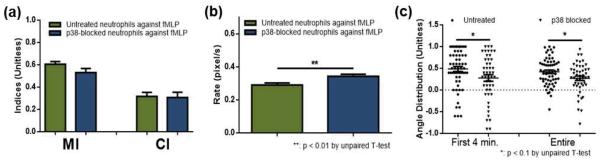
Neutrophil chemotaxis within an fMLP gradient (a) Motility index (MI) and chemotaxis index (CI) of untreated (green) and p38 MAPK-blocked (blue) neutrophils. (b) Average migration rate of untreated (green) and p38 MAPK-blocked (blue) neutrophils. (c) Angle distribution of untreated (circle) and p38 MAPK-blocked (triangle) neutrophils with a value of 1 indicating no deviation from the direction toward fMLP (each point represents angular distribution of a cell and the solid line indicates the average).
As this deviation from the gradient direction seems more significant in the initial 4 minutes (Fig. 1 – c), another hypothesis was made: while pre-expressed p38 MAPK was blocked by inhibitor, the 30 minute period on the device without inhibitor exposure might be long enough for de novo expression of enzymes (p38 MAPK in this case).18, 20 To test such capability of neutrophils, the chemotaxis within the fMLP gradient was compared for neutrophils exposed to inhibitors for enzymes before and throughout assessments. In these experiments, the p38 MAPK inhibitor, SB203580 (5.3 μM) and LY294002, a commercially available inhibitor for PI3K (6.5 μM, Sigma-Aldrich, St. Louis, MO) were used as enzyme inhibitors. 1 – 10 μM LY294002 has shown its effectiveness in PI3K inhibition with no influence on ERK.49–51 In one set of experiments (denoted as “Temporal”), neutrophils were treated with either SB203580 or LY294002, plated onto a device, and then exposed to a 0 – 10 ng/mL fMLP gradient. In the other set of experiments (denoted as “Continuous”), neutrophils were treated with either SB203580 or LY294002, plated onto a device, and then exposed to a 0 – 10 ng/mL fMLP gradient that contained a constant concentration of either 5.3 μM SB203580 or 6.5 μM LY294002. There were no significant differences in the chemotactic behaviour among these two exposure conditions (ESI Fig. S3). This suggests that 30 minutes is not long enough for the cells to newly express sufficient enzyme to influence chemotactic behaviour, and thus, our investigation is not biased by potential resurrection of p38 MAPK upon neutrophil activation.
To further investigate the role of the p38 MAPK-dependent pathway in neutrophil chemotaxis, p38 MAPK-blocked neutrophils were also exposed to competing gradients of fMLP, CXCL8, CXCL2, and LTB4 (denoted as CXCL8-fMLP, CXCL2-fMLP, and LTB4-fMLP throughout this article). Pairwise comparison of neutrophil chemotaxis in these gradients reveals the neutrophil response hierarchy and the potential role of p38 MAPK in providing directional cues to neutrophils. Again, for p38 MAPK inhibition, neutrophils were exposed to 5.3 μM SB203580 before being plated onto the device. To establish a competing gradient of CXCL8 and fMLP, 10 ng/mL CXCL8 was introduced to one inlet while 10 ng/mL fMLP was introduced to the other. (ESI Fig. S1). To be noted, the 0 – 10 ng/mL concentration gradient for each chemoattractant was chosen based on previous study.23 Under this concentration gradient, neutrophils exposed to just CXCL8 yield a similar level of chemotaxis to cells exposed to just fMLP; as such, potential bias caused by different concentration gradients is removed. First, the results demonstrate that the majority of neutrophils are still able to prioritize the fMLP signal under all competing gradients (Fig. 2, ESI Fig. S4 and S5). As shown in Figure 2, the p38 MAPK-blocked cells responding to the CXCL8-fMLP gradient did not result in altered MI and CI when compared to untreated cells. This trend of unaltered MI and CI was observed from all competing gradient conditions (ESI Fig. S4 and S5). As shown in Figure 2, the migration rate of cells was slightly slower in p38 MAPK-blocked neutrophils, compared to untreated cells, under the CXCL8-fMLP gradient (0.2559 ± 0.01417 for p38 MAPK-blocked cells compared to 0.3282 ± 0.01632 for untreated neutrophils, p = 0.0011). Also, p38 MAPK-blocked neutrophils showed more directional deviation in general. Under the CXCL8-fMLP competing gradient, significantly more p38 MAPK-blocked neutrophils hesitated in deciding which signal to follow (0.0002 ± 0.04757 for p38 MAPK-blocked cells compared to 0.2608 ± 0.03826 for untreated cells, p < 0.0001). This trend was consistent among all competing gradient conditions, and was more obvious in the first 4 minutes of each trial than the entire 30 minutes (Fig. 2 – c, ESI Fig. S4 – c and S5 – c). A more important trend is shown in Figure 3. As mentioned, the % population data shows how many cells moved AWAY from the fMLP signal; thus, under competing gradient conditions, this % population data represents how potent the non-fMLP chemoattractant was against fMLP. Untreated neutrophils showed their preference for chemoattractants in the order of CXCL8 > CXCL2 > LTB4 (47.8, 32.4, and 29.7% cells moved away from fMLP) while p38 MAPK-blocked neutrophils had them in the order of CXCL8 > LTB4 > CXCL2 (45.6, 39.5, and 32.6% population of cells moved away from fMLP), indicating a role for p38 MAPK in prioritizing a certain signal.
Fig. 2.
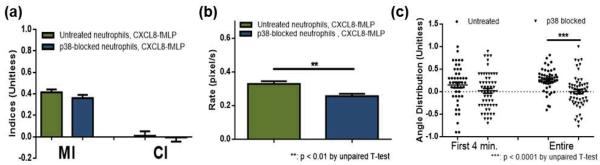
Neutrophil chemotaxis within a CXCL8-fMLP competing gradient (a) Motility index (MI) and chemotaxis index (CI) of untreated (green) and p38 MAPK-blocked (blue) neutrophils. (b) Average migration rate of untreated (green) and p38 MAPK-blocked (blue) neutrophils. (c) Angle distribution of untreated (circle) and p38 MAPK-blocked (triangle) neutrophils with a value of 1 indicating no deviation from the direction of fMLP (each point represents angular distribution of a cell and the solid line indicates the average).
Fig. 3.
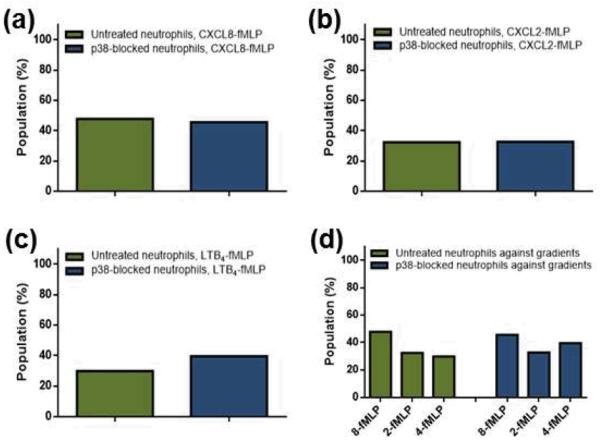
% population of untreated (green) and p38 MAPK-blocked (blue) cells that moved away from the fMLP signal within competing gradients. (a) CXCL8-fMLP gradient. (b) CXCL2-fMLP gradient. (c) LTB4-fMLP gradient. (d) Summary. CXCL8-fMLP, CXCL2-fMLP, LTB4-fMLP gradients are denoted as 8-fMLP, 2-fMLP, and 4-fMLP, respectively.
In our previous study, it was found that fMLP was the most potent chemoattractant among those considered.23 Here, it is clear that, despite the difference in extent, neutrophils without p38 MAPK activity are still capable of prioritizing fMLP over CXCL8, CXCL2, and LTB4. In addition, previous study showed that fMLP was a signalling cue that altered the hierarchical response of neutrophils to CXCL2 and LTB4; CXCL2 was more potent when either of those competes with fMLP while LTB4 was more potent when they compete with each other (where no fMLP is present).23 Such alternating preference from the previous study does not hold in p38-blocked neutrophils; even though they were competing with fMLP, LBT4 was always more potent than CXCL2. The results suggest that the role of p38 MAPK falls in sensing and prioritizing a certain signal rather than the actual action of migration. p38 MAPK-blocked neutrophils under competing gradient conditions had a slower migration rate than untreated cells, likely because neutrophils lost their ability, in part, to interpret surrounding signals.
Adhesion molecules and p38 MAPK
Adhesion of neutrophils to a surface/cell layer is critical in chemotaxis; thus, investigation of CD11b and CD66 on p38 MAPK-blocked neutrophils can provide insight into how p38 MAPK regulates surface marker expression. In this study, surface markers on individual cells were assessed using fluorescently labelled antibodies specific for either CD11b or CD66b, and all reported values in this manuscript are maximum fluorescence intensity from individual cells in arbitrary units (denoted as AUmax). To be noted, throughout this manuscript, control cells indicate cells, whether or not treated with SB203580, exposed to no chemoattractant.
In untreated cells, exposure to the fMLP gradient resulted in down-regulation of CD11b and CD66b expression when compared to control cells (Fig. 4 & Table 2). However, SB203580-treated cells exposed to the fMLP gradient showed unchanged expression of CD11b (p = 0.1804) and up-regulation of CD66b (Fig. 4 & Table 2) when compared to control cells. Similarly, despite the difference in extent, both CD11b and CD66b were down-regulated in untreated cells after exposure to CXCL8 (Fig. 4 & Table 2). In p38 MAPK-blocked cells, on the other hand, exposure to CXCL8 resulted in unchanged CD11b levels (p = 0.1623) and up-regulated CD66b levels (Fig. 4 & Table 2). The same trend was found in the CXCL8-fMLP conditions. In untreated cells, both CD11b and CD66b (Fig. 4 & Table 2) were down-regulated upon exposure to the CXCL8-fMLP gradient. On the other hand, in p38 MAPK-blocked cells, the CXCL8-fMLP gradient resulted in unchanged CD11b levels (p = 0.6673) and up-regulated CD66b levels (Fig. 4 & Table 2). To be noted, treating neutrophils with SB203580 resulted in significant down-regulation of both CD11b and CD66b expression (p < 0.0001 for both CD11b and CD66b). And CD66b expression of p38 MAPK-blocked cells exposed to a chemoattractant gradient is always higher than that of untreated cells (p < 0.0001 for all comparison). These results clearly demonstrate the involvement of p38 MAPK in the regulation of CD11b and CD66b.
Fig. 4.
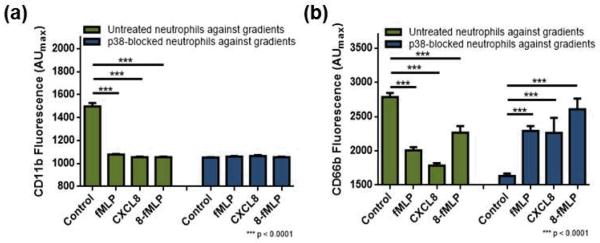
Expression of adhesion molecules after exposure to no gradient, fMLP gradient, CXCL8 gradient, and CXCL8-fMLP competing gradient (denoted as 8-fMLP). (a) CD11b expression in untreated (green) and p38 MAPK-blocked (blue) neutrophils. (b) CD66b expression in untreated (green) and p38 MAPK-blocked (blue) neutrophils.
Table 2.
Fluorescence assessment of surface markers.
| Untreated | p38 MAPK-blocked | |||||||
|---|---|---|---|---|---|---|---|---|
| Control | fMLP | CXCL8 | 8-fMLP | Control | fMLP | CXCL8 | 8-fMLP | |
| CD11b | 1498 ± 28.5 | 1078 ± 5.3 | 1056 ± 4.2 | 1056 ± 4.7 | 1053 ± 3.0 | 1061 ± 5.0 | 1065 ± 8.5 | 1055 ± 3.7 |
| (p < 0.0001) | (p < 0.0001) | (p < 0.0001) | ||||||
| CD66b | 2786 ± 60.3 | 2007 ± 45.1 | 1785 ± 35.0 | 2266 ± 94.3 | 1633 ± 33.9 | 2290 ± 70.6 | 2263 ± 220.1 | 2559 ± 150.8 |
| (p < 0.0001) | (p < 0.0001) | (p < 0.0001) | (p < 0.0001) | (p < 0.0001) | (p < 0.0001) | |||
| FPR2 | 2046 ± 25.7 | 1816 ± 10.1 | 2095 ± 19.3 | 2778 ± 97.1 | 2292 ± 9.4 | 2223 ± 11.9 | 2245 ± 8.8 | 2221 ± 10.3 |
| (p < 0.0001) | (p < 0.0001) | (p < 0.0001) | (p < 0.0001) | (p < 0.0001) | ||||
| BLTR | 1216 ± 6.4 | 1183 ± 7.8 | 1328 ± 29.5 | 1766 ± 100.9 | 1262 ± 10.0 | 1252 ± 24.5 | 1223 ± 9.5 | 1241 ± 10.1 |
| (p < 0.01) | (p < 0.0001) | (p < 0.0001) | (p < 0.01) | |||||
| CXCR1 | 1501 ± 9.1 | 1472 ± 8.6 | 1638 ± 24.5 | 1682 31.2 | 1618 ± 18.3 | 1502 ± 6.6 | 1503 ± 6.6 | 1441 ± 7.8 |
| (p < 0.1) | (p < 0.0001) | (p < 0.0001) | (p < 0.0001) | (p < 0.0001) | (p < 0.0001) | |||
* Maximum fluorescence intensity in arbitrary units (p value from t-test comparing to the control)
Down-regulation of adhesion molecules upon p38 MAPK inhibition may lead to less adhesion of neutrophils, but once they are adhered, they may be better equipped to respond faster. Our observations were indicative of this as more cells washed out of the device at the beginning of experiments in p38 MAPK-blocked cells, but the adherent cells moved faster within a gradient of fMLP. In untreated cells, CD11b levels were highest in control cells and similar between a gradient of fMLP, CXCL8 and a competing gradient of CXCL8-fMLP conditions. Interestingly, on the other hand, CD66b expression levels were in the order of CXCL8-fMLP > fMLP > CXCL8 in untreated cells. This indicates that CXCL8 and fMLP have different impacts on CD66b expression, and that CXCL8 and fMLP have a synergistic impact in regulating CD66b expression. This helps to explain our previous finding that all presenting signals cooperatively contribute to the outcome chemotaxis.23 A similar cooperative effect was found in the p38 MAPK-blocked cells even though the synergistic effect was not as clear as that in untreated cells. The CD66b expression was highest in p38-blocked cells exposed to CXCL8-fMLP. Again, this indicates that CXCL8 and fMLP cooperate to affect neutrophils. The general trend of CD66b expression upon stimulation is down-regulation in untreated cells; however, p38-blocked cells showed significantly up-regulated CD66b expression upon stimulation. Also, upon stimulation, CD11b levels were down-regulated in untreated cells but unchanged in p38 MAPK-blocked cells. This may have allowed the cells to have more time to interpret the surrounding signal, thus, increasing the opportunity for the cells to deviate from its route to a signal, especially when there is more than a single chemoattractant signal present. The migration rate data in which p38 MAPK-blocked cells moved slower than untreated cells under competing gradient conditions support this conclusion. All above demonstrate that p38 MAPK is involved in regulating surface expression of CD11b and CD66b. To consider the effect of spatial variation in chemoattractant concentrations on neutrophil surface receptor expression in competing gradient conditions on neutrophil surface receptor expression, the neutrophils in the main observation channel were analyzed as four separate populations based on their lateral locations. For example, in this investigation, cells in sub-section 2 under a CXCL8-fMLP competing gradient are the cells exposed to medium-high concentration of CXCL8 and medium-low concentration of fMLP. Whether or not neutrophils were treated with SB203580, the results showed variations in surface CD11b and CD66b expression among sub-sections (ESI Fig. S8). The same trend was found from surface FPR2, BLTR, and CXCR1 expressions as well. To be noted, while untreated neutrophils showed variation in surface CD11b expression among sub-sections, p38 MAPK-blocked neutrophils did not show such variations. Only p38 MAPK-blocked neutrophils exposed to a CXCL8-fMLP competing gradient showed variation in CD11b expression among sub-sections. Interestingly, for all surface markers, the trend among sub-sections found to beis statistically different between untreated and p38 MAPK-blocked neutrophils, which is another piece of evidence suggesting p38 MAPK involvement in sensing surrounding chemoattractants.
It has been reported that stimulation of neutrophils induces release of granules filled with adhesion molecules, including CD11b and CD66b;38, 52 however, in our study, untreated neutrophils challenged with chemoattractant gradients showed lower or similar level of those adhesion molecules on their surface. This might be due to 1) CD11b and CD66b being stored in different granules with different threshold for degranulation,17, 36, 38, 52–55 2) our assessments were on neutrophils (both untreated and p38 MAPK-blocked), already adhered to a fibronectin-coated surface,56–58 or 3) our separation of neutrophils from other blood components. In previous studies that have shown up-regulation of these adhesion molecules, stimulation was frequently applied in the presence of other cell type.53–55 It will be of significant interest in future studies to investigate the potential existence of separate signalling pathways for degranulation and distribution of granular contents into the plasma membrane, kinetics of surface expression of those adhesion molecules, or potential communication mechanism of neutrophils with other parties in the immune system.
Surface Receptors and p38 MAPK
Neutrophils, and other cell types as well, interact with signalling molecules through receptors expressed on their plasma membrane, and regulation of surface receptors is another key for cells to manage their functions. Surface expression of receptors FPR2, BLTR, and CXCR1, the specific receptors for the chemoattractants used herein, on p38 MAPK-blocked cells was assessed from individual cells using fluorescently-labelled antibodies and compared to those from untreated cells. Again, control cells indicate cells exposed to no chemoattractant, and all reported values here are maximum fluorescence intensity in arbitrary units (AUmax). While the chemoattractant-bound receptors that have already bound to chemoattractants may not be available for antibody labeling, the control experiments allow the assessment of surface receptors, which are available for further binding, after exposure to chemoattractant gradients. As it will become clear below, our method revealed reveals a clear difference in neutrophils' receptor expression when they are exposed to a competing chemoattractant gradient. In untreated cells, the surface receptors, FPR2, BLTR, and CXCR1, showed decreased expression upon stimulation. Compared to the untreated cells exposed to no chemoattractant signals, cells exposed to the fMLP gradient showed lower expression of all three receptors (Fig. 5 & Table 2). In the case of FPR2 expression, this may not mean actual down-regulation of FPR2 after stimulation as fMLP can bind to this receptor, making the site unavailable for labelling. fMLP exposure of p38 MAPK-blocked cells showed a similar trend in surface receptor expression. FPR2, BLTR, and CXCR1 expressions were generally lower in fMLP-exposed cells than those in control cells (Fig. 5 & Table 2). On the other hand, exposure to the CXCL8 gradient or the CXCL8-fMLP gradient resulted in different trends. First, up-regulation of FPR2, BLTR, and CXCR1 surface expression was observed in untreated cells exposed to CXCL8 (Fig. 5 & Table 2). Exposure of untreated cells to CXCL8-fMLP gradient resulted in, again, up-regulated FPR2, BLTR, and CXCR1 level (Fig. 5 & Table 2) However, exposure of p38 MAPK-blocked cells to the CXCL8 or the CXCL8-fMLP gradient resulted in down-regulation of those surface markers. In p38 MAPK-blocked cells, compared to no chemoattractant condition, surface receptors FPR2, BLTR, and CXCR1 were down-regulated when cells were exposed to the CXCL8 (Fig. 5 & Table 2). Similarly, FPR2, BLTR, and CXCR1 expression of p38 MAPK-blocked cells exposed to CXCL8-fMLP were lower than those in control cells (Fig. 5 & Table 2). These results indicate that p38 MAPK also participates in the regulation of these specific surface receptors upon stimulation.
Fig. 5.
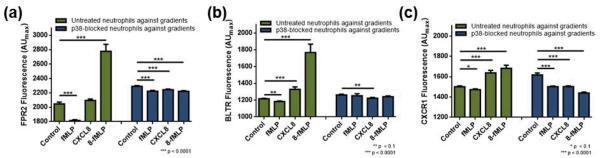
Expression of surface receptors after exposure to no gradient, fMLP gradient, CXCL8 gradient, and a CXCL8-fMLP competing gradient (denoted as 8-fMLP). (a) FPR2 expression in untreated (green) and p38 MAPK-blocked (blue) neutrophils. (b) BLTR expression in untreated (green) and p38 MAPK-blocked (blue) neutrophils. (c) CXCR1 expression in untreated (green) and p38 MAPK-blocked neutrophils.
In untreated cells, exposure to an fMLP gradient resulted in down-regulation of chemoattractant receptors, with the largest effect on FPR2. This is likely due to fMLP binding to the receptor. On the other hand, exposure to CXCL8 or CXCL8-fMLP gradient resulted in up-regulation of FPR2, BLTR and CXCR1 in general. To be noted, BLTR expression in p38-blocked cells did not show as significant a change as that seen in CXCR1 level. The chemotaxis data show that the inhibition of p38 MAPK resulted in stronger favour of neutrophils for LTB4 than CXCL2, and the fluorescence data suggest that this may be in part through diminishing the preference on CXCL2 rather than enhancing the preference on LTB4. Comparison of untreated vs. p38 MAPK-blocked neutrophils responding to fMLP shows higher levels of FPR2, BLTR, and CXCR1 in p38 MAPK-blocked cells (p < 0.01 for FPR2 and BLTR, and p = 0.0185 for CXCR1). On the other hand, p38 MAPK-blocked cells responding to CXCL8 or CXCL8-fMLP resulted in lower levels of FPR2, BLTR, and CXCR1 expression compared to untreated cells (p < 0.01 for all receptors in CXCL8 condition and p < 0.0001 for all receptors in CXCL8-fMLP condition). These indicate that p38 MAPK is involved in the responding mechanism of neutrophils to fMLP and CXCL8 to a different extent. Also, in untreated cells, FPR2, BLTR, and CXCR1 expression was particularly high in the CXCL8-fMLP condition compared to either the CXCL8 or fMLP conditions, indicating a synergistic impact of CXCL8 and fMLP in the regulation of those surface receptors. However, this synergistic impact of CXCL8 and fMLP was unclear in p38 MAPK-blocked cells, indicating potential involvement of p38 MAPK in intracellular signal enhancement. All results demonstrate again that the role of p38 MAPK is more important in sensing/interpreting surrounding signals rather than actual movement in part by regulating surface receptor expression.
Conclusions
This work provides detailed information about the role of the p38 MAPK pathway in regulating neutrophil chemotaxis under the influence of chemoattractants (CXCL8-fMLP, CXCL2-fMLP, and LTB4-fMLP). Using a p38 MAPK inhibitor SB203580, on-chip evaluation of neutrophil chemotaxis revealed neutrophils were still capable of migrating toward fMLP but not as effective as untreated neutrophils. Assessment of surface markers (adhesion molecules CD11b and CD66b and chemoattractant receptors FPR2, BLTR, and CXCR1) suggests the involvement of p38 MAPK in surface expression of those markers, and thus, the critical role of p38 MAPK in interpreting surrounding signals and prioritizing particular signals. All of the results suggest that p38 MAPK is more important in neutrophil chemotaxis signal prioritization than actual movement and it is done in part by regulating surface markers. This research illuminates fundamentals of neutrophil chemotaxis by revealing the important role of p38 MAPK-dependent pathway in neutrophil chemotaxis.
Supplementary Material
Table 1.
Chemotaxis result summary.
| MI | CI | Migration Rate | Angle Distribution | % Population | |
|---|---|---|---|---|---|
| fMLP gradient | = | = | > (p < 0.01) | < (p < 0.1) | > |
| CXCL8-fMLP gradient | = | = | < (p < 0.01) | < (p < 0.0001) | = |
| CXCL2-fMLP gradient | = | = | = | < (p < 0.1) | = |
| LTB4-fMLP gradient | = | = | < (p < 0.1) | = | > |
“<” indicates “lower in p38-blocked cells”, “=” indicates “no significant difference”, and “>” indicates “higher in p38-blocked cells”. * Statistical tests by unpaired T-test, “=” indicates p > 0.1.
Acknowledgement
This work was funded by the National Institutes of Health New Innovator Award (DP2 OD004258-01). Microfluidic devices were fabricated in the Nanofabrication Center (NFC) at the University of Minnesota.
Footnotes
† Electronic Supplementary Information (ESI) available: Experimental procedures, device schematics, and supplemental data. See DOI: 10.1039/b000000x/
Notes and references
- 1.Mathis SP, Jala VR, Lee DM, Haribabu B. J. Immunol. 2010;185:3049–3056. doi: 10.4049/jimmunol.1001031. [DOI] [PubMed] [Google Scholar]
- 2.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CCM, Beck PL, Muruve DA, Kubes P. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 3.Braciale TJ, Sun J, Kim TS. Nat. Rev. Immunol. 2012;12:295–305. doi: 10.1038/nri3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 5.Galdiero MR, Garlanda C, Jaillon S, Marone G, Mantovani A. J. Cell. Physiol. 2013;228:1404–1412. doi: 10.1002/jcp.24260. [DOI] [PubMed] [Google Scholar]
- 6.Houghton AM. Nat. Rev. Cancer. 2013;13:233–245. doi: 10.1038/nrc3477. [DOI] [PubMed] [Google Scholar]
- 7.Vega A, Monteseirin J, Ventura I, Chamorro C, Aroca R, Conde J. Allergy. 2008;63:354–3599. [Google Scholar]
- 8.Monteseirin J. J. Investig. Allergol. Clin. Immunol. 2009;19:340–354. [PubMed] [Google Scholar]
- 9.Neveu WA, Allard JL, Raymond DM, Bourassa LM, Burns SM, Bunn JY, Irvin CG, Kaminsky DA, Rincon M. Respir. Res. 2010;11:28–37. doi: 10.1186/1465-9921-11-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Funamoto S, Meili R, Lee S, Parry L, Firtel RA. Cell. 2002;109:611–623. doi: 10.1016/s0092-8674(02)00755-9. [DOI] [PubMed] [Google Scholar]
- 11.Cuadrado A, Nebreda AR. Biochem. J. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- 12.Ashwell JD. Nat. Rev. Immunol. 2006;6:532–540. doi: 10.1038/nri1865. [DOI] [PubMed] [Google Scholar]
- 13.Liu X, Ma B, Malik AB, Tang H, Yang T, Sun B, Wang G, Minshall RD, Li Y, Zhao Y, Ye RD, Xu J. Nat. Immunol. 2012;13:457–464. doi: 10.1038/ni.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heit B, Liu L, Colarusso P, Puri KD, Kubes P. J. Cell Sci. 2008;121:205–214. doi: 10.1242/jcs.020412. [DOI] [PubMed] [Google Scholar]
- 15.Heit B, Tavener S, Raharjo E, Kubes P. J. Cell Biol. 2002;159:91–102. doi: 10.1083/jcb.200202114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heit B, Robbins SM, Downey CM, Guan Z, Colarusso P, Miller BJ, Jirik FR, Kubes P. Nat. Immunol. 2008;9:743–752. doi: 10.1038/ni.1623. [DOI] [PubMed] [Google Scholar]
- 17.Xu N, Hossain M, Liu L. Mediators of inflammation. 2013;2013:290565–290565. doi: 10.1155/2013/290565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zu Y-L, Qi J, Gilchrist A, Fernandez GA, Vazquez-Abad D, Kreutzer DL, Huang C-K, Sha'afi RI. J. Immunol. 1998;160:1982–1989. [PubMed] [Google Scholar]
- 19.Nick JA, Avdi NJ, Gerwins P, Johnson GL, Worthen GS. J. Immunol. 1996;156:4867–4875. [PubMed] [Google Scholar]
- 20.Cara DC, Kaur J, Forster M, McCafferty D-M, Kubes P. J. Immunol. 2001;167:6552–6558. doi: 10.4049/jimmunol.167.11.6552. [DOI] [PubMed] [Google Scholar]
- 21.Iwata Y, Wada T, Furuichi K, Sakai N, Matsushima K, Yokoyama H, Kobayashi K.-i. J. Amer. Soc. Nephrol. 2003;14:57–67. doi: 10.1097/01.asn.0000037402.83851.5f. [DOI] [PubMed] [Google Scholar]
- 22.Shen YH, Zhang L, Gan Y, Wang X, Wang J, LeMaire SA, Coselli JS, Wang XL. J. Biol. Chem. 2006;281:7727–7736. doi: 10.1074/jbc.M511105200. [DOI] [PubMed] [Google Scholar]
- 23.Kim D, Haynes CL. Anal. Chem. 2012;84:6070–6078. doi: 10.1021/ac3009548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar S, Jiang MS, Adams JL, Lee JC. Biochem. Biophys. Res. Comm. 1999;263:825–831. doi: 10.1006/bbrc.1999.1454. [DOI] [PubMed] [Google Scholar]
- 25.Haddad E, Birrell M, McCluskie K, Ling A, Webber SE, Foster ML, Belvisi MG, B. J. Pharmacol. 2001;132:1715–1724. doi: 10.1038/sj.bjp.0704022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campbell J, Ciesielski CJ, Hunt AE, Horwood NJ, Beech JT, Hayes LA, Denys A, Feldmann M, Brennan FM, Foxwell BMJ. J. Immunol. 2004;173:6928–6937. doi: 10.4049/jimmunol.173.11.6928. [DOI] [PubMed] [Google Scholar]
- 27.van den Blink B, Juffermans NP, ten Hove T, Schultz MJ, van Deventer SJH, van der Poll T, Peppelenbosch MP. J. Immunol. 2001;166:582–587. doi: 10.4049/jimmunol.166.1.582. [DOI] [PubMed] [Google Scholar]
- 28.Coxon P, Rane M, Uriarte S, Powell D, Singh S, Butt W, Chen Q, McLeish K. Cell. Signal. 2003;15:993–1001. doi: 10.1016/s0898-6568(03)00074-3. [DOI] [PubMed] [Google Scholar]
- 29.Matsushima K, Terashima Y, Toda E, Shand F, Ueha S. Inflamm. Regen. 2011;31:11–22. [Google Scholar]
- 30.Reilly IA, Knapp HR, Fitzgerald GA. J. Clin. Pathol. 1988;41:1163–1167. doi: 10.1136/jcp.41.11.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnett ML, Lamb KA, Costello KM, Pike MC. BBA - Mol. Cell Res. 1993;1177:275–282. doi: 10.1016/0167-4889(93)90123-7. [DOI] [PubMed] [Google Scholar]
- 32.Kreisle RA, Parker CW. J. Exp. Medicine. 1983;157:628–632. doi: 10.1084/jem.157.2.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cavicchioni G, Fraulini A, Falzarano S, Spisani S, E. J. Med. Chem. 2009;44:4926–4930. doi: 10.1016/j.ejmech.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 34.Li Jeon N, Baskaran H, Dertinger SKW, Whitesides GM, Van De Water L, Toner M. Nat. Biotech. 2002;20:826–830. doi: 10.1038/nbt712. [DOI] [PubMed] [Google Scholar]
- 35.Lin F, Nguyen CM-C, Wang S-J, Saadi W, Gross SP, Jeon NL. Ann. Biomed. Eng. 2005;33:475–482. doi: 10.1007/s10439-005-2503-6. [DOI] [PubMed] [Google Scholar]
- 36.Anderson SI, Hotchin NA, Nash GB. J. Cell Sci. 2000;113:2737–2745. doi: 10.1242/jcs.113.15.2737. [DOI] [PubMed] [Google Scholar]
- 37.Buyon JP, Philips MR, Merrill JT, Slade SG, Leszczynska-Piziak J, Abramson SB. J. Leukoc. Biol. 1997;61:313–321. doi: 10.1002/jlb.61.3.313. [DOI] [PubMed] [Google Scholar]
- 38.Capodici C, Hanft S, Feoktistov M, Pillinger MH. J. Immunol. 1998;160:1901–1909. [PubMed] [Google Scholar]
- 39.Detmers PA, Zhou DH, Polizzi E, Thieringer R, Hanlon WA, Vaidya S, Bansal V. J. Immunol. 1998;161:1921–1929. [PubMed] [Google Scholar]
- 40.Kutsuna H, Suzuki K, Kamata N, Kato T, Hato F, Mizuno K, Kobayashi H, Ishii M, Kitagawa S. Am. J. Physiol. Cell Physiol. 2004;286:C55–C63. doi: 10.1152/ajpcell.00131.2003. [DOI] [PubMed] [Google Scholar]
- 41.Montecucco F, Bianchi G, Gnerre P, Bertolotto M, Dallegri F, Ottonello L. Ann. Ny. Acad. Sci. 2006;1069:463–471. doi: 10.1196/annals.1351.045. [DOI] [PubMed] [Google Scholar]
- 42.Billadeau DD. Nat. Immunol. 2008;9:716–718. doi: 10.1038/ni0708-716. [DOI] [PubMed] [Google Scholar]
- 43.Sun CX, Downey GP, Zhu F, Koh ALY, Thang H, Glogauer M. Blood. 2004;104:3758–3765. doi: 10.1182/blood-2004-03-0781. [DOI] [PubMed] [Google Scholar]
- 44.Gambardella L, Anderson KE, Jakus Z, Kovacs M, Voigt S, Hawkins PT, Stephens L, Mocsai A, Vermeren S. J. Immunol. 2013;190:381–391. doi: 10.4049/jimmunol.1201330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rafiee P, Heidemann J, Ogawa H, Johnson NA, Fisher PJ, Li MS, Otterson MF, Johnson CP, Pritchard KA, Binion DG. Faseb J. 2004;18:A1014. [Google Scholar]
- 46.Nitto T, Araki Y, Takeda Y, Sendo F. B. J. Pharmacol. 2002;137:353–360. doi: 10.1038/sj.bjp.0704860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee HL, Yi T, Baek K, Kwon A, Hwang HR, Qadir AS, Park HJ, Woo KM, Ryoo HM, Kim GS, Baek JH. J. Cell. Physiol. 2013;228:1076–1086. doi: 10.1002/jcp.24256. [DOI] [PubMed] [Google Scholar]
- 48.Liu XW, Ma B, Malik AB, Tang HY, Yang T, Sun B, Wang G, Minshall RD, Li Y, Zhao Y, Ye RD, Xu JS. Nat. Immunol. 2012;13:457–464. doi: 10.1038/ni.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cowburn AS, Cadwallader KA, Reed BJ, Farahi N, Chilvers ER. Blood. 2002;100:2607–2616. doi: 10.1182/blood-2001-11-0122. [DOI] [PubMed] [Google Scholar]
- 50.Brown WJ, Dewald DB, Emr SD, Plutner H, Balch WE. J. Cell Biol. 1995;130:781–796. doi: 10.1083/jcb.130.4.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fuhler GM, Drayer AL, Vellenga E. Blood. 2003;101:1172. doi: 10.1182/blood.V101.3.1172. [DOI] [PubMed] [Google Scholar]
- 52.Lominadze G, Powell DW, Luerman GC, Link AJ, Ward RA, McLeish KR. Mol. Cell. Proteomics. 2005;4:1503–1521. doi: 10.1074/mcp.M500143-MCP200. [DOI] [PubMed] [Google Scholar]
- 53.Fortunati E, Kazemier KM, Grutters JC, Koenderman L, Van den Bosch VMM. Clin. Exp. Immunol. 2009;155:559–566. doi: 10.1111/j.1365-2249.2008.03791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deree J, Lall R, Melbostad H, Grant M, Hoyt DB, Coimbra R. J. Surg. Res. 2006;133:22–28. doi: 10.1016/j.jss.2006.02.031. [DOI] [PubMed] [Google Scholar]
- 55.Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 56.Ginis I, Tauber AI. Blood. 1990;76:1233–1239. [PubMed] [Google Scholar]
- 57.Zarbock A, Ley K. Microcirculation. 2009;16:31–42. doi: 10.1080/10739680802350104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berton G, Yan SR, Funagalli L, Lowell CA. Int. J. Clin. & Lab. Res. 1996;26:160–177. doi: 10.1007/BF02592978. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.